

Abstract
Tissue localization is a critical determinant of T cell immunity. CD8+ T cells are contact-dependent killers, which requires them to physically be within the tissue of interest to kill peptide–MHC class I-bearing target cells. Following their migration and extravasation into tissues, T cells receive many extrinsic cues from the local microenvironment, and these signals shape T cell differentiation, fate and function. Because major organ systems are variable in their functions and compositions, they apply disparate pressures on T cells to adapt to the local microenvironment. Additional complexity arises in the context of malignant lesions (either primary or metastatic), and this has made understanding the factors that dictate T cell function and longevity in tumours challenging. Moreover, T cell differentiation state influences how cues from the microenvironment are interpreted by tissue-infiltrating T cells, highlighting the importance of T cell state in the context of tissue biology. Here, we review the intertwined nature of T cell differentiation state, location, survival and function, and explain how dysfunctional T cell populations can adopt features of tissue-resident memory T cells to persist in tumours. Finally, we discuss how these factors have shaped responses to cancer immunotherapy.
TOC blurb:
Here, Schenkel and Pauken consider how specific patterns of T cell trafficking and localization in tissue microenvironments shape their immune functions in acute infection and cancer settings. They further consider the relevance of this for the efficacy of immune checkpoint blockade therapy in the clinic.
Introduction
Cancer immunotherapy [G] using checkpoint inhibitors [G] has truly been transformative for clinical cancer care. Inhibitors of cytotoxic T lymphocyte-associated antigen 4 (CTLA4) were approved for use in patients with metastatic melanoma in 2011 and there are now seven FDA-approved inhibitors of the PD-1 pathway for use in more than 25 different late-stage cancer indications1–6. Despite these successes, the majority of patients still do not have long-term durable responses following immune checkpoint blockade and response rates vary greatly depending on cancer type1–6. As such, significant efforts are focused on making these therapies more effective.
A key goal has been to understand how immunotherapy directly impacts immune cell populations present within the tumour microenvironment. However, adaptive immune responses are not initiated or propagated in these tissue sites. Rather, naive T cells [G] are anatomically restricted to secondary lymphoid organs (SLOs) and blood, and must become activated in SLOs prior to going through a tightly regulated, heavily orchestrated process of migration to get into tissues. Consequently, in order to optimize immunotherapy for the treatment of cancer, it is imperative to consider the anatomy of the immune response, how T cell differentiation states can vary from location to location, and how the tissue microenvironment and/or tumour tissue impact the functional status and longevity of T cell populations in these locations.
Decades of work in models of acute infection have honed our understanding of the mechanistic basis of CD8+ T cell responses to pathogens in nonlymphoid tissues (see Box 1). Briefly, following activation, naive T cells give rise to different effector and memory T cell populations that eliminate the initial infection and provide long-term immunity to subsequent infections with the same pathogen. One memory T cell subset of particular relevance due to its emerging roles in cancer responses is the tissue-resident memory (TRM) [G] cell subset, which is permanently retained within tissues7,8. TRM cells are stably maintained in many different locations, as demonstrated in both mice and humans9–12, and upon antigen reactivation they provide superior secondary immunity within the tissue site, driving rapid clearance of reinfecting pathogens13–15.
Box 1: T cell differentiation following priming of naive T cells.
Under optimal priming conditions to foreign antigens, naive T cells become activated and undergo a differentiation process where they develop into highly functional effector T cells [G] capable of migrating to sites of antigen and inflammation to eliminate antigen-bearing target cells7,8. CD8+ T cells kill target cells in a contact-dependent manner, requiring them to migrate to peripheral sites of pathogen entry and replication to perform their effector functions. Because of the necessity to function in diverse tissue microenvironments, effector T cells evolved to efficiently adapt to different types of tissues7,8. If antigen is cleared, memory T cell [G] populations form that retain high recall potential, including the ability to re-acquire effector functions including rapid proliferation and production of effector molecules (including effector cytokines and cytotoxic molecules) upon antigen reencounter. Memory T cells are characterized into subsets including central memory T (TCM) cells [G] , effector memory T (TEM) cells [G], and TRM cells, which are broadly defined based on location, tissue residence, longevity, and plasticity7,8. Because acute infection models drive robust and stable CD8+ TRM cell responses that are highly capable of eliciting robust recall responses upon antigen reencounter, these model systems are instructive for identifying and understanding the optimal conditions for differentiation and effector potential. Moreover, they have been informative to establish the basal requirements for TRM cells to thrive in different organ systems.
If antigen fails to be cleared, which is common during chronic infection and cancer, T cells go through an altered developmental trajectory that leads to T cell exhaustion16,17. Early work characterizing exhausted T (TEX) cells [G] showed that on a population level, TEX progressively lose effector functions (for example, the ability to proliferate, produce effector cytokines, and kill target cells), upregulate exhaustion-associated transcription factors such as TOX, and express high and sustained levels of multiple inhibitory receptors (including PD-1, T cell immunoglobulin mucin 3 (TIM3), lymphocyte activation gene 3 (LAG3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT))16,18. However, despite the notable erosion of effector functions, TEX cells are not functionally inert. Rather, TEX cells retain some residual functionality that contributes to immune responses against chronic pathogens and tumours19,20. Furthermore, this residual effector activity can be functionally boosted using immunotherapy interventions including PD-1 blockade. It should be noted, however, that not all TEX cells respond to PD-1 inhibitors in the same way (see Box 2)21–23. One subset, termed progenitor TEX (TPEX) cells [G], appears to be less dysfunctional, less epigenetically constrained, and more responsive in terms of proliferation to PD-1 inhibitors than other TEX cell subsets22–30. Consequently, intense efforts are currently focused on understanding the ontogeny of TEX cells, how heterogeneity in TEX cell populations relates to their ability to be therapeutically harnessed in chronic disease, and strategies to optimally engage TEX cells to treat chronic diseases (see Box 2).
Box 2: Exhausted T cell heterogeneity – lessons from infection.
Early work from the Wherry group demonstrated heterogeneity within exhausted T (TEX) cell populations in chronic LCMV infection in mice26,27, with critical ramifications for both responses to PD-1 checkpoint blockade26 and longevity of the TEX cell response27. Seminal work in 201623–25,227 further refined the conceptualization of two of the major TEX cell subsets, showing that the transcription factor TCF1 was required for the formation of the precursors of exhausted T (TPEX) cell population. Since these original papers, there has been tremendous efforts to understand the lineage relationships between the TPEX and terminally-exhausted TEX cell subsets, identify key molecular and cellular regulators of these major cell lineages, and define additional functional heterogeneity between these two polar extremes22,60,124,131,228–232.
TPEX cells are generally believed to be the least differentiated of the exhausted subsets, with the greatest reprogramming potential and responsiveness to checkpoint blockade21–23,139,140. Additionally, TPEX cells show different metabolic properties compared to other TEX cell subsets152,233–235, which may aid their reprogramming potential. In TPEX cells, TCF1 is the critical lineage-defining transcription factor, driving expression of Eomes and MYB which reinforce this programme, and are critical for maintaining TPEX cells124,232. Moreover, additional work has demonstrated the critical need for BACH2 expression, and BATF repression to maintain TPEX cells 131,236. TCF1-KLRG1+ effector-like T cells can form early during chronic LCMV infection, and appear to branch off early from the TPEX cell population and do not follow the same differentiation trajectory as canonical TEX cells124. Along the differentiation trajectory that leads to terminally-exhausted TEX cells, TPEX cells give rise to intermediate-like or transitory TEX cell populations135,136,237. Different groups have given different names to each subset. One study described Texprog1, Texprog2, Texint and Texterm, which followed a lineage trajectory237. In another study, a transitory population developed from TPEX cells that expressed CX3CR1 and TIM3, but retained higher levels of granzyme B and lacked the marker CD101135,136. The most terminally-differentiated TEX cell subset is generally thought to express CD101 and high levels of other inhibitory receptors, including TIM3135,136. While there is evidence to suggest that similar subpopulations exist in some cancer types in both mice and humans22,55,56,60,166,216,238 (for example TPEX cells in the lymph node, terminally-exhausted TEX cells in immunologically hot tumours), additional work is needed to understand how much of this TEX cell framework from chronic infection will extrapolate to cancer, and how to best therapeutically harness the T cell subsets present in diverse cancer types.
Once within a tissue, it is imperative that T cells undergo adaptations in response to local cues within the microenvironment8,31. T cells must have a certain degree of plasticity to be able to adapt to these very different tissue microenvironments. The major nonlymphoid organs and organ systems differ substantially from the SLOs where naive T cells are maintained during homeostasis8,31. Each organ system contains unique environmental pressures that are essential for their physiological functions (for example, nutrient intake and digestion in the alimentary tract, conducting electrical signals for pumping blood in the cardiovascular system). Extending this concept to neoplasia, tumours do not exist in a vacuum. Tumours exist, grow, and evolve surrounded by normal tissue cells and stroma, so all of the unique physiological attributes of each host tissue are also present in malignant lesions32. Moreover, the cancer context adds additional challenges such as chronic antigen exposure, chronic inflammation (or lack of inflammation), abnormal vasculature, immunosuppressive cytokines [G] and immunosuppressive leukocyte populations [G], fibrosis [G], nutrient deprivation and hypoxia [G]32. Ultimately, T cells have to cope with all of these environmental barriers to perform their effector functions and there is a pressing need to understand how to overcome these obstacles to maximize the potential of T cell-based therapies for cancer.
In this Review, we will discuss how CD8+ T cell developmental trajectories converge with tissue biology, and how factors such as migration to, localization within, and the ability to sense different types of microenvironments shape T cell responses. We highlight shared principles underlying CD8+ T cell migration and adaptation to tissue microenvironments in the settings of acute infection and cancer, as well as the unique challenges that arise in the cancer setting. Lastly, we consider the relevance of these concepts in terms of responsiveness to checkpoint inhibitors in the clinic. The primary focus of the Review will be on CD8+ T cells, which have been shown to be key drivers of anti-tumour immunity. Although some of the same concepts may extend to CD4+ T cells (for example, activation in lymph nodes and migration to tissues), other concepts may be less generalizable (for instance, issues pertaining to the formation and maintainance of CD4+ TRM cell and CD4+ TEX cell populations), and we refer the reader to other reviews on these topics16,31.
Where it starts — lymphoid tissue
One of the most critical properties underlying the ability of T cells to execute any type of immune response is their migration to where they are needed and adaptation to their new environment. Lymphocyte trafficking is a dynamic, tightly coordinated, and heavily orchestrated process allowing for purposeful cellular locomotion and infiltration into tissues33,34. Successful migration is dependent on T cells having the right selectins [G], chemokine receptors [G], and integrins [G] that are required to enter a tissue, as well as on the tissue expressing the corresponding selectin ligands, chemokines, and integrin ligands, respectively33,34. The necessity for migration is rooted in the fact that naive T cells are anatomically restricted to SLOs and blood7,35. In homeostasis, there is a balancing act that dictates T cell dwell time in SLOs. This process is dependent on signals via CC-chemokine receptor 7 (CCR7), which keeps T cells in the T cell zone, and sphingosine 1 phosphate receptor 1 (S1PR1) [G], which senses the bioactive lipid sphingosine-1 phosphate in efferent lymphatics. Desensitization of CCR7 and re-sensitization of S1PR1 enables T cell migration out of the T cell zone and into the downstream efferent lymphatics36,37.
In order for T cells to leave SLOs and respond to perceived threats in a nonlymphoid tissue, they must undergo antigen-driven activation in SLOs. The process of T cell activation is accompanied by their upregulation of surface proteins (including chemokine receptors, such as CXCR3, CXCR6 and CCR5, and integrins, such as α4β7 and α4β1) that are necessary for trafficking to sites of inflammation, as well as their downregulation of receptors involved in keeping T cells in SLOs8,31. Inflammation and antigen stimulation drive dramatic changes to the ordinary rhythms of lymphocyte trafficking into and out of lymph nodes. Inflammation fundamentally changes the lymph node ultrastructure and causes lymph nodes to swell, via a process that involves CLEC2- and podoplanin-mediated regulation of actinomysin contractility in fibroblastic reticular cells38–40. Newer studies have revealed that sensory neuronal input, which can also regulate lymphocyte trafficking and motility in a β2-adrenergic-dependent manner, increases with inflammatory changes41–44. Migration into the lymph node also changes, with high endothelial venules expressing various inflammatory chemokines, including CCL2 and CXCL945–47. This process recruits numerous additional leukocytes, including inflammatory monocytes, which have recently been shown to be critical regulators of naive and effector T cell egress during inflammation as they become a dominant source of S1P48,49. Many of these lessons likely apply to the tumour-draining lymph node, as lymph node hypertrophy (or swelling) is often seen in pre-clinical mouse models and primary human samples50–53. Indeed, lymph draining from tumours contains cytokines, extracellular vesicles and metabolites, all of which flood into the draining lymphoid tissue and likely affect trafficking and cellular function51,53,54.
Recent work has demonstrated that tumour-specific CD8+ T cells are readily identifiable in the tumour-draining lymph nodes55–59. These tumour-specific CD8+ T cells generally display a TPEX phenotype — for example, they express high levels of TCF1 and retain a high degree of functionality55–59. By contrast, tumour-specific CD8+ T cells in the tumour itself generally display terminally exhausted-like states, including the expression of multiple inhibitory receptors and decreased proliferative potential.55–58,60,61. Importantly, the proliferative cell burst that occurs following PD-1 blockade largely occurs in the TPEX cell population, and not in the terminally-exhausted TEX cell [G] population22,23,60,62. Consequently, the anatomical distribution of TPEX versus terminally-exhausted TEX cells is critically important to consider in the context of cancer immunotherapy, and highlights the notion that the tumour-draining lymph node may serve as a critical reservoir of CD8+ T cells that are capable of producing a more durable effector response following PD-1 blockade. Consistent with this concept, preclinical studies have highlighted the importance of the tumour-draining lymph node for responses to checkpoint blockade57,63. Similarly, it is speculated that the tumour-draining lymph node is important for protective anti-tumour immune responses when checkpoint inhibitors are given in the neoadjuvant setting (see Box 3), since delivering checkpoint inhibitors before primary tumour removal could enhance tumour cell killing and delivery of tumour antigens to the tumour-draining lymph node. However, there is evidence in preclinical models suggesting that the TPEX cell population is sequestered in the tumour-draining lymph node, and is not efficiently recruited into the tumour59,61. These tumour-specific CD8+ T cells persistently express high levels of CD69, although whether this is driven entirely by antigen or by some level of inflammation is unclear59,61. If such T cells are sequestered in the tumour-draining lymph node, developing approaches to release these T cells from the lymph node so that they can be recruited into the tumour may be an effective approach for boosting anti-tumour immunity, particularly since the tumour-specific CD8+ T cells in the lymph node generally appear to be more functional and less exhausted than their corresponding counterparts in the tumour.
Box 3: Neoadjuvant checkpoint blockade.
With the successes of checkpoint blockade in advanced, non-resectable disease1–6, there is interest in using checkpoint inhibitors in earlier stage disease. Here, checkpoint blockade can be given in the adjuvant setting [G] (after surgery), or in the neoadjuvant setting [G] (prior to surgery). Perioperative anti-CTLA4 (ipilimumab) was first used in localized urothelial carcinoma, where safety was demonstrated239. Since this initial work, larger clinical studies have examined the efficacy of neoadjuvant checkpoint inhibitors (targeting PD-1, CTLA4, or both) in several cancer types3,221,240. Studies in resectable early stage NSCLC241–244, triple-negative breast cancer (TNBC)245–247, and locally advanced melanoma248–253 in particular have shown tremendous promise. Neoadjuvant checkpoint-based immunotherapy is now FDA approved for specific indications in combination with chemotherapy, including pembrolizumab plus chemotherapy for clinical stage II or III TNBC (July 2021) and nivolumab plus chemotherapy for early stage NSCLC (March 2022). Considering successes in clinical trials, it is anticipated that this approach will become more common in the clinic.
Despite the clinical successes for neoadjuvant checkpoint blockade, we lack an understanding of why this approach is so effective3,221,240. One proposed mechanism is that an early surge in tumour cell killing can release antigens from the primary tumour to the tumour-draining lymph node, improving T cell activation in the lymph node. This could lead to the generation of a more functional anti-tumour T cell response, and after surgical removal of the primary tumour, the T cells remaining in the patient could serve as a source of protection against either relapse or micrometastatic lesions. A major advantage of harnessing the adaptive immune system to eliminate cancer is the notion that T cells are long lived and can traffic to and survey nearly every tissue in the body if adequately primed. Neoadjuvant checkpoint blockade may provide an effective approach for eliciting a broad acting systemic T cell-mediated immune response3,221,240.
Re-positioning T cells to tissues
Following activation in the tissue-draining lymph node, T cells downregulate CD69, upregulate S1PR1, and exit the lymph node64 (Figure 1). From there, T cells are tasked with disseminating to sites of antigen and inflammation33,34. Activated T cells in draining lymphoid tissue can be imprinted with specific homing molecules that allow for preferential migration to the respective upstream organ (for example, α4β7 and CCR9 regulate T cell migration into the gut, and E-selectin, P-selectin, CCR4 and CCR10 are involved in T cell trafficking in skin)65–69. However, while specific homing molecules have been described for the small intestine and skin, lymphocyte migration does not appear to be driven by organ-specific migratory cues for other tissues. Rather, several integrins and chemokine receptors have been shown to be important for migration towards and into sites of inflammation, including the integrins αLβ2 (which binds ICAMs) and α4β1(which binds VCAM1), and the chemokine receptors CXCR3 (the receptor for CXCL9, CXCL10 and CXCL11) and CCR5 (the receptor for CCL3, CCL4 and CCL5)7,33,34. However, it should be noted that upregulation of the requisite receptors for trafficking is not sufficient to get T cells into a tissue of interest. In order for T cells to successfully migrate into a nonlymphoid organ, that tissue must also produce the requisite chemokines and integrin ligands to attract those T cells70,71. The requirement for T cells to have the right receptors and the nonlymphoid organ to have the right chemokines and/or receptor ligands ensures that T cells do not end up at the wrong place at the wrong time.
Figure 1: Comparison of the development of optimal and suboptimal T cell responses.
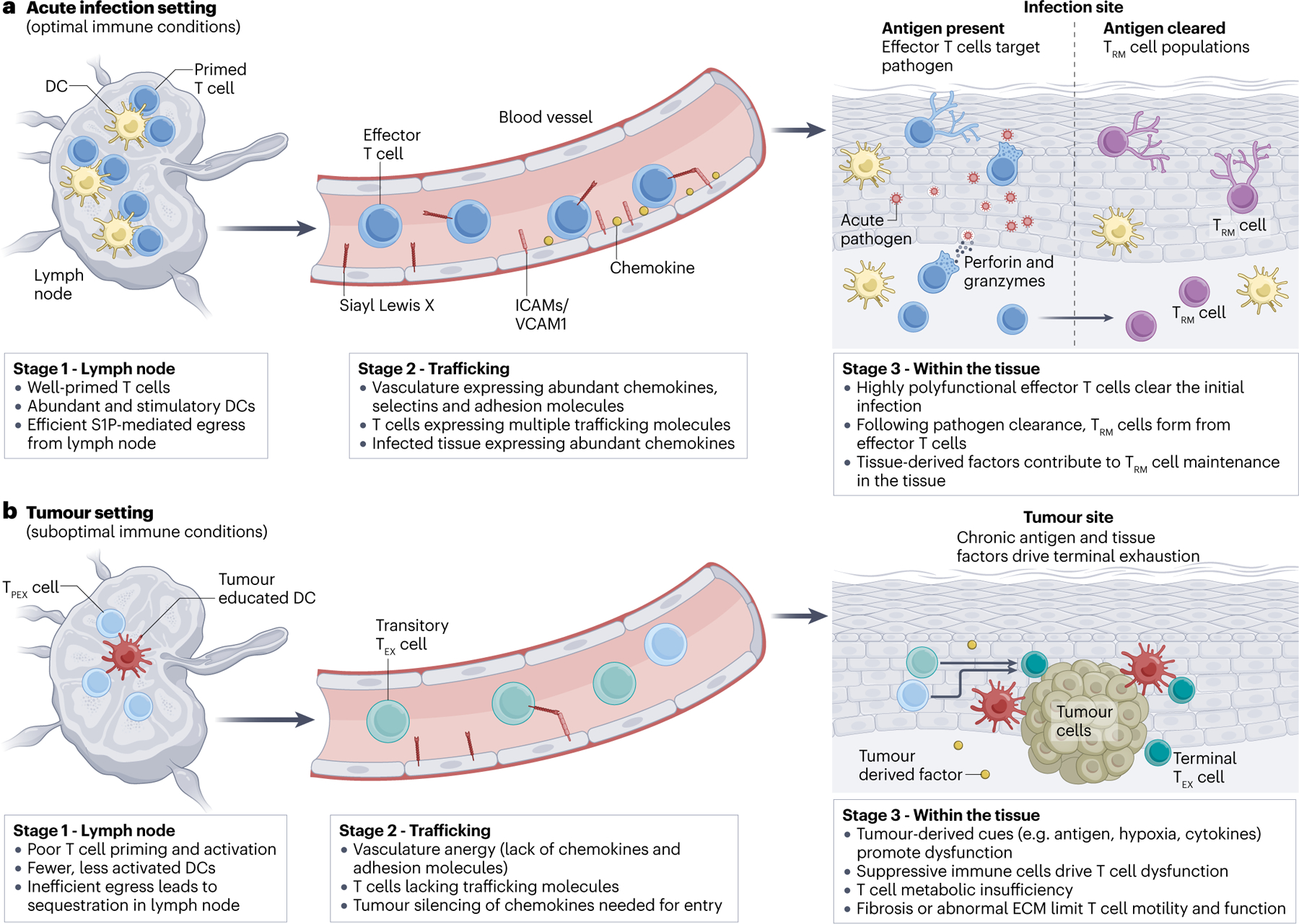
The figure compares the development of a T cell response in (a) an optimal setting (for example, in acute infection) and (b) in a suboptimal setting (for example, in cancer). The major stages of the CD8+ T cell response are depicted, including: stage 1, T cell priming in the tissue-draining lymph node; stage 2, T cell trafficking through blood vessels and extravasation into tissues; and stage 3, T cell adaptation within the tissue microenvironment and their execution of effector functions in tissues. During acute infection (a), T cells are primed in tissue-draining lymph nodes with abundant, stimulatory dendritic cells (DCs), resulting in full effector T cell differentiation and they egress from the lymph node via S1PR1–S1P. Effector T cells enter the vasculature and bind abundant chemokines, selectin ligands, and integrin ligands present on the inflamed vasculature. These effector T cells then extravasate into tissues and are highly polyfunctional, producing inflammatory cytokines (for example, IFNγ, TNF and IL-2) and killing target cells. The concerted efforts of these highly effective, polyfunctional effector T cells results in clearance of the pathogen. Following pathogen clearance, a population of CD8+ TRM cells forms from the effector T cell population and establishes permanent residency within the tissue. TRM cells use molecules such as CD69 and CD103 to stay within the tissue. In the cancer setting (b), CD8+ T cells encounter barriers to successful anti-tumour immunity. In the tumour-draining lymph node, CD8+ T cells can be poorly primed owing to fewer, less-stimulatory DCs. Moreover, some tumour-specific CD8+ T cells become sequestered, failing to go through the normal S1PR1–S1P egress process. In the vasculature, tumour-specific CD8+ T cells can lack the requisite trafficking molecules to efficiently migrate to sites of inflammation. Alternatively, vascular anergy can occur, where blood vessels receive continual VEGF stimulation, causing them to become unable to upregulate inflammatory chemokines (such as CXCL9 and CXCL10) and integrin ligands (such as VCAM1 and ICAM1) that permit leukocyte trafficking and extravasation. Within the tumour, CD8+ T cells are exposed to chronic antigen stimulation, immunosuppressive cytokines and leukocyte populations, hypoxia and/or nutrient deprivation, and fibrosis and/or abnormal extracellular matrix deposition — this limits T cell motility and function. However, exhausted T cells (TEX) cells are not inert. Though functionally not as effective as effector T cells, TEX cells do continue to respond against the tumour, producing some inflammatory cytokines and killing some tumour cells. TEX cell subsets show differential anatomical distribution. Precursors of TEX cells (TPEX) cells are preferentially enriched in the tumour-draining lymph node, while terminally-exhausted TEX cells are more enriched in the tumour itself. While the state of the TEX cells in the blood is still an active area of investigation, it is thought that the blood is enriched in more intermediate-like TEX cell states (such as the CX3CR1+ transitory TEX cell state).
T cell migration into the tumour microenvironment is required for productive responses in cancer (Figure 1). If T cells cannot infiltrate into tumours, they cannot kill cancer cells. While many of the same inflammatory mechanisms, including the secretion of chemokines (such as CXCL9, CXCL10, CCL4 and CCL5) and expression of integrin ligands (ICAMs and VCAM1) can drive T cell recruitment to tumours, numerous barriers exist that often prevent proper T cell accumulation in tumours72–77. Mechanistically, these barriers can be T cell intrinsic (for example, T cells not expressing the right trafficking receptors), or extrinsic (for example, vascular endothelial cells and/or tumour cells not expressing the right ligands and/or chemokines) (Figure 1). One of the key steps for tissue entry is tethering to vascular endothelial cells permeating tissues and extravastating through the endothelial cell layer into the tissue parenchyma. In the setting of cancer, angiogenesis — which is the de novo formation of blood vessels — is common due to the increasing metabolic demands of tumour cells78. New vessel formation and growth are driven by multiple factors, including the production of vascular endothelial growth factor (VEGF) and hypoxia79–82. However, tumoural angiogenesis forms maladapted blood vessels, which are not pruned appropriately, tend to be leaky and suffer from an inability to upregulate inflammatory chemokines and integrin ligands (termed vascular anergy [G]) due to continual VEGF stimulation79–82. These atypical vasculature structures can present a number of challenges for T cell migration, representing a physical barrier against entry (Figure 1).
While the vasculature is a critical gatekeeper for entry into tumours, cancer cells are also able to secrete inflammatory chemokines, including CCL5, CXCL9 and CXCL10, to precipitate leukocyte recruitment into the tumour microenvironment72,76,83. However, tumours can turn off chemokine expression directly through multiple mechanisms, including epigenetic silencing76,84,85 (Figure 1). Moreover, tumour cells can halt T cell trafficking by inhibiting inflammatory pathways (such as type I and type II interferon signalling) that drive chemokine production86–88. Finally, tumour cells can also inhibit secretion of inflammatory chemokines from tumour-infiltrating leukocytes, as has been observed in the context of ovarian cancer, where macrophage production of CXCL9 is blocked by tumor cells76. Collectively, these barriers represent a significant hurdle that must be overcome for T cell infiltration to occur during an anti-tumour immune response (Figure 1). The consequences of these different mechanisms that inhibit T cell migration can lead to immune-excluded or immunologically ‘cold’ microenvironments, which are often associated with poor responses to immunotherapy. Indeed, tumours with positive responses to checkpoint blockade therapy have increased levels of inflammatory chemokines72,89,90. When considering these factors, it is important to be mindful of the dynamic nature of the immune response — early stage disease may exhibit a more productive inflammatory state, better T cell infiltration and a lower degree of T cell exhaustion55,56. Indeed, recent work in autochthonous mouse models of lung adenocarcinoma has demonstrated the progressive loss of T cell recruitment into tumour lesions, and thus the decline of intratumoural T cells with advanced disease55,56. While definitive human studies in multiple cancer types are lacking in this regard, data in pancreatic cancer supports these findings — T cell infiltration is highest in pre-malignant lesions, and decreases as a function of tumour progression91. These alterations are likely due to a multitude of factors, including decreasing levels of T cell stimulation in the tumour draining lymph node, inefficient recruitment and/or extravasation of T cells into tumours, and increasingly unfavorable (that is, immunosuppressive) tumour microenvironments. Additional work will be necessary to define factors that differentiate early and late stage disease.
Adapting is key to survival in tissues
After extravasation [G] into peripheral tissue sites, T cells undergo a broad series of changes that enable them to stay in the new tissue, including an adaptive process that drives numerous transcriptional and epigenetic alterations in order to acclimate to the local microenvironment8,31 (Figure 1). These alterations include the downregulation of cellular machinery that drives tissue egress (including downregulation of S1PR1 and upregulation of CD69)92–95, and the upregulation of adhesion receptors necessary to interact with the local microenvironment (including CD103)94,95. Once within a nonlymphoid tissue, T cells experience numerous cues from their external environment that contribute to shaping their phenotype and function94,95. However, each tissue is different in terms of the environmental constraints placed on residing T cells96–98, and tissues containing malignancies have significantly greater intrinsic barriers for anti-tumour T cell responses. In this section, we will discuss basic principles of T cell retention within tissues and T cell plasticity to adapt to certain microenvironments, drawing parallels between TRM cells and TEX cells in terms of how each of these subsets handles these pressures.
Lessons from TRM cell tissue adaptability
TRM cells that form during acute infection represent an ideal model for studying the T cell adaptations that allow for durable persistence in nonlymphoid tissues and for rapid effector functions upon antigen reactivation94,95. After sensing cognate antigen, CD8+ TRM cells are able to rapidly divide, kill target cells and orchestrate robust and diverse innate and adaptive immune responses in peripheral tissue sites by producing the polyfunctional cytokines IFNγ, TNF and IL-2, collectively resulting in a local and potent anti-pathogen state13,14,99–101 (Box 4).
Box 4: Comparison of shared features between TRM and TEX cell states.
TRM cells that form during acute infection are an ideal example of functional CD8+ T cells in tissues. TRM cells are characterized by their superior ability to mediate host protection against pathogen reencounter, retaining high proliferative capacity, cytotoxicity, and the ability to produce inflammatory cytokines rapidly upon rechallenge (see Box Table)13–15,94,99. These cells are also highly capable of adapting to their tissue microenvironment, are long lived, and retain a high degree of epigenetic plasticity, possessing the ability to re-differentiate into TCM cells or TEM cells upon antigen rechallenge31,9,4,121–123. Notably, the longevity of TRM cells varies depending on the tissue (for example, they are shorter lived in lung and longer lived in skin epidermis). Conversely, TEX cells lose the ability to sustain proliferation, are poorly cytotoxic, and show impaired cytokine production compared to TRM cells (see Box Table)16. TEX cells comprise multiple subsets, with the TPEX cell population retaining higher proliferative potential and showing less signs of dysfunction than the more terminally-exhausted TEX cell subset19,20,23,27. While TEX cells are inferior in host protection compared to TRM cells, they are not inert, and do possess some ability to kill, produce cytokines, and provide some level of host protection. Consequently, TEX cells are important for the host in terms of generating some level of protection in these chronic settings, and provide a therapeutic opportunity to improve outcomes since some TEX cells (for example, TPEX cells) can be at least temporarily reinvigorated to function better22,23,60. Similarly to TRM cells, TEX cells do retain some ability to adapt to their microenvironment. Like TEX cells,TRM cells do express higher levels of inhibitory receptors than other memory subsets.
| Level of characteristic shown in T cell subset | |||
|---|---|---|---|
| Characteristic | Tissue resident memory T cells (TRM) | Precursors of exhausted T cells (TPEX) | Exhausted T cells (TEX) |
| Epigenetic plasticity | High | Medium | Low |
| Proliferative capacity | High | Medium | Low |
| Longevity | High | Medium | Low |
| Antigen-free maintenance | High | Medium | Low |
| Cytokine secretion | High | Medium | Low |
| Cytotoxicity | High | Low | Medium |
| Sensitivity to tissue-derived cues | High | Unknown | Unknown |
| Contribution to host protection | High | Medium | Low |
| Expression of inhibitory receptors | Medium | Medium | High |
| Antigen addiction | Low | Unknown | High |
Concerted efforts to reveal the factors responsible for establishing tissue residency have demonstrated that a variety of cues are critical for the changes that occur in CD8+ T cells. CD69 and CD103 are markers often associated with CD8+ TRM cells during acute infection, and both molecules can play critical roles in retaining these cells in tissues15,94–96,102,103. CD69 antagonizes S1PR1, preventing CD8+ T cells from sensing S1P gradients in the lymphatic vessels draining the tissue that would drive egress104. CD103 is an integrin (also known as αEβ7) that binds the intercellular adhesion molecule E-cadherin, which is particularly important for maintenance of CD8+ T cells in epithelial layers96,102,103,105,106. CD69 and CD103 are not completely faithful markers of CD8+ TRM cells — it is well documented that there can be CD8+ TRM cells lacking surface expression of CD69 and/or CD103, and CD8+ TRM cells can still form in the genetic absence of either CD69 or CD1039,96,107. However, both CD69 and CD103 can play an important role in the retention of cells within certain microenvironments of some tissues, including epithelial layers in the small intestine and skin epidermis96,102,107.
Cues from the tissue can impact expression of both CD103 and CD69 by CD8+ T cells. Studies of CD8+ TRM cells have highlighted a role for the cytokine tumour growth factor β (TGFβ) in regulating CD103 expression92,96,98,103,105,108. TGFβ is synthesized intracellularly and deposited in the extracellular matrix, where it remains in an inactive state unless cleaved109,110. Different molecules and signals can induce cleavage of TGFβ, including proteases, the integrins αvβ6 or αvβ8, reactive oxygen species, thrombospondin-1, or even changes in pH109,110. Moreover, TGFβ signalling can impact a large and broad number of critical host processes, including bone remodelling, wound healing, angiogenesis and fibroblast activation109,110. From a leukocyte perspective, TGFβ is a potent anti-inflammatory cytokine, acting to quell local immune responses109,110. Despite the anti-inflammatory properties of TGFβ, recent work has revealed the critical role it plays in CD8+ TRM cell maintenance in epithelial tissues 92,96,98,103,105,108,111. Elegant work where TGFβRII or CD103 was knocked out in anti-viral CD8+ T cells demonstrated a loss of TRM cells in the skin epidermis and epithelium of the small intestine, while TRM cells in other compartments were spared96,98,103,105,108. Notably, constitutive TGFβ signalling appears to be important, at least in the small intestine epithelium, as blocking active TGFβ after CD8+ TRM cells are established resulted in a loss of TRM as well108. Whether this was due to apoptosis, sloughing off with the epithelium, or migration out of the compartment remains to be determined.
Other cytokines, including IL-33 and type I interferons, have been implicated in establishing CD8+ TRM cells in different peripheral tissue sites. Both cytokines have been shown to regulate the C-type lectin CD69 independently of antigen92,96,112,113. Upregulation of CD69 is a common feature in CD8+ TRM cells. However, CD69 expression is not required for the formation of CD8+ TRM cells, as TRM cells lacking CD69 expression have been documented and CD69-deficient CD8+ T cells are able to establish tissue residency in most peripheral tissues, with the exception of the skin and kidney9,102,107. Additional non-cytokine based cues, including signalling via the aryl hydrocarbon receptor (AhR) [G] and hypoxia, have all been shown to induce phenotypical and transcriptional changes associated with CD8+ TRM cells114,115. Moreover, numerous transcription factors (including HOBIT, BLIMP1, Eomes, T-bet, and RUNX3) have been implicated as being important for the establishment and maintenance of tissue residency111,116,117. However, the respective roles and functions of these transcription factors appears to be somewhat variable. For example, prior work has shown that while downregulation of Eomes and T-bet leads to enhanced enlodgement of TRM cells in the skin epidermis, total knockout results in impaired long-term maintanence due to downregulation of CD122 and diminished IL-15 sensing111. Notably, over expression of either T-bet or Eomes resulted in impaired CD103 expression and reduced TRM cell formation111. On the other hand, expression of HOBIT, BLIMP1, and RUNX3 was required for the formation and maintanence of TRM cells in multiple tissues, including the skin, small intestine, and kidney116–118. Significantly less is known about transcription factor requirements for CD4+ TRM cells. Notably, RUNX3 does not appear to be important for CD4+ TRM cells, demonstrating that there are still significant gaps in our understanding of the molecular drivers responsible for TRM cell formation and longevity, and that these requirements may vary significantly depending on context and cell type118.
While it has become increasingly apparent that CD8+ TRM cells play a dominant role in host protection in nonlymphoid tissues, recent work has highlighted the epigenetic and transcriptional diversity that CD8+ T RM cells adopt across multiple tissues97,102,119,120. However, despite this diversity, CD8+ TRM cells remain relatively plastic, maintaining a poised epigenetic state similar to that seen in TCM cells, which are a population thought to retain the capacity to differentiate into multiple effector T cells subsets in a similar manner to activated naive T cells 121. Consistent with these findings, adoptive transfer of CD8+ TRM cells into naive mice and re-stimulation demonstrates that TRM cells are able to differentiate into effector T cells and to form not only TRM cells, but also TCM and TEM cells, similarly to what is seen with reactivated TCM cells121,122. These findings have been extended in a HOBIT-reporter mouse model, nicely demonstrating CD8+ TRM cell egress and differentiation into functional circulating memory CD8+ T cells123. Some tissues may drive less plastic TRM cells, as may be the case for skin TRM cells that develop after herpes simplex virus infection98. Therefore, despite the numerous tissue-specific alterations that CD8+ TRM cells undergo to adapt to their new environment, these cells remain functional and retain a high degree of plasticity in terms of reprogramming potential. Telologically, TRM cell plasticity may be key with respect to tissue maintanence and functionality. Indeed, tissues are not static, and undergo multiple changes associated with time and environmental factors. Thus, the ability of TRM cells to adapt to changes in the local microenvironment may be dependent on this inherent plasticity. However, this area remains underdeveloped, and additional work is needed to clarify the importance of TRM cell plasticity.
Reprogramming potential of TEX cells
In contrast to CD8+ TRM cells, most CD8+ TEX subsets have an inflexible epigenetic landscape [G] that limits their reprogramming potential and ability to persist in some types of environments (Box 4). TEX cells follow a different developmental trajectory than memory T cells, with the epigenetic landscape of CD8+ T cells that go on to become full fledged TEX cells diverging extremely early from CD8+ T cells that go on to become memory T cells124–129. Indeed, recent work in lymphocytic choriomeningitis virus (LCMV) infection models in mice has demonstrated that significant transcriptional and epigenetic differences emerge as early as the first cell division130. The one TEX cell subset that seems to retain the highest amount of epigenetic plasticity is the TPEX subset21,131. Importantly, some memory properties can be maintained in CD8+ TPEX cells21,125,131,132. Early work demonstrated that if CD8+ T cells were isolated one week after activation in models of chronic infection or tumours and adoptively transferred into antigen-free mice, then they could persist, albeit to a lesser extent than effector CD8+ T cell isolated from acute infection hosts125,126. Moreover, if CD8+ TEX cells are maintained in the context of chronic antigen stimulation for roughly four weeks, TEX cells can no longer persist or adopt functional features associated with bona fide memory T cells125,126. More recent work has demonstrated that CD8+ TPEX cells are more adept at antigen-independent persistence21,131,132. However, while TPEX cells may have more plasticity than other TEX cell subsets, TPEX cells have been shown to adopt a constricted epigenetic posture in chronic infection, even as early as day 5, which appears to limit their differentiation potential21,131. This epigenetic scar persists even after CD8+ TPEX cell transfer into antigen-free hosts, suggesting these changes are permenant. This limited potential to adopt canonical memory-like features is likely multifactorial, but would include a decreased sensitivity to the homeostatic cytokines IL-7 and IL-15128,133,134.
While TEX cells do appear to have a more constrained epigenetic profile, they are not inert, and can be therapeutically leveraged for anti-viral and anti-tumour immunity using several modalities including checkpoint blockade and cytokine stimulation. Although anti-PD-1 monotherapy is able to drive a proliferative burst and effector differentiation in CD8+ TPEX cells in chronic infection and tumours23–25,135,136, it does not fundamentally alter the CD8+ TPEX cell epigenetic state21,22. Thus the efficacy and durability of PD-1-induced functional reinvigoration is limited22,128,137,138. However, more recent work combining anti-PD-1 with IL-2-based therapies have demonstrated improved quantitative and qualitative CD8+ T cell responses in chronic infection and cancer139,140. Moreover, these changes were associated with TEX cell epigenetic alterations, restoring some, but not all epigenetic features associated with effector and memory CD8+ T cells139. These findings have important ramifications for therapeutic modalities aimed at harnessing CD8+ TEX cells to eliminate cancer, suggesting that the boost in anti-tumour immunity will likely be limited if only severely exhausted TEX cells can be targeted.
TEX cells can adopt some TRM-like features
While CD8+ TRM and TEX display significant differences in terms of multipotency, there is increasing evidence, particularly in the context of tumours, that TEX cells may share features of TRM cells that allow them to persist in tissues (Box 4). Indeed, RNA-seq analysis of tumour-infiltrating CD8+ T cells has revealed that CD8+ TEX cells share some transcriptional features observed in CD8+ TRM cells117,141–144. The integrin CD103, which is required to maintain CD8+ TRM cells in the small intestine epithelium and epidermis, is expressed by CD8+ TEX cells in multiple cancer types117,141–146. Moreover, CD103 expression on tumour-infiltrating CD8+ T cells has been shown to be prognostically favourable, igniting an interest in CD103+ TRM-like cells as not only a biomarker for favourable responses in tumours, but also as a possible avenue to explore for immunotherapy117,141–144,147. However, as discussed above, CD103 is regulated by TGFβ, an anti-inflammatory cytokine that is abundantly expressed in the tumour microenvironment148,149. Consequently, it is unclear whether CD103 expression by CD8+ T cells in tumours is simply reflective of a TGFβ-rich microenvironment, or if there is some functional significance to the expression of this marker. Interestingly, different CD8+ T cell populations respond to TGFβ signalling very differently, so the persistence of CD103+ CD8+ T cells in tumours may at least in part reflect the type of T cells capable of surviving in the microenvironment. While TGFβ is able to drive CD103 expression, it only does so in a subset of IL-7R+CD8+ effector T cells; naive or memory CD8+ T cells do not upregulate CD103 expression in response to TGFβ, and KLRG1+ CD8+ effector T cells undergo apoptosis after exposure to TGFβ, demonstrating the importance of T cell differentiation in adapting to different signals61,102,103,150. Recent work in acute infection has demonstrated that TGFβ is able to downregulate the transcription factor TCF1 in the lung, which appeared critical for the formation of TRM cells151. Additionally, work in chronic infection and tumours has demonstrated that TGFβ can play important roles in modulating TPEX cells, including in driving tissue residency within the tumour-draining lymph node, and in modulating mTOR activity and metabolism61,152,153. Taken together, the CD103+ TRM-like population of TEX cells may represent former TCF1+ TEX cells that were capable of responding to TGFβ to adapt to the tissue, rather than undergoing apoptosis. With this perspective in mind, the improved prognosis seen in patients with CD103+ TRM-like TEX cells may be associated with a larger TCF1+ TEX cell response. Indeed, the latter population is known to be important for maintaining the native immune response to tumours and for propagating the immune response in checkpoint blockade22,23,55–57,60,154. Future work developing ways to enable TEX cells to better adapt to their tissue microenvironment in a similar manner to TRM cells may be extremely useful for improving the longevity of T cell responses to tumours.
The tumour–immune microenvironment
Fundamentally, tumours are derived from self. They originate from a wide spectrum of cell types throughout the body. Concordantly, disease manifestation is markedly variable with respect to tumour progression and evolution, the resultant microenvironment, and concomitant immune response. The developmental and evolutionary processes that occur in tumours are markedly complex, but follow a stereotypical progression155. For non-inherited cancers, a transformation event (for example, carcinogen exposure or ageing) occurs that generally produces a genetic alteration, resulting in a driver mutation156,157. Some transformed cells will progress to pre-malignant lesions, which continue to accrue mutations and dysplastic features155–157. After some time, pre-malignant lesions can evolve into primary malignant disease, whereby tumours are restricted to their tissues of origin but invade into new subanatomic compartments155. Finally, after sufficient tumour evolution, some cancers are able to metastasize from their tissue of origin to new tissues, using either the lymph and/or blood as a conduit158–160 (Figure 2). Because of the diverse transcriptional changes that occur over the course of tumour evolution, the immune response, and especially the T cell response, changes considerably over the course of tumour progression155–157. In this section, we will discuss how signals within the tumour microenvironment can shape T cell fate and function. Additionally, we will discuss emerging evidence in the field of metastasis, and how forming a metastatic lesion within certain tissues can lead to poor prognosis in the context of checkpoint blockade.
Figure 2: Immunological challenges associated with metastasis.
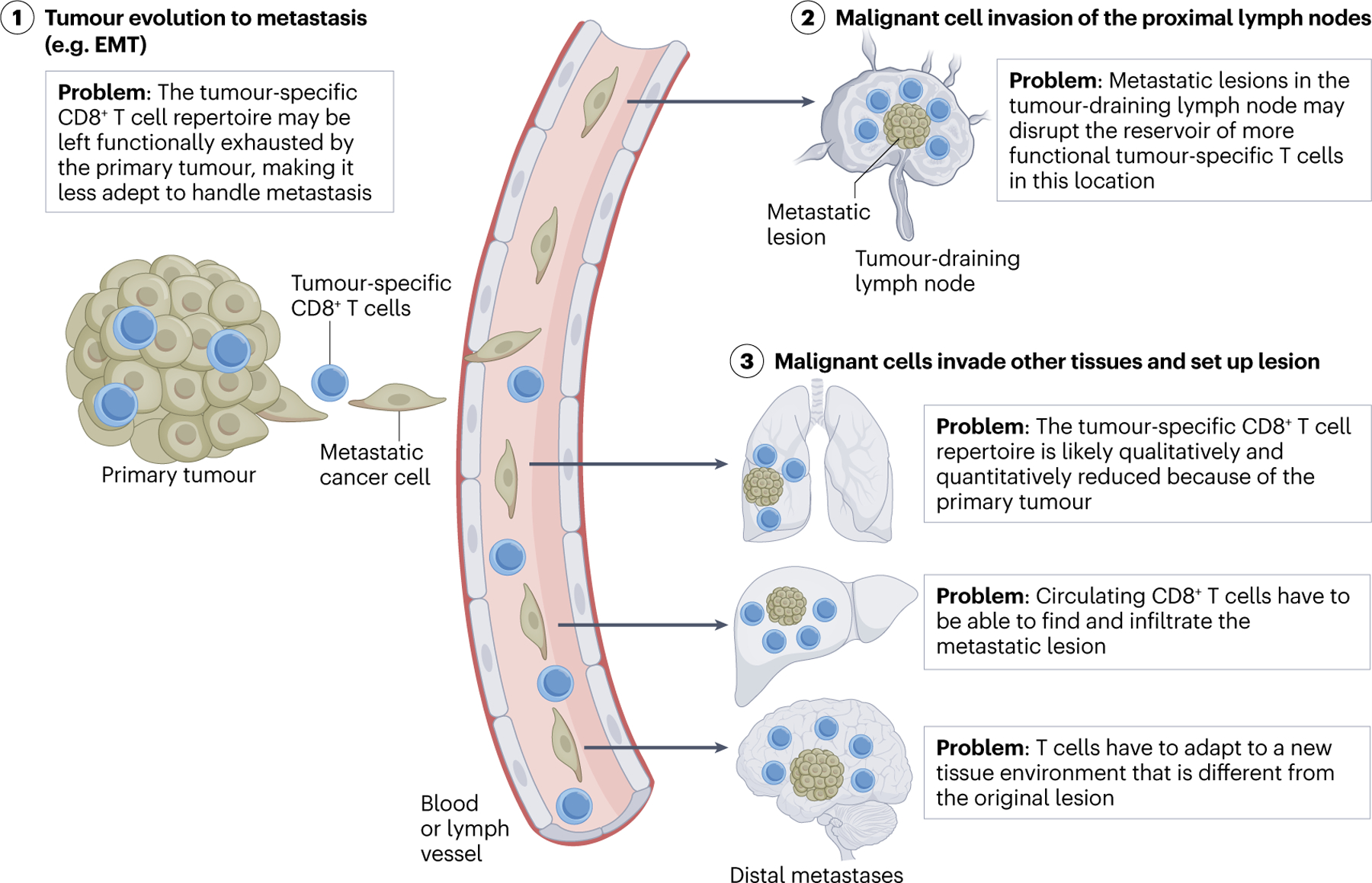
(1) Metastasis is a complex process whereby malignant cells from one location intravasate into blood or lymphatic vasculature, and migrate to another tissue (for example, epithelial to mesenchymal transition (EMT) shown here). A major immunological hurdle for tackling metastasis is that many of the CD8+ T cells in the primary tumour are already exhausted, and may be unable to exit the tissue to search out and destroy the metastatic lesion. (2) Metastases often first localize to the lymph nodes proximal to the primary tumour. The tumour-draining lymph node contains a functional reservoir of tumour-specific CD8+ T cells. How a metastatic lesion in the lymph node impacts this reservoir remains to be determined, but likely will impact the anti-tumour immune response. (3) Malignant cells can also invade other tissues, but must adapt to survive and establish a lesion. CD8+ T cells have to be able to able to find and infiltrate the metastatic lesion in order to execute effector functions. This is compounded by tumour-specific CD8+ T cell repertoire dysfunctionality, and likely due to sequestration of these cells in the primary tumour or tumour-draining lymph node. Thus, there must be circulating tumour-specific CD8+ T cells with the requisite trafficking molecules (on the T cell and vasculature), and if they do infiltrate, they must adapt to yet another tissue microenvironment. It is unclear how plastic or fit the circulating tumour-specific CD8+ T cells must be to overcome these barriers.
Primary tumour environmental challenges
In order to combat the tumour, T cells have to become activated in the tumour-draining lymph node, migrate to the tissue, and adapt to the unique tissue microenvironment containing the tumour (Figure 1). As discussed previously, there are significant hurdles at each of these steps that T cells have to overcome to get into the tumour microenvironment in the first place (Figure 1). Once within the tumour microenvironment, there are a number of additional challenges that T cells have to surmount, including chronic antigen exposure, chronic inflammation (or lack of inflammation), aberrant vasculature, immunosuppressive cytokines, immunosuppressive leukocytes, fibrosis, metabolic constraints and/or nutrient deprivation, and hypoxia32, some of which will be discussed here.
Chronic antigen exposure within the tumour microenvironment is of particular relevance to anti-tumour immunity because of the well described role it plays in the formation of TEX cells. As stated above, once CD8+ T cells progress towards a TEX cell differentiation state, it is incredibly difficult to recover CD8+ T cell function and plasticity125–128,161. Moreover, the development of T cell exhaustion may make dealing with the other factors in the tumour microenvironment more challenging. A significant determinant for how T cell receptor stimulation is perceived by CD8+ T cells is based on the amount of stimulation received and the cell type presenting peptide–MHC class I. Undoubtably, antigen presentation by tumour cells can drive T cell dysfunction18. However, the amount of antigen presented can have significant effects on T cell differentiation — even low levels of antigen expression can drive T cell dysfunction162. While tumours present antigens on MHC class I molecules via the endogenous pathway of antigen processing, other leukocyte populations, namely macrophages and dendritic cells (DCs), are able to cross-present tumour-derived antigens on MHC class I. Macrophages are tasked with maintaining host homeostasis and responding to tissue disruption. In the context of the tumour microenvironment, macrophages often play a role in suppressing the CD8+ T cell response163. In fact, recent work has demonstrated that macrophage–CD8+ T cell interactions tend to be long lived and prevent infiltration and elimination of tumour cells164. This was demonstrated to be due to tumour antigen presentation by macrophages, and it was shown that macrophage antigen presentation to T cells is a strong inducer of T cell dysfunction165. On the other hand, DCs, although far sparser in the tumour, are more likely to promote productive T cell activation in tumours166,167.
Beyond chronic antigen, numerous other factors can dictate the efficacy of the T cell immune response. A substantial barrier to CD8+ T cell immunity is the stroma within tumours. Cancer-associated fibroblasts (CAFs) are a heterogenous group of stromal cells with many significant roles in tumours, but are especially responsible for shaping the local ultrastructure of the tumour. CAFs are derived from local fibroblast precursors168,169, and occupy a variety of cell states within tumours170–172. The fibroblast composition and density of extracellular matrix can be important factors limiting T cell responses173,174. Indeed, in pancreatic cancer in particular, local fibrosis markedly blunts T cell motility within the tumour microenvironment175. Moreover, recent work has highlighted that myofibroblasts alter the extracellular matrix in pancreatic cancer176. Here, it was demonstrated that myofibroblasts secrete a type I collagen homotrimer, consisting of three α1 subunits176. This altered microenvironment changed the local composition of the microbiome and inflammatory chemokines, and preventing formation of type I collagen homotrimers resulted in improved CD8+ T cell responses against tumours176.
Aside from physical constraints in the tumour microenvironment, numerous cytokines are elevated in tumours, including TGFβ, IL-33 and type I IFN. As discussed above, TGFβ and IL-33 can be important for driving a TRM-like phenotype in CD8+ T cells148,149,177. However these cytokines do more than drive direct changes in CD8+ T cell phenotype. TGFβ also modulates CAFs to suppress productive immune responses178,179. Additionally, TGFβ can drive epithelial-to-mesenchymal transition [G] in tumour cells, promoting metastasis180–183. These shifts in tumour cell state matter, as evolution into a mesenchymal or mesenchymal-like state can cause the upregulation of numerous inhibitory receptor ligands (such as PD-L1)184, chemokines and cytokines185 and changes in metabolism186. IL-33, much like TGFβ, can have intrinsic effects on tumour cell state and on CAFs187. It also drives changes in numerous innate and adaptive leukocytes, including mast cells, eosinophils, macrophages, innate lymphoid cells and regulatory T cells188–190. Importantly, eliminating IL-33 or TGFβ results in improved CD8+ T cell responses in multiple different pre-clinical models189,191–193 despite the role of these cytokines in contributing to the establishment of residency and tissue-specific adaptations in CD8+ T cells. These data suggest that the potential benefits of IL-33 and TGFβ on tumour-specific CD8+ T cells may be outweighed by the detrimental effects in terms of modulating immunosuppressive populations and tumour cell biology. Lastly, type I (IFNα/IFNβ) and type II (IFNγ) IFNs have also been shown to be elevated in tumours, though the precise role of these cytokines both in endogenous anti-tumour responses as well as in responses to checkpoint blockade has been somewhat controversial. While acute IFN signalling can have protective anti-tumour effects, chronic IFN signalling can be extremely immunosuppressive, which has been extensively reviewed elsewhere194,195. Determining how to best modulate these cytokines for therapeutic gain remains an active area of investigation, and will likely be heavily context-dependent depending on disease type and stage.
Tumours are exceptional in their ability to metabolically dominate their local microenvironment, stealing resources to grow, proliferate and evolve196. Because of this, the concentration of local metabolites is shifted in the tumour microenvironment as compared to in normal, non-neoplastic tissue [G]196. Moreover, the specific resources used by tumours can be highly dependent on the types of mutations each tumour has (for example, KEAP1 mutation in lung cancer drives a distinct metabolic state)197 and on the tissue the tumours are located in, even where the tumours are of the same origin (for example, pancreatic tumours in the pancreas and those in the subcutaneous space use completely different metabolic pathways)198. These changes inherently matter for T cells. Different T cell subsets all have discrete metabolic needs and requirements. For example, effector T cells are highly dependent on glycolysis, and in the context of a low glucose environment, T cell functionality declines199,200. Moreover, metabolism can divert T cell differentiation state, and can either improve or compound alterations seen in T cell dysfunction201–204. Collectively, tumour metabolism can have significant direct and indirect reprecussions for anti-tumour immunity.
Metastasis – the recipient tissue matters
Metastasis is the process whereby cancer cells migrate away from the primary tumour site, intravasate into either blood or lymphatic vessels, and enter a different tissue to form a new neoplastic lesion158–160 (Figure 2). Metastasis can occur regionally in downstream lymph nodes and adjacent tissues, or can be distal, with tumour cells ending up in a location far away from the original primary tumour158 (Figure 2). Patients with metastasic disease have a significantly worse clinical prognosis, and as such there have been intensive efforts to understand the biology behind metastatic spread158,205,206.
Fundamentally, the biological processes engaged in metastatic versus primary tumours are distinct. In the context of primary tumours, the original transformed cells are derived from the tissue they are initiated in. However, with metastatic spread, one cell type may end up in a tissue lacking that cell type altogether. While metastasis profoundly changes the local microenvironment inhabited by the tumour cells, organ-specific factors are likely to play an important role in shaping the T cell response. Indeed, recent work examining T cells in 21 different tumour types found that T cell states were varied in each tumour type207. However, examining different tumour types that metastasized to the brain demonstrated a much more uniform cell state for T cells across tumour type in the same location, suggesting that the location of the metastatic lesion, rather than the tissue of origin, may be a critical regulator of the T cell response208. Consistent with this concept, when prostate cancer cells were transplanted subcutaneously into the flank or intraosseously into the bone marrow, the resultant T cell response was altered209. Mechanistically, local TGFβ levels were much higher in the bone marrow than in the flank209. Increased TGFβ was sufficient to change the CD4+ T cell response (skewing from a Th1 cell response to a Th17 cell response), and critically, this altered the response to checkpoint blockade therapy209.
A metastatic site that carries additional significance is the lymph node (Figure 2). The lymph node, as alluded to above, represents a safe-haven for tumour-specific CD8+ T cells, and is an important site for lymphocyte homeostasis55–57. By virtue of being downstream of the primary tumour, the tumour-draining lymph node receives a variety of cues including cytokines, metabolites and other factors that can impact lymphatic and endothelial vessel structures, and, by extension, impact T cell recruitment53,210–215. Metastasizing to the lymph node represents a significant immunological problem, as it may disrupt the reservoir of functional tumour-specific CD8+ T cells that are important for maintaining the anti-tumour immune response in the primary tumour lesion210,216. Future studies examining how CD8+ T cell responses in metastatic lesions in diverse tissues differ from one another, and how the tissue site of the metastatic lesion shapes the immune response will be critical.
The impact of metastatic disease on immune responses to checkpoint blockade is currently an active area of investigation. Considering the diversity of potential combinations of factors that are likely to influence response to checkpoint blockade, this is an area that will take significant time and clinical data to disentangle. While it remains unclear how the location of the metastatic lesion contributes to increased resistance to checkpoint blockade therapy, there are common themes that have emerged. For example, the presence of metastatic lesions in the liver is often associated with poor response to checkpoint blockade217. This has been seen in a number of primary cancer types218, including melanoma, non-small cell lung cancer (NSCLC), microsatellite stable (MSS) metastatic colorectal cancer, advanced gastric cancer and advanced colorectal cancer218. Mechanistically, it is speculated that the reason for this poor prognosis is the fact that the liver microenvironment can delete or broadly induce a state of immunological tolerance in liver-infiltrating T cells217. The ability of the liver to induce tolerance has been observed extensively in the transplant setting, where liver transplant patients require less immunosuppression than patients receiving other types of tissue transplants217,219. Recent work in a pre-clinical model utilized a two-tumour approach (one in the flank or lung and one in the liver) to interrogate the impact of tumours in the liver on systemic anti-tumour immunity and on outcomes to PD-1 immunotherapy220. Here, tumour antigens in the liver led to systemic suppression of anti-tumour immunity and reduced efficacy to PD-1 inhibitors. This immune suppression was dependent on regulatory T cells, and reducing regulatory T cell activity using either anti-CTLA4 or an EZH2 inhibitor could promote responsiveness to PD-1 blockade220. Future work extending these findings and further interrogating how metastases to certain organs impacts systemic immunity during PD-1 blockade are warranted to identify potential combination therapies that improve the efficacy of immunotherapy in patients with metastasized tumours.
Concluding remarks
CD8+ T cells are contact-dependent killers. Because the naive T cell repertoire is anatomically restricted to the lymph nodes, spleen and blood, migration is a requisite for all T cell-based immune responses occurring in nonlymphoid sites7,8. Consequently, the ability to localize and adapt to tissue microenvironments is essential for productive CD8+ T cell responses8,31. Once within a tissue, T cells are profoundly shaped by local tissue-derived cues, a process that is important for persistence within the unique physiological constraints of that microenvironment94,95. In cancer, each major step of the T cell response — from productive T cell activation in the tumour-draining lymph node, to migration to the tumour, infiltration of the tumour and adaptation to survive in the tumour microenvironment — represents an opportunity for the tumour to evade host immunity, but also a therapeutic opportunity that can be leveraged clinically (Figure 1). This complexity is amplified in the context of metastasis due to the combinations of primary tumour origin, site of metastatic lesion, current state of the T cell response, and the necessary cues/signals to get T cells to the metastatic lesion, collectively making metastatic disease more difficult to treat clinically (Figure 2).
While immunotherapy has come a long way in terms of curing cancer, additional progress must be made221. Deficiencies in T cell migration and tissue-specific adaptations to thrive in the new microenvironment represent major therapeutic opportunities to augment the efficacy of immunotherapy. Starting from the initiation or generation of a response, better approaches to stimulate CD8+ T cells within the lymph node are needed. These modalities could include: one, newer vaccination-based methodologies, including lipid nanoparticles222–224; two, approaches to release functional tumour-specific CD8+ T cells from the tumour-draining lymph node; and three, approaches that increase migratory DC numbers and their stimulatory capacity within the tumour-draining lymph node, as has been done in pre-clinical models with FLT3L and agonistic CD40 antibody55,225,226. With respect to migration, approaches to surmount or fix vascular anergy may allow for optimal migration of effector T cells into the tumour microenvironment. Significant efforts should also be placed on understanding which T cell differentiation state is best suited to infiltrating and adapting to tumours, and how can T cells be manipulated (for example, through genetic manipulation) to best conform to this cell state. Moreover, efforts to help T cells better adapt and survive within the challenging tumour microenvironment may significantly improve outcomes following immunotherapy. For example, relieving metabolic constraints, nutrient deprivation, and/or hypoxia may improve T cell survival. Additionally, recent work has identified tertiary lymphoid structures (TLS) [G] within tumours, and the presence of these structures is associated with better prognosis and better responses to checkpoint blockade (Box 5). Finally, significant progress to disentangle basic mechanisms of immunological dysfunction in the setting of metastasis, and how to best utilize immunotherapy approaches to overcome these obstacles, will be necessary for future clinical success. Metastatic lesions remain a substantial and often intractable clinical disease, and this area could significantly benefit from reverse translation [G], an approach that takes clinical observations back to the preclinical setting to probe deep mechanism221. Understanding how metastatic lesion location affects the overarching immune response, how to best get T cells to infiltrate metastatic lesions, and what strategies can be utilized to provoke the best immune response possible are all areas needing additional efforts.
Box 5: Tertiary lymphoid structures in cancer.
Emerging work has demonstrated that clusters of leukocytes can be identified in tumours in pre-clinical mouse models and primary human samples. The most common cluster identified is tertiary lymphoid structures (TLS), which are defined by the presence of high endothelial venules and can develop in or around tumours254–258. The degree of organization for TLS is variable, ranging from disorganized (with a mixed B cell and T cell infiltrate) to structures with an established T cell zone, follicles and active germinal centres. TLS have been associated with better clinical prognosis and response to checkpoint blockade therapy255–259. While there are many outstanding questions regarding TLS in cancer, a current hypothesis is that these structures represent areas where T cells can be primed or continuously activated by antigen presenting cells (APCs). Mouse models of viral infection and autoimmunity have demonstrated that a variety of cues result in the genesis of these structures, including lymphotoxin, CXCL12, CXCL13, type I interferon and IL-17260,261. What factor or factors drive TLS formation in tumours is unknown, but recent work has demonstrated that checkpoint blockade may be sufficient to generate TLS262. The other recent TLS-like structure that has been described in tumours consists of proliferative TCF1+CD8+ T cells and MHC class II+ APCs166,263. This co-clustering of TCF1+CD8+ T cells and APCs is prognostically favourable. Patients lacking these co-clusters tend to have poorer T cell infiltration into tumours and progressive disease166. How these structures form, and what their precise role is remains unclear.
A better understanding of the fundamentals of cancer immunology will be key to drive the next generation of immunotherapies. Applying lessons learned from acute infection and pre-clinical models will light the way, and hopefully help to turn the page on cancer fatality.
Key Points:
Productive T cell-mediated immunity is dependent on the ability of T cells to traffic to the site where they are needed and adapt to the new tissue site.
Not all T cells interpret cues from their tissue microenvironment in the same way — T cell differentiation state shapes the way external cues are sensed by the cell.
While most exhausted CD8+ T cell subsets have an inflexible epigenetic framework that limits their adaptability to certain types of microenvironments, they can adopt some resident memory-like features to persist in tissues.
Primary tumours represent aberrant versions of the original host tissue and place strains on T cells, including a high antigen load, an abnormal vasculature, hypoxia, nutrient deprivation and an abnormal extracellular matrix.
Metastasis involves tumour cells from one tissue invading and establishing residence in another tissue, and the physiology of the destination tissue can shape both the new tumour microenvironment and the immune response.
T cell migration and adaptability to different types of microenvironments represent therapeutic opportunities to improve outcomes during cancer immunotherapy.
Acknowledgements
We apologize to colleagues whose work was not cited in our Review due to space constraints. This work was supported by a grant from the US National Institutes of Health (NCI, K08-CA256044 – J.M.S.).
Glossary Terms:
- Adjuvant setting
Refers to therapies that are given after the primary cancer treatment (for example, after major surgery to remove the tumour) and that are intended to keep the cancer from returning.
- Aryl hydrocarbon receptor (AhR)
A ligand-activated transcription factor that integrates environmental, dietary, metabolic and microbial cues within a cell to modulate immune responses in settings of both health and disease. This receptor acts in a ligand-specific, cell-type specific and context-specific manner.
- Cancer immunotherapy
A type of treatment that targets the host immune system to fight cancer.
- Central memory T (TCM) cells
A population of memory T cells that is anatomically restricted to spleen, lymph nodes, and blood, and that uses the same trafficking molecules as naive T cells (namely CCR7, CD62L and LFA1) to circulate through these organs. TCM cells are thought to retain the highest level of plasticity in terms of re-differentiating into other T cell subsets and possess the greatest degree of longevity of the memory subsets.
- Checkpoint inhibitors
Refers to a type of immunotherapy where monoclonal antibodies are used to block major immunological ‘checkpoints’ for immune activation. These checkpoint molecules generally refer to inhibitory receptors expressed by T cells, including CTLA4, PD-1, LAG3, TIM3 and TIGIT, though in some cases the ligand for these receptors (for example, PD-L1) are the target instead of the receptor itself.
- Chemokine receptors
A family of G-protein coupled receptors involved in cellular migration and activation.
- Effector T cells
T cells that have recently encountered antigen and have fully differentiated into an activated state. This differentiation process includes proliferation and acquisition of effector functions, such as inflammatory cytokine production and cytotoxicity.
- Effector memory T (TEM) cells
A population of memory T cells that surveys non-lymphoid tissue and blood following antigen clearance. TEM cells are thought to be a recirculating population, which differentiates them from canonical tissue resident memory T cells, which permanently establish residency in a tissue. TEM cells are generally thought to be less long-lived and less plastic than central memory T cells.
- Epigenetic landscape
Epigenetic regulation refers to the broad set of heritable changes in gene expression that occur independently of changes to the DNA sequence (for example, DNA methylation, histone modifications). The epigenetic landscape refers to the entire set of accessible chromatin regions in a cell, which dictates cell lineage, fate, and effector potential by controlling which genes can actually be expressed.
- Epithelial-to-mesenchymal transition (EMT)
A complex, biological process that allows polarized epithelial cells that normally interact with a basement membrane to convert into a mesenchymal state, enabling enhanced migratory capacity, invasiveness, increased production of extracellular matrix components, and increased resistance to apoptosis. This allows the cell to detach from the basement membrane and migrate away from the epithelial layer. This process occurs during normal embryonic development, tissue generation, organ fibrosis, and wound healing. This process is notably exploited by cancer cells, and is a major pathway involved in tumour invasiveness and metastasis.
- Exhausted T cells (TEX)
A type of T cell dysfunction that is common in chronic infection and cancer. Following activation and differentiation, chronic antigen exposure causes TEX cells to progressively lose effector activity and effector potential, marked by decreased proliferation, cytokine production, and cytotoxicity. TEX cells also express high levels of co-inhibitory receptors and the transcription factor TOX.
- Extravasation
The process of cellular migration from the blood vessels into a tissue.
- Fibrosis
The process by which fibrous connective tissue accumulates in response to tissue injury or damage
- Hypoxia
Refers to a tissue environment where oxygen levels are low
- Immunosuppressive cytokines
Broadly refers to a class of cytokines capable of suppressing or dampening host immune responses. These cytokines are often overexpressed in cancer, and can include IL-10 and TGFβ.
- Immunosuppressive leukocytes
Refers to leukocyte populations that are capable of countering pro-inflammatory immune responses, and often lead to immunotherapy resistance in the context of cancer. These populations include regulatory T cells, myeloid-derived suppressor cells, and some populations of tumour-associated macrophages and neutrophils.
- Integrins
A family of transmembrane receptors that is critical for facilitating cell-cell adhesion and/or cell-extracellular matrix adhesion. Integrins are heterodimers. In humans, there are at least 18 different alpha subunits and 8 different beta subunits, which can heterodimerize to form 24 heterodimers. Integrins bind ligands that are members of the immunoglobulin superfamily.
- Memory T cells
Antigen-experienced T cells that persist long term after antigen clearance. There are multiple subtypes of memory T cells classified broadly based on location, including central memory T cells (restricted to secondary lymphoid organs), effector memory T cells (found recirculating through tissues), and tissue resident memory T cells (permanently retained within a tissue). Memory T cells can re-acquire effector properties upon antigen re-encounter more rapidly than naive T cells.
- Naive T cells
T cells that have not yet become activated by cognate peptide–MHC presented by professional antigen presenting cells. Naive T cells are anatomically restricted to the spleen, lymph nodes, and blood, using the trafficking molecules CCR7 (chemokine receptor binding CCL19 and CCL21), CD62L (selectin binding 6-sulpho sialyl Lewis X oligosaccharides present on high endothelial venules), and LFA1 (an integrin that binds ICAM1) to mediate entry into these sites.
- Neoadjuvant setting
Broadly refers to therapies that are given before the primary cancer treatment (for example, prior to major surgery to remove the tumour).
- Non-neoplastic tissue
A tissue that has not transformed and/or does not contain a tumour
- Progenitor TEX (TPEX) cells
A subset of TEX cells that expresses high levels of the transcription factor TCF1, lower levels of co-inhibitory receptors, and retains higher proliferative capacity than other TEX cell subsets. The TPEX cell subset contains stem-like properties, being able to divide to give raise to more TPEX cells, as well as differentiate into other TEX cell subset including the terminally-exhausted TEX cell subset. This subset preferentially proliferates in response to PD-1 checkpoint blockade.
- Resident memory T (TRM) cells
A population of memory T cells that establishes residency within a given tissue (that is, once it enters, it does not leave). TRM cells have been described in both lymphoid tissue and non-lymphoid tissue. The surface markers CD69 and CD103 have both been associated with TRM cells, though not all TRM cells express these markers.
- Reverse translation
An approach where observations are made from clinical samples that are hypothesis generating, and then those hypotheses are subsequently tested in preclinical mouse models where mechanism can be interrogated.
- Selectins
A family of cell surface adhesion molecules that is important for leukocyte trafficking. Selectins are single chain, transmembrane glycoproteins that bind fucosylated, sialyated, or sulfated ligands.
- Sphingosine-1-phosphate receptor 1 (S1PR1)
A G-protein coupled receptor that binds the phospholipid sphingosine 1-phosphate (S1P). S1PR1 regulates T cell migration between tissues and circulatory fluids. S1PR1 plays a critical role in T cell egress from lymph nodes and tissues by enabling T cells to sense high levels of S1P in efferent lymphatics and blood. S1PR1 is directly antagonized by CD69 at the cell surface, so if CD69 is expressed, T cells fail to up regulate S1PR1 and respond to S1P.
- Terminally-exhausted TEX cells.
A subset of TEX cells that is terminally differentiated, expresses low to no TCF1, and high levels of co-inhibitory receptors including PD-1, TIM3, LAG3, and TIGIT. Terminally-exhausted TEX have poorer proliferative capacity and inflammatory cytokine production than other TEX cell subsets, but do retain a heightened ability to kill target cells. Terminally-exhausted TEX cells cannot differentiate into other TEX cell subsets, and are poorly proliferative in response to PD-1 checkpoint blockade.
- Tertiary lymphoid structures (TLS)
Induced ectopic lymphoid structures that develop in non-lymphoid tissues and/or tumours. TLS are organized aggregates of immune cells that resemble secondary lymphoid organs, but are not encapsulated. TLS are generally associated with inflamed tissues, and have been documented in cancer, autoimmunity, and chronic inflammatory disorders.
- Vascular anergy
A phenomenon that occurs when blood vessels receive continual VEGF stimulation, causing them to become unable to upregulate inflammatory chemokines and integrin ligands to permit leukocyte trafficking
Footnotes
Competing Interests
The authors declare that they have no competing interests.
Peer review information
Nature Reviews Immunology thanks C. Pritzl, E. Teixeiro-Pernas and the other, anonymous, reviewer for their contribution to the peer review of this work.
References
- 1.Sharpe AH & Pauken KE The diverse functions of the PD1 inhibitory pathway. Nature reviews. Immunology, doi: 10.1038/nri.2017.108 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Sharma P & Allison JP The future of immune checkpoint therapy. Science 348, 56–61, doi: 10.1126/science.aaa8172 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Taube JM & Pardoll DM Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367, doi: 10.1126/science.aax0182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355, doi: 10.1126/science.aar4060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pauken KE, Torchia JA, Chaudhri A, Sharpe AH & Freeman GJ Emerging concepts in PD-1 checkpoint biology. Semin Immunol, 101480, doi: 10.1016/j.smim.2021.101480 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vesely MD, Zhang T & Chen L Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annual review of immunology 40, 45–74, doi: 10.1146/annurev-immunol-070621-030155 (2022). [DOI] [PubMed] [Google Scholar]
- 7.Masopust D & Schenkel JM The integration of T cell migration, differentiation and function. Nature reviews. Immunology 13, 309–320, doi: 10.1038/nri3442 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Mueller SN, Gebhardt T, Carbone FR & Heath WR Memory T cell subsets, migration patterns, and tissue residence. Annual review of immunology 31, 137–161, doi: 10.1146/annurev-immunol-032712-095954 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Steinert EM et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749, doi: 10.1016/j.cell.2015.03.031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartolome-Casado R et al. Resident memory CD8 T cells persist for years in human small intestine. J Exp Med 216, 2412–2426, doi: 10.1084/jem.20190414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallett LJ et al. Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes. J Exp Med 217, doi: 10.1084/jem.20200050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snyder ME et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol 4, doi: 10.1126/sciimmunol.aav5581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schenkel JM et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101, doi: 10.1126/science.1254536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ariotti S et al. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105, doi: 10.1126/science.1254803 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Gebhardt T et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nature immunology 10, 524–530, doi: 10.1038/ni.1718 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nature reviews. Immunology 15, 486–499, doi: 10.1038/nri3862 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thommen DS & Schumacher TN T Cell Dysfunction in Cancer. Cancer cell 33, 547–562, doi: 10.1016/j.ccell.2018.03.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philip M & Schietinger A CD8(+) T cell differentiation and dysfunction in cancer. Nature reviews. Immunology 22, 209–223, doi: 10.1038/s41577-021-00574-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pauken KE & Wherry EJ Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276, doi: 10.1016/j.it.2015.02.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kallies A, Zehn D & Utzschneider DT Precursor exhausted T cells: key to successful immunotherapy? Nature reviews. Immunology 20, 128–136, doi: 10.1038/s41577-019-0223-7 (2020). [DOI] [PubMed] [Google Scholar]
- 21. Abdel-Hakeem MS et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nature immunology 22, 1008–1019, doi: 10.1038/s41590-021-00975-5 (2021). Refs 21, 28–30 are recent studies demonstrating that TEX bear epigenetic scars that prevent them from forming functional memory T cells.
- 22.Miller BC et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nature immunology 20, 326–336, doi: 10.1038/s41590-019-0312-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421, doi: 10.1038/nature19330 (2016). Ref 146 was critical for showing that TCF-1+ CD8 T cells were the cells responsible for proliferating after checkpoint blockade therapy
- 24.He R et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–428, doi: 10.1038/nature19317 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Utzschneider DT et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427, doi: 10.1016/j.immuni.2016.07.021 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Blackburn SD, Shin H, Freeman GJ & Wherry EJ Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proceedings of the National Academy of Sciences of the United States of America 105, 15016–15021, doi: 10.1073/pnas.0801497105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225, doi: 10.1126/science.1229620 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hensel N et al. Memory-like HCV-specific CD8(+) T cells retain a molecular scar after cure of chronic HCV infection. Nature immunology 22, 229–239, doi: 10.1038/s41590-020-00817-w (2021). [DOI] [PubMed] [Google Scholar]
- 29.Yates KB et al. Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nature immunology 22, 1020–1029, doi: 10.1038/s41590-021-00979-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tonnerre P et al. Differentiation of exhausted CD8(+) T cells after termination of chronic antigen stimulation stops short of achieving functional T cell memory. Nature immunology 22, 1030–1041, doi: 10.1038/s41590-021-00982-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masopust D & Soerens AG Tissue-Resident T Cells and Other Resident Leukocytes. Annual review of immunology 37, 521–546, doi: 10.1146/annurev-immunol-042617-053214 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salmon H, Remark R, Gnjatic S & Merad M Host tissue determinants of tumour immunity. Nature reviews. Cancer 19, 215–227, doi: 10.1038/s41568-019-0125-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krummel MF, Bartumeus F & Gerard A T cell migration, search strategies and mechanisms. Nature reviews. Immunology 16, 193–201, doi: 10.1038/nri.2015.16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fowell DJ & Kim M The spatio-temporal control of effector T cell migration. Nature reviews. Immunology 21, 582–596, doi: 10.1038/s41577-021-00507-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Girard JP, Moussion C & Forster R HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nature reviews. Immunology 12, 762–773, doi: 10.1038/nri3298 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Pham TH, Okada T, Matloubian M, Lo CG & Cyster JG S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity 28, 122–133, doi: 10.1016/j.immuni.2007.11.017 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shannon LA et al. CCR7/CCL19 controls expression of EDG-1 in T cells. J Biol Chem 287, 11656–11664, doi: 10.1074/jbc.M111.310045 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Astarita JL et al. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nature immunology 16, 75–84, doi: 10.1038/ni.3035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Assen FP et al. Multitier mechanics control stromal adaptations in the swelling lymph node. Nature immunology 23, 1246–1255, doi: 10.1038/s41590-022-01257-4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horsnell HL et al. Lymph node homeostasis and adaptation to immune challenge resolved by fibroblast network mechanics. Nature immunology 23, 1169–1182, doi: 10.1038/s41590-022-01272-5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakai A, Hayano Y, Furuta F, Noda M & Suzuki K Control of lymphocyte egress from lymph nodes through beta2-adrenergic receptors. J Exp Med 211, 2583–2598, doi: 10.1084/jem.20141132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki K, Hayano Y, Nakai A, Furuta F & Noda M Adrenergic control of the adaptive immune response by diurnal lymphocyte recirculation through lymph nodes. J Exp Med 213, 2567–2574, doi: 10.1084/jem.20160723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devi S et al. Adrenergic regulation of the vasculature impairs leukocyte interstitial migration and suppresses immune responses. Immunity 54, 1219–1230 e1217, doi: 10.1016/j.immuni.2021.03.025 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Huang S et al. Lymph nodes are innervated by a unique population of sensory neurons with immunomodulatory potential. Cell 184, 441–459 e425, doi: 10.1016/j.cell.2020.11.028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palframan RT et al. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med 194, 1361–1373, doi: 10.1084/jem.194.9.1361 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kastenmuller W et al. Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity 38, 502–513, doi: 10.1016/j.immuni.2012.11.012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guarda G et al. L-selectin-negative CCR7- effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nature immunology 8, 743–752, doi: 10.1038/ni1469 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Leal JM et al. Innate cell microenvironments in lymph nodes shape the generation of T cell responses during type I inflammation. Sci Immunol 6, doi: 10.1126/sciimmunol.abb9435 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baeyens A et al. Monocyte-derived S1P in the lymph node regulates immune responses. Nature 592, 290–295, doi: 10.1038/s41586-021-03227-6 (2021). Newer study demonstrating the important role inflammatory monocytes play in T cell retention withing inflammed lymph nodes.
- 50.Habenicht LM, Albershardt TC, Iritani BM & Ruddell A Distinct mechanisms of B and T lymphocyte accumulation generate tumor-draining lymph node hypertrophy. Oncoimmunology 5, e1204505, doi: 10.1080/2162402X.2016.1204505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pucci F et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science 352, 242–246, doi: 10.1126/science.aaf1328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz LH et al. Evaluation of lymph nodes with RECIST 1.1. Eur J Cancer 45, 261–267, doi: 10.1016/j.ejca.2008.10.028 (2009). [DOI] [PubMed] [Google Scholar]
- 53.du Bois H, Heim TA & Lund AW Tumor-draining lymph nodes: At the crossroads of metastasis and immunity. Sci Immunol 6, eabg3551, doi: 10.1126/sciimmunol.abg3551 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ubellacker JM et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118, doi: 10.1038/s41586-020-2623-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schenkel JM et al. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1(+) CD8(+) T cells in tumor-draining lymph nodes. Immunity, doi: 10.1016/j.immuni.2021.08.026 (2021). Refs 55–58 are recent studies demonstrating the importance of tumor specific CD8 T cells in the tumor draining lymph node for both maintaining the native immune response in tumors and after checkpoint blockade therapy.
- 56.Connolly KA et al. A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol 6, eabg7836, doi: 10.1126/sciimmunol.abg7836 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dammeijer F et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer cell, doi: 10.1016/j.ccell.2020.09.001 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Huang Q et al. The primordial differentiation of tumor-specific memory CD8(+) T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 185, 4049–4066 e4025, doi: 10.1016/j.cell.2022.09.020 (2022). [DOI] [PubMed] [Google Scholar]
- 59. Molodtsov AK et al. Resident memory CD8(+) T cells in regional lymph nodes mediate immunity to metastatic melanoma. Immunity 54, 2117–2132 e2117, doi: 10.1016/j.immuni.2021.08.019 (2021). Refs 59 and 61 are important studies demonstrating that tumor specific CD8 T cells appear resident and/or sequestered within the tumor draining lymph node.
- 60.Siddiqui I et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110, doi: 10.1016/j.immuni.2018.12.021 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Li G et al. TGF-beta-dependent lymphoid tissue residency of stem-like T cells limits response to tumor vaccine. Nat Commun 13, 6043, doi: 10.1038/s41467-022-33768-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurtulus S et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50, 181–194 e186, doi: 10.1016/j.immuni.2018.11.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fransen MF et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 3, doi: 10.1172/jci.insight.124507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benechet AP et al. T cell-intrinsic S1PR1 regulates endogenous effector T-cell egress dynamics from lymph nodes during infection. Proceedings of the National Academy of Sciences of the United States of America 113, 2182–2187, doi: 10.1073/pnas.1516485113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iwata M et al. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538, doi: 10.1016/j.immuni.2004.08.011 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Johansson-Lindbom B et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med 202, 1063–1073, doi: 10.1084/jem.20051100 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hammerschmidt SI et al. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J Exp Med 205, 2483–2490, doi: 10.1084/jem.20080039 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reiss Y, Proudfoot AE, Power CA, Campbell JJ & Butcher EC CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J Exp Med 194, 1541–1547, doi: 10.1084/jem.194.10.1541 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sigmundsdottir H et al. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nature immunology 8, 285–293, doi: 10.1038/ni1433 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Campanella GS, Medoff BD, Manice LA, Colvin RA & Luster AD Development of a novel chemokine-mediated in vivo T cell recruitment assay. J Immunol Methods 331, 127–139, doi: 10.1016/j.jim.2007.12.002 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shin H & Iwasaki A A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491, 463–467, doi: 10.1038/nature11522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoch T et al. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci Immunol 7, eabk1692, doi: 10.1126/sciimmunol.abk1692 (2022). [DOI] [PubMed] [Google Scholar]
- 73.Spranger S, Dai D, Horton B & Gajewski TF Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer cell 31, 711–723 e714, doi: 10.1016/j.ccell.2017.04.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mikucki ME et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6, 7458, doi: 10.1038/ncomms8458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Galeano Nino JL et al. Cytotoxic T cells swarm by homotypic chemokine signalling. Elife 9, doi: 10.7554/eLife.56554 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dangaj D et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer cell 35, 885–900 e810, doi: 10.1016/j.ccell.2019.05.004 (2019). Refs 76 and 84 demonstrate that tumors epigentically silence inflammatory chemokines to modulate the anti-tumor immune response.
- 77.Woods AN et al. Differential Expression of Homing Receptor Ligands on Tumor-Associated Vasculature that Control CD8 Effector T-cell Entry. Cancer Immunol Res 5, 1062–1073, doi: 10.1158/2326-6066.CIR-17-0190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Palma M, Biziato D & Petrova TV Microenvironmental regulation of tumour angiogenesis. Nature reviews. Cancer 17, 457–474, doi: 10.1038/nrc.2017.51 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Huang Y et al. Improving immune-vascular crosstalk for cancer immunotherapy. Nature reviews. Immunology 18, 195–203, doi: 10.1038/nri.2017.145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P & Griffioen AW Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol 18, 527–540, doi: 10.1038/s41571-021-00496-y (2021). [DOI] [PubMed] [Google Scholar]
- 81.Griffioen AW, Damen CA, Blijham GH & Groenewegen G Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood 88, 667–673 (1996). [PubMed] [Google Scholar]
- 82. Huijbers EJM, Khan KA, Kerbel RS & Griffioen AW Tumors resurrect an embryonic vascular program to escape immunity. Sci Immunol 7, eabm6388, doi: 10.1126/sciimmunol.abm6388 (2022). Newer work demonstrating that vascular anergy in tumors is due to the development of an angiogenesis gene program seen in embryonic development.
- 83.Reschke R et al. Immune cell and tumor cell-derived CXCL10 is indicative of immunotherapy response in metastatic melanoma. J Immunother Cancer 9, doi: 10.1136/jitc-2021-003521 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253, doi: 10.1038/nature15520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nagarsheth N et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer research 76, 275–282, doi: 10.1158/0008-5472.CAN-15-1938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao J et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404 e399, doi: 10.1016/j.cell.2016.08.069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grasso CS et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer cell 38, 500–515 e503, doi: 10.1016/j.ccell.2020.08.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Paschen A, Melero I & Ribas A Central Role of the Antigen-Presentation and Interferon-γ Pathways in Resistance to Immune Checkpoint Blockade. Annual Review of Cancer Biology 6, 85–102, doi: 10.1146/annurev-cancerbio-070220-111016 (2022). [DOI] [Google Scholar]
- 89.Chow MT et al. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512 e1495, doi: 10.1016/j.immuni.2019.04.010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.House IG et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 487–504, doi: 10.1158/1078-0432.CCR-19-1868 (2020). [DOI] [PubMed] [Google Scholar]
- 91.Bernard V et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 2194–2205, doi: 10.1158/1078-0432.CCR-18-1955 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Skon CN et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nature immunology 14, 1285–1293, doi: 10.1038/ni.2745 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Evrard M et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J Exp Med 219, doi: 10.1084/jem.20210116 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mueller SN & Mackay LK Tissue-resident memory T cells: local specialists in immune defence. Nature reviews. Immunology 16, 79–89, doi: 10.1038/nri.2015.3 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Schenkel JM & Masopust D Tissue-Resident Memory T Cells. Immunity 41, 886–897, doi: 10.1016/j.immuni.2014.12.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Casey KA et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol 188, 4866–4875, doi: 10.4049/jimmunol.1200402 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Crowl JT et al. Tissue-resident memory CD8(+) T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nature immunology 23, 1121–1131, doi: 10.1038/s41590-022-01229-8 (2022). Cutting edge single cell RNAseq and ATACseq study demonstrating how different tissue microenvironments drive significant changes in TRM
- 98.Christo SN et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nature immunology 22, 1140–1151, doi: 10.1038/s41590-021-01004-1 (2021). [DOI] [PubMed] [Google Scholar]
- 99.Schenkel JM, Fraser KA, Vezys V & Masopust D Sensing and alarm function of resident memory CD8(+) T cells. Nature immunology 14, 509–513, doi: 10.1038/ni.2568 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beura LK et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nature immunology 19, 173–182, doi: 10.1038/s41590-017-0029-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park SL et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nature immunology 19, 183–191, doi: 10.1038/s41590-017-0027-5 (2018). [DOI] [PubMed] [Google Scholar]
- 102.Mackay LK et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nature immunology 14, 1294–1301, doi: 10.1038/ni.2744 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Sheridan BS et al. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40, 747–757, doi: 10.1016/j.immuni.2014.03.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shiow LR et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544, doi: 10.1038/nature04606 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Zhang N & Bevan MJ Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696, doi: 10.1016/j.immuni.2013.08.019 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cepek KL et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature 372, 190–193, doi: 10.1038/372190a0 (1994). [DOI] [PubMed] [Google Scholar]
- 107.Walsh DA et al. The Functional Requirement for CD69 in Establishment of Resident Memory CD8(+) T Cells Varies with Tissue Location. J Immunol 203, 946–955, doi: 10.4049/jimmunol.1900052 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mohammed J et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nature immunology 17, 414–421, doi: 10.1038/ni.3396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tauriello DVF, Sancho E & Batlle E Overcoming TGFbeta-mediated immune evasion in cancer. Nature reviews. Cancer 22, 25–44, doi: 10.1038/s41568-021-00413-6 (2022). [DOI] [PubMed] [Google Scholar]
- 110.Akhurst RJ & Hata A Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 11, 790–811, doi: 10.1038/nrd3810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mackay LK et al. T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 43, 1101–1111, doi: 10.1016/j.immuni.2015.11.008 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Freeman BE, Hammarlund E, Raue HP & Slifka MK Regulation of innate CD8+ T-cell activation mediated by cytokines. Proceedings of the National Academy of Sciences of the United States of America 109, 9971–9976, doi: 10.1073/pnas.1203543109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kohlmeier JE, Cookenham T, Roberts AD, Miller SC & Woodland DL Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 33, 96–105, doi: 10.1016/j.immuni.2010.06.016 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zaid A et al. Persistence of skin-resident memory T cells within an epidermal niche. Proceedings of the National Academy of Sciences of the United States of America 111, 5307–5312, doi: 10.1073/pnas.1322292111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hasan F, Chiu Y, Shaw RM, Wang J & Yee C Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program. JCI Insight 6, doi: 10.1172/jci.insight.138970 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mackay LK et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463, doi: 10.1126/science.aad2035 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Milner JJ et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257, doi: 10.1038/nature24993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fonseca R et al. Runx3 drives a CD8(+) T cell tissue residency program that is absent in CD4(+) T cells. Nature immunology 23, 1236–1245, doi: 10.1038/s41590-022-01273-4 (2022). [DOI] [PubMed] [Google Scholar]
- 119.Milner JJ et al. Heterogenous Populations of Tissue-Resident CD8(+) T Cells Are Generated in Response to Infection and Malignancy. Immunity 52, 808–824 e807, doi: 10.1016/j.immuni.2020.04.007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kurd NS et al. Early precursors and molecular determinants of tissue-resident memory CD8(+) T lymphocytes revealed by single-cell RNA sequencing. Sci Immunol 5, doi: 10.1126/sciimmunol.aaz6894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fonseca R et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nature immunology 21, 412–421, doi: 10.1038/s41590-020-0607-7 (2020). Refs 121–123 are important studies demonstrating the plasticity of TRM to redifferentiate into effectors, and form TRM, TEM and TCM.
- 122.Masopust D, Vezys V, Wherry EJ, Barber DL & Ahmed R Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol 176, 2079–2083 (2006). [DOI] [PubMed] [Google Scholar]
- 123.Behr FM et al. Tissue-resident memory CD8(+) T cells shape local and systemic secondary T cell responses. Nature immunology 21, 1070–1081, doi: 10.1038/s41590-020-0723-4 (2020). [DOI] [PubMed] [Google Scholar]
- 124.Chen Z et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51, 840–855 e845, doi: 10.1016/j.immuni.2019.09.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Angelosanto JM, Blackburn SD, Crawford A & Wherry EJ Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. Journal of virology 86, 8161–8170, doi: 10.1128/JVI.00889-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Schietinger A et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 45, 389–401, doi: 10.1016/j.immuni.2016.07.011 (2016). Refs 125 and 126 provide functional evidence for the rapid potential change (e.g. the inability to form TMEM) in TEX in tumors and chronic infection.
- 127. Sen DR et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169, doi: 10.1126/science.aae0491 (2016). Refs 127, 128, and 161 were the first studies to demonstrate that TEX are epigentically constrained in chronic infections and tumors.
- 128.Pauken KE et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165, doi: 10.1126/science.aaf2807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Giles JR et al. Shared and distinct biological circuits in effector, memory and exhausted CD8(+) T cells revealed by temporal single-cell transcriptomics and epigenetics. Nature immunology 23, 1600–1613, doi: 10.1038/s41590-022-01338-4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Quezada LK et al. Early transcriptional and epigenetic divergence of CD8+ T cells responding to acute versus chronic infection. PLoS Biol 21, e3001983, doi: 10.1371/journal.pbio.3001983 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Utzschneider DT et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nature immunology 21, 1256–1266, doi: 10.1038/s41590-020-0760-z (2020). [DOI] [PubMed] [Google Scholar]
- 132.Utzschneider DT et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nature immunology 14, 603–610, doi: 10.1038/ni.2606 (2013). [DOI] [PubMed] [Google Scholar]
- 133.Shin H, Blackburn SD, Blattman JN & Wherry EJ Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med 204, 941–949, doi: 10.1084/jem.20061937 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pellegrini M et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 144, 601–613, doi: 10.1016/j.cell.2011.01.011 (2011). [DOI] [PubMed] [Google Scholar]
- 135.Zander R et al. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51, 1028–1042 e1024, doi: 10.1016/j.immuni.2019.10.009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hudson WH et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 51, 1043–1058 e1044, doi: 10.1016/j.immuni.2019.11.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ghoneim HE et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 170, 142–157 e119, doi: 10.1016/j.cell.2017.06.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mognol GP et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proceedings of the National Academy of Sciences of the United States of America 114, E2776–E2785, doi: 10.1073/pnas.1620498114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hashimoto M et al. PD-1 combination therapy with IL-2 modifies CD8(+) T cell exhaustion program. Nature 610, 173–181, doi: 10.1038/s41586-022-05257-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Codarri Deak L et al. PD-1-cis IL-2R agonism yields better effectors from stem-like CD8(+) T cells. Nature 610, 161–172, doi: 10.1038/s41586-022-05192-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ganesan AP et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nature immunology 18, 940–950, doi: 10.1038/ni.3775 (2017). Refs 141 and 142 were some of the original studies correlating TRM-like cell magnitude with improved prognosis
- 142.Savas P et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nature medicine 24, 986–993, doi: 10.1038/s41591-018-0078-7 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Egelston CA et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 4, doi: 10.1172/jci.insight.130000 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Clarke J et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med 216, 2128–2149, doi: 10.1084/jem.20190249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Webb JR, Milne K, Watson P, Deleeuw RJ & Nelson BH Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 434–444, doi: 10.1158/1078-0432.CCR-13-1877 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Duhen T et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9, 2724, doi: 10.1038/s41467-018-05072-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Park SL et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 565, 366–371, doi: 10.1038/s41586-018-0812-9 (2019). [DOI] [PubMed] [Google Scholar]
- 148.Derynck R, Turley SJ & Akhurst RJ TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol 18, 9–34, doi: 10.1038/s41571-020-0403-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Batlle E & Massague J Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 50, 924–940, doi: 10.1016/j.immuni.2019.03.024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sanjabi S, Mosaheb MM & Flavell RA Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 31, 131–144, doi: 10.1016/j.immuni.2009.04.020 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wu J et al. T Cell Factor 1 Suppresses CD103+ Lung Tissue-Resident Memory T Cell Development. Cell Rep 31, 107484, doi: 10.1016/j.celrep.2020.03.048 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Gabriel SS et al. Transforming growth factor-beta-regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity 54, 1698–1714 e1695, doi: 10.1016/j.immuni.2021.06.007 (2021). [DOI] [PubMed] [Google Scholar]
- 153.Hu Y et al. TGF-beta regulates the stem-like state of PD-1+ TCF-1+ virus-specific CD8 T cells during chronic infection. J Exp Med 219, doi: 10.1084/jem.20211574 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zehn D, Thimme R, Lugli E, de Almeida GP & Oxenius A ‘Stem-like’ precursors are the fount to sustain persistent CD8(+) T cell responses. Nature immunology 23, 836–847, doi: 10.1038/s41590-022-01219-w (2022). [DOI] [PubMed] [Google Scholar]
- 155.Umar A, Dunn BK & Greenwald P Future directions in cancer prevention. Nature reviews. Cancer 12, 835–848, doi: 10.1038/nrc3397 (2012). [DOI] [PubMed] [Google Scholar]
- 156.McGranahan N & Swanton C Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628, doi: 10.1016/j.cell.2017.01.018 (2017). [DOI] [PubMed] [Google Scholar]
- 157.Kennedy SR, Zhang Y & Risques RA Cancer-Associated Mutations but No Cancer: Insights into the Early Steps of Carcinogenesis and Implications for Early Cancer Detection. Trends Cancer 5, 531–540, doi: 10.1016/j.trecan.2019.07.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Klein CA Cancer progression and the invisible phase of metastatic colonization. Nature reviews. Cancer 20, 681–694, doi: 10.1038/s41568-020-00300-6 (2020). [DOI] [PubMed] [Google Scholar]
- 159.Pereira ER et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 359, 1403–1407, doi: 10.1126/science.aal3622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brown M et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 359, 1408–1411, doi: 10.1126/science.aal3662 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456, doi: 10.1038/nature22367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Westcott PMK et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat Cancer 2, 1071–1085, doi: 10.1038/s43018-021-00247-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.DeNardo DG & Ruffell B Macrophages as regulators of tumour immunity and immunotherapy. Nature reviews. Immunology 19, 369–382, doi: 10.1038/s41577-019-0127-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Peranzoni E et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proceedings of the National Academy of Sciences of the United States of America 115, E4041–E4050, doi: 10.1073/pnas.1720948115 (2018). Refs 164 and 165 were critical for demonstrating the inhibitory nature of macrophage based antigen presentation on CD8 T cell differentiation and responses.
- 165.Kersten K et al. Spatiotemporal co-dependency between macrophages and exhausted CD8(+) T cells in cancer. Cancer cell 40, 624–638 e629, doi: 10.1016/j.ccell.2022.05.004 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jansen CS et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470, doi: 10.1038/s41586-019-1836-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Broz ML et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer cell 26, 638–652, doi: 10.1016/j.ccell.2014.09.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Arina A et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proceedings of the National Academy of Sciences of the United States of America 113, 7551–7556, doi: 10.1073/pnas.1600363113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Turley SJ, Cremasco V & Astarita JL Immunological hallmarks of stromal cells in the tumour microenvironment. Nature reviews. Immunology 15, 669–682, doi: 10.1038/nri3902 (2015). [DOI] [PubMed] [Google Scholar]
- 170.Davidson S et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep 31, 107628, doi: 10.1016/j.celrep.2020.107628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Foster DS et al. Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer cell 40, 1392–1406 e1397, doi: 10.1016/j.ccell.2022.09.015 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Luo H et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun 13, 6619, doi: 10.1038/s41467-022-34395-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Krishnamurty AT et al. LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 611, 148–154, doi: 10.1038/s41586-022-05272-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lakins MA, Ghorani E, Munir H, Martins CP & Shields JD Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun 9, 948, doi: 10.1038/s41467-018-03347-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cox TR The matrix in cancer. Nature reviews. Cancer 21, 217–238, doi: 10.1038/s41568-020-00329-7 (2021). [DOI] [PubMed] [Google Scholar]
- 176.Chen Y et al. Oncogenic collagen I homotrimers from cancer cells bind to alpha3beta1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer cell 40, 818–834 e819, doi: 10.1016/j.ccell.2022.06.011 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Briukhovetska D et al. Interleukins in cancer: from biology to therapy. Nature reviews. Cancer 21, 481–499, doi: 10.1038/s41568-021-00363-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sahai E et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nature reviews. Cancer 20, 174–186, doi: 10.1038/s41568-019-0238-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bordignon P et al. Dualism of FGF and TGF-beta Signaling in Heterogeneous Cancer-Associated Fibroblast Activation with ETV1 as a Critical Determinant. Cell Rep 28, 2358–2372 e2356, doi: 10.1016/j.celrep.2019.07.092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.David CJ et al. TGF-beta Tumor Suppression through a Lethal EMT. Cell 164, 1015–1030, doi: 10.1016/j.cell.2016.01.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Su J et al. TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577, 566–571, doi: 10.1038/s41586-019-1897-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Oshimori N, Oristian D & Fuchs E TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160, 963–976, doi: 10.1016/j.cell.2015.01.043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Scheel C et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940, doi: 10.1016/j.cell.2011.04.029 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chen L et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 5, 5241, doi: 10.1038/ncomms6241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hsu DS et al. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer cell 26, 534–548, doi: 10.1016/j.ccell.2014.09.002 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Ramesh V, Brabletz T & Ceppi P Targeting EMT in Cancer with Repurposed Metabolic Inhibitors. Trends Cancer 6, 942–950, doi: 10.1016/j.trecan.2020.06.005 (2020). [DOI] [PubMed] [Google Scholar]
- 187.Andersson P et al. Molecular mechanisms of IL-33-mediated stromal interactions in cancer metastasis. JCI Insight 3, doi: 10.1172/jci.insight.122375 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liew FY, Girard JP & Turnquist HR Interleukin-33 in health and disease. Nature reviews. Immunology 16, 676–689, doi: 10.1038/nri.2016.95 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Li A et al. IL-33 Signaling Alters Regulatory T Cell Diversity in Support of Tumor Development. Cell Rep 29, 2998–3008 e2998, doi: 10.1016/j.celrep.2019.10.120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pastille E et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol 12, 990–1003, doi: 10.1038/s41385-019-0176-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Park JH et al. Nuclear IL-33/SMAD signaling axis promotes cancer development in chronic inflammation. EMBO J 40, e106151, doi: 10.15252/embj.2020106151 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li S et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in T(H) cells. Nature 587, 121–125, doi: 10.1038/s41586-020-2850-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen ML et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proceedings of the National Academy of Sciences of the United States of America 102, 419–424, doi: 10.1073/pnas.0408197102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Minn AJ Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol 36, 725–737, doi: 10.1016/j.it.2015.09.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gocher AM, Workman CJ & Vignali DAA Interferon-gamma: teammate or opponent in the tumour microenvironment? Nature reviews. Immunology 22, 158–172, doi: 10.1038/s41577-021-00566-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lyssiotis CA & Kimmelman AC Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol 27, 863–875, doi: 10.1016/j.tcb.2017.06.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sporn MB & Liby KT NRF2 and cancer: the good, the bad and the importance of context. Nature reviews. Cancer 12, 564–571, doi: 10.1038/nrc3278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lau AN et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife 9, doi: 10.7554/eLife.56782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chang CH et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251, doi: 10.1016/j.cell.2013.05.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Menk AV et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep 22, 1509–1521, doi: 10.1016/j.celrep.2018.01.040 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bader JE, Voss K & Rathmell JC Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell 78, 1019–1033, doi: 10.1016/j.molcel.2020.05.034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Reina-Campos M, Scharping NE & Goldrath AW CD8(+) T cell metabolism in infection and cancer. Nature reviews. Immunology 21, 718–738, doi: 10.1038/s41577-021-00537-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Wei J et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476, doi: 10.1038/s41586-019-1821-z (2019). Refs 203 and 204 use CRISPR screens to demonstrate the promise of modulating CD8 T cell metabolism to drive improved anti-tumor immune responses.
- 204.Huang H et al. In vivo CRISPR screening reveals nutrient signaling processes underpinning CD8(+) T cell fate decisions. Cell 184, 1245–1261 e1221, doi: 10.1016/j.cell.2021.02.021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Stuelten CH, Parent CA & Montell DJ Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nature reviews. Cancer 18, 296–312, doi: 10.1038/nrc.2018.15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Joyce JA & Pollard JW Microenvironmental regulation of metastasis. Nature reviews. Cancer 9, 239–252, doi: 10.1038/nrc2618 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zheng L et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474, doi: 10.1126/science.abe6474 (2021). Large, multi-cancer based study demonstrating similarities and differences in tumor infiltrating CD8 T cells in multiple cancer types.
- 208. Sudmeier LJ et al. Distinct phenotypic states and spatial distribution of CD8(+) T cell clonotypes in human brain metastases. Cell Rep Med 3, 100620, doi: 10.1016/j.xcrm.2022.100620 (2022). First demonstration by single cell RNAseq that CD8 T cells infiltrating different types of metastatic tumors in the brain look relatively homogenous, demonstrating that tumor location may play a significant role in T cell programming.
- 209. Jiao S et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell 179, 1177–1190 e1113, doi: 10.1016/j.cell.2019.10.029 (2019). This study demonstrated that prostate tumor cells implanted in different organ had very different looking tumor microenvironments, immune responses, and responses to checkpoint blockade therapy
- 210.Reticker-Flynn NE et al. Lymph node colonization induces tumor-immune tolerance to promote distant metastasis. Cell 185, 1924–1942 e1923, doi: 10.1016/j.cell.2022.04.019 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kataru RP et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 34, 96–107, doi: 10.1016/j.immuni.2010.12.016 (2011). [DOI] [PubMed] [Google Scholar]
- 212.Harrell MI, Iritani BM & Ruddell A Tumor-induced sentinel lymph node lymphangiogenesis and increased lymph flow precede melanoma metastasis. Am J Pathol 170, 774–786, doi: 10.2353/ajpath.2007.060761 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hirakawa S et al. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med 201, 1089–1099, doi: 10.1084/jem.20041896 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Qian CN et al. Preparing the “soil”: the primary tumor induces vasculature reorganization in the sentinel lymph node before the arrival of metastatic cancer cells. Cancer research 66, 10365–10376, doi: 10.1158/0008-5472.CAN-06-2977 (2006). [DOI] [PubMed] [Google Scholar]
- 215.Bekkhus T et al. Remodeling of the Lymph Node High Endothelial Venules Reflects Tumor Invasiveness in Breast Cancer and is Associated with Dysregulation of Perivascular Stromal Cells. Cancers (Basel) 13, doi: 10.3390/cancers13020211 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Rahim MK et al. Dynamic CD8(+) T cell responses to cancer immunotherapy in human regional lymph nodes are disrupted in metastatic lymph nodes. Cell 186, 1127–1143 e1118, doi: 10.1016/j.cell.2023.02.021 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lee JC et al. The Liver-Immunity Nexus and Cancer Immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research 28, 5–12, doi: 10.1158/1078-0432.CCR-21-1193 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chen XJ, Ren A, Zheng L, Zheng ED & Jiang T Pan-Cancer Analysis Identifies Liver Metastases as Negative Predictive Factor for Immune Checkpoint Inhibitors Treatment Outcome. Front Immunol 12, 651086, doi: 10.3389/fimmu.2021.651086 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Benseler V et al. The liver: a special case in transplantation tolerance. Semin Liver Dis 27, 194–213, doi: 10.1055/s-2007-979471 (2007). [DOI] [PubMed] [Google Scholar]
- 220.Lee JC et al. Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci Immunol 5, doi: 10.1126/sciimmunol.aba0759 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sharma P & Allison JP Immune checkpoint therapy: Forging ahead. Science translational medicine 14, eadf2947, doi: 10.1126/scitranslmed.adf2947 (2022). [DOI] [PubMed] [Google Scholar]
- 222.Hou X, Zaks T, Langer R & Dong Y Lipid nanoparticles for mRNA delivery. Nat Rev Mater 6, 1078–1094, doi: 10.1038/s41578-021-00358-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Baharom F et al. Intravenous nanoparticle vaccination generates stem-like TCF1(+) neoantigen-specific CD8(+) T cells. Nature immunology 22, 41–52, doi: 10.1038/s41590-020-00810-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Baharom F et al. Systemic vaccination induces CD8(+) T cells and remodels the tumor microenvironment. Cell 185, 4317–4332 e4315, doi: 10.1016/j.cell.2022.10.006 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hegde S et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer cell 37, 289–307 e289, doi: 10.1016/j.ccell.2020.02.008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lin JH et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med 217, doi: 10.1084/jem.20190673 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wu T et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1, doi: 10.1126/sciimmunol.aai8593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Khan O et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218, doi: 10.1038/s41586-019-1325-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274, doi: 10.1038/s41586-019-1324-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269, doi: 10.1038/s41586-019-1326-9 (2019). [DOI] [PubMed] [Google Scholar]
- 231.Yao C et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nature immunology 20, 890–901, doi: 10.1038/s41590-019-0403-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tsui C et al. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature 609, 354–360, doi: 10.1038/s41586-022-05105-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Scharping NE et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nature immunology 22, 205–215, doi: 10.1038/s41590-020-00834-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ford BR et al. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci Immunol 7, eabj9123, doi: 10.1126/sciimmunol.abj9123 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Feng Q et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun 13, 4981, doi: 10.1038/s41467-022-32521-8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yao C et al. BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8(+) T cells. Nature immunology 22, 370–380, doi: 10.1038/s41590-021-00868-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Beltra JC et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841 e828, doi: 10.1016/j.immuni.2020.04.014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Brummelman J et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med 215, 2520–2535, doi: 10.1084/jem.20180684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Carthon BC et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 2861–2871, doi: 10.1158/1078-0432.CCR-10-0569 (2010). This is the first study to examine the beneficial effects of neoadjuvant immunotherapy in human tumors.
- 240.Mittendorf EA, Burgers F, Haanen J & Cascone T Neoadjuvant Immunotherapy: Leveraging the Immune System to Treat Early-Stage Disease. Am Soc Clin Oncol Educ Book 42, 1–15, doi: 10.1200/EDBK_349411 (2022). [DOI] [PubMed] [Google Scholar]
- 241.Forde PM et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. The New England journal of medicine 378, 1976–1986, doi: 10.1056/NEJMoa1716078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Cascone T et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: the phase 2 randomized NEOSTAR trial. Nature medicine 27, 504–514, doi: 10.1038/s41591-020-01224-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gao S et al. Neoadjuvant PD-1 inhibitor (Sintilimab) in NSCLC. J Thorac Oncol 15, 816–826, doi: 10.1016/j.jtho.2020.01.017 (2020). [DOI] [PubMed] [Google Scholar]
- 244.Altorki NK et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: a single-centre, randomised phase 2 trial. Lancet Oncol 22, 824–835, doi: 10.1016/S1470-2045(21)00149-2 (2021). [DOI] [PubMed] [Google Scholar]
- 245.Mittendorf EA et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet 396, 1090–1100, doi: 10.1016/S0140-6736(20)31953-X (2020). [DOI] [PubMed] [Google Scholar]
- 246.Schmid P et al. Pembrolizumab for Early Triple-Negative Breast Cancer. The New England journal of medicine 382, 810–821, doi: 10.1056/NEJMoa1910549 (2020). [DOI] [PubMed] [Google Scholar]
- 247.Schmid P et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. The New England journal of medicine 386, 556–567, doi: 10.1056/NEJMoa2112651 (2022). [DOI] [PubMed] [Google Scholar]
- 248.Tarhini AA et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PloS one 9, e87705, doi: 10.1371/journal.pone.0087705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Amaria RN et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nature medicine 24, 1649–1654, doi: 10.1038/s41591-018-0197-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Blank CU et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nature medicine 24, 1655–1661, doi: 10.1038/s41591-018-0198-0 (2018). [DOI] [PubMed] [Google Scholar]
- 251.Huang AC et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nature medicine 25, 454–461, doi: 10.1038/s41591-019-0357-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Menzies AM et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nature medicine 27, 301–309, doi: 10.1038/s41591-020-01188-3 (2021). [DOI] [PubMed] [Google Scholar]
- 253.Rozeman EA et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nature medicine 27, 256–263, doi: 10.1038/s41591-020-01211-7 (2021). [DOI] [PubMed] [Google Scholar]
- 254.Joshi NS et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43, 579–590, doi: 10.1016/j.immuni.2015.08.006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Petitprez F et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560, doi: 10.1038/s41586-019-1906-8 (2020). [DOI] [PubMed] [Google Scholar]
- 256.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555, doi: 10.1038/s41586-019-1922-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cabrita R et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565, doi: 10.1038/s41586-019-1914-8 (2020). [DOI] [PubMed] [Google Scholar]
- 258.Gao J et al. Neoadjuvant PD-L1 plus CTLA-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nature medicine 26, 1845–1851, doi: 10.1038/s41591-020-1086-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Vanhersecke L et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer 2, 794–802, doi: 10.1038/s43018-021-00232-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sautes-Fridman C, Petitprez F, Calderaro J & Fridman WH Tertiary lymphoid structures in the era of cancer immunotherapy. Nature reviews. Cancer 19, 307–325, doi: 10.1038/s41568-019-0144-6 (2019). [DOI] [PubMed] [Google Scholar]
- 261.Schumacher TN & Thommen DS Tertiary lymphoid structures in cancer. Science 375, eabf9419, doi: 10.1126/science.abf9419 (2022). [DOI] [PubMed] [Google Scholar]
- 262.Hua Y et al. Cancer immunotherapies transition endothelial cells into HEVs that generate TCF1(+) T lymphocyte niches through a feed-forward loop. Cancer cell, doi: 10.1016/j.ccell.2022.11.002 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Di Pilato M et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 184, 4512–4530 e4522, doi: 10.1016/j.cell.2021.07.015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]