

Abstract
Abstract
Voltage-gated sodium channels are crucial mediators of neuronal damage in ischemic and excitotoxicity disease models. Fenamates have been reported to have anti-inflammatory properties following a decrease in prostaglandin synthesis. Several researches showed that fenamates appear to be ion channel modulators and potential neuroprotectants. In this study, the neuroprotective effects of tolfenamic acid, flufenamic acid, and mefenamic acid were tested by glutamate-induced injury in SH-SY5Y cells. Following this, fenamates’ effects were examined on both the expression level and the function of hNav1.1 and hNav1.2, which were closely associated with neuroprotection, using Western blot and patch clamp. Finally, the effect of fenamates on the expression of apoptosis-related proteins in SH-SY5Y cells was examined. The results showed that both flufenamic acid and mefenamic acid exhibited neuroprotective effects against glutamate-induced injury in SH-SY5Y cells. They inhibited peak currents of both hNav1.1 and hNav1.2. However, fenamates exhibited decreased inhibitory effects on hNav1.1 when compared to hNav1.2. Correspondingly, the inhibitory effect of fenamates was found to be consistent with the level of neuroprotective effects in vitro. Fenamates inhibited glutamate-induced apoptosis through the modulation of the Bcl-2/Bax-dependent cell death pathways. Taken together, Nav1.2 might play a part in fenamates’ neuroprotection mechanism.
Graphic Abstract
Nav1.2 and NMDAR might take part in the neuroprotection mechanism of the fenamates. The fenamates inhibited glutamate-induced apoptosis through modulation of the Bcl-2/Bax-dependent cell death pathways.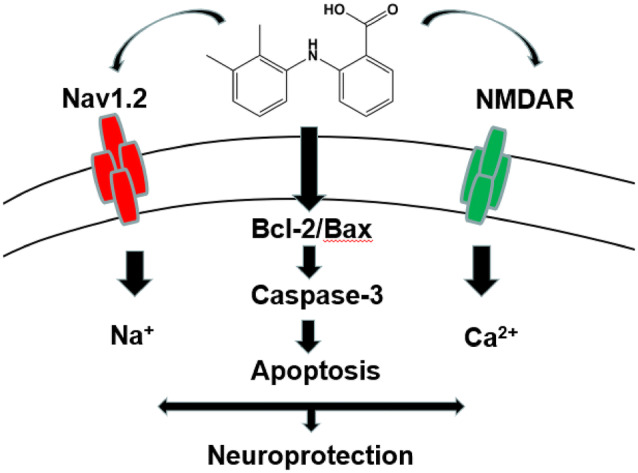
Keywords: Neuroprotection, Fenamates, Voltage-gated sodium channel, Apoptosis
Introduction
Neurons are known to be susceptible to ischemic injury. Glutamate receptors, which are widely distributed in the central nervous system (CNS), are one of the mechanisms of neuron ischemic susceptibility (Rothman 1984; Simon et al. 1984). They can lead to excitotoxic neuro-degenerative changes when over-activated (Olney 1978). Recent research has focused on voltage-gated sodium channel (VGSC-) inhibitors, which may be able to protect the surviving neurons from further destruction.
Nonsteroidal anti-inflammatory drugs (NSAIDs), along with anti-inflammatory, antipyretic, and analgesic activities, have been widely used in clinical as therapeutic drugs. Fenamates (anthranilic acid), tend to be used for the clinical treatment of inflammation and pain (Jiang et al. 2012). These drugs reduce the biosynthesis of prostaglandins by inhibiting the cyclooxygenase (COX) enzyme (Flower et al. 1972; Sanger and Bennett 1979). Recently, several studies have demonstrated the potential neuroprotective effects of fenamates in several experiments. Both flufenamic acid and mefenamic acid are efficacious in NLRP3-dependent rodent models. They have also been shown to have therapeutic effects for both memory loss and Alzheimer's disease in mice (Daniels et al. 2016). It has also been demonstrated that mefenamic acid has a neuroprotective effect against MCAO brain injury in rats (Khansari and Halliwell 2009).
Several approaches for the treatment of neuronal ischemic injury are being developed. Ion-channel inhibitors, particularly sodium channel and calcium channel inhibitors, have been used as approaches under ischemic insult. Fenamates, a family of NSAIDs, may inhibit the COX pathway (Flower 1974). In addition, recent research has demonstrated that fenamates may inhibit several ion channels, including sodium currents in rat dorsal root ganglion (DRG) neurons, rat hippocampal pyramidal neurons, and VGSC subtypes Nav1.7 and Nav1.8 (Sun et al. 2018; Yau et al. 2010; Lee et al. 2003), as well as CaCC and KATP channels (White and Aylwin 1990; Greenwood and Large 1995; Grover et al. 1994).
VGSCs are crucial mediators of neuronal damage in ischemic and excitotoxicity disease models. Fenamates have shown inhibition effects on several ion channels. Furthermore, mefenamic acid has demonstrated neuroprotective effects in several models. Mefenamic acid has the potential to reduce glutamate-induced cell death and reduce infarct volume, total ischemic brain damage and edema (Khansari and Halliwell 2009). However, the relationship between the neuroprotective activity of fenamates and VGSCs has received less research attention.
In this study, the therapeutic potential of tolfenamic acid, flufenamic acid, and mefenamic acid in terms of neuroprotective effects as well as potential mechanisms of action was examined. Using an in vitro model against glutamate-induced cell toxicity, the research demonstrated that mefenamic acid and flufenamic acid showed both neuroprotective effects and inhibitory effects on glutamate-induced apoptosis in SH-SY5Y cells. Inhibition effects on peak currents of Nav1.2 may participate in their neuroprotective mechanism.
Results
Fenamates Ameliorated the Decrease of Neuronal Cell Viability Induced by Glutamate
As detected by the MTT method, 100 μmol/L–100 mmol/L glutamate treatment for 24 and 48 h caused a decrease of cell viability in SH-SY5Y cells (Fig. 1a). In addition, treatment with 10 nmol/L–100 μmol/L mefenamic acid, flufenamic acid, and tolfenamic acid treatment alone did not affect the survival rate of SH-SY5Y cells (Fig. 1b). Therefore, 10 mmol/L glutamate and 1 μmol/L, 10 μmol/L, and 100 μmol/L of the fenamates were selected for cell co-treatment. Data demonstrated that the flufenamic acid and mefenamic acid could ameliorate the decrease of cell viability in a concentration-dependent manner, although tolfenamic acid did not. Deltamethrin, a sodium channel activator, could aggravate the nerve damage by glutamate (Fig. 1c). When glutamate, fenamates and deltamethrin were administered simultaneously, the neuroprotective effects of both mefenamic acid and flufenamic acid were, to some extent, reduced (Fig. 1d, e, f). These results suggested that fenamates’ neuroprotective are related to sodium channels.
Fig. 1.

Fenamates could ameliorate the decrease of neuronal cells viability induced by glutamate. Effect of different concentrations of glutamate (a) and fenamates (b) on cell viability in SH-SY5Y cells. Changes in cell survival rate after 48 h of 10 mmol/L glutamate treatment in the absence or presence of deltamethrin (c). Changes in cell survival rate after 48 h of 10 mmol/L glutamate treatment in the absence or presence of tolfenamic acid (d), flufenamic acid (e), and mefenamic acid (f) at referential doses. Data represent mean ± SEM (n = 12). *P < 0.05, **P < 0.01 (glutamate vs fenamates)
Effects of Fenamates on the Expression of Nav1.1, Nav1.2, and Nav1.6
Nav1.1, Nav1.2, and Nav1.6 are VGSC subtypes closely associated with neuroprotection (Sandalon et al. 2013; Stevens et al. 2013; Shi et al. 2019). Therefore, fenamates effects were determined and compared on the expression of Nav1.1, Nav1.2, and Nav1.6 in glutamate-treated SH-SY5Y cells. According to Western blot analysis, Nav1.1 and Nav1.2 were expressed in SH-SY5Y, while Nav1.6 was not. Cells pre-incubated with fenamates showed similar expression levels of Nav1.1 and Nav1.2 with SH-SY5Y and glutamate-treated groups (Fig. 2).
Fig. 2.

Effects of fenamates on the expression of Nav1.1, Nav1.2, and Nav1.6. Cells were treated with 10 mmol/L glutamate for 48 h with or without treatment with fenamates. Nav1.1, Nav1.2, and Nav1.6 protein expression were determined in the whole-cell extract by Western blot. Nav1.1, Nav1.2, and Nav1.6 protein expression levels were normalized to β-actin. Data represent mean ± SEM (n = 3)
Inhibitory Effects on hNav1.1 and hNav1.2 by Fenamates
In order to explore fenamates’ effects on sodium currents, they were determined on hNav1.1 and hNav1.2, as Nav1.6 was essentially not expressed. Results showed that all three fenamates exhibited inhibitory effects on peak currents of hNav1.2. However, peak currents of hNav1.1 exhibited decreased inhibitory effect by fenamates. The amplitude of peak currents on hNav1.1 could be inhibited to 76, 79, and 84% by 100 μmol/L of mefenamic acid, flufenamic acid, and tolfenamic acid, while the peak currents of hNav1.2 could be inhibited to 28, 60, and 70% by 100 μmol/L of mefenamic acid, flufenamic acid, and tolfenamic acid (Fig. 3). Mefenamic acid, flufenamic acid, and tolfenamic acid inhibited the peak sodium currents in a concentration-dependent manner with calculated IC50 values of 535 μM, 1.80 mM, and 2.01 mM for hNav1.1 and 29.8 μM, 249 μM, and 615 μM for hNav1.2, respectively (Fig. 3c, f). Compared with hNav1.1, the fenamates showed much stronger inhibitory effects on hNav1.2. In addition, the intensity of inhibition of hNav1.2 evoked by the three fenamates was consistent with the intensity of their neuroprotective effects in SH-SY5Y cells.
Fig. 3.

Effects of fenamates on the Na+ peak current of hNav1.1 and hNav1.2. Averaged current traces were obtained from hNav1.1 (a) and hNav1.2 (c) in each group. Current–voltage relationships of hNav1.1 (b) and hNav1.2 (d) were evoked by 100 μmol/L fenamates. Normalized Na+ peak currents were depolarized at − 20 mV after 5 min treatment in absence or presence of 100 nmol/L, 1 μmol/L, 10 μmol/L, and 100 μmol/L of fenamates on hNav1.1 (c) and hNav1.2 (f). Data represent mean ± SEM (n = 6)
Effects of Voltage Dependent Properties on hNav1.1 and hNav1.2 by Fenamates
Fenamates’ effects were tested on the activation, inactivation, and recovery properties of hNav1.1 and hNav1.2. As can be seen in Fig. 4, V1/2 values were shifted by − 10.06, − 10.67, and − 9.15 mV on hNav1.1 and − 6.05, − 8.14, and − 6.55 mV on hNav1.2 in the activation process of 100 μmol/L fenamates. The slope factor of the activation phase evoked by the fenamates rarely changed in either hNav1.1 or hNav1.2 when compared with the control group (Fig. 4a, d). During the inactivation phase, V1/2 values were shifted by − 9.99, − 11.86, and − 14.76 mV on hNav1.1 and − 19.91, − 14.92, and − 5.47 mV on hNav1.2 in the presence of 100 μmol/L fenamates. The slope factor of the inactivation phase evoked by fenamates also rarely changed in either hNav1.1 or hNav1.2 when compared with the control group (Fig. 4b, e). In the recovery phase, 100 μmol/L of the fenamates made it hard to recover on hNav1.2, but essentially did not affect hNav1.1. The time constant of the recovery phase changed with 13.42, 5.65, and 1.66 ms for hNav1.2 (Fig. 4c, f). Parameters of activation, inactivation, and recovery phase are summarized in Tables 1, 2.
Fig. 4.

Effects of fenamates on activation, inactivation, and recovery phases of hNav1.1 and hNav1.2. Normalized conductance of hNav1.1 (a) and hNav1.2 (d) were showed with peak currents converted into conductance in activation curve. Normalized currents of hNav1.1 (b) and hNav1.2 (e) were showed at prescribed potentials. The recovery of Na+ current of hNav1.1 (c) and hNav1.2 (f) were characterizing by I/Imax ratio. Data represent mean ± SEM (n = 6)
Table 1.
Effects of fenamates on activation, inactivation, and recovery properties of hNav1.1 (n = 6)
| Group | Ctrl | 100 μmol/L mefenamic acid | 100 μmol/L flufenamic acid | 100 μmol/L tolfenamic acid | |
|---|---|---|---|---|---|
| Activation | V1/2 (mV) | − 20.29 ± 0.61 | − 29.94 ± 0.84** | − 30.55 ± 0.61** | − 29.03 ± 0.39** |
| k | 6.81 ± 0.57 | 5.94 ± 0.78 | 5.41 ± 0.57 | 4.69 ± 0.38 | |
| Inactivation | V1/2 (mV) | − 46.66 ± 0.37 | − 55.66 ± 0.53** | − 57.53 ± 0.34** | − 60.43 ± 0.25** |
| k | 5.58 ± 0.33 | 6.07 ± 0.48 | 6.53 ± 0.31 | 5.65 ± 0.26 | |
| Recovery | τ (ms) | 3.55 ± 0.50 | 4.60 ± 0.55 | 4.75 ± 0.42 | 5.59 ± 0.51 |
Table 2.
Effects of fenamates on activation, inactivation, and recovery properties of hNav1.2 (n = 6)
| Group | Ctrl | 100 μmol/L mefenamic acid | 100 μmol/L flufenamic acid | 100 μmol/L tolfenamic acid | |
|---|---|---|---|---|---|
| Activation | V1/2 (mV) | − 27.78 ± 0.47 | − 33.41 ± 0.79** | − 35.50 ± 1.06** | − 33.91 ± 0.27** |
| k | 5.54 ± 0.43 | 3.15 ± 0.55* | 7.05 ± 0.95 | 4.34 ± 0.24 | |
| Inactivation | V1/2 (mV) | − 54.60 ± 0.48 | − 74.51 ± 0.26** | − 69.52 ± 0.30** | − 60.07 ± 0.40* |
| k | 6.84 ± 0.34 | 7.21 ± 0.19 | 8.58 ± 0.28 | 5.80 ± 0.36 | |
| Recovery | τ (ms) | 5.36 ± 0.39 | 18.93 ± 1.94** | 11.16 ± 1.66** | 7.17 ± 0.79 |
*P < 0.05; **P < 0.01. (Control vs fenamates; one-way ANOVA)
Inhibitory Effects of Fenamates on Glutamate-Induced Apoptosis in SH-SY5Y Cells
A decrease in the ratio of anti-apoptotic Bcl-2 and pro-apoptotic Bax induces mitochondrial cytochrome c release, which leads to apoptosis. Release of cytochrome activates downstream caspases via cleavage, one of which is caspase 3. Result demonstrated that glutamate treatment downregulated Bcl-2 expression in SH-SY5Y cells, which resulted in the decrease of Bcl-2/Bax ratio when compared to the control group. Mefenamic acid treatment up-regulated Bcl-2 thus increasing the Bcl-2/Bax ratio in comparison to glutamate-treated cells. It was shown that glutamate treatment increased expression levels of cleaved caspase 3 when compared to the control group in investigating of caspase 3. Mefenamic acid treatment decreased caspase 3 cleavage (Fig. 5).
Fig. 5.

Fenamates inhibited glutamate induced apoptosis in SH-SY5Y cells. Bax, Bcl-2, Caspase3, and Cleaved caspase3 protein expression were determined in the whole-cell extract by Western blot (a). The protein expression levels of Bax, Bcl-2, Caspase3, and Cleaved caspase3 were normalized to β-actin (b–e). The Bcl-2/Bax ratio and cleaved caspase3/caspase3 were determined using densitometry and the fold protein expression is represented as a bar graph (f, g). Data represent mean ± SEM (n = 3). *P < 0.05, **P < 0.01 (glutamate vs fenamates)
Annexin V-FITC/PI was used to detect cell apoptosis, which can clearly and accurately observe the ratio of early apoptosis and late apoptosis. As shown in Fig. 6, compared with the control group, the apoptosis rate of cells in the glutamate group increased to 33.3% after 48 h of treatment with 10 mmol/L of glutamate. The apoptosis rate decreased to 14.9%, 20.7%, and 27.2% when 100 μmol/L of mefenamic acid, flufenamic acid and tofenamic acid were administered, which indicated that 100 μmol/L of mefenamic acid and flufenamic acid could inhibit the apoptosis of SH-SY5Y cells induced by glutamate.
Fig. 6.

Fenamates reduced glutamate-induced apoptosis. SH-SY5Y cells were treated with 100 μmol/L of fenamates and 10 mmol/L of glutamate for 48 h. Flow cytometry was used to detect apoptotic cells based on the Annexin V-FITC/PI staining. Data represent mean ± SEM (n = 3). *P < 0.05, **P < 0.01 (glutamate vs fenamates)
Effect of Fenamates on NMDA Receptor-Mediated Currents
In order to examine whether the neuroprotection of fenamates is also associated with the inhibition of NMDA receptors, the NMDA-evoked currents on neurons in rat hippocampus in the presence or absence of fenamates. Results showed that all the three fenamates could inhibit NMDA receptor-mediated currents, and the inhibition rates were 41.56%, 37.33%, and 21.07% respectively at the concentration of 100 μmol/L (Fig. 7). In addition, the intensity of inhibition of NMDA currents evoked by the three fenamates was consistent with the intensity of their neuroprotective effects. This suggests that NMDA receptors also play a role in the neuroprotective effect of the fenamates.
Fig. 7.

The inhibition of NMDA-evoked currents by fenamates. Currents recorded from neuron of the rat hippocampus in response to applications of 100 μmol/L NMDA and 10 μmol/L glycine in the absence or presence of 100 μmol/L mefenamic acid, flufenamic acid, and tolfenamic acid (a). Averaged inhibition rate of NMDA-evoked currents was obtained in each group (b, c). Data represent mean ± SEM (n = 3)
Discussions
Neurons are extremely vulnerable to ischemic injury. In addition, ischemic injury can lead to over-stimulation of glutamate receptors, ion imbalances, and calcium overload in neurons, as well as the generation of reactive oxygen species (ROS), mitochondrial injury, and apoptosis (Santos et al. 1996; Small et al. 1999; Lyden and Wahlgren 2000; Lo et al. 2003). Until now, despite much research, effective treatment has not been identified. However, many studies have shown that sodium channel blockers may be potential neuroprotectants (Waxman 2008).
As is well known, the inhibition of COX means that fenamates are widely used in the clinical treatment of inflammation and pain. Recently, several studies have demonstrated that fenamates have neuroprotective effects. Both mefenamic acid and flufenamic acid are effective in the model of NLRP3-dependent air pouch and peritoneal inflammation in mice, as well as in the model of amyloid β-induced memory loss and Alzheimer's disease in mice (Daniels et al. 2016). Mefenamic acid has the potential to reduce infarct volume, total ischemic brain damage and edema in the middle cerebral artery occlusion (MCAO), as well as improving learning and memory disorders in Aβ1-42-infused Alzheimer’s disease in rats (Khansari and Halliwell 2009). Furthermore, fenamates may inhibit a diversity of ion channels subtypes, such as CaCC, KATP, Nav1.7, and Nav1.8 channels. This means that the inhibition of ion channels is suggested to be a mechanism in the neuroprotective effects of fenamates. However, whether fenamates’ neuroprotective activity is associated with VGSCs has been less studied.
In this study, to examine the neuroprotective effects of fenamates, SH-SY5Y cells were pre-added with 1 μmol/L, 10 μmol/L, and 100 μmol/L of tolfenamic acid, flufenamic acid, and mefenamic acid for one hour, followed by exposure to 10 mmol/L glutamate for 48 h. Both flufenamic acid and mefenamic acid provided protection against glutamate-induced injury in the concentration of 100 μmol/L. Deltamethrin, a sodium channel activator, is able to reduce the neuroprotective effects evoked by the fenamates. This suggests that sodium channel plays an important role in the nerve damage induced by glutamate (Fig. 1).
Nav1.1, Nav1.2, and Nav1.6 are VGSC subtypes mainly expressed in CNS and are closely associated with neuroprotection (Sandalon et al. 2013; Stevens et al. 2013; Shi et al. 2019). The expression of these VGSC subtypes was examined in SH-SY5Y cells. Results showed that Nav1.1 and Nav1.2 were expressed in SH-SY5Y cells, while Nav1.6 was rarely expressed. Following this, the effects of fenamates were determined and compared on the expression level of Nav1.1 and Nav1.2 in glutamate-treated SH-SY5Y cells. Western blot analysis suggested that fenamates did not affect the expression level of Nav1.1 and Nav1.2 in SH-SY5Y cells (Fig. 2).
In order to further examine the effects on the function of these sodium channel subtypes, whole-cell patch clamp was carried out on Nav1.1 and Nav1.2. Data showed that fenamates weakly inhibited peak currents of hNav1.1. However, peak currents of hNav1.2 were significantly inhibited by fenamates, especially mefenamic acid. Meanwhile, no significant difference was found in the inhibitory effects on hNav1.1 between the three fenamates although, as can be seen from the inhibitory effects on hNav1.2, mefenamic acid was much stronger than flufenamic acid, while flufenamic acid was stronger than tolfenamic acid. Therefore, as can be seen from the neuroprotective effects against glutamate-induced injury, mefenamic acid is much stronger than flufenamic acid, and flufenamic acid is stronger than tolfenamic acid. These findings suggest that Nav1.2 may take part in the neuroprotection mechanism of fenamates. Given that the inhibition to Nav1.2 and neuroprotection effect of mefenamic acid was shown to be stronger than flufenamic acid and tolfenamic acid, it may be suggested that the two methyl groups on the benzene ring of mefenamic acid play key roles (Fig. 8). That is to say, relatively minor structural differences may lead to obvious changes in effectiveness and efficacy.
Fig. 8.
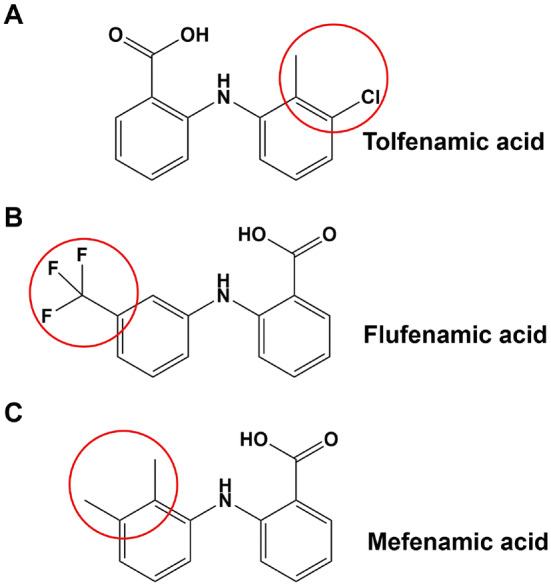
The chemical structures of the three fenamates. The slight structural differences are circled in red
The kinetic characteristics of hNav1.1 registered during fenamates treatment exhibited similar variation, including activation, inactivation, and recovery processes. The three fenamates promoted both the activation and the inactivation of hNav1.1. However, the effects of the fenamates on the kinetic characteristics of hNav1.2 were different. Although the fenamates exhibited similar variation in the activation, the shiftment of inactivation curve of mefenamic acid was larger than that of flufenamic acid and tolfenamic acid (Fig. 4). The promotion of mefenamic acid and flufenamic acid on hNav1.2 inactivation will lead to a decrease in persistent current and may lead to an increase in activation threshold. Considering the strong inhibitory effect of mefenamic acid and flufenamic acid on the peak current of Nav1.2, the promotion of them on inactivation may reduce the overall effect on neuroprotective effect.
It has been reported that mefenamic acid can inhibit N-methyl-d-aspartate (NMDA) receptor-mediated currents (Chen et al. 1998). As a result, the inhibitory effect of the fenamates on NMDA currents was also examined. Findings demonstrated that all three fenamates could inhibit NMDA receptor-mediated currents (Fig. 6). The fenamates inhibit NMDA receptor-mediated currents; however, their inhibitory effect is not very strong at 100 μmol/L. In general, the IC50 values of neuroprotective drugs that mainly act on NMDA receptors are generally low-micromolar concentrations (Florio et al. 2009; Han et al. 2013; Wu and Tymianski 2018). Therefore, taken together, both sodium channels and NMDA receptors play roles in the fenamates’ neuroprotective effects.
We evaluated the effects of fenamates on the hallmarks of apoptosis, such as alteration in the Bcl-2/Bax ratio, and proteolytic cleavage of caspase 3. We found that fenamates inhibited glutamate-induced Bcl-2/Bax ratio increase and caspase 3 cleavage inhibition (Fig. 5), and inhibited the apoptosis of SH-SY5Y cells induced by glutamate (Fig. 6). Therefore, the results demonstrated that fenamates inhibited glutamate-induced apoptosis through the modulation of the Bcl-2/Bax-dependent cell death pathways.
In conclusion, fenamates demonstrated neuroprotection effects against glutamate-induced injury in SH-SY5Y cells. They exhibited decreased inhibitory effects on hNav1.1 when compared with hNav1.2. Correspondingly, the inhibition effects of the fenamates were consistent with levels of neuroprotective effects in vitro, which indicates that Nav1.2 may take part in fenamates’ neuroprotection mechanism. Fenamates inhibited glutamate-induced apoptosis through modulation of the Bcl-2/Bax-dependent cell death pathways.
Materials
Drugs
Tolfenamic acid, flufenamic acid, and mefenamic acid (MedChem Express) were dissolved in DMEM/external solution.
Animals
Animal care was in accordance with the Guidelines for Animal Experimentation of Shenyang Pharmaceutical University and the protocol was approved by the Animal Ethics Committee of the institution. All the cells and tissues of the mice were authorized to scientific purpose.
hNav Cell Lines
CHO (dhFr−) cells were obtained from American Type Culture Collection (ATCC). Transfection cells were transfected using Lipo3000 transfection reagent, and G418 was screened for resistance, and stable expression cell lines were obtained. Human Nav1.1 and Nav1.2 cell lines (SCN1A NM_006920, SCN2A NM_001040142) were cultured in Iscove's modified Dulbecco's medium (IMDM) with 10% (v/v) fetal bovine serum (FBS). Cells were cultured in an atmosphere of 5% CO2 and 37 °C. Cell culture-related reagents were all purchased from Gibco.
Human Neuroblastoma Cells
SH-SY5Y cells, neuroblastoma cell line used in this study obtained from American Type Culture Collection (ATCC), were cultured in Dulbecco's modified eagle medium (DMEM) containing 10% (v/v) FBS, 2 mmol/L l-glutamine. 7 days prior to recording, cells were plated in media containing 1% serum and 10 μM all-trans retinoic acid. Cell culture-related reagents were all purchased from Gibco. All-trans retinoic acid was purchased from Sigma.
Neurons Cultures
Hippocampal cultures were prepared as described previously (Zhang et al. 2003) with some modifications. Briefly, whole brains were isolated from embryonic day 18 Sprague–Dawley rats, and the hippocampi were dissected out and treated with 0.25% trypsin–EDTA at 37 °C for 20 min. The cells were suspended with DMEM containing 20% fetal bovine serum and were plated at about 700,000 cells in 35-mm dishes. Four hours after plating, the medium was replaced by the serum-free Neurobasal medium with 2% B27 supplement and 0.25% glutamine. In order to avoid the growth of glial cell in hippocampal cultures, cytarabine was added. Afterward, half of the medium was changed twice a week.
MTT Assay
The suspension of SH-SY5Y cells was plated in a 96-well plate for a 48 h culture at 37 °C with 5% CO2. Details of MTT assays of the fenamates were as previously described (Zhao et al. 2017). The experiment was repeated 3 times (each time 4 wells/sample), and the mean values were obtained. MTT was purchased from Sigma-Aldrich.
Western Blotting
Total protein extraction was carried out using Total Protein Extraction kit (Kaiji, Nanjing, China). Protein concentration was determined using the BCA Protein Assay Kit (Kaiji, Nanjing, China). Samples containing 30 mg total protein were added to the polyacrylamide gel. Details of Western blotting were as previously described (Zhao et al. 2017). After transferred to a PVDF membrane, blocked with 10% skim milk, they were immersed in diluted primary antibody (Nav1.1 antibody: Abcam, ab24820, 1:1000; Nav1.2 antibody: Abcam, ab65163, 1:1000; Nav1.6 antibody: Abcam, ab65166, 1:1000; Caspase3 antibody: Cell Signaling, 9665S, 1:1000; Cleaved Caspase3 antibody: Cell Signaling, 9664S; Bax antibody: Cell Signaling, 14796S, 1:1000; and Bcl-2 antibody: Cell Signaling, 15071S, 1:1000; β-actin antibody, Invitrogen, MA5-15739, 1:1000) at 4 °C overnight. β-actin was used as an internal reference. Each experiment was repeated three times to obtain the mean values.
Annexin V-FITC/PI Apoptosis Assay
For Annexin V-FITC/PI analysis, the collected cells were resuspended in 200 μL of binding buffer provided with the Annexin V-FITC/PI analysis kit (Thermo Fisher), and 5 μL of Annexin V-FITC and 5 μL of PI were added. Next, the cells were incubated at 4 °C for 1 h in the dark. All labeled cells were analyzed by flow cytometry.
Patch Clamp
hNav1.1 and hNav1.2 cells were cultured in a selection antibiotic-free medium 48 h prior the test. The standard external solution, internal solution, and seal enhancer solution were used (external solutions in mM: 140NaCl, 4KCl, 1MgCl2, 2CaCl2, 5 d-Glucose monohydrate, 10 HEPES/NaOH, pH 7.4; internal solutions in mM: 50CsCl, 10NaCl, 60CsF, 20 EGTA, 10 HEPES/CsOH, pH 7.2; seal enhancer solution in mM: 80NaCl, 3KCl, 35CaCl2, 10MgCl2, 10 HEPES/NaOH, pH 7.4) (24). Patch clamp experiments were conducted with the Port-a-Patch setup (Nanion, Germany) and the EPC10 Amplifier (HEKA, Germany). Patchcontrol HT software (Nanion, Germany) and Patchmaster software (HEKA, Germany) were also used to establish the whole-cell configuration and perform voltage-clamp protocols. All the details of each process were as previously described (Xu et al. 2017; Kahlig et al. 2010).
The NMDA-induced current was recorded via a glass microelectrode filled with internal solution (in mmol/L: K-Gluconate 125, NaCl 5, CaCl2 0.1, EGTA 1, MgCl2 1, Mg-ATP 2, and HEPES 10, pH 7.2). Conventional whole-cell patch clamp technique was adopted for recording the NMDA-induced current in the neurons in an external solution (in mmol/L: NaCl 140, KCl 4, CaCl2 2, EGTA 10, Glucose 5, and HEPES 10, pH 7.4). EPC-10 Quattro (Heka, Germany), Patchmaster (Heka, Germany) and IGOR-Pro (WaveMetrics Inc, USA) were used. The pipette filled with pipette solution had a resistance of 2–3 MΩ. All NMDA currents were recorded while membrane potential was held at − 80 mV for 30 s. In order to record the NMDA current, 100 μmol/L NMDA and 10 μmol/L Glycine were added to stimulate after the voltage holding for 10–20 s. 100 μmol/L NMDA, 10 μmol/L Glycine, and 100 μmol/L fenamates were added for a second stimulate at the same voltage mode. All current responses were elicited at room temperature.
Data Analysis
Data were analyzed by using GraphPad Prism and SPSS. Pooled data were presented as the mean ± SEM. Statistical comparisons were made by using one-way ANOVA.
Acknowledgements
We gratefully acknowledge the financial support from the Innovation Team Project of the Department of Education of Liaoning Province (LT2015010) and the PhD Start-up Fund of Natural Science Foundation of Liaoning Province (2019-BS-231).
Abbreviations
- CNS
Central nervous system
- VGSC
Voltage-gated sodium channel
- NSAIDs
Nonsteroidal anti-inflammatory drugs
- DRG
Dorsal root ganglion
- ROS
Reactive oxygen species
- MCAO
Middle cerebral artery occlusion
- DMEM
Dulbecco's modified Eagle medium
- IMDM
Iscove's modified Dulbecco's medium
- CHO
Chinese hamster ovary
- FBS
Fetal bovine serum
- ATCC
American Type Culture Collection
Author Contributions
All authors contributed to the study conception and design. JS, YX, and XK carried out electrophysiology studies. JS carried out MTT assay. JS, YX, and YS carried out Western blot experiments and Annexin V-FITC/PI analysis. YX and ZW conceived and designed the experiments. JS, MZ, and YX wrote this manuscript, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Compliance with Ethical Standards
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Conflict of interest
The authors declare that they have no conflicts of interest.
Informed Consent
This article does not contain any studies with human participants performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yi-Jia Xu, Email: xyj0922@126.com.
Zhan-You Wang, Email: wangzy@cmu.edu.cn.
References
- Chen Q, Olney JW, Lukasiewicz PD, Almli T, Romano C (1998) Fenamates protect neurons against ischemic and excitotoxic injury in chick embryo retina. Neurosci Lett 242:163–166 [DOI] [PubMed] [Google Scholar]
- Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS, Baldwin AG, Freeman S, Wong R, Latta C, Yu S, Jackson J, Fischer N, Koziel V, Pillot T, Bagnall J, Allan SM, Paszek P, Galea J, Harte MK, Eder C, Lawrence CB, Brough D (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat Commun 7:12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio SK, Loh C, Huang SM, Iwamaye AE, Kitto KF, Fowler KW, Treiberg JA, Hayflick JS, Walker JM, Fairbanks CA, Lai Y (2009) Disruption of nNOS-PSD95 protein-protein interaction inhibits acute thermal hyperalgesia and chronic mechanical allodynia in rodents. Br J Pharmacol 158:494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower RJ (1974) Drugs which inhibit prostaglandin biosynthesis. Pharmacol Rev 26:33–67 [PubMed] [Google Scholar]
- Flower R, Gryglewski R, Herbaczyn′ ska-Cedro K, Vane JR (1972) Effects of anti-inflammatory drugs on prostaglandin biosynthesis. Nat New Biol 238(104):106 [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Large WA (1995) Comparison of the effects of fenamates on Ca-activated chloride and potassium currents in rabbit portal vein smooth muscle cells. Br J Pharmacol 116:2939–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover GJ, D’Alonzo AJ, Sleph PG, Dzwonczyk S, Hess TA, Darbenzio RB (1994) The cardioprotective and electrophysiological effects of cromakalim are attenuated by meclofenamate through a cyclooxygenase-independent mechanism. J Pharmacol Exp Ther 269:536–540 [PubMed] [Google Scholar]
- Han Z, Yang JL, Jiang SX, Hou ST, Zheng RY (2013) Fast, non-competitive and reversible inhibition of NMDA-activated currents by 2-BFI confers neuroprotection. PLoS ONE 8:e64894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zeng B, Chen GL, Bot D, Eastmond S, Elsenussi SE, Atkin SL, Boa AN, Xu SZ (2012) Effect of non-steroidal anti-inflammatory drugs and new fenamate analogues on TRPC4 and TRPC5 channels. Biochem Pharmacol 83:923–931 [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Lepist I, Leung K, Rajamani S, George AL (2010) Ranolazine selectively blocks persistent current evoked by epilepsy-associated Nav1.1 mutations. Br J Pharmacol 161:1414–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khansari PS, Halliwell RF (2009) Evidence for neuroprotection by the fenamate NSAID, mefenamic acid. Neurochem Int 55:683–688 [DOI] [PubMed] [Google Scholar]
- Lee HM, Kim HI, Shin YK, Lee CS, Park M, Song JH (2003) Diclofenac inhibition of sodium currents in rat dorsal root ganglion neurons. Brain Res 992:120–127 [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4:399–415 [DOI] [PubMed] [Google Scholar]
- Lyden P, Wahlgren NG (2000) Mechanisms of action of neuroprotectants in stroke. J Stroke Cerebrovasc Dis 9:9–14 [DOI] [PubMed] [Google Scholar]
- Olney JW (1978) Neurotoxicity of excitatory amino acid. In: McGeer E, Olney JW, McGeer P (eds) Kainic acid as a tool in neurobiology. Raven, New York, pp 95–121 [Google Scholar]
- Rothman SM (1984) Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci 4:1884–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandalon S, Könnecke B, Levkovitch-Verbin H, Simons M, Hein K, Sättler MB, Bähr M, Ofri R (2013) Functional and structural evaluation of lamotrigine treatment in rat models of acute and chronic ocular hypertension. Exp Eye Res 115:47–56 [DOI] [PubMed] [Google Scholar]
- Sanger GJ, Bennett A (1979) Fenamates may antagonize the actions of prostaglandin endoperoxides in human myometrium. Br J Clin Pharmacol 8:479–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MS, Moreno AJ, Carvalho AP (1996) Relationships between ATP depletion, membrane potential, and the release of neurotransmitters in rat nerve terminals. An in vitro study under conditions that mimic anoxia, hypoglycemia, and ischemia. Stroke 27:941–950 [DOI] [PubMed] [Google Scholar]
- Shi HS, Lai K, Yin XL, Liang M, Ye HB, Shi HB, Wang LY, Yin SK (2019) Ca2+-dependent recruitment of voltage-gated sodium channels underlies bilirubin-induced overexcitation and neurotoxicity. Cell Death Dis 10:774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Griffiths T, Meldrum BS (1984) Blockade of N-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science 226:850–852 [DOI] [PubMed] [Google Scholar]
- Small DL, Morley P, Buchan AM (1999) Biology of ischemic cerebral cell death. Prog Cardiovasc Dis 42:185–207 [DOI] [PubMed] [Google Scholar]
- Stevens M, Timmermans S, Bottelbergs A, Hendriks JJ, Brône B, Baes M, Tytgat J (2013) Block of a subset of sodium channels exacerbates experimental autoimmune encephalomyelitis. J Neuroimmunol 261:21–28 [DOI] [PubMed] [Google Scholar]
- Sun JF, Xu YJ, Kong XH, Su Y, Wang ZY (2018) Fenamates inhibit human sodium channel Nav1.7 and Nav1.8. Neurosci Lett 696:67–73 [DOI] [PubMed] [Google Scholar]
- Waxman SG (2008) Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nat Clin Pract Neurol 4:159–169 [DOI] [PubMed] [Google Scholar]
- White MM, Aylwin M (1990) Niflumic and flufenamic acids are potent reversible blockers of Ca2+-activated Cl- channels in Xenopus oocytes. Mol Pharmacol 37:720–724 [PubMed] [Google Scholar]
- Wu QJ, Tymianski M (2018) Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain 11:15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Meng X, Hou X, Sun J, Kong X, Sun Y, Liu Z, Ma Y, Niu Y, Song Y, Cui Y, Zhao M, Zhang J (2017) A mutant of the Buthus martensii Karsch antitumor-analgesic peptide exhibits reduced inhibition to hNav1.4 and hNav1.5 channels while retaining analgesic activity. J Biol Chem 292:18270–18280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau HJ, Baranauskas G, Martina M (2010) Flufenamic acid decreases neuronal excitability through modulation of voltage-gated sodium channel gating. J Physiol 588:3869–3882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S (2003) ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40:971–982 [DOI] [PubMed] [Google Scholar]
- Zhao ZW, Fan XX, Song JJ, Xu M, Chen MJ, Tu JF, Wu FZ, Zhang DK, Liu L, Chen L, Ying XH, Ji JS (2017) ShRNA knock-down of CXCR7 inhibits tumour invasion and metastasis in hepatocellular carcinoma after transcatheter arterial chemoembolization. J Cell Mol Med 21:1989–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]