

Abstract
Seven treatments are approved for Alzheimer’s disease, but five of them only relieve symptoms and do not alter the course of the disease. Aducanumab (Adu) and lecanemab are novel disease-modifying antiamyloid-β (Aβ) human monoclonal antibodies that specifically target the pathophysiology of Alzheimer’s disease (AD) and were recently approved for its treatment. However, their administration is associated with serious side effects, and their use is limited to early stages of the disease. Therefore, drug discovery remains of great importance in AD research. To gain new insights into the development of novel drugs for Alzheimer’s disease, a combination of techniques was employed, including mutation screening, molecular dynamics, and quantum biochemistry. These were used to outline the interfacial interactions of the Aducanumab::Aβ2–7 complex. Our analysis identified critical stabilizing contacts, revealing up to 40% variation in the affinity of the Adu chains for Aβ2–7 depending on the conformation outlined. Remarkably, two complementarity determining regions (CDRs) of the Adu heavy chain (HCDR3 and HCDR2) and one CDR of the Adu light chain (LCDR3) accounted for approximately 77% of the affinity of Adu for Aβ2–7, confirming their critical role in epitope recognition. A single mutation, originally reported to have the potential to increase the affinity of Adu for Aβ2–7, was shown to decrease its structural stability without increasing the overall binding affinity. Mimetic peptides that have the potential to inhibit Aβ aggregation were designed by using computational outcomes. Our results support the use of these peptides as promising drugs with great potential as inhibitors of Aβ aggregation.
Keywords: computational biology, molecular dynamics, aducanumab affinity, monoclonal antibody
Introduction
Alzheimer’s disease (AD) is directly responsible for 60–80% of cases of dementia in older individuals.1 Dementia refers to a significant loss of cognitive abilities beyond the natural neurodegenerative effects of aging.1 As life expectancy has increased over the past centuries, age-related diseases such as cancer, atherosclerosis, diabetes, and Alzheimer’s disease have become a growing concern.2−4 Although advanced age is a risk factor for these diseases, it is particularly critical for AD, where age is the greatest risk factor, surpassing genetics, and family history.1 It was estimated that healthcare costs associated with AD exceeded $345 billion in the United States in 2023, which is twice as much as the cost 10 years ago.1,5 This intensification in healthcare costs is related to the increase in life expectancy and aging, which is the greatest risk factor for AD development. The progression of AD is usually divided into three phases: preclinical AD, mild cognitive impairment due to AD, and dementia due to AD. This disease progresses slowly, resulting in the destruction of neurons, memory impairment, and decreased physical function.5 Decades of research on this neurodegenerative disease have revealed that cognitive decline is caused by a variety of pathological processes, including excessive extracellular aggregation of amyloid-β (Aβ), intracellular neurofibrillary tangles formed by hyperphosphorylation of tau protein, cholinergic dysfunction, excessive glutamatergic stimulation, oxidative stress, and neuroinflammation.6−11
Although AD has a complex pathophysiology, two hypotheses have gained significant evidence: the amyloid cascade hypothesis (ACH)12−14 and the tau hyperphosphorylation hypothesis (THH).15−17 The amyloidogenic pathway results from the sequential cleavage of the amyloid precursor protein (APP) by β-secretase and γ-secretase. β-Secretase cleaves at the N-terminus of APP, while γ-secretase cleaves at its intramembranous domain. The products of β-secretase-mediated cleavage are APP-β and a membrane-bound fragment, which is the target of γ-secretase. The γ-secretase enzyme is responsible for releasing C-terminal fragments and Aβ peptides that contain 38, 40, or 42 amino acid residues.18,19 These Aβ peptides become neurotoxic upon translocation to the extracellular cell and aggregate, progressively forming oligomers, protofibrils, and mature fibrils.20 Some evidence suggest these Aβ aggregates accumulate because the health balance between production and clearance of Aβ is dysregulated in the brain of AD patients.21,22 The THH is based on an imbalance in the degree of tau phosphorylation, which is 3–4 times higher than that found in healthy individuals.23,24 These filaments undergo further aggregation and thickening. In many cases, pathologically hyperphosphorylated tau protein’s ability to bind to tubulin is reduced, leading to a microtubule formation impairment.23,24
Although many efforts are directed toward finding a cure for Alzheimer’s disease, the available treatments have limitations. Five of seven AD treatments approved by the Food and Drug Administration (FDA) can improve symptoms without changing the course of the disease.1 These treatments are based on acetylcholinesterase inhibitors (donepezil, rivastigmine, galantamine), a glutamate receptor antagonist (memantine), and a combination of memantine and donepezil.19 Although these drugs are commonly used in the treatment of Alzheimer’s disease, they cannot interrupt neurodegeneration.19 As a result, numerous clinical trials have been conducted to evaluate potential disease-modifying therapies.25,26
As a result of numerous efforts, two monoclonal antibodies (mAbs) have been approved by the FDA: Aducanumab (Adu)27 and Lecanemab (Lec).28 Adu and Lec were found to have the strongest affinities for Aβ fibrils and protofibrils, respectively.29 These affinities lead to the destabilization of these structures and activate immune system-mediated clearance of Aβ aggregates.29−31 In addition, Donanemab’s clinical trial results recently led to an FDA approval application whose acceptance is expected soon.32 Currently, these drugs are only approved to treat patients with mild cognitive impairment due to AD and mild AD.19 Additionally, the clinical efficacy of Aducanumab lacks solid evidence,33 and the administration of Adu and Lec has resulted in significant occurrences of amyloid-related imaging abnormalities due to edema or effusion, leading to extensive debate in recent years.34−38 Therefore, it is widely agreed that drug discovery remains of major importance in Alzheimer’s disease research.
AD immunotherapy research aims to provide a disease-modifying therapy that improves cognitive functions by clearing aggregates of Aβ and impairing oligomerization and fibrillation in the Aβ cascade.31 Although many anti-Aβ mAbs have failed in clinical trials, Adu and Lec have resulted in a reduced decline in cognitive abilities.26 It is well-established that antibodies directed toward the N-terminus of Aβ elicit superior clinical responses compared to those that bind to central segments or the C-terminus, such as Solanezumab and Crenezumab.39 Compared to other anti-Aβ monoclonal antibodies, Adu has a unique inhibitory mechanism of Aβ aggregation, selectively reducing the secondary nucleation rate of Aβ42 resulting in a significant reduction of Aβ oligomers.31 The FDA’s approval of Adu is a historic landmark, but it has also raised concerns and highlighted the fact that a definitive treatment for Alzheimer’s disease is still far from being developed. Although there are some contradictions, this monoclonal antibody may lead to better cognitive and clinical improvements compared to Donanemab and Lecanemab, which are also Aβ cleaners.40
Aducanumab is a recombinant human immunoglobulin (IgG1) that can cross the blood–brain barrier and promote the clearance of Ab aggregates.41 It has a distinctive selectivity for soluble Ab oligomers and insoluble Aβ fibrils.41 A comparison with gantenerumab, another anti-Aβ mAb, illustrates this distinct selectivity: Adu has Aβ monomer binding affinity that is approximately 100-fold lower than that of gantenerumab, while both antibodies have similar affinities for AB aggregates.42 Clinical trials have shown that Aβ cleaning mediated by Adu occurs in a dose- and time-dependent manner.41 In addition, Adu is the only monoclonal antibody with FDA approval to treat Alzheimer’s disease and a resolved and published three-dimensional structure (see Figure 1).26,42 The elucidated cocrystal structure demonstrates that Adu interacts with the N-terminus of Aβ, which adopts an extended conformation during binding.42 Based on structural and biochemical analyses, the determinant regions responsible for recognizing the epitope formed by Aβ amino acids 3 to 7 are HCDR2, HCDR3, and LCDR1,42 see Figure 1.
Figure 1.
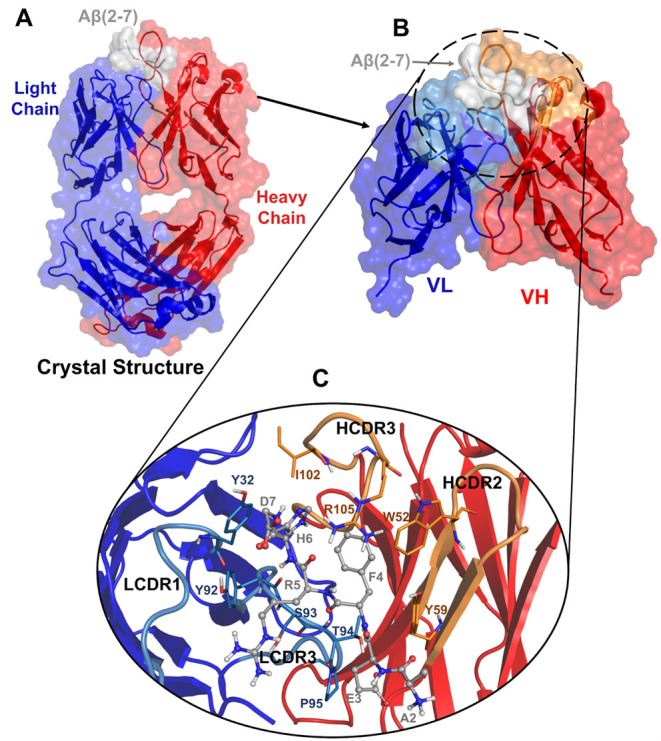
Overview of Aducanumab::Aβ2–7 crystallographic structure with HC’s missing residues modeled. (A) Crystallographic structure of Aducanumab::Aβ2–7 represented in the cartoon with HC, LC, and Aβ2–7 colored in red, blue, and gray. (B) Portion subjected to molecular dynamics simulations corresponding to the variable fragment heavy chain (VH) and variable fragment light chain (VL) bound to Aβ2–7. (C) Illustration of the main contacts on the Aducanumab::Aβ2–7 surface.
From a pharmaceutical viewpoint, it is critical to understand the atomic interactions that are responsible for positive clinical responses. This comprehension is essential for the development and discovery of drugs that can eliminate Aβ aggregates and prevent oligomer/fibril formation. Therefore, designing such a pharmaceutical drug necessitates comprehensive knowledge not only of the Adu::Aβ structure but also of its binding mechanism including dynamics and affinity characterization. The structural data of anti-Aβ mAbs has provided useful information in AD research.42−45 Some studies have outlined mAb::Aβ complexes through molecular dynamics (MD) simulations to gain new insights into the binding mechanism.46−48 On the other hand, quantum biochemistry is a powerful computational method that has been used to study biological complexes49 related to various diseases, including schizophrenia,50 cancer,51,52 arterial hypertension,53 and COVID-19.54 Thus, the implementation of quantum biochemistry coupled with MD is a reliable approach to describe the surface interactions of Alzheimer’s drugs as Aducanumab, the only anti-Aβ drug with both FDA approval and a solved three-dimensional structure.
This work aims to provide a detailed molecular-level description of the surface interactions between Aducanumab and Aβ2–7 (Figure 1). In addition, we investigate mutations that could enhance Adu’s binding affinity to this epitope, and also we design synthetic peptides with a mechanism of action similar to Aducanumab, which can be cheaply synthesized and improved. To accomplish this, we employ computational techniques based on molecular mechanics and quantum mechanics. To the best of our knowledge, this is the first attempt to apply quantum mechanics to study/design drugs in the realm of Alzheimer’s disease immunotherapy treatment. In particular, the computational results allowed the design of mimetic peptides that have potential use in the treatment of Alzheimer’s disease.
Results and Discussion
Screening of Mutations
A set of 23 Adu’s amino acid mutations with potential to increase the affinity between aducanumab and Aβ2–7 was initially obtained. Most of them are LC’s mutations, such as LC-SER91 and LC-SER30, which had the highest numbers of potential single amino acid substitutions identified (Table 1). However, only the LC-S91Y mutation presented three favorable predictive measurements of ΔΔGBind, ΔΔGAffinity, and ΔΔGStability using different methods (Table 1). This initial screening provides a rationale for selecting S91Y as the most promising mutation based on two suspected increases in Adu::Aβ2–7 binding affinity and an indication of improvement in complex stability. Since previous reports have shown that antibody mutations can improve affinity and specificity for epitopes,55,56 this screening was conducted with the aim of identifying potential mutations that could enhance the therapeutic properties of Aducanumab. Thus, the LC-S91Y mutation was inserted into the complex Adu::Aβ2–7 to perform a comparison between wild and mutated complexes in terms of structural stability and binding affinity data using robust computational techniques.
Table 1. Screening of Potential Mutations in Aducanumab’s Chainsa.
| LC mutationb | ΔΔGbindb (kcal/mol) | ΔΔGaffinityc (kcal/mol) | ΔΔGstabilityd (kcal/mol) | HC mutationb | ΔΔGbindb (kcal/mol) | ΔΔGaffinityc (kcal/mol) | ΔΔGstabilityd (kcal/mol) |
|---|---|---|---|---|---|---|---|
| S91W | –0.43 | 0.39 | 0.25 | R105H | –0.2 | 0.95 | 0.03 |
| S91Y | –0.29 | –1.85 | 0.46 | G107F | –0.14 | 0.01 | 0.05 |
| S30W | –0.25 | 0.83 | –0.12 | P108W | –0.12 | 0.76 | –0.21 |
| S91F | –0.25 | 0.18 | 0.46 | P108M | –0.11 | 0.73 | –0.71 |
| S30G | –0.19 | 0.81 | –0.32 | R105F | –0.09 | 0.87 | –0.09 |
| S91I | –0.19 | 0.85 | 0.54 | G107L | –0.09 | 0.04 | 0.37 |
| T94W | –0.14 | 0.49 | 0.03 | R105L | –0.06 | 0.85 | –0.05 |
| Y32W | –0.12 | 0.95 | –0.25 | P108Y | –0.04 | –0.27 | –0.23 |
| S93M | –0.1 | 1.16 | –0.26 | ||||
| S91V | –0.09 | 0.61 | 0.14 | ||||
| S30F | –0.07 | 0.88 | –0.18 | ||||
| S91H | –0.07 | –0.24 | –0.11 | ||||
| S30Y | –0.06 | 0.96 | –0.01 | ||||
| S30H | –0.02 | 0.93 | –0.46 | ||||
| S93L | –0.02 | 1.16 | –0.2 |
The only mutation with three predictive adequate parameters (and its parameters) are underlined.
List of HC and LC mutations provided by BEATMUSIC 1.0 (http://babylone.ulb.ac.be/beatmusic/index.php) with its respectively increase in the affinity measured in kcal/mol.
A second measurement of binding affinity variation (in kcal/mol) assessed in MutaBind2 (https://lilab.jysw.suda.edu.cn/research/mutabind2/research/mutabind2/).
The structural stability variation (in kcal/mol) resultant from mutation calculated by DeepDDG (https://protein.org.cn/ddg.html).
Structural Variations and Fluctuations of Aducanumab::Aβ2–7 Complex
For the simulation time interval of 5–100 ns in the short molecular dynamics, the average root-mean-square deviation (RMSD) fluctuations of all heavy atoms (c-α-ΔRMSD) were 0.83 (0.95 Å) and 0.91 (1.07 Å) for the wild and mutated complexes, respectively (Figure 2). The extended simulations also demonstrated that the mutated complex exhibited the highest ΔRMSD, with fluctuations slightly exceeding those observed in the short MD. The ΔRMSD values for these complexes, calculated using all heavy atoms (C-α), were 0.94 (1.05) and 1.17 (1.24) (Figure 2). The flexibility of aducanumab residues measured by root-mean-square fluctuations (RMSF) values revealed that this structure is predominantly rigid with RMSF < 1 Å, with minor segments having RMSF near 2 Å (Figure 3A,B). The Aβ2–7 residues showed a similar profile, with AβALA2 as the only residue with RMSF near 2 Å in multiple replicas (Figure 3C). The only difference detected between wild and mutated Aducanumab chains was a minor increase in the level of fluctuations of residues close to that of LCTYR91 (Figure 3E).
Figure 2.

RMSD values vs simulation time. RMSD values of the wild complex within short MDs and measurements based on (A) all heavy atoms and (B) C-α atoms. The RMSD variation of mutated aducanumab::Aβ2–7 for (C) all heavy atoms and (D) C-α atoms in short MDs. The RMSD measurements considered (E) all heavy atoms and (F) C-α atoms in long MDs.
Figure 3.

RMSF values calculated from short (MD1-MD6) and long (LMD-Wild and LMD-Mut) simulations of (A–C) wild and (D–F) mutated aducanumab::Aβ2–7 based on C-α atoms.
Moreover, when compared to the wild complex, Aβ2–7 bound to mutated Adu showed higher fluctuations (Figure 3F). In comparison to the fluctuations observed in the Aβ2–7 fragment bound to the wild form of Adu (Figure 3C), the fluctuations in the entire Aβ2–7 fragment bound to the mutated form of Adu (Figure 3F) were found to be higher. Both short molecular dynamics replicas and long molecular dynamics (LMD) corroborated this structural hallmark (Figure 3). This instability occurs mainly in AβALA2, AβGLU3, and AβPHE4 which showed RMSF > 2 Å in simulation MD6 (Figure 3F). However, a similar pattern was also observed in MD5 and the LMD of the mutated complex (LMD-Mut; Figure 3F). Interestingly, the comparison of RMSF measurements from 1 μs LMD simulations also revealed the highest structural fluctuations of the portion AβARG5-AβASP7 of Aβ2–7 bound to the mutated Adu (Figure 3).
The AβGLU3-AβHIS6 portion has RMSF values between 1 and 2 Å in the simulations of the unbound Aβ2–7 fragment (Figure S1), while the same portion has only values below 1 Å when bound to wild Adu (Figure 3C) and few values above 1 Å in the mutated complex (Figure 3F), confirming that this portion is critical for maintaining Aβ2–7 trapped on the aducanumab surface, as previously reported.42 For more information about the structural stability of unbound Aβ2–7 during simulations, see Figures S1 and S2. Moreover, AβARG5 showed a distinct fluctuation pattern: the high fluctuation level in the side chain, while the main chain was rigid (Figure S3).
It was found that LC has more hydrogen bonds with Aβ2–7 than HC in both short and long simulations (Figures S4 and S5). AβGLU3 and AβHIS6 were the residues with the most conserved hydrogen bond contacts with HC (Figure 4), while AβPHE4 and AβHIS6 were predominantly responsible for these contacts with LC (Figure 4). Compared with simulations of the wild complex, the MD5′s and MD6′s hydrogen bond profiles (performed by Aβ2–7 during the simulations) showed that AβALA2 stopped interacting with LC and started establishing strong interactions with HC (Figure 4). LMD simulations demonstrated that this phenomenon is a characteristic feature of mutated complex dynamics, occurring in a cyclic manner. Specifically, AβALA2 forms and subsequently loses hydrogen bonds with LC, before reestablishing them at a later point in time (Figure 4). Moreover, the hydrogen bonds AβPHE4::LC and AβHIS6::LC were not conserved in MD6 after 20 ns (Figure 4). This dynamics profile suggests that the ability to maintain Aβ2–7 trapped and stable within the Aducanumab’s paratope was affected by the single amino acid substitution S91Y in the Aducanumab’s light chain. Despite these changes resulting from LC-S91Y, the mutated system remains as a stable complex and does not undergo extensive conformational changes and has the potential for spontaneous occurrence, as suggested by both its average RMSD values below 2 Å and the ΔRMSD of the order of 1 Å (Figure 2).
Figure 4.

Hydrogen bond profile of the Aβ2–7 residues with the aducanumab (A–H) heavy chain and (I–P) light chain over the entire period of the simulations. Data of simulations (A, I) MD1, (B, J) MD2, (C, K) MD3, (D, L) LMD-Wild, (E, M) MD4, (F, N) MD5, (G, O) MD6, and (H, P) LMD-Mut are represented.
In contrast to the previous work, which performed simulations for complexes containing Aβ2–7, Aβ monomer, and Aβ oligomers,47 our efforts were focused on the crystallographic structure of Aducanumab::Aβ2–7. This Aβ fragment contains the Adu epitope Aβ3–7,42 which makes it a valuable structure for understanding surface interactions. The simulations demonstrated that Aducanumab undergoes minor conformational changes, which is consistent with the findings of Arndt et al. Differences in structural stability between Adu::Aβ2–7 and Mut-Adu::Aβ2–7 complexes can be inferred from the differences between their RMSD and RMSF values. The LC-S91Y mutation appears to cause a slight change in the structural stability of Adu::Aβ2–7. It is worth noting that some of the RMSF values obtained for the wild complex differ slightly from those reported by Frost and Zacharias.
Most simulations indicate that Aβ2–7 is rigid, but one simulation (MD6) of the mutated complex showed a high displacement of AβALA2-AβPHE4, similar to the lack of stabilization in the N-terminus of Aβ2–7 of the crystallographic structure previously reported through MD simulation.47 Although the computational results did not reveal high RMSF values for the wild complex, a 500 ns simulation time of wild Adu::Aβ2–7 previously reported that this displacement in the N-terminus can be part of the recognition of Adu::Aβ2–7.47 In view of this, performing this process in a faster mode can be an interesting feature, as is the case here with the mutated Adu. This modified antibody may potentially offer a novel binding way with the potential to disrupt Aβ aggregates, as previously proposed by distinct theoretical studies that evaluated mutations in proteins.57−59 The divergence between molecular dynamics conducted at varying time scales reiterates that simulation time significantly influences structural stability measurements. Additionally, the force field utilized by Frost and Zacarias differed from the one applied herein, which helped to elucidate why some structural behaviors observed here differed from those reported by them. Nevertheless, the structural descriptors RMSD and RMSF can suggest a minor propensity for Mut-Adu::Aβ2–7 to occur spontaneously, and additional assays are required to determine if LC-S91Y affects the stability and binding affinity of Aβ2–7. It is crucial to acknowledge that these structural descriptors (RMSD and RMSF) were employed to examine the LC-S91Y impact, drawing upon prior studies that effectively utilized theoretical methodologies in disparate biological systems.60,61
Conformational Ensembles and Description of Nonbonded Interactions
The conformational ensembles under scrutiny consist of the final conformations of the wild complex (Wild-FC) named MD1, MD2, MD3, and LMD-Wild, and the final conformations of the mutated complex (Mut-FC) are named MD4, MD5, MD6, and LMD-Mutated (Figure 5A). Additionally, the representative conformations (RC) of the wild-type (MD2/Wild-RC) and mutated (MD6/Mut-RC) complexes were enumerated as #0–#8 (Figure 5B) and #0–#7 (Figure 5C), respectively. Among the simulations of the wild complex, MD2 exhibited the highest RMSD fluctuation, while MD6 showed the highest fluctuation among the mutated complex simulations (Figure 2). As a consequence (and aiming a high conformational flexibility between the representative conformations), the trajectories of these two simulations were used to generate the RC above-mentioned.
Figure 5.

Structural alignment of conformational groups named the (A) final conformations (FC), (B) MD2 (Wild) ensemble, and (C) MD6 (Mut) ensemble.
Compared to the crystallographic conformation, most of the final MD conformations showed a dislocation of AβALA2 directed toward LC (Figure 5A), confirming the hydrogen bond profile that shows a strong interaction between AβALA2 and LC beginning before 20 ns in MD1, MD2, MD3, MD4 and LMD-Wild (Figure 4). However, MD6 and Mut-RC revealed a distinct behavior where AβALA2-AβPHE4 dislocated from LC to HC (Figure 5A–C).
The conformational ensemble structures have RMSD values smaller than 1.2 Å from each other, including those provided by LMD (Figure 6). This structural similarity suggests that short molecular dynamics may be an appropriate approach to obtain Adu::Aβ2–7 binding states with energetic convergence. Although these RMSD values indicate high similarity, some conformations exhibit exclusive nonbonded interactions that are absent in others. For example, there are LCSER93(OG)::AβALA2(N), LCTYR92(OH)::AβARG5(NH1), LCTYR92(OH)::AβARG5(NH2), LCTYR92(OH):: AβASP7(OD2), HCLYS57(NZ)::AβGLU3(OE1), and LCGLN27(OE1)::AβARG5(NH1) hydrogen bonds (Tables S1–S3). The high number of hydrophobic and hydrogen bond contacts conserved among many conformations confirms the rigidity of the Adu::Aβ2–7 complex (Tables S1–S3). To name a few, the hydrophobic interactions HCTRP52::AβPHE4, HCLYS65::GLU3, HCILE102::AβHIS6, HCPRO108::AβHIS6, LCTYR92::AβARG5, and LCSER93::AβPHE4 were detected at least 15 of the 23 Adu::Aβ2–7 conformations. Noteworthy, the hydrophobic interaction HCLYS65::AβGLU3 was not detected within MD6(Mut)-ensemble, suggesting that LC-S91Y directly affects this contact (Tables S1–S3). This mutation also resulted in the loss of three conserved hydrogen bonds: LCTYR92(O)::AβHIS6(N), LCTHR94(OG1)::AβPHE4(N), and LCTHR94(N)::AβPHE4(O) (Tables S1–S3). For more details about surface interactions about these interactions, see Figures S6–S9.
Figure 6.
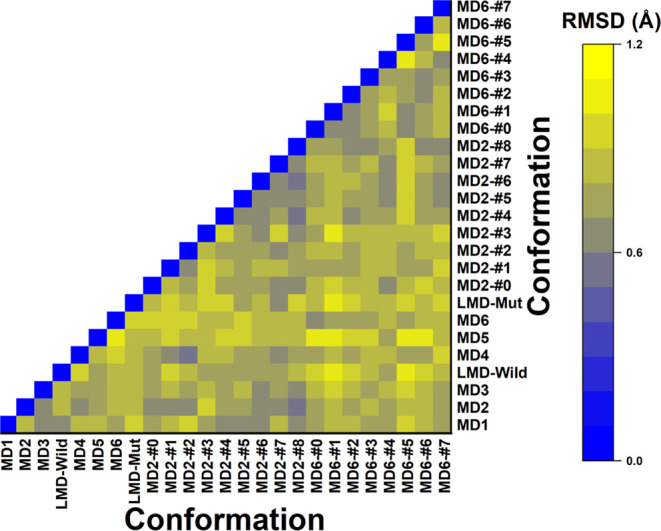
Heatmap of RMSD values between Aducanumab::Aβ2–7 conformations. The caption for the correlation of colors and RMSD values is on the right panel of the graph.
Most of the hydrophobic and hydrogen bond interactions detected here are in accordance with those previously reported by Arndt et al. This abundance of conserved interactions is also consistent with the small differences between conformations with all RMSDs measured within 1.20 Å. However, the conformational ensemble revealed some transient hydrogen bonds that were not previously reported, such as HCARG105::AβASP7, HCASP1::AβALA2, LCGLN27::AβARG5, and LCTYR92:: AβARG5. In agreement with previous reports, this clearly reveals the significance of employing a conformational ensemble approach within the context of molecular dynamics, even for rigid proteins.62
The Aducanumab::Aβ2–7 Quantum Biochemistry
The close contacts (4 Å) between HC and Aβ2–7 are primarily mediated by HCLYS65, HCTRP52, HCPRO108, HCTYR59, and HCARG105, while the LC::Aβ2–7 close contacts (4 Å) are mainly mediated by LCASP1 and the LCDR3 residues LCSER93, LCTHR94, LCPRO95, and LCTYR92 (Figure 7A–D). This suggests that Adu’s heavy chain has more interaction points than its light chain, as discussed previously. Quantum biochemistry calculations also provide more definitive data on the differences between wild and mutated complexes, in terms of the binding affinities between Aducanumab and Aβ2–7. All values that follow are averages of the interaction energies. The interaction energies between HC and Aβ2–7 were −62.42 and −62.65 kcal·mol–1 in wild (MD1-LMD-Wild) and mutated (MD4-LMD-Mut) FC, respectively (Figure 7A,C). The affinity of the RC increased slightly from −48.88 kcal·mol–1 in the wild ensemble to −49.87 kcal·mol–1 in the mutated ensemble (Figure 7E,G). The affinity of LC::Aβ2–7 in wild and mutated FC (RC) were −69.26 (−53.57) and −65.33 (−37.07) kcal·mol–1 (Figure 7B,D,F,H), respectively. Additionally, the FC indicates that the total interaction energy (sum of HC and LC affinities) was not greatly affected by LC-S91Y, while the RC showed a decrease in the total affinity between Adu and Aβ2–7. It is noteworthy that the LMD simulations indicated a tendency toward a reduction in the binding affinity between HC and Aβ2–7 on the mutated complex (Figure 7A–D), which corroborates the findings of crystallographic reports that identified L3 as the most crucial LCDR in maintaining the capture of Aβ2–7, because its interaction was not significantly affected.42 In comparison to the short MD, the long MD demonstrated a significant impact on the affinity of the mutated complex, emphasizing the necessity of long simulations for the validation of mutations in biological systems.
Figure 7.

Sum of individual interaction energies involving the Aducanumab-residue::Aβ2–7-residue contacts up to 8 Å. The profiles of interaction energy of (A) Aducanumab-HC(Wild):: Aβ2–7, (B) Aducanumab-LC(Wild):: Aβ2–7, (C) Aducanumab-HC(Mut)::Aβ2–7, and (D) Aducanumab-LC(Mut):: Aβ2–7 are based on the ensemble of final conformations from MD. The same description of these subsystems (Aducanumab-HC (Wild)/Aducanumab-LC (Wild)/Aducanumab-HC (Mut)/Aducanumab-LC (Mut)::Aβ2–7) considering RC are represented in (E–H), respectively. The residues near lines (A–D) make most of the surface contacts.
The total interaction energies between Adu and Aβ2–7 from their multiple conformations showed significant differences. The convergence of total interaction energies was more successfully achieved for LC::Aβ2–7 than for HC::Aβ2–7 which can be seen by some slight repulsive interactions after 6 Å (Figure 7). Within wild-FC, the difference between MD1 and MD3 interaction energies, the conformations of highest and lowest affinities, was 34.5 kcal·mol–1 which represents ∼28.5% of MD3 affinity (Figure 7A,B). The highest difference found in wild conformations from RC ensemble was −34.00 kcal·mol–1, which represents an increase in ∼40% of the #8 conformation’ affinity (Figure 7E,F). The respective differences found in mutated ensembles from FC (MD4-LMD-Mut) and RC (#0–#7) were: −45.31 kcal·mol–1 (Figure 7C,D) and −23.21 kcal·mol–1 (Figure 7G,H). These differences represent approximately 43.2% and 30.8% of the weaker binding affinity calculated. Moreover, compared to the mutated ensembles, the distributions of total interaction energy values of wild ensembles are more uniform (Figure S10). Additional details concerning the quantum biochemistry calculations performed for each AduResidue::AβResidue studied here are described in Tables S4–S28.
The LC was identified as the most attractive chain for the epitope based on the interaction energies obtained through quantum mechanical principles. This finding corroborates the earlier experimental reports published by Arndt et al., which demonstrated that this chain was responsible for four of the seven hydrogen bonds between Aducanumab and Aβ2–7 detected in the crystallographic solved structure. In contrast, the HC atom established three hydrogen bonds. These outcomes highlight the reliability of quantum biochemistry in the description of biological structural systems.
The role of water molecules in the binding affinity between proteins and distinct molecules63,64 is of critical importance. However, the high cost of computational resources associated with quantum biochemistry has hindered the implementation of explicit water on DFT calculations directed to RC, due to the large number of conformations involved (Figure 7). A comparison of distinct DFT calculations (explicit water/COSMO model) directed only to FC MD1, MD2, MD3, MD4, MD5, and MD6 was also performed to assess the impact of explicit water on the measurement of the interaction energy. The comparison revealed that all interaction energies were lower when explicit water was incorporated into the DFT calculations, indicating that the actual binding affinities of RC are undoubtedly more attractive than those calculated using the COSMO solvation model (Figure 7). This finding is consistent with previous reports emphasizing the crucial role of water networks in protein interactions.63,64 Please refer to Figure S11 for further details.
Aducanumab::Aβ2–7 Energetic Hot Spots and Multiple Conformations
The quantum biochemistry identified three energetic hot spots, H2, H3, and L3, corresponding to complementarity determinant regions HCDR2, HCDR3, and LCDR3, which account for approximately 77% of the total affinity between Adu and Aβ2–7 (Figure 8). This proportion may exceed 80% depending on the structural conformation. HCLYS65, HCARG105, HCTYR59, and HCPRO108 were Aducanumab’s residues predominantly responsible by the HC affinity for Aβ2–7 in FC (RC) with interaction energies of −14.36 (−10.43), −10.95 (−10.82), −10.04 (−7.51), and −7.19 (−6.32) kcal·mol–1 (Figure 8A). Moreover, residues LCTYR92, LCTHR94, LCSER93 and LCASP1 are the most critical to stabilize the Aβ2–7 trapped next to LC in FC (RC) presenting interaction energies of −17.77 (−16.08), −15.08 (−13.18), −13.42 (−9.86), and −9.35 (−4.51) kcal·mol–1 (Figure 8C). For detailed information regarding the quantum biochemistry description of representative conformations, see Table S29.
Figure 8.

Quantum description of energetic hot spots based on final conformations. The energetic profiles of (A, B) HC and (C, D) LC from wild complex and mutated complexes are located at the top and bottom panels, respectively. The residues (of Aβ2–7) colored gray next to the bars are the most critical for each contact.
The observed differences in affinity between wild and mutated complexes can be attributed directly to the three hot spot segments, H2, H3, and L3. The Aducanumab in the wild-FC exhibits an average interaction energy of −17.92 (−29.56) kcal·mol–1 between H2 (H3) and Aβ2–7 (Figure 8A). This value decreased (increased) to −25.15 (−24.69) kcal·mol–1 (Figure 8A,B). The residues mainly responsible for this difference are HCTRP52, HCLYS57, and HCARG105 (Figure 8A,B). The L3 experienced a slight increase in its interaction energy from −50.26 to −49.78 kcal·mol–1 (Figure 8C,D). As predicted in Table 1, the local binding affinity at position 91 improved, with LCSER91 and LCTYR91 having −1.08 and −1.45 kcal·mol–1, respectively (Figure 8C,D). Additionally, LCTYR92 exhibited an enhanced affinity for Aβ2–7, particularly at the conformation extracted from long molecular dynamics (Figure 8C,D). However, compared to the wild complex, nearby residues LCGLN90, LCSER93, LCTHR94, LCPRO95, and LCLEU96 showed decreased affinity for Aβ2–7 in the mutated complex (Figure 8C,D).
One conceivable rationale for this diminished affinity stemming from the substitution of serine with tyrosine is the divergence in the physicochemical attributes of these amino acids. Serine is characterized by a shorter and predominantly hydrophilic side chain (slightly more polar than tyrosine), whereas tyrosine possesses a longer side chain comprising a phenol, which predisposes it to engage in hydrophobic and hydrophilic interactions. Given that the affinity of Aβ2–7 for TYR91 was marginally higher than that for SER91, one potential explanation is that the LC-S91Y mutation may have facilitated novel intramolecular interactions with proximate residues, thereby creating steric hindrance for the Aβ2–7::LC interaction. This could account for the diminished affinity of the L3 region for Aβ2–7.
The AβGLU3-AβPHE4 and AβHIS6-AβASP7 are the hot spots of highest affinities for HC in both wild and mutated complexes (Figure 9). In the wild complex, H3 residues are responsible for the main contacts with AβHIS6-AβASP7, while both H3 and H2 are crucial for the contacts with AβGLU3-AβPHE4. HCLYS65 also makes a critical contribution to the affinity of HC for AβGLU3. Moreover, the AβPHE4-AβHIS6 segment is mainly responsible for the Aβ2–7 affinity for wild-LC, while AβARG5-AβASP7 is the most critical segment for stabilizing the interaction with mutated LC, as shown in Figure 9H. The L3 residues LCTYR92-LCTHR94, with the exception of LCASP1::AβARG5, LCGLN27::AβARG5, and LCTYR32::AβHIS6, are primarily responsible for the energetic contacts between AβPHE4-AβARG5 and wild-LC (Figure 9E). In the mutated complex, LCASP1::AβARG5 and LCGLN27::AβARG5 had minor critical energetic contacts, while new contacts between LCSER30/LCTYR32/LCTYR92 and AβASP7 were energetically important (Figure 9F). According to the RC outlined by quantum biochemistry, the mutated complex showed a decreased affinity for HC in AβALA2 and AβGLU3, while AβHIS6 and AβASP7 presented increased affinity (Figure 9). In comparison to the wild complex, all residues except for AβASP7, showed a decrease in affinity for mutated LC (Figure 9G,H). The mutation LC-S91Y had a deeper impact on AβGLU3 and AβPHE4, as evidenced by the fluctuations in the values of interaction energies within the mutated ensemble (Figure 9B,D,F,H).
Figure 9.

Affinity of Aβ2–7 amino acid residues for (A–D) HC Aducanumab in (A, B) final conformations and (C, D) representative conformations, and for (E–H) LC in (E, F) FC and (G, H) RC. The depicted figure illustrates the interaction energy of Aβ2–7 residues for (A, C) Wild-HC, (E, G) Wild-LC, (B, D) Mut-HC, and (F, H) Mut-LC. The residues (of Aducanumab) in gray next to the bars are the most critical for each contact.
The calculated interaction energies suggest that the LC chain is the most crucial in stabilizing the binding of Aducanumab to the Aβ2–7 surface and the crystallographic study supports this finding,42 as above-discussed. The Adu residues responsible for this binding affinity, in decreasing order of affinity, are as follows: LCTYR92, LCTHR94, LCSER93, HCLYS65, HCARG105, HCTYR59, HCPRO108, and LCASP1. In the mutated complex, the heavy chain plays the main role in binding affinity, causing a change in the order of the most attractive residues to LCTYR92 > HCR105 > HCTYR59 > LCTYR32 > LCSER93 > HCLYS65 > LCTHR94 > HCPRO108. Except for LCASP1, LCTYR32, and HCLYS65, the aforementioned critical residues are part of the groups H2 (HCTYR59), H3 (HCR105 and HCPRO108), and L3 (LCTYR92, LCSER93, and LCTHR94). This result is in accordance with previous structural analysis based on X-ray data, which identified these three complementarity determining regions as essential for confining Aβ2–7 on surface contact.42 It is noteworthy that significant fluctuations in affinities account for up to 40% of the total interaction energy, even when all RMSD values between the Adu::Aβ2–7 conformations analyzed here are lower than 1.20 Å. This emphasizes the importance of outlining the flexibility of this complex using multiple binding conformations.
The quantum mechanics calculations performed here reveal that AβGLU3-AβPHE4 and AβHIS6-AβASP7 are the main contacts with HC, while the binding affinity for LC is mainly mediated by AβPHE4, AβARG5, and AβHIS6. These residues are also responsible for the majority of the conserved hydrophobic, salt bridge, and hydrogen bond interactions through multiple Adu::Aβ2–7 binding conformations. The computational results are in agreement with previous reports that identified AβPHE4 and AβHIS6 as the epitope core42 and the most energetic residues,47 respectively, followed by AβARG5, AβGLU3, AβASP7, and AβALA2.47 The mutation LC-S91Y resulted in structural instability of Aβ2–7, leading to decreased affinities of AβALA2/AβGLU3::HC and AβALA2/AβGLU3/AβPHE4/AβARG5/AβHIS6::LC, and an increased AβHIS6/AβASP7::HC affinity. Although this mutation did not result in an increased affinity, as predicted in the Screening of Mutations section, our computational outcomes suggest a new binding mechanism that can effectively clear Aβ. One conceivable explanation for this diminished affinity is the discrepancy in the physical and chemical characteristics of SER and TYR. The longer side chain of TYR enables greater hydrophobic and polar interactions, which may explain the higher local affinity between Aβ2–7 and LCTYR91 in comparison to LCSER91 and Aβ2–7. However, the new interactions mediated by the long LCTYR91 side chain may also create steric hindrances, which could contribute to the observed decrease in the global affinity of the mutant LC by Aβ2–7.
Compared to the results of Arndt et al., the quantum mechanical analysis revealed a new critical contact mediated by HCLYS65 with an interaction energy of −10.45 kcal·mol–1 based on Wild-RC. The binding affinity measurements showed the following affinity order for Aβ2–7: L3 > H3 > H2 > L1. This is also in agreement with the crystallographic study, which identified LCDR3 as the primary point of Aβ confinement and L1 as a minor contributor.42 The L1 is here represented by LCTYR32 with an interaction energy of −4.94 kcal·mol–1, based on DFT calculations directed toward the Wild-RC.
Design of Mimetic Peptides
The design of mimetic peptides was based on the insights provided by quantum biochemistry calculations, which revealed that three complementarity determining regions are responsible for ∼77% of the affinity of Adu for its epitope. A linear and a cyclic peptide were designed (Figure 10) to mimic the binding capacity of each of the energetic hot spots detected on the chains of aducanumab using a similar approach previously established by Amaral et al.52 The proposed molecules are also based on a substantial body of literature on antibody-like mimetic peptides with proven biological activities and advantageous characteristics.65 Given that all the peptides proposed here are less than 2 kDa, their production costs are considerably lower than those associated with a full monoclonal antibody, which is approximately 150 kDa.66 Moreover, previous research has demonstrated that peptides elicit fewer immunogenic effects and are less toxic than monoclonal antibodies due to their reduced size.66,67
Figure 10.

Three-dimensional representation of mimetic peptides. The (A–D) linear and (E–H) cyclic peptides are shown. Cysteine residues have been added to the C- and N-terminus to make the peptides cyclic and are indicated by ‘’. Dark lime green residues refer to the mutation added to LCDR3. (I–L) The alignments of multiple predicted structures for each linear peptide obtained by PEP-FOLD3 are also shown, where only the green peptide represents the structure directly removed from Aducanumab. Peptides with red and blue names are derived from HC and LC, respectively.
Despite the slightly reduced affinity of the mutant LC for Aβ2–7, the perturbations resulting from this mutation led us to design two additional mimetic peptides, a linear (Figure 10D) and a cyclic (Figure 10H), with a potentially different mechanism.
The linear mimetic peptides based on the hot spots H2, H3, L3, and mutant L3 were named PEP-H2-L, PEP-H3-L, PEP-L3-L1, and PEP-L3-L2, respectively (Figure 10). These peptides correspond to VAL50 to TYR60, GLY101 to TYR109, GLN90 to LEU96 with SER91, and GLN90 to LEU96 with TYR91. The cyclic peptides are composed of the same amino acid residue with the addition of one N- and one C-terminal cysteine, forming a disulfide bond (Figure 10). Except for PEP-H3-L, the predicted structures of linear peptides indicate that these molecules tend to maintain their secondary structures in a conformation very similar to that of their corresponding CDRs on Aducanumab (Figure 10I–L). The amino acid sequences of these peptides and predictions of their physical and chemical properties are indicated in Table 2. The solubility predictions indicate that linear peptides are more soluble than their derivative cyclic peptides, which may be a feature to consider in drug development (Table 2 and Figure S12).
Table 2. Physical and Chemical Properties of Mimetic Peptides.
| peptide | sequence | pIa | molecular weighta | Tm indexb | intrinsic solubility scorec | net charged |
|---|---|---|---|---|---|---|
| Pep-H2-L | VIWFDGTKKYY | 8.40 | 1419.64 | –5.15 | 0.42 | +1 |
| Pep-H2-C | CVIWFDGTKKYYC | 8.02 | 1625.92 | –4.15 | –0.18 | +1 |
| Pep-H3-L | GIGARRGPY | 10.84 | 946.08 | 1.15 | 2.10 | +2 |
| Pep-H3-C | CGIGARRGPYC | 8.96 | 1152.35 | 2.62 | 1.70 | +2 |
| Pep-L3-L1 | QSYSTPL | 5.52 | 794.86 | –6.23 | 1.75 | 0 |
| Pep-L3-C1 | CQSYSTPLC | 5.51 | 1001.14 | –5.72 | 1.06 | 0 |
| Pep-L3-L2 | QYYSTPL | 5.52 | 870.96 | –6.00 | 1.09 | 0 |
| Pep-L3-C2 | CQYYSTPLC | 5.51 | 1077.23 | –5.53 | 0.25 | 0 |
Isoelectric point and molecular mass calculations of peptide sequences using ProtParam (web.expasy.org/protparam/).
Tm index calculated using Tm Predictor (tm.life.nthu.edu.tw/)Tm index >1 indicates Tm above 65 °C; Tm index <0 indicates Tm less than 55 °C; 0 < Tm index <1 represents a Tm value between 55 and 65 °C.
Calculated solubility score on the Chemistry of Health server (www-cohsoftware.ch.cam.ac.uk/index.php/camsolintrinsic) where score >1 indicates good solubility while score <−1 indicates low levels of solubility.
Net charge of peptides was calculated using the Antimicrobial Peptide Calculator and Predictor (aps.unmc.edu/prediction).
The computational predictions of these mimetic peptides showed that those derived from aducanumab’s HC are potentially more stable than the LC derivatives in an intestinal-like environment (Table 3). Predictions of allergenicity, hemolytic, and toxicity potentials showed that all designed peptides are potentially safe in these regards (Table 3). Since Aβ aggregates, the potential target of these molecules, accumulate in the brain of AD patients,14 the potential ability to cross the blood–brain barrier (BBB) was also evaluated, with positive predictions for all peptides (Table 3). Although the peptides have a lower number of cleavage sites susceptible to proteolysis by digestive enzymes than aducanumab, most of them have at least one cleavage site susceptible to the action of chymotrypsin, pepsin, and trypsin (Table 4). The exceptions are PEP-H3-L and PEP-H3-C, which are uniquely resistant to the action of pepsin (Table 4).
Table 3. In Silico Characterization of Features with Biological Importance.
| peptide | half-lifea | stabilitya | antigenic determinantsb | hemolytic potentialc | toxin predictiond | BBB prediction (score)e |
|---|---|---|---|---|---|---|
| Pep-H2-L | 1.91 | high | 0 | nonhemolytic | non-toxic | BBB+ (0.15) |
| Pep-H2-C | 1.75 | high | 0 | nonhemolytic | non-toxic | BBB+ (0.17) |
| Pep-H3-L | 1.08 | high | 0 | nonhemolytic | non-toxic | BBB+ (0.25) |
| Pep-H3-C | 1.18 | high | 0 | nonhemolytic | non-toxic | BBB+ (0.25) |
| Pep-L3-L1 | 0.90 | normal | 0 | nonhemolytic | non-toxic | BBB+ (0.13) |
| Pep-L3-C1 | 0.88 | normal | 0 | nonhemolytic | non-toxic | BBB+ (0.16) |
| Pep-L3-L2 | 0.90 | normal | 0 | nonhemolytic | non-toxic | BBB+ (0.12) |
| Pep-L3-C2 | 0.88 | normal | 0 | nonhemolytic | non-toxic | BBB+ (0.23) |
Prediction of the half-life/stability of the mimetic peptides in the intestine like environment using HLP (https://webs.iiitd.edu.in/raghava/hlp/).
The number of antigenic determinants in each peptide sequence was obtained from Predicting Antigenic Peptides (http://imed.med.ucm.es/Tools/antigenic.pl).
The hemolytic potential of the sequences predicted using HemoPred (http://codes.bio/hemopred/).
The toxicity potential of the peptides was obtained using ToxinPred (http://crdd.osdd.net/raghava/toxinpred/).
The classification of the peptides regarding their potential ability to cross the blood–brain barrier was carried out using the parameters of B3Pred (https://webs.iiitd.edu.in/raghava/b3pred/predict.php).
Table 4. Susceptibility of Mimetic Peptides to the Action of Digestive Proteases.
| peptide | chymotrypsin (high specificity)a | chymotrypsin (low specificity)a | pepsin (pH 1.3)a | pepsin (pH > 2)a | trypsina |
|---|---|---|---|---|---|
| HC-Adu | 20 | 40 | 34 | 54 | 23 |
| LC-Adu | 18 | 37 | 36 | 52 | 18 |
| Pep-H2-L | 4 | 4 | 2 | 4 | 2 |
| Pep-H2-C | 4 | 4 | 2 | 4 | 2 |
| Pep-H3-L | 1 | 1 | 0 | 0 | 2 |
| Pep-H3-C | 1 | 1 | 0 | 0 | 2 |
| Pep-L3-L1 | 1 | 2 | 1 | 3 | 0 |
| Pep-L3-C1 | 1 | 2 | 1 | 3 | 0 |
| Pep-L3-L2 | 2 | 3 | 1 | 4 | 0 |
| Pep-L3-C2 | 2 | 3 | 1 | 4 | 0 |
Number of sites that can be cleaved by each enzyme in each amino acid sequence. These predictions were carried out by ExPASy PeptideCutter (https://web.expasy.org/peptide_cutter/).
Compared to other anti-Aβ mAbs, Aducanumab inhibits the multiplication of aggregates by secondary nucleation of Aβ monomers at the surfaces of existing Aβ fibrils and promotes the removal of oligomers/fibrils via the immune system.31 Its mechanism of action is distinct. However, the use of Aducanumab presents several challenges, including amyloid-related imaging abnormalities (ARIA) caused by edema or effusion, high associated costs, and a limited number of eligible patients.34−37 Since ARIA are employed as an inclusion and exclusion criterion to approve continuous treatment based on monoclonal antibodies,34 it is crucial for the development of new Alzheimer’s disease drugs that have fewer adverse effects and a more favorable benefit-risk profile. As the field of peptide research expands, multiple potential activities are being proposed, including antimicrobial,68,69 anti-inflammatory,70,71 antidiabetic,72,73 and anti-Alzheimer.74,75 The potential anti-Alzheimer peptides that have been previously proposed are typically based on rational design, as exemplified by the bicyclic peptide developed by Ikenoue et al. or extracted from biological sources, such as kefir, by Malta et al. In addition to exhibiting antioxidant and antiacetylcholinesterase properties,75 these peptides demonstrated the capacity to modulate Aβ aggregation (in vitro) and suppress Aβ42 toxicity in Caenorhabditis elegans (in vivo).74
Based on these data and the unique mechanism of action of Aducanumab, it is expected that mimetic peptides based on the interaction energy hot spots H2, H3, and L3, which account for approximately 77% of the Adu affinity of Aβ, have the potential to inhibit this secondary nucleation with reduced cost and low side effects.65,66 In addition to the three mimetic peptides directly extracted from Aducanumab’s chains, a peptide based on mutated L3 was also designed. Four corresponding cyclic peptides were also designed. In addition to the interaction energy, another factor suggesting the potential usability of these peptides is the peptide folding predictions. These predictions revealed that the linear peptides PEP-H2-L, PEP-L3-L1, and PEP-L3-L2 tend to retain their three-dimensional structure, such as that obtained directly from Aducanumab. This is critical because conformational changes have the potential to disrupt the peptide::Aβ interactions.76,77In silico predictions also indicate that all peptide sequences are nonallergenic, nonhemolytic, nontoxic, and BBB+ (positive for crossing the blood–brain barrier). Although all sequences were indicated as potentially susceptible to intestinal proteases, cyclic peptides may be less susceptible to proteases,78 making oral administration a potential option. Moreover, the cyclic peptides that were designed in this study are distinct from those that were previously theoretically obtained by quantum biochemistry. This is due to the fact that cysteines were added to the N- and C-terminal portions of the linear peptides before a conventional end-to-end cyclization method was employed.79
The binding affinity of a drug candidate represents a pivotal parameter in the drug development process, as it has the potential to significantly impact the drug’s efficacy. This is exemplified by the case of certain corticotropin-releasing hormone receptor 1 antagonists.80 This property is typically assessed through a combination of computational and experimental methods.81 This property is typically evaluated through a combination of computational and experimental methods. In this context, and based on the results of quantum analysis, peptides derived from L3 (PEP-L3-L1 and PEP-L3-C1) have been identified as promising candidates. These peptides have been designed to target the most energetic hot spot and, in theoretical terms, are the most promising for forming a peptide::Aβ complex and impairing Aβ aggregation. The HCDR3-based molecules exhibited the most favorable predictive parameters with respect to protease resistance and the capacity to cross the blood–brain barrier. This potential resistance to proteolysis may facilitate the development of orally administered drugs,82 while the ability to cross the BBB is critical to obtaining an anti-Aβ, as its pathological accumulation in Alzheimer’s disease occurs in the brain.14 While these predictions require further experimental validation, they indicate that peptides derived from HCDR2 may be the least promising for further drug development.
Synthetic peptides were previously investigated for their druggability and therapeutic use as antimicrobial agents through in silico analysis.83 These computational results were confirmed by hemolysis and toxicity assays, which validated the reliability of the in silico predictions performed here. A variety of synthetic peptides have demonstrated the ability to bind to Aβ, resulting in modulation or inhibition of Aβ aggregation.74,84−86 Altogether, the quantum mechanical outcomes, in silico predictions, and previous reports of anti-Aβ peptides74,84−86 suggest that it is possible to obtain peptides with a mechanism of action similar to that of Aducanumab. While the synthetic peptides proposed here cannot achieve the typical focus of anti-Aβ immunotherapies, which are based on immune signaling and removal of Aβ aggregates through microglia-mediated phagocytosis, it is believed that they affect the perturbation of the secondary nucleation, mechanism previously linked to the Adu efficacy and specificity.31 Thus, we are confident that these findings suggest that these molecules should have potential applications in the therapy of Alzheimer’s disease. To confirm and measure the peptides’ ability to bind to Aβ and inhibit Aβ fibrilization/aggregation, thioflavin T fluorescence assays,86 and atomic force microscopy74 are being conducted. Peptides containing LC-S91Y will also be evaluated, which may elucidate whether this mutation affects the affinity of LCDR3 for Aβ. Further assays should be performed to confirm the mechanism of action and in vivo efficacy of the peptides.
Conclusions
In summary, only the LC-S91Y mutation was selected for further testing by using more robust computational techniques. Computational outcomes revealed that Aβ2–7 has a minor binding affinity and is structurally less stable when bound to the mutated version of aducanumab. This could suggest a distinct binding mode or simply a minor propensity of Aβ2–7 to remain bound to Adu. Further assays should be conducted to confirm this behavior and to determine whether the affinity really decreases. The results of the classical simulations demonstrated the significance of the LMD in the context of a mutated complex lacking a crystallographic structure. Furthermore, the application of short MD was found to be an effective approach for achieving affinity levels comparable to those obtained through LMD. These findings highlight the potential of short MD simulations as a viable alternative to LMD, offering a more efficient method for studying complex molecular interactions in the study of Adu::Aβ2–7. Quantum biochemistry indicated the presence of three distinct segments (L3, H3, and H2) that could serve as templates for the design of synthetic peptides with a mechanism of action similar to that of Aducanumab. Although a reduction in the affinity of the mutated L3 for Aβ2–7 was observed, this segment was also employed in the design of mimetic peptides for the purpose of comparing the binding capacity of wild-type and mutated L3 derivatives through further experimental procedures. The new insights provided by the computational results indicate that the eight mimetic peptides based on Aducanumab’s energetic hot spots exhibit promising characteristics, suggesting that they may be a promising line of inquiry in further research. In addition, quantum biochemistry and bioinformatic predictive parameters suggest that peptides Pep-H3-L, Pep-H3-C, Pep-L3-L1, and Pep-L3-C1 are the most promising, making them the most appropriate candidates for future in vitro and in vivo testing.
Methods
Preparing Aducanumab::Aβ2–7
The crystallographic structure of Aducanumab::Aβ2–7 (ID 6CO3 - resolution 2.38 Å)42 was obtained from the Protein Data Bank.87 Essential residues that were missing in Aducanumab’s heavy chain (SER139-GLY145) were modeled using SWISSMODEL.88 Playmolecule ProteinPrepare89 carried out the protonation adjustments of the amino acid side chains at pH 7.4. This prepared complex was used for screening mutations (2.2) and molecular dynamics (2.3). For more details about this modeled structure and main surface stabilizing contacts, see Figure 1.
Screening of Mutations
The first step in identifying potential mutations that could increase the affinity of Adu for Aβ2–7 was to conduct an initial screening of potential mutations in the heavy chain (HC) and light chain (LC) using BeAtMuSiC 1.0.90 The most promising mutations, according to BeAtMuSiC energetic parameters, were then scrutinized by DeepDDG91 and MutaBind292 to evaluate potential changes in stability and affinity. DeepDDG utilized a nonintegrative model for its assays, while MutaBind2 predictions were based on a single mutation per run. These three predictive parameters were utilized to select mutations with significant potential to enhance the affinity of Aducanumab for Aβ2–7 without decreasing the stability.
Molecular Dynamics (MD)
In line with a previous theoretical work,47 the Adu’s HC and LC were reduced to GLN1-SER126 and ASP1-THR109, corresponding to a variable fragment heavy chain (HC) and a variable fragment of light chain (LC), to diminish the computational cost during simulations (Figure 1B).47 Moreover, a mutated Adu::Aβ2–7 was created using the selected mutation (LC-S91Y), which was inserted in the Adu::Aβ2–7. Then, two Adu::Aβ2–7 complexes, wild and mutated, were submitted to three short and independent all-atom MD simulations of 100 ns and one long MD of 1 μs using the GROMACS 2023 package.93 The same scheme was repeated for a system containing only Aβ2–7 to compare the structural stability and folding of its bound and unbound states: three independent and short MDs and a single long MD. The force field CHARMM36 was used to set the interatomic potential during simulations.94 Each complex was initially inserted into a water box based on TIP3P CHARMM modified water model95 and neutralized with counterions Na+ and Cl– (0.15 mol·L–1). The steepest descent algorithm was used to minimize the potential energy using the maximum force <10.0 kJ mol–1. An equilibration phase of 2 ns was performed using NVT (1 ns) and NPT (1 ns) ensembles to equilibrate the temperature and pressure of the system. Temperature coupling was set at 300 K through V-rescale thermostat96 and the Parrinello–Rahman barostat97 was configured to stabilize the pressure with a compressibility of 4.5 · 10–5. Subsequently, the position restraints were removed, and the molecular dynamics was performed. This step-by-step procedure was performed in triplicate for each complex. The Particle Mesh Ewald (PME) method was used to define long-range electrostatic interactions.98 Leapfrog integration was employed to integrate the differential equations of motion,99 and the LINCS algorithm was used to reset covalent bonds to their appropriate lengths.100 Furthermore, a time step of 2 fs was used. Finally, the gmx trjconv algorithm was used to extract the initial and final conformations of each MD assay. Also, gmx rmsd and gmx rmsf were used to obtain the RMSD and RMSF data.
Structural Ensemble Generation
Two MD samples were used to generate the conformational ensemble: one from the wild-type and one from the mutant complex. The criterion choice was the level of structural flexibility, where the MD assays with highest deviations and fluctuations have been chosen. Both the initial conformation and the trajectory file of these simulations were used as input to generate the ensemble of representative conformations in EnGens.62 Furthermore, initial data of binding affinity was used to select the amino acid residues to be scrutinized in terms of dihedral angles of the backbone and pairwise distances between residues. The dimensionality reduction was performed through Uniform Manifold Approximation and Projection.101 The structural clustering was carried out using Gaussian Mixture Models102 and the silhouette method103 was performed to achieve the optimal number of representative conformations for each MD assay outlined. The conformations occupying the cluster’s hubs were then extracted and outlined through interactions description (2.5) and quantum biochemistry (2.6). The two ensembles generated were named representative conformations (RCs) of the wild (Wild-RC) and mutated (Mut-RC) complexes.
Nonbonded Interactions Description
Hydrogen bonds and hydrophobic interactions were identified using LigPlot+ version 2.2.104 The distances between the hydrogen (H), acceptor atom (A), and donor atom (D) were analyzed to identify hydrogen bonds, which were confirmed when the distances between H–A and D–A were within the ranges of 2.70 and 3.35 Å, respectively. Hydrophobic interactions were detected based on the fulfillment of minimum and maximum distances of 2.90 and 3.90 Å between amino acid residues of the Aducanumab and Aβ2–7.
Quantum Biochemistry
The final conformation of each molecular dynamics simulation and representative conformations obtained in Aducanumab::Aβ2–7 Energetic Hot Spots and Multiple Conformations section were submitted to the molecular fractionation with conjugated caps (MFCC), splitting the Adu::Aβ2–7 interface contacts into subsystems and decreasing the computational cost of the binding affinity calculations between Adu and Aβ amino acid residues.49−54,105−108 These MFCC calculations are based on DFT and allow us to obtain the interaction energies between residues (Ri and Rj) present in the different protein chains (HC-Adu/LC-Adu and Aβ), as shown in the following scheme:
The initial term of the equation, E(Ci – 1RiCi + 1 + Cj – 1RjCj + 1), delineates the cumulative energy (E) associated with the interaction between residues Ri and Rj and their respective molecular caps. The subsequent component, E(Ci – 1RiCi + 1 + Cj – 1Cj + 1), corresponds to the energy of the system formed by Ri with its molecular caps and the caps of Rj. The third term, E(Ci – 1 Ci + 1 + Cj – 1RjCj + 1), indicates the total interaction energy from the ensemble comprising Rj, its molecular caps, and the Ri caps. In the last part, E(Ci – 1 Ci + 1 + Cj – 1Cj + 1) characterizes the interaction energy inherent to the system exclusively encompassing the molecular caps of both Ri and Rj. Herein, Ci – 1, Ci + 1, Cj – 1, and Cj + 1 symbolize the molecular caps, which encompass residues covalently linked to the amino or carboxyl groups of Ri and Rj, and hydrogen atoms filling the gaps caused by molecular fragmentation.
The quantum biochemistry study conducted energy interaction analyses focusing on noncovalent interactions between Adu (Ri) and Aβ2–7 (Rj) residues within a maximum distance threshold of 8.0 Å. Additionally, the energetic contributions of explicit water molecules positioned within 2.5 Å of these residues (Ri and Rj) were considered in the quantum mechanical calculations of the final conformations. The ensembles of Wild-RC and Mut-RC were outlined without considering the explicit water in their quantum calculations due to the high number of conformations present. In contrast, the RCs were delineated through the employment of the COSMO solvation model, which was utilized to represent the waters in proximity to surface interactions.109 Furthermore, the majority of FC (MD1, MD2, MD3, MD4, MD5, and MD6) were also subjected to DFT calculations employing only the COMO solvation model. This was done to gain insight into the real impact of explicit water in quantum calculations.
Based on a previous study by our research group, it was found that DFT calculations using a dielectric constant of 40 were more suitable for simulating the electrostatic environment around a protein::protein interaction surface than a lower dielectric constant of 4.51 Therefore, a dielectric constant of 40 was used for the DFT calculations carried out in this current study. For each distinct set of final atomic coordinates derived from molecular dynamics simulations, the MFCC scheme was initially applied, followed by Density Functional Theory calculations using the DMOL3 package.110 The DFT calculations carried out by using generalized gradient approximation (GGA) with functional PBE,111 and TS scheme.112 A homemade Phyton-based program was used to straightforwardly determine the interactions between the Adu (Ri) and Aβ2–7 (Rj) residues of interest through DFT calculations.
Design of Aducanumab-Based Mimetic Peptides
The linear and cyclic peptides were designed following an adapted protocol established by Amaral et al.52 The linear and cyclic peptides were designed by using the energetic hot spots of HC and LC as templates. These critical regions were extracted from the first MD assay of each complex, corresponding to the MD1 (wild) and MD4 (mutated) final conformations. The linear peptides correspond precisely to the segments isolated from Adu. Cysteine residues were added to their N- and C-terminus, and a disulfide bond was subsequently formed between them to obtain the cyclic peptides. Thus, each linear peptide has a corresponding cyclic peptide. The cyclic peptides were optimized, and their sterically acceptable structures were predicted using the Dreiding-like force field.113 This force field employs elements, bond orders, number of bonds, and valence to calculate energy in a fast and efficient manner.
In Silico Characterization of Mimetic Peptides
Various computational tools were used to perform sequence-based predictions regarding critical pharmacokinetic characteristics in drug discovery. These predictions are becoming increasingly important for saving time and costs in drug development research.114,115 PEP-FOLD3116 was used to predict potential tridimensional conformations of the linear mimetic peptides by using sOPEP to sort cluster models. The ExPASy ProtParam was used to predict the isoelectric point and molecular weight.117 The Tm Predictor (http://tm.life.nthu.edu.tw/) was used to predict the melting temperature of each peptide. The CamSol method118 and APD3119 were used to calculate the solubility and net charge of the peptides, respectively. The stability of each peptide in an intestinal-like environment was evaluated using the HLP web server.120 Additionally, susceptibility to chymotrypsin, pepsin, and trypsin was assessed using ExPASy PeptideCutter.117 The number of antigenic determinants for the peptide sequences were obtained using Predicting Antigenic Peptides (http://imed.med.ucm.es/Tools/antigenic.html) according to the method of Kolaskar and Tongaonkar.121 HemoPred122 and ToxinPred123 were used to evaluate the hemolytic and toxic potentials of these molecules, respectively. Additionally, the potential capacity to cross the blood–brain barrier was investigated using B3Pred.124
Acknowledgments
H.F.C., V.N.F., and G.M. are class 1A, 1B, and 1C researchers from the Brazilian National Council for Scientific and Technological Development (CNPQ) and acknowledge the financial support they received during the development of this work (contract number 465699/2014-6). This study was also financed in part by the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES) Finance Code 001 (PROEX 88887.711696/2022-00). We would like to thank Dr. Arlindo de Alencar Araripe Noronha Moura for providing access to BioRender, which allowed us to create the Table of Contents illustration. Also, the motivation for this research came from the personal experience of one of the authors, V.N.F., and was carried out in memory of his mother, J. N. Freire, who suffered for 20 years from Alzheimer’s disease.
Glossary
Abbreviations
- AB
amyloid-β
- ACH
amyloid cascade hypothesis
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Adu
aducanumab
- CDR
complementarity determining region
- FC
final conformations
- FDA
Food and Drug Administration
- HC
heavy chain
- HCDR
heavy complementarity determining region
- LC
light chain
- LCDR
light complementarity determining region
- LMD
long molecular dynamics
- Lec
lecanemab
- MD
molecular dynamics
- mAb
monoclonal antibody
- RC
representative conformations
- RMSD
root-mean-square deviation
- RMSF
root-mean-square fluctuations
- THH
tau hyperphosphorylation hypothesis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.4c00453.
Detailed analyses and visualizations supplementing the main text. These include RMSD and RMSF values for molecular dynamics (MD) simulations of wild and mutated Aducanumab::Aβ2–7 complexes (Figures S1–S3), hydrogen bond interactions across multiple MD replicas (Figures S4 and S5), structural illustrations of final conformations (Figures S6 and S7), interaction schematics (Figures S8 and S9), energy distributions and solvation effects (Figures S10 and S11), and peptide solubility predictions (Figure S12). Nonbonded interaction data for various MD simulations are presented in Tables S1–S3. Additionally, details of quantum mechanics calculations are depicted in Tables S4–S29. (PDF)
Author Contributions
V.L.B.F., H.F.C., V.N.F., and G.M. planned, wrote, and edited the paper. V.L.B.F. conducted all molecular dynamics simulations, generated conformational ensembles, performed quantum mechanics calculations, and made in silico predictions under the supervision of V.N.F. and G.M. G.M., V.N.F., E.M.B., and R.F.d.C. provided insights into data interpretation. H.F.C. and V.N.F. were mainly responsible for raising funds to support this work.
The Article Processing Charge for the publication of this research was funded by the Coordination for the Improvement of Higher Education Personnel - CAPES (ROR identifier: 00x0ma614).
The Article Processing Charge for the publication of this research was funded by the Coordination for the Improvement of Higher Education Personnel—CAPES (ROR identifier: 00x0ma614).
The authors declare no competing financial interest.
Supplementary Material
References
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19 (4), 1598–1695. 10.1002/alz.13016. [DOI] [PubMed] [Google Scholar]
- Luo J.; Mills K.; le Cessie S.; Noordam R.; van Heemst D. Ageing, age-related diseases and oxidative stress: What to do next?. Ageing Res. Rev. 2020, 57, 100982 10.1016/j.arr.2019.100982. [DOI] [PubMed] [Google Scholar]
- Rea I. M.; Gibson D. S.; McGilligan V.; McNerlan S. E.; Alexander H. D.; Ross O. A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586 10.3389/fimmu.2018.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Escosura L. P. Health, income, and the preston curve: A long view. Econ. Hum. Biol. 2023, 48, 101212 10.1016/j.ehb.2022.101212. [DOI] [PubMed] [Google Scholar]
- Khan S.; Barve K. H.; Kumar M. S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18 (11), 1106–1125. 10.2174/1570159X18666200528142429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J.; Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. 10.1016/0165-6147(91)90609-V. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T.; Hollister R.; West H.; et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41 (1), 17–24. 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Kása P.; Rakonczay Z.; Gulya K. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1997, 52 (6), 511–535. 10.1016/S0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- Pinky P. D.; Pfitzer J. C.; Senfeld J.; et al. Recent Insights on Glutamatergic Dysfunction in Alzheimer’s Disease and Therapeutic Implications. Neuroscience 2023, 29 (4), 461–471. 10.1177/10738584211069897. [DOI] [PubMed] [Google Scholar]
- Christen Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71 (2), 621S–629S. 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- Heneka M. T.; Carson M. J.; Khoury J. El.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14 (4), 388–405. 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256 (5054), 184–185. 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297 (5580), 353–356. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J.; Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8 (6), 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K.; Carlos A. A.; Chen S.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739 (2–3), 198–210. 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Maccioni R. B.; Farías G.; Morales I.; Navarrete L. The Revitalized Tau Hypothesis on Alzheimer’s Disease. Arch. Med. Res. 2010, 41 (3), 226–231. 10.1016/j.arcmed.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Arnsten A. F. T.; Datta D.; Del Tredici K.; Braak H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17 (1), 115–124. 10.1002/alz.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. M.; Holtzman D. M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179 (2), 312–339. 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Fu L.; Li Q.; et al. Recent Advancements in the Early Diagnosis and Treatment of Alzheimer’s Disease. Adv. Ther. 2023, 6 (11), 181 10.1002/adtp.202300181. [DOI] [Google Scholar]
- Taneja V.; Verma M.; Vats A. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18 (2), 138. 10.4103/0972-2327.144284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildsmith K. R.; Holley M.; Savage J. C.; Skerrett R.; Landreth G. E. Evidence for impaired amyloid β clearance in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5 (4), 33. 10.1186/alzrt187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. J.; Zhou H. D.; Zhou X. F. Clearance of amyloid-beta in Alzheimer’s disease: progress, problems and perspectives. Drug Discovery Today 2006, 11 (19–20), 931–938. 10.1016/j.drudis.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Lloret A.; et al. When Does Alzheimer′s Disease Really Start? The Role of Biomarkers. Int. J. Mol. Sci. 2019, 20 (22), 5536 10.3390/ijms20225536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D. M.; Carrillo M. C.; Hendrix J. A.; et al. Tau: From research to clinical development. Alzheimer’s Dement. 2016, 12 (10), 1033–1039. 10.1016/j.jalz.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Conti Filho C. E.; Loss L. B.; Marcolongo-Pereira C.; et al. Advances in Alzheimer’s disease’s pharmacological treatment. Front. Pharmacol. 2023, 14, 1452 10.3389/fphar.2023.1101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L. K.; Kuan Y. C.; Lin H. W.; Hu C. J. Clinical trials of new drugs for Alzheimer disease: a 2020–2023 update. J. Biomed. Sci. 2023, 30 (1), 83. 10.1186/s12929-023-00976-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. Aducanumab: First Approval. Drugs. 2021, 81 (12), 1437–1443. 10.1007/s40265-021-01569-z. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Lecanemab: First Approval. Drugs. 2023, 83 (4), 359–365. 10.1007/s40265-023-01851-2. [DOI] [PubMed] [Google Scholar]
- Söderberg L.; Johannesson M.; Nygren P.; et al. Lecanemab, Aducanumab, and Gantenerumab — Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20 (1), 195–206. 10.1007/s13311-022-01308-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D. A. Antibody-Mediated Clearance of Brain Amyloid-β: Mechanisms of Action, Effects of Natural and Monoclonal Anti-Aβ Antibodies, and Downstream Effects. J. Alzheimer’s Dis. Rep. 2023, 7 (1), 873–899. 10.3233/ADR-230025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse S.; Scheidt T.; Bernfur K.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020, 27 (12), 1125–1133. 10.1038/s41594-020-0505-6. [DOI] [PubMed] [Google Scholar]
- Kurkinen M. T. Donanemab: Not two without a third. Adv. Clin. Exp. Med. 2023, 32 (10), 1085–1087. 10.17219/acem/172673. [DOI] [PubMed] [Google Scholar]
- Dickson S. P.; Hennessey S.; Nicodemus Johnson J.; Knowlton N.; Hendrix S. B. Avoiding future controversies in the Alzheimer’s disease space through understanding the aducanumab data and FDA review. Alzheimers Res. Ther. 2023, 15 (1), 98. 10.1186/s13195-023-01238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran S. J.; Sawyer R. P. Risk factors in developing amyloid related imaging abnormalities (ARIA) and clinical implications. Front. Neurosci. 2024, 18, 1326784 10.3389/fnins.2024.1326784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann R.; Nixon J.; Love B. L.; Yunusa I. Impacts of FDA approval and Medicare restriction on antiamyloid therapies for Alzheimer’s disease: patient outcomes, healthcare costs, and drug development. Lancet Reg. Health Am. 2023, 20, 100467 10.1016/j.lana.2023.100467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshir S. A.; Aadithsoorya A. M.; Parveen A.; Goh S. S. L.; Hussain N.; Menon V. B. Aducanumab Therapy to Treat Alzheimer’s Disease: A Narrative Review. abate giulia, ed. Int. J. Alzheimers Dis. 2022, 2022, 9343514 10.1155/2022/9343514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampi R. R.; Forester B. P.; Agronin M. Aducanumab: evidence from clinical trial data and controversies. Drugs Context 2021, 10, 1–9. 10.7573/dic.2021-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman D. S.; Hershey L. Implications of the Approval of Lecanemab for Alzheimer Disease Patient Care. Neurology 2023, 101 (14), 610–620. 10.1212/WNL.0000000000207438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D.; Dou F.; Gao J. Development of amyloid beta-directed antibodies against Alzheimer’s disease: Twists and turns. Drug Discovery Ther. 2023, 17 (6), 2023.01215 10.5582/ddt.2023.01215. [DOI] [PubMed] [Google Scholar]
- Qiao Y.; Gu J.; Yu M.; Chi Y.; Ma Y. Comparative Efficacy and Safety of Monoclonal Antibodies for Cognitive Decline in Patients with Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. CNS Drugs 2024, 38 (3), 169–192. 10.1007/s40263-024-01067-2. [DOI] [PubMed] [Google Scholar]
- Sevigny J.; Chiao P.; Bussière T.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537 (7618), 50–56. 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Arndt J. W.; Qian F.; Smith B. A.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8 (1), 6412 10.1038/s41598-018-24501-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi G. A. N.; Hermans S. J.; Parker M. W.; Miles L. A. Molecular basis for mid-region amyloid-β capture by leading Alzheimer’s disease immunotherapies. Sci. Rep. 2015, 5 (1), 9649 10.1038/srep09649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohrmann B.; Baumann K.; Benz J.; et al. Gantenerumab: A Novel Human Anti-Aβ Antibody Demonstrates Sustained Cerebral Amyloid-β Binding and Elicits Cell-Mediated Removal of Human Amyloid-β. J. Alzheimer’s Dis. 2012, 28 (1), 49–69. 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- Ultsch M.; Li B.; Maurer T.; et al. Structure of Crenezumab Complex with Aβ Shows Loss of β-Hairpin. Sci. Rep. 2016, 6 (1), 39374 10.1038/srep39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan M.; Shahzadi S.; Seo S. Y.; Alashwal H.; Zaki N.; Moustafa A. A. Molecular Docking and Dynamic Simulation of AZD3293 and Solanezumab Effects Against BACE1 to Treat Alzheimer’s Disease. Front. Comput. Neurosci. 2018, 12, 34 10.3389/fncom.2018.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C. V.; Zacharias M. From monomer to fibril: Abeta-amyloid binding to Aducanumab antibody studied by molecular dynamics simulation. Proteins 2020, 88 (12), 1592–1606. 10.1002/prot.25978. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Wei G.; Zhao J.; Nussinov R.; Ma B. Computational Investigation of Gantenerumab and Crenezumab Recognition of Aβ Fibrils in Alzheimer’s Disease Brain Tissue. ACS Chem. Neurosci. 2020, 11 (20), 3233–3244. 10.1021/acschemneuro.0c00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque E.; Fulco U. L.; Caetano E.; Freire V.. Quantum Chemistry Simulation of Biological Molecules; Cambridge University Press, 2020. [Google Scholar]
- Zanatta G.; Della Flora Nunes G.; Bezerra E. M.; et al. Two Binding Geometries for Risperidone in Dopamine D3 Receptors: Insights on the Fast-Off Mechanism through Docking, Quantum Biochemistry, and Molecular Dynamics Simulations. ACS Chem. Neurosci. 2016, 7 (10), 1331–1347. 10.1021/acschemneuro.6b00074. [DOI] [PubMed] [Google Scholar]
- Morais P. A.; Maia F. F.; Solis-Calero C.; Caetano E. W. S.; Freire V. N.; Carvalho H. F. The urokinase plasminogen activator binding to its receptor: a quantum biochemistry description within an in/homogeneous dielectric function framework with application to uPA–uPAR peptide inhibitors. Phys. Chem. Chem. Phys. 2020, 22 (6), 3570–3583. 10.1039/C9CP06530J. [DOI] [PubMed] [Google Scholar]
- Amaral J. L.; Santos S. J. M.; Souza P. F. N.; et al. Quantum biochemistry in cancer immunotherapy: New insights about CTLA-4/ipilimumab and design of ipilimumab-derived peptides with high potential in cancer treatment. Mol. Immunol. 2020, 127, 203–211. 10.1016/j.molimm.2020.09.013. [DOI] [PubMed] [Google Scholar]
- Bezerra E. M.; de Alvarenga ÉC.; dos Santos R. P.; et al. Losartan as an ACE inhibitor: a description of the mechanism of action through quantum biochemistry. RSC Adv. 2022, 12 (44), 28395–28404. 10.1039/D2RA04340H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquita F. P.; Noronha Souza P. F.; Aragão D. R.; et al. In silico analysis of ACE2 from different animal species provides new insights into SARS-CoV-2 species spillover. Future Virol. 2023, 18 (6), 359–371. 10.2217/fvl-2022-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie P. H.; Parhami-Seren B.; Wysocki L. J.; Margolies M. N. A single engineered amino acid substitution changes antibody fine specificity. J. Immunol. 1994, 152 (1), 146–152. 10.4049/jimmunol.152.1.146. [DOI] [PubMed] [Google Scholar]
- Ye W.; Liu X.; He R.; et al. Improving antibody affinity through in vitro mutagenesis in complementarity determining regions. J. Biomed. Res. 2022, 36 (3), 155. 10.7555/JBR.36.20220003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldeghi M.; Gapsys V.; de Groot B. L. Accurate Estimation of Ligand Binding Affinity Changes upon Protein Mutation. ACS Cent. Sci. 2018, 4 (12), 1708–1718. 10.1021/acscentsci.8b00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R. Computational studies of protein–drug binding affinity changes upon mutations in the drug target. WIREs Comput. Mol. Sci. 2022, 12 (1), e1563 10.1002/wcms.1563. [DOI] [Google Scholar]
- Kulshreshtha S.; Chaudhary V.; Goswami G. K.; Mathur N. Computational approaches for predicting mutant protein stability. J. Comput.-Aided Mol. Des. 2016, 30 (5), 401–412. 10.1007/s10822-016-9914-3. [DOI] [PubMed] [Google Scholar]
- Khan A.; Gui J.; Ahmad W.; et al. The SARS-CoV-2 B.1.618 variant slightly alters the spike RBD–ACE2 binding affinity and is an antibody escaping variant: a computational structural perspective. RSC Adv. 2021, 11 (48), 30132–30147. 10.1039/D1RA04694B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlowe A. E.; Singh A.; Yingling Y. G. The effect of point mutations on structure and mechanical properties of collagen-like fibril: A molecular dynamics study. Mater. Sci. Eng. C 2012, 32 (8), 2583–2588. 10.1016/j.msec.2012.07.044. [DOI] [Google Scholar]
- Conev A.; Rigo M. M.; Devaurs D.; et al. EnGens: a computational framework for generation and analysis of representative protein conformational ensembles. Brief. Bioinform. 2023, 24 (4), e242 10.1093/bib/bbad242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby J. F.; Hopkins A. P.; Shimizu S.; et al. Water Networks Can Determine the Affinity of Ligand Binding to Proteins. J. Am. Chem. Soc. 2019, 141 (40), 15818–15826. 10.1021/jacs.9b06275. [DOI] [PubMed] [Google Scholar]
- Quiocho F. A.; Wilson D. K.; Vyas N. K. Substrate specificity and affinity of a protein modulated by bound water molecules. Nature 1989, 340 (6232), 404–407. 10.1038/340404a0. [DOI] [PubMed] [Google Scholar]
- Murali R.; Greene M. I. Structure Based Antibody-Like Peptidomimetics. Pharmaceuticals 2012, 5 (2), 209–235. 10.3390/ph5020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van holsbeeck K.; Martins J. C.; Ballet S. Downsizing antibodies: Towards complementarity-determining region (CDR)-based peptide mimetics. Bioorg. Chem. 2022, 119, 105563 10.1016/j.bioorg.2021.105563. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; King G. F.; Adams D. J.; Alewood P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discovery 2021, 20 (4), 309–325. 10.1038/s41573-020-00135-8. [DOI] [PubMed] [Google Scholar]
- Ennaas N.; Hammami R.; Beaulieu L.; Fliss I. Purification and characterization of four antibacterial peptides from protamex hydrolysate of Atlantic mackerel (Scomber scombrus) by-products. Biochem. Biophys. Res. Commun. 2015, 462 (3), 195–200. 10.1016/j.bbrc.2015.04.091. [DOI] [PubMed] [Google Scholar]
- Piotto S. P.; Sessa L.; Concilio S.; Iannelli P. YADAMP: yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39 (4), 346–351. 10.1016/j.ijantimicag.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Daroit D. J.; Brandelli A. In vivo bioactivities of food protein-derived peptides – a current review. Curr. Opin. Food Sci. 2021, 39, 120–129. 10.1016/j.cofs.2021.01.002. [DOI] [Google Scholar]
- Guha S.; Majumder K. Structural-features of food-derived bioactive peptides with anti-inflammatory activity: A brief review. J. Food Biochem. 2019, 43 (1), e12531 10.1111/jfbc.12531. [DOI] [PubMed] [Google Scholar]
- Mojica L.; Gonzalez de Mejia E.; Granados-Silvestre MÁ.; Menjivar M. Evaluation of the hypoglycemic potential of a black bean hydrolyzed protein isolate and its pure peptides using in silico, in vitro and in vivo approaches. J. Funct. Foods 2017, 31, 274–286. 10.1016/j.jff.2017.02.006. [DOI] [Google Scholar]
- Rivero-Pino F.; Espejo-Carpio F. J.; Guadix E. M. Antidiabetic Food-Derived Peptides for Functional Feeding: Production, Functionality and In Vivo Evidences. Foods 2020, 9 (8), 983. 10.3390/foods9080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T.; Aprile F. A.; Sormanni P.; et al. A rationally designed bicyclic peptide remodels Aβ42 aggregation in vitro and reduces its toxicity in a worm model of Alzheimer’s disease. Sci. Rep. 2020, 10 (1), 15280 10.1038/s41598-020-69626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malta S. M.; Batista L. L.; Silva H. C. G.; et al. Identification of bioactive peptides from a Brazilian kefir sample, and their anti-Alzheimer potential in Drosophila melanogaster. Sci. Rep. 2022, 12 (1), 11065 10.1038/s41598-022-15297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser V. J.; Gómez J.; Freire E. The enthalpy change in protein folding and binding: Refinement of parameters for structure-based calculations. Proteins 1996, 26 (2), 123–133. 10.1002/(SICI)1097-0134(199610)26:2<123::AID-PROT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Gromiha M. M.; Selvaraj S. Inter-residue interactions in protein folding and stability. Prog. Biophys. Mol. Biol. 2004, 86 (2), 235–277. 10.1016/j.pbiomolbio.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Ji X.; Nielsen A. L.; Heinis C. Cyclic Peptides for Drug Development. Angew. Chem., Int. Ed. 2024, 63 (3), e202308251 10.1002/anie.202308251. [DOI] [PubMed] [Google Scholar]
- Góngora-Benítez M.; Tulla-Puche J.; Albericio F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114 (2), 901–926. 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- Hoare S. R. J.; Fleck B. A.; Williams J. P.; Grigoriadis D. E. The importance of target binding kinetics for measuring target binding affinity in drug discovery: a case study from a CRF1 receptor antagonist program. Drug Discovery Today 2020, 25 (1), 7–14. 10.1016/j.drudis.2019.09.011. [DOI] [PubMed] [Google Scholar]
- Kairys V.; Baranauskiene L.; Kazlauskiene M.; Matulis D.; Kazlauskas E. Binding affinity in drug design: experimental and computational techniques. Exp. Opin. Drug Discovery 2019, 14 (8), 755–768. 10.1080/17460441.2019.1623202. [DOI] [PubMed] [Google Scholar]
- Kong X. D.; Moriya J.; Carle V.; et al. De novo development of proteolytically resistant therapeutic peptides for oral administration. Nat. Biomed. Eng. 2020, 4 (5), 560–571. 10.1038/s41551-020-0556-3. [DOI] [PubMed] [Google Scholar]
- Oliveira J. T. A.; Souza P. F. N.; Vasconcelos I. M.; et al. Mo-CBP3-PepI, Mo-CBP3-PepII, and Mo-CBP3-PepIII are synthetic antimicrobial peptides active against human pathogens by stimulating ROS generation and increasing plasma membrane permeability. Biochimie 2019, 157, 10–21. 10.1016/j.biochi.2018.10.016. [DOI] [PubMed] [Google Scholar]
- Takahashi T.; Mihara H. Peptide and Protein Mimetics Inhibiting Amyloid β-Peptide Aggregation. Acc. Chem. Res. 2008, 41 (10), 1309–1318. 10.1021/ar8000475. [DOI] [PubMed] [Google Scholar]
- Goyal D.; Shuaib S.; Mann S.; Goyal B. Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19 (2), 55–80. 10.1021/acscombsci.6b00116. [DOI] [PubMed] [Google Scholar]
- Lu X.; Brickson C. R.; Murphy R. M. TANGO-Inspired Design of Anti-Amyloid Cyclic Peptides. ACS Chem. Neurosci. 2016, 7 (9), 1264–1274. 10.1021/acschemneuro.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M. The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46 (W1), W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Rosell G.; Giorgino T.; De Fabritiis G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57 (7), 1511–1516. 10.1021/acs.jcim.7b00190. [DOI] [PubMed] [Google Scholar]
- Dehouck Y.; Kwasigroch J. M.; Rooman M.; Gilis D. BeAtMuSiC: prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 2013, 41 (W1), W333–W339. 10.1093/nar/gkt450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H.; Wang J.; He L.; Qi Y.; Zhang J. Z. DeepDDG: Predicting the Stability Change of Protein Point Mutations Using Neural Networks. J. Chem. Inf. Model. 2019, 59 (4), 1508–1514. 10.1021/acs.jcim.8b00697. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Chen Y.; Lu H.; et al. MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience 2020, 23 (3), 100939 10.1016/j.isci.2020.100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; et al. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8 (9), 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126 (1), 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52 (12), 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103 (19), 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Fincham D. Leapfrog Rotational Algorithms. Mol. Simul. 1992, 8 (3–5), 165–178. 10.1080/08927029208022474. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18 (12), 1463–1472. 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- Trozzi F.; Wang X.; Tao P. UMAP as a Dimensionality Reduction Tool for Molecular Dynamics Simulations of Biomacromolecules: A Comparison Study. J. Phys. Chem. B 2021, 125 (19), 5022–5034. 10.1021/acs.jpcb.1c02081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klem H.; Hocky G. M.; McCullagh M. Size-and-Shape Space Gaussian Mixture Models for Structural Clustering of Molecular Dynamics Trajectories. J. Chem. Theory Comput. 2022, 18 (5), 3218–3230. 10.1021/acs.jctc.1c01290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw P. J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. 10.1016/0377-0427(87)90125-7. [DOI] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51 (10), 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Gao A. M.; Zhang D. W.; Zhang J. Z. H.; Zhang Y. An efficient linear scaling method for ab initio calculation of electron density of proteins. Chem. Phys. Lett. 2004, 394 (4–6), 293–297. 10.1016/j.cplett.2004.06.137. [DOI] [Google Scholar]
- He X.; Zhang J. Z. H. A new method for direct calculation of total energy of protein. J. Chem. Phys. 2005, 122 (3), 031103 10.1063/1.1849132. [DOI] [PubMed] [Google Scholar]
- Dantas D. S.; Oliveira J. I. N.; Lima Neto J. X.; et al. Quantum molecular modelling of ibuprofen bound to human serum albumin. RSC Adv. 2015, 5 (61), 49439–49450. 10.1039/C5RA04395F. [DOI] [Google Scholar]
- da Costa R. F.; Freire V. N.; Bezerra E. M.; et al. Explaining statin inhibition effectiveness of HMG-CoA reductase by quantum biochemistry computations. Phys. Chem. Chem. Phys. 2012, 14 (4), 1389–1398. 10.1039/C1CP22824B. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, (5), 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Delley B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113 (18), 7756–7764. 10.1063/1.1316015. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Tkatchenko A.; Scheffler M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102 (7), 073005 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- Mayo S. L.; Olafson B. D.; Goddard W. A. DREIDING: a generic force field for molecular simulations. J. Phys. Chem. A 1990, 94 (26), 8897–8909. 10.1021/j100389a010. [DOI] [Google Scholar]
- Shaker B.; Ahmad S.; Lee J.; Jung C.; Na D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851 10.1016/j.compbiomed.2021.104851. [DOI] [PubMed] [Google Scholar]
- Agoni C.; Olotu F. A.; Ramharack P.; Soliman M. E. Druggability and drug-likeness concepts in drug design: are biomodelling and predictive tools having their say?. J. Mol. Model. 2020, 26 (6), 120. 10.1007/s00894-020-04385-6. [DOI] [PubMed] [Google Scholar]
- Lamiable A.; Thévenet P.; Rey J.; Vavrusa M.; Derreumaux P.; Tufféry P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44 (W1), W449–W454. 10.1093/nar/gkw329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31 (13), 3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sormanni P.; Vendruscolo M. Protein Solubility Predictions Using the CamSol Method in the Study of Protein Homeostasis. Cold Spring Harb. Perspect. Biol. 2019, 11 (12), a033845 10.1101/cshperspect.a033845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44 (D1), D1087–D1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Singla D.; Rashid M.; Raghava G. P. S. Designing of peptides with desired half-life in intestine-like environment. BMC Bioinform. 2014, 15 (1), 282. 10.1186/1471-2105-15-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaskar A. S.; Tongaonkar P. C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276 (1–2), 172–174. 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- Win T. S.; Malik A. A.; Prachayasittikul V.; S Wikberg J. E.; Nantasenamat C.; Shoombuatong W. HemoPred: a web server for predicting the hemolytic activity of peptides. Future Med. Chem. 2017, 9 (3), 275–291. 10.4155/fmc-2016-0188. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Kapoor P.; Chaudhary K.; Gautam A.; Kumar R.; Raghava G. P. S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS One 2013, 8, e73957 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Patiyal S.; Dhall A.; Sharma N.; Raghava G. P. S. B3Pred: A Random-Forest-Based Method for Predicting and Designing Blood–Brain Barrier Penetrating Peptides. Pharmaceutics 2021, 13 (8), 1237. 10.3390/pharmaceutics13081237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.