

Abstract
EGFR mutations are a major prognostic factor in lung adenocarcinoma. However, current detection methods require sufficient samples and are costly. Deep learning is promising for mutation prediction in histopathological image analysis but has limitations in that it does not sufficiently reflect tumor heterogeneity and lacks interpretability. In this study, we developed a deep learning model to predict the presence of EGFR mutations by analyzing histopathological patterns in whole slide images (WSIs). We also introduced the EGFR mutation prevalence (EMP) score, which quantifies EGFR prevalence in WSIs based on patch‐level predictions, and evaluated its interpretability and utility. Our model estimates the probability of EGFR prevalence in each patch by partitioning the WSI based on multiple‐instance learning and predicts the presence of EGFR mutations at the slide level. We utilized a patch‐masking scheduler training strategy to enable the model to learn various histopathological patterns of EGFR. This study included 868 WSI samples from lung adenocarcinoma patients collected from three medical institutions: Hallym University Medical Center, Inha University Hospital, and Chungnam National University Hospital. For the test dataset, 197 WSIs were collected from Ajou University Medical Center to evaluate the presence of EGFR mutations. Our model demonstrated prediction performance with an area under the receiver operating characteristic curve of 0.7680 (0.7607–0.7720) and an area under the precision‐recall curve of 0.8391 (0.8326–0.8430). The EMP score showed Spearman correlation coefficients of 0.4705 (p = 0.0087) for p.L858R and 0.5918 (p = 0.0037) for exon 19 deletions in 64 samples subjected to next‐generation sequencing analysis. Additionally, high EMP scores were associated with papillary and acinar patterns (p = 0.0038 and p = 0.0255, respectively), whereas low EMP scores were associated with solid patterns (p = 0.0001). These results validate the reliability of our model and suggest that it can provide crucial information for rapid screening and treatment plans.
Keywords: EGFR, whole‐slide image analysis, deep learning in histopathology, multiple‐instance learning
Introduction
Epidermal growth factor receptor (EGFR) is a crucial gene for cellular regeneration and survival, and it plays key roles in tissue repair and maintaining the cellular microenvironment [1, 2, 3, 4]. However, mutations in the EGFR gene are linked to various cancers, including non‐small cell lung cancer (NSCLC), gallbladder cancer, and glioblastoma [5, 6, 7]. Therefore, EGFR is an important target for cancer treatment. In particular, the development and clinical application of EGFR tyrosine kinase inhibitors (EGFR‐TKIs) have significantly improved the survival and clinical outcomes of NSCLC patients with EGFR mutations. Consequently, EGFR‐TKIs are recommended as the standard first‐line treatment for patients with these mutations [8, 9]. It is crucial to rapidly identify patients with mutations to administer the appropriate treatment in a timely manner. Although next‐generation sequencing (NGS) has enabled relatively fast mutational genomic diagnostics [10], challenges such as difficulty in obtaining sufficient samples and high costs persist [11].
In contrast, machine‐learning‐based whole slide image (WSI) analysis has shown promise in various medical fields [12, 13, 14, 15, 16, 17, 18] and has demonstrated satisfactory performance in predicting mutational genomics [16, 19, 20, 21]. Predicting mutational genomics using WSI is time‐ and cost‐effective and, because it utilizes previously collected samples, it has the potential to be an effective alternative to mutational genomic testing.
In machine learning, a multiple‐instance learning (MIL) approach is employed for WSI analysis [22]. MIL is a type of weakly supervised learning method that trains patch‐level prediction models using slide‐level labels. This enables the learning of the spatial heterogeneity of tumors and the prediction of slide‐level probabilities by aggregating patch‐level predictions. MIL is known to be effective for WSI analysis [23, 24, 25, 26] and has been utilized to successfully predict the mutational genomics of various cancer types. For instance, MIL‐based approaches have been applied to predict EGFR mutations in lung adenocarcinoma [20, 21], genetic alterations in gastric cancer [27], and mutation status in breast carcinoma [28]. Moreover, a hierarchical deep MIL model to predict gene mutations in bladder cancer has been reported [19]. These studies demonstrate the potential of MIL in capturing the spatial heterogeneity of tumors and predicting clinically relevant mutations from histopathology images across various cancer types.
MIL can learn patch‐level probabilities through weakly supervised learning using only slide‐level labels. We hypothesized that, by utilizing this feature, we could obtain the probability of EGFR mutation in the entire tumor region and calculate the EGFR prevalence in WSI. To validate our hypothesis, we collected histopathological patterns (lepidic, acinar, solid, papillary, and micropapillary) from pathology reports and analyzed the allele frequencies obtained through DNA mutation testing, indirectly measuring the significance of the predicted region. The proposed method effectively addresses the limitations of previous studies [20, 21], which provided only binary predictions of EGFR mutation status, by predicting the prevalence of EGFR mutations.
Materials and methods
Data collection
We retrospectively selected data from patients with histopathologically confirmed lung adenocarcinoma who underwent EGFR mutation testing (PCR or NGS) at four large independent institutions: Hallym University Medical Center (HUMC) (n = 145), Inha University Hospital (INHA) (n = 129), Chungnam National University Hospital (CNUH) (n = 397), and Ajou University Medical Center (AJMC) (n = 197) from 2012 to 2022. We collected one WSI per patient, totaling 868 WSIs. Detailed information on the cohort and the relationship between clinical data and EGFR mutations is presented in Table 1, and detailed information on the WSIs is shown in Table 2.
Table 1.
Details of cohort
| Clinical characteristics | AJMC (n = 197) | HUMC (n = 145) | CNUH (n = 397) | INHA (n = 129) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EGFR wild‐type (n = 69) | EGFR mutant (n = 128) | p | EGFR wild‐type (n = 80) | EGFR mutant (n = 65) | p | EGFR wild‐type (n = 235) | EGFR mutant (n = 162) | p | EGFR wild‐type (n = 67) | EGFR mutant (n = 62) | p | ||
| Gender | Female | 26 | 85 | 0.0001 | 29 | 41 | 0.0023 | 85 | 104 | 0.0001 | 24 | 45 | 0.0001 |
| Male | 43 | 43 | 51 | 24 | 150 | 58 | 43 | 17 | |||||
| Smoking status | Y | 52 | 107 | 0.2272 | 53 | 53 | 0.0605 | 97 | 92 | 0.0032 | 32 | 51 | 0.0001 |
| N | 17 | 21 | 27 | 12 | 128 | 70 | 35 | 11 | |||||
| Family history | Y | 66 | 126 | 0.477 | 74 | 59 | 0.736 | 209 | 128 | 0.0101 | 49 | 46 | 0.9493 |
| N | 3 | 2 | 5 | 6 | 26 | 34 | 18 | 16 | |||||
| Age (mean) | 66.09 ± 9.94 | 64.10 ± 9.78 | 0.0941 | 65.15 ± 11.07 | 66.34 ± 12.51 | 0.3934 | 67.19 ± 9.34 | 66.37 ± 9.433 | 0.1505 | 63.17 ± 10.94 | 62.98 ± 12.01 | 0.4727 | |
| Height (mean) | 162.69 ± 8.09 | 158.78 ± 9.15 | 0.0007 | 161.40 ± 8.51 | 159.21 ± 7.33 | 0.0404 | 162.01 ± 8.73 | 158.65 ± 8.28 | 0.0001 | 163.17 ± 7.07 | 157.89 ± 7.85 | 0.0003 | |
| Weight (mean) | 62.85 ± 10.68 | 59.37 ± 10.93 | 0.0195 | 62.84 ± 10.67 | 59.58 ± 9.51 | 0.0334 | 64.50 ± 10.32 | 60.77 ± 10.24 | 0.0001 | 65.04 ± 11.64 | 62.23 ± 9.39 | 0.1813 | |
| Stage | I | 44 | 101 | 0.0832 | 46 | 41 | 0.2866 | 189 | 133 | 0.3522 | 41 | 46 | 0.2664 |
| II | 11 | 14 | 7 | 7 | 26 | 18 | 8 | 8 | |||||
| III | 11 | 8 | 12 | 12 | 10 | 2 | 16 | 7 | |||||
| IV | 3 | 5 | 15 | 5 | 10 | 9 | 2 | 1 | |||||
The clinicopathological characteristics of cohorts of lung adenocarcinoma patients collected from four institutions, AJMC, HUMC, CNUH, and INHA, are summarized according to EGFR mutations. The statistical significance of differences between groups was evaluated by the chi‐square test or t‐test.
Table 2.
Details of WSIs
| AJMC | HUMC | CNUH | INHA | ||
|---|---|---|---|---|---|
| Number of patients | 197 | 145 | 397 | 129 | |
| Number of WSIs | 197 | 145 | 397 | 129 | |
| Scanning magnification | ×20 | 197 | 126 | 69 | 129 |
| ×40 | 0 | 19 | 328 | 0 | |
| Microns per pixel | 0.5025 ± 0.0003 | 0.4695 ± 0.0847 | 0.2479 ± 0.0027 | 0.5000 ± 0.0 | |
| Width | 52,138 ± 7,377 | 58,413 ± 23,900 | 82,714 ± 7,562 | 46,961 ± 8,096 | |
| Height | 40,922 ± 6,179 | 42,932 ± 16,835 | 114,027 ± 19,933 | 56,232 ± 10,464 | |
| Machine | Aperio | 197 | 145 | 397 | 129 |
| Number of patches (1,024 × 1,024 pixel, ×20 magnification) | Tumor | 140,857 | 77,805 | 213,844 | 79,628 |
| Non‐tumor | 39,981 | 25,176 | 140,585 | 50,438 | |
This table describes the details of the whole slide images (WSIs).
To further validate the developed model, we collected histopathological pattern information from pathology reports (n = 197) and allele frequency data from NGS results (n = 68) in the AJMC cohort. The predominant histopathological pattern was defined as a pattern (lepidic, acinar, solid, papillary, or micropapillary) occupying more than 50% of the WSI, as described by the pathologist in the pathology report. The AJMC cohort was chosen as the test dataset because it provided additional information for evaluating the predictive performance of the model from multiple perspectives. The Institutional Review Board (IRB) of Ajou University Hospital approved this study (AJOUIRB‐MDB‐2022‐249). Furthermore, the requirement for informed consent from all participants was waived by the IRB because of the retrospective nature of this study. All methods were performed in accordance with the Declaration of Helsinki.
Preprocessing
The collected WSIs were scanned using Aperio equipment. All WSIs were standardized to ×20 magnification using the Lanczos filter [29] to ensure uniform cell and tissue sizes and then divided into fixed‐size (1,024 × 1,024 pixels) patch images for use as input images for MIL.
To develop and evaluate the tumor region segmentation model, the tumor regions were annotated by experienced pathologists specializing in lung cancer diagnosis and pathological examination. Pathologists with over 3 years of clinical experience meticulously labeled the boundaries of the cancerous tissue by using the polygon tool in the open‐source software QuPath [30].
The patches were divided into non‐tumor and tumor regions, and background areas that were not tissue regions were excluded using Otsu's adaptive thresholding method [31] as per prior studies [32]. Variations in the tissue staining and scanning processes between medical centers can occur. Methods such as stain normalization [33] and Macenko normalization [34] have been proposed to address this, but we proposed RandStainNA [35], which showed more diverse staining enhancement. The method described above was applied to the patch image for learning. We used ViT‐B/14, a variant of the vision transformer (ViT), to extract features from the divided patch images. This model was pretrained using the DINO algorithm on the ImageNet dataset [36]. DINO is an unsupervised learning technique that uses self‐distillation to train models and is commonly used with ViT models. It has also been successfully applied to various medical image analysis tasks. For instance, Wessels et al [37] employed a self‐supervised ViT pretrained with DINO to predict survival from histopathology images in renal cell carcinoma, thereby demonstrating its effectiveness in capturing prognostic information. Similarly, Li et al [38] utilized a DINO‐pretrained ViT for the weakly supervised histopathological image analysis of primary brain tumors, showcasing its ability to learn discriminative features from limited labeled data. These studies highlight the potential of DINO‐pretrained ViT models in extracting meaningful representations from complex medical images.
In this study, the pretrained ViT‐B/14 model with all frozen parameters was used to extract high‐dimensional features from the patch images. The input image size was down‐sampled to 518 × 518 pixels to fit the ViT model. The extracted features were used for subsequent analyses, including the prediction of EGFR mutations. The detailed workflow from WSI extraction to patch image feature extraction is illustrated in Figure 1.
Figure 1.
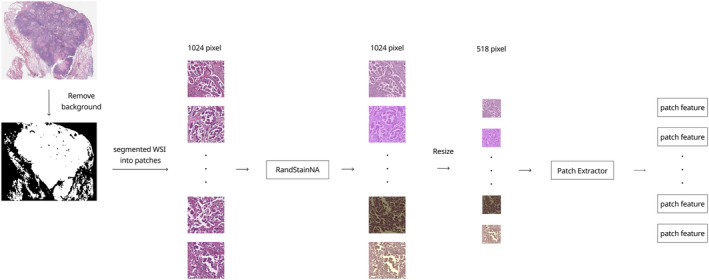
Preprocessing of WSIs. Mimetic diagram illustrating the preprocessing process of WSI data. Series of steps: extraction of patch images from the original WSI, RandStainNA, and feature extraction. The ViT‐B/14 DINO model was used for feature extraction.
Tumor region segmentation
To analyze EGFR mutations in the WSIs, we first performed tumor region segmentation. The model was trained using the WSSS4LUAD dataset [39]. WSSS4LUAD is a publicly available dataset specifically designed for the weakly supervised semantic segmentation of lung adenocarcinoma histopathology images. It consists of WSIs annotated with patch‐level labels for tumor and non‐tumor regions and is therefore appropriate for tumor region segmentation. We classified the patch images into tumor and non‐tumor regions using a binary classification model. The model used for the classification was ViT‐B/14.
MIL with patch‐masking strategy
For predicting EGFR mutations, we used the MIL model, specifically the dual‐stream MIL (DSMIL) model. The DSMIL consists of a two‐stream architecture. The first stream identifies the critical instance that is expected to be most related to EGFR mutations among the patches. The second stream calculates the attention score by measuring the distance between each instance and the critical instance, thereby evaluating the importance of each instance. This approach is robust even when the proportion of positive patches is low, thereby addressing the data imbalance issues [22]. Considering the need to accurately detect a small number of patches related to EGFR mutations, the DSMIL model was selected for this study. The structure of the DSMIL used in this study is shown in Figure 2.
Figure 2.
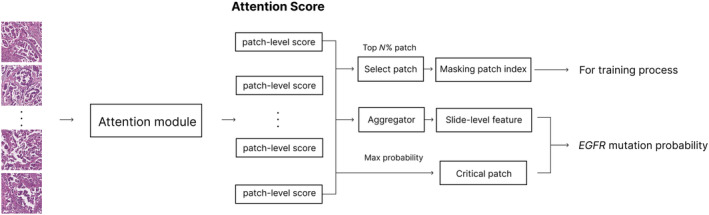
MIL architecture. Structure of dual‐stream multiple‐instance learning (DSMIL) model used for predicting EGFR mutations in this study. DSMIL learns features of the patch with the highest probability of EGFR mutation and aggregates slide‐level features based on the attention score. During the training phase, the teacher model performs masking based on the predicted score.
We also adopted masked hard‐instance mining [40] to train the model on various histopathological patterns exhibiting EGFR mutations. This training strategy uses two models: teacher and student. The teacher model first calculates the patch‐level attention score, which indicates the relevance of each patch for the EGFR mutation. The student model receives masked patches as input and learns various histopathological patterns. Figure 3 illustrates the process of masking the top N% of patches based on the attention scores, focusing the training on the more challenging and diverse histopathological patterns that are less indicative of EGFR mutations.
Figure 3.
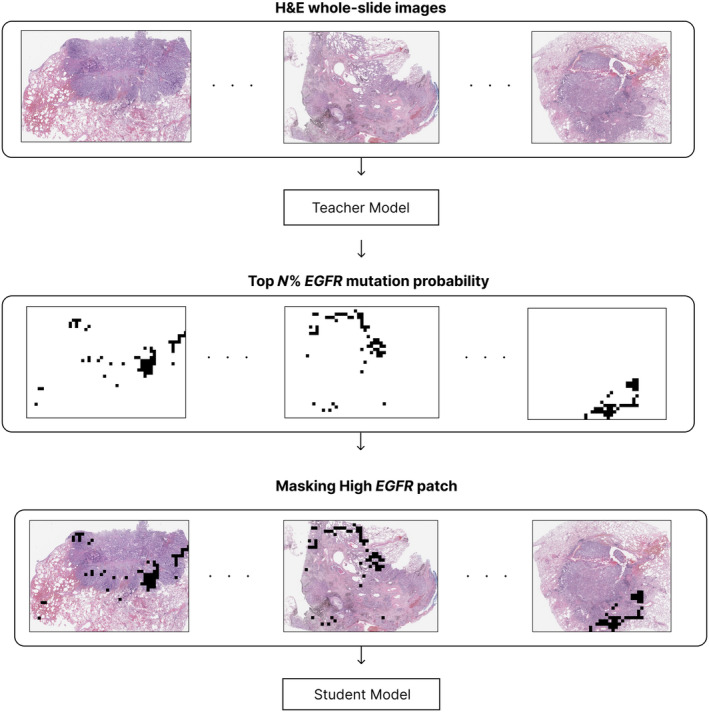
Attention‐based masking. Process of masking the top N% of patches with high EGFR mutation rate. By masking these patches, the model can focus on learning various patterns, including the easy‐to‐distinguish features of EGFR mutation.
However, the fixed‐ratio patch‐masking method can lead to the loss of common and easily classified histopathological patterns of EGFR mutations during the early stages of training. Therefore, we introduced a patch‐masking scheduler that dynamically adjusts the masking ratio as training progresses. This allows the model to learn the characteristics of tissues with high EGFR mutation presence early in the training and gradually increases the masking ratio to learn various tissue patterns as the training continues. This process enables the model to initially learn the easier histopathological features and then the more complex EGFR patterns as the training progresses. The teacher model was used only during the training process and not during the inference process. The training and inference processes using the proposed model are shown in Figure 4.
Figure 4.
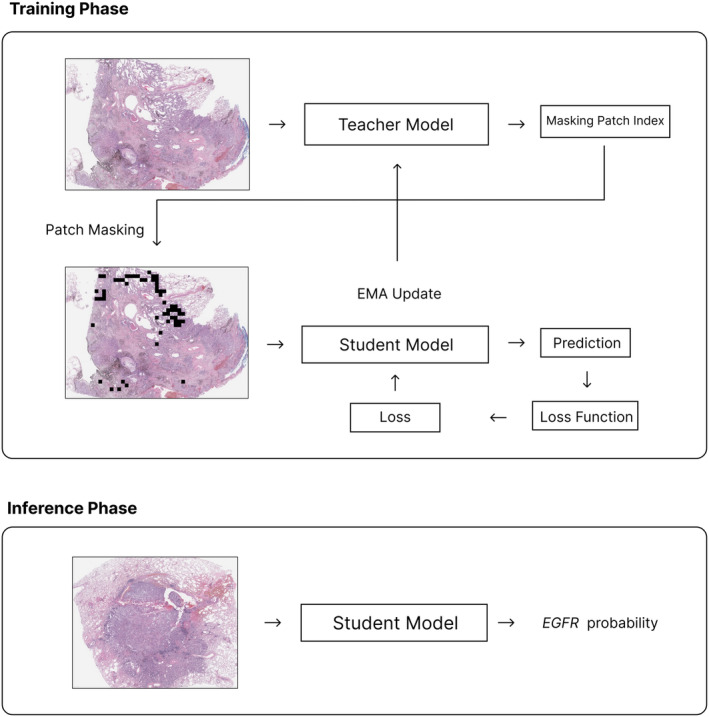
Training and inference process. Process of learning and reasoning using MIL and patch‐masking strategies. A teacher model is utilized for learning various patterns during the training phase and is not involved in the reasoning process.
Artificial intelligence‐based EGFR mutation prevalence score in tumor areas
In the process of learning the presence of EGFR mutations at the slide level, the MIL model learns the prediction probabilities at the patch level. We proposed an artificial intelligence (AI)‐based EGFR mutation prevalence (EMP) score that quantifies the prevalence of EGFR mutation in the tumor region of WSIs. This score is the ratio of the sum of the EGFR mutation probability values predicted by the model at the patch level to the total number of patches in the tumor region, as shown in the formula below:
where represents the total number of patches and represents the score of the th patch among the patches where the presence of EGFR mutation is predicted. By dividing the sum of the prediction probabilities by the total number of patches , we obtained the EMP score, which represents the prevalence of tumor regions containing EGFR mutations in the WSIs.
Results
Tumor region classification performance
The tumor region classification model, trained using the WSSS4LUAD dataset, was evaluated using expert annotations in our cohort. In the training data (CNUH, INHA, and HUMC, n = 868), the model achieved an area under the receiver operating characteristic curve (AUROC) of 0.9048 (0.8826–0.9210) and an F1 score of 0.7628 (0.7482–0.7871). In the test data (AJMC, n = 197), the model achieved an AUROC of 0.8922 (0.8714–0.9224) and an F1 score of 0.7632 (0.7486–0.7822) (Table 3). For EMP, following common practice in medical image analysis [41, 42], only patch images with a tumor prediction probability above 0.5 were used. If no patches in a WSI had a probability above 0.5, we sequentially applied lower thresholds, and no WSIs had tumor patches with a probability below 0.3.
Table 3.
Performance of tumor classification
| Metric | Tumor classification | |
|---|---|---|
| Training cohort (HUMC, INHA, CNUH) | Test cohort (AJMC) | |
| AUROC | 0.9048 (0.8826–0.9210) | 0.8922 (0.8714–0.9224) |
| F1 score | 0.7628 (0.7482–0.7871) | 0.7632 (0.7486–0.7822) |
| Accuracy | 0.8321 (0.8126–0.8488) | 0.8251 (0.8018–0.8432) |
A performance table of tumor/non‐tumor region classification models learned using the WSSS4LUAD dataset. Indicators such as AUROC, F1 score, and accuracy were used in the training datasets (HUMC, INHA, and CNUH) and the test/verification datasets (AJMC).
Performance of deep learning model in predicting EGFR mutation
In this study, we proposed a patch‐masking scheduler learning strategy that allows the model to learn easy features related to EGFR presence early in training and more difficult features as the training progresses. Our model achieved an AUROC of 0.7680 (0.7607–0.7720) and an area under the precision‐recall curve of 0.8391 (0.8326–0.8430) in the AJMC cohort (n = 197), outperforming traditional MIL methods (Table 4).
Table 4.
Performance of MIL models in EGFR mutation prediction
| Ours | MHIM | DSMIL | ABMIL | |
|---|---|---|---|---|
| AUROC | 0.7680 (0.7607–0.7720) | 0.7441 (0.7381–0.7508) | 0.7210 (0.7170–0.7302) | 0.7021 (0.6982–0.7094) |
| AUPRC | 0.8391 (0.8326–0.8430) | 0.8098 (0.7983–0.8124) | 0.7904 (0.7862–0.8022) | 0.7622 (0.7584–0.7712) |
Table comparing the performance of multiple‐instance learning (MIL) models developed for EGFR mutation prediction. AUROC and AUPRC values of the proposed model (Ours) and existing methods (MHIM, DSMIL, and ABMIL) are presented.
AUPRC, area under the precision‐recall curve.
Evaluating EMP score in tumor areas
We quantitatively measured the AI‐based EMP score, which represents the prevalence of EGFR in the WSIs based on patch‐level predictions. We evaluated the significance of the EMP score by comparing it with the variant allele frequency (VAF) of EGFR mutations using NGS and predominant histopathological patterns.
Correlation between EMP score and allele frequency
Spearman's correlation analysis was performed to compare the relationship between the EMP score and VAF in tumor regions (n = 64). The two variables showed a correlation of 0.3708 (p = 0.00679), indicating that higher EMP in tumor tissue corresponded to a higher frequency of EGFR mutations in DNA, despite not showing a strong correlation.
We also compared the results based on clinically significant EGFR mutation subtypes, exon 19 deletion (exon 19 del) and p.L858R, known for their distinct histopathological patterns [43, 44] and treatment responses [45]. Using fewer larger patches (1,024 × 1,024 pixel patches from ×20 magnification), exon 19 del (n = 22) showed a correlation of 0.59184 (p < 0.00371), whereas exon 21 p.L858R (n = 30) did not show any correlation (0.1704, p = 0.3689). Conversely, using numerous smaller patches (512 × 512 pixel patches from ×20 magnification), p.L858R showed a correlation of 0.4705 (p = 0.00877), but exon 19 del did not (0.0412, p = 0.8553) (Table 5). Figure 5 visually presents the EMP score as a heatmap based on patch‐level prediction probabilities, showing that higher VAF scores tend to correspond to larger regions of EGFR mutation presence.
Table 5.
Spearman correlation between VAF and EMP score
| 512 x 512 pixel patches | 1024 x 1024 pixel patches | |||
|---|---|---|---|---|
| Coefficient | p value | Coefficient | p value | |
| Total | 0.3038 | 0.0285 | 0.3708 | 0.0067 |
| p.L858R | 0.4705 | 0.0087 | 0.1704 | 0.3689 |
| exon 19 del | 0.0412 | 0.8553 | 0.5918 | 0.0037 |
Correlation between the variant allele frequency (VAF) and the EMP score of EGFR mutations by Spearman correlation analysis. The differences according to patch size (512 x 512 / 1024 x 1024 pixels) and EGFR mutation subtype are shown.
Figure 5.
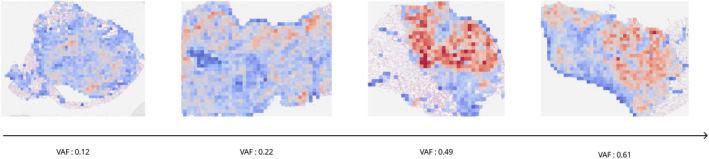
EGFR prevalence heatmaps (EMP scores) and VAFs. Heat maps showing the probability of EGFR mutation predicted by the AI model for each WSI. They visually represent that higher VAF scores correspond to more areas predicted to be EGFR mutation positive.
EMP score and histopathological pattern analysis
Table 6 presents the results of the chi‐square test analysis of the histopathological patterns of the WSIs and the presence of EGFR mutations. EGFR‐positive samples showed a significant relationship with the lepidic pattern (p = 0.0479), and EGFR wild‐type samples showed a significant relationship with the solid pattern (p = 0.0047), which is consistent with previous studies [46]. We divided the samples into low and high groups based on the median EMP score and analyzed the predominant histopathological patterns (Table 7). A solid pattern was observed significantly more in the group with low EMP scores (p = 0.0001). Papillary (p = 0.0038) and acinar (p = 0.0255) patterns were observed significantly more in the group with high EMP scores.
Table 6.
Clinical analysis based on predominant growth pattern and EGFR mutation
| Predominant growth pattern | EGFR wild‐type (n = 69) | EGFR mutation (n = 128) | p value | |
|---|---|---|---|---|
| Lepidic | N | 68 | 115 | 0.0479 |
| Y | 1 | 13 | ||
| Solid | N | 53 | 118 | 0.0047 |
| Y | 16 | 10 | ||
| Acinar | N | 28 | 36 | 0.1049 |
| Y | 41 | 92 | ||
| Papillary | N | 64 | 120 | 1 |
| Y | 5 | 8 | ||
| Micropapillary | N | 66 | 125 | 0.7291 |
| Y | 3 | 3 | ||
Results of distribution difference analysis by chi‐square test of predominant histopathological growth patterns (lepidic, solid, acinar, papillary, and micropapillary) according to the presence or absence of EGFR mutations.
Table 7.
Predominant growth pattern and EMP score
| Predominant growth pattern | EMP low | EMP high | p value | |
|---|---|---|---|---|
| Lepidic | N | 92 | 91 | 0.7967 |
| Y | 7 | 7 | ||
| Solid | N | 76 | 95 | 0.0001 |
| Y | 23 | 3 | ||
| Acinar | N | 40 | 24 | 0.0255 |
| Y | 59 | 74 | ||
| Papillary | N | 98 | 86 | 0.0038 |
| Y | 1 | 12 | ||
| Micropapillary | Y | 96 | 95 | 0.6876 |
| N | 3 | 3 | ||
Comparison results of the high/low group distribution of the EGFR mutation prevalence (EMP) score according to each histopathological growth pattern. The EMP score is an index that quantifies the prevalence of EGFR mutations in the WSI predicted by the AI model.
Discussion
In this study, we developed an MIL‐based deep learning model to analyze the prevalence of EGFR mutations in WSIs of patients with lung adenocarcinoma. Several studies have confirmed that specific histopathological patterns appear in tumors that contain EGFR mutations. In 2009, Ninomiya et al reported that micropapillary patterns were associated with EGFR mutations [47]. In 2014, Kadota et al reported that the lepidic‐predominant group was associated with EGFR mutations (p < 0.011), whereas papillary‐ and solid‐predominant tumors were associated with wild‐type EGFR [46]. More recently, in 2021, Saito et al reported that the pure micropapillary nests group was associated with EGFR mutations, whereas the small solid nests group was associated with wild‐type EGFR [48]. These examples suggest the possibility of predicting EGFR mutations through the histopathological pattern analysis of WSIs. Furthermore, previous reports [49, 50] on the close association between histopathological patterns of EGFR mutations and the therapeutic effects of EGFR‐TKIs further highlight the importance of histopathological pattern studies using WSIs.
However, tumor heterogeneity is a major challenge when predicting mutations using WSIs. Not all tumor cells contain the same genetic alterations [51], and EGFR mutations are only observed in a subset of tumor cells [52]. Existing MIL models are limited in that they focus on learning patches with strong features based on attention scores [22] and fail to learn diverse histopathological patterns [46, 47, 48, 49, 50] arising from tumor heterogeneity. To address this problem, we introduced a masking strategy based on patch probabilities [40] during the learning process. We advanced the existing patch‐masking learning technique using a patch‐masking scheduler strategy that dynamically adjusts the masking ratio according to the learning progress rather than using a fixed masking ratio. Through this, our model focuses on learning the features of patches with a high probability of EGFR mutation presence in the early stages of learning and gradually includes patches with lower probabilities as learning progresses. This strategy enables the model to comprehensively learn diverse and heterogeneous histopathological features associated with EGFR mutations.
In MIL, extracting diverse features at the patch level is crucial, and convolutional neural network (CNN) models are primarily used [53, 54, 55, 56]. However, CNNs have the limitation of primarily focusing on local features within a patch, while ignoring the global context and relative positional information of the cells [57, 58, 59]. Recently, ViTs [60] have gained attention as alternatives to CNNs. ViTs extract features by reflecting cell‐to‐cell interactions through a self‐attention mechanism and position encoding [58, 61]. Furthermore, ViTs can generate high‐level representations without additional fine‐tuning through unsupervised learning techniques such as DINO [36].
We demonstrate the effectiveness of our methodology by utilizing a large‐scale dataset collected from a multi‐institutional cohort. Prior to predicting EGFR mutations, we developed a model to classify tumor and non‐tumor regions in WSIs. Through this process, the EGFR mutation prediction model considers only the patch images predicted as tumors for EGFR mutation prediction. This two‐stage approach minimizes the influence of non‐tumor tissues on WSI analysis and allows the model to focus on learning the characteristics of the tumor region. The developed model showed superior performance to existing MIL‐based models (Table 4). This suggests that the patch‐masking scheduler strategy is effective for our dataset. Future studies with larger cohorts, including more lepidic cases, would be needed to further investigate the relationship between the lepidic subtype and EMP score predicted by the AI model.
When the allele frequency of EGFR mutations is low, other pathways are more likely to be involved in tumor pathogenesis. Therefore, at the treatment strategy development stage, it is necessary to test for alterations in other druggable targets in addition to EGFR. Previous studies have focused primarily on predicting the presence of EGFR mutations. However, our study predicted the EMP score, that is, the degree of EMP, using an AI model. By utilizing this, medical professionals can predesign the scope of genetic testing for patients, preventing unnecessary medical expenses. With future technological advancements, personalized treatment strategies can be offered to patients.
Our study has some limitations. First, we trained the model using only Korean lung cancer tissue data, which does not reflect the characteristics of diverse races and regions. Because the histopathological patterns of lung cancer may vary according to race and region, validation using data from other countries and races is necessary. However, our study demonstrated some degree of generalizability using data collected from four large institutions.
Second, we confirmed that the deep learning model identified EGFR mutation regions differently depending on the EGFR subtype (exon 19 del, p.L858R) and size and number of patches. This suggests that each EGFR subtype has distinct histopathological characteristics that may manifest differently depending on patch size. To elucidate the clinical significance of these findings, future studies will focus on classifying EGFR mutation subtypes based on the histopathological features differentially identified by the deep learning model at various patch size levels. Third, we measured the prevalence ratio of EGFR mutations in the entire tumor region and compared it with VAF and histopathological patterns. However, considering the intratumoral genetic heterogeneity, it is important to examine the distribution of mutations at the single‐cell level. A comparison of the genetic profiles measured at the individual cell level by single‐cell sequencing with the regions of EGFR mutation presence predicted by our model could further strengthen the utility of the model. However, the application of current single‐cell sequencing technologies to large‐scale analyses is difficult owing to cost and technical limitations. Nevertheless, we anticipate that our study, which utilizes deep learning to explore tumor heterogeneity at the single‐cell level, will serve as a foundation for future research in this area.
In conclusion, the proposed method has great potential for mutation prediction from WSIs using DL. The prediction of EGFR mutation status demonstrates the possibility of its use as a rapid and primary screening tool in clinical settings, particularly in scenarios where tissue samples for molecular testing are unavailable. Additionally, the method used to measure the prevalence of EGFR mutations in the entire tissue has the potential to establish prognosis and treatment plans for patients with EGFR mutant tumors. The learning methods and models we propose are effective in considering various aspects of genomic analysis in WSIs and can be utilized in various WSI analyses.
Author contributions statement
JHP, JH and JR conceived and designed the analysis. JHP, JHyL, SHK, C‐HK, J‐SC, JHeL, LK, JWC, DP, SK, I‐SP, SHH, ES and JR collected the data. JHP and JH contributed to data or analysis tools. JHP performed the analysis. JHP and JH wrote the paper. JH and JR edited the manuscript.
Acknowledgements
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant numbers: HR21C1003 and HR22C1734). This work was supported by research fund of Ajou University Medical Center (2023).
No conflicts of interest were declared.
Contributor Information
Jin Roh, Email: jin.roh@aumc.ac.kr.
Jaesung Heo, Email: nahero@ajou.ac.kr.
Data availability statement
The datasets generated and/or analyzed in this study are available from the corresponding author upon reasonable request.
References
- 1. Lazcanoiturburu N, García‐Sáez J, González‐Corralejo C, et al. Lack of EGFR catalytic activity in hepatocytes improves liver regeneration following DDC‐induced cholestatic injury by promoting a pro‐restorative inflammatory response. J Pathol 2022; 258: 312–324. [DOI] [PubMed] [Google Scholar]
- 2. Fontugne J, Wong J, Cabel L, et al. Progression‐associated molecular changes in basal/squamous and sarcomatoid bladder carcinogenesis. J Pathol 2023; 259: 455–467. [DOI] [PubMed] [Google Scholar]
- 3. Pastorino GA, Sheraj I, Huebner K, et al. A partial epithelial‐mesenchymal transition signature for highly aggressive colorectal cancer cells that survive under nutrient restriction. J Pathol 2024; 262: 347–361. [DOI] [PubMed] [Google Scholar]
- 4. Gonzalez‐Sanchez E, Vaquero J, Caballero‐Diaz D, et al. The hepatocyte epidermal growth factor receptor (EGFR) pathway regulates the cellular interactome within the liver fibrotic niche. J Pathol 2024; 263: 482–495. [DOI] [PubMed] [Google Scholar]
- 5. O'Leary C, Gasper H, Sahin KB, et al. Epidermal growth factor receptor (EGFR)‐mutated non‐small‐cell lung cancer (NSCLC). Pharmaceuticals (Basel) 2020; 13: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura T, Nishikawa Y, Shiokawa M, et al. ELF3 suppresses gallbladder cancer development through downregulation of the EREG/EGFR/mTOR complex 1 signalling pathway. J Pathol 2023; 261: 28–42. [DOI] [PubMed] [Google Scholar]
- 7. Liu F, Hon GC, Villa GR, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell 2015; 60: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park K, Tan E‐H, O'Byrne K, et al. Afatinib versus gefitinib as first‐line treatment of patients with EGFR mutation‐positive non‐small‐cell lung cancer (LUX‐Lung 7): a phase 2B, open‐label, randomised controlled trial. Lancet Oncol 2016; 17: 577–589. [DOI] [PubMed] [Google Scholar]
- 9. Fu K, Xie F, Wang F, et al. Therapeutic strategies for EGFR‐mutated non‐small cell lung cancer patients with osimertinib resistance. J Hematol Oncol 2022; 15: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li T, Kung HJ, Mack PC, et al. Genotyping and genomic profiling of non‐small‐cell lung cancer: implications for current and future therapies. J Clin Oncol 2013; 31: 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rusch M, Nakitandwe J, Shurtleff S, et al. Clinical cancer genomic profiling by three‐platform sequencing of whole genome, whole exome and transcriptome. Nat Commun 2018; 9: 3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murchan P, Ó'Brien C, O'Connell S, et al. Deep learning of histopathological features for the prediction of tumour molecular genetics. Diagnostics (Basel) 2021; 11: 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Comes MC, Fucci L, Mele F, et al. A deep learning model based on whole slide images to predict disease‐free survival in cutaneous melanoma patients. Sci Rep 2022; 12: 20366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dehkharghanian T, Bidgoli AA, Riasatian A, et al. Biased data, biased AI: deep networks predict the acquisition site of TCGA images. Diagn Pathol 2023; 18: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dolezal JM, Wolk R, Hieromnimon HM, et al. Deep learning generates synthetic cancer histology for explainability and education. NPJ Precis Oncol 2023; 7: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo B, Li X, Yang M, et al. A robust and lightweight deep attention multiple instance learning algorithm for predicting genetic alterations. Comput Med Imaging Graph 2023; 105: 102189. [DOI] [PubMed] [Google Scholar]
- 17. Jiang S, Suriawinata AA, Hassanpour S. MHAttnSurv: multi‐head attention for survival prediction using whole‐slide pathology images. Comput Biol Med 2023; 158: 106883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pan Y, Sheng W, Shi L, et al. Whole slide imaging‐based deep learning to predict the treatment response of patients with non‐small cell lung cancer. Quant Imaging Med Surg 2023; 13: 3547–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan R, Shen Y, Zhang X, et al. Histopathological bladder cancer gene mutation prediction with hierarchical deep multiple‐instance learning. Med Image Anal 2023; 87: 102824. [DOI] [PubMed] [Google Scholar]
- 20. Pao JJ, Biggs M, Duncan D, et al. Predicting EGFR mutational status from pathology images using a real‐world dataset. Sci Rep 2023; 13: 4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang Y, Ma S, Xiao W, et al. Predicting EGFR gene mutation status in lung adenocarcinoma based on multifeature fusion. Biomed Signal Process Control 2023; 84: 104786. [Google Scholar]
- 22. Ilse M, Tomczak J, Welling M. Attention‐based deep multiple instance learning. In Proceedings of the 35th International Conference on Machine Learning. Jennifer D, Andreas K, (ed). Proceedings of Machine Learning Research, 2018; 2127‐2136.
- 23. Quellec G, Cazuguel G, Cochener B, et al. Multiple‐instance learning for medical image and video analysis. IEEE Rev Biomed Eng 2017; 10: 213–234. [DOI] [PubMed] [Google Scholar]
- 24. Shao Z, Bian H, Chen Y, et al. TransMIL: transformer based correlated multiple instance learning for whole slide image classification. In: Proceedings of the 35th International Conference on Neural Information Processing Systems. Curran Associates Inc., 2021; 164. [Google Scholar]
- 25. Li B, Li Y, Eliceiri KW. Dual‐stream multiple instance learning network for whole slide image classification with self‐supervised contrastive learning. Conf Comput Vis Pattern Recognit Workshops 2021; 2021: 14318–14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu MY, Williamson DFK, Chen TY, et al. Data‐efficient and weakly supervised computational pathology on whole‐slide images. Nat Biomed Eng 2021; 5: 555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jang HJ, Lee A, Kang J, et al. Prediction of genetic alterations from gastric cancer histopathology images using a fully automated deep learning approach. World J Gastroenterol 2021; 27: 7687–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qu H, Zhou M, Yan Z, et al. Genetic mutation and biological pathway prediction based on whole slide images in breast carcinoma using deep learning. NPJ Precis Oncol 2021; 5: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duchon CE. Lanczos filtering in one and two dimensions. J Appl Meteorol Climatol 1979; 18: 1016–1022. [Google Scholar]
- 30. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017; 7: 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khened M, Kori A, Rajkumar H, et al. A generalized deep learning framework for whole‐slide image segmentation and analysis. Sci Rep 2021; 11: 11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Otsu N. A threshold selection method from gray‐level histograms. IEEE Trans Syst Man Cybern 1979; 9: 62–66. [Google Scholar]
- 33. Boschman J, Farahani H, Darbandsari A, et al. The utility of color normalization for AI‐based diagnosis of hematoxylin and eosin‐stained pathology images. J Pathol 2022; 256: 15–24. [DOI] [PubMed] [Google Scholar]
- 34. Macenko M, Niethammer M, Marron JS, et al. A method for normalizing histology slides for quantitative analysis. In IEEE International Symposium on Biomedical Imaging: From Nano to Macro, 2009; 1107–1110.
- 35. Shen Y, Luo Y, Shen D, et al. RandStainNA: learning stain‐agnostic features from histology slides by bridging stain augmentation and normalization. In: Medical Image Computing and Computer Assisted Intervention – MICCAI 2022, Wang L, Dou Q, Fletcher PT, et al. (Eds). Springer Nature Switzerland: Cham, 2022; 212–221. [Google Scholar]
- 36. Oquab M, Darcet T, Moutakanni T. et al. DINOv2: learning robust visual features without supervision. arXiv preprint arXiv 2023; abs/2304.07193.
- 37. Wessels F, Schmitt M, Krieghoff‐Henning E, et al. A self‐supervised vision transformer to predict survival from histopathology in renal cell carcinoma. World J Urol 2023; 41: 2233–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Z, Cong Y, Chen X, et al. Vision transformer‐based weakly supervised histopathological image analysis of primary brain tumors. iScience 2023; 26: 105872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han C, Pan X, Yan L, et al. WSSS4LUAD: grand challenge on weakly‐supervised tissue semantic segmentation for lung adenocarcinoma. arXiv preprint arXiv 2022; abs/2204.06455.
- 40. Tang W, Huang S, Zhang X, et al. Multiple instance learning framework with masked hard instance Mining for Whole Slide Image Classification. In Proceedings of the IEEE/CVF International Conference on Computer Vision (ICCV), 2023.
- 41. Coudray N, Ocampo PS, Sakellaropoulos T, et al. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat Med 2018; 24: 1559–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Campanella G, Hanna MG, Geneslaw L, et al. Clinical‐grade computational pathology using weakly supervised deep learning on whole slide images. Nat Med 2019; 25: 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li WQ, Cui JW. Non‐small cell lung cancer patients with ex19del or exon 21 L858R mutation: distinct mechanisms, different efficacies to treatments. J Cancer Res Clin Oncol 2020; 146: 2329–2338. [DOI] [PubMed] [Google Scholar]
- 44. Boukansa S, Benbrahim Z, Gamrani S, et al. Correlation of epidermal growth factor receptor mutation with major histologic subtype of lung adenocarcinoma according to IASLC/ATS/ERS classification. Cancer Control 2022; 29: 10732748221084930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang H, Zhu M, Li Y, et al. Association between EGFR exon 19 or exon 21 mutations and survival rates after first‐line EGFR‐TKI treatment in patients with non‐small cell lung cancer. Mol Clin Oncol 2019; 11: 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kadota K, Yeh YC, D'Angelo SP, et al. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am J Surg Pathol 2014; 38: 1118–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ninomiya H, Hiramatsu M, Inamura K, et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas: significance of the micropapillary pattern and the hobnail cell type. Lung Cancer 2009; 63: 235–240. [DOI] [PubMed] [Google Scholar]
- 48. Saito R, Ninomiya H, Okumura S, et al. Novel histologic classification of small tumor cell nests for lung adenocarcinoma with prognostic and etiological significance: small solid nests and pure micropapillary nests. Am J Surg Pathol 2021; 45: 604–615. [DOI] [PubMed] [Google Scholar]
- 49. Miller VA, Kris MG, Shah N, et al. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non–small‐cell lung cancer. J Clin Oncol 2004; 22: 1103–1109. [DOI] [PubMed] [Google Scholar]
- 50. Ebright MI, Zakowski MF, Martin J, et al. Clinical pattern and pathologic stage but not histologic features predict outcome for bronchioloalveolar carcinoma. Ann Thorac Surg 2002; 74: 1640–1646; discussion 1646‐1647. [DOI] [PubMed] [Google Scholar]
- 51. Dagogo‐Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018; 15: 81–94. [DOI] [PubMed] [Google Scholar]
- 52. Guo L, Chen Z, Xu C, et al. Intratumoral heterogeneity of EGFR‐activating mutations in advanced NSCLC patients at the single‐cell level. BMC Cancer 2019; 19: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen C‐L, Chen C‐C, Yu W‐H, et al. An annotation‐free whole‐slide training approach to pathological classification of lung cancer types using deep learning. Nat Commun 2021; 12: 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pisula JI, Datta RR, Valdez LB, et al. Predicting the HER2 status in oesophageal cancer from tissue microarrays using convolutional neural networks. Br J Cancer 2023; 128: 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanavati F, Toyokawa G, Momosaki S, et al. Weakly‐supervised learning for lung carcinoma classification using deep learning. Sci Rep 2020; 10: 9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang S, Zhu Y, Yu L, et al. RMDL: recalibrated multi‐instance deep learning for whole slide gastric image classification. Med Image Anal 2019; 58: 101549. [DOI] [PubMed] [Google Scholar]
- 57. Ayana G, Dese K, Dereje Y, et al. Vision‐transformer‐based transfer learning for mammogram classification. Diagnostics (Basel) 2023; 13: 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen RJ, Chen C, Li Y, et al. Scaling vision transformers to gigapixel images via hierarchical self‐supervised learning. In 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2022; 16123–16134.
- 59. Dosovitskiy A, Beyer L, Kolesnikov A, et al. An image is worth 16x16 words: transformers for image recognition at scale. arXiv preprint arXiv 2020; abs/2010.11929.
- 60. Alexey D, Lucas B, Alexander K, et al. An image is worth 16x16 words: transformers for image recognition at scale. In International Conference on Learning Representations, 2021.
- 61. Kang M, Song H, Park S, et al. Benchmarking self‐supervised learning on diverse pathology datasets. In 2023 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2023; 3344–3354.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed in this study are available from the corresponding author upon reasonable request.