

Abstract
Since no definitive treatment has been suggested for diffuse traumatic brain injury (TBI), and also as the effect of exercise has been proven to be beneficial in neurodegenerative diseases, the effect of endurance exercise on the complications of TBI along with its possible neuroprotective mechanism was investigated in this study. Our objective was to find out whether previous endurance exercise influences brain edema and neurological outcome in TBI. We also assessed the probable mechanism of endurance exercise effect in TBI. Rats were randomly assigned into four groups of sham, TBI, exercise + sham and exercise + TBI. Endurance exercise was carried out before TBI. Brain edema was assessed by calculating the percentage of brain water content 24 h after the surgery. Neurological outcome was evaluated by obtaining veterinary coma scale (VCS) at − 1, 1, 4 and 24 h after the surgery. Interleukin-1β (IL-1β), total antioxidant capacity (TAC), malondialdehyde (MDA), protein carbonyl and histopathological changes were evaluated 24 h after the surgery. Previous exercise prevented the increase in brain water content, MDA level, histopathological edema and apoptosis following TBI. The reduction in VCS in exercise + TBI group was lower than that of TBI group. In addition, a decrease in the level of serum IL-1β and the content of brain protein carbonyl was reported in exercise + TBI group in comparison with the TBI group. We suggest that the previous endurance exercise prevents brain edema and improves neurological outcome following diffuse TBI, probably by reducing apoptosis, inflammation and oxidative stress.
Keywords: Brain injury, Malondialdehyde, Interleukin-1β, Apoptosis, Protein carbonyl, Oxidative stress, Inflammation
Introduction
Each year, 10 million people worldwide suffer from traumatic brain injury (TBI) that leads to hospitalization or death. Cognitive, physical and psychological impairments are seen among TBI survivors (Ansari et al. 2008). Cellular and biochemical events, leading to secondary brain damage in TBI, contribute to long-term disabilities (Jacotte-Simancas et al. 2015). The processes of secondary brain damage such as the formation of free radicals and the activation of inflammatory processes play roles in delayed neuronal dysfunction and death (Khaksari et al. 2018b). Accordingly, increased level of oxidative stress markers has been found in cerebrospinal fluid (CSF) after human and animal TBI (Opii et al. 2007; Varma et al. 2003).
The production of free radicals at the initial time of brain damage (3 to 72 h) causes oxidative damage and impairment of synaptic function after the injury (Ansari et al. 2008; Singh et al. 2006). The increase in free radicals (Khaksari et al. 2018a) and impairment of antioxidant mechanisms (Hall et al. 2004) play essential roles in the secondary events induced by TBI. Traumatic neuroinflammation may be a link between the mechanism of acute injury and chronic neurodegeneration (Ramlackhansingh et al. 2011). Inflammation and oxidative stress result in long-term structural changes and progressive practical defects (Byrnes et al. 2012b).
Trauma to head initiates responses of both the neuronal degeneration and the neuronal restoration, whereby the relative balance between these activities determines the extent of delayed neurological disorders (Adkins et al. 2006; Carmichael 2006). Evidence suggests that these responses are modulated by exercise due to an increment in neurotrophic factors leading to increased neuronal plasticity, and anti-inflammatory and anti-apoptotic agents (Itoh et al. 2011; Mota et al. 2012). The exercise has been propounded to adjust inflammatory responses in animals and humans (Petersen and Pedersen 2005). In recent years, neuroprotective effects of exercise have been studied and shown as an intervention in healthy elder people and people with neurological disorders (Hindin and Zelinski 2012; Smith et al. 2010). The benefits of exercise have been shown in animal models of stroke (Hicks et al. 1998). Exercise exerts beneficial effects on cortical function through neurogenesis and angiogenesis (Cotman et al. 2007).
Physical activity can reduce neuronal loss and inflammation and facilitate post-injury recovery (Zhao et al. 2014). Better outcomes following exercise have been reported in rodents with TBI (Kim et al. 2010; Wu et al. 2013). However, the improvement in neurological outcome has not been observed following exercise in some TBI research (Piao et al. 2013; Silva et al. 2013). Clinical studies of the effect of exercise on TBI have led to the contradictory results (Bland et al. 2011). The acute exercise exerted after TBI exacerbates neuronal damage by inducing greater energy consumption (Griesbach et al. 2012). Therefore, performing exercise immediately after TBI may produce detrimental outcomes.
Although pre-injury exercise for humans may not be the most effective prophylactic strategy since the time of injury cannot be predicted (Vaynman and Gomez-Pinilla 2005), regular exercise may have preventive potential as it induces metabolic changes that can modulate the response of organs involved in metabolism like liver and brain, which ultimately lead to the prevention of cell damage and disabilities after TBI (da Silva Fiorin et al. 2016). The evidence has demonstrated that oxidative and inflammatory cascades in TBI alter signal transduction in peripheral organs and CNS (Bayır et al. 2007; Lima et al. 2013). Also, inflammatory reaction in the liver in response to brain injury might contribute to the inflammatory cascades of the brain. Thus, peripheral inhibition of inflammation may reduce the inflammatory cascades in the brain (Soltani et al. 2015). A cross-sectional study has suggested that regular exercise could have a protective effect in diseases associated with systemic inflammation (Petersen and Pedersen 2005). It is believed that the favorable changes in the hepatic antioxidant and anti-inflammatory status induced by previous exercise may have a prophylactic effect on the early inflammatory and oxidative response after TBI (Hoene and Weigert 2010; Sun et al. 2010). Therefore, people who undertake regular exercise may experience less damage and disability following TBI compared to others.
It has been demonstrated that regular exercise before the brain ischemia produces prophylactic effects on brain damage after ischemia (Endres et al. 2003). In a study, mice that had intense exercise on treadmill before middle cerebral artery occlusion (MCAO) showed a decline in MCAO-induced infarction volume and an increase in NO production, which resulted in increased blood flow without altering oxidative stress (Arrick et al. 2014). Previous exercise reduces lipid and protein peroxidation and levels of IL-1β and TNF-α cytokines and also increases the level of IL-10 anti-inflammatory cytokine in fluid percussion injury (FPI), (Lima et al. 2009), which probably lead to improved motor function after injury (Carvalho et al. 2005; Mota et al. 2012). Previous exercise has a protective effect against cell loss in the hippocampus of FPI animals (Castro et al. 2017). Beneficial effect of exercise in TBI is exerted by upregulation of hippocampus BDNF (Griesbach et al. 2009). Although exercise before TBI may not be the most effective treatment for humans (Vaynman and Gomez-Pinilla 2005), it may have prophylactic effects on brain injury.
It has been proposed that performing exercise is useful after TBI (Ang and Gomez-Pinilla 2007) and may have a prophylactic role in TBI (Lima et al. 2009). However, the prophylactic effect of endurance exercise on deleterious consequences of diffuse TBI has not been studied so far, and only few studies have been conducted on neuroprotective mechanisms of previous endurance exercise in TBI. The current study aimed to examine the prophylactic effect of eight-week treadmill exercise on male rates with diffuse TBI and its probable mechanisms. For this reason, the effect of previous treadmill exercise on brain edema and neurological outcome following TBI was assessed by obtaining brain water content and veterinary coma scale (VCS) 24 h after the injury. Meanwhile, biochemical factors associated with the inflammation [interleukin-1beta (IL-1β)] and oxidative stress (lipid and protein peroxidation and total antioxidant capacity), as well as factors related to histopathological injury (edema and apoptosis), were evaluated to investigate the probable neuroprotective mechanisms of previous exercise.
Materials and Methods
Study Subjects
Mature male Wistar rats (weighing 180–210 g) were purchased from animal center of Afzalipour Medical College of Kerman University of Medical Sciences to be used in this study. The animals were kept in an air-conditioned room at 22–25 °C in a 12 h light and 12 h dark cycle. Food and water were available to the animals during the study. The study was executed in accordance with the guidelines for animal experimental protocols of Kerman University of Medical Sciences and the internationally accepted principles for animal use and care (EU Directive of 2010; 010/63/EU). The research protocol was approved by the ethics committee of Kerman University of Medical Sciences (No. EC/KNRC/94-420).
Study Groups
Twenty-four animals were randomly assigned into two main groups (n = 12 per group) of true traumatic brain injury (TBI group) and false traumatic brain injury (sham group). The animals were further divided into two exercise groups (exercise + TBI and exercise + sham groups).
Model of Diffuse TBI
The moderate diffuse TBI was induced using Marmarou’s weight drop model (Marmarou et al. 1994) as previously explained (Soltani et al. 2016). First, the animal was anesthetized with a mixture of 2% isoflurane/66% N2O/33% O2 and a midline incision was made in the scalp. Then, a steel disc (10 mm in diameter, 3-mm thick) was fixed between bregma and lambda sutures by polyacrylamide glue. The animal was positioned on a piece of foam under the device and a 250 g weight was dropped on the disc from 2-m distance. Then, the animal was immediately attached to a respiratory pump (TSE animal respirator, Germany) until it began spontaneous respiration in its cage. Only animals with the VCS score of 8–12 immediately after TBI were selected for the study. The sham animal received all stages of TBI induction except the falling weight part.
Protocol of Treadmill Exercise
Treadmill exercise was carried out for 8 weeks, 5 days per week until the day before the TBI surgery (Arrick et al. 2014). For the first 5 days, the exercise was performed for duration of 10 min per day and speed of 20 m/min on a flat surface. During the next 5 days, exercise duration was increased by 10 min each day to reach the duration of 60 min with the speed of 25 m/min on a sloped surface. During the next 3 days, the speed (25 m/min) and duration (60 min/day) of exercise remained constant, but the surface had a 5% slope. Finally, for the remainder of the experimental protocol, the speed (25 m/min) and duration (60 min) remained the same, and the incline increased to 10%.
Assessment of Brain Edema
Edema was assessed by calculating the brain water content for each rat (O’Connor et al. 2005). The anesthetized rat was decapitated 24 h after the surgery, and its brain was removed and dissected into left and right hemispheres. The left hemisphere was immediately stored at − 70 °C for other investigations. The right hemisphere was placed in a pre-weighed vial. Each vial was weighed (wet weight of brain) and placed in an incubator (Memmert, Germany) at 100 °C for 48 h and then was reweighed (dry weight of brain). The brain water content (%) was calculated by [(wet wt-dry wt)/wet wt]*100.
Evaluation of Neurological Outcome
The neurological outcome was evaluated according to VCS as described previously (Soltani et al. 2009). The VCS was reported as a score of 3 to 15 that was the sum of three scores of motor situation (ranging 1–8), eye situation (1–4) and respiration situation (1–3). Higher score indicated a better neurological outcome, and lower score reflected a worse neurological outcome. In the present study, the outcome was reported 1 h before the surgery, and 1, 4 and 24 h after the surgery.
Sample Collection
Blood samples were taken from the jugular vein of anesthetized animal 24 h after the surgery. The blood samples collected with ethylenediaminetetraacetic acid (EDTA) were centrifuged, and obtained plasma was used to determine the ferric reducing ability of plasma (FRAP). Non-heparinized blood samples were centrifuged, and the serum was immediately used to assess interleukin-1beta (IL-1β), malondialdehyde (MDA) and protein carbonyl groups. Collected samples were frozen and stored for biochemical experiments.
The left hemisphere of brain was weighed and homogenized by T-PERTM tissue protein extraction reagent (500 mg tissue per 2 mL of the reagent) with 0.5% Triton- × 100, 150 mM NaCl, 50 mM tris and a protease inhibitor cocktail (Taupin et al. 1993). Homogenate was stirred by a shaker (Behdad, Iran) for 90 min and then was centrifuged at 4000 g (4 °C) for 15 min. Then, the supernatant was collected to determine MDA and protein carbonyl content in the brain. The MDA and protein carbonyl levels were measured in the brain tissue as they did not show any changes in the serum.
Assay of Serum IL-1β Level
An equal amount of protein from each sample was used for the assay of serum IL-1β level after protein content of the supernatant was determined by BCA protein assay reagent kit (Pierce) (Taupin et al. 1993). ELISA kit for IL-1 (sensitivity range 31.2–2000 pg/mL) was purchased from R&D systems (Minneapolis, Minnesota, USA), and the manufacturer’s protocols were followed. Cytokine level was evaluated in duplicate. The concentration of cytokine was reported as picograms (pg) of antigen per milliliter (mL) in the supernatant.
Assay of Plasma Total Antioxidant Capacity (TAC)
Total antioxidant power was evaluated by measuring the ferric reducing ability of plasma (FRAP) to withstand the oxidative effects of reactive species generated according to Benzie and Strain (Benzie and Strain 1996). This method measures the reduction in ferric-tripyridyltriazine (Fe III-TPTZ) complex to ferrous (Fe II)-TPTZ formed by colorimetric method.
The FRAP reagent was prepared by mixing acetate buffer (300 mM, pH = 3.6), 2, 4, 6-Tripyridyl-sTriazine (TPTZ) (10 mM in 40 mM HCl) and FeCl3 (20 mM) in a 10:1:1 ratio (v:v:v). Then, reagent and plasma were mixed (100:1 ratio) and incubated at 37 °C for 10 min. The reduction in Fe3+-TPTZ complex to a colored Fe2+-TPTZ complex was read at 593 nm against a blank reagent (1 mL FRAP reagent + 10 mL distilled water). The results were reported as micromoles (µM) of FRAP per mL serum.
Determination of Lipid Peroxidation Level in the Serum and Brain
Malondialdehyde (MDA) is a well-established indicator of lipid peroxidation. It was evaluated by the thiobarbituric acid reactive substance (TBARS) assay (Ohkawa et al. 1979). In this method, sample was incubated with 0.8% thiobarbituric acid solution, acetic acid buffer (pH = 3.2) and 8% sodium dodecyl sulfate solution at 95 °C for 1 h. Then, the color reaction of mixture was read at 532 nm. The results were reported as µM of MDA per mL supernatant or serum.
Determination of Protein Oxidation Level in the Serum and Brain
Protein carbonyl content is an indicator of protein oxidation, which was assessed by the method of Yan et al. (1995). Each sample was diluted to contain 750–800 μg/mL of protein, and 1 mL aliquot was mixed with 0.2 mL of 2,4-dinitrophenylhydrazine (DNPH, 10 mM) or 0.2 mL HCl (2 M). After incubation at room temperature for 1 h in a dark environment, 0.6 mL of denaturing buffer (150 mM sodium phosphate buffer, pH = 6.8, containing 3% SDS), 1.8 mL of heptane (99.5%) and 1.8 mL of ethanol (99.8%) were added, mixed for 40 s and centrifuged for 15 min. Then, the isolated protein was washed twice by 1 mL of ethyl acetate/ethanol 1:1 (v/v) and suspended in 1 mL of denaturing buffer. Each DNPH sample was read at 370 nm against the corresponding HCl sample (blank). The total carbonylation was calculated using a molar extinction coefficient of 22,000 M−1 cm−1 that was reported as nanomoles (nM) per milligram (mg) protein in supernatant or serum.
Histopathological Assay
Brain samples of anaesthetized animals (50 mg/kg, thiopental) were removed 24 h after the TBI, fixed in 10% buffered formaldehyde, sliced into Sections (5 µm) with microtome automatically (LEICA, Germany) and stained with hematoxylin–eosin. The brain injury was evaluated in light microscope by two pathologists who were blinded to the animal group. The apoptosis was scored as scant (0), mild (1), moderate (2) and severe (3). The severity of apoptosis was graded using a microscope with a magnification of 10 × in the following manner: Scant; when apoptosis was hardly seen in a high power field. Mild; when apoptosis was seen in a high power field. Moderate; when apoptosis was seen in more than one field. Severe; when apoptosis was seen in almost all of the high power fields. Brain edema was considered for the spaces that were seen in the extracellular spaces and separated parenchymal cells. It was graded by a microscope with a magnification of 10 × in the following manner: mild (when edema was < 10%), moderate (when edema was 10–50%) and severe (when edema was > 50%).
Statistics
The results were presented as mean ± SEM. The normality of variables was checked using Shapiro–Wilk’s W test. A mixed design analysis of variance was used to evaluate the interaction between the times of VCS and the groups. Greenhouse–Geisser correction was used when sphericity was rejected. Also, the VCS was analyzed by one-way analysis of variance (ANOVA) as with other variables of the study. Tukey’s test was used for post hoc analysis, and P < 0.05 was considered as the level of significance.
Results
Exercise Prevented the Development of Brain Edema Following TBI
The effect of exercise on brain edema by determining brain water content 24 h post-TBI is shown in Fig. 1. Brain water content in TBI group (79.48 ± 0.1%) was higher than the sham (78.22 ± 0.09%) and exercise + sham (77.65 ± 0.34%) groups (p < 0.01, p < 0.001, respectively). The exercise prevented the increase in brain water content following TBI (exercise + TBI group, 78.03 ± 0.17%) in comparison with TBI group (p < 0.01).
Fig. 1.
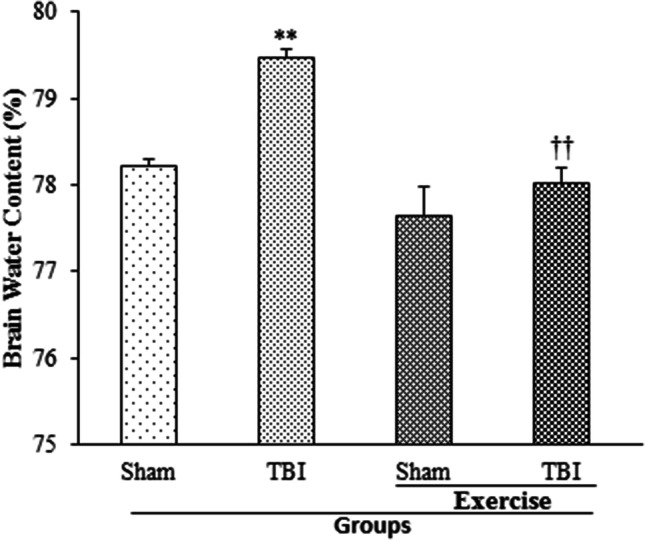
Brain water content (%) in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are shown as mean ± SEM. **p < 0.01 compared to sham group; ††p < 0.01 compared to TBI group. TBI traumatic brain injury
Neurological Outcome Improved Following TBI in Rates with the History of Exercise
The alteration in the neurological outcome by evaluating VCS at − 1, 1, 4 and 24 h post-TBI in the presence of previous treadmill training is shown in Fig. 2. Before the trauma, there was no statistical difference in VCS between the groups. A decrease in VCS appeared in TBI group (7.33 ± 0.33, 9.5 ± 0.34, 12 ± 0.26, respectively) compared to that of sham and exercise + sham groups (15 ± 0) at the evaluation times post-TBI (p < 0. 001). Also, VCS reduced in exercise + TBI group following TBI (9 ± 0.26, 11.83 ± 0.31, 14 ± 0, respectively) compared to the sham and exercise + sham groups (p < 0. 001). However, the decrease in VCS in exercise + TBI group was lower than the TBI group (p < 0. 001).
Fig. 2.

Veterinary coma scale (VCS) in exercised male rats at − 1, 1, 4 and 24 h post-TBI (n = 6 in each group). Data are shown as mean ± SEM. ***p < 0.001 compared to sham group at 1, 4 and 24 h after TBI; ###p < 0.001 compared to exercise + sham group at 1, 4 and 24 h after TBI; †††p < 0.001 compared to TBI group at 1, 4 and 24 h after TBI. TBI traumatic brain injury
Serum Level of IL-1β Reduced Following TBI in Rates with the History of Exercise
Serum level of IL-1β 24 h post-TBI in rates with the history of exercise is shown in Fig. 3. An increase in IL-1β level was discerned in TBI group (129.73 ± 2.37 pg/mL) in comparison with that of the sham group (122.13 ± 0.83 pg/mL; p < 0.05). However, a decrease in IL-1β level was reported in exercise + TBI group (119.42 ± 1.97 pg/mL) compared to TBI group (p < 0.01).
Fig. 3.
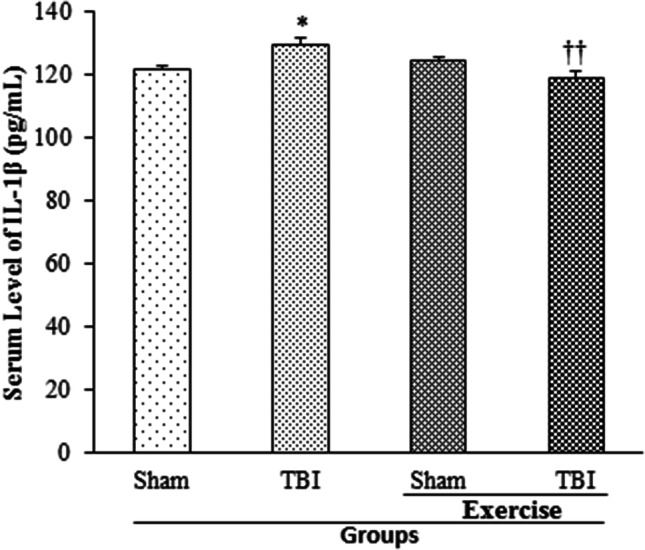
Serum level of IL-1β in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are presented as mean ± SEM. *p < 0.05 compared to sham group; ††p < 0.01 compared to TBI group. TBI traumatic brain injury
Exercise Did Not Influence the Serum Level of TAC Following TBI
The effect of exercise on serum level of TAC, which was determining by FRAP level 24 h post-TBI, is shown in Fig. 4. FRAP level decreased in TBI group (431.32 ± 9.93 µM/mL) in comparison with the sham group (520.1 ± 29.72 µM/mL; p < 0.01). Meanwhile, a decrease in FRAP level was observed in the exercise + sham (355.9 ± 11.76 µM/mL) and exercise + TBI (413.4 ± 12.3 µM/mL) groups compared to the sham group (p < 0.001, p < 0.01, respectively). The FRAP level of exercise + sham group was lower than that of TBI group (p < 0.05).
Fig. 4.
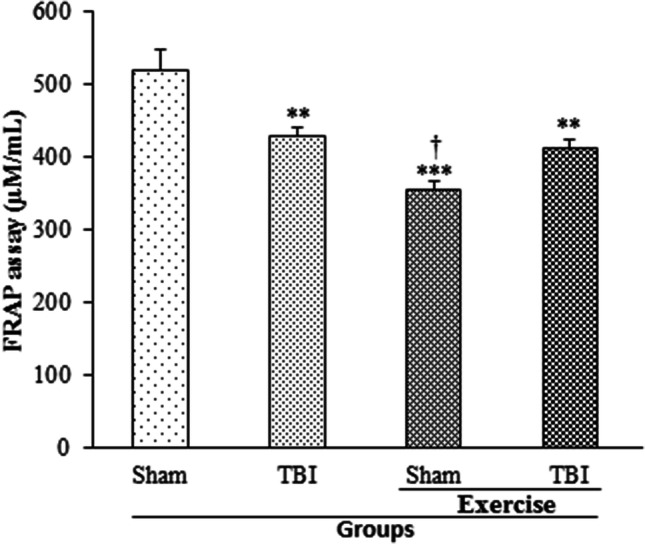
Serum level of FRAP in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are presented as mean ± SEM. **p < 0.01 compared to sham group; ***p < 0.001 compared to sham group. TBI traumatic brain injury, FRAP ferric reducing antioxidant power
Exercise Did Not Alter Lipid Peroxidation Index in Serum Following TBI
The alteration in lipid peroxidation index of serum determined by MDA level 24 h post-TBI in rats with exercise preconditioning is illustrated in Fig. 5. The MDA level of serum did not change due to TBI. An increase in MDA level of serum was observed in exercise + sham group (1025.72 ± 55.67 µM/mL) in comparison with the TBI group (853.85 ± 53.62 µM/mL; p < 0.05). The MDA level of serum was the same among exercise + TBI (981.55 ± 18.18 µM/mL) and sham groups.
Fig. 5.

Serum level of MDA in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are presented as mean ± SEM. TBI traumatic brain injury, MDA malondialdehyde
Previous Exercise Reduced Protein Peroxidation Index in Serum Following TBI
The effect of exercise on protein peroxidation index in serum following TBI determined by protein carbonyl level 24 h post-TBI is presented in Fig. 6. The protein carbonyl level of serum was not affected by TBI. Protein carbonyl level of serum declined by previous exercise in TBI-induced rats (0.52 ± 0.01 nM/mg) and in sham rats (0.59 ± 0.02 nM/mg protein) in comparison with the TBI group (0.72 ± 0.04 nM/mg; p < 0.05). Although protein carbonyl level of serum in exercise + sham group did not differ from sham group (0.67 ± 0.03 nM/mg), it was lower in the exercise + TBI group than the sham group (p < 0.05).
Fig. 6.
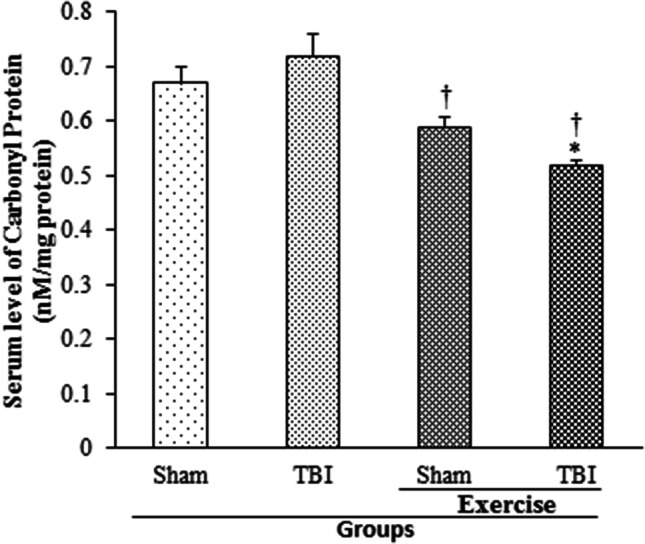
Serum level of protein carbonyl in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are shown as mean ± SEM. †p < 0.05 compared to TBI group. TBI traumatic brain injury
Exercise Impeded Brain Lipid Peroxidation Following TBI
The amount of brain lipid peroxidation 24 h post-TBI in rats with the history of exercise is shown in Fig. 7. An increase in MDA level was reported in TBI group (454.6 ± 45.07 µM/mL) in comparison with the sham (299.1 ± 11.58 µM/mL) and exercise + sham (335.99 ± 19.5 µM/mL) groups (p < 0.01, p < 0.05, respectively). Previous exercise impeded the increase in MDA level following TBI in the exercise + TBI group (355.84 ± 12.73 µM/mL) compared to TBI group (p < 0.05).
Fig. 7.
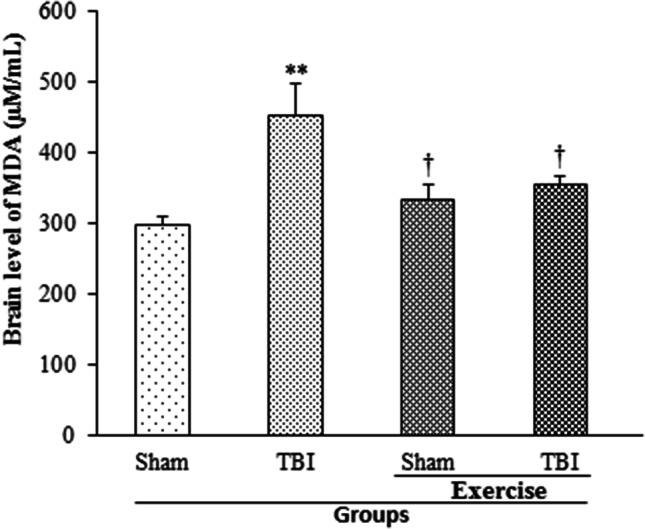
Brain level of MDA in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are presented as mean ± SEM. **p < 0.01 compared to sham group; †p < 0.05 compared to TBI group. TBI traumatic brain injury, MDA malondialdehyde
Previous Exercise Decreased Brain Protein Peroxidation Following TBI
The effect of exercise on brain protein peroxidation 24 h post-TBI is presented in Fig. 8. Protein carbonyl level increased following TBI (6.37 ± 0.53 nM/mg protein) in comparison with the sham group (2.4 ± 0.17 nM/mg protein; p < 0.001). Exercise increased protein carbonyl level (6.1 ± 0.63 nM/mg protein) in comparison with the sham group (p < 0.001), but it decreased the protein carbonyl level following TBI compared to TBI and exercise + sham (3.98 ± 0.19 nM/mg protein) groups (p < 0.01, p < 0.05, respectively).
Fig. 8.

Brain level of protein carbonyl in exercised male rats, 24 h post-TBI (n = 6 in each group). Data are shown as mean ± SEM. ***p < 0.001 compared to sham group; ††p < 0.01 compared to TBI group; #p < 0.05 compared to exercise + sham group. TBI traumatic brain injury
Exercise Prevented the Induction of Histopathological Brain Edema and Apoptosis Following TBI
The effect of exercise on brain edema and apoptosis measured by histopathological evaluation 24 h post-TBI is shown in Fig. 9. Histopathological edema and apoptosis scores in TBI group (2.5 ± 0.22, 2 ± 0.2; respectively) were higher than that the sham (0.83 ± 0.31, 0.83 ± 0.17; respectively), exercise + sham (1.3 ± 0.21, 1 ± 0.1; respectively) and exercise + TBI (1.67 ± 0.21, 1.33 ± 0.21; respectively) groups (p < 0.01, p < 0.05, p < 0.05, respectively). These scores were the same among exercise groups and sham group (Fig. 10).
Fig. 9.

Histopathological changes in exercised male rats, 24 h post-TBI with magnification of 400 X. a Normal brain tissue in sham group. b Edema (thin white arrow) and apoptosis (thick black arrows) in TBI group. c Normal brain tissue in exercise + sham group. d Decrease in apoptosis and brain edema in exercise + TBI group. TBI traumatic brain injury
Fig. 10.

Histopathological brain edema (a) and apoptosis (b) in the brain of exercised male rats, 24 h post-TBI (n = 6 in each group). Data are shown as mean ± SEM. **p < 0.01 compared to sham group; ††p < 0.01 compared to TBI group; †p < 0.05 compared to TBI group. TBI traumatic brain injury
Discussion
Despite extensive research, no successful treatment for TBI has been reported so far. Our study was conducted as the first research to investigate the effect and the probable mechanism of previous endurance exercise on brain edema and neurological outcome following diffuse TBI. This study had the following main findings: (1) Previous endurance exercise prevented the development of brain edema and apoptosis following TBI. (2) Previous endurance exercise improved neurological outcome post-TBI. (3) Serum IL-1β level reduced following TBI in rates with the history of exercise. (4) Previous exercise prevented lipid peroxidation and decreased protein oxidation in brain following TBI. (5) Serum TAC level did not change following TBI in rates with the history of exercise.
The existence of brain edema and neurological disturbance following TBI in the present study has also been reported in our previous study (Soltani et al. 2015). Endurance exercise prevented the increase in brain water content following TBI. Also, reduction in VCS following TBI was small in rates with the history of exercise. Reduction in brain edema and improvement in neurological outcome have been reported in rats with ischemic stroke that exercised on a treadmill (Nishioka et al. 2016). Also, reduction in brain edema and post-ischemic infarction following stroke have been reported in exercised rats (Stummer et al. 1994). Exercise preconditioning reduces brain edema and neurological disorders in MCAO (Ding et al. 2006; Taylor et al. 2015). Some studies do not report better neurological outcome after cerebral injury in exercised group (Piao et al. 2013; Silva et al. 2013), which could be attributed to the time, type, severity and duration of exercise.
Brain edema plays a role in neurological deterioration (Battey et al. 2014). Physical exercise may improve neurological outcomes by preventing the formation of brain edema at the first place.
Pathological pathways responsible for neuronal death in TBI primarily include excitotoxicity, free radical production, inflammation and apoptosis. Protective mechanisms that spontaneously initiate in TBI include the expression of anti-inflammatory cytokines, growth factors (GFs) and endogenous antioxidants (Leker and Shohami 2002). Inflammation and oxidative stress contribute to the creation of progressive structural alterations and functional defects (Byrnes et al. 2012a). We speculated the inflammation and oxidative stress-related factors to be associated in the beneficial effects of treadmill exercise on TBI. An augmentation in IL-1β level of serum post-TBI was found similar to a study (Khaksari et al. 2010). A decrease in serum IL-1β level was observed in TBI rats with the history of exercise in this study that confirms other reports related to this subject (Mota et al. 2012; Petersen and Pedersen 2005). Pro-inflammatory cytokines aggregation (e.g. IL-1β), neutrophil infiltration and diminution of anti-inflammatory cytokines (e.g. IL-10) are suppressed in the presence of previous exercise in animals with FPI, which may mediate the improvement in motor function (Mota et al. 2012; Petersen and Pedersen 2005). Delayed voluntary exercise for four weeks reduced IL-1β level and increased IL-10 and IL-6 levels after the TBI (Piao et al. 2013). The athletes did not show any enhancement in their serum level of IL-1β after repetitive head trauma in competition compared to pre-trauma period (Di Battista et al. 2016).
Cytokines stimulate the expression of MMP-3 and MMP-9, partly by effecting nuclear factor-kappa-B (NF-κB) gene transcription (Cheng et al. 2006). The imbalance between metalloproteinase 9 (MMP-9) and TIMP-1 (tissue inhibitors of metalloproteinase-1), and also the disruption of occludin (a substrate of MMP-9) could cause a disruption in blood–brain barrier (BBB) (Rosenberg et al. 1998; Zhang et al. 2013). The loss of BBB integrity during MCAO is an early event that contributes to the initiation of inflammatory activity, edema formation and ultimately poor outcomes (Khan et al. 2012). Three days of treadmill exercise after MCAO suppressed the ischemia-induced upregulation of MMP-9 and downregulation of TIMP-1 as well as occludin (Zhang et al. 2013). The palliation of BBB permeability associated with a reduction in brain edema was observed in animals with pre-ischemic exercise (Guo et al. 2008). Exercise also attenuated inflammation by decreasing activation of microglia, astrocytosis and expression of pro-inflammatory cytokines, such as IL-1β and tumor necrosis factor-alpha (TNF-α) (Yang and Rosenberg 2011). Therefore, exercise may protect BBB morphology by inhibiting accumulation of pro-inflammatory cytokine and thus declining brain edema formation in TBI. This latter hypothesis needs additional research for confirmation.
In the present study, TBI raised oxidant markers (e.g. MDA and protein carbonyl), but lowered antioxidant markers (e.g. TAC), which correspond with the results of studies conducted by others (de Castro et al. 2017; Hall et al. 2004). Since the brain has a high potential for oxidative damage with its low antioxidant capacity (Floyd and Hensley 2002), oxidant markers were examined both in serum and in brain; however, antioxidant markers were assessed only in serum as IL-1β. Although an increment in levels of MDA and protein carbonyl in brain was revealed after TBI, there was no alteration in serum levels of these factors possibility due to insufficient permeability of BBB caused by moderate and diffuses TBI. In a human assay containing serum biomarkers, the serum level of MDA was not defined as a blood-based biomarker predicting the presence of intracranial injury demonstrated by initial brain CT scanning (Sharma et al. 2017). Also, the majority of studies have assessed oxidative stress-related biomarkers in brain rather than in serum (Dong et al. 2017; Jia et al. 2017). However, patients with mild TBI showed an increase in serum level of MDA, unlike protein carbonyl. It seems that some serum biomarkers could not be used as evaluation criteria in all cases of TBI.
Exercised rats displayed elevated serum levels of MDA contrary to decreased serum levels of protein carbonyl and TAC, both in the presence and absence of TBI. This finding suggests that exercise could induce systemic oxidative stress as reported in another study (Shi et al. 2007). The ROS produced by exercise can be harmful in some tissues but can also stimulate adaptive responses to oxidative stress including the upregulation of antioxidant genes expression (Bouzid et al. 2014). From the one hand, mitochondria are both sources of oxidant production and targets of oxidants, and on the other hand, regular exercise causes an increase in O2 consumption and mitochondrial biogenesis (Boveris and Navarro 2008), so previous physical training may result in ROS generation in some periods (Soustiel and Sviri 2007).
In another part of the current study, a decrease in brain protein carbonyl content was observed following TBI in rates with the history of exercise. Also, exercise impeded the increase in brain MDA level following TBI. However, the previous exercise did not influence serum level of FRAP after the TBI. Six weeks of previous swimming training reduced the level of MDA and protein carbonyl associated with Na + ,K + -ATPase dysfunction induced by TBI (Lima et al. 2009). According to existing evidence, oxidative stress may inhibit Na + , K + -ATPase activity leading to neurological dysfunction in TBI (da Silva Fiorin et al. 2016). Oxidative stress, the most important component of secondary injury cascade in TBI, is an imbalance in the ratio of harmful reactive oxygen and nitrogen species and useful antioxidants defense enzymes (Radak et al. 2013). It seems that treadmill training in this study developed systemic oxidative stress as an adaptive response and prevented local oxidative stress in diffuse TBI. The findings related to the effects of treadmill training on brain oxidant and antioxidant factors in rats with TBI may be due to adaptive responses of endurance exercise (Packer et al. 2008).
Irisin, an exercise-induced neuroprotective myokine, is decreased in cerebral ischemia (Kuloglu et al. 2014). Irisin is also expressed in the brain, the skeletal muscles and the heart (Dun et al. 2013), and its blood level increases following acute exercises (Löffler et al. 2015). A negative correlation was found between plasma irisin levels and plasma pro-inflammatory cytokines levels (Li et al. 2017). Irisin administration reduced brain infarct volume, brain edema, neurological deficits, neuroinflammation and oxidative stress after cerebral ischemia (Li et al. 2017). The improvement in neurological function and the reduction in neuroinflammation suggested by physical exercise were largely weakened with the administration of neutralizing antibody in cerebral ischemia. Thus, irisin has a key role in the beneficial effects of exercise on the brain (Farshbaf et al. 2016). The neuroprotective effects of irisin may be mediated by the activation of Akt and ERK1/2 (Li et al. 2017). It is presumed that an increase in irisin after treadmill training modifies oxidative stress and inflammation-related biomarkers resulting in partial debarment or diminution in TBI-induced brain edema and neurological deficits. This hypothesis will be considered in a future investigation.
The cooperative roles of aquaporin 4 (AQP4) and sodium–hydrogen exchanger 1 (NHE1) have been suggested in the genesis of brain edema following stroke (Nishioka et al. 2016). NHE1 enhances the intracellular osmotic pressure by increasing the influx of Na + leading to water influx via AQP4 in glial cells. Treadmill training before MCAO has been reported to reduce AQP4 and NHE1 expression in the brain, possibility through a moderate increase in corticosterone level exerted by both gluco- and mineralocorticoid receptors; therefore, it prohibits the formation of brain edema (He et al. 2014). Thus, physical exercise may prevent the development of brain edema post-TBI partly by attenuating AQP4 and NHE1 expression. This hypothesis requires further investigation.
Histopathological evaluation of brain tissue in current research indicated that apoptosis is not induced in exercised rats following TBI and edema. The pathological results are confirmed by an animal study that showed rats exercised prior to CCI (controlled cortical impact) had better neurological and motor outcomes associated with reduced apoptosis markers and increased anti-apoptotic markers (Zhao et al. 2014).
Conclusion
Previous exercise prevented the development of brain water content and improved the neurological outcome following TBI. Neuroprotective effect of previous exercise in TBI may be mediated by modulating inflammation, oxidative stress and apoptosis. Consequently, some athletes may be protected against TBI. It is assumed that an increase in protective molecules created by physical exercise before TBI reduces the immediate and late detrimental outcomes of injury. However, this hypothesis requires more research for confirmation. One of the limitations of study was the use of VCS to evaluate neurological outcome, as this index is beneficial in short-term evaluation.
Acknowledgments
The authors would like to thank Dr. Shiebani and Dr. Pardakhti for their help and support in this study.
Author Contributions
MK directed the project and carried out the interpretations. NS and MH carried out neurobehavioral and brain edema evaluations. GHE and MI assessed biochemical and histopathological agents. ZS directed the project, carried out the data analysis and interpretations, and prepared the manuscript.
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest.
Ethical Approval
The study was executed in accordance with the guidelines for animal experimental protocols of Kerman University of Medical Sciences and the internationally accepted principles for animal use and care (EU Directive of 2010; 010/63/EU). The research protocol was approved by the ethics committee of Kerman University of Medical Sciences (No. EC/KNRC/94-420). The animals were maintained in an air-conditioned room at 22–25 °C in a 12 h light and 12 h dark cycle. Also, food and water were available to the animals during the study.
Informed Consent
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adkins DL, Boychuk J, Remple MS, Kleim JA (2006) Motor training induces experience-specific patterns of plasticity across motor cortex and spinal cord. J Appl Physiol 101(6):1776–1782 [DOI] [PubMed] [Google Scholar]
- Ang E, Gomez-Pinilla F (2007) Potential therapeutic effects of exercise to the brain. Curr Med Chem 14(24):2564–2571 [DOI] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW (2008) Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radical Biol Med 45(4):443–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrick DM, Yang S, Li C, Cananzi S, Mayhan WG (2014) Vigorous exercise training improves reactivity of cerebral arterioles and reduces brain injury following transient focal ischemia. Microcirculation 21(6):516–523 [DOI] [PubMed] [Google Scholar]
- Battey TW, Karki M, Singhal AB et al (2014) Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke 45(12):3643–3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayır H, Kagan VE, Clark RS et al (2007) Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem 101(1):168–181 [DOI] [PubMed] [Google Scholar]
- Benzie IF, Strain JJ (1996) The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 239(1):70–76 [DOI] [PubMed] [Google Scholar]
- Bland DC, Zampieri C, Damiano DL (2011) Effectiveness of physical therapy for improving gait and balance in individuals with traumatic brain injury: a systematic review. Brain Inj 25(7–8):664–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzid MA, Hammouda O, Matran R, Robin S, Fabre C (2014) Low intensity aerobic exercise and oxidative stress markers in older adults. J Aging Phys Act 22(4):536–542 [DOI] [PubMed] [Google Scholar]
- Boveris A, Navarro A (2008) Brain mitochondrial dysfunction in aging. IUBMB Life 60(5):308–314 [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI (2012a) Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation 9(1):43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI (2012b) Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation 9:43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST (2006) Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol 59(5):735–742 [DOI] [PubMed] [Google Scholar]
- Carvalho JF, Masuda MO, Pompeu FA (2005) Method for diagnosis and control of aerobic training in rats based on lactate threshold. Comp Biochem Physiol A 140(4):409–413 [DOI] [PubMed] [Google Scholar]
- Castro MRT, Ferreira APdO, Busanello GL et al (2017) Previous physical exercise alters hepatic profile of oxidative-inflammatory status and limits the secondary brain damage induced by severe TBI in rats. J Physiol 595(17):6023–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Zhang L et al (2006) Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med 12(11):1278 [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC, Christie L-A (2007) Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci 30(9):464–472 [DOI] [PubMed] [Google Scholar]
- da Silva Fiorin F, de Oliveira Ferreira AP, Ribeiro LR et al (2016) The impact of previous physical training on redox signaling after traumatic brain injury in rats: a behavioral and neurochemical approach. J Neurotrauma 33(14):1317–1330 [DOI] [PubMed] [Google Scholar]
- de Castro MRT, de Oliveira Ferreira AP, Busanello GL et al (2017) Previous physical exercise alters the hepatic profile of oxidative-inflammatory status and limits the secondary brain damage induced by severe traumatic brain injury in rats. J Physiol 595(17):6023–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Battista AP, Rhind SG, Richards D, Churchill N, Baker AJ, Hutchison MG (2016) Correction: altered blood biomarker profiles in athletes with a history of repetitive head impacts. PLoS ONE 11(10):e0164912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Li J, Yao W, Rafols J, Clark J, Ding Y (2006) Exercise preconditioning upregulates cerebral integrins and enhances cerebrovascular integrity in ischemic rats. Acta Neuropathol 112(1):74 [DOI] [PubMed] [Google Scholar]
- Dong N, Diao Y, Ding M, Cao B, Jiang D (2017) The effects of 7-nitroindazole on serum neuron-specific enolase and astroglia-derived protein (S100β) levels after traumatic brain injury. Exp Ther Med 13(6):3183–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dun SL, Lyu R-M, Chen Y-H, Chang J-K, Luo JJ, Dun NJ (2013) Irisin-immunoreactivity in neural and non-neural cells of the rodent. Neuroscience 240:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Gertz K, Lindauer U et al (2003) Mechanisms of stroke protection by physical activity. Ann Neurol 54(5):582–590 [DOI] [PubMed] [Google Scholar]
- Farshbaf MJ, Ghaedi K, Megraw TL et al (2016) Does PGC1α/FNDC5/BDNF elicit the beneficial effects of exercise on neurodegenerative disorders? NeuroMol Med 18(1):1–15 [DOI] [PubMed] [Google Scholar]
- Floyd RA, Hensley K (2002) Oxidative stress in brain aging: implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23(5):795–807 [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Gomez-Pinilla F (2009) Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res 1288:105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Tio DL, Vincelli J, McArthur DL, Taylor AN (2012) Differential effects of voluntary and forced exercise on stress responses after traumatic brain injury. J Neurotrauma 29(7):1426–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Cox B, Mahale S et al (2008) Pre-ischemic exercise reduces matrix metalloproteinase-9 expression and ameliorates blood–brain barrier dysfunction in stroke. Neuroscience 151(2):340–351 [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC (2004) Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma 21(1):9–20 [DOI] [PubMed] [Google Scholar]
- He Z, Wang X, Wu Y et al (2014) Treadmill pre-training ameliorates brain edema in ischemic stroke via down-regulation of aquaporin-4: an MRI study in rats. PLoS ONE 9(1):e84602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks RR, Boggs A, Leider D et al (1998) Effects of exercise following lateral fluid percussion brain injury in rats. Restor Neurol Neurosci 12(1):41–47 [PubMed] [Google Scholar]
- Hindin SB, Zelinski EM (2012) Extended practice and aerobic exercise interventions benefit untrained cognitive outcomes in older adults: a meta-analysis. J Am Geriatr Soc 60(1):136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoene M, Weigert C (2010) The stress response of the liver to physical exercise. Exerc Immunol Rev 16:163 [PubMed] [Google Scholar]
- Itoh T, Imano M, Nishida S et al (2011) Exercise inhibits neuronal apoptosis and improves cerebral function following rat traumatic brain injury. J Neural Transm 118(9):1263–1272 [DOI] [PubMed] [Google Scholar]
- Jacotte-Simancas A, Costa-Miserachs D, Coll-Andreu M, Torras-Garcia M, Borlongan CV, Portell-Cortés I (2015) Effects of voluntary physical exercise, citicoline, and combined treatment on object recognition memory, neurogenesis, and neuroprotection after traumatic brain injury in rats. J Neurotrauma 32(10):739–751 [DOI] [PubMed] [Google Scholar]
- Jia L, Wang F, Gu X et al (2017) Propofol postconditioning attenuates hippocampus ischemia-reperfusion injury via modulating JAK2/STAT3 pathway in rats after autogenous orthotropic liver transplantation. Brain Res 1657:202–207 [DOI] [PubMed] [Google Scholar]
- Khaksari M, Soltani Z, Shahrokhi N, Moshtaghi G, Asadikaram G (2010) The role of estrogen and progesterone, administered alone and in combination, in modulating cytokine concentration following traumatic brain injury. Can J Physiol Pharmacol 89(1):31–40 [DOI] [PubMed] [Google Scholar]
- Khaksari M, Rajizadeh MA, Bejeshk MA et al (2018a) Does inhibition of angiotensin function cause neuroprotection in diffuse traumatic brain injury? Iran J Basic Med Sci 21(6):615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaksari M, Soltani Z, Shahrokhi N (2018b) Effects of female sex steroids administration on pathophysiologic mechanisms in traumatic brain injury. Trans Stroke Res 9(4):393–416 [DOI] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Sakakima H et al (2012) The inhibitory effect of S-nitrosoglutathione on blood–brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J Neurochem 123(s2):86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D-H, Ko I-G, Kim B-K et al (2010) Treadmill exercise inhibits traumatic brain injury-induced hippocampal apoptosis. Physiol Behav 101(5):660–665 [DOI] [PubMed] [Google Scholar]
- Kuloglu T, Aydin S, Eren MN et al (2014) Irisin: a potentially candidate marker for myocardial infarction. Peptides 55:85–91 [DOI] [PubMed] [Google Scholar]
- Leker RR, Shohami E (2002) Cerebral ischemia and trauma—different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Rev 39(1):55–73 [DOI] [PubMed] [Google Scholar]
- Li D-J, Li Y-H, Yuan H-B, Qu L-F, Wang P (2017) The novel exercise-induced hormone irisin protects against neuronal injury via activation of the Akt and ERK1/2 signaling pathways and contributes to the neuroprotection of physical exercise in cerebral ischemia. Metabolism 68:31–42 [DOI] [PubMed] [Google Scholar]
- Lima FD, Oliveira MS, Furian AF et al (2009) Adaptation to oxidative challenge induced by chronic physical exercise prevents Na + , K + -ATPase activity inhibition after traumatic brain injury. Brain Res 1279:147–155 [DOI] [PubMed] [Google Scholar]
- Lima FD, Stamm DN, Della-Pace ID et al (2013) Swimming training induces liver mitochondrial adaptations to oxidative stress in rats submitted to repeated exhaustive swimming bouts. PLoS ONE 8(2):e55668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler D, Müller U, Scheuermann K et al (2015) Serum irisin levels are regulated by acute strenuous exercise. J Clin Endocrinol Metab 100(4):1289–1299 [DOI] [PubMed] [Google Scholar]
- Marmarou A, Foda MAA-E, Wvd Brink, Campbell J, Kita H, Demetriadou K (1994) A new model of diffuse brain injury in rats: part I: pathophysiology and biomechanics. J Neurosurg 80(2):291–300 [DOI] [PubMed] [Google Scholar]
- Mota BC, Pereira L, Souza MA et al (2012) Exercise pre-conditioning reduces brain inflammation and protects against toxicity induced by traumatic brain injury: behavioral and neurochemical approach. Neurotox Res 21(2):175–184 [DOI] [PubMed] [Google Scholar]
- Nishioka R, Sugimoto K, Aono H et al (2016) Treadmill exercise ameliorates ischemia-induced brain edema while suppressing Na +/H + exchanger 1 expression. Exp Neurol 277:150–161 [DOI] [PubMed] [Google Scholar]
- O’Connor CA, Cernak I, Vink R (2005) Both estrogen and progesterone attenuate edema formation following diffuse traumatic brain injury in rats. Brain Res 1062(1):171–174 [DOI] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95(2):351–358 [DOI] [PubMed] [Google Scholar]
- Opii WO, Nukala VN, Sultana R et al (2007) Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J Neurotrauma 24(5):772–789 [DOI] [PubMed] [Google Scholar]
- Packer L, Cadenas E, Davies KJ (2008) Free radicals and exercise: an introduction. Free Radical Biol Med 44(2):123–125 [DOI] [PubMed] [Google Scholar]
- Petersen AMW, Pedersen BK (2005) The anti-inflammatory effect of exercise. J Appl Physiol 98(4):1154–1162 [DOI] [PubMed] [Google Scholar]
- Piao C-S, Stoica BA, Wu J et al (2013) Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol Dis 54:252–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak Z, Zhao Z, Koltai E, Ohno H, Atalay M (2013) Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid Redox Signal 18(10):1208–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ et al (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70(3):374–383 [DOI] [PubMed] [Google Scholar]
- Rosenberg G, Estrada E, Dencoff J (1998) Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 29(10):2189–2195 [DOI] [PubMed] [Google Scholar]
- Sharma R, Rosenberg A, Bennett ER, Laskowitz DT, Acheson SK (2017) A blood-based biomarker panel to risk-stratify mild traumatic brain injury. PLoS ONE 12(3):e0173798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Wang X, Yamanaka T, Ogita F, Nakatani K, Takeuchi T (2007) Effects of anaerobic exercise and aerobic exercise on biomarkers of oxidative stress. Environ Health Prev Med 12(5):202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LFA, Hoffmann MS, Gerbatin RdR et al (2013) Treadmill exercise protects against pentylenetetrazol-induced seizures and oxidative stress after traumatic brain injury. J Neurotrauma 30(14):1278–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED (2006) Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab 26(11):1407–1418 [DOI] [PubMed] [Google Scholar]
- Smith PJ, Blumenthal JA, Hoffman BM et al (2010) Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med 72(3):239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltani Z, Khasksari M, Shahrokhi N, Nakhaei N, Shaibani V (2009) Effect of combined administration of estrogen and progesterone on brain edema and neurological outcome after traumatic brain injury in female rats. Iran J Endocrinol Metab 10(6):629–638 [Google Scholar]
- Soltani Z, Khaksari M, Jafari E, Iranpour M, Shahrokhi N (2015) Is genistein neuroprotective in traumatic brain injury? Physiol Behav 152:26–31 [DOI] [PubMed] [Google Scholar]
- Soltani Z, Khaksari M, Shahrokhi N et al (2016) Effect of estrogen and/or progesterone administration on traumatic brain injury-caused brain edema: the changes of aquaporin-4 and interleukin-6. J Physiol Biochem 72(1):33–44 [DOI] [PubMed] [Google Scholar]
- Soustiel JF, Sviri GE (2007) Monitoring of cerebral metabolism: non-ischemic impairment of oxidative metabolism following severe traumatic brain injury. Neurol Res 29(7):654–660 [DOI] [PubMed] [Google Scholar]
- Stummer W, Weber K, Tranmer B, Baethmann A, Kempski O (1994) Reduced mortality and brain damage after locomotor activity in gerbil forebrain ischemia. Stroke 25(9):1862–1869 [DOI] [PubMed] [Google Scholar]
- Sun L, Shen W, Liu Z, Guan S, Liu J, Ding S (2010) Endurance exercise causes mitochondrial and oxidative stress in rat liver: effects of a combination of mitochondrial targeting nutrients. Life Sci 86(1):39–44 [DOI] [PubMed] [Google Scholar]
- Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F (1993) Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion: influence of pre-and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol 42(2):177–185 [DOI] [PubMed] [Google Scholar]
- Taylor JM, Montgomery MH, Gregory EJ, Berman NE (2015) Exercise preconditioning improves traumatic brain injury outcomes. Brain Res 1622:414–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma S, Janesko KL, Wisniewski SR et al (2003) F2-isoprostane and neuron-specific enolase in cerebrospinal fluid after severe traumatic brain injury in infants and children. J Neurotrauma 20(8):781–786 [DOI] [PubMed] [Google Scholar]
- Vaynman S, Gomez-Pinilla F (2005) License to run: exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehabilitation Neural Repair 19(4):283–295 [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F (2013) Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience 248:655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L-J, Traber MG, Packer L (1995) Spectrophotometric method for determination of carbonyls in oxidatively modified apolipoprotein B of human low-density lipoproteins. Anal Biochem 228(2):349–351 [DOI] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA (2011) Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42(11):3323–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Zhang Y, Zhang J et al (2013) Early exercise protects against cerebral ischemic injury through inhibiting neuron apoptosis in cortex in rats. Int J Mol Sci 14(3):6074–6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Sabirzhanov B, Wu J, Faden AI, Stoica BA (2014) Voluntary exercise preconditioning activates multiple anti-apoptotic mechanisms and improves neurological recovery after experimental traumatic brain injury. J Neurotrauma. 10.1089/neu.2014.3739 [DOI] [PMC free article] [PubMed] [Google Scholar]