

Abstract
Hearing loss is the third most prevalent chronic health condition affecting older adults. Age-related hearing loss affects one in three adults over 65 years of age and is caused by both extrinsic and intrinsic factors, including genetics, aging, and exposure to noise and toxins. All cells possess antioxidant defense systems that play an important role in protecting cells against these factors. Reduced nicotinamide adenine dinucleotide phosphate (NADPH) serves as a co-factor for antioxidant enzymes such as glutathione reductase and thioredoxin reductase and is produced by glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, isocitrate dehydrogenase 1 (IDH1) or malic enzyme 1 in the cytosol, while in the mitochondria, NADPH is generated from mitochondrial transhydrogenase, glutamate dehydrogenase, malic enzyme 3 or IDH2. There are three isoforms of IDH: cytosolic IDH1, and mitochondrial IDH2 and IDH3. Of these, IDH2 is thought to be the major supplier of NADPH to the mitochondrial antioxidant defense system. The NADP+/NADPH and NAD+/NADH couples are essential for maintaining a large array of biological processes, including cellular redox state, and energy metabolism, mitochondrial function. A growing body of evidence indicates that mitochondrial dysfunction contributes to age-related structural or functional changes of cochlear sensory hair cells and neurons, leading to hearing impairments. In this review, we describe the current understanding of the roles of NADPH and IDHs in cochlear mitochondrial antioxidant defense and aging.
Keywords: inner ear, cochlea, mitochondrial dysfunction, oxidative stress, isocitrate dehydrogenase, antioxidant defense
1. Introduction
1.1. Mitochondria and ROS
Mitochondrial DNA (mtDNA) mutations have been hypothesized to contribute to aging and age-related disorders because of their critical role in energy metabolism (Balaban et al., 2005; Finkel and Holbrook, 2000; Kujoth et al., 2007; Someya and Prolla, 2010; Wallace, 2005). Most of the chemical energy needed to drive biochemical reactions is produced by the mitochondria. However, it is estimated that during this process ~90% of intracellular unstable reactive oxygen species (ROS) are generated as a by-product (Balaban et al., 2005; Finkel and Holbrook, 2000; Wallace, 2005). Both superoxide and the hydroxyl radical are extremely unstable ROS (Beckman and Ames, 1998; Finkel and Holbrook, 2000). Hydrogen peroxide is freely diffusible and long-lived. Of the sites along the electron transport chain, Complex I (NADH dehydrogenase), Complex II (succinate dehydrogenase), and Complex III (ubiquinone-cytochrome c reductase) are thought to generate superoxide. Under normal metabolic environments, Complex III is thought to be the major site of superoxide production.
1.2. Mitochondrial antioxidant defense system
It is thought that the mitochondrial antioxidant defense systems do not keep pace with the age-related increase in mitochondrial ROS production, and that during aging, the balance between the mitochondrial antioxidant defense and ROS production shifts progressively toward a more pro-oxidant state (Rebrin and Sohal, 2008). To protect the mitochondria from the damaging effects of ROS imbalance, the antioxidant defense systems interact to convert ROS into less toxic forms (Balaban et al., 2005; Finkel and Holbrook, 2000; Halliwell and Gutteridge, 2007; Mari et al., 2009). For example, mitochondrial superoxide dismutase 2 (SOD2) converts superoxide into hydrogen peroxide. The mitochondrial glutathione and thioredoxin antioxidant defense systems then decompose hydrogen peroxide into water (Fig. 1). Within the glutathione antioxidant system, GSR (glutathione reductase) depends on NADPH to reduce oxidized glutathione (GSSG) to reduced glutathione (GSH). In the thioredoxin system, TXNRD2 (thioredoxin reductase 2) depends on NADPH to reduce oxidized thioredoxin 2 (TXN2) to reduced TXN2. In the mitochondria, NADPH is generated from mitochondrial transhydrogenase (NNT), glutamate dehydrogenase (GLUD1), malic enzyme 3 (ME3) or isocitrate dehydrogenase 2 (IDH2) (Dang and Su, 2017; Reitman and Yan. 2010; Plaitakis et al., 2017; Arkblad et al., 2002; Francisco et al., 2022) (Fig. 2). Thus, NADPH plays an important role in both glutathione and thioredoxin antioxidant defense systems, and the NADP+/NADPH couple is essential for maintaining a large array of biological processes, including cellular redox state, and energy metabolism, mitochondrial function (Xiao et al., 2017). In this review, we describe the current understanding of the roles of NADPH and IDH in cochlear mitochondrial antioxidant defense and aging in laboratory animals and humans.
Fig 1.
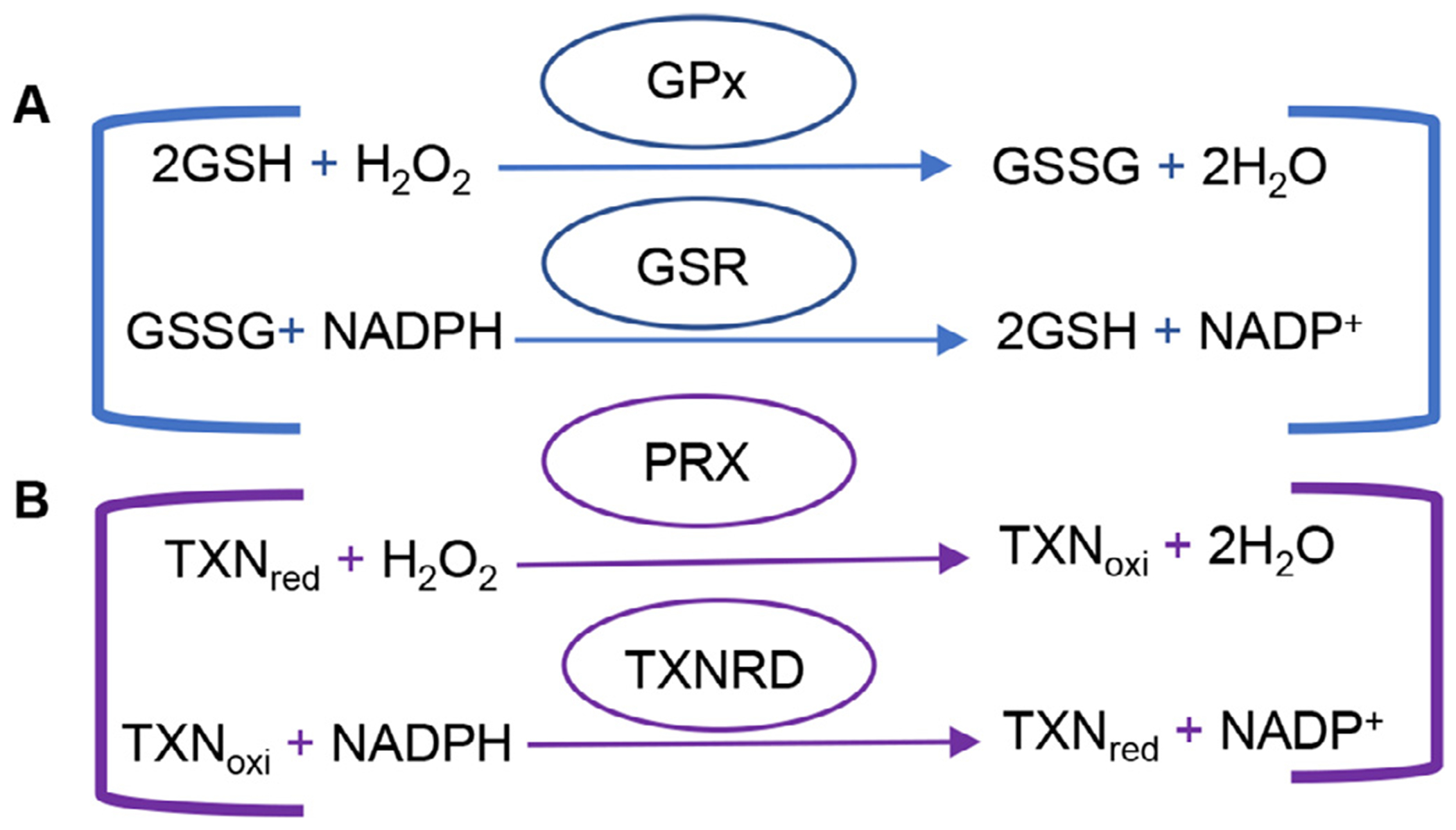
Glutathione and thioredoxin antioxidant defense systems. (A) Reduced glutathione functions in quenching radicals, maintaining thiol groups, serving as a co-enzyme for glutathione peroxidase and maintaining cellular redox homeostasis. GPx reduces H2O2 to H2O while oxidizing GSH to GSSG. GSR then uses NADPH as a co-factor to form -SH groups in proteins that interact with the reduction of GSSG to GSH. (B) Thioredoxin reduces H2O2 and protein disulfide, in protein repair and DNA synthesis, regulating transcription factors and apoptosis, and in immunomodulation. PRX reduces H2O2 to H2O while oxidizing reduced thiore-doxin. NADPH-dependent TXNRD then catalyzes the reduction of oxidized thioredoxin. GPx=glutathione peroxidase, GSH = reduced glutathione, GSSG = oxidized glutathione, GSR = glutathione reductase, NADP = reduced nicotinamide adenine dinucleotide phosphate, PRX = peroxiredoxin, TXNRD = thioredoxin reductase
Fig 2.
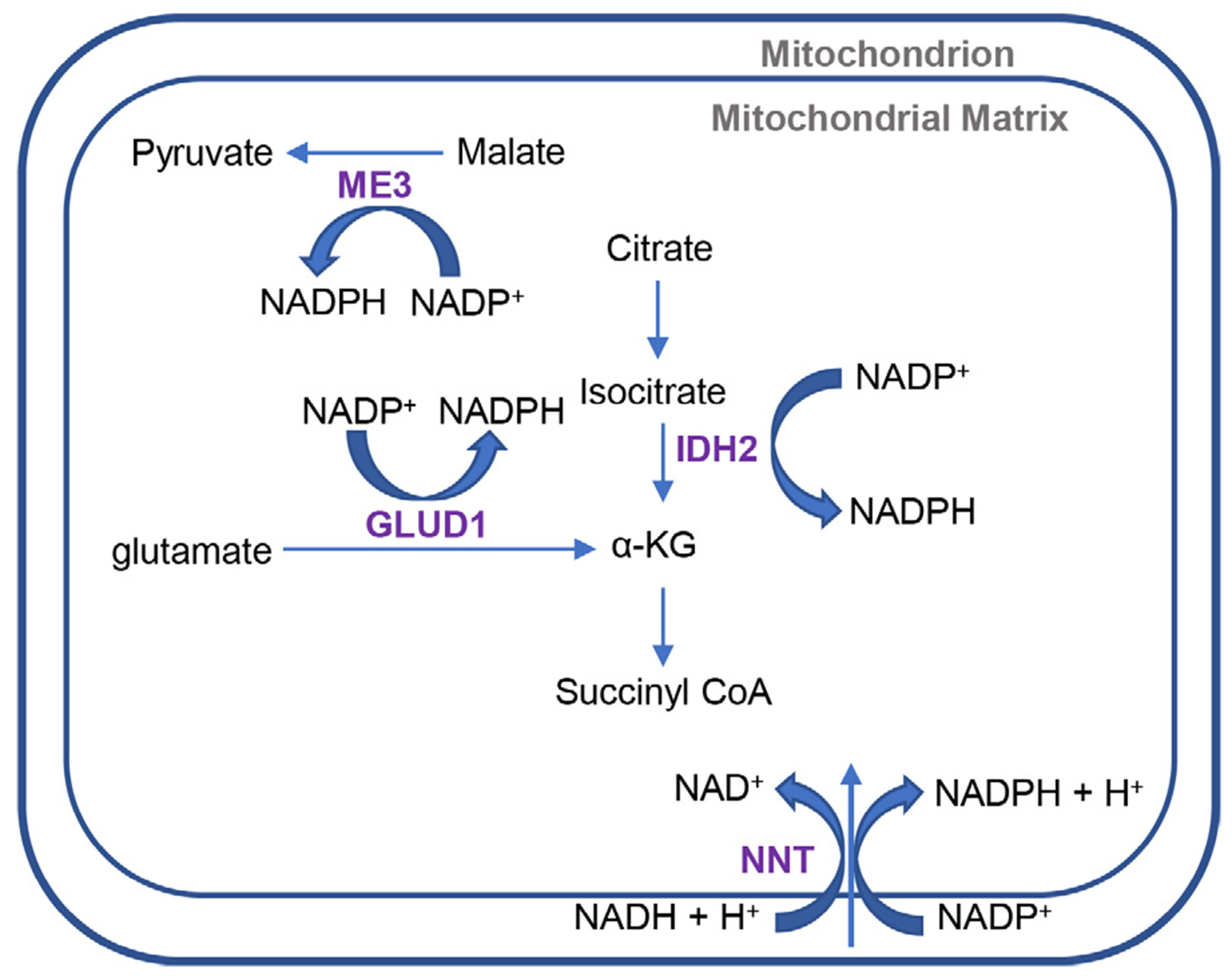
Mitochondrial production of NADPH. IDH2, ME3, NNT, and GLUD1 are all mitochondrial enzymes that produce NADPH. GLUD1 catalyzes the oxidative deamination of glutamate to alpha-ketoglutarate and ammonia and convert NADP+ to NADPH. NNT is an inner mitochondrial membrane protein that functions as a proton pump and couples hydride transfer between NADH and NADP+ to proton translocation across the inner mitochondrial membrane. ME3 is localized to the mitochondria and catalyzes the decarboxylation of malate to pyruvate and convert NADP+ to NADPH. IDH2 catalyzes the decarboxylation of isocitrate to α-KG and converts NADP+ to NADPH. IDH2 = isocitrate dehydrogenase 2, ME3 = malic enzyme 3, NNT = nicotinamide nucleotide transhydrogenase, GLUD1 = glutamate dehydrogenase, α-KG = α-ketoglutarate, NADP+ = oxidized nicotinamide adenine dinucleotide phosphate, NADPH = reduced nicotinamide adenine dinucleotide phosphate
2. Roles of NADPH and IDH in Antioxidant Defense
2.1. NADPH and antioxidant defense
NADPH is the phosphorylated form of NADH and is formed through de novo synthesis from NAD+ by NAD+ kinases or from NADP+ by NADPH producing enzymes (Pollack et al., 2007; Ying, 2008; Xiao et al., 2017). NADPH provides reductive equivalents for a variety of cellular reactions including the biosynthesis of fatty acids, cholesterol, neurotransmitters and nucleotides, and in the cytochrome P450 system that functions in the metabolism of toxic compounds (Berg et al., 2002). NADPH also plays critical roles for both the cytosolic and mitochondrial glutathione and thioredoxin antioxidant defense systems (Fig. 1) (Ying, 2008). Under normal physiological conditions, the NADP pool largely remains in its reduced state in order to be readily available for its use by antioxidant defense systems (Pollack et al., 2007). In the cytosol, NADPH is produced by isocitrate dehydrogenase 1 (IDH1), malic enzyme 1 (ME1), glucose-6-phosphate dehydrogenase (G6PD), and 6-phosphogluconate dehydrogenase (6PGD). In the mitochondria, NADPH is produced by IDH2, ME3, GLUD1 and NNT (Fig. 2).
2.2. IDH and mitochondrial antioxidant defense
In the cytosol, ME1 catalyzes the reversible oxidative decarboxylation of malate to pyruvate and NADP+ to NADPH for fatty acid biosynthesis (Ying, 2008). G6PD is the first and rate limiting enzyme of the pentose phosphate pathway and catalyzes the conversion of glucose-6-phosphate to 6-phosphogluconate and NADP+ to NADPH. G6PD is thought to be the major supplier of NADPH for the cytosolic antioxidant defense system and is regulated by the cytosolic NADPH/NADP+ ratio in response to the production of ROS (Nkhoma et al., 2009). 6PGD catalyzes the conversion of 6-phosphogluconate to ribulose 5-phosphate and NADP+ to NADPH in the pentose phosphate pathway.
In the mitochondria, ME3 catalyzes the decarboxylation of malate to pyruvate and converts NADP+ to NADPH. GLUD1 catalyzes the reversible metabolism of glutamate to α-KG and ammonia and NADP+ to NADPH (Plaitakis et al., 2017). Mitochondrial NNT is an inner mitochondrial membrane (IMM) protein that functions as a proton pump and catalyzes the hydride transfer between NADH and NADP+, using the energy from the proton translocation to produce NADPH (Arkblad et al., 2002; Francisco et al., 2022). The C57BL/6J mouse strain has a null mutation of the Nnt gene that influences diet-induced obesity (Nicholson et al., 2010;), but his mutation is not present in the C57BL/6NJ substrain (Kane et al., 2017). Johnson and co-workers have shown previously that C57BL/6 and C57BL/6NJ mice have identical hearing loss onset times and rates of progression, indicating that Nnt deficiency does not accelerates age-related hearing loss (Kane et al., 2017). Cytosolic IDH1 and mitochondrial IDH2 and IDH3 are the three isoforms of isocitrate dehydrogenase (IDH). Although all three isoenzymes catalyze the conversation of isocitrate to α-ketoglutarate (α-KG), IDH1 and IDH2 convert NADP+ to NADPH while IDH3 converts NAD+ to NADH (Fig. 3) (Dang and Su, 2017; Reitman and Yan. 2010). IDH1 is involved in lipid metabolism, glucose sensing, and cytosolic antioxidant defense (Reitman and Yan 2010).
Fig 3.

IDH isoforms. The isocitrate dehydrogenases (IDH1, IDH2 and IDH3) catalyze the oxidative decarboxylation of isocitrate to α-KG. IDH3 is localized to the mitochondria and serves as an intermediate within the Tricarboxylic Acid (TCA) cycle. IDH3 converts NAD+ to NADH, while IDH2 convert NADP+ to NADPH in the mitochondrion. IDH1 is localized to the cytosol convert NADP+ to NADPH. IDH1 = cytosolic isocitrate dehydrogenase 1, IDH2 = mitochondrial isocitrate dehydrogenase 2, IDH3 = mitochondrial isocitrate dehydrogenase 3, NAD+ = oxidized nicotinamide adenine dinucleotide, NADH = reduced nicotinamide adenine dinucleotide, NADP+ = oxidized nicotinamide adenine dinucleotide phosphate, NADPH = reduced nicotinamide adenine dinucleotide phosphate
Of these mitochondrial NADPH-producing enzymes, IDH2 is thought to be a major source of NADPH for mitochondrial GSR and TXNRD2 (Dang and Su, 2017; Reitman and Yan. 2010; Jo et al., 2001). In agreement with this view, overexpression of Idh2 increases resistance to oxidative stress (Jo et al., 2001) in mouse NIH3T3 fibroblast cells. In rat hearts, Idh2 deficiency results in early onset of cardiac hypertrophy and increased levels of oxidative stress (Benderdour et al., 2004). In human IMR-90 fibroblasts, IDH2 knockdown increases levels of ROS, decreases NADPH/total NADP ratio, and GSH levels in the mitochondria (Kil et al., 2006), while overexpression of Idh2 increases resistance to oxidative stress due to increased NADPH levels in human HEK293 kidney cells (Someya et al., 2010). Collectively, these reports show a crucial role of IDH2 as the major source of NADPH for the mitochondrial antioxidant system in both rodents and humans.
3. Mitochondrial dysfunction and hearing loss
3.1. Mitochondrial DNA mutations and hearing loss
A large number of genetic syndromes associated with hearing loss are due to defects in mitochondria (Chinnery et al., 2000; Fischel-Ghodsian, 2003; Kokotas et al., 2007; Someya and Prolla, 2010), suggesting that cochlear cells, including hair cells, spiral ganglion neurons, and stria vascularis cells, are exquisitely sensitive to energy metabolism disturbances. In support of this idea, increases of deletions, point mutations, or both in mtDNA were observed in human archival temporal bone samples from patients with age-related hearing loss (ARHL) (Bai et al., 1997). The 4977-bp mtDNA deletion was more frequently found in the archival temporal bones from patients with ARHL compared to those with normal hearing. Moreover, in archival temporal bones from patients with ARHL, specific point mutations in the mitochondrial COX2 (mitochondrially encoded cytochrome c oxidase II) gene were more frequently found compared to those with normal hearing (Fischel-Ghodsian et al., 1997). Additionally, our group (Kim, M.J. et al., 2019) has shown that mtDNA deletions accumulate with age in the inner ears of CBA/CaJ mice, a well-established model of ARHL.
The most common genetic defect observed in individuals with mitochondrial diseases are point mutations or deletions (Krishnan, 2008) and over 100 different mtDNA deletions have been identified in individuals with mitochondrial diseases (MITOMAP, 2019). Previous literature has documented that a common symptom in individuals with inherited mtDNA mutations is hearing loss (Chinnery et al., 2000; Fischel-Ghodsian, 2003; Kokotas et al., 2007; Xing et al., 2007): Kearns-Sayre syndrome (KSS), a sporadic mitochondrial disorder caused by a 4977-bp deletion, includes progressive sensorineural hearing loss (Chinnery et al., 2000; Fischel-Ghodsian, 2003; Kokotas et al., 2007; Taylor and Turnbull, 2005). Additionally, numerous studies report mtDNA deletions in aged postmitotic tissues such as brain and in neurodegenerative diseases, including Parkinson’s disease (Bender et al., 2006; Chen et al., 2011; Copeland and Longley, 2014; Kauppila et al., 2017; Kraytsberg et al., 2006; Krishnan et al., 2008; Schon et al., 2012; Schon and Przedborski, 2011; Taylor and Turnbull, 2005). Individuals with KSS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), and myoclonic epilepsy with ragged red fibers (MERRF) may have hearing loss as a clinical feature of the genetic disorder (Chinnery, 2000). Neurogenic weakness, ataxia, and retinitis pigmentosa (NARP), diabetes insipidus, diabetes mellitus, optic atrophy, Wolfram syndrome, and Pearson syndrome are all caused by a large spectrum of mtDNA mutations and hearing loss is not always but is frequently a symptom in individuals with these disorders (Kokotas et al., 2007).
The only active DNA polymerase in the mitochondria is mitochondrial DNA polymerase gamma (POLG). Previous reports demonstrated that in mice carrying a mutator allele of mitochondrial Polg, accumulation of mtDNA mutations led to reduced lifespan, osteoporosis, and early onset of hearing loss (Kujoth et al., 2007; Someya et al., 2008; Someya and Prolla, 2010), providing direct evidence for mtDNA mutations in the development of hearing loss. Compared to age-matched control mice, young mitochondrial mutator mice exhibited a 500-fold increase in mtDNA point mutations in brain and heart tissues (Vermulst et al., 2008) and a 238-fold increase in mtDNA point mutation frequencies in the inner ears (Kim, M.J. et al., 2019). Additionally, middle-aged mtDNA mutator mice displayed significant cochlear spiral ganglion neuron loss and early-onset AHL (Kim, M.J. et al., 2019; Kujoth et al., 2005; Niu et al., 2007; Someya et al., 2008). In humans, several mutations in POLG have been identified as a cause of Alper’s syndrome and deafness (Mancuso et al., 2004; Kujoth et al., 2007; Someya and Prolla, 2010). Together, these studies strongly suggest that mtDNA mutations and subsequent mitochondrial dysfunction contribute to the development of hearing loss.
3.2. Mitochondrial antioxidant enzymes and hearing loss
Previous reports suggest that mitochondrial antioxidants play an important role in protection of hearing. Catalase (CAT) is one of the most energy-efficient antioxidant enzymes found in cells (Halliwell and Gutteridge, 2007). It converts H2O2 into water and oxygen: each catalase molecule can decompose millions of H2O2 molecules every second, indicating the importance of this antioxidant enzyme in protecting the cells against ROS. Catalase is primarily localized in the peroxisomes although catalase expression is also observed in the cytosol and mitochondria. Schriner and co-workers (Schriner et al., 2005) have shown that mice overexpressing human mitochondrial catalase (MCAT) had extended median and maximum lifespans, while median lifespan was slightly extended in mice overexpressing peroxisome catalase (PCAT). Overexpression of catalase in nuclei did not show a significant extension of median nor maximum life span. Importantly, in centenarians, CAT activity was found to be significantly higher in red blood cells (Klapcinska et al., 2000). Our group (Someya et al., 2009) has shown previously that young MCAT mice display normal hearing compared to age-matched WT mice; however, middle-aged MCAT mice display significantly lower ABR thresholds at low, middle and high frequencies than those of middle-aged WT mice. Furthermore, middle-age MCAT mice exhibited reduced oxidative DNA damage and loss of spiral ganglion neurons and hair cells in the cochlea. In agreement with these results, middle-age C57BL/6 mice displayed increased oxidative nuclear DNA damage (Someya et al., 2009), while middle-age CBA/J mice displayed increased oxidative protein damage in the cochlea (Jiang et al., 2007). Collectively, these studies suggest that enhancing CAT activity in cochlear mitochondria may protect cochlear hair cells and slow the development of ARHL.
Coenzyme Q10 is an essential component of the mitochondrial electron transport chain and acts as a mitochondrial antioxidant (Sohal and Forster, 2007). Guastini and colleagues (Guastini et al., 2011) have shown that patients with ARHL had significantly improved pure tone audiometric thresholds at 1000, 2000, 4000, and 8000 Hz when treated with coenzyme Q10 . In mice, supplementation with coenzyme Q10 delayed the onset of age-related hearing loss at high frequencies in mice (Someya et al., 2009).
Glutathione peroxidase 1 (GPX1) plays an important role in mitochondrial antioxidant defense by decomposing H2O2 into water (Halliwell and Gutteridge, 2007; Mari et al., 2009). Ohlemiller and colleagues found that Gpx1−/− mice exhibited significantly greater ABR threshold shift compared to WT mice after noise exposure (Ohlemiller et al., 2000). Nose-exposed Gpx1−/− mice also had greater loss of sensory hair cells compared to WT mice. In agreement with these results, C57BL/6 mice exhibited elevated cochlear ROS levels following acute noise exposure (Ohlemiller et al., 1999), while increased hair cell loss was found in the cochlea of mice lacking Sod1 when compared to WT mice (McFadden et al., 1999). Therefore, a decline in GPX1 activity in the mitochondria may lead to increased levels of ROS, which in turn result in mitochondrial dysfunction, leading to cochlear cell loss and associated hearing loss.
GSH acts as the major small molecule antioxidant in cells and the glutathione antioxidant system is one of the major antioxidant defense systems in the mitochondria (Anderson, 1998; Halliwell and Gutteridge, 2007; Mari et al., 2009). Alpha-lipoic acid and N-acetylcysteine are thiol compounds that have been shown to reduce mitochondrial ROS production (Banaclocha, 2001; Hart, et al., 2004; Palaniappan and Dai, 2007). In rats, supplementation with alpha-lipoic acid slowed the development of ARHL (Seidman et al., 2000). Guinea pigs treated with GSH displayed increased hair cell survival and decreased noise-induced temporary threshold shifts (Ohinata et al., 2000), while treatment with N-acetyl cysteine reduced loss of cochlear outer hair cells and reduced levels of noise-induced hearing loss (NIHL) (Wu et al., 2010). In mice, supplementation with alpha-lipoic acid or N-acetylcysteine delayed the onset of ARHL at the high frequency (Someya et al., 2009; Ahn et al., 2008). In humans, treatment with N-acetyl cysteine significantly reduced noise-induced temporary threshold shift in male workers with glutathione transferase mu1 (GSTM1) and glutathione transferase theta 1 (GSST1) polymorphisms (Lin et al., 2010). Taken together, these reports support the idea that mitochondrial antioxidants may protect cochlear cells and slow the progression of ARHL.
4. Mitochondrial IDH and hearing loss
A growing body of evidence suggests that mitochondrial IDH2 plays an important role in protection against oxidative stress as well as hearing loss. A previous study has shown that homozygous mutations in Idh2 resulted in a 41% increase in heart size, extensive damage to the heart, and cardiac dysfunction (Ku et al., 2015). This was associated with a 55% decrease in ATP production and mitochondrial dysfunction in the heart. In young mouse kidney, loss of Idh2 resulted in decreased NADPH levels, increased oxidative damage markers, and greater kidney damage after ischemia-reperfusion compared to wild-type mice (Han et al., 2017). Calorie restriction (CR) is known to extend lifespan and delays a variety of age-related diseases in multiple species (Weindruch and Walford, 1988; Sohal and Weindruch, 1996; Someya et al., 2007; Colman et al., 2009). In mice, CR increased mitochondrial NADPH levels and IDH2 activities in the brain, liver, and inner ear tissues (Someya et al., 2010). In contrast, Idh2 KO mice on a high fat diet exhibited increased weight gain and decreased mitochondrial function and increased accumulation of ROS in brown adipose tissue (Lee et al., 2020). A recent study (Kim, Y.R. et al., 2019) found profound hearing loss in 10-month-old C57BL/6N; Idh2−/− mice with hair cell and spiral ganglion neuron degeneration. The authors also demonstrated that supplementation of MitoQ an analog of ubiquinone, protected cochlear explants from H2O2. In rats, brain tissue homogenates 120 hours after severe traumatic brain injury had significantly decreased gene expressions of the tricarboxylic acid cycle genes, including Idh2, Idh3, Odh (α-ketoglutarate dehydrogenase), Scs (succinyl-CoA synthetase), Sdh (succinate dehydrogenase), Me2 and Me3 (Lazzarino et al, 2019). Our group has recently shown that loss of Idh2 resulted in decreased NADPH redox state and mitochondrial TXNRD2 activity in inner ears of young Idh2 KO mice (White et al., 2018). Loss of Idh2 also increased oxidative DNA damage, apoptotic cell death, and profound loss of spiral ganglion neurons and hair cells in the cochlea, and accelerated age-related hearing loss in mice on the CBA/CaJ background. Furthermore, in HEI-OC1 mouse auditory cells, knockdown of Idh2, resulted in a decline in cell viability and mitochondria oxygen consumption. Collectively, these results along with the previous reports suggest the importance of IDH2 function in the supply of NADPH for the mitochondrial antioxidant defense system and a crucial role of IHD2 in the protection of cochlear hair cells and neurons under normal physiological conditions as well as during aging.
5. Conclusion
The National Institute on Aging and Deafness and Other Communication Disorders (NIDCD) estimates that 50% of Americans over 75 years of age and 33% of Americans between the ages of 65 and 74 have hearing loss (Oyler, 2012). The WHO estimates that by 2050 more than 700 million persons will have some form of disabling hearing loss (WHO, 2021). It is estimated that if left unaddressed, the impact of hearing loss will cost $980 billion. Because it is projected that nearly 72.1 million people will be over 65 years of age by 2030, the number of older adults with ARHL is expected to rise dramatically and hence, AHL is expected to become a major health care problem. Currently, cochlear implants and hearing aids are the only treatments for ARHL.
ARHL is defined as the loss of hearing that gradually occur in the elderly (Gates and Mills, 2005) that is primarily caused by aging and or the accumulation of extrinsic and intrinsic damage (Liu and Yan, 2007). It is characterized by impaired temporal resolution, central auditory processing deficits, and difficulty understanding speech in noise (Yamasoba et al., 2013). ARHL is typically associated with the progressive loss of sensory hair cells, synaptic loss, spiral ganglion neurons, and or cells of the stria vascularis (Yamasoba et al., 2007). However, the precise molecular mechanisms underlying ARHL still remain unclear since ARHL is thought to be a multifactorial condition resulting from the interaction of numerous causes including aging, exposure to noise and ototoxic chemicals, genetics, epigenetic variables, comorbidities, and lifestyle (Yamasoba et al., 2013).
A growing body of evidence supports a central role for mitochondrial dysfunction in progressive hearing impairments (Biousse and Newman, 2001; Chinnery et al., 2000; Fischel-Ghodsian, 2003; Gorman and Taylor, 2011; Hudson and Chinnery, 2006; Kenney et al., 2010; Kokotas et al., 2007; Kujoth et al., 2007; McKechnie et al., 1985; Someya and Prolla, 2010). Because mitochondrial NADPH acts as a substrate for mitochondrial antioxidant enzymes, mitochondrial IDH2 is the major supplier of NADPH for the mitochondrial antioxidant defenses, and the NADP+/NADPH couple is essential for maintaining a large array of biological processes, including cellular redox state mitochondrial function (Xiao et al., 2017), a decline in mitochondrial NADPH production and IDH2 activity likely contributes to the development of age-related disease, including ARHL. In agreement with this idea, Szu and co-workers have shown that decreased levels of IDH1 and IHD2 were observed in old mouse brain (Guo et al., 2020). In summary, this report reviewed what has been learned about the roles of mitochondrial NADPH and IDH2 in mitochondrial function, mitochondrial dysfunction, and hearing loss. These findings have significantly advanced the field of cochlear mitochondrial function and dysfunction.
Footnotes
CRediT authorship contribution statement
Karessa White: Writing – original draft, Writing – review & editing. Shinichi Someya: Writing – review & editing, Supervision, Project administration, Funding acquisition.
Data Availability
No data was used for the research described in the article.
References
- Ahn JH, Kang HH, Kim TY, Shin JE, Chung JW, 2008. Lipoic acid rescues DBA mice from early-onset age-related hearing impairment. Neuroreport 19 (13), 1265–1269. doi: 10.1097/WNR.0b013e328308b33800001756-200808270-00004, [pii]. [DOI] [PubMed] [Google Scholar]
- Anderson ME, 1998. Glutathione: an overview of biosynthesis and modulation. Chem. Biol. Interact 111, 1–14. -112Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9679538 . [DOI] [PubMed] [Google Scholar]
- Arkblad EL, Egorov M, Shakhparonov M, Romanova L, Polzikov M, Rydström J, 2002. Expression of proton-pumping nicotinamide nucleotide transhydrogenase in mouse, human brain and C. elegans. Comp. Biochem. Physiol. B Biochem. Mol. Biol 133 (1), 13–21. [DOI] [PubMed] [Google Scholar]
- Bai U, Seidman MD, Hinojosa R, Quirk WS, 1997. Mitochondrial DNA deletions associated with aging and possibly presbycusis: a human archival temporal bone study. Am. J. Otol 18 (4), 449–454. Retreieved from https://www.ncbi.nlm.nih.gov/pubmed/9233484 . [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T, 2005. Mitochondria, oxidants, and aging. Cell 120 (4), 483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Banaclocha MM, 2001. Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med. Hypotheses 56 (4), 472–477. doi: 10.1054/mehy.2000.1194. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN, 1998. The free radical theory of aging matures. Physiol. Rev 78 (2), 547–581. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9562038 . [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Moprris CM, Taylor GA, Reeve AK, Perry RH, Turnbull DM, 2006. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet 38 (5), 515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Benderdour M, Charron G, Comte B, Ayoub R, Beaudry D, Foisy S, Des-Rosiers C, 2004. Decreased cardiac mitochondrial NADP+-isocitrate dehydrogenase activity and expression: a marker of oxidative stress in hypertrophy development. Am. J. Physiol.-Heart Circulat. Physiol 287 (5), H2122–H2131. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L, & Clarke ND (2002). Biochemistry. ed. Michelle Julet: New York. [Google Scholar]
- Biousse V, Newman NJ, 2001. Neuro-ophthalmology of mitochondrial diseases. Semin. Neurol 21 (3), 275–291. doi: 10.1055/s-2001-17945. [DOI] [PubMed] [Google Scholar]
- Chen T, He J, Shen L, Fang H, Nie H, Jun T, Bai Y, 2011. The mitochondrial DNA 4,977-bp deletion and its implication in copy number alteration in colorector cancer. BMC Med. Genet 12, 8. doi: 10.1186/1471-2350-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, 2000. Primary mitochondrial disorders overview. Gene Rev.. [Google Scholar]
- Chinnery PF, Elliott C, Green GR, Rees A, Coulthard A, Turnbull DM, Griffiths TD, 2000. The spectrum of hearing loss due to mitochondrial DNA defects. Brain 123 (1), 82–92. doi: 10.1093/brain/123.1.82, Pt. [DOI] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Weindruch R, 2009. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WC, Longley MJ, 2014. Mitochondrial genome maintenance in health and disease. DNA Repair (Amst.) 19, 190–198. doi: 10.1016/j.dnarep.2014.03010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, Su SM, 2017. Isocitrate dehydrogenase mutation and R-2-hydroxyglutarate: from basic discovery to therapeutics development. Ann. Rev. Biochem 86 (1), 305–331. doi: 10.1146/annurev-biochem-061516-044732. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ, 2000. Oxidants, oxidative stress and the biology of ageing. Nature 408 (6809), 239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Fischel-Ghodsian N, Bykhovskaya Y, Taylor K, Kahen T, Cantor R, Ehrenman K, Keithley E, 1997. Temporal bone analysis of patients with presbycusis reveals high frequency of mitochondrial mutations. Hear. Res 110 (1), 147–154. doi: 10.1016/s0378-5955(97)00077-4, -2. [DOI] [PubMed] [Google Scholar]
- Fischel-Ghodsian N, 2003. Mitochondrial deafness. Ear Hear. 24, 303–313. [DOI] [PubMed] [Google Scholar]
- Francisco A, Figueira TR, Castilho RF., 2022. Mitochondrial NAD(P)+ transhydrogenase: from molecular features to physiology and disease. Antioxid. Redox. Signal 36 (13), 864–884. doi: 10.1089/ars.2021.0111, -15. [DOI] [PubMed] [Google Scholar]
- Gates GA, Mills JH, 2005. Presbycusis. Lancet North Am. Ed 366 (9491), 1111–1120. [DOI] [PubMed] [Google Scholar]
- Gorman GS, Taylor RW, 2011. Mitochondrial DNA abnormalities in ophthalmological disease. Saudi. J. Ophthalmol 25 (4), 395–404. doi: 10.1016/j.sjopt.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guastini L, Mora R, Dellepiane M, Santomauro V, Giorgio M, Salami A, 2011. Water-soluble coenzyme Q10 formulation in presbycusis: long-term effects. Acta Otolaryngol. 131 (5), 512–517. doi: 10.3109/00016489.2010.539261. [DOI] [PubMed] [Google Scholar]
- Guo X, Park JE, Gallart-Palau X, Sze SK, 2020. Oxidative damage to the TCA Cycle enzyme MDH1 dysregulates bioenergetic enzymatic activity in the aged murine brain. J. Proteome Res 19 (4), 1706–1717. doi: 10.1021/acs.jproteome.9b00861. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge J, 2007. Free Radicals in Biology and Medicine. Oxford University Press Inc., New York. [Google Scholar]
- Han SJ, Jang HS, Noh MR, Kim J, Kong MJ, Kim JI, Park KM (2017). Mitochondrial NADP + -dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol, 28(4), 200–1215 (2017). doi: 10.1681/ASN.2016030349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AM, Terenghi G, Kellerth JO, Wiberg M, 2004. Sensory neuroprotection, mitochondrial preservation, and therapeutic potential of N-acetyl-cysteine after nerve injury. Neuroscience 125 (1), 91–101. doi: 10.1016/j.neuroscience.2003.12.040S0306452203009539, [pii]. [DOI] [PubMed] [Google Scholar]
- Jiang H, Talaska AE, Schacht J, Sha SH, 2007. Oxidative imbalance in the aging inner ear. Neurobiol. Aging 28 (10), 1605–1612. doi: 10.1016/j.neurobiolaging.2006.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Huh TL, 2001. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J. Biol. Chem 276 (19), 16168–16176. [DOI] [PubMed] [Google Scholar]
- Kenney MC, Atilano SR, Boyer D, Chwa M, Chak G, Chinichian S, Udar NS, 2010. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest. Ophthalmol. Vis. Sci 51 (8), 4289–4297. doi: 10.1167/iovs.09-4778. [DOI] [PubMed] [Google Scholar]
- Kil IS, Huh TL, Lee YS, Lee YM, Park W., 2006. Regulation of replicative senescence by NADP+ -dependent isocitrate dehydrogenase. Free Radical. Biol. Med 40 (1), 110–119 . [DOI] [PubMed] [Google Scholar]
- Kim MJ, Haroon S, Chen GD, Ding D, Wanagat J, Liu L, Someya S, 2019. Increased burden of mitochondrial DNA deletions and point mutations in early-onset age-related hearing loss in mitochondrial mutator mice. Exp. Gerontol 125, 110675. doi: 10.1016/j.exger.2019.110675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YR, Jeong-In B, Kim SH, Kim MA, Lee H, Ryu N, Kim UK, 2019. Therapeutic potential of the mitochondria-targeted antioxidant MitoQ mitochondrial-ROS induced sensorineural hearing loss caused by Idh2 deficiency. Redox. Biol 20, 544–555. doi: 10.1016/j.redox.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapcinska B, Derejczyk J, Wieczorowska-Tobis K, Sobczak A, Sadowska-Krepa E, Danch A, 2000. Antioxidant defense in centenarians (a preliminary study). Acta Biochim. Pol 47 (2), 281–292. [PubMed] [Google Scholar]
- Kokotas H, Petersen MB, Willems PJ, 2007. Mitochondrial deafness. Clin. Genet 71 (5), 379–391. doi: 10.1111/j.1399-0004.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavsteva E, McKee AC, Geula C, Kowall NW, Khrapko K, 2006. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet 38 (5), 518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Turnbull DM, 2008. What causes mitochondrial DNA deletions in human cells? Nat. Genet 40 (3), 275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Ku HJ, Ahn Y, Lee JH, Park KM, Park JW, 2015. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radical. Biol. Med 80, 84–92. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Prolla TA, 2005. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309 (5733), 481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Bradshaw PC, Haroon S, Prolla TA, 2007. The role of mitochondrial DNA mutations in mammalian aging. PLos Genet. 3 (2), e24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarino G, Amorini AM, Signoretti S, Musumeci G, Lazzarino G, Caruso G, Belli A, 2019. Pyruvate dehydrogenase and tricarboxylic acid cycle enzymes are sensitive targets of traumatic brain injury induced metabolic derangement. Int. J. Mol. Sci 20 (22), 5774. doi: 10.3390/ijms20225774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Go Y, Kim DY, Lee SH, Kim OH, Jeon YH, Im SS, 2020. Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochdonria from brown adipose tissue. Exp. Mol. Med 52 (6), 238–252. doi: 10.1038/s12276-020-0451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Wu JL, Shih TS, Tsai PJ., Sun YM, Ma MC, Guo YL, 2010. N-Acetylcysteine against noise-induced temporary threshold shift in male workers. Hear. Res 269 (1), 42–47. doi: 10.1016/j.heares.2010.07.005S0378-5955(10)00332-1, -2[pii]. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Yan D, 2007. Ageing and hearing loss. J. Pathol 211 (2), 188–197. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Filosto M, Bellan M, Liguori R, Montagna P, Baruzzi A, Carelli V, 2004. POLG mutations causing ophthalmoplegia, sensorimotor polyneuropathy, ataxia, and deafness. Neurology 62 (2), 316–318. doi: 10.1212/wnl.62.2.316. [DOI] [PubMed] [Google Scholar]
- Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC, 2009. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox. Signal 11 (11), 2685–2700. doi: 10.1089/ARS.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Reaume AG, Flood DG, Salvi RJ, 1999. Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol. Aging 20 (1), 1–8. doi: 10.1016/s0197-4580(99)00018-4. [DOI] [PubMed] [Google Scholar]
- McKechnie NM, King M, Lee WR, 1985. Retinal pathology in the Kearns-Sayre syndrome. Br. J. Ophthalmol 69 (1), 63–75. doi: 10.1136/bjo.69.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITOMAP. (2019). A human mitochondrial genome database. Retrieved from https://www.mitomap.org/MITOMAP
- Niu X, Trifunovic A, Larsson NG, Canlon B, 2007. Somatic mtDNA mutations cause progressive hearing loss in the mouse. Exp. Cell. Res 313 (18), 3924–3934. doi: 10.1016/j.yexcr.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Nicholson A, Reifsnyder PC, Malcolm RD, Lucas CA, MacGregor GR, Zhang W, Leiter EH, 2010. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity (Silver Spring) 8 (10), 1902–1905. doi: 10.1038/oby.2009.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E, 2009. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol. Dis 42, 267–278. doi: 10.1016/j.bcmd.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Ohinata Y, Yamasoba T, Schacht J, Miller JM, 2000. Glutathione limits noise-induced hearing loss. Hear. Res 146 (1), 28–34. doi: 10.1016/S0378-5955(00)00096-4, -2[pii]. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, McFadden SL, Ding DL, Lear PM, Ho YS, 2000. Targeted mutation of the gene for cellular glutathione peroxidase (Gpx1) increases noise-induced hearing loss in mice. J. Assoc. Res. Otolaryngol 1 (3), 243–254. doi: 10.1007/s101620010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Wright JS, Dugan LL, 1999. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol. Neurootol 4 (5), 229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- Oyler A, 2012. The American hearing loss epidemic: Few of 46 million with hearing loss seek treatment. The ASHA Leader 5–7. [Google Scholar]
- Palaniappan AR, Dai A, 2007. Mitochondrial ageing and the beneficial role of alpha-lipoic acid. Neurochem. Res 32 (9), 1552–1558. doi: 10.1007/s11064-007-9355-4. [DOI] [PubMed] [Google Scholar]
- Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C, 2017. The glutatmate dehydogenase pathway and its roles in cell and tissue biology in health and disease. Biology 6 (1), 11. doi: 10.3390/biology6010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack N, Niere M, Ziegler M, 2007. NAD kinase levels control the NADPH concentration in human cells. J. Biol. Chem 282 (46), 33562–33571. [DOI] [PubMed] [Google Scholar]
- Rebrin I, Sohal RS, 2008. Pro-oxidant shift in glutathione redox state during aging. Adv. Drug. Deliv. Rev 60 (13), 1545–1552. doi: 10.1016/j.addr.2008.06.001, -14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Yan H, 2010. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst 102 (13), 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman MD, Khan MJ, Bai U, Shirwany N, Quirk WS, 2000. Biologic activity of mitochondrial metabolites on aging and age-related hearing loss. Am. J. Otol 21 (2), 161–167. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10733178. [DOI] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M, 2012. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet 13 (12), 8768–8890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S, 2011. Mitochondria: the next (neurode) generation. Neuron 70 (6), 1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Rabinovitch PS, 2005. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308 (5730), 1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Forster MJ, 2007. Coenzyme Q oxidative stress and aging. Mitochondrion 7, S103–S111. doi: 10.1016/j.mito.2007.03.006S1567-7249(07)00063-3, Suppl [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R, 1996. Oxidative stress, caloric restriction, and aging. Science 273 , 59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yamasoba T, Kujoth GC, Pugh TD, Weindruch R, Tanokura M, Prolla TA, 2008. The role of mtDNA mutations in the pathogenesis of age-related hearing loss in mice carrying a mutator DNA polymerase gamma. Neurobiol. Aging 29 (7), 1080–1092. doi: 10.1016/j.neurobiolaging.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Prolla TA, 2010. Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech. Ageing Dev 131 (7), 480–486. doi: 10.1016/j.mad.2010.04.006, -8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Xu J, Kondo K, Ding D, Salvi RJ, Yamasoba T, Prolla TA, 2009. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl. Acad. Sci. U. S. A 106 (46), 19432–19437. doi: 10.1073/pnas.0908786106, [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yamasoba T, Weindruch R, Prolla TA, Tanokura M, 2007. Caloric restriction suppresses apoptotic cell death in the mammalian cochlea and leads to prevention of presbycusis. Neurobiol. Aging 28, 1613–1622. doi: 10.1016/j.neurobiolaging.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Prolla TA, 2010. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143 (5), 802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM, 2005. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet 6 (5), 389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA, 2008. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet 40 (4), 392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Wallace DC, 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL, 1988. The Retardation of Aging and Disease by Dietary Restriction. Charles C Thomas Publishing, LTD, Springfield, IL. [Google Scholar]
- White K, Kim MJ, Han C, Park HJ, Ding D, Boyd K, Someya S, 2018. Loss of IDH2 accelerates age-related hearing loss in male mice. Sci. Rep 8 (1), 5039. doi: 10.1038/s41598-018-23436-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2021). Deafness and hearing loss. Retrieved from https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss
- Wu HP, Hsu CJ, Cheng TJ, Guo YL, 2010. N-acetylcysteine attenuates noise-induced permanent hearing loss in diabetic rats. Hear. Res 267 (1), 71–77. doi: 10.1016/j.heares.2010.03.082S0378-5955(10)00194-2, -2[pii]. [DOI] [PubMed] [Google Scholar]
- Xing G, Chen Z, Cao X, 2007. Mitochondrial rRNA and tRNA and hearing function. Cell Res. 17 (3), 227–239. doi: 10.1038/sj.cr.7310124. [DOI] [PubMed] [Google Scholar]
- Yamasoba T, Someya S, Yamada C, Weindruch R, Prolla TA, Tanokura M, 2007. Role of mitochondrial dysfunction and mitochondrial DNA mutations in age-related hearing loss. Hear. Res 226, 185–193. doi: 10.1016/j.heares.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Yamasoba T, Lin FR, Someya S, Kashio A, Sakamoto T, Kondo K, 2013. Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hear. Res 303, 30–38. doi: 10.1016/j.heares.2013.01.021 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, 2008. NAD + /NADH and NADP + /NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signaling 10 (2), 179–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.