

Abstract
A group of conserved hydrophobic residues faces the interior of the coiled-coil-like structure within the N-terminal domain of the human immunodeficiency virus type 1 (HIV-1) capsid protein (CA). It has been suggested that these residues are important for maintaining stable structure and functional activity. To investigate this possibility, we constructed two HIV-1 clones, in which Trp23 or Phe40 was changed to Ala. We also constructed a third mutant, D51A, which has a mutation that destroys a salt bridge between Pro1 and Asp51. All three mutants are replication defective but produce virus particles. Mutant virions contain all of the viral proteins, although the amount and stability of CA are decreased and levels of virion-associated integrase are reduced. The mutations do not affect endogenous reverse transcriptase activity; however, the mutants are blocked in their ability to initiate reverse transcription in infected cells and no minus-strand strong-stop DNA is detected. The defect in reverse transcription is associated with striking defects in the morphology of mutant virus cores, as determined by transmission electron microscopy. Our data indicate that the mutations made in this study disrupt CA structure and prevent proper maturation of virus cores. We propose that this results in a defect in core stability or in an early postentry event preceding reverse transcription.
The Gag polyprotein of human immunodeficiency virus type 1 (HIV-1) is essential for virus assembly and release and, in fact, is the only viral protein that is required (reviewed in references 9, 43, 49, and 55). During or shortly after the budding of virus particles, Gag is cleaved by the viral protease to yield the mature structural proteins, i.e., matrix (MA, p17), capsid (CA, p24), nucleocapsid (NC, p7), and p6 (25, 36; reviewed in references 9, 48, and 49). Gag processing induces a structural rearrangement of the individual proteins in a process known as “maturation,” which results in the conversion of immature particles to mature, infectious virions. For example, upon cleavage at the MA-CA junction, refolding at the N terminus of CA leads to the formation of a new β-hairpin/helix structure that is stabilized by formation of a buried salt bridge between the charged amino terminus of Pro1 and the carboxylate of Asp51 (20, 51). These rearrangements are associated with a dramatic morphological change that transforms the immature spherical, electron-lucent core to one that is electron dense and conical (18, 22, 23, 51; for earlier citations, see references 43, 49, and 55). In addition, the nucleic acid chaperone activity of mature NC (reviewed in reference 41) catalyzes refolding of genomic RNA to form a thermostable dimer (6, 14, 15).
The interior of the viral core consists of a ribonucleoprotein complex containing genomic RNA; the tRNA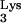 primer; NC; the viral enzymes, protease, reverse transcriptase (RT), and integrase (IN); Nef; and Vpr (1, 31, 53). In the mature virion, the RNA-protein complex is surrounded by a shell formed by the CA protein (reviewed in references 19, 49, and 55). Approximately 1,500 copies of CA are present in each particle (50). The HIV-1 CA protein has 231 residues and consists of two independent folding domains: an N-terminal domain (residues 1 to 145) (16, 20, 38) and a C-terminal domain (residues 151 to 231) (17). Interestingly, in a recent study on Rous sarcoma virus CA mutants, evidence suggesting that there are functional interdomain interactions is presented (2b).
primer; NC; the viral enzymes, protease, reverse transcriptase (RT), and integrase (IN); Nef; and Vpr (1, 31, 53). In the mature virion, the RNA-protein complex is surrounded by a shell formed by the CA protein (reviewed in references 19, 49, and 55). Approximately 1,500 copies of CA are present in each particle (50). The HIV-1 CA protein has 231 residues and consists of two independent folding domains: an N-terminal domain (residues 1 to 145) (16, 20, 38) and a C-terminal domain (residues 151 to 231) (17). Interestingly, in a recent study on Rous sarcoma virus CA mutants, evidence suggesting that there are functional interdomain interactions is presented (2b).
Analysis by nuclear magnetic resonance spectroscopy (20) and X-ray crystallography (38) revealed that the N-terminal region is composed of five long α-helices (I, II, III, IV, and VII [20] or A, B, C, D, and G [38]) and two shorter helices (V and VI [20] or E and F [38]), two β-hairpins, and an exposed proline-rich loop (Fig. 1). On the basis of a multiple-sequence alignment, Momany et al. (38) identified a group of conserved hydrophobic residues that faces the interior of the coiled-coil-like structure formed by the five long α-helices, and it was suggested that these residues are important for maintaining the stability of the structure (38). The proline-rich loop is the binding site of host cellular protein cyclophilin A (CypA) (16), a peptidyl-prolyl cis-trans isomerase that is incorporated into virions and that is required for production of infectious virions (8, 45).
FIG. 1.

Ribbon representation showing a front view of the N-terminal core domain of HIV-1 CA. The β-hairpin structure, CypA binding loop, and seven α-helices are labeled (20, 38). Note that the helices are numbered as described by Gitti et al. (20). The three residues mutated in this study, W23, F40, and D51, are located in helices I, II, and III, respectively. The diagram was generated with Molscript (32).
The N-terminal domain of CA plays a major role in the early steps of the virus life cycle. Interestingly, linker insertions and deletions in this region (5, 39, 40, 52) are compatible with efficient assembly of virus particles, and as many as 56 residues in this domain can be deleted without an adverse effect on particle formation (52). However, mutations in the N-terminal domain often affect proper virion core formation and virus replication (5, 7, 39, 40, 51, 52). Indeed, aberrant core morphology is consistently associated with loss of infectivity (5, 7, 39, 51), although the converse is not necessarily true. (For example, CA mutants with substitutions in residues in the proline-rich CypA binding loop are poorly infectious but exhibit normal core morphology [3, 7, 54].) Some studies have also shown that formation of abnormal cores is associated with a defect in reverse transcription in infected cells (7, 39). However, whether the defect represents a block in the very first step in the process, i.e., synthesis of minus-strand strong-stop DNA [(−) SSDNA], was not tested. Defects in reverse transcription postentry have also been described for Rous sarcoma virus (4a) and Moloney murine leukemia virus (MLV) (2a) CA mutants.
The goal of the present work was to investigate the impact of specific N-terminal CA mutations on the ability of HIV-1 virions to undergo reverse transcription in the infected cell and the possible correlation with virus core assembly. Our approach was to make single alanine substitutions rather than deletions or insertions, since point mutations might be less likely to result in drastic perturbation of CA secondary and tertiary structures. Mutations were made in two aromatic residues, Trp23 (helix I; Fig. 1) and Phe40 (helix II; Fig. 1), which are among the conserved hydrophobic residues identified by Momany et al. (38) (see above). For comparison, we also constructed another mutant, D51A. This mutant can no longer form the Pro1-Asp51 salt bridge and was previously shown to produce noninfectious particles having an abnormal core (51); however, the ability of the D51A mutant to make viral DNA during infection was not assessed.
Here, we demonstrate that all three mutants produce essentially normal levels of virus particles but are unable to replicate in single-cycle and long-term culture assays. These particles also exhibit abnormal core structures, as determined by transmission electron microscopy. Although the endogenous RT activity of the mutants is not impaired, none of the viral DNA products normally made during reverse transcription, including (−) SSDNA, can be detected in cells infected with these mutants. Taken together, our findings indicate that assembly of virions with aberrant core structures leads to a defect in initiation of reverse transcription and loss of infectivity. We conclude that aromatic residues Trp23 and Phe40 and acidic residue Asp51 are required for formation of functional virus cores.
MATERIALS AND METHODS
Materials.
Competent STBL2 cells were obtained from Life Technologies Inc. (Rockville, Md.). [35S]cysteine (500 to 1,000 Ci/mmol) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, Mo.).
Plasmids.
The plasmids used for this work were the HIV-1 infectious proviral clone pNL4-3 (2) and env-negative subclone pNL4-3KFS, which has a frameshift mutation in the env coding region (10, 13). The parent pNL4-3KFS clone is sometimes referred to as “wild type” since the CA domain in Gag has the wild-type sequence. The envelope vector is amphotropic MLV envelope glycoprotein expression plasmid pSV-A-MLVenv (33).
Cells.
293T, HeLa, and HeLa CD4-LTR/β-gal indicator cells (29) (CD4-positive HeLa indicator cells) were maintained in Dulbecco's modified Eagle's medium; H9 and CEM (12D7) CD4-positive T-lymphocyte cell lines were cultured in RPMI 1640 medium. All media contained 10% fetal bovine serum, 2 mM glutamine, and antibiotics.
Mutagenesis and cloning.
The strategy for introducing mutations into the CA domain of Gag in pNL4-3 (2) or pNL4-3KFS (10, 13) by PCR-based site-directed mutagenesis has been described previously (44). Specific details on the procedures used to construct the W23A, F40A, and D51A mutations are available upon request. To avoid undesirable deletion or recombination events, plasmid DNAs were propagated in STBL2 cells at 30°C. The sequences of all CA mutants were confirmed by sequencing performed by the Biopolymer Lab (University of Maryland, College Park, Md).
Transfection and infection procedures.
Transfection of 293T and HeLa cells was performed by the calcium phosphate precipitation method as described previously (12). To generate virus particles capable of only a single round of replication, cells were cotransfected with parent or mutant pNL4-3KFS clones (10, 13) and pSV-A-MLVenv (33).
Replication kinetics were determined over an 8-week period using 5 × 106 CEM (12D7) cells transfected with 5 μg of pNL4-3 wild-type (2) or mutant DNA in the presence of 0.7 mg of DEAE-dextran/ml. The presence of virus particles in the supernatant fluids was determined by assay of exogenous RT activity (11). For single-cycle infections, cells were infected with pseudotyped virus particles, which were normalized for the amount of exogenous RT activity (106 cpm). Infectivity was measured in the MAGI assay, as described previously (29).
Endogenous RT assay.
Virions obtained by transfection of cells with pNL4-3KFS clones (10, 13) were normalized for exogenous RT activity (106 cpm) and were then pelleted at 14,000 × g for 90 min. The pellets were resuspended in 30 μl of a mixture containing the reaction components specified by Guo et al. (24) and were incubated for 16 h at 37°C. The samples were purified, denatured in 0.1 N NaOH at 37°C for 1 h, and subjected to electrophoresis in a 1% denaturing agarose gel (20 mM NaOH, 1 mM EDTA). The gels were dried and exposed to X-ray film.
For PCR analysis of the DNA products made in the endogenous reactions, the assay conditions were the same as those described above, except that all four deoxyribonucleoside triphosphates (dNTPs) (unlabeled) were added at a final concentration of 0.4 mM. Reaction products were diluted 1:1,000, and 1 μl of each dilution was used for PCR, as described below.
PCR analysis of reverse transcription in acutely infected cells.
Supernatant fluids from 293T cells cotransfected with pNL4-3KFS parent or mutant clones (10, 13) and pSV-A-MLVenv (33) were normalized for exogenous RT activity and were used to infect H9 cells (5 × 106 cpm [RT activity] per 106 H9 cells) for 3 h at 37°C. Cells were washed three times in phosphate-buffered saline (PBS), resuspended in 6 ml of growth medium, and incubated for 20 h at 37°C. After being washed with PBS, the cells were treated with 100 U of DNase I for 1 h at 37°C in the presence of 10 mM MgCl2. Proteinase K digestion and DNA extraction were performed using a QIAamp blood and tissue kit (Qiagen), as directed by the manufacturer. The DNA samples were treated with DpnI, which digests methylated plasmid DNA but which does not degrade unmethylated, newly synthesized viral DNA (56).
To eliminate any virus particles remaining on the cell surface, which could lead to contamination of the cell lysate with virion DNA, several changes in the previous procedure were made: (i) prior to infection, virus particles were incubated with 10 U of DNase I and 10 mM MgCl2 at 37°C for 30 min; (ii) CD4-positive HeLa indicator cells (rather than a suspension culture of H9 cells) were used for infection; and (iii) the cells were treated with a trypsin-EDTA solution to detach any bound virus particles (34) and were then washed two times with PBS before preparation of the cell lysate and extraction of DNA.
Five hundred nanograms of each DNA preparation was used as the template for PCR with PCR SuperMix (Life Technologies). All samples were screened for contaminating pNL4-3 DNA by PCR amplification (3). None of the samples was found to contain contaminating plasmid DNA. To normalize for the quantity of total cellular DNA present in each sample, human glyceraldehyde-3-phosphate dehydrogenase (hGAPDH) DNA was amplified with forward primer JL416 (nucleotides [nt] 446 to 465) and reverse primer JL417 (nt 654 to 636) (46).
Primers used to amplify viral DNA synthesized during the different steps of reverse transcription are as follows (note that the nucleotide positions correspond to the numbering in pNL4-3 [2]): (i) (−) SSDNA, forward primer JL405 (nt 454 to 477) and reverse primer JL406 (nt 635 to 612); (ii) DNA made after minus-strand transfer, forward primer JL407 (nt 9028 to 9051) and reverse primer JL408 (nt 9307 to 9288); (iii) 3′ end of minus-strand DNA, forward primer JL409 (nt 652 to 675) and reverse primer JL410 (nt 816 to 796); (iv) plus-strand strong-stop DNA [(+) SSDNA], forward primer JL405 and reverse primer JL411 (nt 653 to 635); (v) DNA made after plus-strand transfer, forward primer JL405 and reverse primer JL412 (nt 701 to 683); (vi) full-length double-stranded linear DNA, forward primer JL434 (nt 251 to 274) and reverse primer JL412. Two-long-terminal-repeat (2-LTR) circular DNA was detected by “nested” PCR with outer primer set JL405 and JL433 (reverse primer JL433, nt 9369 to 9349), followed by amplification with inner primer set JL413 and JL408 (forward primer JL413, nt 597 to 621). PCR products were separated by electrophoresis in nondenaturing 7.5% polyacrylamide gels. The gels were stained with Vistra Green (Molecular Dynamics), and the amount of DNA in each sample was quantified by PhosphorImager analysis. Under our conditions, ≥500 or 1,000 copies of (−) SSDNA and other DNA intermediates, respectively, can be detected.
Analysis of mutant and wild-type viral proteins.
The procedures used for metabolic labeling of transfected HeLa cells with [35S]Cys, preparation of cell and virion lysates, and immunoprecipitation of labeled viral proteins and conditions for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) have been described previously (12, 26).
For Western blot analysis, the Western-Star chemiluminescence detection system (TROPIX Inc., Bedford, Mass.) was used. The following primary antibodies were used: rabbit anti-HIV CA sera (kindly provided by A. Ono, National Institute of Allergy and Infectious Diseases, National Institutes of Health [NIH], Bethesda, Md.), rabbit anti-HIV IN and RT peptide antisera (30), and AIDS patient sera (obtained from the NIH AIDS Research and Reference Reagent Program). The secondary antibodies were alkaline phosphatase-conjugated anti-rabbit immunoglobulin G (IgG) and anti-human IgG (TROPIX Inc.).
Analysis of viral core morphology by transmission electron microscopy.
Forty-eight hours posttransfection, 293T and HeLa cells transfected with pNL4-3KFS (10, 13) clones were washed twice with PBS and then fixed in situ with 1% paraformaldehyde and 3% glutaraldehyde for 2 h. The cells were scraped off the plate, further fixed with 2% OsO4 for 2 h, and then dehydrated with a graded series of ethanol dilutions ranging from 30 to 100% before they were embedded in Epon resin. Thin sections were counterstained with 1% lead citrate and 2% uranyl acetate before being examined by transmission electron microscopy.
RESULTS
Alanine substitution mutations in the N-terminal domain of CA.
A group of conserved hydrophobic residues faces the interior of the coiled-coil-like structure formed by the five long α-helices in the N-terminal domain of CA (38). To evaluate the role of these hydrophobic residues in HIV-1 replication, we made single alanine substitutions in two aromatic residues, Trp23 (helix I, Fig. 1) and Phe40 (helix II, Fig. 1), and introduced these mutations (i.e., W23A and F40A) into the CA domain of Gag in the pNL4-3 infectious clone of HIV-1 (2) and env-negative subclone pNL4-3KFS (10, 13). We also constructed a mutant in which Asp51 was changed to alanine (D51A). In earlier work (20, 51), it was shown that, in the mature CA protein, the carboxylate of Asp51 forms a buried salt bridge with the charged amino terminus of Pro1. The D51A mutation destroys this salt bridge and unfolds the β-hairpin at the N terminus of CA.
The CA mutations do not impair virus particle production.
To investigate the effect of the mutations on virus production and release, HeLa cells or 293T cells were cotransfected with the pNL4-3KFS clones and the pSV-A-MLVenv expression plasmid. Virus particle production was determined by measuring the amount of RT activity present in the supernatant fluids of the transfected cells (11). None of the mutations had a major effect on virus release (Table 1). High-speed sedimentation of the supernatant fluids of transfected cells indicated that the RT activities could be recovered in the pellet fraction and are therefore associated with virus particles (data not shown).
TABLE 1.
Phenotypes of HIV-1 wild-type and mutant virions
| Virus | Virus production (%)a ± 1 SD | Infectivity by:
|
Presence of:
|
|||
|---|---|---|---|---|---|---|
| Replication kineticsb | MAGIc (%) | Endogenous RTd | Viral DNA in cellse | Cone-shaped coref | ||
| WTg | 100 | + | 100 | + | + | + |
| W23A | 71 ± 12 | − | <0.5 | + | − | − |
| F40A | 59 ± 16 | − | <0.5 | + | − | − |
| D51A | 99 ± 22 | − | <0.5 | + | − | − |
Virus production was assayed as RT activity in the supernatant of transfected cells. Values were converted to percentages of wild-type level for each of nine independent transfections and were averaged.
Viral infectivity was assayed by measuring RT activity in the supernatant of infected CEM cells.
Infectivity in the MAGI assay in a single round of infection. The numbers of blue cells were normalized to the RT activity of input virions and are reported as percentages of wild type.
Endogenous RT activity was assayed as described in Materials and Methods.
Virions obtained from transfected 293T cells were used to infect CD4-positive HeLa indicator cells, which were then lysed. Viral DNA was amplified by PCR.
Cone-shaped viral cores were detected by transmission electron microscopy in thin sections of cells producing virus.
WT, wild type.
Composition of wild-type and mutant proteins in lysates of infected cells and in virions.
Once it was established that virus particles are released into the supernatants (Table 1), it was of interest to determine whether the W23A, F40A, and D51A mutations affect the expression and incorporation of viral proteins. To approach this question, we transfected HeLa cells with the pNL4-3 wild-type or mutant clones and then metabolically labeled the proteins with [35S]Cys; cell- and virion-associated viral proteins were immunoprecipitated with AIDS patient serum and separated by gel electrophoresis (12, 26).
The results indicated that the protein patterns for the different mutants were largely similar to the wild-type viral protein profile (Fig. 2). As shown in Fig. 2A, the cell lysates all contained unprocessed Pr160gag-pol and Pr55gag, the Pr41gag cleavage intermediate, uncleaved envelope glycoprotein gp160, envelope protein gp120, and mature CA and MA proteins. Thus, the mutations introduced into CA did not interfere with the expression of viral proteins. Note, however, that with the exception of mature CA the proteins were expressed to a slightly greater extent for the mutant samples.
FIG. 2.

Analysis of mutant and wild-type (WT) cell- and virion-associated proteins. HeLa cells were transfected with the indicated pNL4-3 molecular clones (2). (A and B, top) Detection of [35S]Cys-labeled cell- and virion-associated proteins, respectively. Lysates were immunoprecipitated with AIDS patient serum and separated by SDS-PAGE in 10% gels; radioactive bands were visualized by fluorography. The positions of the precursor and mature viral proteins are indicated. (B, bottom) Detection of unlabeled HIV-1 CA bands by Western blot analysis. Unlabeled viral lysates were subjected to SDS-PAGE in a 10% minigel and were then analyzed by Western blotting (see Materials and Methods). Blots were sequentially probed with primary rabbit antibodies against HIV-1 CA and with secondary anti-rabbit IgG antibodies; the protein bands were visualized by chemiluminescence. Note that, since a minigel was used for the Western blot analysis, the CA and CA-related bands were not separated as well as in the gel of the virion-associated immunoprecipitated proteins (B, top).
Analysis of virus particles (Fig. 2B, top) showed that mutant virions contained levels of gp120 comparable to those in wild-type particles. This demonstrates that the CA mutations did not inhibit incorporation of the envelope protein into virions. In addition, processing of the precursor proteins occurred normally since mature CA, RT, IN, and MA having the expected sizes were also present in mutant particles. However, the mutants, especially W23A and F40A, had reduced levels of IN and CA proteins, compared with the wild type. Direct analysis of the proteins without prior immunoprecipitation gave the same results, indicating that reduced immunoreactivity is not responsible for the decrease in the amounts of CA and IN in mutant virions (data not shown).
All of the mutant samples also exhibited three bands migrating ahead of CA. Independent Western blot analysis with the anti-CA antibody confirmed that, unlike the band corresponding to MA (Fig. 2B, top), the two other smaller bands are related to CA (Fig. 2B, bottom) and are probably fragments produced by degradation of CA. A similar finding was made in the earlier work on the D51A mutant (51). These results indicate that the CA protein present in mutant virions is less stable than that in wild-type particles. The identities of the p32 and p66 proteins as IN and RT, respectively, were confirmed by Western blot analysis with anti-IN and anti-RT peptide sera (30); no IN-related bands smaller than p32 were detected (data not shown).
Endogenous RT activity is not inhibited by the three mutations in CA.
To further characterize mutant virions and examine the functionality of the encapsidated RT protein, we measured endogenous RT activity in virions disrupted with 0.1% Nonidet P-40 (24). The results presented in Fig. 3A indicate that both wild-type and mutant particles synthesized viral DNAs ranging in size from ∼0.4 to ∼2.5 kb, which appear as a smear in a denaturing agarose gel. A smear of DNA products is typically observed in HIV endogenous RT assays (21, 28) and contrasts with analogous MLV reactions, where distinct species of full-length minus-strand and linear double-stranded DNA products can be easily detected (37, 42). The data also indicate that a somewhat higher level of total radioactive products was synthesized in the mutant samples.
FIG. 3.

Endogenous RT activities of pNL4-3KFS wild-type (WT) and mutant virions. (A) 32P-labeled viral DNA products made by wild-type and mutant virus particles. Incubation conditions and methods for the analysis of the DNA products by electrophoresis in a denaturing agarose gel are given in Materials and Methods. 32P-labeled λ HindIII size markers are shown on the left. (B) PCR analysis of unlabeled DNA products made by mutant and wild-type virus particles. The DNA products were amplified using appropriate primer sets, as described in Materials and Methods. The negative control (Mock) is a reaction mixture containing the reaction buffer but not virus. One nanogram of pNL4-3KFS DNA (10, 13) was used as a positive control. The size of each PCR product is shown on the right; the specific product amplified is indicated on the left.
To identify individual DNA intermediates synthesized in the endogenous reactions, we performed PCR analysis of unlabeled wild-type and mutant samples (Fig. 3B). DNA products corresponding to (−) SSDNA, DNA made after minus-strand transfer, (+) SSDNA, and DNA made after plus-strand transfer were detected in wild-type and mutant reactions. In all cases, no full-length linear DNA was observed, suggesting that the endogenous reverse transcription reactions were not very efficient. (Note that in the control reaction with pNL4-3KFS DNA [Fig. 3B, right lane] and in cells infected with wild-type virus [Fig. 4A], full-length linear DNA could be readily detected.)
FIG. 4.

PCR analysis of viral DNA in infected cells. Virus stocks obtained by cotransfection of 293T cells with pNL4-3KFS (10, 13) molecular clones and pSV-A-MLVenv (33) were normalized for RT activity and used to infect H9 cells (A) and CD4-positive HeLa indicator cells (29) (B). Isolation of viral DNA from each cell line including trypsin treatment to remove bound virions from the HeLa cells, PCR analysis, and electrophoretic separation of the products are described in Materials and Methods. To control for the amount of total DNA in each sample, hGAPDH sequences (46) were amplified from the same samples used to analyze viral DNA products. The size of each PCR product is shown on the right; the specific product amplified is indicated on the left. Mock, DNA from uninfected cells; WT, wild type.
The results of the endogenous assays indicate that the CA mutants contain functional RT. In addition, the data show that genomic RNA and the tRNA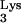 primer are encapsidated in mutant virions and are functional in reverse transcription.
primer are encapsidated in mutant virions and are functional in reverse transcription.
Replication kinetics and single-cycle infectivity of wild-type and mutant virions.
Thus far, we have shown that the CA mutants produce virus particles that exhibit exogenous and endogenous RT activities and contain all of the viral proteins normally found in HIV-1 virions. One indication that mutant virions may not completely resemble wild-type particles comes from the observation that mutant CA proteins are reduced in amount and stability and the level of integrase is decreased (Fig. 2B). It was therefore important to determine whether the mutants are infectious.
To assay the mutants for infectivity in long-term culture experiments, CEM (12D7) cells were transfected with pNL4-3 wild-type and mutant clones. As expected, the wild-type virus replicated to high levels (Table 1), with virus production peaking on day 7 posttransfection and then declining with subsequent host cell death (data not shown). In contrast, there was no evidence of viral replication with any of the mutant viruses for up to 60 days in tissue culture (Table 1; data not shown).
The infectivity of pNL4-3KFS pseudotyped virions was tested in the MAGI assay, which involves a single round of replication (29). In accord with the long-term culture experiment, none of the CA mutants showed any detectable infectivity (Table 1). To confirm these results, the W23A and D51A mutations were inserted into an HIV-1 molecular clone modified to express luciferase following infection (27). Consistent with the MAGI data, the single-cycle luciferase assay showed that luciferase expression could not be detected in the mutant samples (data not shown).
Thus, using three different assays, we could establish that the CA mutations we have made lead to a complete loss of infectivity (i.e., a reduction of at least 3 log units relative to wild type). The data indicate that the loss of infectivity is not affected by the receptor that is utilized for viral entry, since the results were the same regardless of whether virus entry was mediated by the HIV-1 or MLV Env glycoproteins.
The CA mutations impair the synthesis of viral DNA in newly infected cells.
On the basis of the results from single-cycle infectivity assays, it appears as if the CA mutants are blocked at an early stage in virus replication. However, these assays cannot define which early step (e.g., reverse transcription, nuclear import, or integration) is affected. To approach this question, we used PCR to measure the amounts of viral DNA intermediates synthesized in newly infected H9 cells. To avoid the possibility that the Gag mutations might disrupt HIV-1 Env function at the level of virus entry and to increase the sensitivity of the assay, the infections were performed with pNL4-3KFS pseudotyped virions.
The primer sets used for this analysis were specific for the replication intermediates that are synthesized during the early and late stages of reverse transcription as well as for two-LTR circular DNA. Although this DNA is not a precursor to integrated DNA (reviewed in reference 4), it is found in the nuclei of infected cells after synthesis of linear double-stranded viral DNA; thus, the presence of the circular DNA is used as a marker for import of full-length viral DNA into the nucleus (4).
The results of the PCR analysis are shown in Fig. 4. Figure 4A shows that the wild-type sample contained products corresponding to all of the intermediates that are normally synthesized during reverse transcription. In contrast, the samples from mutant virus-infected cells showed small amounts of (−) SSDNA and trace amounts of elongated minus-strand DNA and (+) SSDNA but no elongated plus-strand DNA, full-length DNA, or 2-LTR circular DNA. The identity of the (−) SSDNA product was confirmed by Southern blot hybridization (data not shown). Analysis of serially diluted samples indicated that the (−) SSDNA product present in cells infected with the mutants was at least 20- to 50-fold less abundant than that present in wild-type virus-infected cells (data not shown). These quantitative differences are not due to differences in the amounts of total DNA in the samples, since amplification of the hGAPDH cellular gene gave equivalent amounts of product in each case.
Since a low level of reverse transcription can occur in HIV-1 virions prior to infection (35, 47, 57, 58), it is entirely possible that the small amounts of (−) SSDNA detected in mutant virus-infected cells represent DNA synthesized in virus particles that adhered to the cell surface at the time the cells were lysed and not DNA synthesized following infection. To address this issue, CD4-positive HeLa indicator cells were infected with wild-type and mutant viruses and were then trypsinized prior to extraction of DNA. Under these conditions, PCR analysis showed that (−) SSDNA and DNA made after minus-strand transfer were easily detected in DNA from wild-type virus-infected cells, whereas neither of these two products could be detected in samples from mutant virus-infected cells (Fig. 4B). These results demonstrate that the small amounts of DNA products detected in mutant samples from untreated cells (Fig. 4A) are derived from virions and are not the result of de novo DNA synthesis in infected cells.
Thus, our findings indicate that virions bearing mutations that substitute alanine for W23, F40, and D51 residues in the CA domain of Gag are unable to initiate reverse transcription in the infected cell. This defect is unrelated to any effect of the mutations on RT itself, since mutant and wild-type particles exhibit similar levels of endogenous (Fig. 3) and exogenous (Table 1) RT activities.
Electron-microscopic analysis shows aberrant morphology of mutant virions.
Since some studies have indicated a link between viral DNA synthesis postentry and proper maturation of the viral core (7, 39), it was of great interest to examine the effect of the three CA mutations on virion morphology and core structure. 293T and HeLa cells transfected with the pNL4-3KFS clones were examined by transmission electron microscopy (Fig. 5). An example of a wild-type particle with the typical cone-shaped core is shown in Fig. 5A; particles with centric or acentric cores were also present in the wild-type virus population (Fig. 5B). (An acentric core is defined as an electron-dense, round core that is pushed up against a small region of the viral membrane; a centric core is also round and electron dense but is centrally located in the particle). Mutants D51A (Fig. 5C and D) and F40A (Fig. 5E) exhibited only centric or acentric cores and no conical cores. The absence of a conical core in D51A virions has also been noted by von Schwedler et al. (51). Analysis of the W23A particles revealed gross abnormalities (Fig. 5F). No particles with a defined core could be detected; in addition, W23A particles had unusual shapes, with a layer of what is probably the Gag protein lining the inner surface of the viral membrane. Figure 5F shows one of the abnormal particles budding from the cell membrane.
FIG. 5.

Electron micrographs of wild-type and CA mutant viral particles. (A and B) Wild type; (C and D) D51A; (E) F40A; (F) W23A. Scale bars, 100 nm (all panels).
To determine whether the phenotypes shown in Fig. 5 are typical for the types of core structures found in the various virus populations, a quantitative estimate was performed. Groups of virus particles produced in 293T and HeLa cells, numbering from 100 to 200 particles (wild-type and D51A) or approximately 60 particles (F40A), were counted, and the distribution of the different viral core phenotypes was measured (Table 2). The wild-type virus population contained similar numbers of particles with centric and acentric cores and a relatively small, but significant, number of cone-shaped cores. In contrast, for mutants D51A and F40A, no cone-shaped cores could be detected, and, in both cases, the majority of the particles contained acentric cores. It is important to note that the relative amounts of the various types of particles were very similar in both 293T and HeLa cells. This was also true of theW23A particles, which appeared as large “blebs” in both cell lines.
TABLE 2.
Electron-microscopic analysis of HIV-1 wild-type and mutant virionsa
| Virus | Cell type | No. of particles | No. of particles whose cores are:
|
||
|---|---|---|---|---|---|
| Cone shaped | Centric | Acentric | |||
| WTb | 293T | 196 | 10 (5)c | 100 (51) | 86 (44) |
| HeLa | 120 | 18 (15) | 60 (50) | 42 (35) | |
| D51A | 293T | 176 | 0 | 16 (9) | 160 (91) |
| HeLa | 120 | 0 | 16 (13) | 104 (87) | |
| F40A | 293T | 56 | 0 | 16 (29) | 40 (71) |
| HeLa | 64 | 0 | 14 (22) | 50 (78) | |
For mutant W23A, no particles with a visible core could be detected.
WT, wild type.
Numbers in parentheses are percentages of the total number of particles.
The data obtained from electron-microscopic analysis demonstrate that the CA mutations under investigation interfere with proper assembly of virion cores and maturation of viral particles.
DISCUSSION
In the present study, our goal was to investigate the effect of single-alanine-substitution mutations of several conserved residues in the N-terminal domain of the HIV-1 CA protein on the ability of virions to undergo reverse transcription in the infected cell. The mutated amino acids included (i) Trp23 and Phe40, two representative members of a group of highly conserved hydrophobic residues (Fig. 1) (38) that has been suggested to be important for maintaining the stability of the CA structure and functional activity, and (ii) Asp51, another highly conserved residue that forms a buried salt bridge with Pro1 in the mature CA protein (20, 51). Here, we report that, while the mutations have little effect on in vitro RT activities, the mutants are severely impaired in the initiation of viral DNA synthesis in infected cells and do not make detectable amounts of viral DNA products, including (−) SSDNA.
To correlate the RT results with other parameters of viral infection, we also examined (i) virus particle production, (ii) infectivity, (iii) viral proteins present in lysates of infected cells and in virions, (iv) endogenous reverse transcription, and (v) virion core morphology. The three mutations do not have a major effect on production of virus particles (Table 1), but mutant virions are not infectious in single-cycle and long-term infectivity assays (Table 1). These findings are consistent with what has been observed for other N-terminal domain mutants and support the conclusion that this domain is not involved in particle release but is required for infectivity (5, 7, 40, 51, 52).
The mutants express all of the normal intracellular and virion-associated viral proteins (Fig. 2). The amount of Pr55gag is increased in lysates of mutant virus-infected cells, but the amount of intracellular CA is reduced. Similarly, reduced amounts of CA are present in mutant particles. The decrease in CA is correlated with lowered stability of the mutant CA proteins, as shown by the appearance of CA-related bands that are smaller than p24 (Fig. 2B) (51). Reduction in the level of CA protein has also been found in studies of other HIV-1 N-terminal CA mutants (5, 7, 40). The finding that mutant particles contain decreased amounts of IN (Fig. 2B) is surprising and has not been previously reported for virions with N-terminal CA mutations. Although no evidence for IN degradation was detected by Western blot analysis (data not shown), it is possible that the CA mutations may destabilize the core, thereby leading to a loss or instability of IN.
Examination of mutant virions, produced in either HeLa or 293T cells, by transmission electron microscopy indicated that they exhibit a phenotype that differs from that of wild-type particles. The population of wild-type virions contained a significant number of cone-shaped cores as well as approximately equal proportions of centric and acentric cores (Fig. 5A and B; Table 2); the appearance of conical or round cores depends on whether the plane of sectioning was vertical or horizontal. In contrast, no conical cores could be detected in any of the mutant virus samples (Table 2). We found that the majority of F40A and D51A particles have an acentric core, with only a small percentage containing centric cores (Fig. 5C, D, and E; Table 2) (51). Mutant W23A has an unusual phenotype: these virions seem unable to assemble recognizable mature particles and instead produce immature virions of varied size and shape, which appear to have a protein (presumably Gag) lining the inner surface (Fig. 5F). This is a rather striking effect on virus assembly, particularly since it results from mutation of a single residue.
Based on studies performed thus far, two different classes of N-terminal CA mutants can be distinguished. For example, previous reports on the properties of virions with mutations in the CypA loop (3, 7) indicate that these particles contain normal cores but are defective in reverse transcription. Although the mutants synthesize some viral DNA in infected cells, including in most cases full-length DNA, the amount is reduced with respect to that made by wild-type virus; moreover, no 2-LTR circular DNA can be detected, possibly because the full-length DNA product cannot enter the nucleus or the ends are abnormal and cannot be amplified.
Other N-terminal CA mutants that have defects in reverse transcription and in core condensation and that are more similar to the class of mutants in this study have been described. For example, in work on N-terminal CA linker insertion mutants, it was found that virions containing only round cores do not synthesize 2-LTR circular DNA (39). However, since synthesis of early DNA intermediates was not evaluated, it is not possible to determine at which step reverse transcription was blocked. Another example is a P1L mutant that lacks a cone-shaped core and that does not synthesize a vif DNA product, indicative of partial elongation of minus-strand DNA; synthesis of (−) SSDNA was not measured (7). In view of the fact that Pro1 forms a buried salt bridge with Asp51 (20, 51), it is not surprising that P1L virions have a phenotype similar to that of D51A (Fig. 4 and 5) (51).
Here, we demonstrate that the W23A, F40A, and D51A mutants, which do not produce any particles with conical cores, are profoundly compromised in their ability to initiate reverse transcription in infected cells (Fig. 4B). Thus, by treating infected cells with trypsin to remove virions adhering to the cell surface, we could clearly establish that none of the mutants makes any detectable (−) SSDNA (compare Fig. 4A and B). We have also made five additional mutants with alanine substitutions at the conserved hydrophobic residues (38) including L20A (helix I), L52A (helix III), M55A (helix III), V59A (helix III), and L138A (helix VII). Preliminary results indicate that these mutants exhibit abnormal core structures and are also unable to initiate reverse transcription postinfection (our unpublished observations).
It should be noted that, while all of the alanine substitution mutants we have made are blocked in synthesis of (−) SSDNA in infected cells (Fig. 4B) (our unpublished observations), they make somewhat larger amounts of total DNA than wild-type virions in endogenous assays (Fig. 3A) (our unpublished observations). This may reflect a more “open” core structure in these mutants, which could allow more efficient penetration of dNTPs into the particle.
It is of interest to speculate on the explanation for the phenotype of the mutants described in this study. Taken together, our findings are consistent with a defect in core stability or in a postentry step that precedes reverse transcription. An investigation of the stability of the mutant cores has been initiated. An alternate possibility that there is a defect in viral entry appears to be less likely: Thus, to assay DNA synthesis in infected cells, virions pseudotyped with an MLV envelope were used (Fig. 4); this route bypasses the normal gp120-dependent mode of HIV-1 entry.
In conclusion, we have demonstrated a crucial connection between the ability of virus particles to carry out reverse transcription in infected cells and virion core morphology. We propose the existence of classes of N-terminal CA mutants which differ in the severity of individual mutations with respect to core structure and the extent to which DNA synthesis is inhibited. Two such classes can already be identified. The particular class of mutants analyzed in this and some other (7, 39) studies has an extreme phenotype. In this case, a profound deficiency in reverse transcription postentry appears to be predictive of a prior defect in core maturation during virus assembly.
ACKNOWLEDGMENTS
We thank Kunio Nagashima for expert assistance with the electron microscopy, Brenda Soto for outstanding help with cloning, Akira Ono for a gift of anti-CA serum, Zhonglin Yang for generating the ribbon diagram illustrating the structure of the N-terminal CA domain, and Alan Rein for critical reading of the manuscript.
This work was supported in part by funds from the National Institutes of Health Intramural AIDS Targeted Antiviral Program awarded to J.G.L.
REFERENCES
- 1.Accola M A, Öhagen Å, Göttlinger H G. Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6gag. J Virol. 2000;74:6198–6202. doi: 10.1128/jvi.74.13.6198-6202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adachi A, Gendelman H E, Koenig S, Folks T, Willey R, Rabson A, Martin M A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Alin K, Goff S P. Amino acid substitutions in the CA protein of Moloney murine leukemia virus that block early events in infection. Virology. 1996;222:339–351. doi: 10.1006/viro.1996.0431. [DOI] [PubMed] [Google Scholar]
- 2b.Bowzard J B, Wills J W, Craven R C. Second-site suppressors of Rous sarcoma virus CA mutations: evidence for interdomain interactions. J Virol. 2001;75:6850–6856. doi: 10.1128/JVI.75.15.6850-6856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braaten D, Franke E K, Luban J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol. 1996;70:3551–3560. doi: 10.1128/jvi.70.6.3551-3560.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown P O. Integration. In: Coffin J M, Hughes S H, Varmus H E, editors. Retroviruses. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1997. pp. 161–203. [PubMed] [Google Scholar]
- 4a.Cairns T M, Craven R C. Viral DNA synthesis defects in assembly-competent Rous sarcoma virus CA mutants. J Virol. 2001;75:242–250. doi: 10.1128/JVI.75.1.242-250.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorfman T, Bukovsky A, Öhagen Å, Höglund S, Göttlinger H G. Functional domains of the capsid protein of human immunodeficiency virus type 1. J Virol. 1994;68:8180–8187. doi: 10.1128/jvi.68.12.8180-8187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Y X, Copeland T D, Henderson L E, Gorelick R J, Bosche W J, Levin J G, Rein A. HIV-1 nucleocapsid protein induces “maturation” of dimeric retroviral RNA in vitro. Proc Natl Acad Sci USA. 1996;93:7577–7581. doi: 10.1073/pnas.93.15.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzon T, Leschonsky B, Bieler K, Paulus C, Schröder J, Wolf H, Wagner R. Proline residues in the HIV-1 NH2-terminal capsid domain: structure determinants for proper core assembly and subsequent steps of early replication. Virology. 2000;268:294–307. doi: 10.1006/viro.1999.0178. [DOI] [PubMed] [Google Scholar]
- 8.Franke E K, Yuan H E, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 9.Freed E O. HIV-1 Gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 10.Freed E O, Delwart E L, Buchschacher G L, Jr, Panganiban A T. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci USA. 1992;89:70–74. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freed E O, Englund G, Martin M A. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol. 1995;69:3949–3954. doi: 10.1128/jvi.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freed E O, Martin M A. Evidence for a functional interaction between the V1/V2 and C4 domains of human immunodeficiency virus type 1 envelope glycoprotein gp120. J Virol. 1994;68:2503–2512. doi: 10.1128/jvi.68.4.2503-2512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freed E O, Martin M A. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol. 1995;69:1984–1989. doi: 10.1128/jvi.69.3.1984-1989.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu W, Gorelick R J, Rein A. Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J Virol. 1994;68:5013–5018. doi: 10.1128/jvi.68.8.5013-5018.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu W, Rein A. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J Virol. 1993;67:5443–5449. doi: 10.1128/jvi.67.9.5443-5449.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamble T R, Vajdos F F, Yoo S, Worthylake D K, Houseweart M, Sundquist W I, Hill C P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 17.Gamble T R, Yoo S, Vajdos F F, von Schwedler U K, Worthylake D K, Wang H, McCutcheon J P, Sundquist W I, Hill C P. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 18.Ganser B K, Li S, Klishko V Y, Finch J T, Sundquist W I. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 19.Gelderblom H R. Assembly and morphology of HIV: potential effect of structure on viral function. AIDS. 1991;5:617–637. [PubMed] [Google Scholar]
- 20.Gitti R K, Lee B M, Walker J, Summers M F, Yoo S, Sundquist W I. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- 21.Goncalves J, Korin Y, Zack J, Gabuzda D. Role of Vif in human immunodeficiency virus type 1 reverse transcription. J Virol. 1996;70:8701–8709. doi: 10.1128/jvi.70.12.8701-8709.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross I, Hohenberg H, Huckhagel C, Kräusslich H G. N-terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J Virol. 1998;72:4798–4810. doi: 10.1128/jvi.72.6.4798-4810.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross I, Hohenberg H, Wilk T, Wiegers K, Grättinger M, Müller B, Fuller S, Kräusslich H G. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 2000;19:103–113. doi: 10.1093/emboj/19.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo J, Henderson L E, Bess J, Kane B, Levin J G. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J Virol. 1997;71:5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson L E, Bowers M A, Sowder II R C, Serabyn S A, Johnson D G, Bess J W, Jr, Arthur L O, Bryant D K, Fenselau C. Gag proteins of the highly replicative MN strain of human immunodeficiency virus type 1: posttranslational modifications, proteolytic processings, and complete amino acid sequences. J Virol. 1992;66:1856–1865. doi: 10.1128/jvi.66.4.1856-1865.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang M, Orenstein J M, Martin M A, Freed E O. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol. 1995;69:6810–6818. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiernan R E, Ono A, Englund G, Freed E O. Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J Virol. 1998;72:4116–4126. doi: 10.1128/jvi.72.5.4116-4126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiernan R E, Ono A, Freed E O. Reversion of a human immunodeficiency virus type 1 matrix mutation affecting Gag membrane binding, endogenous reverse transcriptase activity, and virus infectivity. J Virol. 1999;73:4728–4737. doi: 10.1128/jvi.73.6.4728-4737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klutch M, Woerner A M, Marcus-Sekura C J, Levin J G. Generation of HIV-1/HIV-2 cross-reactive peptide antisera by small sequence changes in HIV-1 reverse transcriptase and integrase immunizing peptides. J Biomed Sci. 1998;5:192–202. doi: 10.1007/BF02253469. [DOI] [PubMed] [Google Scholar]
- 31.Kotov A, Zhou J, Flicker P, Aiken C. Association of Nef with the human immunodeficiency virus type 1 core. J Virol. 1999;73:8824–8830. doi: 10.1128/jvi.73.10.8824-8830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraulis P J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 33.Landau N R, Page K A, Littman D R. Pseudotyping with human T-cell leukemia virus type 1 broadens the human immunodeficiency virus host range. J Virol. 1991;65:162–169. doi: 10.1128/jvi.65.1.162-169.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levin J G, Grimley P M, Ramseur J M, Berezesky I K. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J Virol. 1974;14:152–161. doi: 10.1128/jvi.14.1.152-161.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lori F, di Marzo Veronese F, de Vico A L, Lusso P, Reitz M S, Jr, Gallo R C. Viral DNA carried by human immunodeficiency virus type 1 virions. J Virol. 1992;66:5067–5074. doi: 10.1128/jvi.66.8.5067-5074.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mervis R J, Ahmad N, Lillehoj E P, Raum M G, Salazar F H R, Chan H W, Venkatesan S. The gag gene products of human immunodeficiency virus type 1: alignment within the gag open reading frame, identification of posttranslational modifications, and evidence for alternative gag precursors. J Virol. 1988;62:3993–4002. doi: 10.1128/jvi.62.11.3993-4002.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Messer L I, Currey K M, O'Neill B J, Maizel J V, Jr, Levin J G, Gerwin B I. Functional analysis of reverse transcription by a frameshift pol mutant of murine leukemia virus. Virology. 1985;146:146–152. doi: 10.1016/0042-6822(85)90062-5. [DOI] [PubMed] [Google Scholar]
- 38.Momany C, Kovari L C, Prongay A J, Keller W, Gitti R K, Lee B M, Gorbalenya A E, Tong L, McClure J, Ehrlich L S, Summers M F, Carter C, Rossmann M G. Crystal structure of dimeric HIV-1 capsid protein. Nat Struct Biol. 1996;3:763–770. doi: 10.1038/nsb0996-763. [DOI] [PubMed] [Google Scholar]
- 39.Reicin A S, Ohagen A, Yin L, Hoglund S, Goff S P. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J Virol. 1996;70:8645–8652. doi: 10.1128/jvi.70.12.8645-8652.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reicin A S, Paik S, Berkowitz R D, Luban J, Lowy I, Goff S P. Linker insertion mutations in the human immunodeficiency virus type 1 gag gene: effects on virion particle assembly, release, and infectivity. J Virol. 1995;69:642–650. doi: 10.1128/jvi.69.2.642-650.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rein A, Henderson L E, Levin J G. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem Sci. 1998;23:297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 42.Rothenberg E, Smotkin D, Baltimore D, Weinberg R A. In vitro synthesis of infectious DNA of murine leukaemia virus. Nature. 1977;269:122–126. doi: 10.1038/269122a0. [DOI] [PubMed] [Google Scholar]
- 43.Swanstrom R, Wills J W. Synthesis, assembly, and processing of viral proteins. In: Coffin J M, Hughes S H, Varmus H E, editors. Retroviruses. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1997. pp. 263–334. [PubMed] [Google Scholar]
- 44.Tang S, Collier A J, Elliott R M. Alterations to both the primary and predicted secondary structure of stem-loop IIIc of the hepatitis C virus 1b 5′ untranslated region (5′UTR) lead to mutants severely defective in translation which cannot be complemented in trans by the wild-type 5′UTR sequence. J Virol. 1999;73:2359–2364. doi: 10.1128/jvi.73.3.2359-2364.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh C T, Sodroski J, Göttlinger H G. Functional association of cyclophilin A with HIV-1 virions. Nature. 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 46.Tokunaga K, Nakamura Y, Sakata K, Fujimori K, Ohkubo M, Sawada K, Sakiyama S. Enhanced expression of a glyceraldehyde-3-phosphate dehydrogenase gene in human lung cancers. Cancer Res. 1987;47:5616–5619. [PubMed] [Google Scholar]
- 47.Trono D. Partial reverse transcripts in virions from human immunodeficiency and murine leukemia viruses. J Virol. 1992;66:4893–4900. doi: 10.1128/jvi.66.8.4893-4900.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner B G, Summers M F. Structural biology of HIV. J Mol Biol. 1999;285:1–32. doi: 10.1006/jmbi.1998.2354. [DOI] [PubMed] [Google Scholar]
- 49.Vogt V M. Retroviral virions and genomes. In: Coffin J M, Hughes S H, Varmus H E, editors. Retroviruses. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1997. pp. 27–69. [PubMed] [Google Scholar]
- 50.Vogt V M, Simon M N. Mass determination of Rous sarcoma virus virions by scanning transmission electron microscopy. J Virol. 1999;73:7050–7055. doi: 10.1128/jvi.73.8.7050-7055.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Schwedler U K, Stemmler T L, Klishko V Y, Li S, Albertine K H, Davis D R, Sundquist W I. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 1998;17:1555–1568. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang C T, Barklis E. Assembly, processing, and infectivity of human immunodeficiency virus type 1 Gag mutants. J Virol. 1993;67:4264–4273. doi: 10.1128/jvi.67.7.4264-4273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Welker R, Hohenberg H, Tessmer U, Huckhagel C, Kräusslich H G. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J Virol. 2000;74:1168–1177. doi: 10.1128/jvi.74.3.1168-1177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiegers K, Rutter G, Schubert U, Grättinger M, Kräusslich H G. Cyclophilin A incorporation is not required for human immunodeficiency virus type 1 particle maturation and does not destabilize the mature capsid. Virology. 1999;257:261–274. doi: 10.1006/viro.1999.9669. [DOI] [PubMed] [Google Scholar]
- 55.Wills J W, Craven R C. Form, function, and use of retroviral Gag proteins. AIDS. 1991;5:639–654. doi: 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Wu X, Liu H, Xiao H, Conway J A, Hehl E, Kalpana G V, Prasad V, Kappes J C. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J Virol. 1999;73:2126–2135. doi: 10.1128/jvi.73.3.2126-2135.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zack J A, Haislip A M, Krogstad P, Chen I S. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J Virol. 1992;66:1717–1725. doi: 10.1128/jvi.66.3.1717-1725.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H, Zhang Y, Spicer T, Henrard D, Poiesz B J. Nascent human immunodeficiency virus type 1 reverse transcription occurs within an enveloped particle. J Virol. 1995;69:3675–3682. doi: 10.1128/jvi.69.6.3675-3682.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]