

Abstract
In this community effort, we compare measurements between 34 laboratories from 19 countries, utilizing mixtures of labelled authentic synthetic standards, to quantify by mass spectrometry four clinically used ceramide species in the NIST (National Institute of Standards and Technology) human blood plasma Standard Reference Material (SRM) 1950, as well as a set of candidate plasma reference materials (RM 8231). Participants either utilized a provided validated method and/or their method of choice. Mean concentration values, and intra- and inter-laboratory coefficients of variation (CV) were calculated using single-point and multi-point calibrations, respectively. These results are the most precise (intra-laboratory CVs ≤ 4.2%) and concordant (inter-laboratory CVs < 14%) community-derived absolute concentration values reported to date for four clinically used ceramides in the commonly analyzed SRM 1950. We demonstrate that calibration using authentic labelled standards dramatically reduces data variability. Furthermore, we show how the use of shared RM can correct systematic quantitative biases and help in harmonizing lipidomics. Collectively, the results from the present study provide a significant knowledge base for translation of lipidomic technologies to future clinical applications that might require the determination of reference intervals (RIs) in various human populations or might need to estimate reference change values (RCV), when analytical variability is a key factor for recall during multiple testing of individuals.
Subject terms: Predictive markers, Lipidomics
Here, the authors compared measurements between 34 laboratories from 19 countries, to quantify by mass spectrometry four ceramides of clinical relevance in human blood plasma Standard Reference Materials. The main goals were to evaluate concordance obtained in a large inter-laboratory trial and to report absolute concentrations of four circulating lipids in a publicly available standard.
Introduction
Mass spectrometry (MS) is a powerful technology that allows the qualitative and quantitative analysis of circulating metabolites, including lipids, in blood plasma1. Challenges in the translation of MS-based analytical methods stem from technical aspects (e.g., dependency on stringent and consistent sampling procedures, reproducibility between different laboratories, control of bias and noise)2–4. Beyond that, introducing and adopting new MS-based workflows into existing fields will require additional efforts, such as demonstrating cost-benefit effectiveness, scale, and pertinent turn-around times from sampling to reporting. In the case of clinical applications, demonstration of utility to individuals and populations will be required in addition to economic benefit5.
Once established, through future efforts by the scientific community, reference intervals (RI, aka reference ranges) will be an important tool for the convincing communication of lipidomic measurements and clinical adoption of laboratory-developed tests (LDT) within established clinical practices. RI represent lower and higher concentration boundaries of analytes, lipids, and metabolites, in the case of lipidomics and metabolomics, respectively. Clinicians use and rely on established RI for interpretation of laboratory results, medical diagnosis, and evaluation of treatment options for patients within a given reference group (e.g., total cholesterol, LDL-cholesterol RI according to different sex, age, and ethnicity). Appropriate standardization, which is the focus of this manuscript, is a pre-requisite for (i) establishment and (ii) transportability of RI.
This inter-laboratory and cross-platform study is an extension of a community-initiated effort that started in 2017 and is now hosted by the International Lipidomics Society. As detailed and recommended in the initial proposal, reporting of molar concentrations of individual lipids by different laboratories should be regarded as a first step in harmonizing plasma lipidomics. The main goals of this study were to: (i) evaluate the degree of accuracy and concordance obtained in a large inter-laboratory trial using the same shared samples and custom-tailored calibrant materials; (ii) highlight technical issues contributing to variability and technical outliers to avoid in future; (iii) document as precisely as possible the absolute concentrations of four circulating lipids in a publicly available standard reference material and (iv) lay the foundation for determination of MS-based lipidomic RI in diverse human populations across the world in a standardized fashion.
The evaluation of utility of population RIs for the interpretation of lipidomic results is an important step that needs evaluation. If the analyte shows low individuality and the value dispersion in each individual covers most of the dispersion among individuals, then the RI value is a useful parameter6. However, for analytes showing high individuality the use of RI is not recommended, while monitoring longitudinal changes of concentrations by the reference change values (RCV) application is a better strategy. The RCV is defined as the change needed between two serial results from the same individual to be statistically significantly different7. The RIs are descriptive of a specific population, derived from a reference distribution (usually 95% interval) and are different from the decision limits above or below which a specific medical decision is recommended. And because we must consider that, for many analytes, a continuous variation may have diagnostic significance, the concept of RCV is useful, although not much used in everyday practice.
Why ceramides? The initial group of investigators involved in the human blood plasma guidelines initiative8 identified ceramides, among other circulating lipids, as a particularly well-suited test case for a number of reasons including: (i) Disease association: Ceramides are associated with major adverse cardiovascular events in individuals with coronary artery disease, Type 2 Diabetes, renal impairment, and age9–13 as well as major depressive disorders14. (ii) Clinical utility: Ceramides are used clinically to identify the risk of adverse cardiovascular events within 5 years15,16. (iii) Actionability: Circulating ceramide levels can be modified by long-term lifestyle interventions17–19, and the sphingolipid synthetic pathway can be targeted therapeutically (e.g. via several naturally occurring enzyme inhibitors and derivatives thereof such as Fingolimod)20,21. (iv) Genetic heritability: Ceramides display among the highest reported heritability values of blood lipids in different populations, and accordingly, sphingolipids are strongly influenced by ethnicity22. (v) Technical considerations: Analyte stability, detectable concentration levels in the circulation with currently used analytical platforms, and availability of standards were additional important considerations.
Results
The main goal of this community effort was to compare MS measurements between a large number of laboratories (n = 34) using the (i) same human blood plasma reference materials (four NIST plasma RMs)23, utilizing (ii) the same synthetic standards (a mixture of deuterated ceramides at a pre-defined concentration prepared by Avanti Polar Lipids), and (iii) to report absolute concentrations of four ceramide species (Cer 18:1;O2/16:0, Cer 18:1;O2/18:0, Cer 18:1;O2/24:0 and Cer 18:1;O2/24:1) in the different RMs.
The choice of the NIST SRM 1950 as one of the RMs was deliberate given its commercial accessibility offering widespread access and utility. Consequently, this material has been used in numerous lipidomic and metabolomic community-driven studies24–29. Furthermore, NIST SRM 1950 is a relatively well-characterized standard reference material (e.g., sterol and fatty acid concentrations are certified by NIST). It is increasingly being adopted in the lipidomic and metabolomic communities for inter-laboratory comparisons and as a shared reference material for human plasma studies30–32. The other three RMs23 used in this ring trial allowed us to test different matrix effects which might be useful in future clinical research33. As ceramides are implicated in cardio-metabolic diseases, we included pooled samples from populations with hypertriglyceridemia (hTAG) and diabetes (DB), respectively. Ethnicity is another factor affecting plasma sphingolipid levels34. For this reason, we also analysed the Young African American (YAA) blood plasma as one of the RMs.
To help standardize experimental steps between laboratories experienced in lipidomic analysis and those newer to this field, a “Standard” operating protocol (referred to as SOP throughout, see Supplementary Information) was prepared by the study coordinators, according to a published quantitative method35. This SOP included detailed, step-by-step procedures covering ceramide extraction from human blood plasma, preparation of external calibration curves using deuterated and non-deuterated synthetic standards, and use of MRM-based LC-MS/MS for measurement on triple quadrupole instruments. A template to design the sample analytical sequence and data reporting in a standardized format was also included (see the SOP in Supplementary Information). To minimize variability-associated procedures, peak areas were chosen as the raw data report format rather than concentration values derived by normalization to internal standards. The structured data reports facilitated integration with a specially designed pipeline for automated downstream processing, including conversion of peak areas to concentration values. The pipeline was designed to simplify reproducibility of both tabular and graphical results. The structured reporting format contained sections to report peak areas for all four ceramides and their labelled counterparts in columns, with three replicates each. The report rows contained the peak areas of all dilution steps of both calibration line samples for multi-point calibration, the areas for every aliquot of each RM, the areas for each concentration step of the QC samples, and finally for the ceramide peak areas measured in the matrix and total blank samples (see the SOP in the Supplementary Information for the definitions of different sample types). The calibration lines were calculated using R’s ‘lm‘ method, regressing the ceramide area to labelled standard ratio on the known concentration, weighted by 1/x2, following the method used by Kauhanen et al.35. We then evaluated the average of the calibration lines for each lab to calculate the adjusted concentrations from the reported areas of each ceramide and corresponding labelled standard within the QC and RM samples (Supplementary Fig. 1). As a commonly adopted procedure in lipidomics, the concentration was also calculated using a single-point calibration, based on the ratio of each ceramide and its corresponding labelled standard, multiplied by the known internal standard concentration. The individual concentrations were then combined and enriched with lab-specific metadata, e.g., the methodology and MS-platform used, to calculate CV values for the inter-lab comparison.
The participating laboratories were also invited to use their preferred protocol (from extraction to detection to the type of instrument used) with which they might be more familiar, either as an alternative or an addition to the SOP (referred to as OTHER below). However, the same set of synthetic standards were to be used in either case. At the end of the study, we collected 23 datasets generated with the SOP method and 15 with OTHER methods. The latter included a combination of different techniques, such as nano or capillary-LC, flow injection analysis (FIA), different types of LC columns and gradients, as well as different detection methods (Table 1). The breadth of different approaches allowed us to observe the influence of a prescribed methodological procedure (SOP) in comparison to performing the measurements in different ways (OTHER) and, importantly, the effect that a common set of carefully adjusted standards might have on the quantitation performances when different methodologies are used. All the final annotated data are available as Supplementary Data.
Table 1.
Analytical methods and platforms
| Method | SOP | OTHER | ||
|---|---|---|---|---|
| MS platform | QQQ/QTRAP | QQQ/QTRAP | QTOF | Orbitrap |
| Number Datasets | 23 (23 RP) | 10 (8 RP, 2 FIA) | 3 (2 RP, 1 SFC) | 3 (2 RP, 1 FIA) |
| Sum | 23 | 16 | ||
SOP refers to the method recommended by the study authors, OTHER to methods chosen by the individual laboratories. The number in brackets indicates the number for each chromatographic/injection method (RP Reversed Phase, FIA Flow Injection Analysis, SFC Supercritical Fluid Chromatography). Other variations present in OTHER may include type of extraction, nano/capLC, LC duration (7–50 min), column chemistries and MS source settings. Platforms: QQQ = triple quadrupole, QTRAP = ion trap, QTOF = quadrupole time-of-flight.
It should be noted that for simplicity, since all four ceramides share the same sphingoid base moiety, i.e., 18:1;O2, we will refer to Cer 18:1;O2/16:0 as Cer16, Cer 18:1;O2/18:0 as Cer18, Cer 18:1;O2/24:0 as Cer24:0, and Cer 18:1;O2/24:1 as Cer24:1 in the text, but always according to the updated LIPID MAPS nomenclature in the displays and tables36. In addition, and again for simplicity, we limit the report of the results in the next section to those obtained for NIST SRM 1950 plasma. A separate paragraph will discuss the comparison of ceramide concentrations between the four RMs.
Before estimating consensus values for the concentration of the ceramides in SRM 1950, we analysed the data contributed by all the participant laboratories (Fig. 1). This allowed us to compare the reproducibility of the results between different methods and instruments, starting from the same plasma sample and internal standard mixtures and excluding possible effects derived from different ways of estimating concentration values. Please note that the results shown in Fig. 1 and Supplementary Fig. 2 represent the full dataset, with outlier removal thresholds based on Tukey’s 1.5 × IQR fences37 represented and determined separately for each of the four ceramides and quantification methods, i.e., single vs. multi-point calibration. The outliers filtering resulted in the exclusion of 4 (10% of the total contributions) for Cer16 and Cer24, and 8 sets (21%) for Cer18 and Cer24:1, respectively, when considering multi-point calibration (Supplementary Table 1 and Supplementary Data). Outliers were not represented in Figs. 2 and 3 and in specific Supplementary Figs. (please refer to the corresponding legends).
Fig. 1. Consensus concentrations of ceramides in NIST SRM 1950 across all study participants.

Concentrations of the 4 reported ceramides in NIST SRM 1950. Each circle corresponds to the mean of 1 to 6 replicate measurements from a vial of NIST SRM 1950 shipped to each of the participating laboratories. The error bars depict the ±1 × SD of 3 injection replicates, which in some cases is smaller than the diameter of the plotted circle or absent if only 1 measurement was made. Horizontal bars represent the mean values of the mean measured concentration per NIST vial. The outer green dotted lines indicate used for as cut-off values for outlier removal. The mean ±2 × SD (dashed grey lines) are based on data after outlier removal. Molar concentrations were calculated using the external calibration curve (multi-point calibration). SOP (red) and OTHER (blue) refer to the Standard vs Other methods, respectively. Source data are provided as Source Data File.
Fig. 2. Comparison of single-point and multi-point calibrations.

Molar concentrations derived from single-point vs. multi-point calibration based quantifications. The diagonal line indicates equal results between single-point (horizontal axis) and multi-point (vertical axis) calibration-based quantification. Outlier measurements were removed for each ceramide species in each reference plasma (see Fig. 1 and text). Each circle corresponds to the mean value reported by one laboratory (n = 6). NIST SRM 1950 (SRM, dark blue); hypertriglyceridemic (hTAG, red), diabetic (DB, light orange), and Young African American (YAA, light blue), which are part of NIST RM 8231. Source data are provided as Source Data File.
Fig. 3. Concentration values of ceramides in NIST SRM 1950 and the suite of human plasma RM contained in RM 8231.

Concentrations of the 4 ceramides in different NIST reference plasma samples. Each circle corresponds to the values reported by one laboratory. P values are based on paired two-tailed t-tests of complete datasets. The horizontal bars correspond to the mean ±1 × SD of all shown data per group (number of data points indicated below each group). Outlier measurements were removed for ceramide species in each reference plasma (see Fig. 1 and text). SOP (red) and OTHER (blue) refer to the Standard vs Other methods, respectively. Source data are provided as Source Data File.
The medians of the intra-lab CVs ranged from 3.7 to 4.2% for the concentration of the four ceramides when using the multi-point calibration, confirming good reproducibility between the measurements acquired by the individual participants (Fig. 1, Table 2). The level of concordance of the measurements between the different contributors was estimated by the inter-lab CVs, ranging from 9 % to 14% when excluding outliers, and 25% to 31% for the unfiltered dataset (Table 2 and Supplementary Table 1). The concordance of the results between the participants was not affected by the use of SOP or OTHER procedures, as the values generated from these two groups were not significantly different (P > 0.05) for any of the analytes in all RMs (Supplementary Table 2 and Fig. 1). Considering the disparity between the number of contributions obtained with low (quadrupole/linear ion traps, n = 33) and high resolution (Orbitrap and Time of flight, n = 6, Table 1) instruments, we are not reporting any statistics for this comparison, although we did not find significant differences between these two sets of results. In addition, the CV did not show any obvious dependency on the signal intensity of the measured molecules, except for measurements acquired via FIA, where the sensitivity of the detection was lower for low abundant ceramides (e.g., Cer18 in Fig. 1, Lab ♯22). Collectively, these results demonstrate that precise and concordant (low intra- and inter-lab CVs, respectively) values can be obtained independently of different methods and MS platforms in many laboratories and establish the concentrations based on calibration curve quantification for the four ceramides in SRM 1950 as 0.244 ± 0.006 µmol/L for Cer 18:1;O2/16:0 (mean ± SEM; n = 35), 0.0835 ± 0.0017 µmol/L for Cer 18:1;O2/18:0 (n = 31), 2.42 ± 0.04 µmol/L for Cer 18:1;O2/24:0 (n = 35) and 0.855 ± 0.013 µmol/L Cer 18:1;O2/24:1 (n = 31) (Table 1).
Table 2.
Consensus concentrations of the 4 measured ceramides in the 4 NIST reference materials
| Matrix | Ceramide | n (SP) (SOP/OTH) | n (MP) (SOP/OTH) | μmol/L (SP) mean | μmol/L (SP) SEM | μmol/L (MP) mean | μmol/L (MP) SEM | Inter-lab %CV (SP) | Inter-lab %CV (MP) | Intra-lab %CV (MP) |
|---|---|---|---|---|---|---|---|---|---|---|
| SRM | Cer 18:1;O2/16:0 | 33 (21/12) | 35 (21/14) | 0.3507 | 0.0056 | 0.2438 | 0.0057 | 9.2 | 13.8 | 4.2 |
| Cer 18:1;O2/18:0 | 33 (22/11) | 31 (21/10) | 0.0553 | 0.0009 | 0.0835 | 0.0017 | 9.8 | 11.6 | 4.2 | |
| Cer 18:1;O2/24:0 | 35 (20/15) | 35 (20/15) | 2.5670 | 0.0494 | 2.4116 | 0.0413 | 11.4 | 10.1 | 3.7 | |
| Cer 18:1;O2/24:1 | 32 (21/11) | 31 (20/11) | 0.8397 | 0.0221 | 0.8554 | 0.0130 | 14.9 | 8.5 | 3.7 | |
| hTAG | Cer 18:1;O2/16:0 | 35 (22/13) | 35 (20/15) | 0.4019 | 0.0086 | 0.2888 | 0.0062 | 12.7 | 12.8 | 5.8 |
| Cer 18:1;O2/18:0 | 34 (22/12) | 31 (20/11) | 0.0652 | 0.0015 | 0.0995 | 0.0020 | 13.5 | 11.0 | 5.6 | |
| Cer 18:1;O2/24:0 | 35 (20/15) | 36 (21/15) | 3.9250 | 0.0892 | 3.7641 | 0.0765 | 13.4 | 12.2 | 4.2 | |
| Cer 18:1;O2/24:1 | 34 (21/13) | 33 (20/13) | 1.2207 | 0.0400 | 1.2784 | 0.0267 | 19.1 | 12.0 | 4.7 | |
| DB | Cer 18:1;O2/16:0 | 33 (22/11) | 35 (21/14) | 0.3088 | 0.0060 | 0.2161 | 0.0052 | 11.2 | 14.3 | 5.5 |
| Cer 18:1;O2/18:0 | 35 (22/13) | 32 (21/11) | 0.0631 | 0.0014 | 0.0966 | 0.0020 | 13.3 | 11.8 | 5.0 | |
| Cer 18:1;O2/24:0 | 35 (20/15) | 36 (21/15) | 2.5252 | 0.0498 | 2.4013 | 0.0475 | 11.7 | 11.9 | 3.5 | |
| Cer 18:1;O2/24:1 | 35 (21/14) | 33 (21/12) | 0.8498 | 0.0308 | 0.8663 | 0.0173 | 21.4 | 11.5 | 4.4 | |
| YAA | Cer 18:1;O2/16:0 | 34 (21/13) | 34 (20/14) | 0.2127 | 0.0048 | 0.1450 | 0.0039 | 13.1 | 15.6 | 6.3 |
| Cer 18:1;O2/18:0 | 33 (22/11) | 34 (21/13) | 0.0331 | 0.0007 | 0.0494 | 0.0016 | 12.7 | 18.6 | 5.6 | |
| Cer 18:1;O2/24:0 | 34 (20/14) | 35 (21/14) | 1.7312 | 0.0318 | 1.6485 | 0.0375 | 10.7 | 13.4 | 3.5 | |
| Cer 18:1;O2/24:1 | 33 (21/12) | 33 (21/12) | 0.5333 | 0.0181 | 0.5518 | 0.0135 | 19.5 | 14.0 | 4.6 |
SOP and OTH refer to the standard and OTHER methods, respectively. n indicates the number of datasets that were included in the calculation of the variables given in this table after removal of outliers of the single-point (SP) or multi-point (MP) calibration, respectively, using Tukey’s 1.5 × IQR method (see text). The total number of datasets before removal was 39 (26 SOP/13 OTHER). Intra-lab %CV corresponds to the median of individual intra-lab %CVs.
Estimation of ceramides concentrations in SRM 1950 by single-point internal standard vs. multi-point calibration curve
To generate reference values and absolute concentrations for the four ceramides in SRM 1950 plasma, we chose to quantify the analytes in two different ways, namely (i) via the use of calibration curves (multi-point, Fig. 1 and Supplementary Fig. 1) and (ii) by using the labelled internal standards (single-point calibration in a verified linear range, Supplementary Fig. 2). While the second approach is common when performing large lipidomic studies utilizing a small set of internal standards representative of each of the lipid classes measured and their concentrations, the use of a calibration curve built with both endogenous and labelled molecules is characteristic of targeted approaches. It should aid in accurate quantitation, minimizing the errors that could occur when using a single-point calibration. As both methods are currently part of lipidomic workflows, we tried to clarify if, in this specific case, a significant benefit was conferred by either of them. As previously mentioned, we used only the dataset without outliers for this comparison. This exercise aimed to estimate if the two procedures generate the same results with the same variability between laboratories. When considering all the participating laboratories independently of the analytical method used (SOP vs. OTHER methods), we obtained inter-lab CVs for concentrations derived using single-point normalization of 9.2% for Cer16:0, 9.8% for C18:0, 11.4% for C24:0, and 14.9% for C24:1, respectively, while the corresponding CVs for concentrations derived using the multi-point calibration (external calibration curve) were 13.8%, 11.6%, 10.1% and 8.5% (Table 2).
When comparing the mean absolute concentrations determined via single- or multi-point calibration, we observed different values for Cer16 and Cer18, but not Cer24:0 and Cer24:1 (Fig. 2, Table 2). In the case of Cer16 we note a 1.4-fold higher concentration (for single-point) when compared with the multi-point calibration results. The opposite is true for Cer18. We do not have a conclusive explanation for this discrepancy other than a possible disagreement in amount between labelled and unlabelled standards in the case of Cer16 and Cer18, also observed in the calibration curves prepared in bovine serum albumin solution (Supplementary Fig. 1). Given the excellent agreement between single-point and multi-point quantifications in the case of Cer24:0 and Cer24:1, it can be argued that the absolute levels of endogenous Cer24:0 and Cer24:1 in NIST SRM 1950 are close to 2.48 and 0.85 µmol/L (average of single-point and multi-point calibration), respectively, unless both unlabelled and labelled ceramide standards employed for calibration were affected by the same concentration bias.
Inter-laboratory CVs of SOP-based results were marginally lower than those obtained by OTHER methods, collectively confirming that appropriately titrated authentic labelled standards significantly reduce variability, irrespective of the degree of standardization of the procedures.
Comparison of ceramides levels in different reference materials
The concentration values of the four ceramides were obtained for each of the RMs (i.e., SRM 1950 and the three materials within RM 8231) with the two different procedures described above. Also here, we removed outliers for each RM and ceramide species, using the Tukey’s 1.5×IQR approach as previously described. As expected, the values generated by single-point and multi-point calibration for Cer24:0 and Cer24:1 fall along the y = x line of equality (5% lower and 3% higher, respectively, when using multi-point compared to single-point calibration), while Cer16 and Cer18 show a clear bias away from the y = x line, with 30.1% lower and 51.5% higher values, respectively (Fig. 2 and Table 2). The mean concentrations of all ceramide species were elevated in the RM 8231-2 (hypertriglyceridemic, hTAG) when compared with SRM 1950, most strikingly for Cer24:0 and Cer24:1 (1.56 fold and 1.47 fold, respectively) confirming previous results of 1.5-2 fold changes from the same comparison using different MS-platforms23,33. These results are consistent with many independent human plasma lipidomic studies that reported a positive correlation between TAG and ceramides.
Genetic and environmental conditions such as diet and exercise influence circulating sphingolipids, including ceramides. Ceramides display high genetic variability compared to other circulating lipids38,39 and vary across different ethnicities, age and sex40. The levels of all four ceramide species are significantly lower in RM 8231-3 (Young African American, YAA, Fig. 3, Supplementary Table 1), again in agreement with previous characterizations of these RM23,33, thus calling for determination of stratified reference intervals for applications in clinical testing. The concentrations of all four ceramides in the diabetic RM (RM 8231-1) were not significantly different from SRM 1950, consistent with previous studies23,33. We did not observe any consistent difference in inter-lab variation of concentration values between the four RM (Fig. 3, Table 2 and Supplementary Table 1). This suggests that the measurements had comparable concordance among the laboratories for different matrices, despite their potentially different properties, e.g., the hTAG plasma sample being of different viscosity and YAA having lower ceramide concentrations, compared to SRM 1950.
Comparison with previous inter-laboratory and/or cross MS-platform studies measuring ceramides in NIST SRM 1950
We next compared our results to previous inter-laboratory studies using MS platforms which reported concentration values of ceramides in NIST SRM 195024,27,29,33. A summary of the comparison is presented in Fig. 4. Note that in this figure we included all datasets from our study, i.e., no outliers were removed, as in other published datasets. Details of the previous trials and this current study are tabulated in Supplementary Table 3, i.e., year of publication, usage of internal standards, type of extraction and MS-platforms, and the number of participating laboratories. As we only measured four endogenous molecules, the most distinguishing feature of our study is the use of authentic labelled standards for each of the quantified analytes. Furthermore, the choice of extraction protocol and MS platforms in our study were at the discretion of the participants compared with previous trials, with the notable exception of reference24. The current study is also the largest based on the number of participating laboratories and returned datasets. We would also like to highlight and acknowledge that several laboratories participated in more than one of these trials, underlying the broad common efforts put forward by our communities to advance the field through improvement and harmonization of procedures.
Fig. 4. Comparison of concentration values obtained in this and previous studies covering ceramides by MS-platforms in NIST SRM 1950.

Comparison of inter-laboratory and/or cross-platform trials reporting ceramide concentrations in NIST SRM 1950. Each open circle corresponds to the reported value from one laboratory in each of the respective studies referenced by first author and year of publication. (sp) refers to a single, standardized protocol and platform employed by all laboratories while (mp) denotes studies in which multiple protocols and platforms were used. The red boxes and numbers indicate median ±MAD (mean absolute deviation) and the number of participating laboratories, respectively. Depending on the precise nature of the study and/or reporting laboratory, concentrations were either reported with sphingoid-base and fatty acyl chain details (e.g. Cer 18:1;O2/16:0) or as sum compositions (i.e. Cer 34:1;O2). In the latter case, isobaric ceramide species other than the 4 ceramides targeted in this study may therefore have contributed to the reported concentrations. Details of the individual studies are presented in Supplementary Table 3. No outlier removal has been applied for any of the studies. Source data are provided as Source Data File.
We compared the ceramide concentration values and their variability across the five studies wherever possible. For example, the distinct cores within the original LIPID MAPS consortium reported results in a lipid class-specific fashion, thus limiting the laboratory reporting sphingolipids/ceramides to n = 1, without an option to calculate inter-laboratory CVs27. Another complicating factor in the comparison of diverse studies is the precise identity of the reported analytes. For example, three out of the five studies reported to the levels of lipid species identities (i.e., Cer 18:1;O2/16:0) while others were based on sum composition (e.g., Cer 34:1;O2) due to technological limitations. Finally, the mean absolute deviations (MAD) in our study are among the smallest despite the large number of participants and diverse methods and MS platforms utilized to generate the results.
Use of authentic vs. class-specific internal standards for calibration
The inclusion of authentic labelled standards, prepared to approximate the levels of endogenous ceramides in human plasma (Supplementary Fig. 1), was an early design choice of our trial. It is grounded in the belief that appropriately matching each analyte with its closest stable-isotope derivative would overcome complications, such as different extraction efficiencies, ionization and fragmentation properties, chain-length dependencies, lack of co-elution in reversed-phase LC. To evaluate the relevance of this choice, we normalized the peak areas returned for each endogenous ceramide species (from each participant laboratory) with either the authentic standard (i.e., the corresponding stable isotope derivative) or one of the other three available labelled standards with differing fatty acid moieties (Fig. 5). For this analysis we excluded the outlier datasets (based on authentic internal standard) as described before (Fig. 1). In the case of the endogenous concentrations of Cer24:0 we obtained 2.41 ± 0.24 μmol/L (mean ± SD) when using the authentic internal standard for normalization, i.e., D7-Cer24:0, and 3.07 ± 1.86 μmol/L when normalizing with the D7-Cer16 internal standard, corresponding to a 1.27-fold difference in absolute concentrations. In addition, and importantly, the corresponding variabilities were also significantly affected by the choice of the IS and resulted in inter-lab CVs of 10% and 61%, respectively, when using the authentic labelled analyte or not. Similar disparities were found in other permutations of this procedure when testing the use of different labelled standards (Supplementary Fig. 6).
Fig. 5. Authentic vs non authentic internal standard normalization and its effects on quantification for SRM 1950 RM.
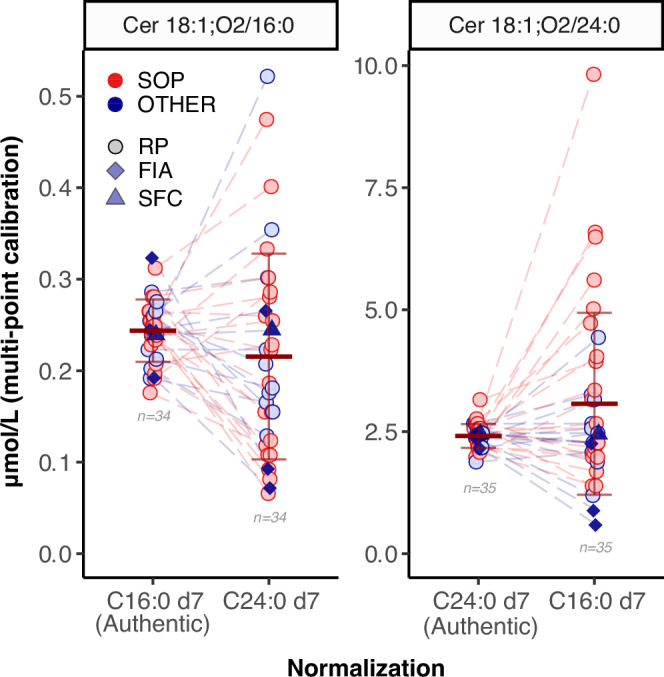
Authentic vs non authentic internal standard normalization and its effects on bias and variability. The horizontal bars correspond to the mean and ±1 × SD of all shown data per group (number of data points indicated for each group). Data originating from the same laboratory are connected by dashed lines. SOP (red) and OTHER (blue) refer to the Standard vs other methods, respectively. Outliers in the authentic internal standard groups have been removed in all groups for each ceramide species (see Fig. 1 and text). Additionally, dataset 11 was excluded for Cer16:0 as it had 15x higher concentrations than the second highest data point for C24:0 d7 normalization.
Recalibration of RMs using SRM 1950 as shared reference
The use of shared reference samples can correct systematic method-specific quantitative biases and help in harmonising lipidomic data30,31. Harmonization with shared materials is quite accepted in clinical settings, where accurate results over time and location are achieved by standardising measurements and establishing traceability to a reference system41–45. Since NIST SRM 1950 is commercially available and one of the most popular references for human plasma, we harmonised the data between all participants by using the following procedure: the four ceramides concentration values obtained from the different RMs (hTAG, DB and YAA) by each participating laboratory were normalised by the mean concentration of the corresponding ceramide obtained by the same lab for SRM 1950, according to the following formula (i).
| i |
where the sample’s measured concentration is divided by the mean concentration of SRM 1950 replicates measured by the corresponding lab. This unitless ratio is rescaled by multiplying by the concentration assigned to the SRM1950, which corresponds to the consensus value established in this study.
After this recalibration, the inter-laboratory variability (inter-lab CV) decreased for all the reference materials, with the number of outliers significantly reduced (Fig. 6 and Supplementary Figs. 4–6). For the DB RM, inter-lab CVs were reduced 5-fold for all four ceramide species. For hTAG and YAA RM the CV value decreased 2-fold. However, the final mean values did not show any significant difference after harmonization. Although it delivers final concentration values relative to the shared reference (SRM 1950), this approach improves comparability between measurements generated with different methods or instruments. The differences between the normalised inter-lab CV values for the various SRMs possibly reflect the influence of matrix effects. Similarly, matrix effects of individual plasma samples in clinical studies will affect the reported concentrations, something that may need to be investigated further. While we consider the use of authentic labelled standards essential to yield quantitative results with low inter-laboratory and cross-platform CV (above), we show here that adding this normalisation approach can reduce systematic errors significantly. Even rather severe outlier values (e.g., #34) are realigned with the rest of the participants when using this approach. This last example illustrates well that for better transparency and for future applications, we suggest reporting not only the harmonized concentration values but the ones generated before and after recalibration.
Fig. 6. Re-calibration of RM results using SRM 1950 as a shared reference materials.

Concentration of the 4 reported ceramides in NIST RM 8231-1 (DB) plasma before and after re-calibration using the NIST SRM 1950 concentration. Each circle corresponds to one measurement from one NIST DB sample shipped to each of the participating laboratories before (None) and after (SRM 1950) re-calibration with the SRM1950 concentration of the corresponding laboratory (see text). The horizontal bars correspond to the mean ±1 × SD of all shown data per group (number of data points indicated for each group). This plot contains all datasets before and after re-calibration, no outlier removal has been performed.
Discussion
Our results demonstrate that, independent of the pre-analytical steps, separation method and type of mass spectrometer used for measurements, our community was able to provide reproducible data from a targeted lipidomic approach, when provided normalization procedures using corresponding labelled standards46,47. This inter-laboratory and cross-platform trial yielded excellent reproducibility, albeit only for a small number of analytes. This good reproducibility was achieved even if we did not recommend any system suitability test procedures to optimize the performances of the analytical systems prior to analysis. We highlight the importance of using labelled standards authentic to the endogenous analyte of interest to improve the accuracy of the reported concentrations in a sample and reduce the variability associated with such measurements (Fig. 5, Supplementary Fig. 3). The reasons for the findings illustrated in Fig. 5, where increased variability between participants is associated with the use of class-specific standards instead of authentic ones, were inconclusive. However, one could consider matrix effects (e.g., at different retention times and different in-source ionization efficiency), as well as fragmentation conditions/efficiencies for the different analytes, to exert a relevant effect. Some of these factors could possibly be excluded by using HILIC/SFC separations or FIA, allowing co-elution of the analyte with the surrogate standard, improving on the homogeneity of matrix effects with co-elution, and by the optimization of the ionization, transport, and fragmentation of each single lipid species, although the number of such datasets in our study was too small to draw a definitive conclusion. In this study, most analytes were fragmented with the same or very comparable collision energies despite displaying different chain lengths. In the absence of authentic labelled standards, it has been shown for different lipid species that accurate quantitation requires the use of individual response factors, determined for each molecular species48. Structural features (chain length and number of double bonds) and coelution with other analytes can influence ionization and in-source fragmentation, when present, which also depends on the individual setup of the mass spectrometer used.
A second notable aspect included in this work is the comparison of single-point calibration vs. standard curves to generate final concentration values. While the use of calibration curves is an accepted requirement in clinical chemistry, many research laboratories rely on single-point calibration after testing for linearity under the experimental conditions used. Specifically, we compared the reference values generated with the two methodologies and their associated CV. While the variability (CV) between participants was comparable, when using the two methods we did not obtain the same concentration values for Cer16 and Cer18 (Fig. 2). This might be because in our calibration standard mix for Cer16 and Cer18 equimolar solutions of labelled and non-labelled standards did not produce identical areas. This might point to a deuterium effect during ionization and fragmentation or small differences in the concentrations of standards. This was noticed during the calibration curve measurement and affected the calculated final values. We can also highlight that according to the use of fit-for-purpose calibration curves, we would now design differently the concentration intervals for the standards used to build the calibration curves used here. After measuring the highest and lowest values in the reference materials and in order to increase the accuracy of the results, a dilution series spanning a smaller concentration range, and with a median close to the value we report here for each ceramide, would be a preferable choice.
Analysis of outliers
We found concordant final concentrations (inter-laboratory CV < 30%) between participants. However, to find potential causes of variability and improve future studies, we dedicated some attention to understanding the most divergent values. We decided to classify “outliers” based on Tukey’s 1.5× IQR fences, corresponding to approximately 3× SD37,49. This outlier removal method was used to filter the dataset before calculating the average consensus concentrations of the four ceramides in NIST SRM 1950; see also Fig. 1. As we systematically reported results based on both single-point and multi-point calibrations, we noticed that some of the outliers resulting from the use of the single-point calibration were ‘corrected’ and re-aligned to the average value when using multi-point calibration instead (e.g., #04 and #20; Fig. 1, Supplementary Fig. 2). However, as the opposite was observed in specific cases (#17), we conclude that while in principle the use of a calibration curve can help generate more accurate values by developing a response function in the expected range of concentrations of the analytes in unknown samples, sometimes it might also introduce variations due to more tedious protocols. In addition, some measurements were identified as outliers with both calibration methods. When considering the outliers, we did not notice any specific bias towards datasets generated with the recommended SOP or OTHER methods and in terms of the instrument used. We next carefully inspected several of these outliers (Fig. 1). We contacted participants to manually check raw data to exclude possible errors due to peak integration and/or calculations. This in-depth analysis provided satisfactory explanations for some of the original discrepancies, while we can highlight several useful observations for others. As an example, several participants returned higher values for Cer18 and Cer24:1 when compared to the average of all measurements. These values were generated using a preferred method that included acquisition on a high-resolution instrument, in MS1 only, and identification based on accurate mass of the precursor ion, retention time and co-elution with the labelled standard. It is possible that when measuring the intact mass of ceramides in high-resolution MS (but without sufficient chromatographic separation, e.g., via HILIC or reversed phase with short gradients) without using any correction during quantitation, the resulting peak area might include contributions from isomeric or isobaric compounds present in solution or generated during ionization. With chromatographic conditions unable to separate possible isomers of the target analytes (such as Cer 16:1;O2/20:0 or Cer 18:2;O2/24:0), a partial contribution to the signal of Cer18 and Cer24:1 would have been included with the reported value. When we independently estimated the levels of Cer 16:1;O2/20:0 or Cer 18:2;O2/24:0 in SRM 1950 plasma by LC-MRM, we confirmed the existence of a substantial contribution (10–15% and 30%, respectively) to the MS1 signal of Cer18 and Cer 24:1. After this was taken into account, the values could be aligned with the rest of the participants.
Another source of error was due to copy/paste mistakes (e.g., swapping values between two different ceramide species when generating calibration curves or preparing the final data report). These are quite common errors that lead to the recommendation of using automated procedures, even for data management. At the same time, an outlier characterized by significantly lower values for all ceramides (#34) was likely the result of the erroneous addition of internal standard volumes. In this case, all analytes showed 3-fold lower values compared to the average of participants.
Although each individual outlier value was examined and possible reasons were discussed with the corresponding participants, we could not clarify all the cases reported. Possible factors could include LC-related suppression by coeluting analytes and ionization factors that are MS platform-dependent and that we described earlier in this section, even considering that isotopically labelled analogues are the best solution to counteract matrix effects50.
Two laboratories were not able to receive plasma samples of human origin, but only organic lipid extracts thereof. As a result, we had the opportunity to evaluate extract stability for this kind of analysis. Human plasma lipid extracts were prepared in Singapore, following the procedure described in the SOP, and were subsequently shipped to the respective participants. Upon package arrival, one of the recipients noticed that some of the vials containing the lipid extracts were not properly sealed any longer, with significant portions being spilled resulting in smaller volumes of samples available. Nevertheless, it was decided to proceed with the analysis as planned. Interestingly, the results from both participants receiving extracts instead of plasma fully aligned with the mean of the other values. This underscores again the importance of adding isotopically labelled authentic standards in such complex mixtures at the beginning of sample processing to compensate for possible variations during the entire procedure.
Value of this study for the development of reference materials and calibration standards
NIST SRM 1950 (“Metabolites in Frozen Human Plasma”) was prepared from 100 individuals representing the demographic diversity in the United States. A primary utilization purpose was to serve as a reference material of ‘normal’ human blood plasma for validation studies. The original three layers of content characterization provided by NIST are (i) certified values, (ii) reference values and (iii) information values, in decreasing order of metrological uncertainty (https://www-s.nist.gov/srmors/certificates/1950.pdf). For example, mass concentrations of cholesterol and total glycerides (as triolein) are certified values (category of highest accuracy confidence) determined by isotope dilution gas chromatography mass spectrometry after hydrolysis and derivatization. Levels of other lipid-related metabolites (e.g., fatty acids, vitamin D) are combinations of certified and reference values. We show here that generating these values can have a big impact on the normalization of results obtained with different methods and instruments by different laboratories and that harmonization using a shared RM, in addition to the calibration with authentic standards, decreases significantly inter-lab variability. We are aware that this might not be yet a perfect solution to solve the reproducibility issues in the field and that more investigations are needed to clarify other aspects of this process, such as the commutability of the SRM 1950 for specific lipid measurements intended “as the equivalence of the mathematical relationships between the results of different measurement procedures for a reference material and for representative samples from healthy and diseased individuals”51. However, we think that this approach is valuable in decreasing systematic biases between platforms and our data support this. Our community is also aware that there are limitations when using SRM 1950 for harmonization, for example its cost and possible limited availability in the future. As alternatives, either a cheaper laboratory-specific Long Term Reference sample or a complex mixture of labelled lipids could be considered, as explained elsewhere30.To serve the expanding research communities in metabolomics with reference materials representing different analytical purposes, populations, and diseases, NIST has initiated the development of new lines of RM (e.g., system suitability, human milk, proteomics among others). RM 8231 is a package of three pooled human plasma materials including RM 8231-1 (DB), RM 8231-2 (hTAG), and RM 8231-3 (YAA)23. Unlike SRM 1950, the above RM 8231s are prepared from a smaller number of individuals (n = 11, n = 11, and n = 16, respectively) and are not documented with certified or reference values to the level of SRM 1950. As mentioned earlier, a notable development over the past decade is the expansion of community efforts aimed at standardization. Collaboration between metrological institutions and interest groups in Quality Assurance and Quality Control is a promising future opportunity for the co-development of RMs, their characterization, validation and eventually certification32, (https://lipidomicssociety.org/interest_groups/reference-materials-and-biological-reference-ranges/). Here we added value to these efforts by providing the most precise publicly available absolute concentration values reported to date for four clinically used ceramides, not only in the commonly used SRM 1950 but also the other RMs as well.
The standards used in this study originated from the same synthetic batch. A single vial containing these four ceramides is commercially available as Deuterated Ceramide LIPIDOMIX® Mass Spec Standard (Avanti Polar Lipids, Cat. #330713). However, the relative concentrations of #330713 differ from those used in this study. It can be expected that our results will encourage the development of precise standard mixtures similar to the one used within this study, as well as future generation of standards, including other stable isotope derivatives of endogenous species.
Relevance for the utility of ceramides in clinical testing
It should be noted that this study constituted a technical phase to address inter-laboratory and cross-platform variability rather than biological variations. However, it forms the basis for the latter to be addressed in future initiatives (e.g., RI in populations or RCV in individuals). Recently, Ramos and colleagues at the Mayo Clinic measured the concentration of these four ceramides in 24 healthy individuals and showed that the variability of ceramide levels at different time points is minimal within individuals52. Moreover, when comparing the absolute concentrations of ceramides in these 24 individuals with the results of our study, we notice a good agreement. Establishing reference values in RMs representative of specific disease conditions was also a propaedeutic exercise for the next phase of our study. These could be from cohorts collected for different studies worldwide, including bio-banked samples. As it is clear from Fig. 3, each specific condition and plasma matrix will require dedicated attention to generate characteristic concentration values that will advance the understanding of the role that ceramides may play in a range of human health and disease conditions.
In summary
This study represents the largest, most targeted public inter-laboratory and cross-platform ring trial for distinct ceramides in human plasma.
Standardization of the analytical process helps decreasing variability, but it is not a fundamental requirement to obtain concordant results across multiple laboratories, instruments, and operators.
We report mean absolute concentration values rather than medians or means of medians. The research and medical community can utilize these reference values as quality control and a quantitative benchmark for ceramides in the reference materials.
Usage of authentic stable isotope labelled standards for each metabolite of interest reduces variability in inter-laboratory comparisons.
Recalibration with a shared reference material decreases inter-laboratory variability even further.
Comparison of pooled plasma samples provides an estimation of biological variation that can be expected between healthy individuals and those with hyperlipidemia as well as an approximation of inter-ethnic differences.
Despite the highly systematic approach we cannot rule out potential biases of our results from the ‘true’ endogenous ceramide concentrations in human plasma. These remain unresolved and depend on details of preparation procedures of the RM and accuracy of the quality and quantity of the utilized calibration standards.
Methods
Study design
Coordinators
The role of the coordination team within the reference materials and biological reference range interest group of the International Lipidomic Society (https://lipidomicssociety.org/interest_groups/reference-materials-and-biological-reference-ranges/ which included Federico Torta, Nils Hoffmann, Robert Ahrends, John Bowden, Kim Ekroos, Markus Wenk) was to design, plan and execute the study in consultation with its partners (Avanti Polar Lipids, National Institute of Standards and Technology- NIST) and participants (other co-authors of the article listed in alphabetical order). Notable activities in the coordination group were establishing a diverse network of participants, negotiating agreements with institutions and companies that supported the project, preparing recommended protocols and forms for return of results in a standardized format, overseeing the distribution of samples, answering questions from participants and keeping them informed about the progress of the study, and, finally, consolidating data and disseminating results, including this report.
Partners
This project was conducted with the essential support of NIST, which made available the plasma reference materials (RMs), and Avanti Polar Lipids, which prepared customized solutions of labelled and non-labelled standards for all study participants. Avanti furthermore helped with the logistics of the sample (both standards and NIST RMs) distribution.
Samples
A set of samples including four different human plasma RMs and, separately, pre-mixed solutions of ceramide standards (deuterated and non-deuterated) was shipped to all participants. Exceptions were countries with restrictions on import of human plasma. In these cases, the corresponding RM samples were extracted at the Singapore Lipidomics Incubator, National University of Singapore, according to the Standard Operating Protocol (SOP) and distributed to the participants as final lipid extracts in polypropylene tubes.
Each laboratory was responsible for sample preparation, instrument setup, data collection and raw data analysis. No requirements were set in terms of instrument performances before starting the experiments. Raw data (chromatographic peak areas or signal intensities in case of flow injection analysis) were submitted to the coordinators that assigned a random numerical identifier to each lab and redistributed the anonymized data to the data centre (Forschungszentrum Jülich and University of Vienna) for quality check and analysis. Each laboratory was free to choose between the SOP prepared by the organizers and based on published data35 (19 participants) or performing the analysis according to a method of choice (OTHER, 12 participants) or using both the SOP and OTHER methods (3 participants). OTHER methods differed from the SOP in many ways, as they were based on the favourite protocol of each lab that performed the OTHER (preferred) approach. Variations included the use of different solvents for ceramide extraction, different chromatographic conditions (column type and gradient, direct infusion) and different detection methods by mass spectrometry (MRM vs full scan at high resolution, for example). The laboratories were informed of the identity of the sample types, and all agreed to remain blinded to laboratory IDs (other than their own) throughout data acquisition and publication of the results. Identifying information other than the laboratory ID was also removed from the report forms before data processing, integration and visualization were performed.
Standard protocol and reporting forms
An SOP (included as Supplementary Information) describing the procedures for extraction, with six independent extractions for each material, each extract analysed in triplicates, and absolute quantification of Cer 18:1;O2/16:0, Cer 18:1;O2/18:0, Cer 18:1;O2/24:0 and Cer 18:1;O2/24:1 in human plasma using LC-MS/MS, in multiple reaction monitoring (MRM) mode, was sent to all participants, together with the samples to be analysed. Please see the SOP for more details on samples, calibration curve, quality control (QC) and blanks preparation. In addition, two Excel files for reporting of the results (one for the SOP and one for OTHER method) were prepared by the coordinators and sent to all participants (Supplementary Information).
Human plasma samples
SRM 1950 Metabolites in Frozen Human Plasma and candidate RM 8231 Frozen Human Plasma Suite for Metabolomics (hypertriglyceridemic, diabetic, and African American plasma samples) are reference materials produced by NIST. All NIST plasma samples were collected after informed consent under approved IRB protocols reviewed by the NIST Human Subjects Protection Office. Details on the preparation of SRM 1950 have been previously reported23,53,54. Briefly, Bioreclamation, Inc. (Hicksville, NY) collected whole blood using lithium heparin as the anticoagulant from 100 donors (1:1 male to female ratio; 77% White, 12% African American or Black, 4% Asian, 2% American Indian or Alaskan Native, 5% “other”) between 40 and 50 years of age after an overnight fast and a 72-h abstention from medication. Plasma was obtained by centrifuging the collected blood at 8000 × g for 25 min at 4 °C. Plasma from each donor was thawed once and subsequently blended and aliquoted into individual vials to produce the SRM 1950. The diabetic (DB) plasma material (RM 8231-1) was created by Solomon Park Research Laboratories, Inc. from a pool of five female and six male donors (55% White, 36% African American or Black, 9% Asian) between 34 and 68 years of age after an overnight fast, each meeting the specified ranges for glucose (>126 mg/dL) and triacylglycerols (<150 mg/dL). The hypertriglyceridemic (hTAG) plasma material (RM 8231-2) was also created by Solomon Park Research Laboratories, Inc. (Kirkland, WA) from a pool of 11 male donors (55% White, 36% African American or Black, 9% Hispanic) between 31 and 72 years of age after an overnight fast, each meeting the NIST-specified ranges for glucose (<100 mg/dL) and triacylglycerols (>300 mg/dL). Plasma for both the hypertriglyceridemic and diabetic materials were obtained by centrifuging the respective collected blood at 1718 × g for 10 min, followed by additional centrifugation at 4300 × g. Lithium heparin was used as the anticoagulant, and plasma from each donor was thawed once and blended to make the respective donor pools before aliquoting into individual vials. For the Young African American (YAA) plasma (RM 8231-3), Bioreclamation Inc. (Westbury, NY) collected whole blood using dipotassium ethylenediaminetetraacetic acid (K2-EDTA) as the anticoagulant from 16 donors (1:1 male to female ratio) between 20 and 25 years of age after an overnight fast. The 16 units of blood were subsequently thawed and pooled by Solomon Park Research Laboratories in the same manner as the hypertriglyceridemic and diabetic plasma materials. NIST plasma aliquots were thawed, pooled and re-aliquoted for this project. Thawing time was minimized and all pooling and aliquoting was performed under inert nitrogen flow using a robotic enclosed system and the samples were returned to the freezer quickly (the entire activity was approximately 1 h). Aliquots were stored at −80 °C until shipped on dry ice to each laboratory. A lipid extract was shipped to those participants that were not able to receive human plasma materials. Details are described in the next sections. Since the scope of this trial was not to compare biofluids such as plasma vs serum55 or establish the influence of the type of anticoagulant56 on the measured ceramide levels, we only report the measurement of the materials described in this section.
Lipid standards and generation of lipid mixtures
Stable isotope labelled internal standards (referred in the text also as ISTD or simply internal standards) were synthesized and provided by Avanti Polar Lipids as a mixture solution in ethyl acetate: 2-propanol (EtOAc:IPA) 2:8 (v/v) at 99% purity. The concentration value of each standard (0.125 μmol/L Cer 18:1;O2[D7]/16:0, 0.050 μmol/L, Cer 18:1;O2[D7]/18:0, 1.500 μmol/L Cer 18:1;O2[D7]/24:0, 0.500 μmol/L Ce r18:1;O2[D7]/24:1, Lot #792537-01-010) was chosen based on previously measured levels of the corresponding endogenous analytes35. Non-labelled standards (2 μmol/L Cer 18:1;O2/16:0, 2 μmol/L Cer 18:1;O2/18:0, 20 μmol/L Cer 18:1;O2/24:0, 20 μmol/L Cer 18:1;O2/24:1, Lot #792536-01-010) were also synthesized and provided by Avanti Polar Lipids as a mixture in EtOAc:IPA 2:8 (v/v) at 99% purity and were used to build a calibration curve through serial dilutions of the starting mixture as described in the SOP included as Supplementary Information. Avanti used quantitative proton nuclear magnetic resonance (QHNMR) to determine potency value and concentration of the products. During this process, the solvent from a known volume of each component is removed under nitrogen gas or by centrifugal evaporation. Approximately 10 mg of NIST traceable QHNMR internal standard are added to each along with 1 mL of deuterated solvent. Samples are analysed by a Bruker 400 MHz NMR spectrometer, with cryoprobe, using a validated quantitative proton method. For data interpretation, the integral response of the internal standard is used to calibrate the response of each component so that an accurate concentration is determined. The variance of this method is 2%. Standard mixtures were formulated by first creating stock solutions of each individual component and quantified through QHNMR (variability of the concentrations were determined to be −/+2% during method validation), then diluted to the final concentrations using glass pipetting with accuracy −/+0.1% of the intended volume. Individual components of unlabelled and deuterated ceramides were then identified via nominal mass measurement using a QQQ MS. In addition, isotopic purity was determined using a ratio (including isotopic correction) of the fully labelled species to the incompletely labelled species via Q-TOF MS. As part of a Quality Assurance process, we would recommend that the users should always analyse the pure commercial standards in full scan mode to at least estimate the purity of the labelled compounds before using them for analysis. Our strategy to improve the accuracy of quantitation involved the use of a pre-mixed solution containing a fixed amount of each of the four corresponding deuterated (D7) ceramides. This solution was prepared as a single batch by Avanti Polar Lipids after close consultation with the trial coordinators and distributed as aliquots of the same batch to all participants. This strategy was followed to increase the reproducibility of the absolute concentration values measured by the participants, that were not dependent on potential variations between different lots of internal standard solutions. The stock concentration of each component was determined by QHNMR so that the mixture was formulated at the desired concentrations. The subsequently formulated mixture was then analysed via high resolution, accurate mass measurement by Q-TOF MS. As the four ceramides species are present at different concentrations in human plasma, the group of coordinators established expected average concentration values for each of them before the preparation of the pooled batch. These values, ranging from 0.050 to 1.5 μmol/L, were derived from a previous publication35. From this and the fact that the range for the ceramide concentrations is small and linear, it can be expected that single-point calibration was acceptable for absolute quantitation. However, as it is widely accepted in analytical chemistry, and recommended by the international guidelines for analyte quantitation, to prepare a series of standards at different concentrations and build a multi-point calibration curve, a second type of pre-mixed solution consisting of the non-deuterated versions of the four ceramides for the preparation of an external calibration curve was prepared by the provider at higher concentration levels (2 and 20 μmol/L, respectively) and distributed to the participants. Due to the presence of endogenous lipids in human plasma and unavailability of lipid-free plasma matrix, calibration line standards were prepared in 5% bovine serum albumin (no specific type was required). As previously reported, curves in plasma and albumin were shown to have identical slopes. Both solutions were kept at −20 °C, equilibrated at room temperature and sonicated 10 min before use.
Data reporting, processing and analysis
All data reports were initially checked manually for completeness and conformance with the requested reporting format. Deviations were recorded and corrected, where apparent (e.g., an inverse order of concentration steps for calibration lines or deviation from the reporting format structure). Remarks by participants concerning potential issues during sample preparation or measurement steps were recorded and associated to the specific dataset that was manually examined for a potential bias. Two sets of calibration line model parameters were calculated for each of the four ceramides per dataset, using a 1/x2 weighted linear model between the expected concentration and the ratio of unlabelled and labelled ceramide. This model was chosen because of the large range of concentrations measured and because the absolute variation is usually larger for higher concentrations; we therefore tried to limit the error at the bottom of the curve by weighting the data inversely with the concentration, as reported previously. LoD and LoQ concentrations were calculated as averages of the standard error of the intercept of the calibration lines of the STD 6 samples (lowest concentration of the calibration line samples), divided by the slope of the respective calibration line and multiplied by 3.3 (LoD) and 10 (LoQ), due to the lack of consistent Matrix Blanks57,58. Both values were then normalized by the slope of the corresponding calibration curve. Final concentrations for each ceramide were calculated using the averaged output of the two corresponding calibration line models. As a common procedure in lipidomics, for each dataset and each sample, single-point calibrated concentrations were calculated from the ratio of the corresponding unlabelled and labelled ceramide, multiplied with the internal standard concentration. To compare different normalizations, adjusted concentrations were calculated using calibration curves built with either the authentic or non-authentic labelled internal standards in combination with the authentic non-labelled standard, i.e. Cer 16:0 and Cer 24:0, respectively.
Outlier removal
Calculation of consensus concentrations, inter-lab CVs and data in Figs. 1, 2, 3, 5 and 6 were based on outlier-filtered datasets. The outlier filtering was applied using Tukey’s 1.5× IQR (inter quantile range) method, which corresponds to approximately a mean ± 3 × SD37,49. For each lipid species, matrix and quantification approach (single and multi-point calibration), datasets from laboratories with average concentrations below Q1 (1st quantile) – 1.5 × IQR, or above Q3 + 1.5 × IQR were excluded.
Calculations and visualizations
All calculations were performed in R (version 4.4.1) using R packages targets (1.2.2), tidyverse (version 2.0.0), ggbeeswarm (0.7.2) and ggh4x (0.2.8)59–61.
All raw and processed datasets and the R pipeline used to process, calculate, and plot the data are available from the GitHub repository (https://github.com/lifs-tools/ils-ceramide-ring-trial). A detailed list of all R packages and their versions is available in the ‘renv.lock‘ file within the same repository. The GitHub repository version used to prepare the data and figures for this accepted manuscript was archived to Zenodo (https://zenodo.org/doi/10.5281/zenodo.10081970).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
1,2 Work in the MRW laboratory is supported by grants from the National University of Singapore via the Life Sciences Institute (LSI) and A*STAR IAF-ICP I1901E0040. 4 N.H. acknowledges support by de.NBI and the de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) and ELIXIR-DE (Forschungszentrum Jülich and W-de.NBI-001, W-de.NBI-004, W-de.NBI-008, W-de.NBI-010, W-de.NBI-013, W-de.NBI-014, W-de.NBI-016, W-de.NBI-022). 5 Bundesministerium für Bildung, Wissenschaft und Forschung” BMBFW within the project DigiOmics4Austria. 6 Work in the SALB laboratory is supported by grants from the Canadian Institutes of Health Research and the Canadian Consortium on Neurodegeneration and Aging CAN-CIHR 163902 and PJT 183604. 7,8,55,56 H.T. was supported by JSPS KAKENHI (21K18216 to Hiroshi T.), the National Cancer Centre Research and Development Fund (2020-A-9, H.T.), and JST ERATO Grant (JPMJER2101 to H.T.). 9 This research is partly supported by AMED-BINDS (JP23ama121055), JST A-STEP (JPMJTR204J), JST-NBDC (JPMJND2305), and the MEXT Cooperative Research Project Program, Medical Research Center Initiative for High Depth Omics, and CURE:JPMXP1323015486 for the Medical Institute of Bioregulation, Kyushu University. 10 Work in MetaToul (Toulouse metabolomics & fluxomics facilities, www.mth-metatoul.com) part of the French National Infrastructure for Metabolomics and Fluxomics is supported by MetaboHUB-ANR-11-INBS-0010 and Inserm (French National Institut for Research in Health). 13 Work at the University of Manchester supported in part by grants from the British Heart Foundation (PG/2019/34923) and BBSRC (BB/W006022/1). 16 Work at University of Pardubice was supported by grant No. 21-20238S (Czech Science Foundation) and ERC Adv grant No. 101095860 (European Research Council). 19 Work in the MTS department is supported by the MetaboHUB infrastructure (ANR-11-INBS-0010 grant). 21 BBSRC for funding to the Babraham Institute and to Michael Wakelam’s memory for his life of contribution to the lipidomics world. 23 Mass spectrometeric analysis was performed at the Turku Metabolomics Centre with the support of Biocenter Finland. 24,25,26 This publication is based upon work from COST Action EpiLipidNET, Pan-European Network in Lipidomics and Epilipidomics (CA19105; https://www.epilipid.net), supported by COST (European Cooperation in Science and Technology).” 30 M.G. is an early-stage researcher supported by the H2020 ITN consortium ArthritisHeal (#812890). M.G. was partially supported by the NWO XOmics project #184.034.019. 31 Nanyang Assistant Professorship, Lee Kong Chian School of Medicine, Nanyang Technological University. 36 Supported by grants from the National Natural Science Foundation of China (21934006 and 22074144). 48 Ser Cymru Project Sepsis grant funded by Welsh Government/EU-ERDF (VJT, VBO). 51,52 Supported by the Tohoku Medical Megabank Project from MEXT, the Japan Agency for Medical Research and Development (AMED; under grant numbers JP20km0105001 and JP21tm0124005) and KAKENHI Grant Number JP23H02625 [D.S.]. 54 The Christian Doppler Laboratory for Innovative Gut Health Concepts of Livestock has been funded by the Austrian Federal Ministry for Digital and Economic Affairs, the Austrian National Foundation for Research, Technology and Development as well as BIOMIN Holding GmbH, which is part of DSM. The authors also thank Marco Reiter for technical support. The participants would like to acknowledge Clay Davis from the National Institute of Standards and Technology. B.B. would like to acknowledge Christian Luechtenborg for his contribution to this project.
Author contributions
All authors contributed to the work presented in this paper for the experimental part, data analysis and in assembling the manuscript.
Peer review
Peer review information
Nature Communications thanks Karolina Sulek and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
All raw and processed datasets and the R pipeline used to process, calculate, and plot the data are available from the GitHub repository (https://github.com/lifs-tools/ils-ceramide-ring-trial). The GitHub repository version used for this accepted manuscript was archived to Zenodo under the 10.5281/zenodo.10081970. All mass spectrometry raw data generated by the participants were deposited to Zenodo under the 10.5281/zenodo.12632989. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Federico Torta, Nils Hoffmann, Bo Burla.
Contributor Information
Kim Ekroos, Email: kim@lipidomicsconsulting.com.
Robert Ahrends, Email: robert.ahrends@univie.ac.at.
Markus R. Wenk, Email: mwenk@hbku.edu.qa
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52087-x.
References
- 1.Züllig, T., Trötzmüller, M. & Köfeler, H. C. Lipidomics from sample preparation to data analysis: a primer. Anal. Bioanal. Chem.412, 2191–2209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Köfeler, H. C. et al. Quality control requirements for the correct annotation of lipidomics data. Nat. Commun.12, 1–4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Köfeler, H. C. et al. Recommendations for good practice in MS-based lipidomics. J. Lipid Res.62, 100138 (2021). [DOI] [PMC free article] [PubMed]
- 4.Liebisch, G. et al. Lipidomics needs more standardization. Nat. Metab.1, 745–747 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Meikle, T. G., Huynh, K., Giles, C. & Meikle, P. J. Clinical lipidomics: realizing the potential of lipid profiling. J. Lipid Res.62, 100127 (2021). [DOI] [PMC free article] [PubMed]
- 6.Defining, Establishing and Verifying Reference Intervals in the Clinical Laboratory: Approved Guideline (Clinical and Laboratory Standards Institute, Wayne, Pa, 2010).
- 7.Harris, E. K. & Yasaka, T. On the calculation of a ‘reference change’ for comparing two consecutive measurements. Clin. Chem.29, 25–30 (1983). [PubMed] [Google Scholar]
- 8.Burla, B. et al. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res.10.1194/jlr.S087163 (2018). [DOI] [PMC free article] [PubMed]
- 9.Bikman, B. T. A role for sphingolipids in the pathophysiology of obesity-induced inflammation. Cell. Mol. Life Sci.69, 2135–2146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boon, J. et al. Ceramides Contained in LDL Are Elevated in Type 2 Diabetes and Promote Inflammation and Skeletal Muscle Insulin Resistance. Diabetes62, 401–410 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haus, J. M. et al. Plasma Ceramides Are Elevated in Obese Subjects With Type 2 Diabetes and Correlate With the Severity of Insulin Resistance. Diabetes58, 337–343 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havulinna, A. S. et al. Circulating Ceramides Predict Cardiovascular Outcomes in the Population-Based FINRISK 2002 Cohort. Arteriosclerosis Thrombosis Vasc. Biol.36, 2424–2430 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Nicholson, R. J., Holland, W. L. & Summers, S. A. Ceramides and Acute Kidney Injury. Seminars Nephrol.42, 151281 (2022). [DOI] [PMC free article] [PubMed]
- 14.Schumacher, F. et al. Ceramide levels in blood plasma correlate with major depressive disorder severity and its neutralization abrogates depressive behavior in mice. J. Biological Chem.298, 102185 (2022). [DOI] [PMC free article] [PubMed]
- 15.Laaksonen, R. et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J.37, 1967–1976 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poss, A. M. et al. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J. Clin. Invest130, 1363–1376 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandropoulou, I. et al. Ceramides in Autoimmune Rheumatic Diseases: Existing Evidence and Therapeutic Considerations for Diet as an Anticeramide Treatment. Nutrients15, 229 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrard, J. et al. How Ceramides Orchestrate Cardiometabolic Health—An Ode to Physically Active Living. Metabolites11, 675 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skácel, J., Slusher, B. S. & Tsukamoto, T. Small Molecule Inhibitors Targeting Biosynthesis of Ceramide, the Central Hub of the Sphingolipid Network. J. Med. Chem.64, 279–297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park, T.-S., Rosebury, W., Kindt, E. K., Kowala, M. C. & Panek, R. L. Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol. Res.58, 45–51 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Blaho, V. A. Druggable sphingolipid pathways: Experimental models and clinical opportunities. In Druggable lipid signaling pathways (ed. Kihara, Y.) 101–135 (Springer International Publishing, Cham, 2020). 10.1007/978-3-030-50621-6_6. [DOI] [PubMed]
- 22.McGurk, K. A. et al. Heritability and family-based GWAS analyses of the N-acyl ethanolamine and ceramide plasma lipidome. Hum. Mol. Genet.30, 500–513 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aristizabal-Henao, J. J., Jones, C. M., Lippa, K. A. & Bowden, J. A. Nontargeted lipidomics of novel human plasma reference materials: hypertriglyceridemic, diabetic, and African-American. Anal. Bioanal. Chem.412, 7373–7380 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Bowden, J. A. et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950–Metabolites in Frozen Human Plasma[S]. J. Lipid Res.58, 2275–2288 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange, M. & Fedorova, M. Evaluation of lipid quantification accuracy using HILIC and RPLC MS on the example of NIST® SRM® 1950 metabolites in human plasma. Anal. Bioanal. Chem.412, 3573–3584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misra, B. B. & Olivier, M. High Resolution GC-Orbitrap-MS Metabolomics Using Both Electron Ionization and Chemical Ionization for Analysis of Human Plasma. J. Proteome Res.19, 2717–2731 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quehenberger, O. et al. Lipidomics reveals a remarkable diversity of lipids in human plasma 1 [S]. J. Lipid Res.51, 3299–3305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siskos, A. P. et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem.89, 656–665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson, J. W. et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem.91, 14407–14416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Triebl, A. et al. Shared reference materials harmonize lipidomics across MS-based detection platforms and laboratories. J. Lipid Res.61, 105–115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chocholoušková, M. et al. Intra-laboratory comparison of four analytical platforms for lipidomic quantitation using hydrophilic interaction liquid chromatography or supercritical fluid chromatography coupled to quadrupole - time-of-flight mass spectrometry. Talanta231, 122367 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Lippa, K. A. et al. Reference materials for MS-based untargeted metabolomics and lipidomics: a review by the metabolomics quality assurance and quality control consortium (mQACC). Metabolomics18, 24 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghorasaini, M. et al. Cross-Laboratory Standardization of Preclinical Lipidomics Using Differential Mobility Spectrometry and Multiple Reaction Monitoring. Anal. Chem.93, 16369–16378 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammad, S. M. et al. Race disparity in blood sphingolipidomics associated with lupus cardiovascular comorbidity. PLOS ONE14, e0224496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kauhanen, D. et al. Development and validation of a high-throughput LC–MS/MS assay for routine measurement of molecular ceramides. Anal. Bioanal. Chem.408, 3475–3483 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Liebisch, G. et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res.61, 1539–1555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tukey, J. W. Exploratory Data Analysis (Addison-Wesley Publishing Company, 1977).
- 38.Cadby, G. et al. Heritability of 596 lipid species and genetic correlation with cardiovascular traits in the Busselton Family Heart Study [S]. J. Lipid Res.61, 537–545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabassum, R. et al. Genetic architecture of human plasma lipidome and its link to cardiovascular disease. Nat. Commun.10, 4329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beyene, H. B. et al. High-coverage plasma lipidomics reveals novel sex-specific lipidomic fingerprints of age and BMI: Evidence from two large population cohort studies. PLoS Biol.18, e3000870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vesper, H. W., Myers, G. L. & Miller, W. G. Current practices and challenges in the standardization and harmonization of clinical laboratory tests1223. Am. J. Clin. Nutr.104, 907S–912S (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myers, G. L. & Miller, W. G. The International Consortium for Harmonization of Clinical Laboratory Results (ICHCLR) - A Pathway for Harmonization. EJIFCC27, 30–36 (2016). [PMC free article] [PubMed] [Google Scholar]
- 43.Diepeveen, L. E. et al. Provisional standardization of hepcidin assays: creating a traceability chain with a primary reference material, candidate reference method and a commutable secondary reference material. Clin. Chem. Lab. Med.57, 864–872 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Pickens, C. A. et al. Harmonizing Newborn Screening Laboratory Proficiency Test Results Using the CDC NSQAP Reference Materials. Int. J. Neonatal Screen.6, 75 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harmonization.net. The International Consortium for Harmonization of Clinical Laboratory Results,https://www.harmonization.net/. [PMC free article] [PubMed]
- 46.Schoeny, H. et al. A combined flow injection/reversed-phase chromatography–high-resolution mass spectrometry workflow for accurate absolute lipid quantification with 13 C internal standards. Analyst146, 2591–2599 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Lehmann, W. D. A timeline of stable isotopes and mass spectrometry in the life sciences. Mass Spectrom. Rev.36, 58–85 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Höring, M., Ejsing, C. S., Hermansson, M. & Liebisch, G. Quantification of Cholesterol and Cholesteryl Ester by Direct Flow Injection High-Resolution Fourier Transform Mass Spectrometry Utilizing Species-Specific Response Factors. Anal. Chem.91, 3459–3466 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Hickman, P. E. et al. Choice of Statistical Tools for Outlier Removal Causes Substantial Changes in Analyte Reference Intervals in Healthy Populations. Clin. Chem.66, 1558–1561 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Berg, T. & Strand, D. H. 13C labelled internal standards—A solution to minimize ion suppression effects in liquid chromatography–tandem mass spectrometry analyses of drugs in biological samples? J. Chromatogr. A1218, 9366–9374 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Vesper, H. W., Miller, W. G. & Myers, G. L. Reference materials and commutability. Clin. Biochem. Rev.28, 139–147 (2007). [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos, P. et al. The Biological Variability of Plasma Ceramides in Healthy Subjects. J. Appl. Lab. Med.7, 863–870 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Phinney, K. W. et al. Development of a Standard Reference Material for Metabolomics Research. Anal. Chem.85, 11732–11738 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simón-Manso, Y. et al. Metabolite Profiling of a NIST Standard Reference Material for Human Plasma (SRM 1950): GC-MS, LC-MS, NMR, and Clinical Laboratory Analyses, Libraries, and Web-Based Resources. Anal. Chem.85, 11725–11731 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Wolrab, D., Chocholoušková, M., Jirásko, R., Peterka, O. & Holčapek, M. Validation of lipidomic analysis of human plasma and serum by supercritical fluid chromatography–mass spectrometry and hydrophilic interaction liquid chromatography–mass spectrometry. Anal. Bioanal. Chem.412, 2375–2388 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Wolrab, D. et al. Determination of one year stability of lipid plasma profile and comparison of blood collection tubes using UHPSFC/MS and HILIC-UHPLC/MS. Analytica Chim. Acta1137, 74–84 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Currie, L. A. Nomenclature in evaluation of analytical methods including detection and quantification capabilities (IUPAC Recommendations 1995). Pure Appl. Chem.67, 1699–1723 (1995). [Google Scholar]
- 58.Magnusson, B. & Örnemark, U. The Fitness for Purpose of Analytical Methods - A Laboratory Guide to Method Validation and Related Topics (Eurachem, 2014).
- 59.Clarke, E., Sherrill-Mix, S. & Dawson, C. Ggbeeswarm: Categorical Scatter (Violin Point) Plots. https://CRAN.R-project.org/package=ggbeeswarm (2023).
- 60.van den Brand, T. Ggh4x: Hacks for ‘Ggplot2’. https://CRAN.R-project.org/package=ggh4x (2023).
- 61.Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw.4, 1686 (2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed datasets and the R pipeline used to process, calculate, and plot the data are available from the GitHub repository (https://github.com/lifs-tools/ils-ceramide-ring-trial). The GitHub repository version used for this accepted manuscript was archived to Zenodo under the 10.5281/zenodo.10081970. All mass spectrometry raw data generated by the participants were deposited to Zenodo under the 10.5281/zenodo.12632989. Source data are provided with this paper.