

Abstract
The enthalpies of formation of brominated C3-C4 hydrocarbons were critically evaluated using experimental data sources ranging from classical thermodynamics methods to modern high-precision mass spectrometry and reported in a time span of over a century. The experimental data were used in conjunction with the results of modern high-level ab initio calculations. To facilitate quantitative analysis, a recently developed local coupled cluster-based computational protocol was extended to organic compounds containing univalent Br. Several erroneous data sources were identified in a course of the study. Possible reasons of the inconsistency between the values recommended by CODATA and Active Termochemical Tables for HBr in the gas and aqueous solution were discussed. The most up-to-date recommendations based on the comprehensive analysis of collected information are provided for 23 brominated hydrocarbons. For several compounds under consideration, the recommended values were previously lacking, while improved values and uncertainties were obtained for those with existing recommendations.
Graphical Absract
Introduction
Bromine-containing organic compounds have a wide range of practical applications. They are used as fire retardants, fumigants and biocides, dyes, pharmaceuticals, and intermediates in organic synthesis,1,2 although some of these uses are being phased out due to environmental and health concerns. Knowledge of accurate thermochemical data, especially the enthalpies of formation, are important to facilitate the informed process design and practical applications.
The experimental enthalpies of formation for CHON molecules are normally determined from combustion experiments in a static bomb.3 Extension to bromine-containing compounds requires a rotating-bomb calorimeter and addition of a reducing agent to quantitatively transform into .4 For relatively small brominated molecules, other techniques have also been widely used. They include reaction (hydrogenation, hydrobromination, bromination, etc.) calorimetry, chemical equilibrium studies in the liquid and gas phases, and mass-spectrometric methods. Overall, the experimental values are available for slightly over 100 compounds.
Kolesov and Papina reviewed the experimental enthalpies of formation of halogenated methanes5 and ethanes.6 Brominated compounds were also critically evaluated in general compilations such as those of Cox and Pilcher 7 and Pedley.8 These sources primarily considered the experimental data, although group-contribution estimates were used in some of them. The enthalpies of formation of brominated and compounds have also been reviewed using high-level ab initio computations.9–12 Introduction of high-level computations to the data analysis and evaluation is beneficial for organoelement compounds because of experimental challenges in the associated thermochemical experiments. However, we are not aware of any high-level ab initio studies involving larger molecules as they were historically hindered by high computational costs.
In this work, the ab initio protocol proposed by us earlier13,14 and used to compute for about 400 CHON-containing species15 is parameterized for brominated compounds and its uncertainty is estimated. The experimental data for the enthalpies of formation and vaporization for and brominated compounds are collected. The data are evaluated using the ab initio protocol and the best estimates of these quantities are recommended. Finally, the experimental challenges are discussed and the erroneous data in the literature are identified.
Computations
The computations were performed according to the aLL5 protocol described in Ref. 14. The vibrational frequencies were calculated with the hybrid density-functional theory, B3LYP-D3(BJ)/def2-TZVP. Separate scaling factors were used for calculation of zero-point vibrational energies (ZPVE) and thermal correction to the enthalpy . For the former, the scaling factor was 0.990. For the latter, they were 0.960 for hydrogen stretches, and 0.985 for all other frequencies. The statistical thermodynamic calculations followed the rigid-rotor/harmonic oscillator approximation. The geometries for the coupled-cluster calculations were optimized with the density-fitted (resolution-of-identity) second-order Møller-Plesset perturbation theory (DF-MP2). The single-point energy calculations were performed with the 2016 version of local of Kállay et al.16,17 In both calculations, aug-cc-pVQZ basis set was used. More details regarding the protocol can be found in the original publication.14 DFT calculations were performed with the Gaussian 16 software,18 DF-MP2 was done with the Psi4 package (version 1.5),19,20 and was carried out with the MRCC suite (release of February 9, 2019).21,22 All correlated calculations used the frozen-core approximation.
For the molecules exhibiting multiple conformations, the conformational search was performed using the procedure adopted from our previous studies (e.g., Refs 14,23–25). In this study, it was updated to include, in addition to the MMFF9426 force field-based generators, the CREST framework27 that uses the recent semi-empirical tight-binding model.28
The standard entropy was found for each conformer using the rigid rotor/harmonic oscillator model described above. Enthalpy of formation for a given compound was computed as the Gibbs-energy average over the conformer population. The difference between this model and the one with hindered tops is discussed in the Supporting Information. For the brominated compounds considered in this work, the potential error introduced by this model is significantly lower than the expected uncertainty of the predicted values.
The enthalpies of formation at were obtained using the equation:13,14,24
| (1) |
where is the total electronic energy, ZPVE is the zero-point vibrational energy, and is the thermal correction from . The summation in the last term of Eq. 1 is performed over all atomic types present in the compound; is the ith type count, and is the type-specific constant. The following values, obtained previously,14,24 were used: , saturated or aromatic , unsaturated , and .
The expanded uncertainties (0.95 level of confidence) of the ab initio values were found as
| (2) |
The procedure to calculate have been described previously,14 ,24 and was determined in this study as described below.
The above protocol has been shown to be accurate and efficient for numerous neutral molecules.14,24,29,30 In this work, analysis of mass spectrometry data also necessitates predictions for hydrocarbon ions. Tests for the methylium ion indicated that, in its original form, Eq. 1 does not predict the experimental within the expected uncertainty of about . To improve the accuracy for ions, the complete-basis-set (CBS) version of the protocol25 was used and the core-valence (CV) correlation was explicitly considered. The CBS extrapolation was based on the computations with aug-cc-pVNZ basis sets. For the Self-Consistent Field (SCF) contribution, the Karton-Martin modification31 of Jensen’s extrapolation formula32 was used:
| (3) |
Where
| (4) |
and .
The LCCSD correlation contribution was extrapolated using
| (5) |
which, to a large extent, is based on empirical observations (see, e.g., Ref. 33). For the CBS version of the protocol, , saturated or aromatic , unsaturated .25
CV correlation energies were obtained as a difference between the all-electron and frozen-core results using the regular calculations carried out with CFOUR v2.1.34 The total energies were extrapolated to the CBS limit using Eqs. (3) and (5) with . For a set of neutral molecules including saturated, unsaturated, and aromatic hydrocarbons (Table 1), the CV correlation energy per carbon was found to be nearly constant, . This result is consistent with the additivity approximation for the contributions not explicitly included in Eq. 1, as implied in the original protocol.14 In other words, this contribution is already included in the effective enthalpies of the carbon atoms derived from the data for neutral compounds. When evaluating the enthalpy of formation for an ion with Eq. 1, the estimated CV contribution for a neutral compound, , was subtracted from the result and the explicitly computed was added instead.
Table 1:
Core-valence correlation energies for hydrocarbons calculated by the CBS extrapolation of the CCSD(T)/cc-pwCVNZ () energies
| Name | ||
|---|---|---|
| ethane | 314.42 | 157.21 |
| propane | 471.46 | 157.15 |
| butane | 628.52 | 157.13 |
| 2-methylpropane | 628.55 | 157.14 |
| cyclohexane | 942.38 | 157.06 |
| ethylene | 314.21 | 157.11 |
| propene | 471.49 | 157.16 |
| 2-methylpropene | 628.68 | 157.17 |
| acetylene | 314.55 | 157.28 |
| benzene | 943.15 | 157.19 |
| Average: | 157.16 ± 0.12 |
Experimental data
In this section, a general characterization of the experimental techniques is provided. Details on particular measurements and the corresponding references are discussed in the subsequent sections. Typically, the main source of the formation enthalpies for brominated organic compounds in the condensed state is a rotating-bomb combustion calorimetry.4 However, this is not the case for the group considered here: three other techniques provided the largest fraction of thermochemical data. Reaction calorimetry has been used to obtain the values in the gas and liquid phases. Typically, the molar enthalpies of the studied reactions (e.g., hydrogenation, bromination, hydrobromination) are at least an order of magnitude lower than the combustion energies of the corresponding brominated derivatives. The repeatability-based uncertainty close to (comparable to that of combustion calorimetry) is often obtained with the instrument not as sophisticated as a rotating-bomb calorimeter (see, e.g., Refs. 7,35). As demonstrated below for 1- and 2-bromopropanes, these uncertainty estimates are not always adequate.
Temperature-dependent equilibrium constants can be used to derive the reaction enthalpies. This is accomplished by fitting the data in vs. coordinates (the Second-Law method7). A more sophisticated form such as the Clarke-Glew equation can also be used.36 Alternatively, if the standard entropies or Gibbs energy functions of the reaction participants are known, the reaction enthalpies can be found using the Third-Law method. The equilibrium data are considered here if they can be used to derive for a brominated compound, i.e. when the values for all other reaction participants are available. Chemical equilibria have been studied for multiple brominated compounds both in the liquid and gas phases, mainly for isomerization. The uncertainties reported in the original publications were found to be acceptable for most systems analyzed in this work.
Thermochemical information for small molecules can also be obtained from high-precision mass spectrometry, particularly the variations of the Threshold PhotoElectron PhotoIon COincidence (TPEPICO) spectroscopy (see, e.g., Ref. 37). In this approach, an RBr molecule is ionized by monochromatic light and the ion is formed; in this context, R is the Markush-style notation for a generic functional group. Next, the dissociation limit at leading to and Br is determined relative to the energy of the initial molecule. is well-established.38,39 If is available, the enthalpy of formation of RBr can be derived. The resulting value is typically accurate within several . The uncertainty is often not rigorously defined in the studies of this type. We assume the reported uncertainties to be a reasonable estimate of the expanded uncertainties for 0.95 level of confidence.
The enthalpies of vaporization can be determined either calorimetrically or from the temperature-dependent vapor pressures . If the vapor-pressure data are used, the quantity is derived first:
| (6) |
This quantity is proportional to the enthalpy of vaporization to the saturated vapor:
| (7) |
where is the compressibility factor change in the process. At low pressures, is close to unity. At ambient pressures, this quantiy can be approximated with the following equation:
| (8) |
where is the second virial coefficient and is the molar volume of the liquid. If this correction is required, the second virial coefficients are found using either the available experimental data evaluated by the NIST ThermoData Engine (TDE)40 or the estimates by the Pitzer-Curl method41. The density of the condensed phase is also evaluated by TDE.
To find the standard enthalpy of vaporization, , a correction for the gas non-ideality should be applied:
| (9) |
Equations (6) to (9) were derived and discussed in detail by Majer et al.42 Deviations of the non-ideality corrections at used in this work from those reported in the compilation of Majer and Svoboda43 do not exceed (Table S3 of Supporting Information).
To reduce the standard enthalpies of vaporization from the average temperature of the measurements to the reference temperature , three levels of approximation are possible.
(i) If the experimental heat capacities are available from to , the following equation is used:
| (10) |
For all compounds considered in this work, the computed ideal-gas heat capacities can be calculated at any temperature of practical interest using the statistical thermodynamic model described above.
(ii) If the liquid-phase heat capacity is only available at a single temperature (typically, ), then Eq. (10) transforms into
| (11) |
(iii) If no liquid-phase heat capacities can be found in the literature, the correlation by Chickos et al.44 is used in the form
| (12) |
The expanded uncertainty (0.95 level of confidence) for Eq. (12) has been estimated to be .44
Results and discussion
Discrepancy analysis for the reference enthalpies of formation of
Before proceeding any further, the discrepancy between the existing recommendations for the standard enthalpies of formation of and needs to be addressed because these values affect most of evaluation results in this work. The present values from CODATA38 and 39 differ by for both, which substantially exceeds their stated uncertainties of and , respectively. Thermochemical networks were used in both cases. for the dissociated hydrogen bromide HBr(aq, diss.) which is equivalent to (aq) recommended by CODATA was derived by simultaneous regression of data for multiple aqueous ions. The gas-phase value was then obtained primarily using the enthalpy of the HBr dissolution in water reported by Vanderzee and Nutter.45 In the ATcT network, the gas-phase ionization energies were introduced but some enthalpies considered by CODATA were omitted. The critical quantity here is the enthalpy of the gas-phase reaction at derived using the spectroscopic data from Refs. 46–48. Also, the standard Gibbs energies and enthalpies for the ammonium bromide decomposition considered in the NIST-JANAF Tables49 were added. The data from both compilations with the highest impact on the resulting enthalpies of formation were analyzed to find a possible cause of the noted inconsistency. The processes involving bromine species considered by CODATA and their enthalpies are listed in Table 2; the results were verified against the original publications. Some of them are used in ATcT as well. Among listed, the processes 71, 75, 78, 81, and 82 require no further comments.
Table 2:
Experimental standard enthalpies of processes involving and HBr at the reference temperature considered by CODATAa,b
| Eq. No.38 | Process | Reference | |
|---|---|---|---|
| 71 | 51.92 ± 0.45 | 50 | |
| 72 | , diss. , diss. | See text | 51,52 |
| 73 | , diss. , diss. | 91.54 ± 0.64 | 53,54 |
| 75 | , diss. | 85.144 ± 0.060 | 45 |
| 78 | , diss. | 120.37 ± 0.80c | 55,56 |
| 79 | 0.83 ± 0.20 | 57 | |
| 80 | 2.17 ± 0.20d | 58 | |
| 81 | , diss. | 86.01 ± 1.00c | 59 |
| 82 | aq,diss. | 240.94 ± 0.15 | 56,60 |
The following reference values at recommended by CODATA38 were used in the regression: , , , , , and
Values obtained by the weighted least-squares regression: , , and
Obtained from the standard Gibbs energy using the reference values from footnote
assumed incorrect in this work
The enthalpy of reaction 72 was obtained from a thermodynamic cycle involving oxidation with and .51,52 To close the cycle, the corrections up to had to be applied to the measured enthalpies. Using the enthalpies of dilution from the NBS Tables,61 the reaction enthalpy of can be obtained instead of the original value of . The differences are mainly caused by changes in the values for the concentrated and acid solutions used in the cycle. The NBS table on bromine was prepared in 1964. If the dilution enthalpies of HBr recommended by Vanderzee et al.62 in 1974 are used instead, the enthalpy becomes . The other problem is that a degree of dissociation of weak acids and decreases if a strong acid is present. The enthalpies of dissociation of these acids are close to and, therefore, this factor, ignored in the original publications, is expected to introduce an additional uncertainty. Because of the described issues, the enthalpy of reaction 72 should not be used as a reference value.
The enthalpy of reaction 73 is derived from a thermodynamic cycle involving oxidation with and 53,54 Unlike reaction 72, the calorimetric measurements were conducted with dilute solutions. The authors of the original works applied both the dilution and dissociation corrections. The revised enthalpy of reaction 73 is more negative than the CODATA value. The updated enthalpies of dilution of the acids were taken from the NBS Tables.61 For the dissociation correction, of 63 was used and the ionic activity coefficients were assumed to be equal to those of the aqueous HCl and 64 at the same ionic strength. Also, the correction to infinite dilution was applied.
The enthalpies of chemically identical processes 79 and 80 differ by while their uncertainties are . To make these results consistent, either the uncertainties should be increased or one of the values should be rejected. If the uncertainties of these values are both increased to , the weighted least-square fit of the data in Table 2 leads to , which does not help resolving the discrepancy. In the calorimetric measurements for both processes, liquid bromine was dissolved in dilute . The temperature rise was relatively small, (1 to 10) mK, in both works. Despite differences in the calorimeter design and materials, there is no obvious reason to reject either of these results.
On the other hand, the authors of Ref. 58 used the same instrument and technique to measure the enthalpies of solution of bromine in and .65 The final molality of was between . The experimental data vary within and exhibit irregular changes with the solvent compositions and bromine molality. To check validity of the reported dependences, modeling of the equilibria between , and using the equilibrium constants from the NBS Tables61 was performed in this work. The activity coefficients of the anions were cancelled in the equilibrium constants, and the activity coefficient of was assumed to be equal to unity. For a given molality, the predicted dissolution enthalpies change by less than between different solvent compositions. For a given solvent composition, it varies with the amount of by less than . These inconsistencies suggest that potential problems may also exist with the enthalpy of process 80. Considering the above, the value of Wu et al. (process 79) appears more credible.
If entry 80 is rejected, (HBr(aq, diss.) becomes , close to the ATcT value of . Therefore, for the purposes of this work, we adopt the current ATcT recommendations for the species in question. However, future work on further refinement of the current recommendations and analysis of the available data is likley to be needed. For example, the enthalpy of reaction 82 obtained66 with an alternative treatment of the electrochemical data from Ref. 60 differed from that in the original publication by , which an order of magnitude higher than its uncertainty.
Determination of the effective enthalpy of a bromine atom
The recent study of Bross et al.12 focused on determination of the enthalpies of formation via total atomization energies suggested increased importance of contributions beyond for small bromine- and iodine-containing hydrocarbons. In particular, the second-order spin-orbit coupling, normally ignored for the first- and second-row species, was found to be important for the accurate description of total atomization energies. In our case, Eq. 1 implies that the combined contributions beyond as well as the core-valence correlation, spin-orbit, and relativistic contributions to the atomization energies can be reasonably described as a sum of atomic contributions. To verify this assumption, we used the results of Bross et al. for , and bromo derivatives of methane, ethene, and ethyne.12 The additive (i.e., linear with respect to atomic counts) equation
| (13) |
was found to closely approximate the sum of these contributions (Table 3) with the largest absolute deviation of and the standard error of . As seen, the results of Bross et al.12 are indeed consistent with the additivity assumption14 for the contributions beyond for compounds containing univalent Br, and we proceed to extending this protocol to brominated organic compounds.
Table 3:
| Compound | CASRN | HOC | CV | DBOC | PP | SO | ||
|---|---|---|---|---|---|---|---|---|
| bromomethane | 74-83-9 | 0.0 | 6.4 | 0.2 | −0.5 | −14.5 | −8.4 | 0.2 |
| dibromomethane | 74-95-3 | −0.3 | 8.1 | 0.2 | −0.3 | −28.6 | −20.9 | 0.2 |
| tribromomethane | 75-25-2 | −0.8 | 9.9 | 0.2 | −0.1 | −42.5 | −33.4 | 0.1 |
| tetrabromomethane | 558-13-4 | −1.6 | 11.9 | 0.1 | 0.0 | −56.1 | −45.7 | −0.1 |
| bromoethyne | 593-61-3 | −0.1 | 11.8 | 0.3 | −1.0 | −14.9 | −4.0 | −0.6 |
| dibromoethyne | 624-61-3 | −0.5 | 13.0 | 0.2 | −0.9 | −29.0 | −17.2 | 0.0 |
| bromoethene | 593-60-2 | −0.1 | 11.6 | 0.3 | −1.1 | −14.9 | −4.2 | 0.0 |
| (Z)-1,2-dibromoethene | 590-11-4 | −0.5 | 13.3 | 0.3 | −0.9 | −29.0 | −16.8 | 0.0 |
| 1,1-dibromoethene | 593-92-0 | −0.5 | 13.3 | 0.3 | −0.9 | −28.9 | −16.7 | −0.2 |
| tribromoethene | 598-16-3 | −1.0 | 14.8 | 0.3 | −0.7 | −43.0 | −29.6 | 0.2 |
| tetrabromoethene | 79-28-7 | −1.8 | 16.8 | 0.2 | −0.6 | −56.9 | −42.3 | 0.3 |
| hydrogen bromide | 10035-10-6 | 0.1 | 2.0 | 0.1 | 0.3 | −14.2 | −11.7 | −0.6 |
| dibromine | 7726-95-6 | 1.7 | 1.5 | 0.0 | 0.2 | −27.7 | −24.3 | −0.5 |
| methane | 74-82-8 | 0.1 | 5.3 | 0.3 | −0.8 | −0.4 | 4.5 | 0.0 |
| dihydrogen | 1333-74-0 | 0.0 | 0.0 | 0.2 | 0.0 | 0.0 | 0.2 | 0.1 |
All computed results are taken from Ref. 12;
HOC, higher-order correlation beyond CCSD(T); CV, core-valence correlation; DBOC, diagonal Born-Oppenheimer correction; PP, difference in energy between the ECP results with the awCVQZ-PP basis and the DK results with the awCVQZ basis set with all outer-core electrons correlated; SO, spin-orbit interaction; , sum of all contributions; , deviation of Eq. 13 from .
The data for the auxiliary compounds used in the following sections are compiled in Table 4.
Table 4:
Standard enthalpies of formation for the liquid phase and the ideal gas for auxiliary compounds at the reference temperature a
| Compound | CASRN | Referencea | ||
|---|---|---|---|---|
| hydrogen atom | 12385-13-6 | 217.998 ± 0.006 | 38 | |
| water | 7732-18-5 | −(285.83 ± 0.04) | −(241.83 ± 0.04) | 38 |
| carbon dioxide | 124-38-9 | −(393.51 ± 0.13) | 38 | |
| bromine atom | 10097-32-2 | 111.85 ± 0.06 | 39 | |
| hydrogen bromide | 10035-10-6 | −(35.69 ± 0.13) | 39 | |
| 10035-10-6 | 39,61 | |||
| dibromine | 7726-95-6 | 0 | 30.91 ± 0.11 | 38 |
| hydrogen iodide | 10034-85-2 | 26.47 ± 0.04 | 39 | |
| iodine bromide | 7789-33-5 | 40.80 ± 0.14 | 67 | |
| ethane | 74-98-6 | −(93.6 ± 2.5) | 8 | |
| propane | 74-98-6 | −(120.9 ± 2.5) | 8 | |
| butane | 106-97-8 | −(147.3 ± 2.5) | 8 | |
| 2-methylpropane | 75-28-5 | −(154.2 ± 1.2) | 8 | |
| 2-methylbut-2-ene | 75-28-5 | −(41.7 ± 1.0) | 8 | |
| propene | 115-07-1 | 4.0 ± 2.5 | 20.02 ± 0.18 | 8; 39 |
| but-1-ene | 106-98-9 | −20.8 ± 1.0 | 0.19 ± 0.34 | 8; 39 |
| Compound | CASRN | Referenceb | ||
|
| ||||
| propan-1-ol | 71-23-8 | −(255.11 ± 0.24) | 39 | |
| allyl alcohol | 107-18-6 | −(169.5 ± 1.6) | −(124.5 ± 1.3) | 40; 68 |
| difluoromethane | 75-10-5 | −(452.3 ± 1.0) | 69 | |
| trifluoromethane | 75-46-7 | −(696.6 ± 2.8) | 69 | |
| tetrafluoroethylene | 116-14-3 | −(673.2 ± 3.0) | 24 | |
| trichloromethane | 67-66-3 | −(134.1 ± 2.5) | 70 | |
| 2-chloropropane | 75-29-6 | −(144.9 ± 1.3) | 8 | |
For hydrocarbons, only the sources different from ATcT v.1.13039 are listed.
Sources of for the liquid and gas phases (if different) are separated by semicolon.
To use the ab initio protocol described above, the effective enthalpy of a bromine atom, , needs to be quantified. Ideally, the compounds included in the training set should have the values based on multiple consistent results from independent sources, which is not possible in the case of brominated compounds. Thus, a different approach was taken, as follows.
We were able to derive the gas-phase enthalpies of formation for about 80 molecular compounds containing , or F atoms in addition to univalent Br. All available experimental results were evaluated without the use of ab initio results to obtain a single value for each compound. Out of all data, the values with repeatability-based expanded uncertainties under per bromine atom were selected. Uncertainties of this type represent the lower limit of this quantity. Potential data problems were detected using the ab initio results at the next step. The compounds with inconsistent data from different laboratories as well as with significant conformational ambiguity were excluded. These selection criteria decreased the number of candidates to 37.
The values were found for each of the remaining compounds using Eq. 1. Since the distribution was found to significantly deviate from the normal one (Fig. 1), its median value was used as a preliminary estimate of the effective enthalpy. The observed distribution, 27 out of 37 values lie within (−2.0 to of the median value. This domain was selected for further use. For the considered dataset, this is similar to the using of data between the fifteenth and eighty-fifth percentiles. Detailed discussion of the outliers and the entire dataset of brominated compounds is a subject of future work and is not covered here. The obtained dataset (Table 5) contained three inorganic, fifteen aliphatic, and nine aromatic compounds. The production effective enthalpy was found by averaging their effective enthalpies.
Figure 1:
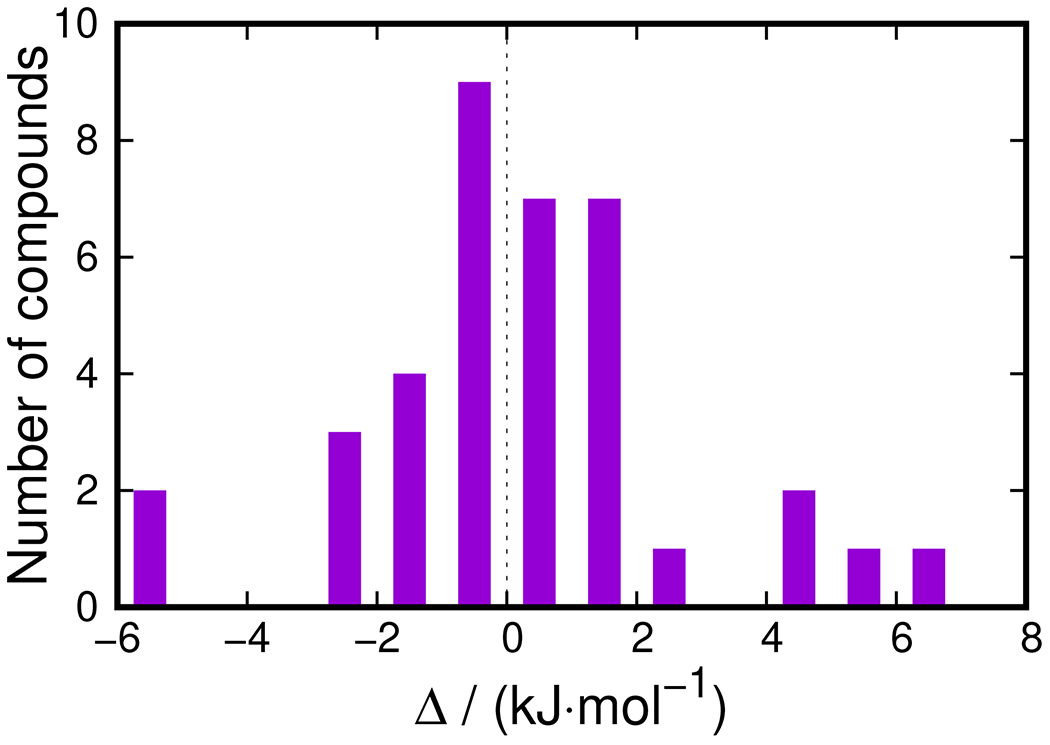
Distribution of compounds with respect to the deviation of the effective enthalpy of , calculated from their experimental gas-phase from the median value for the preliminary set of compounds.
Table 5:
Experimental standard enthalpies of formation for the ideal gas for the training set at the reference temperature and deviation of the effective enthalpies calculated for each compound from the average value
| Compound | CASRN | Referencea | ||
|---|---|---|---|---|
| Inorganic compounds | ||||
| hydrogen bromide | 10035-10-6 | −35.7 ± 0.1 | 39 | −0.4 |
| bromine fluoride | 13863-59-7 | −58.85 ± 1.0 | 67 | −0.9 |
| dibromine | 7726-95-6 | 30.9 ± 0.1 | 38 | 1.9 |
| Aliphatic compounds | ||||
| bromotrifluoromethane | 75-63-8 | −648.4 ± 1.9 | 7,72–74 | −1.0 |
| bromodifluoromethane | 1511-62-2 | −425.6 ± 1.2 | 75 | 1.6 |
| tribromomethane | 75-25-2 | 57.0 ± 3.1 | 76,77b; 78 | 0.1 |
| bromomethane | 74-83-9 | −36.4 ± 0.2 | 7,79–81; 82 | 0.4 |
| 1,2-dibromotetrafluoroethane | 124-73-2 | −803.2 ± 4.1 | 50 | −0.6 |
| 1,2-dibromoethane | 106-93-4 | −38.4 ± 1.3 | 7,83 | −0.3 |
| bromoethane | 74-96-4 | −62.0 ± 0.7 | 7,79,84–87; 88–90 | −0.3 |
| 3-bromopropene | 106-95-6 | 46.1 ± 2.3 | 91,92; 90 | −1.9 |
| 1,2-dibromopropane | 78-75-1 | −(72.1 ± 0.9) | 83 | −0.3 |
| 1,3-dibromopropane | 109-64-8 | −61.6 ± 0.9) | 93; 94–96 | −1.2 |
| 1-bromobutane | 109-65-9 | −105.8 ± 1.0 | 7,97,98; 90,94,99,100 | −0.4 |
| 2-bromobutane | 78-76-2 | −119.2 ± 0.3 | 7,101–103; 94,100 | 1.3 |
| 1-bromo-2-methylpropane | 78-77-3 | −113.5 ± 1.3 | 92,104; 90,94 | −0.8 |
| 2-bromo-2-methylpropane | 507-19-7 | −131.7 ± 1.1 | 7,105,106 | 0.0 |
| 2,3-dibromo-2-methylbutane | 594-51-4 | −138.4 ± 2.0 | 7,83 | 1.8 |
| Aromatic compounds | ||||
| 2-bromophenol | 95-56-7 | −77.5 ± 2.0 | 107; 107 | 1.4 |
| 4-bromophenol | 106-41-2 | −67.7 ± 1.5 | 107; 107,108: 109 | 0.1 |
| 2,6-dibromophenol | 608-33-3 | −48.0 ± 2.2 | 110; 110 | 1.0 |
| 4-bromobenzoic acid | 586-76-5 | −271.8 ± 2.0 | evaluated in Ref. 71c | 0.3 |
| 2-bromomethoxybenzene | 578-57-4 | −39.1 ± 2.1 | 111; 111 | −1.9 |
| 3-bromomethoxybenzene | 2398-37-0 | −47.9 ± 2.1 | 111; 111 | −0.1 |
| 4-bromomethoxybenzene | 104-92-7 | −47.8 ± 2.2 | 111; 111 | 1.5 |
| 4’-bromoacetophenone | 99-90-1 | −59.6 ± 1.3 | 112; 112,113 | −0.5 |
| 2-bromofluorene | 1133-80-8 | 208.7 ± 2.3 | 114; 114,115 | 0.5 |
Sources of combustion or reaction energies used to derive the condensed-phase and sources of enthalpies of sublimation or vaporization are separated by semicolon.
Gas-phase value
Could not be corrected with respect to the updated because of insufficient information in some original publications noted by Chirico et al. 71
The experimental enthalpies of vaporization of brominated compounds are collected in Table 6. The experimental and computed enthalpies of formation are given in Table 7.
Table 6:
Experimental enthalpies of vaporization and derived standard enthalpies of vaporization at the reference temperature for brominated hydrocarbonsa
| range | Methodb | Eq. | Year | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| Propane and propene derivatives | ||||||||
| 1-bromopropane (106-94-5) | ||||||||
| 342.0 | 342.0 | 29.9 ± 0.3 | C | 32.3 ± 0.3 | 11 | 48 ± 2116 | 1931 | Mathews and Fehlandt40,90,99 |
| 298.2 | 298.2 | C | 32.0 ± 0.2 | 1966 | Wadsö40,90,100 | |||
| 322.3 to 352.4 | C | 32.4 ± 0.1 | 11 | 48 ± 2116 | 1977 | Svoboda et al.40,90 | ||
| 302.1 to 343.5 | E | 32.1 ± 0.1 | 1977 | Svoboda et al.40,90 | ||||
| N/A | E | 32.2 | 1985 | Radnai et al.40,117 | ||||
| 32.2 ± 0.1 | ||||||||
| 2-bromopropane (75-26-3) | ||||||||
| 331.8 | 331.8 | 28.4 ± 0.3 | C | 30.4 ± 0.3 | 11 | 50 ± 2118 | 1931 | Mathews and Fehlandt40,90,99 |
| 298.2 | 298.2 | C | 30.3 ± 0.2 | 1966 | Wadsö40,90,100 | |||
| 297.4 to 333.1 | E | 30.7 ± 0.9 | 1974 | van Diemen et al.40,119 | ||||
| 304.6 to 337.7 | C | 30.3 ± 0.1 | 11 | 50 ± 2118 | 1977 | Svoboda et al.40,90 | ||
| 299.7 to 331.4 | E | 30.3 ± 0.1 | 1977 | Svoboda et al.40,90 | ||||
| 30.3 ± 0.1 | ||||||||
| 1,2-dibromopropane (78-75-1) | ||||||||
| 312.8 to 402.4 | E | 42.5 ± 0.8 | 1975 | Pisarev et al. 120–122 | ||||
| 298.2 | 298.2 | 41.7 ± 0.1 | C | 41.7 ± 0.1 | 1980 | Varushchenko et al. 123 | ||
| 41.7 ± 0.1 | ||||||||
| 1,3-dibromopropane (109-64-8) | ||||||||
| 298.2 | 298.2 | 47.5 ± 0.1 | C | 47.5 ± 0.1 | 1968 | Wadsö94 | ||
| N/A | N/A | 47.2 ± 1.0 | E | 47.2 ± 1.0 | 1978 | Varushchenko et al.95 | ||
| 308.2 to 338.2 | 48.1 ± 0.5 | C | 48.1 ± 0.5124 | 1992 | Svoboda et al.96 | |||
| 47.5 ± 0.1 | ||||||||
| 3-bromopropene (106-95-6) | ||||||||
| 297.7 to 343.0 | 318.3 | E | 32.2 ± 0.3 | 1977 | Svoboda et al. 40,90 | |||
| 318.0 to 340.7 | 329.5 | C | 32.6 ± 0.6 | 11 | 41 ± 30 | 1977 | Svoboda et al. 40,90 | |
| 32.3 ± 0.3 | ||||||||
| Monobromobutanes | ||||||||
| 1-bromobutane (109-65-9) | ||||||||
| 372.6 | 372.6 | 32.5 ± 0.3 | C | 37.0 ± 0.3 | 11 | 54 ± 1125 | 1931 | Mathews and Fehlandt 40,99 |
| 298.2 | 298.2 | C | 36.7 ± 0.3 | 1966 | Wadsö 40,100 | |||
| 298.2 | 298.2 | C | 36.7 ± 0.1 | 1968 | Wadsö 40,94 | |||
| 322.3 to 366.4 | C | 37.0 ± 0.1 | 1977 | Svoboda et al. 40,90 | ||||
| 36.9 ± 0.1 | ||||||||
| 2-bromobutane (78-76-2) | ||||||||
| 298.2 | 298.2 | 34.4 ± 0.2 | C | 34.4 ± 0.2 | 1966 | Wadsö 40,100 | ||
| 298.2 | 298.2 | 34.5 ± 0.1 | C | 34.5 ± 0.1 | 1968 | Wadsö 40,94 | ||
| 34.5 ± 0.1 | ||||||||
| 1-bromo-2-methylpropane (78-77-3) | ||||||||
| 298.2 | 298.2 | 34.9 ± 0.1 | C | 35.0 ± 0.1 | 1968 | Wadsö 40,94 | ||
| 329.8 to 365.5 | C | 34.8 ± 0.1 | 11 | 54 ± 2118 | 1977 | Svoboda et al. 40,90 | ||
| 305.8 to 363.0 | E | 34.3 ± 0.4 | 1977 | Svoboda et al. 40,90 | ||||
| 34.9 ± 0.1 | ||||||||
| 2-bromo-2-methylpropane (507-19-7) | ||||||||
| see text | S | 30.9 ± 0.3 | 1951 | Bryce-Smith and Howlett 40,126 | ||||
| 298.2 | 298.2 | 31.8 ± 0.1 | C | 31.9 ± 0.1 | 1968 | Wadsö 40,94 | ||
| 295.2 to 323.2 | 308.7 | 31.4 ± 0.5 | S | 32.1 ± 0.5 | 11 | 51 ± 2118 | 1969 | Chóliz Calero et al. 40,127 |
| 31.9 ± 0.1 | ||||||||
| Dibromobutanes | ||||||||
| 1,2-dibromobutane (533-98-2) | ||||||||
| 273.2 to 333.0 | 301.3 | 45.7 ± 2.5 | S | 45.9 ± 2.5 | 11 | 56 ± 30 | 1941 | Lister 128 |
| 331.32 to 426.0 | E | 46.0 ± 0.7 | 1975 | Pisarev et al. 120,122 | ||||
| 46.0 ± 0.6 | ||||||||
| 1,3-dibromobutane (107-80-2) | ||||||||
| 354.8 to 466.2 | E | 49.4 ± 0.9 | 1980 | Varushchenko et al. 123 | ||||
| 1,4-dibromobutane (110-52-1) | ||||||||
| 305.2 to 470.7 | see text | 51.7 ± 1.5 | 12 | 55 ± 30 | 1947 | Stull 129 | ||
| 298.2 | 298.2 | 53.1 ± 0.1 | C | 53.1 ± 0.1 | 1968 | Wadsö 94 | ||
| 53.1 ± 0.1 | ||||||||
| 1,2-dibromo-2-methylpropane (594-34-3) | ||||||||
| 327 to 422 | 378.5 | 29 ± 1 | M | 32 ± 2 | 12 | 58 ± 30 | 1927 | Coffin and Maass 130 |
| 298.2 | 298.2 | 43.3 ± 0.1 | C | 43.3 ± 0.1 | 1974 | Sunner and Wulff 131 | ||
| 43.3 ± 0.1 | ||||||||
| 1,3-dibromo-2-methylpropane (28148-04-1) | ||||||||
| 287.2 to 447.8 | see text | 46.4 ± 1.5 | 12 | 58 ± 30 | 1947 | Stull 129 | ||
The excluded data are italicized, the evaluation results are in bold
C, calorimetry; E, ebulliometry; S, static method; M, manometric measurements
Table 7:
Experimental standard enthalpies of formation for the liquid phase and the ideal gas , experimental standard enthalpies of vaporization or sublimation , and the computed enthalpies of formation for the ideal gas ; calc) for brominated hydrocarbons at the reference temperature a
| Method | range | Year | Reference | ||||
|---|---|---|---|---|---|---|---|
| Bromo- and dibromopropanes | |||||||
| 1-bromopropane (106-94-5) | |||||||
| R | 367 | −76.8 ± 0.7 | 1950 | Lacher et al. 132 | |||
| R | 401 | −89.9 ± 1.4 | 1957 | Lacher et al. 133 | |||
| C | 298.2 | −(123.5 ± 1.4) | 1961 | Bjellerup 7,102 | |||
| E | 423.9 to 514.0 | −(83.6 ± 1.2) | 1964 | Rozhnov and Andreevskii 134 | |||
| R | 523 | −(83.8 ± 0.6) | 1965 | Davies et al. 135 | |||
| E | 273.2 to 303.2 | −(84.0 ± 1.6) | 1974 | Alenin et al. 136,137 | |||
| −(116.0 ± 0.5) | 32.2 ± 0.1 | −(83.8 ± 0.5) | −(85.6 ± 2.6) | ||||
| 2-bromopropane (75-26-3) | |||||||
| R | 367 | −(99.7 ± 1.0) | 1950 | Lacher et al. 132 | |||
| R | 401 | −(100.7 ± 0.7) | 1957 | Lacher et al. 133 | |||
| C | 298.2 | −(128.2 ± 1.8) | 1961 | Bjellerup 7,102 | |||
| E | 393.8 to 447.5 | −(97.7 ± 3.4) | 1963 | Rozhnov and Andreevskii 138 | |||
| R | 523 | −(95.6 ± 1.0) | 1965 | Davies et al. 135 | |||
| MS | 0 | −(95.9 ± 0.8) | 2010 | Stevens et al. 139 | |||
| MS | 0 | −(94.9 ± 1.5) | 2010 | Stevens et al. 139 | |||
| −(126.3 ± 0.7) | 30.3 ± 0.1 | −(95.9 ± 0.7) | −(96.6 ± 2.6) | ||||
| 1,1-dibromopropane (598-17-4) | |||||||
| E | 373 to 543 | −(93.9 ± 1.0) | 1974 | Izmailov et al. 140,141 | |||
| −(93.9 ± 1.0) | −(53.9 ± 2.7) | ||||||
| 1,2-dibromopropane (78-75-1) | |||||||
| R | 355 | −(72.1 ± 0.9) | 1938 | Conn et al. 7,83 | |||
| −(113.8 ± 0.9) | 41.7 ± 0.1 | −(72.1 ± 0.9) | −(72.7 ± 2.7) | ||||
| 1,3-dibromopropane (109-64-8) | |||||||
| E | 273 to 403 | −(109.1 ± 0.9) | 1971 | Sharonov and Rozhnov 93 | |||
| −(109.1 ± 0.9) | 47.5 ± 0.1 | −(61.6 ± 0.9) | −(64.0 ± 2.7) | ||||
| 2,2-dibromopropane (594-16-1) | |||||||
| E | 397.7 to 446.2 | −(70.4 ± 3.0) | 1971 | Sharonov and Rozhnov 142 | |||
| −(70.4 ± 3.0) | −(68.3 ± 2.7) | ||||||
| C3H5Br compounds | |||||||
| (Z)-1-bromopropene (590-13-6) | |||||||
| MS | 0 | 38.4 ± 2.7 | 2012 | Borkar et al. 143 | |||
| 38.4 ± 2.7 | 39.1 ± 2.6 | ||||||
| (E)-1-bromopropene (590-15-8) | |||||||
| MS | 0 | 40.4 ± 2.6 | 2012 | Borkar et al. 143 | |||
| 40.4 ± 2.6 | 41.1 ± 2.6 | ||||||
| 2-bromopropene (557-93-7) | |||||||
| MS | 0 | 34.5 ± 2.7 | 2012 | Borkar et al. 143 | |||
| 34.5 ± 2.7 | 32.9 ± 2.6 | ||||||
| 3-bromopropene (106-95-6) | |||||||
| R | 298.2 | 15.0 ± 2.6 | 1949 | Gellner and Skinner 91 | |||
| R | 308.2 | 10.6 ± 4.4 | 1981 | Holm 92 b | |||
| 13.8 ± 2.3 | 32.3 ± 0.3 | 46.1 ± 2.3 | 44.2 ± 2.6 | ||||
| bromocyclopropane (4333-56-6) | |||||||
| R | 308.2 | See text | 1981 | Holm 92,144 b | |||
| MS | 0 | See text | 2012 | Borkar et al. 143 | |||
| 79.6 ± 2.6 | |||||||
| Bromobutanes | |||||||
| 1-bromobutane (109-65-9) | |||||||
| C | 298.2 | −(143.1 ± 1.3) | 1961 | Bjellerup 7,97 | |||
| E | 298.2 to 513.7 | −(105.2 ± 1.4) | 1968 | Peshchenko and Andreevskii 98 | |||
| −(142.6 ± 1.0) | 36.9 ± 0.1 | −(105.8 ± 1.0) | −(106.2 ± 2.6) | ||||
| 2-bromobutane (78-76-2) | |||||||
| R | 367 | −(119.3 ± 0.6) | 1952 | Lacher et al. 7,101 | |||
| R | 373 | −(119.7 ± 0.5) | 1952 | Lacher et al. 7,101 | |||
| R | 373 | −(119.0 ± 0.6) | 1952 | Lacher et al. 7,101 | |||
| C | 298.2 | −(154.4 ± 1.3) | 1961 | Bjellerup 7,97 | |||
| E | 405.2 to 433.0 | −(118.5 ± 0.6) | 1968 | Peshchenko and Andreevskii 103 | |||
| −(153.7 ± 0.3) | 34.5 ± 0.1 | −(119.2 ± 0.3) | −(117.9 ± 2.6) | ||||
| 1-bromo-2-methylpropane (78-77-3) | |||||||
| R | 308.2 | −(151.6 ± 4.4) | 1981 | Holm 92 b | |||
| E | 273.2 to 526.2 | −(148.1 ± 1.3) | 1974 | Nesterova and Rozhnov 104 | |||
| −(148.4 ± 1.3) | 34.9 ± 0.1 | −(113.5 ± 1.3) | −(114.3 ± 2.6) | ||||
| 2-bromo-2-methylpropane (507-19-7) | |||||||
| E | 375.0 to 531.7 | −(131.9 ± 2.1) | 1937 | Kistiakowsky and Stauffer 7,105 | |||
| E | 372.9 to 483.6 | −(131.7 ± 1.3) | 1957 | Howlett 7,106 | |||
| −(163.7 ± 1.1) | 31.9 ± 0.1 | −(131.7 ± 1.1) | −(131.7 ± 2.6) | ||||
| bromocyclobutane (4399-47-7) | |||||||
| R | 308.2 | −(7.9 ± 4.4) | 1981 | Holm 92 b | |||
| −(7.9 ± 4.4) | 32.7 ± 2.6 | ||||||
| 1,2-dibromobutane (533-98-2) | |||||||
| R | 355 | −(93.5 ± 1.7) | 1938 | Conn et al. 7,83 | |||
| R | 300 | −(133.4 ± 2.7) | 1941 | Lister 128 | |||
| C | 298.2 | −(146.2 ± 2.3) | 1961 | Bjellerup 7,97 | |||
| −(142.9 ± 4.5) | 46.0 ± 0.6 | −(96.9 ± 3.3) | −(94.6 ± 2.7) | ||||
| 1,3-dibromobutane (107-80-2) | |||||||
| E | 275.2 to 553.2 | −(149.2 ± 3.4) | 1972 | Nesterova et al. 145 | |||
| −(149.2 ± 3.4) | 49.4 ± 0.9 | −(99.8 ± 3.5) | −(97.7 ± 2.7) | ||||
| 1,4-dibromobutane (110-52-1) | |||||||
| E | 275.2 to 553.2 | −(141.3 ± 3.4) | 1972 | Rozhnov et al. 146 | |||
| −(141.3 ± 3.4) | 53.1 ± 0.1 | −(88.2 ± 3.4) | −(86.2 ± 2.7) | ||||
| meso-2,3-dibromobutane (5780-13-2) | |||||||
| E | 275.2 to 553.2 | −(150.2 ± 3.4) | 1972 | Nesterova et al. 145 | |||
| −(102.5 ± 2.7) | |||||||
| dl-2,3-dibromobutane (598-71-0) | |||||||
| E | 275.2 to 553.2 | −(151.9 ± 3.4) | 1972 | Nesterova et al. 145 | |||
| −(103.3 ± 2.7) | |||||||
| 1,2-dibromo-2-methylpropane (594-34-3) | |||||||
| R | 298.2 | −(156.6 ± 0.6) | 1974 | Sunner and Wulff 131 | |||
| −(156.6 ± 0.6) | 43.3 ± 0.1 | −(113.3 ± 0.6) | −(107.8 ± 2.7) | ||||
| 1,3-dibromo-2-methylpropane (28148-04-1) | |||||||
| E | 273.2 to 553.2 | −(147.8 ± 0.6) | 1975 | Rozhnov et al. 147 | |||
| −(147.8 ± 0.6) | 46.4 ± 1.5 | −(101.4 ± 1.6) | −(93.0 ± 2.7) | ||||
The excluded outliers are italicized, the evaluated experimental results are in bold
See Table 8
Reaction calorimetry through organic magnesium bromides
The majority of data sources considered here have measurements of the enthalpy-related properties for the target compounds as their main objectives. In some cases, however, additional useful information can be obtained as a by-product of seemingly unrelated data such as reaction calorimetry for inorganic or organometallic systems.125,148
Holm92,144 reported the enthalpies of two reactions involving organic magnesium bromides RMgBr in diethyl ether , which he described as
| (14) |
| (15) |
The experiments were carried out at . The concentration of RMgBr was between (0.3 and 0.6) . A sum of reactions 14 and 15,
| (16) |
can be used to obtain the value for RBr, if and ) are known. Normally, the latter value is not available, and it is reasonably assumed that . Also, the heat-capacity change in reaction 16 cannot be reliably estimated and is ignored.
We found in at using the available experimental data for five organic bromides (Table 8) to be . Deviations of the individual values from the average did not exceed . The values for 2-bromopropane and isomeric bromobutanes were derived from independent sources, as described below. The references for bromoethane are given in Table 5.
Table 8:
Enthalpies of reactions 14 and ,92,144 standard enthalpies of formation in the liquid phase at of brominated compounds RBr and the corresponding hydrocarbons 8 as well as derived enthalpies of formation of
| RBr | CASRN(RBr) | |||||
|---|---|---|---|---|---|---|
| Reference compoundsa | ||||||
| bromoethane | 74-96-4 | −231.0 | −299.2 | −(93.6 ± 2.5) | −(90.1 ± 0.7) | −563.0 |
| 2-bromopropane | 75-26-3 | −210.0 | −305.9 | −(120.9 ± 2.5) | −(126.3 ± 0.7) | −557.6 |
| 1-bromobutane | 109-65-9 | −233.9 | −292.5 | −(147.3 ± 2.5) | −(142.6 ± 1.0) | −558.0 |
| 2-bromobutane | 78-76-2 | −211.7 | −305.9 | −(147.3 ± 2.5) | −(153.7 ± 0.3) | −560.3 |
| 2-bromo-2-methylpropane | 507-19-7 | −209.2 | −306.7 | −(154.2 ± 1.2) | −(163.7 ± 1.1) | −561.7 |
| Average | −(560.1 ± 2.1) | |||||
| Compounds with derived | ||||||
| 1-bromo-2-methylpropane | 78-77-3 | −237.3 | −289.1 | −(154.2 ± 1.2) | −(151.6 ± 4.4) | |
| bromocyclopropane | 4333-56-6 | −281.6 | −282.8 | 35.8 ± 0.5b | c | |
| bromocyclobutane | 4399-47-7 | −223.4 | −289.1 | 3.9 ± 0.4b | −(7.4 ± 4.4) | |
| 3-bromopropene | 106-95-6 | −271.0 | −259.4 | 4.0 ± 2.5 | 10.6 ± 4.4 | |
Solid-liquid equilibrium with the equilibrium solid phase and metastable liquid-liquid equilibrium have been reported for 149–151 at . If SLE was reached in the considered experiments, should be present in both phases. According to the reported solubilities,149,151 about a half of the formed bromide would precipitate in the experiments with the lower RMgBr concentration of . The final solution also contained non-polar hydrocarbons that are anticipated to decrease the salt solubility. Therefore, is expected to be primarily composed of solid . This suggestion is consistent with the fact that, as mentioned above, the values obtained with different reactants agree within . The uncertainty of reported by Holm appears to be a reasonable estimate for the expanded uncertainty of the enthalpies of formation obtained using this procedure.
Using the value obtained as described above, the enthalpies of formation were derived for four organic bromides (Table 8). For bromocyclopropane, the enthalpy of reaction 14 reported in the earlier paper144 is missing in the author’s thesis152 and a more recent paper.92 reported in the 1981 paper92 significantly deviates from the value of obtained here using the published reaction enthalpies. The latter, in turn, is inconsistent with the ab initio results. Therefore, the enthalpy of formation of bromocyclopropane is excluded from further analysis.
Brominated Propanes
1- and 2-bromopropanes are liquids readily available at high purity. Despite that, it took significant efforts to obtain a consistent set of thermochemical data for these compounds. They were studied by combustion calorimetry by Bjellerup.102 The enthalpies of vaporization required to obtain the gas-phase values are primarily based on the calorimetric and ebulliometric results.90,100 The gas-phase enthalpies of hydrobromination of cyclopropane and propene resulting in 1- and 2-bromopropanes, respectively, were reported by Lacher et al.132 Later, they repeated the measurements to improve the former value.133 The enthalpies of hydrogenation of both bromopropanes in the gas phase were also determined at the same laboratory.135 For both compounds, the enthalpies of formation derived from the hydrogenation study deviate from the earlier results by several , which exceeds their combined uncertainty. The equilibrium of the 2-bromopropane dehydrobromination in the temperature range of (394 to 447) K was studied by Rozhnov and Andreevskii.138 The resulting gas-phase values for 2-bromopropane vary within a reasonable range, . The results can be further refined, as demonstrated below.
The enthalpy of formation of 2-bromopropane can also be derived from photoionization studies. In these experiments, the 2-propylium ion is formed. Since 2000, the enthalpy of formation of this ion has been determined using different variations of the PEPICO technique.139,153,154 As demonstrated by Bodi et al.,155 the experiments of Baer et al. 153 were affected by formation of an adduct with argon and should not be used. Stevens et al. 139 suggested that the 2-chloropropane ionization energy reported by Brooks et al. 154 was too low by because of improper calibration.
In Table 9, the experimental enthalpies of formation of carbocations are compared with their ab initio counterparts. The values of some ions have been obtained from the photoionization studies for appropriate hydrocarbons. The values for 1-propylium and propen-2-ylium cations are based on the ones for 2-propylium and allylium, respectively, and should not be treated as independent entries in this context.
Table 9:
Standard enthalpies of formation in the ideal-gas state of ions at and calculated in this work and reported in the literature and thermal enthalpy of these ions a
| Ion | CASRN | Reference compound | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Calc. | Exp. | ||||||||
| hydronium | 12408-02-5 | 1531.3 | 0 | 6.20 | 0.0 | 1529.3 | 1528.1 | 39 | |
| methylium | 14531-53-4 | 1093.8 | 81.2 | 9.98 | 2.68 | 1100.3 | 1099.35 ± 0.05 | 156 | |
| ethylium (ethenium) | 14936-94-8 | 903.5 | 158.1 | 11.00 | 1.25 | 917.0 | 915.5 ± 1.3 | 87 b | |
| 1-propylium (cyclopropanium) | 19252-52-9 | 838.5 | 236.4 | 14.49 | 1.36 | 858.1 | 856.8 ± 0.8 | 39 b | |
| 2-propylium | 19252-53-0 | 806.9 | 228.7 | 15.32 | 0.62 | 825.0 | 822.8 ± 0.3 | 139 | |
| 823.8 ± 1.4 | 139 | ||||||||
| vinylium (ethynium) | 14604-48-9 | 1114.2 | 89.3c | 11.28d | 0.89 | 1119.3 | 1120.4 ± 1.9 | 157 | |
| 1119.7 | 158 b | ||||||||
| propen-2-ylium | 50457-57-3 | 985.6 | 164.5 | 14.67 | 0.31 | 995.5 | 997 | 159 b | |
| allylium (propen-3-ylium) | 1724-44-3 | 951.8 | 177.5 | 14.67 | 2.26 | 965.8 | 966.8 ± 2.4 | 159 | |
| 176.8 | 964.6e | 160 | |||||||
Enthalpies of formation are based on the ion (stationary electron) convention
Derived using experimental and ab initio results
To improve quality of the computations, ZPVE was calculated using the unscaled harmonic frequencies and anharmonic constant matrix found using the CCSD(T)-based composite method as described in Ref. 161
Using unscaled VPT2 frequencies reported in Ref. 161
Calculated using the HEAT-345(Q) protocol
The deviation of the computed value for 2-propylium from the experimental result obtained by propane ionization is larger than those of the hydronium, methylium, and ethylium cations. The enthalpy of formation of 2-propylium can also be obtained from the dissociation onset energies of 2-haloalkanes, particularly the chloro or iodo derivatives. To find propylium), accurate enthalpies of formation of the halogenated compounds are required. The uncertainty of (2-iodopropane), 8 is too large for this purpose. The enthalpy of formation of 2-chloropropane,8 at corresponding to at , is based on multiple consistent experimental values. The (2-propylium) values obtained from the dissociation onset energies of 2-chloropropane139,154 are also shown in Table 9. These values are closer to the ab initio result.
Ionization of 2-bromopropane was studied in Ref. 139. We obtained its enthalpy of formation using the results both for propane and 2-chloropropane. Once the ionization results are added to the thermochemical data (Table 7), it becomes clear that both hydrobromination studies of Lacher et al. 132,133 yield values which are too negative and should be excluded. The recommended gas-phase value is obtained by weight-averaging of the remaining data. The only liquid-phase was converted to the gas-phase value using the vaporization enthalpy derived in Table 6.
For 1-bromopropane, the variation of the values, , is significantly worse than that for the 2-bromo isomer. The equilibrium data for the reaction 1-bromopropane -bromopropane134,136,163 are available. Mayo and Dolnick163 reported the liquid-phase equilibrium compositions at T = (299, 343, and 523) K. However, the latter temperature is above the estimated critical temperature of 517 K for 2-bromopropane.23 Uncertainty of the enthalpy derived from the remaining two data points would be significant and these results are not considered in the further discussion.
The gas-phase equilbrium was studied in the temperature range of (423.9 to 514.0) K.134 The reaction enthalpy was found by the least-squares fit of the vs. data to be at . This value is reduced to using the thermal enthalpies from the ideal-gas model of this work to be 1.0) . The equilibrium in the liquid phase was stated to be studied at T = (273.2, 303.2, 333.2, 533.2, and 583.2) K. 136 As discussed above, the latter two temperatures are too high for the liquid phase and only three points at the lower temperatures are used here. The value at is derived from these data. The temperature correction to is estimated to be negligible. The corresponding gas-phase value, , can be found using Table 6. These two isomerization enthalpies are combined with of 2-bromopropane selected as described above to obtain two additional experimental values for the 1-bromo isomer. As in the case of 1-bromopropane, the hydrobromination results of Lacher et al.132,133 are found to be outliers. Unexpectedly, the value based on the combustion calorimetry study102 also appears anomalous. The other experimental values are weight-averaged to obtain the best estimate of for 1-bromopropane. The enthalpies of formation derived from the experimental data and their computational counterparts for both bromopropanes differ by less than .
The gas-phase enthalpy of formation of 1,2-dibromopropane is based on the enthalpy of the gas-phase bromination of propene reduced to . The enthalpy of vaporization is mainly based on the calorimetric value reported by Varushchenko et al. 123
For 1,3-dibromopropane, two calorimetric94,96 and one ebulliometric95 values of the vaporization enthalpy are available. Svoboda et al.96 reported the values in the temperature range of (308 to 338) K. The heat-capacity change at vaporization of follows from the linear fit of the dependence. However, the at can be obtained from the statistical thermodynamic calculations for the gas phase and the experimental liquid heat capacity.124 Therefore, the uncertainty of the value of Svoboda et al. is larger than the reported expanded uncertainty of . It was estimated to be . The recommended enthalpy of vaporization is primarily based on the calorimetric value of Wadsö.94
The liquid-phase equilibria of 1,2-dibromopropane with the 1,1- and 1,3-isomers was studied by Rozhnov et al.93,140,141 The eqilibrium with the 1,1-isomer was studied in the temperature range of (373 to 543) K. The enthalpy of this isomer was found to be (19.9 higher than that of the 1,2-derivative. We are not aware of any heat-capacity data for 1,1-dibromopropane, but the heat capacities at of the other two isomers of dibromopropane are indistinguishable within their uncertainty.118,124 Therefore, the enthalpy difference between 1,1- and 1,2-dibromopropanes is expected to insignificantly change with temperature. The enthalpy of formation of 1,1-dibromopropane is found as a sum of that for 1,2-dibromopropane and the above isomerization enthalpy. No vaporization enthalpy data is available for the 1,1-isomer. The use of initio value for the gas phase yields a reasonable value of about for its enthalpy of vaporization.
The 1,3-dibromopropane -dibromopropane equilibrium in the liquid phase was studied at . The enthalpy change was found to be . As mentioned above, it is nearly independent of temperature. The enthalpy of formation of the 1,3-isomer in the liquid phase is found here using this value and the enthalpy of formation of 1,2-dibromopropane. The same authors142 also studied the equilibrium of the 2,2-dibromopropane dehydrobromination into 2-bromopropene in the gas phase. The obtained at becomes at . The gas-phase enthalpy of formation of this compound was found using -bromopropene derived below. The results for bromocyclopropane are discussed in the next section.
The computed of 1,2-dibromopropane is more negative than the experimental one. This difference increases to for 1,3-dibromopropane and becomes negative for the 2,2-dibromo isomer.
Brominated Propenes and Cyclopropane
Gellner and Skinner 91 found the enthalpy of the 3-bromopropene hydrolysis reaction
| (17) |
to be (presumably) at room temperature close to . The enthalpy of formation is derived in this work assuming the excess enthalpies of the aqueous ethanol solutions and allyl alcohol in (ethanol + water) are small relative to the resulting uncertainty. The gas-phase enthalpy of formation of allyl alcohol is found from its enthalpy of hydrogenation68 reduced to and (propan-1-ol(g)). To obtain the liquid-phase value, the critically evaluated vaporization enthalpy,40 , is used. The values for ethanol and allyl alcohol (prop-2-en-1-ol) are listed in Table 4.
The liquid-phase enthalpy of formation of 3-bromopropene can also be derived from the calorimetric data of Holm92 as described above. The best estimate of this quantity is obtained by weight-averaging of these two values. The standard enthalpy of vaporization of 3-bromopropene is determined from the calorimetric and ebulliometric measurements by Svoboda et al.90 The calculated gas-phase deviates from its experimental counterpart by .
Borkar et al.143 studied ionization of five compounds using the imaging PEPICO spectroscopy. Four of these compounds, -, and 3-bromopropenes and bromocyclopropane, ultimately dissociated into the allylium cation. 2-bromopropene was found to form propen-2-ylium. The reported enthalpies of formation of all these compounds were based on of the allylium cation determined by Shuman et al. 159 The initio values for the vinylium and allylium cations are about lower than the experimental counterparts (Table 9). Our value for the allylium cation is also in a good agreement difference) with the HEAT-345(Q) value reported recently.160 About half of the difference is due to ZPVE, which was supposedly found at the theory level in the cited paper. Both computed values agree with the experimental enthalpy of formation within its uncertainty.
To derive the values for the neutral species, Borkar et al. 143 generated a thermochemical network containing two domains tied by the PEPICO data. The first domain included the brominated neutral species and the second one consisted of the allylium and propen-2-ylium cations. The enthalpies of formation were found by minimizing the error function, which contained the differences squared with respect to isomerization energies computed with initio methods and the experimental dissociation onset energies. To derive the absolute enthalpies of formation, the network was anchored to the values for allyl cation 159 and bromine atom.49 Based on the subsequent analysis, the experimental value for bromocyclopropane was shown to be an outlier. The observed appearance energy was assumed to correspond to the overall barrier to its dissociation.
In this work, the gas-phase enthalpies of formation were first obtained using the experimental dissociation energy onsets, (allyllium) from Ref. 159, and the statistical thermodynamic model adopted here. of gaseous 3-bromopropene at derived above using the thermochemical results91,92 is lower than the spectroscopic one, , by . The computed values for other bromopropenes are also systematically lower than the spectroscopic results by (4 to 6) . On the other hand, our calculations support the spectroscopic enthalpies of the isomerization of bromopropenes (i.e., relative enthalpies). This discrepancy slightly exceeds the combined uncertainties of the spectroscopic and computed values. The distribution of the deviations, however, suggests a possible systematic error in the ionization data.
To circumvent the suspected systematic error, we chose to use 3-bromopropene as a reference compound with the difference between the enthalpies of formation of the 2-propenyl and allyl cations at , taken from the results reported in Table 9. The enthalpies of formation of the isomers at were found using the equation:
| (18) |
where is the dissociation onset energy, is the thermal correction to enthalpy, and is equal to for 2-bromopropene and otherwise. For 2-bromo- and both 1-bromopropenes, the resulting difference between the computed and experimental values does not exceed . For bromocyclopropane, the computed value is more credible.
Further verification of the chosen evaluation can be derived from the available equilibrium studies in the liquid164 and gas165 phases. For the isomerization reaction of and -1-bromopropenes in the liquid phase, 164 and at are consistent with and , respectively, calculated in this work for the ideal gas. Abell and Adolf165 determined the equilibrium constant of the 3-bromopropene isomerization into a mixture of the 1-bromopropene isomers as well as the one for the reaction between -1- and -1-bromopropenes. The data provided in tables and graphs in the original source are not fully identical and we use the tabulated values for further analysis. The enthalpy difference between -1- and -1-bromopropenes is found from the vs. dependence to be at , compared to the mass-spectrometric, , and ab initio, , values. The fractions of each bromopropene isomer in the equilibrum mixture can also be found using the reported constants. The equilibrium constants for the 3-bromopropene isomerization into each 1-bromopropene determined from these compositions are used to calculate the corresponding enthalpy changes. The (E)-1-bromopropene enthalpy is found to be lower than that of 3-bromopropene by at the same temperature. This enthalpy difference at is from the mass-spectrometric results143 and from the ab initio calculations.
Monobromobutanes
The enthalpies of formation of 1- and 2-bromobutanes in the liquid phase were determined by combustion calorimetry.97 Their enthalpies of vaporization have been reported by multiple authors. Because of the very large number of publications for vapor pressure and enthalpy of vaporization of 1-bromobutane, only the calorimetric works were considered here. The gas-phase values for 2-bromobutane derived from the hydrobromination enthalpies of butene isomers,101 the equilibrium of the 2-bromobutane decomposition to but-1-ene,103 and the combustion data lie within . This is a remarkable result for an equilibrium study considering its relatively narrow temperature range of 28 K. The recommended value was obtained by weight-averaging of these results.
Alenin et al.136,137 reported the constants of the 1-bromobutane -bromobutane equilibrium in the liquid phase at and 583. These temperatures are close to the estimated critical temperature of 1-bromobutane, .23 Therefore, these results are not used here. Peshchenko and Andreevskii98 reported the constants of this equilibrium in the liquid phase at and in the gas phase in a range of temperatures of . At the average temperature, . It becomes at if one uses the ideal-gas model of Paulechka et al.125 for both isomers. The derived gas-phase enthalpy of formation of 2-bromobutane was combined with the isomerization enthalpy to obtain an additional experimental (1-bromobutane) in the gas phase. The recommended value for 1-bromobutane in the gas phase was obtained as the weight-average of this result and the one from combustion calorimetry adjusted for vaporization.
The gas-phase enthalpies of formation of 2-bromo-2-methylpropane were obtained from two consistent works on the equilibrium hydrobromination of 2-methylpropene.105,106 The vaporization enthalpy of 2-bromo-2-methylpropane can be obtained from the results of two tensimetric works126,127 and a calorimetric one.94 Bryce-Smith and Howlett reported the vapor pressures at . We fit these results with a Clarke-Glew-type equation using three adjustable parameters. The data at were found to deviate from the smooting curve by . Outside this interval, the deviations were systematically positive and reached . Therefore, the results in the shorter interval were used to derive the tabulated enthalpy of vaporization. The recommended value is based on two consistent experimental results.94,127
For the fourth bromobutane isomer, 1-bromo-2-methylpropane, two data sources are available. Nesterova and Rozhnov104 reported the liquid-phase constants for the equilibrium between 1- and 2-bromo-2-methylpropanes over a wide temperature range. The enthalpy of the former isomer relative to the latter was found to be at the average temperature of the measurements of 366.8 K. With the use of their heat capacities in the liquid phase at ,118 this difference became at . A sum of the liquid-phase for 2-bromo-2-methylbutane and this difference gives of the 1-bromo isomer. The other value was found from the reaction calorimetry with alkylmagnesium bromides of Holm,92 as described above. The results from both sources agree within their uncertainties. Therefore, the final value for the liquid phase was set to be their weighted average.
The computed gas-phase values for monobromobutanes show excellent agreement with the experimental data (within ).
For bromocyclobutane, the experimental liquid-phase is the only available data point,92 and there is no vaporization data for the quantitative comparison with the ideal-gas ab intio value.
Dibromobutanes
For this group, there are two key compounds whose enthalpies can be derived from calorimetric measurements, 1,2-dibromobutane83,97,128 and 1,2-dibromo-2-methylpropane.131 The liquid-phase isomerization data of Nesterova et al.145–147,166,167 is the only available source of information that can be used to derive the enthalpies of formation of other isomers. The equlibrium constants were reported in the temperature range of (275 to 553) K. The isomerization enthalpies were found from the slope of the experimental vs. dependences and were assumed to be independent of temperature.
Bjellerup studied 1,2-dibromobutane by combustion calorimetry.97 The enthalpy of the liquid-phase bromination of but-1-ene in which this dibromobutane was formed was measured by Lister.128 Conn et al.83 determined the enthalpies of the gas-phase bromination of but-1-ene as well as - and -but-2-enes. In the case of but-1-ene, 1,2-dibromobutane is expected to be a product. For the but-2-enes, a mixture of meso- and -2,3-dibromobutanes of an unknown composition should be formed. The authors of the original publication incorrectly stated that these reactions are stereospecific and reported the reaction enthalpies for the specific 2,3-dibromobutane isomers.
The enthalpies of vaporization of 1,2-dibromobutane determined from the tensimetric128 and ebulliometric120,121 measurements are in good agreement. The gas-phase enthalpies of formation derived from the above data are inconsistent and vary from to . The result of Lister, deviates from the ab initio value by . Considering the demonstrated performance of the computational method, the value of Lister 128 was rejected. Two other gas-phase values are and .102 There is no obvious reason to give a preference to either of them. At the same time, the uncertainty for at least one of the results is far too low. The best estimate of the experimental gas-phase was made by averaging these two values. The uncertainty was estimated as half of their difference. This result also agrees well with the corresponding initio value.
To our knowledge, the only source of vaporization data for 1,3-dibromobutane is an ebulliometric study by Varushchenko et al. 123 The enthalpy of vaporization was derived by the authors from their unpublished vapor-pressure data. This involved a long-range ( extrapolation based exclusively on these results. Therefore, the real uncertainty of this value can be substantially higher. The vaporization enthalpy of 1,4-dibromobutane was determined in a calorimeter by Wadsö.94 This quantity can also be derived from the smoothed vapor pressures reported by Stull. 129 The value tabulated in this work is estimated from two lowest data points at . Deviation of this value from the calorimetric one allows one to estimate its uncertainty to be about . We are not aware of any experimental vaporization data for 2,3-dibromobutanes.
The computed enthalpies of formation of dibromobutanes of normal structure are in good agreement with the experimental values.
Sunner and Wulff 131 determined the enthalpy of formation of 1,2-dibromo-2-methylpropane from the thermochemistry of the addition of bromine to 2-methylpropene in and the vaporization enthalpy of the dibromide. Coffin and Maass130 reported the temperature-dependent vapor pressures of this compound. The vaporization enthalpy found from these results is about lower than the calorimetric value and too low relative to the results for similar compounds. The enthalpy of formation of liquid 1,3-dibromo-2-methylpropane is found as a sum of of 1,2-dibromo-2-methylpropane discussed above and the enthalpy difference between the isomers, 147 The vaporization enthalpy of this compound is derived from the smoothed vapor-pressure data tabulated by Stull129 at and 313.2) K. Its uncertainty is taken to be the same as for -dibromobutane. For both dibromo-2-methylpropanes, the computed are significantly less negative than the experimental values. The differences are and for the 1,2-dibromo and 1,3-dibromo isomers, respectively. The reason for these deviations cannot be determined given the available information and further experimental investigations would be beneficial.
For the isomeric dibromobutanes, the differences between the experimental and computed values are unexpectedly large considering the demonstrated performance of the ab initio procedure for the other brominated compounds. Therefore, one can suggest that the real uncertainties of these experimental results are higher than those reported by the authors. Uncertainties of the experimental enthalpies of formation for normal dibromobutanes are larger than those of the computed values because of the large uncertainty for 1,2-dibromobutane. Therefore, we recommend using the computed gas-phase enthalpies of formation for all dibromobutanes.
Finally, based on the discussion above, the recommended thermochemical properties of the considered brominated hydrocarbons are compiled in Table 10.
Table 10:
Experimental standard enthalpies of formation for the liquid phase and the ideal gas , the computed enthalpies of formation for the ideal gas (g; calc) and corresponding recommended values for brominated hydrocarbons at the reference temperature
| Compound (CASRN) | |||||
|---|---|---|---|---|---|
| Experimental | ab initio | Recommended | |||
| Bromo- and dibromopropanes | |||||
| 1-bromopropane (106-94-5) | −(116.0 ± 0.5) | −(83.8 ± 0.5) | −(85.6 ± 2.6) | −(116.0 ± 0.5) | −(83.8 ± 0.5) |
| 2-bromopropane (75-26-3) | −(126.3 ± 0.7) | −(95.9 ± 0.7) | −(96.6 ± 2.6) | −(126.3 ± 0.7) | −(95.9 ± 0.7) |
| 1,1-dibromopropane (598-17-4) | −(93.9 ± 1.0) | −(53.9 ± 2.7) | −(93.9 ± 1.0) | −(53.9 ± 2.7) | |
| 1,2-dibromopropane (78-75-1) | −(113.8 ± 0.9) | −(72.1 ± 0.9) | −(72.7 ± 2.7) | −(113.8 ± 0.9) | −(72.1 ± 0.9) |
| 1,3-dibromopropane (109-64-8) | −(109.1 ± 0.9) | −(61.6 ± 0.9) | −(64.0 ± 2.7) | −(109.1 ± 0.9) | −(61.6 ± 0.9) |
| 2,2-dibromopropane (594-16-1) | −(70.4 ± 3.0) | −(68.3 ± 2.7) | −(68.3 ± 2.7) | ||
| C3H5Br compounds | |||||
| (Z)-1-bromopropene (590-13-6) | 38.4 ± 2.7 | 39.1 ± 2.6 | 38.4 ± 2.7 | ||
| (E)-1-bromopropene (590-15-8) | 40.4 ± 2.6 | 41.1 ± 2.6 | 40.4 ± 2.6 | ||
| bromopropene (557-93-7) | 34.5 ± 2.7 | 32.9 ± 2.6 | 34.5 ± 3.8 | ||
| 3-bromopropene (106-95-6) | 13.8 ± 2.3 | 46.1 ± 2.3 | 44.2 ± 2.6 | 13.8 ± 2.3 | 46.1 ± 2.3 |
| bromocyclopropane (4333-56-6) | 79.6 ± 2.6 | 79.6 ± 2.6 | |||
| Bromobutanes | |||||
| 1-bromobutane (109-65-9) | −(142.6 ± 1.0) | −(105.8 ± 1.0) | −(106.2 ± 2.6) | −(142.6 ± 1.0) | −(105.8 ± 1.0) |
| 2-bromobutane (78-76-2) | −(153.7 ± 0.3) | −(119.2 ± 0.3) | −(117.9 ± 2.6) | −(153.7 ± 0.3) | −(119.2 ± 0.3) |
| 1-bromo-2-methylpropane (78-77-3) | −(148.4 ± 1.3) | −(113.5 ± 1.3) | −(114.3 ± 2.6) | −(148.4 ± 1.3) | −(113.5 ± 1.3) |
| 2-bromo-2-methylpropane (507-19-7) | −(163.7 ± 1.1) | −(131.7 ± 1.1) | −(131.7 ± 2.6) | −(163.7 ± 1.1) | −(131.7 ± 1.1) |
| bromocyclobutane (4399-47-7) | −(7.4 ± 4.4) | 32.7 ± 2.6 | −(7.4 ± 4.4) | 32.7 ± 2.6 | |
| 1,2-dibromobutane (533-98-2) | −(142.9 ± 3.4) | −(96.9 ± 3.3) | −(94.6 ± 2.7) | −(140.6 ± 2.8) | −(94.6 ± 2.7) |
| 1,3-dibromobutane (107-80-2) | −(149.2 ± 3.4) | −(99.8 ± 3.5) | −(97.7 ± 2.7) | −(147.0 ± 2.9) | −(97.7 ± 2.7) |
| 1,4-dibromobutane (110-52-1) | −(141.3 ± 3.4) | −(88.2 ± 3.4) | −(86.2 ± 2.7) | −(139.3 ± 2.7) | −(86.2 ± 2.7) |
| meso-2,3-dibromobutane (5780-13-2) | −(150.2 ± 3.4) | −(102.5 ± 2.7) | −(150.2 ± 3.4) | −(102.5 ± 2.7) | |
| dl-2,3-dibromobutane (598-71-0) | −(151.9 ± 3.4) | −(103.3 ± 2.7) | −(151.9 ± 3.4) | −(103.3 ± 2.7) | |
| 1,2-dibromo-2-methylpropane (594-34-3) | −(156.6 ± 0.6) | −(113.3 ± 0.6) | −(107.8 ± 2.7) | −(151.1 ± 2.7) | −(107.8 ± 2.7) |
| 1,3-dibromo-2-methylpropane (28148-04-1) | −(147.8 ± 0.6) | −(101.4 ± 1.6) | −(93.0 ± 2.7) | −(139.3 ± 3.1) | −(93.0 ± 2.7) |
Summary
A comprehensive investigation of the enthalpies of formation for and brominated hydrocarbons was conducted. Thorough review of available experimental data was combined with the computational results of the efficient high-level ab initio protocol developed previously and extended to Br-containing organic compounds in this work. As a part of this evaluation, an extensive analysis of the experimental enthalpies of vaporization was also carried out. Overall, the presented evaluation allowed establishing a consensus between the experimental results collected using different methods during the period of over a century as well as the modern computational chemistry methods. Availability of an accurate computational method capable of efficient treatment of moderate-sized molecules was shown to be essential for data evaluation: given a quite common lack of multiple independent experimental measurements, the computations provide the only means for data validation.
As a result of the present efforts, we have produced recommendations for the enthalpies of formation, in the liquid and gas phases, for 23 brominated compounds containing 3 or 4 carbon atoms. The recommendations substantially improve upon the existing values appearing in the reference literature7,8 as well as cover new cases for which no values were recommended previously. The analysis also revealed a number of likely-erroneous measurements that should be treated with caution in the future studies. Finally, further experimental investigations are recommended for dibromobutanes and bromocycloalkanes and for which the presently available data are not sufficient to yield unambiguous recommendations.
Supplementary Material
Acknowledgement
This article is a contribution of NIST, and is not subject to copyright in the United States for the authors. Trade names are provided only to specify procedures adequately and do not imply endorsement by the National Institute of Standards and Technology. Similar products by other manufacturers may be found to work as well or better. The authors declare no competing financial interest. The authors are grateful to Dr. András Bödi of the Paul Scherrer Institute for the provided information on the PEPICO experiments. We also thank an anonymous reviewer whose comments have lead to reevaluation of CODATA’s recommendations for HBr.
Footnotes
Supporting Information Available
Supporting information includes the listing of all compounds for which the computations were performed along with computed , and derived . The geometries for all compounds optimized with B3LYP-D3(BJ)/def2-TZVP and DF-MP2/aug-cc-pVQZ (inclusive of all considered conformations) are also provided. Additional tests of the effect of hindered rotor treatment for internal rotations on are documented.
References
- (1).Ioffe D; Kampf A Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Ltd, 2011. [Google Scholar]
- (2).Saikia I; Borah AJ; Phukan P Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev 2016, 116, 6837–7042. [DOI] [PubMed] [Google Scholar]
- (3).Rossini FD, Ed. Experimental Thermochemistry; Interscience Publishers: New York, 1956; Vol. 1. [Google Scholar]
- (4).Smith L; Bjellerup L In Experimental Thermochemistry. Measurement of Heats of Reaction; Rossini FD, Ed.; Interscience Publishers, Inc.: New York, NY, USA, 1956; Vol. 1; Chapter 9, pp 205–220. [Google Scholar]
- (5).Kolesov VP Thermochemistry of Halogenomethanes. Russ. Chem. Rev 1978, 47, 599–613. [Google Scholar]
- (6).Kolesov VP; Papina TS Thermochemistry of Haloethanes. Russ. Chem. Rev 1983, 52, 425–439. [Google Scholar]
- (7).Cox JD; Pilcher G Thermochemistry of Organic and Organometallic Compounds; Academic Press: London and New York, 1970. [Google Scholar]
- (8).Pedley JB Thermochemical Data and Structures of Organic Compounds; Thermodynamics Research Center: College Station, TX, 1994. [Google Scholar]
- (9).Oren M; Iron MA; Burcat A; Martin JML Thermodynamic Properties of and Bromo Compounds and Radicals. A Relativistic ab Initio Study. J. Phys. Chem. A 2004, 108, 7752–7761. [Google Scholar]
- (10).Dávalos JZ; Notario R; Cuevas CA; Oliva JM; Saiz-Lopez A Thermochemistry of Halogen-Containing Organic Compounds with Influence on Atmospheric Chemistry. Comput. Theor. Chem 2017, 1099, 36–44. [Google Scholar]
- (11).Jorgensen KR; Cadena M Theoretical Study of Bromine Halocarbons: Accurate Enthalpies of Formation. Comput. Theor. Chem 2018, 1141, 66–73. [Google Scholar]
- (12).Bross DH; Bacskay GB; Peterson KA; Ruscic B Active Thermochemical Tables: Enthalpies of Formation of Bromo- and Iodo-Methanes, Ethenes and Ethynes. J. Phys. Chem. A 2023, 127, 704–723. [DOI] [PubMed] [Google Scholar]
- (13).Paulechka E; Kazakov A Efficient DLPNO-CCSD(T)-Based Estimation of Formation Enthalpies for C-, H-, O-, and N-Containing Closed-Shell Compounds Validated Against Critically Evaluated Experimental Data. J. Phys. Chem. A 2017, 121, 4379–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Paulechka E; Kazakov A Efficient Estimation of Formation Enthalpies for Closed-Shell Organic Compounds with Local Coupled-Cluster Methods. J. Chem. Theory Comput 2018, 14, 5920–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kazakov A; Paulechka E Ideal-Gas Thermodynamic Properties for Organic Compounds Containing Up to 7 C, O, or N Atoms. https://data.nist.gov/pdr/lps/ark:/88434/mds2-2561, accessed: October 20, 2023.
- (16).Nagy PR; Samu G; Kállay M An Integral-Direct Linear-Scaling Second-Order Møller–Plesset Approach. J. Chem. Theory Comput 2016, 12, 4897–4914. [DOI] [PubMed] [Google Scholar]
- (17).Nagy PR; Kállay M Optimization of the linear-scaling local natural orbital method: Redundancy-free triples correction using Laplace transform. J. Chem. Phys 2017, 146, 214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H et al. Gaussian 16 Revision B.01 2016; Gaussian Inc., Wallingford, CT. [Google Scholar]
- (19).Parrish RM; Burns LA; Smith DGA; Simmonett AC; DePrince AE; Hohenstein EG; Bozkaya U; Sokolov AY; Remigio RD; Richard RM et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput 2017, 13, 3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Smith DGA; Burns LA; Simmonett AC; Parrish RM; Schieber MC; Galvelis R; Kraus P; Kruse H; Remigio RD; Alenaizan A et al. Psi4 1.4: Opensource software for high-throughput quantum chemistry. J. Chem. Phys 2020, 152, 184108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kállay M; Nagy PR; Mester D; Rolik Z; Samu G; Csontos J; Csóka J; Szabó PB; Gyevi-Nagy L; Hégely B et al. The MRCC program system: Accurate quantum chemistry from water to proteins. J. Chem. Phys 2020, 152, 074107. [DOI] [PubMed] [Google Scholar]
- (22).Kállay M; Rolik Z; Csontos J; Nagy P; Samu G; Mester D; Csóka J; Szabó B; Ladjánszki I; Szegedy L et al. MRCC, a quantum chemical program suite. http://www.mrcc.hu, 2019.
- (23).Kazakov A; Muzny CD; Diky V; Chirico RD; Frenkel M Predictive Correlations Based on Large Experimental Datasets: Critical Constants for Pure Compounds. Fluid Phase Equilib. 2010, 298, 131–142. [Google Scholar]
- (24).Paulechka E; Kazakov A Critical Evaluation of the Enthalpies of Formation for Fluorinated Compounds Using Experimental Data and High-Level Ab Initio Calculations. J. Chem. Eng. Data 2019, 64, 4863–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Paulechka E; Kazakov A Efficient Ab Initio Estimation of Formation Enthalpies for Organic Compounds: Extension to Sulfur and Critical Evaluation of Experimental Data. J. Phys. Chem. A 2021, 36, 8116–8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Halgren TA Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem 1996, 17, 490–519. [Google Scholar]
- (27).Pracht P; Bohle F; Grimme S Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys 2020, 22, 7169–7192. [DOI] [PubMed] [Google Scholar]
- (28).Bannwarth C; Ehlert S; Grimme S GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- (29).Kazakov A; Paulechka E; Chirico RD Reconciliation of Experimental and Computed Thermodynamic Properties for Methyl-Substituted 3-Ring Aromatics. Part 1:9-Methylanthracene. J. Chem. Eng. Data 2022, 67, 1834–1850. [Google Scholar]
- (30).Kazakov A; Paulechka E; Chirico RD Reconciliation of Experimental and Computed Thermodynamic Properties for Methyl-Substituted 3-Ring Aromatics. Part 2: 3-Methylphenanthrene. J. Chem. Eng. Data 2022, 67, 1851–1863. [Google Scholar]
- (31).Karton A; Martin JML Comment on: “Estimating the Hartree—Fock Limit from Finite Basis Set Calculations” [Jensen F (2005) Theor Chem Acc 113:267]. Theor. Chem. Acc 2006, 115, 330–333. [Google Scholar]
- (32).Jensen F. Estimating the Hartree–Fock Limit from Finite Basis Set Calculations. Theor. Chem. Acc 2005, 113, 267–273. [Google Scholar]
- (33).Halkier A; Helgaker T; Jørgensen P; Klopper W; Koch H; Olsen J; Wilson AK Basis-Set Convergence in Correlated Calculations on , and . Chem. Phys. Lett 1998, 286, 243–252. [Google Scholar]
- (34).Matthews DA; Cheng L; Harding ME; Lipparini F; Stopkowicz S; Jagau T-C; Szalay PG; Gauss J; Stanton JF Coupled-cluster techniques for computational chemistry: The CFOUR program package. J. Chem. Phys 2020, 152, 214108. [DOI] [PubMed] [Google Scholar]
- (35).Rogers DW Heats of Hydrogenation; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2006; Chapter 1, pp 1–22. [Google Scholar]
- (36).Clarke ECW; Glew DN Evaluation of Thermodynamic Functions from Equilibrium Constants. Trans. Faraday Soc 1966, 62, 539–547. [Google Scholar]
- (37).Baer T; Tuckett RP Advances in Threshold Photoelectron Spectroscopy (TPES) and Threshold Photoelectron Photoion Coincidence (TPEPICO). Phys. Chem. Chem. Phys 2017, 19, 9698–9723. [DOI] [PubMed] [Google Scholar]
- (38).Cox JD; Wagman DD; Medvedev VA CODATA Key Values for Thermodynamics; Hemisphere Publishing Corp.: New York, 1989. [Google Scholar]
- (39).Ruscic B; Bross DH Active Thermochemical Tables (ATcT) values based on ver. 1.130 of the Thermochemical Network (2022). http://atct.anl.gov, accessed: October 16, 2023.
- (40).NIST ThermoData Engine Version 10.4 - Pure Compounds, Binary Mixtures and Reactions. NIST Standard Reference Database 103b, Gaithersburg, MD, 2022. https://www.nist.gov/mml/acmd/trc/thermodata-engine/srd-nist-tde-103b, accessed: June 24, 2022. [Google Scholar]
- (41).Pitzer KS; Curl RF The Volumetric and Thermodynamic Properties of Fluids. III. Empirical Equation for the Second Virial Coefficient. J. Am. Chem. Soc 1957, 79, 2369–2370. [Google Scholar]
- (42).Majer V; Svoboda V; Pick J Heats of Vaporization of Fluids; Elsevier: New York, 1989. [Google Scholar]
- (43).Majer V; Svoboda V Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation; Blackwell Science: Oxford, U.K., 1985. [Google Scholar]
- (44).Chickos JS; Hosseini S; Hesse DG; Liebman JF Heat Capacity Corrections to a Standard State: A Comparison of New and Some Literature Methods for Organic Liquids and Solids. Struct. Chem 1993, 4, 271–278. [Google Scholar]
- (45).Vanderzee CE; Nutter JD Heats of Solution of Gaseous Hydrogen Chloride and Hydrogen Bromide in Water at 25°. J. Phys. Chem 1963, 67, 2521–2523. [Google Scholar]
- (46).Norling F. Über die Bandenspektren der ionisierten Halogenwasserstoffe. I. Bromwasserstoffbanden. Z. Phys 1935, 95, 179–188. [Google Scholar]
- (47).Bayes KD; Haugh MJ Predissociation and Dissociation Energy of HBr+. J. Phys. Chem 1971, 75, 1472–1476. [Google Scholar]
- (48).Penno M; Holzwarth A; Weitzel K-M State Selective Predissociation Spectroscopy of Hydrogen Bromide Ions (HBr+) via the 2Σ+ ←2 πi(i=1/2, 3/2) Transition. J. Phys. Chem. A 1998, 102, 1927–1934. [Google Scholar]
- (49).NIST-JANAF Thermochemical Tables 1985 Version 1.0 NIST Standard Reference Database 13, Gaithersburg, MD, 1986. https://janaf.nist.gov/, accessed: September 22, 2022. [Google Scholar]
- (50).Lacher JR; Casali L; Park JD Reaction Heats of Organic Halogen Compounds. V. The Vapor Phase Bromination of Tetrafluoroethylene and Trifluorochloroethylene. J. Phys. Chem 1956, 60, 608–610. [Google Scholar]
- (51).Johnson WH; Sunner S The Heats of Solution and Oxidation of Sulfur Dioxide. Acta Chem. Scand 1963, 17, 1917–1924. [Google Scholar]
- (52).Johnson WH; Ambrose JR Heat of Oxidation of Aqueous Sulfur Dioxide With Gaseous Chlorine. J. Res. Natl. Bur. Stand 1963, 67A, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Bjellerup L; Sunner S; Wadsö I The Heat of Oxidation of Arsenious Oxide to Arsenic Oxide in Aqueous Solution. Acta Chem. Scand 1957, 11, 1761–1765. [Google Scholar]
- (54).Sunner S; Thorén S The Heat of Oxidation of Aqueous Arsenious Oxide with Gaseous Chlorine. Acta Chem. Scand 1964, 18, 1528–1532. [Google Scholar]
- (55).Jones G; Baeckström S The Standard Potential of the Bromine Electrode. J. Am. Chem. Soc 1934, 56, 1524–1528. [Google Scholar]
- (56).Hetzer HB; Robinson RA; Bates RG Standard Electromotive Force of the Cell from . J. Phys. Chem 1962, 66, 1423–1426 [Google Scholar]
- (57).Wu C-H; Birky MM; Hepler LG Thermochemistry of Some Bromine and Iodine Species in Aqueous Solution. J. Am. Chem. Soc 1963, 67, 1202–1205. [Google Scholar]
- (58).Vasil’ev VP; Kozlovskii EV; Kunin BT Standard Thermodynamic Characteristics of Molecular Bromine and Triatomic Bromine Ions in an Aqueous Solution. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol 1973, 16, 365–368. [Google Scholar]
- (59).Haase R; Naas H; Thumm H Experimentelle Untersuchungen über das thermodynamische Verhalten konzentrierter Halogenwasserstoffsäuren. Z. Phys. Chem 1963, 37, 210–229. [Google Scholar]
- (60).Mussini T; Faita G Thermodynamic Functions of Aqueous Hydrobromic Acid at Various Concentrations and Temperatures. Ric. Sci 1966, 36, 175–182. [Google Scholar]
- (61).Wagman DD; Evans WH; Parker VB; Schumm RH; Halow I; Bailey SM; Churney KL; Nuttall RL The NBS tables of chemical thermodynamic properties. Selected values for inorganic and and organic substances in SI units. J. Phys. Chem. Ref. Data 1982, 11, Suppl. 2, 1–392. [Google Scholar]
- (62).Vanderzee CE; Nutter JD; Rodenburg WW; Rodenburg MLN; King DL Enthalpies of Dilution and Related Thermodynamic Properties of Aqueous Hydrobromic Acid. J. Chem. Eng. Data 1974, 19, 320–323. [Google Scholar]
- (63).Goldberg RN; Kishore N; Lennen RM Thermodynamic Quantities for the Ionization Reactions of Buffers. J. Phys. Chem. Ref. Data 2002, 31, 231–370 [Google Scholar]
- (64).Hamer WJ; Wu Y Osmotic Coefficients and Mean Activity Coefficients of Uniunivalent Electrolytes in Water at 25°C. J. Phys. Chem. Ref. Data 1972, 1, 1047–1100. [Google Scholar]
- (65).Vasil’ev VP; Kunin BT; Kozlovskii EV Standard Enthalpies of Formation of Bromine in Aqueous Solutions of Potassium Bromide and Sodium Bromide. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol 1972, 15, 696–699. [Google Scholar]
- (66).Ramette RW; Palmer DA Thermodynamics of Tri- and Pentabromide Anions in Aqueous Solution. J. Solution Chem 1986, 15, 387–395. [Google Scholar]
- (67).Gurvich LV; Veits IV; Alcock CB Thermodynamic Properties of Individual Substances; Hemisphere Publishing Corp.: New York, 1992; Vol. 1. [Google Scholar]
- (68).Dolliver MA; Gresham TL; Kistiakowsky GB; Smith EA; Vaughan WE Heats of Organic Reactions. VI. Heats of Hydrogenation of Some Oxygen-containing Compounds. J. Am. Chem. Soc 1938, 60, 440–450 [Google Scholar]
- (69).Neugebauer CA; Margrave JL The Heats of Formation of and . J. Phys. Chem 1958, 62, 1043–1048 [Google Scholar]
- (70).Manion JA Evaluated Enthalpies of Formation of the Stable Closed Shell C1 and C2 Chlorinated Hydrocarbons. J. Phys. Chem. Ref. Data 2002, 31, 123–172. [Google Scholar]
- (71).Chirico RD; Kazakov A; Bazyleva A; Diky V; Kroenlein K; Emel’yanenko VN; Verevkin SP Critical Evaluation of Thermodynamic Properties for Halobenzoic Acids Through Consistency Analyses for Results from Experiment and Computational Chemistry. J. Phys. Chem. Ref. Data 2017, 46, 023105 [Google Scholar]
- (72).Corbett P; Tarr AM; Whittle E Vapour-Phase Bromination of Fluoroform and Methane. Trans. Faraday Soc 1963, 59, 1609–1618. [Google Scholar]
- (73).Coomber JW; Whittle E Bond Dissociation Energies from Equilibrium Studies. Part 1. and . Trans. Faraday Soc 1967, 63, 608–619. [Google Scholar]
- (74).Lord A; Goy CA; Pritchard HO The Heats of Formation of Trifluoromethyl Chloride and Bromide. J. Phys. Chem 1967, 71, 2705–2707 [Google Scholar]
- (75).Okafo EN; Whittle E Bond Dissociation Energies from Equilibrium Studies. Part 5. – The Equilibria and . Determination of and . J. Chem. Soc. Faraday Soc. 1 1974, 70, 1366–1375. [Google Scholar]
- (76).Papina TS; Kolesov VP; Golovanova YG Standard Enthalpy of Formation of Bromoform. Zh. Fiz. Khim 1982, 56, 2711–2714. [Google Scholar]
- (77).Shuman NS; Zhao LY; Boles M; Baer T; Sztáray B Heats of Formation of , and Their Fragment Ions Studied by Threshold Photoelectron Photoion Coincidence. J. Phys. Chem. A 2008, 112, 10533–10538 [DOI] [PubMed] [Google Scholar]
- (78).Laynez J; Wadsö I Enthalpies of Vaporization of Organic Compounds. IX. Some Halogen Substituted Hydrocarbons and Esters. Acta Chem. Scand 1972, 26, 3148–3152 [Google Scholar]
- (79).Fowell P; Lacher JR; Park JD Reaction Heats of Organic Compounds. Part 3. – Heats of Hydrogenation of Methyl Bromide and Ethyl Bromide. Trans. Faraday Soc 1965, 61, 1324–1327. [Google Scholar]
- (80).Adams GP; Carson AS; Laye PG Thermochemistry of Reductions Caused by Lithium Aluminium Hydride. Part 4. – Heat of Formation of Methyl Bromide. Trans. Faraday Soc 1966, 62, 1447–1449. [Google Scholar]
- (81).Song Y; Qian X-M; Lau K-C; Ng CY High-Resolution Energy-Selected Study of the Reaction : Accurate Thermochemistry for the , I) System. J. Chem. Phys 2001, 115, 4095–4104. [Google Scholar]
- (82).Egan CJ; Kemp JD Methyl Bromide. The Heat Capacity, Vapor Pressure, Heats of Transition, Fusion and Vaporization. Entropy and Density of the Gas. J. Am. Chem. Soc 1938, 60, 2097–2101 [Google Scholar]
- (83).Conn JB; Kistiakowsky GB; Smith EA Heats of Organic Reactions. VII. Addition of Halogens to Olefins. J. Am. Chem. Soc 1938, 60, 2764–2771. [Google Scholar]
- (84).Lane MR; Linnett JW; Oswin HG A Study of the and Equilibria. Proc. Royal Soc. A 1953, 216, 361–374. [Google Scholar]
- (85).Ashcroft SJ; Carson AS; Carter W; Laye PG Thermochemistry of Reductions Caused by Lithium Aluminium Hydride. Part 3. – The C-Halogen Bond Dissociation Energies in Ethyl Iodide and Ethyl Bromide. Trans. Faraday Soc 1965, 61, 225–229. [Google Scholar]
- (86).Baer T; Song Y; Liu J; Chen W; Ng CY Pulsed Field Ionization-Photoelectron Photoion Coincidence Spectroscopy with Synchrotron Radiation: The Heat of Formation of the Ion. Faraday Discuss. 2000, 115, 137–145. [DOI] [PubMed] [Google Scholar]
- (87).Borkar S; Sztáray B Self-Consistent Heats of Formation for the Ethyl Cation, Ethyl Bromide, and Ethyl Iodide from Threshold Photoelectron Photoion Coincidence Spectroscopy. J. Phys. Chem. A 2010, 114, 6117–6123. [DOI] [PubMed] [Google Scholar]
- (88).Zmaczyński A. Recherches Ébullioscopiques et Tonométriques Comparatives de 8 Substances Organiques Étalons. J. Chim. Phys. Phys. Chim. Biol 1930, 27, 503–517. [Google Scholar]
- (89).Kolossowsky N; Alimow A Détermination des chaleurs latentes internes de vaporisation des liquides. II Mémoire. Bull. Soc. Chim. Fr 1934, 1, 877–880 [Google Scholar]
- (90).Svoboda V; Majer V; Veselý F; Pick J Heats of Vaporization of Alkyl Bromides. Collect. Czech. Chem. Commun 1977, 42, 1755–1760. [Google Scholar]
- (91).Gellner OH; Skinner HA 244. Dissociation Energies of Carbon–Halogen Bonds. The Bond Strengths Allyl–X and Benzyl–X. J. Chem. Soc 1949, 1145–1148. [Google Scholar]
- (92).Holm T. Thermochemical Bond Dissociation Energies of Carbon–Magnesium Bonds. J. Chem. Soc. Perkin Trans 2 1981, 464–467. [Google Scholar]
- (93).Sharonov KG; Rozhnov AM Equilibrium of Isomerization of 1,2-Dibromopropane to 1,3-Dibromopropane. Zh. Org. Khim 1971, 7, 2022–2024. [Google Scholar]
- (94).Wadsö I. Heats of Vaporization of Organic Compounds. II. Chlorides, Bromides, and Iodides. Acta Chem. Scand 1968, 22, 2438–2444. [Google Scholar]
- (95).Varushchenko RM; Pisarev VV; Rozhnov AM Enthalpies of Evaporation of Some Mono- and Dihaloalkanes. Termodinam. Organ. Soedin 1978, 7, 75–80. [Google Scholar]
- (96).Svoboda V; Kubeš V; Basařová P Enthalpies of Vaporization and Cohesive Energies of 1,1,2,2-Tetrachloro-1,2-difluoroethane, 1,2-Dibromoethane, 1-Bromo-2-chloroethane, 1,3-Dibromopropane, and 1,4-Dibromo-2,3-dichloro-1,1,2,3,4,4-hexafluorobutane. J. Chem. Thermodyn 1992, 24, 555–558. [Google Scholar]
- (97).Bjellerup L. Heats of Combustion and Formation of the 1-Bromoalkanes from through . Acta Chem. Scand 1961, 15, 231–241. [Google Scholar]
- (98).Peshchenko AD; Andreevskii DN Isomerization Equilibrium of Normal Monobromobutanes. II. Vestsi Akad. Navuk BSSR, Ser. Khim. Navuk 1968, (2), 122–123. [Google Scholar]
- (99).Mathews JH; Fehlandt PR The Heats of Vaporization of Some Organic Compounds. J. Am. Chem. Soc 1931, 53, 3212–3217. [Google Scholar]
- (100).Wadsö I. Heats of Vaporization for a Number of Organic Compounds at 25°C. Acta Chem. Scand 1966, 20, 544–552. [Google Scholar]
- (101).Lacher JR; Billings TJ; Campion DE; Lea KR; Park JD Vapor Phase Heats of Hydrobromination of the Isomeric Butenes. J. Am. Chem. Soc 1952, 74, 5291–5292. [Google Scholar]
- (102).Bjellerup L. On the Accuracy of Heat of Combustion Data Obtained with a Precision Moving-Bomb Calorimetric Method for Organic Bromine Compounds. Acta Chem. Scand 1961, 15, 121–140. [Google Scholar]
- (103).Peshchenko AD; Andreevskii DN Equilibrium of the 2-Bromobutane Conversion Reaction. 2-Bromobutane Decomposition. I. Vestsi Akad. Navuk BSSR, Ser. Khim. Navuk 1968, (1), 88–93. [Google Scholar]
- (104).Nesterova TN; Rozhnov AM Isomerization of Isostructural Monobromobutanes. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol 1974, 17, 556–558 [Google Scholar]
- (105).Kistiakowsky GB; Stauffer CH The Kinetics of Gaseous Addition of Halogen Acids to Isobutene. J. Am. Chem. Soc 1937, 59, 165–170. [Google Scholar]
- (106).Howlett KE The Use of Equilibrium Constants to Calculate Thermodynamic Quantities. Part III. Equilibria in the System tert.-Butyl Bromide–isoButene–Hydrogen Bromide. J. Chem. Soc 1957, 2834–2836. [Google Scholar]
- (107).Ribeiro da Silva MAV; Lobo Ferreira AIMC Gas Phase Enthalpies of Formation of Monobromophenols. J. Chem. Thermodyn 2009, 41, 1104–1110. [Google Scholar]
- (108).Almeida ARRP; Monte MJS Crystalline and Liquid Vapour Pressures of the Four -Monohalophenols: A Thermodynamic Study of Their Phase Transitions. J. Chem. Thermodyn 2013, 65, 150–158. [Google Scholar]
- (109).Parsons GH; Rochester CH; Wood CEC Effect of 4-Substitution on the Thermodynamics of Hydration of Phenol and the Phenoxide Anion. J. Chem. Soc. B 1971, 533–536. [Google Scholar]
- (110).Lobo Ferreira AIMC; Ribeiro da Silva MAV Thermochemical Study of Three Dibromophenol Isomers. J. Chem. Thermodyn 2011, 43, 227–234. [Google Scholar]
- (111).Ribeiro da Silva MAV; Lobo Ferreira AIMC Experimental and Computational Study on the Molecular Energetics of Monobromoanisole Isomers. J. Chem. Thermodyn 2009, 41, 499–505. [Google Scholar]
- (112).Amaral LMPF; Ribeiro da Silva MAV Calorimetric Study of Bromoacetophenone Isomers. J. Chem. Thermodyn 2014, 78, 254–259. [Google Scholar]
- (113).Almeida ARRP; Monte MJS The Influence of the Halogen Atoms and Acetyl Group on Vapour Pressures and Related Properties of the -Haloacetophenones. J. Chem. Thermodyn 2016, 92, 118–125. [Google Scholar]
- (114).Oliveira JASA; Santos AFLOM; Ribeiro da Silva MDMC; Monte MJS Thermodynamic Properties of Bromine Fluorene Derivatives: An Experimental and Computational Study. J. Chem. Thermodyn 2015, 89, 134–141. [Google Scholar]
- (115).Fu J; Suuberg EM Thermochemical Properties and Phase Behavior of Halogenated Polycyclic Aromatic Hydrocarbons. Environ. Toxicol. Chem 2012, 31, 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Chorażewski M; Góralski P Heat Capacities of 1-Chloroalkanes and 1-Bromoalkanes within the Temperature Range from 284.15 K to 353.15 K. A Group Additivity and Molecular Connectivity Analysis. J. Chem. Eng. Data 2005, 50, 619–624. [Google Scholar]
- (117).Radnai G; Bottini S; Rasmussen P; Fredenslund A Vapor-Liquid Equilibrium for Binary Systems with Alkyl Bromides. AIChE Symp. Ser 1985, 81, 102–120. [Google Scholar]
- (118).Shehatta I. Heat Capacity at Constant Pressure of Some Halogen Compounds. Thermochim. Acta 1993, 213, 1–10. [Google Scholar]
- (119).van Diemen AJG; Houtepen CJM; Stein HN Thermodynamic Properties of 2-Propanol + 2-Bromopropane Mixtures. J. Chem. Thermodyn 1974, 6, 805–814. [Google Scholar]
- (120).Pisarev VV; Rozhnov AM; Varushchenko RM; Sarkisov AG Saturated Vapor Pressure and Heats of Vaporization of Dihaloalkanes. I. 1,2-Dibromopropane, 1,2-Dibromobutane, and 1,2-Dibromopentane. Zh. Fiz. Khim 1975, 49, 2464–2465. [Google Scholar]
- (121).Pisarev VV; Rozhnov AM; Varushchenko RM; Sarkisov AG Saturated Vapor Pressure and Heats of Vaporization of Dihaloalkanes. I. 1,2-Dibromopropane, 1,2-Dibromobutane, and 1,2-Dibromopentane; Deposited in VINITI, No. 1670–75, 1975. [Google Scholar]
- (122).Varushchenko RM; Puchkova GA; Druzhinina AI Vapor Pressure and Vaporization Enthalpies of 1,2-Dibromo-n-alkanes . Zh. Fiz. Khim 1993, 67, 897–900. [Google Scholar]
- (123).Varushchenko RM; Pisarev VV; Kotel’nikova TA Enthalpies and Entropies of Vaporization of Some Haloalkanes. Zh. Fiz. Khim 1980, 54, 2666–2668. [Google Scholar]
- (124).Góralski P; Tkaczyk M Heat Capacity of -Bromochloroalkanes and -Dibromoalkanes: Their Dependence on the Hydrocarbon Chain Length and Temperature (285.15 to 355.15) K. J. Chem. Eng. Data 2012, 57, 790–795. [Google Scholar]
- (125).Paulechka YU; Kabo GJ; Blokhin AV; Firaha DS Thermodynamics of Ionic Liquid Precursors. 1-Bromobutane and Its Isomers. J. Chem. Eng. Data 2011, 56, 4891–4899. [Google Scholar]
- (126).Bryce-Smith D; Howlett KE The Preparation and Properties of tert-Butyl Bromide. J. Chem. Soc 1951, 1141–1142. [Google Scholar]
- (127).Chóliz Calero D; Mínguez Valle M; Gutiérrez Losa C Estudio Termodinamico del Sistema 2-Cloro, 2-Metilpropano 2-Bromo, 2-Metilpropano. Rev. Acad. Cienc. Exactas, Fís.-Quím. Nat. Zaragoza 1969, 24, 137–158. [Google Scholar]
- (128).Lister MW Heats of Organic Reactions. X. Heats of Bromination of Cyclic Olefins. J. Am. Chem. Soc 1941, 63, 143–149. [Google Scholar]
- (129).Stull DR Vapor Pressure of Pure Substances. Organic and Inorganic Compounds. Ind. Eng. Chem 1947, 39, 517–540. [Google Scholar]
- (130).Coffin CC; Maass O The Preparation and Physical Properties of Isobutylene. Trans. Royal Soc. Can 1927, 21, 1–10. [Google Scholar]
- (131).Sunner S; Wulff CA The Enthalpy of Formation of 1,2-Dibromo-2-Methylpropane. J. Chem. Thermodyn 1974, 6, 287–292. [Google Scholar]
- (132).Lacher JR; Walden CH; Lea KR; Park JD Vapor Phase Heats of Hydrobromination of Cyclopropane and Propylene. J. Am. Chem. Soc 1950, 72, 331–333. [Google Scholar]
- (133).Lacher JR; Kianpour A; Park JD Reaction Heats of Organic Halogen Compounds. X. Vapor Phase Heats of Hydrobromination of Cyclopropane and Propylene. J. Phys. Chem 1957, 61, 1124–1125 [Google Scholar]
- (134).Rozhnov AM; Andreevskii DN Bromopropane Isomerizaion. Neftekhimiya 1964, 4, 111–118. [Google Scholar]
- (135).Davies J; Lacher JR; Park JD Reaction Heats of Organic Compounds. Part 4. – Heats of Hydrogenation of n- and iso-Propyl Bromides and Chlorides. Trans. Faraday Soc 1965, 61, 2413–2416. [Google Scholar]
- (136).Alenin VI; Rozhnov AM; Nesterova TN; Strizhkov VN Equilibrium of the Isomerization of Monobromolkanes in the Liquid Phase; Deposited in VINITI, No. 2107–74, 1974. . [Google Scholar]
- (137).Alenin VI; Rozhnov AM; Nesterova TN; Strizhkov VN Equilibrium of the Isomerization of Monobromolkanes in the Liquid Phase. Zh. Fiz. Khim 1974, 48, 2893. [Google Scholar]
- (138).Rozhnov AM; Andreevskii DN Dehydrobromination of Isopropyl Bromide. Neftekhimiya 1963, 3, 405–412. [Google Scholar]
- (139).Stevens WR; Bodi A; Baer T Dissociation Dynamics of Energy Selected, Propane, and Ions by iPEPICO: Accurate Heats of Formation of , and . J. Phys. Chem. A 2010, 114, 11285–11291. [DOI] [PubMed] [Google Scholar]
- (140).Izmailov VD; Rozhnov AM; Sharonov KG Equilibrium of Isomerization of 1,1-Dibromoderivatives of Ethane and Propane. Zh. Fiz. Khim 1974, 48, 491. [Google Scholar]
- (141).Izmailov VD; Rozhnov AM; Sharonov KG Equilibrium of Isomerization of 1,1-Dibromoderivatives of Ethane and Propane; Deposited in VINITI, No. 6986–73, 1973. [Google Scholar]
- (142).Sharonov KG; Rozhnov AM Dehydrobromination of 2,2-Dibromopropane. Izv. Vyssh. Uchebn. Zaved. Khim. Khim. Tekhnol 1971, 14, 389–393. [Google Scholar]
- (143).Borkar S; Sztáray B; Bodi A Dissociating Ions: Almost All Roads Lead to the Allyl Cation. Int. J. Mass Spectrom 2012, 330–332, 100–108. [Google Scholar]
- (144).Holm T. Thermochemistry of Grignard [R]eagents. Enthalpies of Formation of Alkylmagnesium Bromides and of Alkyl Bromides. J. Organomet. Chem 1973, 56, 87–93. [Google Scholar]
- (145).Nesterova TN; Rozhnov AM; Kovaleva TV Study of Isomerization Equilibrium for 1,2-, 1,3-, and 2,3-Dibromobutanes; Deposited in VINITI, No. 4988–72, 1972. [Google Scholar]
- (146).Rozhnov AM; Nesterova TN; Kovaleva TV A Study of the Isomerization of the Dibromobutanes. I. The Isomerization of 1,2-, 1,3-, and 1,4-Dibromobutanes. Zh. Org. Khim 1972, 8, 1560–1564. [Google Scholar]
- (147).Rozhnov AM; Nesterova TN; Mukovnina GS Equilibrium of Isomerization of Dibromoalkanes. II. Isomerization of Dibromobutanes of Iso Structure. Zh. Fiz. Khim 1975, 49, 2842–2846. [Google Scholar]
- (148).Paulechka E; Kazakov A Enthalpy of formation of aqueous hydrofluoric acid: Revision needed? J. Chem. Thermodyn 2020, 142, 105999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Menshutkin BN Über die Ätherate des Brom- und Jodmagnesiums. Erste Abhandlung: Über die Diätherate des Brom- und Jodmagnesiums. Z. Anorg. Chem 1906, 49, 34–45. [Google Scholar]
- (150).Menshutkin BN Über die Ätherate des Brom- und Jodmagnesiums. Erste Abhandlung. II Über das Monoätherat des Brommagnesiums. Z. Anorg. Chem 1906, 49,207–212. [Google Scholar]
- (151).Rowley HH Physical Studies of Non-Aqueous Solvates. I. The Solubility of Magnesium Bromide in Ethyl Ether. J. Am. Chem. Soc 1936, 58, 1337–1341. [Google Scholar]
- (152).Holm T. Thermokemiske og Reaktionskinetiske Undersøgelser af Grignard-og Alkyllithiumreagenser. Ph.D. Thesis, Danmarks Tekniske Højskole, 1976. [Google Scholar]
- (153).Baer T; Song Y; Ng CY; Liu J; Chen W The Heat of Formation of 2- and Proton Affinity of Determined by Pulsed Field Ionization-Photoelectron Photoion Coincidence Spectroscopy. J. Phys. Chem. A 2000, 104, 1959–1964. [DOI] [PubMed] [Google Scholar]
- (154).Brooks A; Lau K-C; Ng CY; Baer T The Appearance Energy from 2-Iodopropane and 2-Chloropropane Studied by Threshold Photoelectron Photoion Coincidence. Eur. J. Mass Spectrom 2004, 10, 819–827. [Google Scholar]
- (155).Bodi A; Shuman NS; Baer T On the Ionization and Dissociative Photoionization of Iodomethane: a Definitive Experimental Enthalpy of Formation of . Phys. Chem. Chem. Phys 2009, 11, 11013–11021. [DOI] [PubMed] [Google Scholar]
- (156).Chang Y-C; Xiong B; Bross DH; Ruscic B; Ng CY A Vacuum Ultraviolet Laser Pulsed Field Ionization-Photoion Study of Methane : Determination of the Appearance Energy of Methylium from Methane with Unprecedented Precision and the Resulting Impact on the Bond Dissociation Energies of and . Phys. Chem. Chem. Phys 2017, 19, 9592–9605. [DOI] [PubMed] [Google Scholar]
- (157).Stockbauer R; Inghram MG Threshold Photoelectron–Photoion Coincidence Mass Spectrometric Study of Ethylene and Ethylene-. J. Chem. Phys 1975, 62, 4862–4870. [Google Scholar]
- (158).Lago AF; Baer T A Photoelectron Photoion Coincidence Study of the Vinyl Bromide and Tribromoethane Ion Dissociation Dynamics: Heats of Formation of , and . J. Phys. Chem. A 2006, 110, 3036–3041. [DOI] [PubMed] [Google Scholar]
- (159).Shuman NS; Stevens WR; Lower K; Baer T Heat of Formation of the Allyl Ion by TPEPICO Spectroscopy. J. Phys. Chem. A 2009, 113, 10710–10716. [DOI] [PubMed] [Google Scholar]
- (160).Genossar N; Changala PB; Gans B; Loison J-C; Hartweg S; Martin-Drumel M-A; Garcia GA; Stanton JF; Ruscic B; Baraban JH Ring-Opening Dynamics of the Cyclopropyl Radical and Cation: the Transition State Nature of the Cyclopropyl Cation. J. Am. Chem. Soc 2022, 144, 18518–18525. [DOI] [PubMed] [Google Scholar]
- (161).Fortenberry RC; Huang X; Crawford TD; Lee TJ Quartic Force Field Rovibrational Analysis of Protonated Acetylene, , and Its Isotopologues. J. Phys. Chem. A 2014, 118, 7034–7043. [DOI] [PubMed] [Google Scholar]
- (162).Lacher JR; Lea KR; Walden CH; Olson GG; Park JD Reaction Heats of Organic Fluorine Compounds. III. The Vapor Phase Heats of Hydrobromination of Some Simple Fluoroölefins. J. Am. Chem. Soc 1950, 72, 3231–3234. [Google Scholar]
- (163).Mayo FR; Dolnick AA Rearrangement of Alkyl Halides. J. Am. Chem. Soc 1944, 66, 985–990. [Google Scholar]
- (164).Sharonov KG; Rozhnov AM Isomerization of 1-Bromo-1-Propenes. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol 1971, 14, 557–560. [Google Scholar]
- (165).Abell PI; Adolf PK HBr Catalyzed Photoisomerization of Allyl Halides. J. Chem. Thermodyn 1969, 1, 333–338. [Google Scholar]
- (166).Nesterova TN; Rozhnov AM; Migacheva EG Equilibria of Isomerisation of Optical Forms of 2,3-Dibromobutane. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol 1970, 13, 1300–1302. [Google Scholar]
- (167).Nesterova TN; Rozhnov AM; Borodenko NA Study of Isomerization Equilibrium of 2,2- and 2,3-Dibromobutanes. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol 1973, 16, 1216–1218. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.