

Abstract
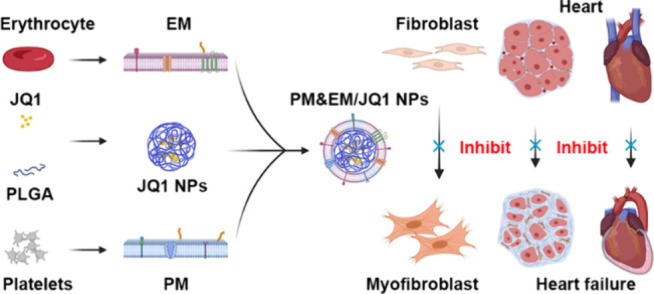
Cardiac fibrosis is a prevalent pathological process observed in the progression of numerous cardiovascular diseases and is associated with an increased risk of sudden cardiac death. Although the BRD4 inhibitor JQ1 has powerful antifibrosis properties, its clinical application is extremely limited due to its side effects. There remains an unmet need for effective, safe, and low-cost treatments. Here, we present a multifunctional biomimetic nanoparticle drug delivery system (PM&EM nanoparticles) assembled by platelet membranes and erythrocyte membranes for targeted JQ1 delivery in treating cardiac fibrosis. The platelet membrane endows PM&EM nanoparticles with the ability to target cardiac myofibroblasts and collagen, while the participation of the erythrocyte membrane enhances the long-term circulation ability of the formulated nanoparticles. In addition, PM&EM nanoparticles can deliver sufficient JQ1 with controllable release, achieving excellent antifibrosis effects. Based on these advantages, it is demonstrated in both pressures overloaded induced mouse cardiac fibrosis model and MI-induced mouse cardiac fibrosis that injection of the fusion membrane biomimetic nanodrug carrier system effectively reduced fibroblast activation, collagen secretion, and improved cardiac fibrosis. Moreover, it significantly mitigated the toxic and side effects of long-term JQ1 treatment on the liver, kidney, and intestinal tract. Mechanically, bioinformatics prediction and experimental validation revealed that PM&EM/JQ1 NPs reduced liver and kidney damage via alleviated oxidative stress and mitigated cardiac fibrosis via the activation of oxidative phosphorylation activation. These results highlight the potential value of integrating native platelet and erythrocyte membranes as a multifunctional biomimetic drug delivery system for treating cardiac fibrosis and preventing drug side effects.
Keywords: platelet membrane, erythrocyte membrane, hybrid membrane, nanotherapy, cardiac fibrosis, heart failure
Introduction
Cardiac fibrosis is a characteristic feature of numerous chronic cardiovascular diseases, ultimately leading to heart failure (HF).1 Although heart failure resulting from severe cardiac fibrosis can be treated through heart transplantation, the scarcity of donors and the extremely low success rate of transplantation significantly constrain the treatment options for cardiac fibrosis.2 Consequently, there exists an urgent demand for the development of safe, effective, and affordable targeted therapies that alleviate the burden of heart disease on patients and healthcare systems.
Previous studies have highlighted the epigenetic reader protein bromodomain-containing protein 4 (BRD4) as a promising target for cancer treatment. Additionally, the BET bromine domain inhibitor JQ1 has demonstrated a therapeutic effect in mice with HF resulting from long-term stress overload or extensive anterior myocardial infarction (MI).3 JQ1 exhibited strong therapeutic potential in treating and reversing fibrotic diseases.4 However, the therapeutic effect of JQ1 is hampered by its relatively narrow therapeutic window (i.e., 50% effective dose/50% lethal dose, ED50/LD50). When administered orally or intravenously, JQ1 can cause a broad systemic effect by inhibiting bromodomain and extraterminal domain (BET) proteins, which is antifibrotic but may cause systemic multisystem damage.5,6 Therefore, there is an urgent need for a targeted drug delivery system that enhances JQ1 accumulation in fibrosed cardiac tissue while minimizing its toxicity.
Recently, a class of drug nanocarriers, fabricated by combining synthetic nanoparticle cores with biologically derived membrane coatings, has been reported.7,8 These biomimetic cell membrane-coated nanoparticles harden the versatility and complexity of natural cell membranes, which play crucial roles in specific functions, particularly at biological interfaces. By transfer of the entire cell membrane to the surface of the nanoparticle, all biologically relevant surface fractions are preserved. These include features that may be utilized for immune evasion and targeting, which are highly desirable for the development of targeted drug delivery systems.9,10 Furthermore, cell-specific functions that are known to exist but have not been adequately characterized at a basic biological level can be exploited. These nanocarriers were pioneers in using erythrocytes (red blood cells) to prolong blood residency time, and these cell membrane-coated nanoparticles serve as immunomodulatory markers with the same density as the original erythrocytes, such as CD47.11 Beyond erythrocytes, other cell types, including platelets, white blood cells, cancer cells, stem cells, and even bacteria, possess their properties as membrane materials.10,12,13
While cell membrane coating represents a powerful approach to enhancing the therapeutic value of nanoparticles, additional functionality may be required depending on the intended application.8,14 For example, although erythrocyte membrane-coated nanoparticles can circulate for extended periods of time, the addition of targeted ligands can further improve their localization to desired sites, such as areas affected by fibrosis. This article describes a method that expands the repertoire of nanocarrier functions by simultaneously incorporating functional membranes from multiple cell types.15 This approach involves the fusion of native cell membranes from different origins and is a simple but effective method that can be used to create nanoparticles capable of executing increasingly complex tasks in biologically relevant environments. Specifically, by combing the functions of human-origin platelets and erythrocytes, the resulting platelet membrane- erythrocytes membrane complex (PM&EM) retained the functions of each cell type when coated onto nanoparticles. Furthermore, after encapsulation of an antifibrosis agent JQ1, the resulting drug-loaded nanoparticles (PM&EM NPs, Table S1) achieved excellent targeting ability and an antifibrotic effect in mouse models of pressure overload-induced and MI-induced cardiac fibrosis. Toxicity investigation of JQ1 in other organs showed that these PM&EM NPs effectively reduced JQ1 accumulation in the liver and kidney, attenuating the adverse effects. We anticipated that this biomimetic nanoparticle delivery system holds great promise as an effective and safe strategy for treating cardiac fibrosis in clinical settings (Figure 1).
Figure 1.

Schematic design of PM&EM/JQ1 NP-mediated delivery of JQ1 to myofibroblast for the treatment of heart failure. (a) Schematic illustration of the preparation of PM&EM/JQ1 NPs. (b) Schematic diagram of intravenous injection of PM&EM/JQ1 NPs to the heart failure mice.
Results and Discussion
Preparation and Characterization of PM&EM/JQ1 NPs
Platelet membranes were selected as ideal coating membranes due to their rich array of functional proteins (e.g., glycoproteins IIb/IIIa and VI), which can effectively target collagen and collagen-secreting activated fibroblasts.16 Moreover, previous studies have reported that erythrocyte membrane-coated nanoparticles can circulate in the bloodstream for extended periods.17 Thus, in this study, the functions of human erythrocytes and platelets were combined to fabricate platelet-erythrocyte hybrid membrane-coated nanoparticles loaded with JQ1 (PM&EM/JQ1 NPs).
To prepare PM&EM/JQ1 NPs, JQ1-loaded PLGA nanoparticles (JQ1 NPs) were first prepared, after which the membranes of platelets and erythrocytes were extracted using the hypotonic lysis method. In this study, platelet and erythrocyte membranes were fused at a protein dry weight ratio of 4:1 using ultrasound, which has been shown to maintain efficient membrane function after fusion of these membranes.18
To monitor the fusion process, a dye pair with Förster resonance energy transfer (FRET) property, DiO and Dil, was used to label the PM&EM NPs,15 followed by fusion with unlabeled erythrocyte membranes. After the addition of the erythrocyte membrane, the fluorescence intensity at 555 nm (Dil) decreased (Figure 2a), indicating that the platelet membrane had fused with the erythrocyte membrane, weakening the fluorescence signal of Dil (the acceptor dye). Furthermore, the platelet membranes and erythrocyte membranes were labeled with DiO and DiD respectively, and coated onto DiI fluorescent dye-loaded PLGA nanoparticles. Dynamic light scattering (DLS) analysis showed that following the hybrid membrane coating step, the diameter of the measured nanoparticles increased from ≈182.6 to ≈195.0 nm, which corresponds to the thickness of the coated biological bilayer cell membranes (Figure 2b). Zeta potentials of the platelet membrane vesicles, erythrocyte membrane vesicles, JQ1 nanoparticles, and PM&EM/JQ1 NPs were −25.73 ± 1.54, – 35.1 ± 1.96, – 16.83 ± 0.544, and −27.8 ± 1.45 mV, respectively, and the zeta potential of the PM&EM/JQ1 NPs was between that of the platelet membrane vesicles and the erythrocyte membrane vesicles (Figure2c), indicating successful fusion of the platelet membranes with the erythrocyte membranes. Transmission electron microscopy (TEM) images further confirmed the “core-shell” structure of the PM&EM/JQ1 NPs (Figure 2d). Under confocal laser scanning microscopy (CLSM), these three raw materials showed almost no overlay when they were only mixed physically, while an effective overlap was observed when assembled by ultrasonic mixing (Figure 2e). Together, these results demonstrate the successful coating of the PLGA cores with the extracted platelet and erythrocyte membranes.
Figure 2.

Preparation and characterization of PM&EM/JQ1 NPs. (a) The lipid membrane is labeled with FRET dye for DiO and Dil and then fused with the platelet membrane. The fluorescence intensity was recorded. (b) The particle size was measured by dynamic light scattering (DLS) (mean ± SD, n = 3). (c) Zeta potential distribution (mean ± SD, n = 3 independent experiments) of platelet membrane vesicles, erythrocyte membrane vesicles, JQ1 NPs, and PM&EM/JQ1 NPs. (d) Representative TEM image of PM&EM/JQ1 NPs. Scale bar = 100 nm. (e) CLSM image of PM&EM/JQ1 NPs. Two membranes and a physical mixture of PLGA coated with DiI fluorescein were used as controls. Scale bar = 20 μm. (f) Principal component analysis (PCA) and the 2D score plots display repertoires of PM vesicles, EM vesicles, and PM&EM vesicles. Each point represents a sample, and ellipses represent 95% confidence regions (mean ± SD, n = 3 independent experiments). (g) Classification of PM&EM/JQ1 NPs proteins by the biological process. (h) Classification of PM&EM/JQ1 NP proteins by cellular component. (i) Heatmap of protein levels from PM&EM NPs, PM NPs, and EM NPs. (mean ± SD, n = 3). (j) Protein composition of PM&EM/JQ1 NPs was shown by SDS-PAGE analysis. (k) Analysis of PM&EM/JQ1 NPs through Western Blot to achieve targeting and immune evasion of key protein receptors.
The protein composition of the platelet and erythrocyte membrane vesicles was determined by 3D-data independent acquisition (DIA). Principal component analysis (PCA) is a mathematical procedure used to reduce the dimensionality of data by transforming the data into principal components through an orthogonal transformation, thereby preserving the majority of the variance in the data set.19 PCA results indicated that PM&EM hybrid vesicles exhibited distinct differences compared to both PM vesicles and EM vesicle groups (Figure 2f). Figure S1 shows specific protein counts for PM vesicles and EM vesicles compared to PM&EM vesicles, highlighting the up-regulated and down-regulated proteins. By searching the UniProt database, a total of 4872 membrane proteins were identified in PM&EM hybrid vesicles. A functional classification of the identified proteins based on the biological process (Figure 2g) involved in localization (613), cellular component organization biogenesis (98), single-organism process (60), positive regulation of biological process (36), multicellular organismal process (23), cellular process (8), response to stimulus (8), metabolic process (5), biological regulation (3), muti-organism process (2), negative regulation of biological process (2), and others (71). The membrane proteins were also classified according to cellular component (Figure 2h): membrane-enclosed lumen (1000), organelle (549), extracellular region (255), cell junction (248), synapse (133), supramolecular complex (99), macromolecular complex (76), cell part (60), organelle part (27), other organism parts (23), extracellular region part (18), membrane (15), nucleid (8), supramolecular fiber (2), cell component (2238), and others (61). Heatmap analysis of the levels of 523 proteins revealed notable differences between PM vesicles and PM&EM vesicles and 2771 between EM and PM&EM vesicles. Encouragingly, the hybrid membrane inherited most of the proteins from both the platelet and erythrocyte membranes (Figure 2i). Ponceau S staining further confirmed that the PM&EM/JQ1 NPs contained most of the cell membrane proteins, similar to those in platelet and erythrocyte membrane vesicles, while the PLGA-encapsulated JQ1 did not show the expected band of protein from cell membranes (Figure2j). To further investigate whether PM&EM/JQ1 NPs can retain the key physiological characteristics of platelets and erythrocytes, Western blotting was used to analyze membrane proteins targeting collagen (glycoproteins IIb/IIIa and VI) and membrane proteins involved in immune evasion (CD47). As expected, following the derivation and fusion of the membranes, the PM&EM/JQ1 NPs were found to contain both platelets and erythrocyte membrane proteins (Figure 2k). These findings demonstrate the successful fusion of platelet and erythrocyte membranes, preserving the key physiological characteristics of the source platelets and erythrocytes.
By scanning the full spectrum of JQ1, it can be found that the largest difference in absorption peaks between JQ1 and DMSO occurred at 320 nm (Figure S2a). The drug loading rate (LE) and encapsulation rate (EE) of PM&EM/JQ1 NPs were 3.58 ± 0.27 and 37.15 ± 2.9%, respectively. To study the release kinetics of JQ1 from nanoparticles, we placed the JQ1 NPs and PM&EM/JQ1 NPs in 20% fetal bovine serum (FBS). After 62 h incubation, the measured JQ1 release rates were 76.31 ± 4.94 and 67.19 ± 0.65%, suggesting that most of JQ1 can be released at the site of fibrosis after injection (Figure S2b). In addition, DLS was employed to measure the particle size of PM&EM/JQ1 NPs in water and a medium containing 10% FBS at room temperature for 72 h. The findings indicated a trend toward decreased particle size both in water and 10% FBS, suggesting potential instability of PM&EM/JQ1 NPs during the incubation process (Figure S2c).
Targeting and Immune Evasion In Vitro
The ability of PM&EM nanoparticles to target collagen in vitro was examined to determine whether the biomimetic nanoparticles can retain the advantages of both erythrocyte and platelet membranes. After 4 h of coincubation with collagen in vitro, CLSM showed that platelet-erythrocyte hybrid membrane-coated nanoparticles loaded with DiI (PM&EM/DiI NPs) adhered to collagen more efficiently than DiI NPs (Figure 3a).
Figure 3.

Targeted delivery and immune evasion performance of PM&EM/JQ1 NPs. (a) After 4h incubation with in vitro collagen, the ability of collagen to adhere to DiI NPs, PM&EM/JQ1 NPs, gray is collagen, red is encapsulated DiI fluorescent molecules, scale bar = 10 μm. (b) Fibroblasts and their collagen production stimulated by angiotensin II, green for type I collagen, blue for nuclei; scale bar = 20 μm. (c) After 4 h incubation of activated fibroblasts (stimulated with angiotensin II), cell adhesion, uptake of DiI nanoparticles, PM&EM/JQ1 NPs scale bar = 25 μm. (d) FC analysis of activated fibroblasts after 4 h incubation of DiI NPs, PM&EM/JQ1 NPs, and DiI-positive fibroblasts as a percentage of total fibroblasts (mean ± SD, n = 4 independent experiments). (e) RAW 264.7 cells were coincubated with DiI NPs and PM&EM/JQ1 NPs nanoparticles for 4 h and then subjected to laser confocal analysis, blue for nuclei, green for lysosomes, red for encapsulated DiI fluorescent molecules, scale bar = 25 μm. (f) DiI-positive macrophages as a percentage of total macrophages after coincubation with DiI-labeled nanoparticles for 4 h (mean ± SD, n = 3 independent experiments). ***p < 0.001, one-way ANOVA, Tukey’s multiple comparison test.
Mouse embryonic fibroblasts stimulated with angiotensin II can mimic cardiac fibroblast activation after HF. The targeting effect of PM&EM/DiI NPs on these activated fibroblasts (myofibroblast) was then evaluated. In detail, after stimulation of fibroblasts with angiotensin II, there was a significant increase in the expression of collagen I (Figure 3b). After incubating DiI NPs and PM&EM/DiI NPs with activated fibroblasts for 4 h, the nanoparticles aggregated within cells. The number of nanoparticles within myofibroblast was much higher in the PM&EM/DiI NP group (Figure 3c). Further flow cytometry (FC) analysis confirmed that myofibroblasts exhibited approximately 30% higher uptake efficiency for PM&EM/DiI NPs compared to DiI NPs (Figure 3d). These results suggest that our hybrid membrane-encapsulated nanoparticles have enhanced adhesion and affinity for myofibroblast.
Next, we tested the ability of PM&EM/DiI NPs to evade cellular uptake by macrophages in vitro. After 4 h of incubation with RAW 264.7 cells, cellular uptake of PM&EM/DiI NPs was monitored by CLSM. As shown in Figure 3e, fewer PM&EM/DiI NPs were observed within macrophages compared to PM&EM/DiI nanoparticle-treated and PLGA nanoparticle-treated cells. Furthermore, LysoTracker staining revealed that most of the internalized DiI nanoparticles were localized within lysosomes. These results suggest that CD47 proteins, derived from erythrocyte membranes, effectively prevented PM&EM/DiI NPs from being phagocytosed by macrophages (Figure 3f).
Biosafety Assignment of PM&EM Nanoparticles
To assess the hemocompatibility of biological materials, it is important to evaluate their blood compatibility. In this study, we employed an in vitro direct contact method to detect the hemolysis of PLGA nanoparticles and PM&EM NPs. As shown in Figure S3a, the results indicated that after incubating the PLGA nanoparticles and PM&EM NPs with blood, there were no abnormal phenomena or particle sediment in the supernatant, suggesting that the red blood cells were not destroyed (Figure S3b).
To assess the biological effect of PM&EM NPs, we incubated them with fibroblasts, endothelial cells (ECs), smooth muscle cells (SMCs), and macrophages, along with PLGA nanoparticles. The cytotoxicity of these nanoparticles was then investigated using the Cell Counting Kit-8 (CCK-8) assay. The results, as shown in Figure S3c–f, indicated that there was no significant difference between the different treatment groups and the untreated group, suggesting good cytocompatibility of PM&EM NPs.
Moreover, clinical biochemistry analysis demonstrated that PLAG nanoparticles and PM&EM NPs did not affect liver and kidney functions, as evidenced by normal levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and serum creatinine (CRE) (Figure S4a–d). Furthermore, testing of mouse serum cytokines (TNF-α, IL1β, IL6, and CCL2) revealed no significant difference between the treatment groups, indicating that our drug delivery system does not cause obvious side effects in vivo (Figure S5a–d).
H&E staining was performed on pathological sections of various organs. After comparing the H&E-stained sections of different main organs from MI-induced HF mice. we found no significant difference between the control group and different treatment groups (Figure S6). This suggests high biocompatibility of PLGA nanoparticles and PM&EM nanoparticles after long-term treatment in MI-induced HF mice.
In this part, we used MI-induced HF mice. Mice are sacrificed on the 30th day after MI. This is also a drawback that we did not conduct more long-term safety studies here due to constraints.
Targeting Research In Vivo
Next, we investigated the in vivo targetability of PM&EM-coated nanoparticles to the site of cardiac fibrosis in a pressure overload-induced HF mouse model. The adult mice were subjected to transverse aortic constriction (TAC) surgery. The same numbers of DiI NPs and PM&EM/DiI NPs were injected intravenously into the pressure overload mice. After 24 h, heart tissue was collected for fluorescence imaging. In the hearts of the DiI NP group, faint fluorescence signals were observed, and the amount of DiI dye was significantly higher in PM&EM/DiI NPs than in DiI NPs In addition, DiI was mainly distributed in the infarcted area, with little enrichment in the remote region (Figure4a). Further, we also measured the accumulation of PM&EM NPs in the liver and kidney. The fluorescence of the PM&EM/DiI NP group was also significantly lower than that of the DiI NP group in the kidney and liver tissue sections of mice (Figure 4b). In addition, we also detected fluorescence signals of DiD in other major organs 24 h after intravenous injection, mainly in the liver and kidneys. This may reflect the clearance of the mononuclear phagocyte system (MPS) in the liver and kidney when nanoparticles are directly injected into the bloodstream. Interestingly, unlike the enrichment of the DiD fluorescence signal in the heart, the DiD NP group exhibited a large amount of fluorescence signal, while the amount of DiD dye in PM&EM/DiD NPs was significantly reduced (Figure 4c–e). This finding indicates that PM&EM NPs have better targetability to fibrotic hearts without accumulating excessively in other tissues, such as the liver and kidney. This selective targeting may be related to glycoproteins IIb/IIIa and VI acquired from the platelet membranes on PM&EM NPs and CD47 inherited from the erythrocyte membranes. For testing the release rates and stability of NPs under physiological conditions, after the tail vein of the mouse was injected with DiD-labeled NPs, we took the whole blood of the mouse at different time points (0, 0.5, 1, 2, 4, 8, 12, 24, and 48 h) for fluorescence intensity detection. The results showed that compared with the DiD NP group, the PM&EM/DiD NP group had stronger sustained release ability and long circulation ability (Figure S7).
Figure 4.

PM&EM-NP-mediated delivery to the heart failure mice and biodistribution in other organs. (a) CLSM images of different DiI-labeled nanoparticles at fibrotic sites of the heart in the heart failure mice at 24 h postintravenous injection, scale bar = 5 μm, 20 μm. (b) CLSM images of different DiI-labeled nanoparticles in kidney and liver of the heart failure mice at 24 h postintravenous injection, scale bar = 15 μm. (c) Representative ex vivo fluorescence images of DiD fluorescence dye accumulated in different organs at 24 h postintravenous injection of DiD-labeled nanoparticles. (d) Heatmap of DiD fluorescence dye in different organs (n = 3). (e) Quantitative data of DiD fluorescence dye in the heart (mean ± SD, n = 3) **p < 0.01, one-way ANOVA, Tukey’s multiple comparison test.
Evaluation of Treatment Effects in the TAC-Induced HF Mouse Model
Given that PM&EM nanoparticles were observed to accumulate in fibrotic lesions in mice with TAC-induced HF, we then explored the role of PM&EM/PLGA nanoparticles in treating cardiac fibrosis and improving HF after JQ1 loading. To evaluate the therapeutic efficacy of each formulation (free JQ1, JQ1 NPs, and PM&EM/JQ1 NPs), we first established a model of pressure overload in mice (Figure 5a), based on previous treatment regimens with appropriate improvements. On postoperative day 18, which is the time point when severe cardiac hypertrophy and left ventricular dysfunction are established in this model, mice were randomized to receive daily tail vein injections of 50 mg/kg of bare JQ1 or the various types of nanoparticle formulations. These treatments were continued until postoperative day 45.3
Figure 5.

Evaluation of the therapeutic effect of novel drug-loaded systems in TAC-induced cardiac insufficiency in mice. (a) Experimental protocol and dosing protocol of pre-established TAC-induced mouse stress overload model. (b) Representative plot of mouse cardiac ultrasound 45 days after construction of the TAC model. (c) Left ventricular ejection fraction quantified by echocardiography. (d) Left ventricular minor axis contraction (mean ± SD, n = 6). (e) Quantification of fibrotic area in the sham group, TAC model group, and TAC with different treatment group (mean ± SD, n = 6). (g) Representative cross sections from remote LV stained with Sirius red; scale bar = 100 μm. (f) Statistical plot of changes in cardiomyocytes area stained with wheat germ agglutinin (WGA) (mean ± SD, n = 6) and (h) representative LV cross-section stained with WGA, scale bar = 20 μm. *p < 0.05, **p < 0.01, and ***p < 0.001, one-way ANOVA, Tukey’s multiple comparison test.
A cardiac ultrasound was performed on day 45 of the TAC to assess heart function and contractile capacity. As shown in Figure 5b–d, cardiac function and left ventricular contractility improved in mice treated with bare JQ1, JQ1 NPs, and PM&EM/JQ1 NPs but not in untreated mice. PM&EM/JQ1 NPs significantly improved the ejection fraction and left ventricular short-axis shortening rate in mice with TAC-induced cardiac fibrosis, and cardiac function increased from 48.9 ± 5.16 and 49 ± 5.16 to 58.49 ± 5.5% compared to free JQ1 and JQ1 NPs. The rate of shortening of the left ventricular minor axis also increased from 24.25 ± 3.09 and 24.25 ± 3.09 to 30.37 ± 3.44%. These findings suggest that the prognosis of TAC-induced HF was significantly better in mice that received PM&EM/JQ1 NPs than in those that received free JQ1 or JQ1 NPs.
Sirius red staining was performed on cardiac tissue to assess the degree of cardiac fibrosis. The results showed that the free JQ1 and JQ1 NPs were able to improve the degree of fibrosis in mice to some extent. However, injection of PM&EM/JQ1NPs significantly reduced the collagen content by 6.23 ± 1.18% (Figure 5e,g). PM&EM/JQ1 NPs were more effective in reducing the degree of fibrosis in the heart than free JQ1 or JQ1 nanoparticles.
The process of cardiac fibrosis is often accompanied by hypertrophy of cardiomyocytes, which typically indicates a worse prognosis.20 Assessment of the size of cardiomyocytes by wheat germ agglutinin staining revealed that, compared with the sham surgery group, cardiomyocyte size increased by an average of 1.84 ± 0.23 times in the mice with TAC-induced fibrosis in the free JQ1 or JQ1 NP groups. However, it increased by only 1.15 ± 0.02 times in the mice treated with PM&EM/JQ1 NPs (Figure 5f,h). Therefore, PM&EM/JQ1 NPs were more effective in reducing hypertrophy of cardiomyocytes than free JQ1 or JQ1 NPs.
Evaluation of Treatment Effects in the MI-induced HF Mouse Model
The TAC model is commonly used to study HF and cardiac fibrosis. However, it is important to note that the acute onset of severe hemodynamic overload in this model does not accurately represent the clinical situation typically encountered in human patients.21 On the other hand, MI is a frequent cause of pathological cardiac remodeling and HF in humans.22 Furthermore, animal models of HF after MI have become the main avenue for exploration of the role of PM&EM/JQ1 NPs in the treatment of cardiac fibrosis and their ability to improve HF. Referencing a previous regimen and making appropriate improvements, we established an MI-induced mouse model of HF by administering 25 mg/kg of free JQ1 or JQ1 encapsulated in the various nanoparticle formulations via the tail vein on postoperative day 6 for 6 days (i.e., the early postoperative period). The dosage was then gradually increased to 50 mg/kg per day until postoperative day 30 (Figure6a).
Figure 6.

Evaluation of the therapeutic effect of novel drug-loaded systems in MI-induced cardiac insufficiency in mice. (a) Experimental protocol and dosing protocol of MI-induced mouse cardiac insufficiency model. (b) Representative plot of mouse cardiac ultrasound 30 days after the construction of the MI model. (c) Left ventricular ejection fraction quantified by echocardiography. (d) Left ventricular minor axis contraction (mean ± SD, n = 6). (e) Quantification of fibrotic area in the sham group, MI model group, and MI with different treatment group. (g) Representative cross sections from remote LV stained with Masson; scale bar = 100 μm. (f) Statistical plot of changes in cardiomyocytes area stained with WGA (mean ± SD, n = 6) and (h) representative LV cross-section stained with WGA, scale bar = 20 μm. *p < 0.05, **p < 0.01, and ***p < 0.001, one-way ANOVA, Tukey’s multiple comparison test.
Consistent with the findings in the stress overload model, all three treatments (free JQ1, JQ1 NPs, and PM&EM/JQ1 NPs) improved cardiac function and left ventricular contractility in mice with MI-induced cardiac fibrosis. However, the treatment effect was more pronounced in the mice that received PM&EM/JQ1NPs (Figure 6b–d). The body weights of the mice in both TAC and MI experiments indicated that no significant body weight abnormalities occurred in any of the treatment groups (Figure S8a,b). Furthermore, these nanoparticles also achieved better results in terms of the degree of cardiac fibrosis and the cardiomyocyte size (Figure 6e–h). These findings demonstrate that PM&EM/JQ1 NPs could inhibit and improve HF after large-scale anterior MI and had a significantly better therapy effect.
Mechanisms of PM&EM/JQ1 NPs for Cardiac Fibrosis Therapy
PM&EM/JQ1 NPs have demonstrated excellent therapeutic efficacy in the treatment of TAC-induced cardiac fibrosis and MI-induced cardiac fibrosis. While some previous reports have suggested that JQ1 treatment of TAC-induced cardiac fibrosis may be achieved through an essential MEOX1-dependent transcriptional switch that governs cellular plasticity in the fibroblast compartment,4 it is important to note that the acute episodes of severe hemodynamic overload in this model do not represent a common clinical situation in humans. Instead, myocardial infarction (MI) is a very common cause of pathological cardiac remodeling and HF in humans. The potential mechanisms and therapeutic targets of JQ1 and PM&EM/JQ1 NPs in MI-induced HF remain to be further studied.
To further investigate the potential mechanisms of how PM&EM/JQ1 NPs ameliorate myocardial infarction-induced cardiac fibrosis, we performed both RNA-seq and ATAC-seq on infarcted cardiac tissue. Compared with the free JQ1 treatment group, 1757 genes were down-regulated and 777 genes were up-regulated in the PM&EM/JQ1 NPs treatment group in RNA-seq (Figure 7a). Pathway enrichment of these up- and down-regulated genes revealed that the pathways upregulated by the PM&EM/JQ1 NPs were mainly focused on oxidative phosphorylation, while among the down-regulated pathways, ECM-receptor interaction was the most significant (Figure 7b–d). Further heatmaps highlighted the core genes that exhibited high variability in these two top pathways (Figure 7e,f). ATAC-seq revealed that PM&EM/JQ1 NPs down-regulated 216 peaks and up-regulated 24 peaks compared with free JQ1 therapy group (Figure 7g). Plcg1 (phospholipase C gamma 1) and Aplp2 (amyloid precursor- like protein 2) were identified as the potential transcriptional regulators. Reproduction_Feeding and Neurohormone signaling, Cell adhesion_Amyloid proteins, and Inflammation_IL-2 signaling were the three pathways with the most significant downward adjustments (Figure 7h,i, Figure S8c). In addition, further validation was performed based on the results of RNA-seq and ATAC-seq analyses. We found that PM&EM NPs have a stronger cardioprotective function due to higher levels of cardiac oxidative phosphorylation. We performed WB testing on heart tissues injected with PM&EM/JQ1 NPs and found that the five protein complexes (complex I, complex II, complex III, complex IV, and complex V) responsible for oxidative phosphorylation were more expressed in the PM&EM/JQ1 NP group (FigureS9).
Figure 7.

Potential mechanisms and therapeutic targets for PM treatment of myocardial infarction-induced cardiac fibrosis. (a) Volcano plots show the differentially expressed genes (DEGs) between the PM&EM/JQ1 NPs and free JQ1 in bulk RNA-seq data sets. (b) Top five upregulated (red) and downregulated (green) KEGG pathways by normalized enrichment score (NES) identified by GSEA in PM&EM/JQ1 NPs. (c) GSEA-based KEGG analysis of Oxidative phosphorylation. (d) GSEA-based KEGG analysis of the ECM-receptor interaction. (e) Heatmap showing the genes of Oxidative phosphorylation. Heatmap scale is a Z score. (f) Heatmap showing the genes of the Oxidative ECM-receptor interaction. Heatmap scale is a Z score. (g) Heatmap of BRD4- bound enhancers in free JQ1 and PM&EM/JQ1 NPs therapy heart covering 3kb upstream and downstream of the enhancer summit, where enhancers are grouped by increased or decreased BRD4 binding in PM&EM/JQ1 NPs therapy heart compared to free JQ1 therapy. (h) ATAC-seq from the fibrosis sites of MI mosue with free JQ1 and PM&EM/JQ1 NPs therapy at the Plcg1 locus. (i) Down-regulated pathway enrichment analysis of PM&EM/JQ1 NPs therapy relative to Free JQ1 in ATACseq.
Biosafety Induced by JQ1
The PM&EM NPs investigated in this study were found to have enhanced efficacy, but their effect on organs other than the heart was unclear. To determine the status of the major organs (liver, kidney, gastrointestinal tract) after different treatments, we stained the liver and kidneys with hematoxylin-eosin, and the intestines with Alcian blue, which characterizes goblet cells.5 Our observations revealed that significant pathological changes occurred in the liver, kidneys, and intestines when 50 mg/kg of free JQ1 or JQ1 nanoparticles were injected via the tail vein for 30 days. However, there were no obvious differences in lesions between the group treated with PM&EM/JQ1NPs and the control group (Figure 8a). Biochemical analysis showed that ALT, AST, BUN, and CRE levels were normal in the sham, MI-Control, and PM&EM/JQ1 NP groups. However, these levels were significantly elevated in the free JQ1 and JQ1 NP groups. (Figure S10) These findings indicate that long-term use of free JQ1 and JQ1 NPs for the treatment of cardiac fibrosis could impair the biological functions of the liver and kidney. In contrast, administration of PM&EM/JQ1 NPs would reduce toxicity, as they have decreased accumulation in liver and kidney tissue. This also readily reflects the ability of PM&EM NPs to deliver more JQ1 to the fibrotic regions of the heart and reduce accumulation in liver and kidney tissues. Due to the increased local concentration and sustained release of JQ1 in the fibrotic regions of the heart, cardiac fibrosis can be ameliorated more effectively.
Figure 8.

Biosafety analysis of JQ1, JQ1 NPs and PM&EM/JQ1 NPs. (a) Representative cross section from liver and kidney stained with H&E and intestines with Alcian blue, scale bar = 100 μm. (b) Venn diagrams of JQ1, nephrotoxicity, gastrointestinal toxicity, and hepatoxicity related gene from GENCARDS. (c) GO pathway enrichment analysis (biological process, BP) of the common gene from different groups. (d) Immunofluorescence staining of mitochondria (Mito, red) and reactive oxygen species (ROS, green) in the kidney and liver. Nuclei were counterstained with DAPI (blue); scale bar = 20 μm.
A comprehensive analysis was conducted to explore the potential mechanisms underlying liver, kidney, and intestinal damage caused by the long-term use of JQ1. A total of 981 JQ1-related genes, 745 genes related to liver toxicity, 681 genes related to renal toxicity, and 7423 genes related to intestinal toxicity were extracted from GENECARDS (https://www.genecards.org/). These data sets were imported into Hiplot (https://hiplot.com.cn/home/index.html). Forty-four common genes were identified and displayed in Venn diagrams (Figure 8b). In order to explore the potential pathways in which these common genes are involved, GO pathway enrichment analysis was performed using the clusterProfiler R package. Biological Process (BP) mainly involves response to oxidative stress, cellular response to oxidative stress, cellular response to chemical stress, and response to reactive oxygen species. Molecular function (MF) mainly includes DNA-binding transcription factor binding, RNA polymerase II-specific DNA-binding transcription factor binding, DNA-binding transcription activator activity, and RNA polymerase II-specific functions. These findings show the toxicity of JQ1 in the liver, kidney, and gastrointestinal tract (Figure 8c, Figure S11a,b).
To further confirm this, these genes were then introduced into Metascape (http://metascape.org/) for GO enrichment analysis (a false discovery rate of <0.01 is considered significantly enriched). The highest levels of enrichment were also observed in the response to the oxidative stress signaling pathway, which is mainly involved in oxidative stress-related and apoptosis-related dysfunctions. Based on the false discovery rate and hit genes, the top 10 GO entry pathways were selected and are shown in Figure S11c. The key regulatory molecules are also shown in Figure S11d.
Finally, the results of pathway enrichment showed that JQ1 can effectively aggravate oxidative stress and apoptosis in murine liver and kidney tissues, which may be the mechanism for the relatively mild hepatic and renal toxicity of these nanoparticles. For further verification, the reactive oxygen species (ROS) test and tunnel test were performed on the liver and kidney of mice in those treatment groups. Compared with the sham and MI-control group, the free JQ1 and JQI NP treatment group significantly increased ROS and apoptosis cells. However, PM&EM/JQ1 NPs inhibited these side effects, indicating that PM&EM NPs effectively reduced JQ1 accumulation in liver and kidney tissues. (Figure 8d and Figure S12).
In addition, we detected the expression of BRD4 in the main organs of mice, including heart, liver, and kidney, and found that the expression was significantly increased in the TAC model of heart tissue, while JQ1 inhibited the protein level of BRD4 to a certain extent. In liver and kidney tissues, there was little change in BRD4 expression (Figure S13).
Conclusions
In our study, we have developed PM&EM NPs that coassemble with natural platelet and erythrocyte membranes, leveraging the advantages of both membranes to target fibrosis in a sustained manner in vivo. Our study demonstrated that PM&EM nanoparticles efficiently adhered to collagen fibers and reduced macrophage phagocytosis in vitro. We further investigated the therapeutic effect of this hybrid system in mouse models of TAC-induced and MI-induced HF. Following long-term intravenous injection of PM&EM/JQ1 NPs, heart function was significantly improved, the degree of cardiac fibrosis was significantly reduced, and the JQ1 toxicity to organs other than the heart was effectively attenuated in mice. In addition, we employed multiomics techniques to investigate nanoloaded drug delivery systems. We thoroughly characterized PM&EM/JQ1 NPs by proteomics and directly demonstrated that PM can inherit functional membrane proteins from platelet and erythrocyte membranes. Furthermore, we used RNA-seq and ATAC-seq to predict core pathways, core genes, and core transcription factors involved in the mechanism of PM&EM/JQ1 NPs in ameliorating cardiac fibrosis. Our study reveals that PM&EM nanoparticles exhibit enhanced cardioprotection, which is attributed to elevated cardiac oxidative phosphorylation. Notably, Western blot analysis of heart tissues treated with PM&EM/JQ1 nanoparticles showed upregulated expression of the five protein complexes (complexes I, II, III, IV, and V) involved in oxidative phosphorylation in the PM&EM/JQ1 nanoparticle group.
Future research should focus on biofilm fusion, as the rational and effective merging of diverse biofilms can significantly enhance the multifunctional properties of targeted drug delivery systems. Such investigations will provide valuable insights into drug delivery strategies after MI. Our work introduces a hybrid biomimetic drug delivery system that integrates native platelet and erythrocyte membranes to enhance the functionality of nanoparticles. This system was designed for combining other cell membranes and even cell membrane hybrids with engineered artificial lipid membranes to form biomimetic nanoparticles for diverse biomedical applications.
Currently, there are many other emerging targeted therapies, such as chimeric antigen receptor T-cell immunotherapy (CAR-T), monoclonal antibody therapy, etc. Problems such as difficulty in obtaining raw materials and high prices have limited their wide application in clinical treatment. However, the development of nanotargeted therapy, especially biomimetic nanomaterials, will greatly improve the status quo of targeted therapy, promote the effect of drug-targeted therapy, and greatly reduce drug side effects. Although the study demonstrated significant preclinical efficacy, the path to clinical translation remains uncertain. For large-scale production, stability and regulatory approval of these complex nanoparticles also remain potential challenges. The acquisition of biomimetic cell membranes still needs to be extracted from living organisms, and the development of artificial cell membranes will promote large-scale production.
Experimental Section
Materials and Cells
The antihuman GP IBa antibody was obtained from ProMab Biotechnology (CA, USA), while the antihuman GP IaIIa antibody was acquired from ABclonal Biotechnology (Shanghai, China). The antihuman GP VI antibody was purchased from AbBox Biotechnology (Suzhou, China), and the antihuman CD47 antibody was obtained from Santa Cruz Biotechnology (USA). Additionally, we used the antimouse Collagen I antibody, which was purchased from ABclonal Biotechnology (Shanghai, China).
JQ1, a chemical compound, was obtained from Selleck (USA). Dimethyl sulfoxide (DMSO) and wheat germ agglutinin (WGA) were acquired from Sigma (USA). PLGA (50:50; MW = 90,000), a type of polymer, was purchased from Dalian Meilun Biotechnology in China. The fluorescent dyes 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine,4-chlorobenzenesulfonate Salt (DiD), and 6-diamidino-2-phenylindole (DAPI) were obtained from Beyotime (China). Masson’s trichrome, hematoxylin, and eosin were purchased from Servicebio (Wuhan, China). Ponceau S, cell counting kit 8, and BCA protein assay kit were purchased from Biosharp (China). Mouse TNF-α, IL-1β, and IL-6 ELISA kits were purchased from LunchangshuoBiotech (Xiamen, China) and ELK Biotechnology (Wuhan, China). The mouse CCL2 ELISA kit was purchased from Fankew (Shanghai, China). LysoTracker Deep Red was purchased from Thermo Fisher (USA). YF 488-annexin tunel apoptosis kits (US EVERBRIGHT, Suzhou, China)
The Murine RAW 264.7 and NIH/3T3 cell lines were purchased from ZQXZ Biotechnology in Shanghai, China, while the Murine smooth muscle cell line was obtained from Beinart Biotech in Beijing, China. HUVECs were purchased from Science Cell in Carlsbad, CA.
Human Platelet Isolation and Membrane Extraction
Fresh blood was collected with 5 mM EDTA and centrifuged at 100g for 20 min. The supernatant was retained, and blood cells were separated. The platelet-rich plasma (PRP) was obtained. PRP was centrifuged at 800g for 20 min. The supernatant is discarded, and the precipitate is platelets. A PBS solution containing 1 mM EDTA and protease inhibitor tablets was used to resuspend the platelet pellet. Further, approximately 3 × 109/1.5 mL of platelets are aliquoted and can be mixed with 1 mg of PLGA. Platelet membrane vesicles (PMV) were obtained using a previously described method of repeated freeze–thaw cycles.12 To obtain PMV, aliquots of platelet suspension were frozen at −80 °C, thawed at room temperature, and then centrifuged at 4,000g for 3 min. The resulting pellet was resuspended in PBS and sonicated for 5 min using a Fisher Scientific FS30D water bath sonicator at a frequency of 42 kHz and 100 W power. The presence of PMV was confirmed through dimensional measurements using dynamic light scattering and morphological examination by transmission electron microscopy (TEM).
Isolation of Red Blood Cells and Membrane Extraction
Human type O blood is treated with 1.5 mg/mL EDTA and erythrocyte isolation is performed approximately 16 h after blood collection. Collect red blood cells by centrifugation at 720g for 10 min and wash twice using prechilled PBS solution (containing 1 mm EDTA, 50 μm leutin, and 1 μg/mL aprotinin). The resulting red blood cell membrane is suspended in a quadruple volume of 0.2 mm EDTA 2Na deionized water to induce membrane rupture. After 60 min of low permeability solubilization, the sample was centrifuged at 20,000g for 20 min and the red blood cell membrane. The low permeability dissolution step described above was repeated once. The erythrocyte membrane was washed using a prechilled PBS solution (pH 7.4 containing 50 μm leucin peptide and 1 μg/mL aprotinin) until the supernatant was colorless. All of the above operations are performed at 4 °C to maintain the structural and functional stability of the membrane proteins. Fresh erythrocyte membranes are stored at 4 °C and used within 6 h.
Verification of Membrane Fusion
Platelet and erythrocyte membranes were fused at a protein dry weight ratio of 4:1 using ultrasound at a frequency of 42 kHz and 100 W power. To determine the fusion process between platelet and red blood cell membranes, we employed the FRET method. Specifically, we added DiO and DiI FRET dye pairs (0.1%, w/w) to the platelet membrane and then introduced the red blood cell membrane to the DiO- and DiI-labeled platelet membranes. The resulting mixture was sonicated and extruded, and the fluorescence spectrum of a sample was measured at 400 nm by using a Tecan fluorescence spectrometer.
To visualize the fusion between platelet and erythrocyte cell membranes, we labeled the platelet membranes with DiO and the erythrocyte cell membranes with DiD, following the fusion methods described above. As a control, we also prepared a physical mixture of the two membranes. The resulting samples were imaged by using laser confocal microscopy (CLSM) from Leica.
Total protein of PM&EM/JQ1 NPs was validated using SDS-PAGE. The specific proteins GP IIbIIIa, GP Ibα, GP VI, CD47 were then characterized by Western blot.
Protein Identification and Classification
We used 3D-DIA based on mass spectrometry (MS) to perform proteomic analysis of the protein composition of PM&EM NPs, as previously described. Briefly, the samples were dissolved in lysis buffer containing 8 M urea, 100 mM Tris-HCL (Sigma, MO, USA), pH 8.5, and 1% protease inhibitor cocktail (Biyuntian Biotechnology Co., Shanghai, CN). The resulting mixture was sonicated and centrifuged at 15,000g at 4 °C for 15 min to remove the sediment. Protein digestion was performed using the FASP method with Trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 (Sigma, MO, USA). Data Independent Acquisition (DIA) analysis was conducted on a Q Exactive HF mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an EASY-nLC 1200 system (Thermo Fisher Scientific). Forty-five variable DIA windows were set for DIA acquisition, and identification and quantification were performed using Spectronaut 17.4 (Biognosys, Schlieren, Switzerland) with the directDIA model. The DIA raw files were searched against the human fasta database (from UniProt) to generate a spectral library using BGS factory settings. All results were filtered by a Q value cutoff of 0.01 (corresponding to an FDR of 1%). The P-value estimator was performed by a kernel density estimator.
JQ1 Released
The sample solution was incubated in a dialysis bag (Sigma, MWCO 100 kDa) containing PBS at pH 7.4 and maintained at 37 °C. At a specific time point, the solution inside the bag was collected, and the amount of JQ1 was measured using a spectrometer (Tecan) with an absorption wavelength of 300 nm. JQ1NPs were prepared and used as a control. The LE and EE were calculated as follows
where M(JQ1) is the mass of colchicine loaded in the NPs, M(PLGA) is the mass of polymer in the formulation, and M(added) is the mass of JQ1 added
Biocompatibility of PM&EM Nanoparticles
In vitro biocompatibility of PM&EM NPs was determined using RAW264.7, mouse embryonic fibroblasts, ECs, and SMCs. Cells are seeded separately in 96-well plates and cultured overnight. Add different concentrations of PM&EM NPs and incubate for another 24 or 48 h. Cell viability was determined using a cell counting kit (Beyotime, China).
Blood Compatibility Tests
To evaluate the hemocompatibility of PLGA nanoparticles and PM&EM NPs, 1 mL of rat blood was mixed with 1.25 mL of 0.9% sodium chloride solution. Then, 0.1 mL of the diluted whole blood was added to the PLGA nanoparticles or PM&EM NPs solution (5 mL, 1 mg/mL) and incubated at 37 °C for 1 h. The mixture was then centrifuged at 3000 rpm for 5 min, and the absorbance of the supernatant was measured at 540 nm to determine the hemoglobin released from the lysed red blood cells.
Methods of Animal Experimentation
All animal experiments were conducted in compliance with the Guidelines for the Protection and Use of Laboratory Animals of the National Institutes of Health and were approved by the Ethical Review Committee of Beijing Anzhen Hospital.
For this study, male C57BL/6J mice (8 weeks old) were obtained from Beijing Huafukang Technology Co., Ltd. The mice were housed under controlled temperature and humidity conditions in a pathogen-free environment with a 12 h light/dark cycle. They were provided with free access to standard laboratory rodent feed and water. During surgery, the mice were placed on a temperature-controlled small animal operating table to maintain their body temperature.
TAC-induced chronic heart failure model: In the aortic arch coarctation (TAC) model, open heart surgery is performed after anesthetized mice are treated using pentobarbital. A 7–0 silk thread and a 27-gauge needle were used to contract the aortic arch between the left common carotid artery and the brachiocephalic trunk artery in mice, as described in a previous article.23 On day 18 after TAC surgery, mice are randomly divided into 4 groups (n = 6). Free JQ1 group, JQ1 NP group, and PM&EM/JQ1 NP group, dissolved in 0.9% sodium chloride solution for injection, containing JQ1 at a dose of 50 mg/kg/day, lasted until the 42nd postoperative day; the TAC-Con group was injected with the same volume of normal saline; and the Sham group was set up with sham surgery. It was treated until postoperative day 45 to perform an ultrasound of the mouse heart, and the mouse was sacrificed under anesthesia.
Model of post-MI heart failure: after inducing anesthesia in mice using isoflurane, adjust the anesthetic concentration and make the mouse continuously inhaled, permanent ligation of the proximal left anterior descending coronary artery (LAD) at the left atrial margin using a 7–0 silk thread. Mice are randomly divided into 4 groups (n = 6) on postoperative day 6. For the free JQ1 group, JQ1 NP group, and PM&EM/JQ1 NP group intraperitoneal injection, the dose containing JQ1 was 25 mg/kg/day, and the dose of JQ1 was increased to 50 mg/kg/day on the 15th postoperative day and continued until the 27th postoperative day, the MI-Con group was injected with the same volume of normal saline, and the Sham group was set up with sham surgery. Mouse heart ultrasound is performed until day 30 postoperatively, and mice are sacrificed under anesthesia.
Echocardiography
After hair removal at the heart site of the mouse, 1–2% inhaled isoflurane was anesthetized, supine on a physiological information detection platform, and imaged using the Vevo high-resolution imaging system. Images were acquired in B mode on the left ventricular minor axis, and the left ventricular central section was measured in two dimensions to obtain the thickness and inner diameter of the anteroposterior wall during the diastolic and systolic periods of the left ventricle.
Organizational Analysis
Cardiac tissue is collected, PBS washed, fixed with 4% paraformaldehyde fixative solution, and sliced after conventional dehydration embedding. The cross-sectional area of cardiomyocytes was determined using WGA staining, and the degree of cardiac fibrosis in the TAC heart failure model and the post-MI heart failure model was assessed using Sirius red and Mason staining, respectively. Quantitative analysis of the cardiomyocyte cross-sectional area and cardiac fibrosis area using ImageJ.
In Vitro Imaging
Mice are injected with 100ul DiI-labeled NPs and PM&EM/NPTs solution through the tail vein and anesthetized at predetermined time points after injection, and fluorescence signal distribution was detected using in vivo imaging systems. After 24 h, mouse organs were removed and IVIS spectroscopic imaging was performed to assess the distribution of fluorescence signals in different organs.
RNA-Seq
Bulk RNA sequencing was performed using the Illumina sequencing platform of Novogene Co. Ltd. (Beijing, China). Raw reads were aligned to the mm10 reference genome using HISAT2 software with default paired-end settings. Transcripts were assembled using StringTie, after being sorted by the SAM tools. Differential expression analysis was performed by the R package DESeq2, according to fold change ≥2.0 and the adjusted p-value of ≤0.05. Gene Set Enrichment Analysis (GSEA) (http://software.broadinstitute.org/gsea) and pathway gene set Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp/kegg/) were used to implement differentially expressed genes set enrichment analysis.
ATAC-seq
Nucleic acid suspensions were subjected to incubation with a transposition mix containing a transposase enzyme. The transposase enzyme penetrated the nuclei and selectively fragmented the DNA in regions with open chromatin. At the same time, adapter sequences were attached to the ends of the DNA fragments. After the transposition reaction at 37 °C for 30 min, the resulting products were purified using a MiniElute DNA Kit from Qiagen, amplified according to a previously published protocol,24 and sequenced using an Illumina HiSeqTM 4000 platform by Gene Denovo Biotechnology Company in Guangzhou, China.
Construction of Pathways
JQ1 and main organ toxicity-related genes were retrieved from GeneCards (https://www.genecards.org/). The Venn diagram, which shows common genes, was designed in Hiplot (https://hiplot.com.cn/home/index.html). The common gene profiles were analyzed by using the Bioconductor packet. R and Metascape (http://metascape.org/) were used for GO enrichment analysis. The interactions of target genes were analyzed using Metascape.
Statistical Analysis
All experimental results were statistically analyzed using GraphPad Prim (version 8.02), which was performed using one-way ANOVA, and each result was expressed as the mean ± standard deviation. The difference significance level is set to *p < 0.05, ** p < 0.01, *** p < 0.001.
Acknowledgments
The authors are very grateful to Dr. Yi Xin (Beijing Anzhen Hospital Affiliated to Capital Medical University), Dr. Rui Lin (Capital Medical University), and Dr. Yanyan Gong (Tianjin Medical University) for scientific advice. The authors want to thank Professor Shaokun Shu and his team (Peking University) for the assistance of ATAC-seq analysis. The authors also want to thank the staff of PROMAB National Center for Protein Sciences (Beijing) and Beijing Institute of Lifeomics for some technique support. We thank Ryan Chastain-Gross, Ph.D., from Liwen Bianji (Edanz) (www.liwenbianji.cn/) for editing the English text of a draft of this manuscript. This work was supported by the National Key R&D Program of China (2021YFA0805100), the National Natural Science Foundation of China (82270499), and Beijing Municipal Health Commission (11000024T000002830987).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.4c04814.
(Figures S1–S13) Protein counts in PM, EM, and PM&EM vesicles, JQ1 spectra, JQ1 release profiles, size change tendency of PM&EM/JQ1 NPs, biocompatibility and clearance of PLGA nanoparticles and PM&EM NPs, body weights of animal models, validation of OXPHOS complexes, GO analysis of JQ1 side effects, tunnel staining, and Western blots of BRD4; (Tables S1 and S2) illustrated explanations of the main abbreviations and abbreviations in this manuscript (PDF)
Author Contributions
§ Y.L. and J.Y. contributed equally to this article.
The authors declare no competing financial interest.
Notes
This work has been previously submitted to Research Square (https://www.researchsquare.com/). Y.L.; J.Y.; C.C.; W.C.; W.C.; J.M.; J.D.; Y.W. Platelet and Erythrocyte Membranes Coassembled Biomimetic Nanoparticles for Heart Failure Treatment. Posted 10 Oct, 2023. 10.21203/rs.3.rs-3326363/v1. Research Square. This work is licensed under a CC BY 4.0 License. URL (accessed Oct 10, 2023).
Supplementary Material
References
- McLellan M. A.; Skelly D. A.; Dona M. S. I.; Squiers G. T.; Farrugia G. E.; Gaynor T. L.; Cohen C. D.; Pandey R.; Diep H.; Vinh A.; et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142 (15), 1448–1463. 10.1161/CIRCULATIONAHA.119.045115. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chen J.; Shearer G. C.; Chen Q.; Healy C. L.; Beyer A. J.; Nareddy V. B.; Gerdes A. M.; Harris W. S.; O’Connell T. D.; Wang D. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation 2011, 123 (6), 584–593. 10.1161/CIRCULATIONAHA.110.971853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou S.; Mendez S. R.; Feinman J.; Mitrani L. R.; Fuster V.; Mangiola M.; Moazami N.; Gidea C.. Heart transplantation: advances in expanding the donor pool and xenotransplantation. Nature reviews. Cardiology 2024. DOI: 2125. 10.1038/s41569-023-00902-1. [DOI] [PubMed] [Google Scholar]
- Duan Q.; McMahon S.; Anand P.; Shah H.; Thomas S.; Salunga H. T.; Huang Y.; Zhang R.; Sahadevan A.; Lemieux M. E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Science translational medicine 2017, 9 ( (390), ). DOI: 10.1126/scitranslmed.aah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexanian M.; Przytycki P. F.; Micheletti R.; Padmanabhan A.; Ye L.; Travers J. G.; Gonzalez-Teran B.; Silva A. C.; Duan Q.; Ranade S. S.; et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 2021, 595 (7867), 438–443. 10.1038/s41586-021-03674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre E. J.; McDaniel K. F.; Albert D. H.; Mantena S. R.; Plotnik J. P.; Wilcox D.; Zhang L.; Bui M. H.; Sheppard G. S.; Wang L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578 (7794), 306–310. 10.1038/s41586-020-1930-8. [DOI] [PubMed] [Google Scholar]
- Schreiber J.; Liaukouskaya N.; Fuhrmann L.; Hauser A. T.; Jung M.; Huber T. B.; Wanner N.. BET Proteins Regulate Expression of Osr1 in Early Kidney Development. Biomedicines 2021, 9 ( (12), ). DOI: 1878. 10.3390/biomedicines9121878. [DOI] [PMC free article] [PubMed] [Google Scholar]; Andrieu G.; Belkina A. C.; Denis G. V. Clinical trials for BET inhibitors run ahead of the science. Drug discovery today. Technologies 2016, 19, 45–50. 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pang Y.; Bai G.; Zhao J.; Wei X.; Li R.; Li J.; Hu S.; Peng L.; Liu P.; Mao H. The BRD4 inhibitor JQ1 suppresses tumor growth by reducing c-Myc expression in endometrial cancer. Journal of translational medicine 2022, 20 (1), 336. 10.1186/s12967-022-03545-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Rao L.; Yao H.; Wang Z.; Ning P.; Chen X. Engineering Macrophages for Cancer Immunotherapy and Drug Delivery. Advanced materials (Deerfield Beach, Fla.) 2020, 32 (40), e2002054 10.1002/adma.202002054. [DOI] [PubMed] [Google Scholar]; Li Y.; Wang J.; Xie J. Biomimetic nanoparticles targeting atherosclerosis for diagnosis and therapy. Smart Med. 2023, 2 (3), e20230015 10.1002/SMMD.20230015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wang J.; Xie J. Biomimetic nanoparticles targeting atherosclerosis for diagnosis and therapy. Smart Med. 2023, 2 (3), e20230015 10.1002/SMMD.20230015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Che J.; Chang L.; Guo M.; Bao X.; Mu D.; Sun X.; Zhang X.; Lu W.; Xie J. CD47- and Integrin α4/β1-Comodified-Macrophage-Membrane-Coated Nanoparticles Enable Delivery of Colchicine to Atherosclerotic Plaque. Adv. Healthcare Mater. 2022, 11 (4), e2101788 10.1002/adhm.202101788. [DOI] [PubMed] [Google Scholar]
- Cui J.; Wang M.; Zhang W.; Sun J.; Zhang Y.; Zhao L.; Hong Z.; Li D.; Huang Y. X.; Zhang N.; et al. Enhancing insulin sensitivity in type 2 diabetes mellitus using apelin-loaded small extracellular vesicles from Wharton’s jelly-derived mesenchymal stem cells: a novel therapeutic approach. Diabetology & metabolic syndrome 2024, 16 (1), 84. 10.1186/s13098-024-01332-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang K.; Qin X.; Li T.; Qiu J.; Yin T.; Huang J.; McGinty S.; Pontrelli G.; Ren J.; et al. Biomimetic Nanotherapies: Red Blood Cell Based Core-Shell Structured Nanocomplexes for Atherosclerosis Management. Advanced science (Weinheim, Baden-Wurttemberg, Germany) 2019, 6 (12), 1900172. 10.1002/advs.201900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C. M.; Fang R. H.; Wang K. C.; Luk B. T.; Thamphiwatana S.; Dehaini D.; Nguyen P.; Angsantikul P.; Wen C. H.; Kroll A. V.; et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 2015, 526 (7571), 118–121. 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che J.; Najer A.; Blakney A. K.; McKay P. F.; Bellahcene M.; Winter C. W.; Sintou A.; Tang J.; Keane T. J.; Schneider M. D.; et al. Neutrophils Enable Local and Non-Invasive Liposome Delivery to Inflamed Skeletal Muscle and Ischemic Heart. Advanced materials (Deerfield Beach, Fla.) 2020, 32 (48), e2003598 10.1002/adma.202003598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wang Y.; Xia Z.; Xie Y.; Ke D.; Song B.; Mu D.; Yu R.; Xie J. Noninvasive platelet membrane-coated Fe3O4 nanoparticles identify vulnerable atherosclerotic plaques. Smart Med. 2024, 3 (2), e20240006 10.1002/SMMD.20240006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che J.; Sun L.; Shan J.; Shi Y.; Zhou Q.; Zhao Y.; Sun L. Artificial Lipids and Macrophage Membranes Coassembled Biomimetic Nanovesicles for Antibacterial Treatment. Small (Weinheim an der Bergstrasse, Germany) 2022, 18 (26), e2201280 10.1002/smll.202201280. [DOI] [PubMed] [Google Scholar]
- Song Y.; Huang Z.; Liu X.; Pang Z.; Chen J.; Yang H.; Zhang N.; Cao Z.; Liu M.; Cao J.; et al. Platelet membrane-coated nanoparticle-mediated targeting delivery of Rapamycin blocks atherosclerotic plaque development and stabilizes plaque in apolipoprotein E-deficient (ApoE(−/−)) mice. Nanomedicine: nanotechnology, biology, and medicine 2019, 15 (1), 13–24. 10.1016/j.nano.2018.08.002. [DOI] [PubMed] [Google Scholar]
- Dai J.; Wu M.; Wang Q.; Ding S.; Dong X.; Xue L.; Zhu Q.; Zhou J.; Xia F.; Wang S.; et al. Red blood cell membrane-camouflaged nanoparticles loaded with AIEgen and Poly(I: C) for enhanced tumoral photodynamic-immunotherapy. National science review 2021, 8 (6), nwab039. 10.1093/nsr/nwab039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang K.; Li T.; Maruf A.; Qin X.; Luo L.; Zhong Y.; Qiu J.; McGinty S.; Pontrelli G.; et al. Macrophage membrane functionalized biomimetic nanoparticles for targeted anti-atherosclerosis applications. Theranostics 2021, 11 (1), 164–180. 10.7150/thno.47841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi H.; Williams L. J. Principal component analysis. Wiley Interdiscip. Rev.: Comput. Stat. 2010, 2 (4), 433–459. 10.1002/wics.101. [DOI] [Google Scholar]
- Kivelä R.; Hemanthakumar K. A.; Vaparanta K.; Robciuc M.; Izumiya Y.; Kidoya H.; Takakura N.; Peng X.; Sawyer D. B.; Elenius K.; et al. Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling. Circulation 2019, 139 (22), 2570–2584. 10.1161/CIRCULATIONAHA.118.036099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunwald E. Research advances in heart failure: a compendium. Circulation research 2013, 113 (6), 633–645. 10.1161/CIRCRESAHA.113.302254. [DOI] [PubMed] [Google Scholar]
- Pfeffer J. M.; Pfeffer M. A.; Braunwald E. Hemodynamic benefits and prolonged survival with long-term captopril therapy in rats with myocardial infarction and heart failure. Circulation 1987, 75 (1 Pt 2), 149–155. [PubMed] [Google Scholar]
- Anand P.; Brown J. D.; Lin C. Y.; Qi J.; Zhang R.; Artero P. C.; Alaiti M. A.; Bullard J.; Alazem K.; Margulies K. B.; et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154 (3), 569–582. 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J. D.; Giresi P. G.; Zaba L. C.; Chang H. Y.; Greenleaf W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10 (12), 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.