

Abstract
This case report presents a 38‐year‐old male patient who, after a febrile infection, developed super‐refractory status epilepticus and multiorgan failure, and died in 2 weeks despite the best possible intensive care. Autopsy revealed findings suggestive of hemophagocytic lymphohistiocytosis (HLH). This case shows that a rare immunological cause such as HLH may cause febrile infection‐related epilepsy syndrome (FIRES), and complications of intensive care can mask the physiological and laboratory changes in HLH.
Plain Language Summary
This case report presents a 38‐year‐old man who, after a febrile infection, developed intractable epileptic activity requiring intensive care treatment. During the intensive care, the patient showed signs of multiple organ damage and died in 2 weeks despite the best possible treatment. Autopsy revealed findings suggestive of hemophagocytic lymphohistiocytosis (HLH), which is a rare immune system regulation disorder leading to persistent inflammatory state and organ damages. This case shows that an immunological disorder like HLH may underlie treatment resistant fever‐related epileptic seizures.
Keywords: febrile infection‐related epilepsy syndrome, hemophagocytic lymphohistiocytosis, immunological epilepsy, super‐refractory status epilepticus
Key points.
Hemophagocytic lymphohistiocytosis (HLH) is a rare hyperinflammatory condition causing fever, splenomegaly, cytopenias, neurological symptoms, and in severe cases multiorgan damage.
The possibility of HLH should be ruled out in patients with febrile infection‐related refractory status epilepticus (FIRES).
Intensive care treatment, sepsis, or underlying malignancy or rheumatoid disease can mimic symptoms and laboratory findings of HLH.
1. INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a rare but life‐threatening, systemic hyperinflammatory disorder characterized by dysregulated cytotoxic immune activity resulting in multiorgan failure and sometimes neurological manifestations like seizures and ataxia. 1 The condition can be caused by mutations in genes regulating immune activity, or by infection, immune compromise, malignancy, drug‐induced hypersensitivity syndromes, or rheumatoid disease. 1 Widely used HLH‐2004 diagnostic criteria require a demonstrated relevant genetic defect or five out of eight clinical features including fever, splenomegaly, cytopenia affecting at least two cell lines, hypertriglyceridemia and/or hypofibrinogenemia, HLH finding in bone marrow, lymph nodes or spleen, high ferritin ≥500 mg/L, high soluble interleukin 2 receptor (sIL‐2R) ≥2400 U/mL, and low or absent NK‐cell activity. 1
Febrile infection‐related epilepsy syndrome (FIRES) is a catastrophic epileptic encephalopathy in which a previously healthy person develops refractory status epilepticus after a febrile illness. 2 , 3 , 4 The cause is unknown but dysregulated inflammation in the central nervous system is a recognized feature in FIRES. 5 We describe here an adult patient with FIRES‐like course of the disease in whom postmortem investigation revealed pathology referring to systemic hemophagocytic lymphohistiocytosis.
2. CASE REPORT
The patient, a 38‐year‐old male, was diagnosed with focal epilepsy at the age of 24 years. He had seizures with déjà vu, feeling of falling and right‐sided tingling. At the time of epilepsy diagnosis, left temporal arachnoid cyst was detected in brain MRI, and EEG showed left frontotemporal slow‐wave disturbance. He had been seizure‐free for years with lamotrigine 100 mg twice a day.
In 2019, he was admitted to emergency department (ED) due to recurrent generalized tonic–clonic seizures (GTCS) with no recovery of consciousness between seizures. He had respiratory symptoms and fever up to 39°C for 5 days. Before arrival to ED, he received i.v. midazolam and levetiracetam (2000 mg) and was intubated and set to propofol‐sedation by emergency medical services. Initial brain CT was normal. Blood leukocytes were 8.5 × 109/l (ref 3.4–8.2 × 109/L) and CRP 19 mg/L. CSF leukocyte count was four (ref 0–5 × 106/L) and protein level 628 mg/L (ref 150–450 mg/L). During a sedation break at ED, he had a GTCS and was then set to propofol‐induced burst‐suppression. Antiseizure medication was enhanced, and acyclovir and ceftriaxone were started (Figure 1). Next day, despite sedation at burst‐suppression level, patient had several electrographic‐generalized seizures without any clinical signs. He also developed acute kidney failure (creatinine 493 μmol/L), lactatemia, high potassium levels, and CRP rise to 263 mg/L. Kidney failure was interpreted as propofol‐related infusion syndrome (PRIS) and propofol was changed to thiopental. On third day, he had gas exchange problems and hemodialysis was started due to kidney failure.
FIGURE 1.
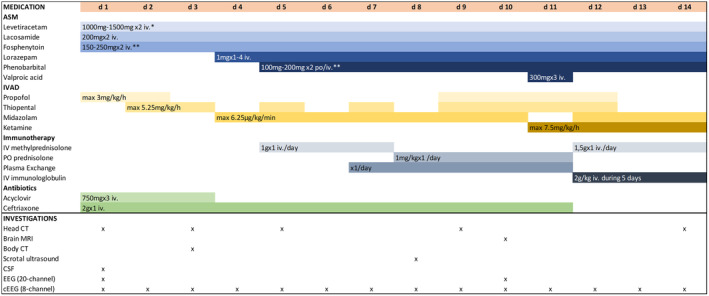
Medications with posology and investigations during the 14 days of hospitalization. ASM, Antiseizure medication; cEEG, Continuous EEG; CSF, Cerebral spinal fluid; CT, Computer tomography; EEG, Electroencephalography; IVAD, Intravenous anesthetic drug; MRI, Magnetic resonance imaging. *posology adjusted based on the kidney function, **posology adjusted based on the blood concentration.
Brain MRI with gadolinium contrast was performed on 10th day and it showed normal findings except for the left temporal arachnoid cyst, which did not compress surrounding structures. No inflammatory or ischemic changes were observed, and hippocampi were normal (Figure S1).
From the day one, the continuous EEG recording showed generalized, 1–5‐min seizures despite well‐attained burst‐suppression level (Figure 2). Ictal activity was extremely treatment resistant. Seizures started even from isoelectric EEG and occurred 5–10 times per hour and increased up to 30/h when weaning anesthetics. Seizures were mainly spontaneous but occasionally stimulus induced, and part of the seizures had a subtle facial twitching as a clinical manifestation.
FIGURE 2.
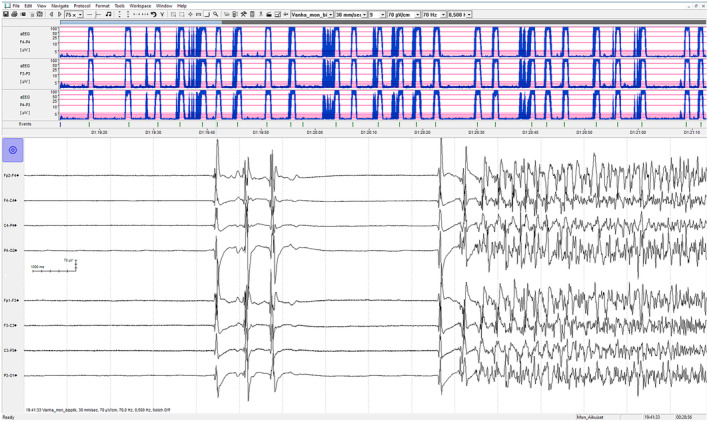
An example of a typical cEEG‐monitoring view showing generalized spike and wave discharges from an isoelectric EEG. Amplitude‐integrated EEG (aEEG), in the upper part of the figure, shows a condensed 2‐h period of monitoring with 25 generalized electrographical seizures.
Routine microbiological, autoimmune, and malignancy investigations were negative (Table S1). Streptococcus A antigen test from pharynx was positive, but culture was negative. Also, serum antistreptolysin and Streptococcal DNase B antibodies were negative speaking against streptococcal infection.
Anemia (Hb 84 g/L, ref 134–167 g/L) and thrombocytopenia (96 × 109/l, ref 150–360 × 109/L) developed during the treatment. Blood leukocyte level varied from normal to mildly elevated. Neutrophils remained normal but lymphopenia 0.37 × 109/l (ref 1.3–3.6 × 109/l) was detected. He also developed coagulopathies including constantly high FIDD (1.8–5.5 mg/L, ref <0.5 mg/L) and hyperfibrinogenemia up to 6.1 g/L which later turned into hypofibrinogenemia (1.6 g/L, ref 2–4 g/L). Liver enzymes were normal or mildly elevated; ALAT 89 U/L (normal <50 U/L), ASAT 135 U/L (normal ≤45 U/L), AFOS, and total bilirubin were normal. NH4 ion was normal and urea mildly elevated, mainly between 12 and 13 mmol/L reaching once a peak value 18.4 mmol/L (ref 3.2–8.1 mmol/L). Ferritin was not measured. Serum triglycerides were 1.83 mmol/L (normal <1.7 mmol/L). Limited cytokine testing was available: mildly elevated sIL2‐R 676 kU/L (normal 160–620 kU/L), elevated IL‐6 25.4 ng/L (normal <5.9 ng/L), elevated IL‐8179 pg/mL (normal <15 pg/mL), and normal TNF‐α 6.5 ng/L (normal <8.1 ng/L).
Methylprednisolone 1000 mg i.v. for 3 days was started on day five, followed by five times of plasmapheresis. As no clinical improvement was observed, another 3 days corticosteroid pulse treatment was given combined with intravenous immunoglobulin 2 g/kg administered over next 5 days (Figure 1). Wide spectrum of antiseizure medication (ASM) was administered and also concomitant anesthetics were used in order to control seizures (Figure 1). Despite these measures, seizures persisted, and the patient developed an increasing need for vasopressors and paralytic ileus, which restricted enteral ASM selection and introduction of ketogenic diet. On day 13, a decision was made to insert a deep brain stimulator and start transcranial magnetic stimulation. In addition, tocilizumab or rituximab were considered. Before starting these treatments, the patient developed recurrent asystole and died on 14th day after admission to hospital.
Autopsy revealed end‐stage tubular necrosis in the kidneys, congestion and edema in the lungs, and hemophagocytic lymphohistiocytosis in the bone marrow (Figure 3D). Histological examination of the brain showed pyknotic neurons cortically in the watershed areas and in the basal ganglia referring to anoxic‐ischemic damage. Mild lymphocytic infiltration was observed in the meninges and perivascularly, particularly in the basal forebrain region and in the parietal lobe, and immunohistochemical staining highlighted a few CD3‐positive T cells in these areas (Figure 3B,D). Furthermore, enhanced microgliosis in particular in the basal nucleus of Meinert region was seen in IBA‐1 immunohistochemistry (Figure 3C). As often seen in cases with epilepsy, mild mossy fiber sprouting was detected in dentate granular cells of hippocampi in MAP2 immunohistochemistry (Figure 3A). C4d complement staining was negative.
FIGURE 3.
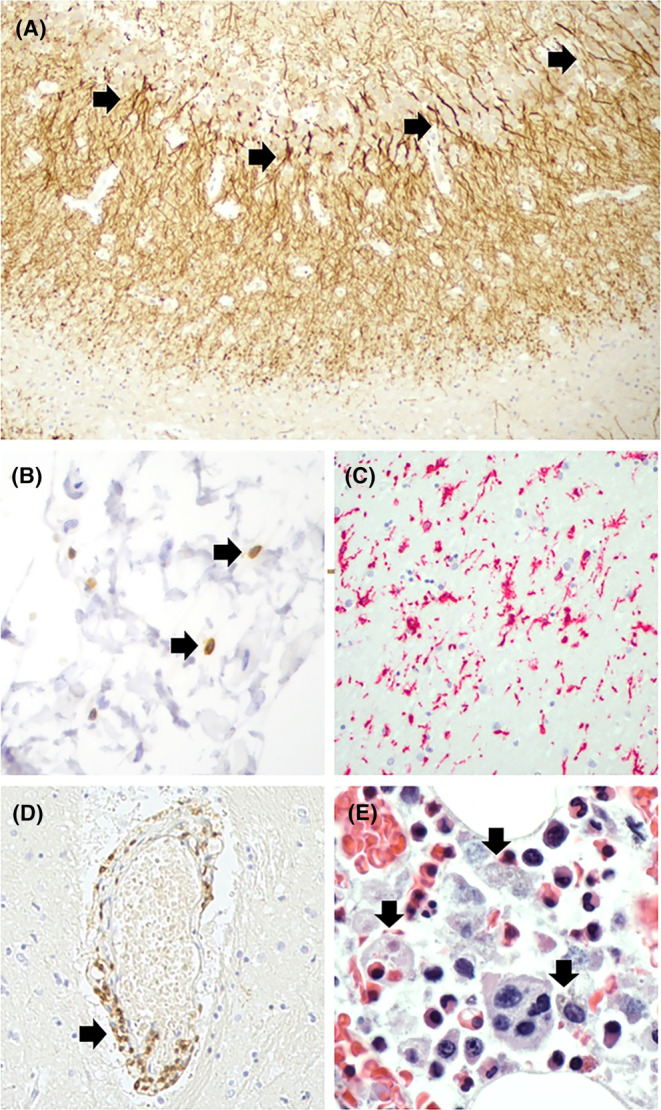
Histopathological findings at the autopsy. (A) Mild mossy fiber sprouting (arrows) in the inner molecular layer of the hippocampus. MAP2 immunohistochemistry 100× magnification. (B) A few T cells (arrows) in the meninges in the basal forebrain region. CD3–CD20 double immunohistochemistry, 400× magnification. (C) Reactive microglial cells in the basal nucleus of Meinert region. IBA‐1 immunohistochemistry, 200× magnification. (D) Mild perivascular lymphocytic infiltration (arrow) in basal forebrain region. CD3–CD20 double immunohistochemistry, 400× magnification. (E) Abundant hemophagocytic cells (arrows) in the bone marrow, containing lysing cell remnants and hemosiderin. Hematoxylin–eosin staining, 1000× magnification, oil objective.
3. DISCUSSION
Our case highlights the importance of identifying the rare but treatable causes of FIRES.
HLH is a rare, life‐threatening immunological syndrome characterized by dysregulated function of cytotoxic lymphocytes and macrophages, resulting in cytokine‐mediated tissue injury and multiorgan failure. 1 Diagnostics is challenging and often delayed due to diverse and unspecific symptoms resulting from dysfunction of various organ systems. 6 Neurological manifestations are reported in 30%–73% of patients with HLH. 7 HLH‐2004 diagnostic criteria were originally developed to recognize children with HLH for clinical treatment trials and these criteria were extended to adults though there are notable differences in the etiology and the diagnostic parameters in adult and children populations. 6 Infections, malignancies, and rheumatoid disorders are the most common triggers in adults while genetic defects are rarely detected. 6 None of the diagnostic criteria alone is specific for HLH and underlying etiological disease, for example, malignancy or rheumatoid disorder can cause similar laboratory or imaging findings as HLH. In intensive care surroundings, a challenge is that HLH can manifest with a phenotype indistinguishable from sepsis or multiple organ failure, and medications, extracorporeal life support, or secondary infections can modify the diagnostic parameters. 6 Further, due to gradual progression of the symptoms, it is common that not all the five criteria are filled in the early stages of the condition. 6
An expert opinion of treatment of HLH in adults has recommended that HLH should be suspected in a critically ill patient with persistent fever, cytopenias, and organomegaly, particularly in cases of sepsis and sepsis‐like syndromes, or evolving multiorgan failure. 6 Our patient met only three of the HLH‐2004 diagnostic criteria (persistent fever, cytopenias, and bone marrow histology, which was detected postmortem). In our patient, the renal insufficiency that developed in first 24 h after admission to hospital and a sepsis‐like clinical picture without detected infective agent also supports the possibility of HLH. Due to intractable status epilepticus, large doses of ASM and anesthetics were used concomitantly, and changes in laboratory parameters, constant hypotonia, and renal failure were therefore interpreted as being related to medications and intensive care. The patient was not suspected to have HLH during the 14 days of treatment and the possibility of HLH was introduced first postmortem. Therefore, not all diagnostic tests were performed (e.g., ferritin, NK‐cell). Also, EBV infection, a common trigger for HLH, was not tested, and the possibility of rheumatoid disorder was not thoroughly excluded, although rheumatoid etiology seems unlikely considering acutely developed symptoms and negative family and symptom history for rheumatoid disorders.
Neuropathological analysis showed only a few lymphocytes perivascularly and meningeally, but instead microglial activation, supporting the role of inflammatory drivers of innate immunity for the refractory status epilepticus in the patient. Generally, neuropathological findings in HLH can be unspecific ranging from mere meningeal or perivascular infiltration of lymphocytes and macrophages to wide hemophagocytic findings, hemorrhagia, and necrosis. 8 In the mildest form, the meningeal involvement can be the only neuropathological finding and due to short and aggressive course of the disease, it is possible that wider histological changes were not yet developed in our patient. 8 Neuropathological findings of FIRES are often reported under an umbrella term NORSE (New‐onset refractory status epilepticus) consisting of patients with sudden intractable status epilepticus without known predisposing factor. Findings have been unspecific including mainly gliosis, neuronal loss, microglial activation, and rarely perivascular T‐cell infiltrations reflecting mainly persistent epileptic activity but only sparse inflammatory findings in the brains. 9 , 10 , 11 , 12 Microglial activation is known to play an important role in epileptogenesis, 13 and interestingly, a recent study showed decreased expression of CX3CR1, a chemokine receptor related to regulation of phagocytic activity in NORSE patients. 14 As overstimulation of macrophages is a core pathophysiological mechanism in HLH, a dysregulation of phagocytic activity might contribute to manifestation of HLH and severe status epilepticus in some patients.
Brain MRI with nondiagnostic findings is a common finding in the early stages of FIRES and is in line with mild neuropathological findings of our patient. 15 Also, EEG remained unspecific though it is notable that seizures were extremely treatment resistant as often is in FIRES patients. 16 Seizures were electrographically and clinically different compared to patient's prior epilepsy and thus refers to independent epileptogenetic process rather than escalation of patient's prior epilepsy.
HLH has previously been described in juvenile patients and one adult patient with FIRES. 2 , 3 Although our patient had an epilepsy diagnosis before developing a fulminant status epilepticus, his illness followed the pattern of FIRES. It is clear that epilepsy, managed with one antiseizure medication with years of seizure‐freedom, does not explain the violent course of the disease leading to death in 2 weeks. In such cases, a previous diagnosis of epilepsy should not delay rapid diagnostic and treatment measures for the immunological disorder. As Kam et al. 3 showed in their case, etoposide‐based chemotherapy (in their case combined with intrathecal methotrexate, dexamethasone pulses, and anakinra) can have dramatic effect on seizure control in recognized HLH cases. Our patient received common first‐line immunotherapies but no chemotherapy or interleukin antagonists such as anakinra or tocilizumab, which also have nowadays growing evidence in the treatment of FIRES. 3
4. CONCLUSIONS
This case introduces one potential etiology of FIRES that neurologist and intensive care unit specialists should be aware of and rule out already during early stages of treatment. As with our patient, high infection parameters, liver dysfunction, coagulopathy, or other signs of multiorgan failure, can easily be mistaken for as complications of intensive care treatment. As infection is an important trigger for both FIRES and HLH in adult patients, further studies to examine shared pathophysiological mechanisms in these conditions are needed.
FUNDING INFORMATION
This study did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors.
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose.
ETHICS STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1.
ACKNOWLEDGMENTS
We thank Antti Kinnunen, clinical neurophysiologist, MD, in Helsinki University Hospital for reevaluation of EEG data and producing EEG image for the article.
Haanpää A, Kämppi L, Kantonen J, Myllykangas L, Laakso SM, Forss N. Hemophagocytic lymphohistiocytosis in an adult patient with super‐refractory status epilepticus. Epilepsia Open. 2024;9:1962–1967. 10.1002/epi4.13026
REFERENCES
- 1. Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34(4):101515. [DOI] [PubMed] [Google Scholar]
- 2. Farias‐Moeller R, Lafrance‐Corey R, Bartolini L, Wells EM, Baker M, Doslea A, et al. Fueling the FIRES: Hemophagocytic lymphohistiocytosis in febrile infection‐related epilepsy syndrome. Epilepsia. 2018;59(9):1753–1763. [DOI] [PubMed] [Google Scholar]
- 3. Kam I, Prentice D, Kho LK, Dharsono F. Inflammatory epilepsy (FIRES) and haemophagocytic lymphohistiocytosis (HLH): an adult case. BMJ Case Rep. 2023;16(1):e252637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meenakshi‐Sundaram S, Sankaranarayanan M, Jeyaraman M, Ayyappan C, Karthik SN, Pandi S. Super refractory status in a case of febrile infection‐related epilepsy syndrome due to hemophagocytic lymphocytic histiocytosis. Epilepsia Open. 2021;6(1):22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kothur K, Bandodkar S, Wienholt L, Chu S, Pope A, Gill D, et al. Etiology is the key determinant of neuroinflammation in epilepsy: elevation of cerebrospinal fluid cytokines and chemokines in febrile infection‐related epilepsy syndrome and febrile status epilepticus. Epilepsia. 2019;60(8):1678–1688. [DOI] [PubMed] [Google Scholar]
- 6. La Rosée P, Horne A, Hines M, Von Bahr GT, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465–2477. [DOI] [PubMed] [Google Scholar]
- 7. Horne A, Wickström R, Jordan MB, Yeh EA, Naqvi A, Henter J‐I, et al. How to treat involvement of the central nervous system in Hemophagocytic Lymphohistiocytosis? Curr Treat Options Neurol. 2017;19(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klein C, Kleinschmidt‐Demasters BK, Liang X, Stence N, Tuder RM, Moore BE. A review of neuropathological features of familial and adult Hemophagocytic Lymphohistiocytosis. J Neuropathol Exp Neurol. 2019;78(3):197–208. [DOI] [PubMed] [Google Scholar]
- 9. Van Baalen A, Häusler M, Boor R, Rohr A, Sperner J, Kurlemann G, et al. Febrile infection–related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia. 2010;51(7):1323–1328. [DOI] [PubMed] [Google Scholar]
- 10. Hanin A, Cespedes J, Huttner A, Strelnikov D, Gopaul M, Distasio M, et al. Neuropathology of new‐onset refractory status epilepticus (NORSE). J Neurol. 2023;270(8):3688–3702. [DOI] [PubMed] [Google Scholar]
- 11. Suchdev K, Kupsky WJ, Mittal S, Shah AK. Histopathology of new‐onset refractory status epilepticus (NORSE) in adults. Seizure. 2021;93:95–101. [DOI] [PubMed] [Google Scholar]
- 12. Kramer U, Chi C‐S, Lin K‐L, Specchio N, Sahin M, Olson H, et al. Febrile infection‐related epilepsy syndrome (FIRES): pathogenesis, treatment, and outcome. Epilepsia. 2011;52(11):1956–1965. [DOI] [PubMed] [Google Scholar]
- 13. Yu C, Deng XJ, Xu D. Microglia in epilepsy. Neurobiol Dis. 2023;185:106249. [DOI] [PubMed] [Google Scholar]
- 14. Hanin A, Zhang L, Huttner AJ, Plu I, Mathon B, Bielle F, et al. Single‐cell transcriptomic analyses of brain parenchyma in patients with new‐onset refractory status epilepticus (NORSE). Neurol Neuroimmunol Neuroinflamm. 2024;11(4):e200259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wickstrom R, Taraschenko O, Dilena R, Payne ET, Specchio N, Nabbout R, et al. International consensus recommendations for management of new onset refractory status epilepticus including febrile infection‐related epilepsy syndrome: statements and supporting evidence. Epilepsia. 2022;63(11):2840–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sculier C, Gaspard N. New‐onset refractory status epilepticus and febrile infection‐related epilepsy syndrome. Curr Opin Neurol. 2023;36(2):110–116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.