

Abstract
All-trans retinoic acid (ATRA), the biologically active form of vitamin A, is instrumental in regulating the patterning and specification of the vertebrate embryo. Various animal models demonstrate adverse developmental phenotypes following experimental retinoid depletion or excess during pregnancy. Windows of vulnerability for altered skeletal patterning coincide with early specification of the body plan (gastrulation) and regional specification of precursor cell populations forming the facial skeleton (cranial neural crest), vertebral column (somites), and limbs (lateral plate mesoderm) during early organogenesis. A common theme in physiological roles of ATRA signaling is mutual antagonism with FGF signaling. Consequences of genetic errors or environmental disruption of retinoid signaling include stage- and region-specific homeotic transformations to severe deficiencies for various skeletal elements. This review derives from an annex in Detailed Review Paper (DRP) of the OECD Test Guidelines Programme (Project 4.97) to support recommendations regarding assay development for the retinoid system and the use of resulting data in a regulatory context for developmental and reproductive toxicity (DART) testing.
Keywords: retinoid signaling, developmental toxicity, skeletal development
1. Introduction
Development of the skeleton commences during gastrulation with genes that pattern the distribution and proliferation of mesenchymal cells from cranial neural crest, somite-derived sclerotomes, and lateral plate mesoderm. During organogenesis, mesenchymal cells condense at sites of future skeletal elements and subsequently differentiate to chondroblasts or osteoblasts to form cartilages and bones (Olsen, Reginato et al. 2000).
Research from over 60 years has led to current understanding of retinoid signaling as a critical player in spatial patterning of the major body axes (e.g., anterior-posterior, dorsal-ventral, right-left) and temporal processes underlying the specification of individual skeletal rudiments (e.g., cranio-facial bones during neurodevelopment; segmentation of the vertebral column during somitogenesis; proximal-distal determination of the appendicular skeleton during limb-bud outgrowth). Common themes and diverse strategies have emerged from experimental models across multiple vertebrate species leading to substantial understanding of retinoid metabolism, transport and homeostasis during developmental and reproductive processes (Ghyselinck and Duester 2019). Elucidating Adverse Outcome Pathways (AOPs) for each system (craniofacial, vertebral, appendicular) is a long-term vision, although specific examples can be invoked in the near-term to demonstrate how diverse information is practically integrated to inform regulatory test method development (or gaps that need to be filled to get there).
Retinoid pathway assays are expected to facilitate early screening of chemicals for tiered testing of developmental toxicity and to enhance existing test guidelines (e.g., OECD TG 414 prenatal developmental toxicity). This scoping review derives from an annex in Detailed Review Paper (DRP) of the OECD Test Guidelines Programme (Project 4.97) that is intended to support recommendations regarding assay development to determine retinoid system toxicants for developmental and reproductive toxicity (DART). Specific examples presented here address potential Adverse Outcome Pathways (AOPs) that can be elucidated to explain chemical effects on skeletal development (in vivo) and link the relevant molecular initiating events (MIEs) to in vitro assays for alternative testing and high-throughput screening (HTS) platforms of the retinoid signaling pathway.
2. Assessing prenatal developmental toxicity
Current developmental toxicity testing for regulatory purposes adheres largely to protocols suggested in 1966 involving the administration of test compound to pregnant laboratory animals. The skeleton is routinely examined in standard developmental toxicity bioassays (e.g., OECD 414) and has proven to be sensitive to a wide variety of chemical agents (Knudsen, Martin et al. 2009, Theunissen, Beken et al. 2016). The fetal skeleton develops with >200 individual bones anatomically comprising an axial skeleton (vertebral column, ribs, skull) and paired appendages (upper, lower extremities). Skeletal phenotypes observed in human populations and animal studies that have been attributed to adverse drug or chemical effects during pregnancy include supernumerary bones, misshapen or reduced bones, missing bones and delayed ossification. The latter is a common observation in fetal examination and is sometimes considered a transitory variation, rather than a permanent abnormality in guideline animal studies (Solecki, Rauch et al. 2015).
Multiple fetal anomalies occur in vitamin A deficient animals as well as in retinoic acid receptor gene ‘knockout’ mice, indicating that all-trans retinoic acid (ATRA), an active metabolite of vitamin A, performs essential functions during normal development (Kam, Deng et al. 2012, Rhinn and Dolle 2012). Generally, work on retinoid biology in the skeleton is anchored to phenotypes that reflect distinct biological processes across three skeletal domains: viscerocranium (facial skeleton), postcranial axial skeleton (vertebral column), and appendicular skeleton (upper and lower extremities). Craniofacial development is closely linked to early brain development, where the facial skeleton is largely derived from cranial neural crest (CNC) cells emigrating from the anterior neural tube. Segmentation of the vertebral column has its origin in sclerotome cells that migrate medially from the somites around the neural tube and elsewhere to form the ribs and sternum. Although a developing individual may be vulnerable to disruption of retinoid signaling at later gestational stages (Williams, Kondo et al. 2009, Zuo and Wan 2017, Conserva, Anelli et al. 2019), this review will focus on the embryonic period where skeletal patterning is vulnerable to disruption of retinoid signaling.
3. Overview of the retinoid signaling pathway
Retinoid signaling has a conserved ancestry from gastropods to humans (Bushue and Wan 2010). Recent publications suggest that signaling by ATRA may be an ancestral feature of bilaterians rather than a chordate innovation; however, there is still no conclusive evidence showing that a retinoid is required for development of non-chordates (Ghyselinck and Duester 2019).
ATRA is a metabolic derivative from dietary vitamin A, existing in isomers (9-cis, 13-cis, all trans). Apart from retinal, which is involved solely in the visual cycle, in mammals ATRA is the best known endogenous active metabolite of vitamin A and is considered the only endogenous ligand for retinoic acid receptors (RARs) (Krezel, Ruhl et al. 2019) (Blaner 2001). The indispensable developmental role of retinoids was first recognized by experimental studies showing severe eye reduction defects in pigs born to maternal vitamin A deficiency (VAD) (Hale 1935), and as a teratologic experimental model in rats showing over 90% ocular and urogenital anomalies, 50% diaphragmatic hernias, and 17% congenital heart defects (Wilson, Roth et al. 1953).
During pregnancy, vitamin A ingested by the mother conveys to the placenta via the blood, where it circulates bound to the retinol-binding protein (RBP) and freely enters the embryonic circulation. The main, central vitamin A store available for the rest of the organism is the maternal liver where it is mobilized as needed (Blaner, Li et al. 2016). In peripheral cells, vitamin A is locally stored in ester forms generated through transesterification by lecithin retinol acyltransferase (the most potent vitamin A-esterifying enzyme) and liberated via retinyl ester hydrolases (Teletin, Vernet et al. 2017). Unlike endocrine hormones, the active ligand is not produced by a specific gland but instead bioactivated (dehydrogenases) and degraded (oxidases) locally (Mey 2017, Ghyselinck and Duester 2019).
Cells can initiate ATRA synthesis from maternal retinol by a two-step oxidation pathway catalyzed by retinol dehydrogenase (e.g., RDH10) and retinaldehyde dehydrogenase (e.g., RALDH) (Chatzi, Cunningham et al. 2013). An early site of ATRA production in the mouse embryo is the presomitic mesoderm on gestational day (GD) GD 7.5 (plug day GD 0). ATRA formation is buffered in part by reversion of retinaldehyde to retinol, a reaction catalyzed by at least one enzyme (DHRS3) that interacts with RDH10 (Kam, Shi et al. 2013). ATRA is rapidly degraded to inactive forms by cytochrome P450 monooxygenases, resulting in a short (~1 h) half-life (Shimozono, Iimura et al. 2013). The relevant CYP family includes three isoforms (Cyp26a1, Cyp26b1, Cyp26c1) that differ in substrate preferences for ATRA, 9-cisRA, and 13-cisRA (Isoherranen and Zhong 2019). The regional patterns of RDH10/ RALDH2 and CYP26A/B/C expression set up ATRA morphogen gradients that restrict signaling to short-range paracrine or autocrine kinematics (Teletin, Vernet et al. 2017).
Once inside the cell, ATRA’s signal is transduced by specific nuclear receptors (RARs) through genomic (canonical) or non-genomic cascades (Mey 2017). Just as most nuclear hormone receptors, RARs exhibit a modular structure composed of 6 conserved domains (designated A-F), wherein the highly conserved ‘C’ domain confers sequence-specific DNA binding (Bastien and Rochette-Egly 2004). In mouse, the canonical genomic response is mediated by one of three RAR isotypes (RARα, RARβ, RARγ) that heterodimerize with rexinoid receptors (RXRα, RXRβ, RXRγ) to transactivate (or repress) genes harboring a DNA sequence known as the RA response element (RARE). Most RARE sites consist of two hexameric motifs, 5’-(A/G)G(G/T)TCA-3’ arranged as palindromes, direct repeats (DR), or inverted repeats (IR) (Balmer and Blomhoff 2005). The classical DR for RAR/RXR binding has a 5-nucleotide spaced direct repeat (referred to as DR5); however, RAR/RXR heterodimers also bind to direct repeats separated by 1 nucleotide (DR1) or 2 nucleotides (DR2). These RAREs bind RAR/RXR with specific polarities. For example, at DR1 elements for the mouse Crbp2 gene (5’-AGGTCA c AGTTCA-3’) the 5’-half site is recognized by RAR and the RAR/RXR heterodimer acts as a transcriptional repressor. In contrast, at DR5 elements for the mouse Cyp26a1 gene (5’-AGTTCA cccaa AGTTCA-3’), RAR occupies the 3’-half site and the RAR/RXR heterodimer acts as a transcriptional activator (Zhang, Wang et al. 2015). Progressively fewer spacers may favor RXR heterodimers with other nuclear receptors (e.g., TR, VDR, and PPAR) (Mangelsdorf 1994, Zhang, Wang et al. 2015), and some RAR/RXR heterodimer-occupied sites in embryoid bodies or F9 embryonal carcinoma cells show non-canonical half-site nucleotide spacing (Moutier, Ye et al. 2012). RARE binding complexes with nuclear receptor coactivator (NCOA) or nuclear receptor corepressor (NCOR) to activate or repress, respectively, gene expression. Absent liganding the RAR/RXR complex is associated with histone deacetylase (HDAC)-containing complexes tethered through corepressors that dissociate upon RAR liganding, allowing the recruitment of coactivators (Bastien and Rochette-Egly 2004).
An important question is what happens in the nucleus after liganded RARs have recruited the transcription machinery. RAR-chromatin immunoprecipitation studies have reported 13,000–15,000 potential RAREs in the mouse genome; however, most are not likely functional as over 500 genes are known to be regulated by ATRA based on ligand involvement, receptor dimerization, DNA binding, and the resulting transcriptional modulation of the gene (Ghyselinck and Duester 2019). It is important to note that RAR/RXR transcriptional control is subject to further regulation by phosphorylation. This may occur in response to G-protein coupled receptors (GPCRs) acting through second messengers (e.g., cyclic AMP, calcium), or receptor tyrosine kinase (RTK) signals acting through downstream kinases (e.g., phosphatidylinositol 3-kinase, PI3K; extracellular signal-related kinase, ERK; mitogen-activated protein kinase, MAPK). Site-specific phosphorylation of RARs modulates cofactor recruitment associated with the general transcription machinery (Bastien and Rochette-Egly 2004). Finally, a non-genomic mechanism of RAR signaling has been characterized during neurogenesis leading to rapid activation of the PI3K and MAPK pathways without new transcription or protein synthesis (Al Tanoury, Piskunov et al. 2013, Evans and Mangelsdorf 2014, Khatib, Marini et al. 2019).
4. Altered ATRA signaling
Several lines of study have established functional evidence for retinoid signaling during pregnancy and development. One line of experimentation addresses the developmental consequences of retinoid deficiency, caused either by (i) dietary deficiency in vitamin A, (ii) inhibition of ATRA synthesis by functional inactivation of genes encoding retinaldehyde or alcohol dehydrogenases, (iii) administration of RALDH inhibitors, (iv) deletion of genes encoding RARs, or (v) administration of RAR antagonists. These manipulations are interesting from a phylogenetic-ontogenetic perspective because they eventually provide information on the physiological functions of ATRA during normal skeletal development (Ghyselinck and Duester 2019). The second line of experimentation addresses an increase in retinoid signaling with teratogenic effects that can be observed after (i) administration of pharmacological doses of vitamin A or its derivatives (natural and synthetic retinoids), (ii) inactivation of genes coding for CYP26 enzymes, or (iii) exposure to CYP26 (and related) chemical inhibitors. Teratogenic effects resulting from systemic administration of exogenous retinoids to embryos, or from increase of endogenous retinoid levels through genetic manipulations, do not necessarily reflect the physiological roles of endogenous ATRA in normal development. For example, the teratogenic effect of excess ATRA on lumbosacral truncation is transduced by RARγ, the function of which is dispensable for normal development of lumbosacral vertebrae (Lohnes, Kastner et al. 1993).
In evaluating the embryonic origins of defects related to retinoid disruption of skeletal development, this annex focuses first on the mammalian (e.g. rodent) models, then data from other animal models (chick, zebrafish, xenopus) and finally what is known in humans or can be extrapolated to humans. The PubMed Abstract Sifter (Baker, Knudsen et al. 2017) returned a catalogue of 5,903 publication records based on Medical Subject Heading (MeSH) curation with the query “(vitamin A or retinoid or retinol or retinal or retinoic acid or tretinoin) and (embryo or fetus) and (development)” (accessed January 2020) broadly annotated for retinoids, embryos, and development and was sifted for review by specific terms in the article’s title, abstract or keywords appropriate to skeletal domains.
5. ATRA signaling in craniofacial development
Endogenous ATRA is essential for development of the facial bones and branchial arches. Craniofacial malformations induced by retinoid excess, including those of Cyp26(−/−) null mutant mice, have been linked to disruption of craniofacial mesenchyme primarily affecting the formation of bones in the midface. It may also be the case for the malformation of bones derived from the caudal branchial arches under conditions of ‘functional ATRA deficiency’ in RAR null mutant mice. Two migratory cell lineages populate the embryonic head: cranial neural crest (CNC) cells, and cells from the paraxial mesoderm (Noden and Trainor 2005). Their distinct migratory streams have been shown in transgenic mice carrying an X-Gal neural crest cell lineage reporter (Wnt1-Cre/R26R) along with DiI-labeling to reveal mesodermal cells (Jiang, Iseki et al. 2002). In mouse, bones of the facial skeleton (viscerocranium) derive primarily from CNC precursors whereas those of the cranial vault (neurocranium) derive from both migratory lineages, depending on the bone. The frontal bone, for example, is primarily of CNC origin whereas the occipital bone is primarily mesodermal.
Regional differences in CNC sensitivity to retinoid signaling reflect their position in the developing anterior neural tube. As to the situation of functional ATRA deficiency, it is well established that ATRA controls anteroposterior patterning of the hindbrain through at least the 5- to 11-somite stage, notably through controlling homeobox genes (Begemann and Meyer 2001) (Mark, Ghyselinck et al. 2006) (Dupe and Pellerin 2009). CNC cells initially form at the level of the midbrain and hindbrain (Figure 1). They start migrating from the neuroepithelium at the 5-somite stage (~GD 8.5 in the mouse) to populate the frontonasal process and branchial arches. CNC cells arising furthest anteriorly form frontonasal structures (e.g., frontal bone). Hindbrain CNCs from the more rostral segments populate the 1st and 2nd branchial arches and the postotic (caudal) cells of the 3rd – 6th branchial arches (Morriss-Kay, Ruberte et al. 1993) (Dupe and Pellerin 2009).
Figure 1. Retinoid signaling during morphogenesis of the craniofacial skeleton.
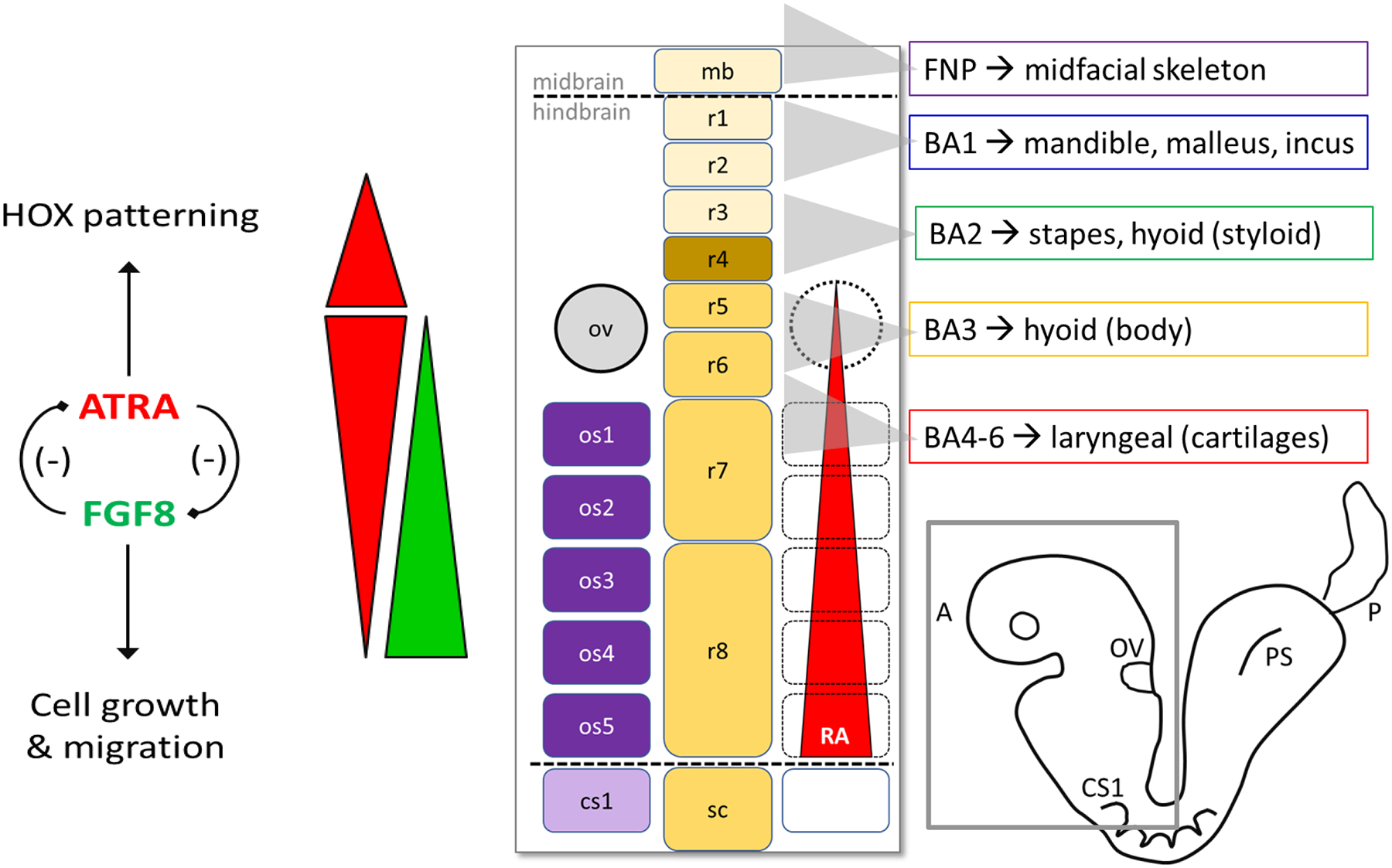
Functional inactivation of Rdh10 in mouse manifests in severe anterior defects (facial malformation, ear and eye deficiency, and loss of forelimb) due to inability of the embryo to metabolically convert retinol → retinoic acid (Rhinn, Schuhbaur et al. 2011). ATRA and FGF8 signals depicted by red and green bars, respectively. Endogenous ATRA is required, but at different threshold levels, for normal development of the midface and branchial arches. Positional information of premigratory hindbrain CNC cells is determined by threshold ATRA levels coming from the paraxial mesoderm/occipital somites. ATRA signaling through RAR/RXR 'posteriorizes' the hindbrain and is essential for specification of rhombomeres r5–r8 (5- to 11-somite stage). ATRA also specifies pharyngeal endoderm, which in turn secretes factors that create an environment permissive to (but not required for) postotic CNC migration. Abbreviations: ov, otic vesicle; os, occipital somite 1–5; cs1, first cervical somite; r1–r8, rhombomeres 1–8; FNP, frontonasal process; BA, branchial arches 1–6; PS, primitive streak. Dotted lines indicate midbrain/hindbrain and hindbrain/spinal cord junctions.
The impact of endogenous retinoid signaling on hindbrain patterning is stage-dependent and determined in part by the threshold response to local retinoid concentrations (Begemann and Meyer 2001). This connection has been demonstrated with a pan-RAR antagonist (BMS493) in the chick, but the concept applies to functional retinoid deficiency in mouse as well. RAR antagonism at the 5–6 somite stage resulted in anteriorization of rhombomeres r5 – r8, presumably by blocking the ‘posteriorizing’ influence of retinoid signaling. BMS493 progressively loses the capacity to invoke posterior regression as the embryo advances to the 11-somite stage. This effect can be phenocopied by different combinatorial RAR null mutant backgrounds. Retinoid deficiency during this window (GD8.5 in mouse and week 4 of human gestation) would, therefore result in misprogramming the positional information of premigratory CNC cells destined to populate the caudal branchial arches.
CNC cells emigrating from the hindbrain region follow a pattern of segmentation that is sensitive to different threshold levels of ATRA along the anteroposterior axis (Begemann and Meyer 2001). ATRA is locally generated in the paraxial mesoderm (somites) of which the occipital somites 1- to 5 are in physical register with rhombomeres r7 to r8. Rhombomere r4 is a conserved signaling center for FGF8 production and therefore may contribute to regulation of the ATRA gradient from the otic vesicle (low end) to the caudal extent of rhombomere r8 (high end) (Figure 1).
Unique programming and integration of CNCs within the frontonasal, maxillary and mandibular prominences generate skeletal structures in the midface and rostral branchial regions of the embryo, but only malformations of bones/cartilages derived from the more caudal branchial arches may eventually be accounted by altered positional information of premigratory CNC cells with functional retinoid deficiency (Wendling, Ghyselinck et al. 2001). There is, however, evidence that the pharyngeal endoderm, not CNC cells (or their precursors in the hindbrain neurectoderm) is the primary target for ATRA signaling. Treatment of mouse embryos with BMS493 induced a lack of caudal branchial arches and altered the paths of postotic CNC cell migration. Like in the chick embryo, this treatment was effective only during a narrow window of development that does not coincide with the period of postotic CNC cell migration. Thus, migrating CNC cells destined to populate the caudal branchial arches may not represent primary targets of ATRA action (Begemann and Meyer 2001). However, BMS493-altered endodermal expression of patterning genes, indicating that ATRA signaling is required to specify the pharyngeal endoderm, which provides a permissive environment for CNC migration through secretion of specific paracrine factors (Mark, Ghyselinck et al. 2006).
Embryos from oviparous species can obtain retinaldehyde by metabolizing carotenoids stored in the yolk; however, mammalian embryos rely on vitamin A (retinol) transferred from the maternal circulation and locally convert it to retinaldehyde. In mice, RALDH2 expression appears in the craniofacial region during neurulation (Ang, Deltour et al. 1996). The first step in tissue-specific regulation of ATRA synthesis (RDH10) was identified in an ENU-mutagenesis screen as being responsible for a spectrum of abnormalities reminiscent of ATRA-deficiency phenotypes (Sandell, Sanderson et al. 2007). Developmentally, Rdh10 is expressed in regions of active retinoid signaling and colocalizes with Raldh2 expression (Cammas, Romand et al. 2007). Functional inactivation of Rdh10 (Rhinn, Schuhbaur et al. 2011) disrupted endogenous ATRA synthesis and caused severe craniofacial defects, demonstrating the requirement of RDH10 for ATRA synthesis and downstream function. In another Rdh10(−/−) murine line, craniofacial defects were rescued by exogenous retinaldehyde on GD 7–9 confirming the requirement for retinol metabolism in anterior patterning (Chatzi, Cunningham et al. 2013). Although three cytosolic isoforms are expressed during organogenesis (Raldh1, Raldh2, Raldh3), only RALDH2 is indispensable for normal development (Niederreither, Fraulob et al. 2002). Raldh2(−/−) mouse embryos kept alive with maternal ATRA supplementation GD 7.5 to 8.5 showed rudimentary development of the 3rd-6th branchial arches (Niederreither, Vermot et al. 2003). In zebrafish, the ‘neckless’ mutation (nls) inactivates RALDH2 leading to truncation of anterior structures in a manner that can be partially rescued with exogenous retinoid, suggesting the phenotype results from a primary defect in endogenous ATRA signaling (Begemann, Schilling et al. 2001). Citral (3,7-dimethyl-2,6-octadienal), an inhibitor of retinol and retinaldehyde dehydrogenases (as well as other alcohol and aldehyde dehydrogenases), alleviated the teratogenic effects of exogenous retinol on neural crest cells (Schuh, Hall et al. 1993). Taken together, these findings demonstrate the importance of endogenous ATRA synthesis for proper anterior patterning of the early embryo.
The appropriate regulation of ATRA homeostasis also depends on its breakdown. A novel cytochrome P450 (P450RA, later identified as CYP26A1) was identified that specifically metabolized biologically active retinoids to 5,8-epoxy retinoids but did not act on retinol or retinal; this enzyme was proposed to help establish ATRA gradients in the gastrulating embryo (Fujii, Sato et al. 1997). Three functionally-redundant paralogs of the CYP26 gene family (Cyp26a1, Cyp26b1, Cyp26c1) expressed at the anterior end of the gastrulating mouse embryo cooperatively regulate anterior-posterior patterning of the neural tube. Whereas functional inactivation of Cyp26c1(−/−) did not appear to affect mouse embryonic development, double knockouts for both Cyp26a1(−/−) and Cyp26c1(−/−) fail to produce migratory CNC cells in the prospective forebrain and midbrain presumably due to excessive ATRA accumulation (Uehara, Yashiro et al. 2007) where it is normally maintained at low threshold concentrations for retinoid signaling (Figure 1). In contrast, Cyp26b1(−/−) mice do not display abnormalities in hindbrain segmentation but instead show phenotypes linked to defective formation of Meckel’s cartilage as a consequence of misdirected migration of hindbrain CNC to locations outside of the 1st branchial arch (Maclean, Dolle et al. 2009). In the gastrulating mouse embryo, Cyp26b1 expression is first observed at GD 8.0 in prospective rhombomeres r3 and r5, then expands to rhombomeres r5 and r6 by GD 9.5 (MacLean, Abu-Abed et al. 2001). As noted earlier, CNC cells start migrating from the preotic hindbrain (r3-r5) at the 5-somite pair stage (~GD 8.5) and follow a pattern of segmentation that is sensitive to different threshold levels of ATRA (Begemann and Meyer 2001). Loss of CYP26B1 function in r3/5 (GD 8.0) and r5/6 (GD 8.5) could be expected to elevate ATRA at a critical time before posterior regression of signaling activity ceases around the 11-somite stage (Figure 1).
Defects in hindbrain segmentation, CNC programming and migration, and craniofacial abnormalities show the importance of ‘retinoid homeostasis’ in patterning the anterior neuraxis during gastrulation-neurulation. Severe craniofacial defects result from loss of endogenous ATRA production (Rdh10 deficiency), whereas caudal defects follow loss of ATRA breakdown (Cyp26a1 deficiency) (Figure 1). Furthermore, retinoid homeostasis in early embryos is dependent on the localized expression of various retinoid binding proteins. For example, early mouse embryos cultured at a presomitic or early somite stage, when deprived of retinol by yolk-sac injection of antisense oligodeoxynucleotides to knockdown the retinol binding protein (RBP), displayed cranial neural tube dysmorphogenesis in a manner that was rescued with exogeneous ATRA; this is consistent with a disruption of precursor retinol to retinoid target tissue (Bavik, Ward et al. 1996). And, in the GD8.5 mouse embryo ATRA and its congeners co-localize with a CRABP-immunoreactive protein (CRABP I) at the transitional zone between surface ectoderm and neuroepithelium, from where neural crest cells emanate (Dencker, Gustafson et al. 1991).
The crucial role of ATRA in patterning mesenchymal structures derived from the neural crest cells that migrate through, or populate, the frontonasal process and branchial arches raises the important question of what role is played by the retinoid nuclear receptors. RAR and RXR families of nuclear receptors each comprise three subtypes (alpha, beta and gamma) that have been individually knocked out in the mouse. Defects displayed by RAR-singular null mutant mice are confined to a small subset of the tissues normally expressing these receptors, or are not observable (Mark, Ghyselinck et al. 1998). Furthermore, singular RAR deficient mice do not display VAD defects, whereas compound RAR mutants die in utero or at birth from severe developmental defects that, aside from the 1st branchial arch, include a spectrum of malformations belonging to the fetal VAD-induced syndrome (Lohnes, Mark et al. 1995). This suggests functional redundancy in the cellular retinoid response (Mark, Ghyselinck et al. 2009), although morpholino knockdown of RARs in zebrafish show the need for all subtypes in patterning the rhombomeres (Linville, Radtke et al. 2009).
Rara-, Rarb-, or Rarg null and compound mutants also exhibit congenital abnormalities that were not described in Hale’s and Warkany’s classical VAD studies, including defects of the neurocranium and viscerocranium. This occurrence of non-VAD defects is most probably accounted for by the difficulty to achieve, by dietary deprivation, a state of severe VAD compatible with pregnancy. In fact, almost all these non-VAD defects have been subsequently replicated in rodent embryos deficient in vitamin A, but supplemented with ATRA; or, that lack RALDH3 (which specifically display defects in the nasal ethmoturbinates and agenesis of the choana) and treated with synthetic retinoids having RAR antagonistic activities (Mark, Ghyselinck et al. 2006). Craniofacial defects that are strikingly similar to those observed in RAR knockout mice are also present in rat embryos treated with a specific inhibitor of retinaldehyde dehydrogenases, namely Win 18,446 (a bis(dichloroacetyl) diamine) (Taleporos, Salgo et al. 1978), as well as in mice lacking Rdh10 (refer to Figure 1). Therefore, dysregulation of RARs can explain the adverse effects resulting from altered endogenous ATRA levels on craniofacial development.
To overcome the early embryonic lethality of compound RAR mutants, mutant mice have been generated in which RARs were specifically ablated in the neural crest lineage using somatic mutagenesis. Ablation of all three RARs in the neural crest lineage did not affect their specification and migration, nor the formation of the branchial arches (Dupe and Pellerin 2009). Finally, mice carrying targeted knock-in mutations of the corepressor Silencing Mediator of Retinoid and Thyroid hormone receptor (SMRT) display defects in CNC-derived structures and posterior homeotic transformations of axial vertebrae; SMRT-dependent repression of RAR signaling can modify the Hox code via epigenetic marking (Hong, Fang et al. 2018).
It is worth noting that RARs have been instrumental to the evolution of the cranial skeleton (Mark, Ghyselinck et al. 2009). In addition to the dramatic craniofacial skeletal deficiencies affecting Rara/g-null mutants, subtle defects which often alter the shape of an individual skeletal element are observed in several Rar-null mice: a cartilaginous or osseous connection between the incus (middle ear bone) and the alisphenoid bone (e.g., the pterygoquadrate element); a cartilage separating the trigeminal ganglion from the brain (the pila antotica); and an agenesis of the rostral ethmoturbinate and maxillary sinus. The pterygoquadrate element and the pila antotica, which were lost during evolution from reptiles to mammals, represent atavistic features. Ethmoturbinate bones and maxillary sinus are typical mammalian features not present in reptiles, and their agenesis in Rar-null mutants also reflects an atavistic condition. As such, the presence of atavistic features in Rar-null mutants supports the notion that changes in temporal or spatial patterns of Rar expression provided a general mechanism for modifying the number and shape of individual cranial skeletal elements during vertebrate evolution.
6. Craniofacial teratogenesis
Many teratogenic effects described for exogenous retinoids in laboratory animal models and humans reflect alterations to tissues derived from cranial neural crest cells (CNCs) (Williams, Mear et al. 2004). Clinical observations following isotretinoin (13-cisRA, Accutane) exposure during pregnancy in humans (Irving, Willhite et al. 1986, Pratt, Goulding et al. 1987) and nonhuman primates (Yip, Kokich et al. 1980) have shown a spectrum of malformations including craniofacial defects linked to hypoplasia of the 1st (mandibular) and 2nd (hyoid) branchial arches. Isotretinoin has a low affinity for RARs and RXRs relative to ATRA but may be converted intracellularly to more active metabolites. Similar branchial arch defects can also be invoked with exogenous ATRA in pregnant rodents (hamsters, rats, mice) via delayed or disorganized patterns of cell migration and excessive cell death in the CNC (Lorente and Miller 1978, Wiley, Cauwenbergs et al. 1983, Sulik, Cook et al. 1988, Granstrom and Kullaa-Mikkonen 1990, Webster and Ritchie 1991, Wise, Xue et al. 2010). These findings from teratological observations showed that CNC cells were a likely target in dysmorphogenesis of the craniofacial skeleton linked to retinoid excess.
A direct, quantitative effect of retinoids on facial morphogenesis has been demonstrated in experimental models monitoring CNC cell functions prior to endochondral or perichondral differentiation. Most of this work is from two lines of investigation, one using controlled-release bead carriers soaked with retinoids and applied to different locations of the developing chick embryo, and the other with in vitro (whole embryo culture, WEC) studies on rodent embryos. Carrier beads soaked with ATRA (or the stable synthetic retinoid TTNPB) applied to the facial primordia of stage 20 chick embryos induced frontonasal dysmorphogenesis in a dose-dependent and time variable manner; these embryos lacked the upper beak whereas the lower beak was normal (Wedden and Tickle 1986). Exposing facial mesenchyme cultured from different regions of the chick embryo (e.g., frontonasal mass, mandibular mesenchyme) under micromass conditions conducive to cartilage differentiation showed no regional specificity to retinoid inhibition, indicating that retinoids do not produce the specific facial defect by directly interfering with cartilage differentiation (Wedden, Lewin-Smith et al. 1987). Furthermore, fragments of the frontonasal mass give rise to typical upper-beak structures (e.g., central rod of cartilage) when transplanted to the wing bud and the extent of cartilage is dependent on inclusion of the surface ectoderm. Heterotypic tissue recombinations (retinoid-ectoderm × control mesoblast versus control ectoderm × retinoid mesoblast) pinpointed frontonasal mesenchyme as the sensitive CNC population, which in situ is dependent on epithelial interactions for upper beak dysmorphogenesis (Wedden 1987). Specific effects on the upper beak and outgrowth and cartilage differentiation in the frontonasal mass with 13-cis linked to disruption of CNCs (but not spinal neural crest cells) by decreasing cell-substratum adhesion in vitro (Smith-Thomas, Lott et al. 1987). Direct exposure of GD 8 mouse embryos to ATRA (0.1 μM) or 13-cisRA (2 μM) in WEC within 6 h led to a dramatic reduction of CNC cell migration to the first and second branchial arches, where the CNC cells either did not leave the neuroepithelium or aggregated nearby due to alterations in the cell surface (Goulding and Pratt 1986, Pratt, Goulding et al. 1987). CNC cells destined to the second branchial arch are most sensitive in rat embryos exposed to threshold levels of 13-cisRA (Webster and Ritchie 1991). The dysmorphogenesis observed upon direct exposure of chick and rodent embryos to retinoids is consistent with the teratological phenotypes observed in vivo and confirm the CNC cell population as the likely target of retinoid teratogenesis.
Early mouse embryos exposed to exogenous ATRA on GD 7.5 – 8.0, just before differentiation of the cranial neural plate and hindbrain segmentation, showed a shortened hindbrain on GD 9 lacking metameric patterning (Morriss-Kay, Murphy et al. 1991). This dysmorphology correlated with ‘posteriorization’ of the hindbrain with respect to Hox-2.9 (normally confined to rhombomere 4) at the expense of Krox-20 (rhombomere 3) (some Krox-20 positive CNC cells that emigrated from the hindbrain remained close to the neural tube) (Morriss-Kay, Murphy et al. 1991). With exogenous ATRA other CNC cells migrated incorrectly to yield ectopic differentiation, such as Meckel’s cartilage in maxillary region (Morriss-Kay, Ruberte et al. 1993).
Similar to mouse, early rat embryos on GD 9.5 exposed to 0.1 μM ATRA in WEC for 6 h displayed reduced size and shape of the first and second branchial arches due to altered regional identity of the hindbrain crest cells (Lee, Osumi-Yamashita et al. 1995). Therefore, although CNC cells form most of the facial skeleton and are primary target cells in the retinoid-induced craniofacial defects, their reprogramming may occur prior to emigration from the neuroepithelium. This implies a critical effect on neural tube patterning that is realized only after CNC cells migrate from the segmented neuroepithelium but prior to reaching their destination to form neural crest-derived skeletal elements. In this regard, endogenous retinoids have been shown to function as ‘posteriorizing factors’ on rat hindbrain development with the caudal hindbrain region being most sensitive to retinoid insufficiency (White, Highland et al. 2000). In Xenopus, CNC cells also acquire positional information in the neural tube prior to migration. A 30 min exposure to supraphysiological concentrations of ATRA at the gastrula stage caused a dose-dependent truncation of the body axis in tadpoles, resulting in progressive loss of anterior structures between 0.5 μM and 5.0 μM, to complete loss of the head at 10.0 μM (Papalopulu, Clarke et al. 1991).
Microarray analysis of the mouse neural crest at 6- to 48 h following in vitro exposure to teratogenic (1 μM) levels of ATRA revealed that more than one-third of all differentially expressed genes participated in pathways linked to developmental regulation over time (6- to 48 h), including canonical and noncanonical WNT signaling pathways, cell adhesion and cell cycle regulation (Williams, Mear et al. 2004). Those findings are consistent with the notion that retinoid signaling reprograms genomic determinants of migratory neural crest function. In the chick embryo, premigratory neural crest cells have limited programming about the lower jaw whereas the upper jaw and facial midline are specified later by local tissue interactions. A transcriptomic analysis of the maxillary prominence 16 h after respecification by RA-beads showed effects on the retinoid, BMP and WNT signaling pathway, as well as cross-talk with Noggin, a diffusible extracellular signal that antagonizes BMP4 and activates the retinoid pathway (Nimmagadda, Buchtova et al. 2015). Msx 1 and Msx 2 transcripts are rapidly down-regulated in upper beak primordia (where outgrowth is inhibited by ATRA) but remain largely unchanged in lower beak primordia (where outgrowth is unaffected) (Brown, Robertson et al. 1997). Both homeobox genes function in the transcriptional regulation of craniofacial development and are likely mediators of the adverse consequences of exogenous retinoid signaling on cleft palate through upstream fibroblast growth factor (FGF) signaling that up-regulates Msx1/2 expression in the maxillary prominence; both Msx1/2 are down-regulated by the application of ATRA to the maxillary mesenchyme (Shimomura, Kawakami et al. 2015) as is Fgf4 in the frontonasal mesenchyme following teratogenic doses of RA (Munoz-Sanjuan, Cooper et al. 2001). Taken together, these findings point to crosstalk between the retinoid system and BMP-, WNT- and FGF-signaling on the craniofacial mesenchyme largely derived from CNC cells.
Interestingly, ATRA exposure in mice at GD 8.5 caused homeotic transformation of the lower jaw into upper jaw-like structures preceded by down-regulation of Fgf8 expression in the 1st branchial arch ectoderm as an indication of reorganization of mandibular arch reprogramming (Abe, Maeda et al. 2008). Endothelin-1 (EDN1) is one of the primary signals that establish the identities of CNC cells within the mandibular portion of the first branchial arch: mice lacking the Edn1 gene or its cognate receptor (Ednra) display homeotic transformation of the lower jaw to an upper jaw like phenotype like that seen with ATRA exposure (Clouthier, Garcia et al. 2010). Perhaps EDN1 is one of the aforementioned paracrine factors secreted by the pharyngeal endoderm to provide a permissive environment for CNC migration (Mark, Ghyselinck et al. 2006). Interestingly, recent teratogenic experiments using ATRA in excess have led to similar conclusions with regards to CNC cells colonizing the 1st branchial arch (Vieux-Rochas, Coen et al. 2007). A single dose of 25 mg/kg ATRA administered to pregnant mice by gavage at GD 8.5–8.75 resulted in a pattern of craniofacial defects affecting most of the 1st branchial arch. The developmental stage at which ATRA treatment resulted in craniofacial malformations equates to embryos of 9 to 14 somites and corresponds to the window of time during which CNC cells from rhombomeres r1 and r2 reach the 1st branchial arch. This evidence showed that ATRA acts on the signaling epithelium of the 1st branchial arch, gradually reducing the expression of EDN1 and FGF8 (Vieux-Rochas, Coen et al. 2007).
7. ATRA signaling in vertebral development
Trunk organization in Vertebrata involves the establishment of a metameric primary body axis leading to formation of the neural tube and paraxial mesoderm (somites). ATRA signaling participates in the organization of both systems (neural tube, vertebral column) although only the vertebral system is considered here. The onset of ATRA signaling in the mouse embryo, based on expression of RDH10, RALDH2, is GD 7.5; however, ectopic or excessive retinoid signaling can disrupt three morphogenetic processes organizing the trunk during early gastrulation (GD 6.5) and well into organogenesis (GD 9.5).
(i). Mesoderm segmentation:
ATRA signaling determines the size of the somite precursors forming from the presomitic mesoderm (PSM) and their right-left alignment in the trunk region. This occurs at the somite ‘determination front’ from the early headfold stage (GD 8.0) through hindlimb initiation stage (~GD 9.5 in mouse) and covers the ‘pharyngula stage’ when the body plan is very similar across diverse Vertebrate species. For example, Raldh2(−/−) mouse embryos that completely lack ATRA activity in the paraxial mesoderm form trunk somites approximately half their normal size ((Rhinn and Dolle 2012) and often exhibit fewer somites on one side (Kaiser, Merrill et al. 2003).
(ii). Axial elongation:
a somite pair is added to the early mouse embryo every 2 hours in rostral (e.g., cervical) to caudal (e.g., sacral) order drawing cells from a posterior growth zone in the ‘nodal region’ at the anterior extent of the primitive streak. This occurs continuously in the pharyngula-stage embryo (GD 7.5 to 8.0) and later in the tail bud region (GD 8.0 through at least GD 10.0). ATRA signaling plays a permissive role in this process by opposing FGF8 and Wnt signaling. For example, Cyp26a1(−/−) mouse embryos that lack the capacity to break down endogenous ATRA may display caudal regression due to premature cessation of posterior elongation caused by excessive retinoid signaling (Rhinn and Dolle 2012).
(iii). Regional identity:
ATRA signaling is involved in the early determination of individual vertebral identities through co-regulation of Hox gene expression (together with FGF8 and other signals) (Lohnes, Mark et al. 1994, Kaiser, Merrill et al. 2003). Four clusters of Hox genes (Hoxa - Hoxd) occur in mammals, individually numbered 1–13 by their sequential position (3’ → 5’) in each genomic cluster. Precise control of ATRA thresholds in the presomitic mesoderm (PSM) and nodal region is critical for proper regulation of Hox genes that specify regional identify in the somite precursors. Alterations in ATRA produce skeletal phenotypes that ultimately reflect defects in anterior-posterior patterning and Hox regulation.
Somitogenesis:
somites form by epithelialization from whorls of precursor cells in the PSM (somitomeres) and are continuously added to the caudal end of the left and right somite columns (Wilson, Olivera-Martinez et al. 2009). The so-called ‘determination front’ is controlled by a clock-and-wavefront scenario. In this model, a periodic signal controls timing of the nascent somite from its somitomere through lateral inhibition by of the Notch-Delta pathway in crosstalk with an ATRA-FGF8 signaling. ATRA is highly concentrated in newly formed somites via RALDH2 expression and dampens FGF8 signaling via repressive interaction with RARE in the Fgf8 regulatory region that, in turn, is maintained by posterior Wnt-8a and Wnt-3a signals (Aulehla and Pourquie 2010) (Cunningham, Brade et al. 2015) (Hubaud and Pourquie 2014) (Cunningham, Kumar et al. 2015). Consequently, WNT-FGF signals primarily set the posterior boundary of a newly formed somite in conjunction with the segmentation clock and ATRA plays a permissive role by suppressing the posterior FGF8 wavefront (Figure 2).
Figure 2. Retinoid signaling in metameric organization of the paraxial mesoderm during somitogenesis.
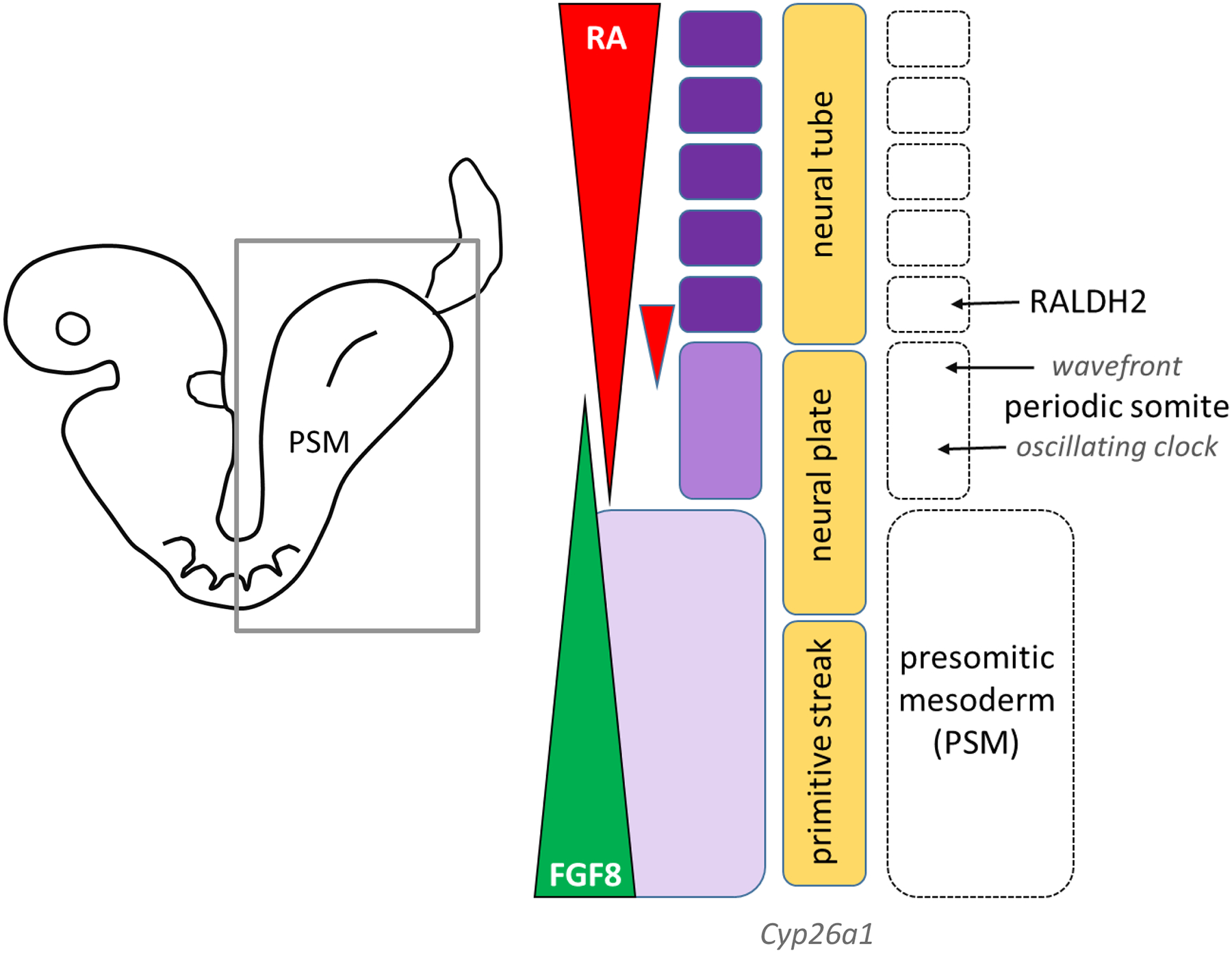
A molecular oscillator (clock) delivers a periodic signal controlling somite production from the presomitic mesoderm (PSM). During axis elongation the signal is displaced posteriorly by a system of traveling signaling gradients (wavefront) that depends on RALDH2 in newly formed somites (rostral) and FGF8 coming posteriorly (Iimura, Denans et al. 2009, Rhinn and Dolle 2012, Shimozono, Iimura et al. 2013). ATRA antagonizes the FGF8-mediated growth front (based on (Strate, Min et al. 2009). Cyp26a1 expression is highest posteriorly; functional inactivation manifests as severe posterior defects in the mouse (loss of hindlimb, caudal regression) due to premature cessation of posterior elongation (Rhinn and Dolle 2012). ATRA signaling modulates somite size in the trunk region, but not the tail region. ATRA thresholds control bilateral symmetry of the left and right somite columns and determine vertebral identity through RARE-dependent Hox genes (gastrulation). Retinoid excess disrupts PSM growth and caudal extension in the posterior region.
A key aspect of this signaling front pertains to the nature of ATRA and FGF8 distribution in the PSM (Deschamps and van Nes 2005). Sources of Fgf8 expression include the node region of the primitive streak during gastrulation and the tail bud post-gastrulation (Iimura, Denans et al. 2009). FGF8 is released into the extracellular milieu as a diffusible signal that facilitates cell growth and migration. It also induces and maintains Cyp26a1 expression for ATRA breakdown. As epiblast cells migrate through the anterior primitive streak and into the PSM, ATRA coming from the paraxial mesoderm suppresses Fgf8 expression and consequently the FGF8 gradient and CYP26A1 activity wane at the determination front. Although the non-peptidic structure renders it difficult to directly image and map ATRA gradients, it is generally accepted that differential expression of RALDH2 and CYP26A1 produce ATRA thresholds that elicit specific cellular behaviors. Questions remain as to the precise ATRA distribution and topological range in the PSM and how, as short-range paracrine signals ATRA and FGF8 can interact with the determination front (Cunningham and Duester 2015).
To address the dynamics of diffusible ATRA gradients during somitogenesis, different zebrafish reporter lines have been generated to express from a family of genetically-encoded probes engineered to respond to different ATRA thresholds (Shimozono, Iimura et al. 2013). That study engineered the ligand-binding domain of mouse RARs to incorporate cyan-emitting and yellow-emitting variants of Aequorea green fluorescent protein (GFP). Using the principle of fluorescence resonance energy transfer (FRET), alterations in the conformation of the ligand-binding domain of the RAR in response to ATRA binding converted into changes from blue to yellow fluorescence that could be mapped by live-cell imaging. Three such reporter lines were generated in which variations in the RAR ligand-binding domain affinities to ATRA were adjusted to produce a yellow FRET signal in response to low (dissociation constant, K’d = 2 nM), medium (K’d = 4 nM), or high (K’d = 50 nM) intracellular ATRA concentration and with negligible sensitivity to retinol or retinaldehyde. These studies revealed a two-tailed linear ATRA gradient across the head-to-tail axis of the zebrafish embryo at the 5-somite stage. The bimodal gradient showed a steady, symmetrical rise and fall of ATRA peaking 6 nM in the mid-trunk and declining to <1 nM anteriorly (head) and posteriorly (tail). Computer simulation of gradients dynamics during growth and development, based on simple diffusion of ATRA (10 μm2 per sec) over a signaling region of 200 μm, created a linear gradient in about 10 min (Shimozono, Iimura et al. 2013). Importantly, these findings support a conventional ‘source-sink’ arrangement established by cell-specific expression of raldh2 and cyp26 isoforms at a much faster time scale than that of embryonic growth, indicating stability of the gradient.
In mouse, the RALDH2 expression domain is strong posterior in the early headfold stage (presomitic) embryo and becomes restricted to the paraxial and lateral mesoderm as rostral expansion advances to the 5-somite stage (Hochgreb, Linhares et al. 2003). Although the ATRA signal generated by RALDH2 in the somitic mesoderm normally travels throughout the trunk mesoderm (Molotkova, Molotkov et al. 2005), the mutual antagonism between ATRA-FGF8 may only be realized over short distances of a few cell diameters at the periodic somitic wavefront (Mallo, Vinagre et al. 2009).
Furthermore, coordination between neural and mesodermal patterning is important to align spinal-vertebral segments. For example, at the head-trunk transition of the early embryo, anatomical segments specified in the posterior hindbrain (e.g., rhombomere 7) and anterior spinal cord (e.g., cervical segment 1) must align with those for the occipital bone of the neurocranium and first vertebra (atlas). ATRA signaling coordinates axial position of neural structures relative to paraxial mesoderm at the head-trunk transition in zebrafish (Lee and Skromne 2014). Like in mouse (Kaiser, Merrill et al. 2003), this synchrony entails mutual antagonism with Wnt, FGF (and/or Notch-Delta) signaling pathways (Kawakami, Raya et al. 2005). Bipotent axial stem cells generate both lineages (Mallo, Vinagre et al. 2009).
An evolutionary scenario has been described for the recruitment of ATRA into the somitic gene regulatory network. Amphioxus, the closest living invertebrate relative of the vertebrates, has a persistent notochord, segmental axial musculature, a dorsal hollow nerve cord that can be posteriorized by exogenous ATRA. This species lacks neural crest cells (Escriva, Holland et al. 2002) but expresses neural crest genes, used for other purposes, that have been co-opted into neural crest formation in higher chordates (Yu, Meulemans et al. 2008). Anterior-posterior patterning in Amphioxus, like vertebrate species, is regulated by Hox genes and a posterior FGF signal; however, unlike vertebrate species, this species develops relatively simple somites, has no PSM, and disruption of ATRA signaling does not affect somitogenesis (Bertrand, Aldea et al. 2015). As such, mutual antagonism between ATRA-FGF8 signaling in higher chordates was concurrent with the advent of the PSM and a more complex control of somitogenesis and specialization of somites along the body axis. While some of the control mechanisms governing ATRA-mediated patterning are well conserved between vertebrates and invertebrate chordates, such as amphioxus, less is known about its roles in non-chordates (e.g., echinoderms). A putative RARE (DR5 element) has been found in close proximity to the Hox1 gene in sea urchin suggesting that at least some components of the ATRA signaling cascade were probably already present in the last common ancestor of deuterostomes (Marlétaz et al. 2006).
Axial elongation:
posterior elongation extends the developing trunk posteriorly through a mesodermal stem cell growth zone maintained caudally at the node of the primitive streak at the pharyngula-stage embryo (GD 7.5 to 8.0) and later in the tail bud region (GD 8.0 through at least GD 10.0) (Figure 3). Posterior elongation is at least partly dependent on an ATRA-depleted environment because Cyp26a1(−/−) null mouse embryos that are deficient in the capacity to degrade ATRA display severe truncation of posterior structures (Rhinn and Dolle 2012). ATRA signaling plays a permissive role in this axial elongation by opposing FGF8 and Wnt signaling. ATRA-sensitive anterior transformations of axial vertebrate have been also been reported in mice genetically deficient in growth differentiation factor 11 (GDF11), a member of the TGF-β superfamily that is involved in axial patterning. The Gdf(−/−) phenotype includes caudal regression from diminished Cyp26a1 expression in the tailbud and can be rescued with the pan RAR antagonist, AGN 193109 (Lee, McPherron et al. 2010). In zebrafish embryos, cyp26a1 expression is further regulated by the miR-19 family microRNAs during vertebral axis formation; miR-19, in turn, is suppressed ATRA and the adverse consequences of ATRA on somitogenesis can be phenocopied with antisense morpholinos that ablate miR-19 function (Franzosa, Bugel et al. 2013). Hyperossification of axial bones and fusions of vertebral primordia have been reported in zebrafish embryos rendered cyp26b1-deficient by genetic mutation (stocksteif) or by exposure to R115866, a pharmacological inhibitor of CYP26B1 expressed in osteoblast cells (Laue, Janicke et al. 2008, Spoorendonk, Peterson-Maduro et al. 2008).
Figure 3. Ontogeny of Hox-mediated axial patterning and its regulation by retinoid signaling.
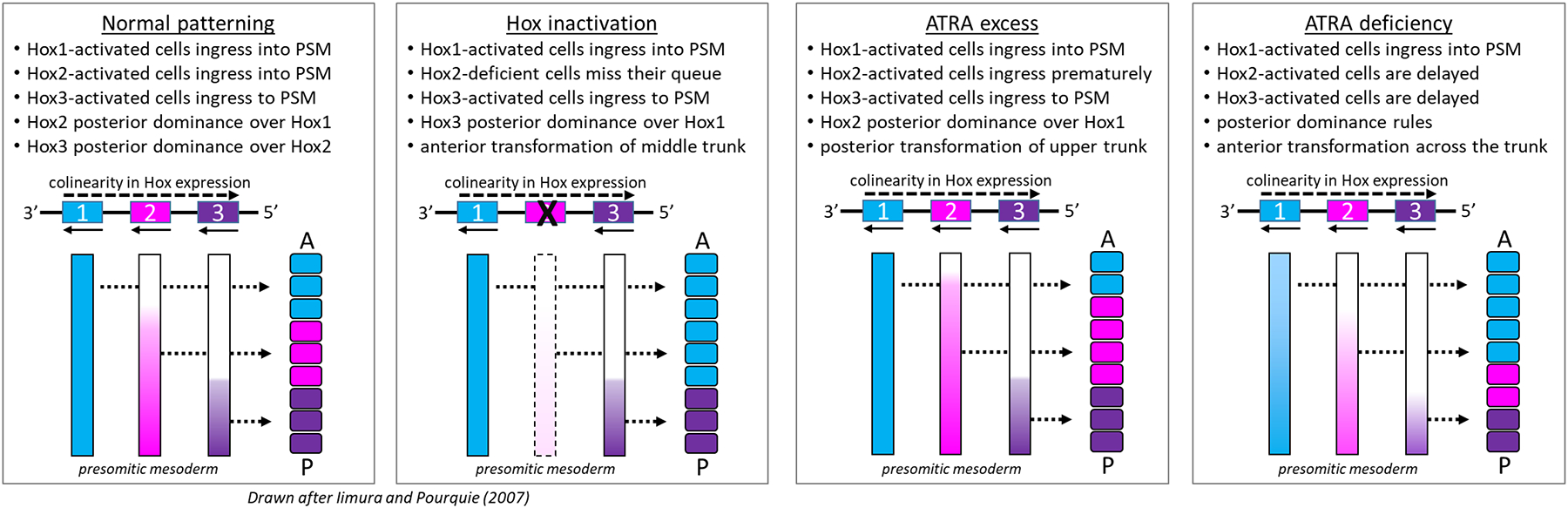
Hox-patterning is decoded during somitogenesis in spatial and temporal waves of transcription to determine relative positions in the vertebral column at which the paralogs are expressed during development [from (Luo, Rhie et al. 2019). Emergence of the somitic column is depicted from PSM based on the ‘posterior dominance’ model [redrawn from (Iimura and Pourquie 2007). Somite colors reflect chromosomally linked Hox genes (simple representation of only 3 genes) in temporal colinear expression. Newly formed somites are morphologically similar across the trunk but are fixed with regards to future vertebral identity. Normal patterning (left) and phenotype with Hox gene inactivation (right) and potential consequences of ATRA excess imposed during late gastrulation (GD 7.3 mouse) and ATRA deficiency.
Similarly, ATRA and FGF8 signaling influence posterior axial development in Xenopus at multiple points of interaction (Shiotsugu, Katsuyama et al. 2004). Experimental suppression of FGF8 signaling was shown to downregulate RALDH2, CYP26, and RARα2 expression whereas a constitutively active RARα2 rescued downstream Hox gene expression (XCAD3, HOXB9). These findings suggest that anteroposterior patterning of the primary body axis is driven by a series of mutually interactive feedback loops among FGF, Wnt, RAR, signals in concert with Hox expression. The constellation of Hox expression foreshadows the type of vertebra that forms in during anterior-posterior patterning and specification of vertebral identity (Diez del Corral and Storey 2004), mediated by the caudal (CDX1/2/4) family of Hox regulatory genes (Deschamps and van Nes 2005).
Vertebral specification:
ATRA produces concentration-dependent defects on regional phenotypes of the vertebral segments that reflect regulation of Hox gene expression patterns in the PSM. The critical biology begins as the bilaminar disc embryo (epiblast, hypoblast) reorganizes into three primary germ layers (ectoderm, mesoderm, endoderm) that will give rise to all tissues in the body. This process, gastrulation, commences in mouse at the egg cylinder stage (GD 6.5) when cells from the posterior region of the epiblast converge on the midline and undergo epithelial-mesenchymal transition (EMT) to form a rod-shaped ‘primitive streak’ that advances anteriorly (Williams, Burdsal et al. 2012). The streak persists as a conduit through which cells undergoing EMT pass. It reaches the distal tip of the egg cylinder proper at GD 7.5. Mesodermal cells so induced now express RALDH2; consequently, the mouse embryo first acquires its capacity to generate ATRA production between the late primitive streak stage (GD 7.2) and early headfold stage (GD 7.7) (Deschamps and van Nes 2005). CYP26A1 is already highly expressed by virtue of FGF8 signaling from the underlying hypoblast. Rostral expansion is characterized by a nodal growth zone that adds tissue to the caudal end of the embryo proper from GD 7.7 through axis elongation stages.
Hox gene expression (e.g., Hoxb1) is first observed in the posterior streak by GD 7.2; however, this does not concern the future paraxial structures because fate maps show these cells contribute to the extraembryonic mesoderm. Rather, epiblast cells migrating through anterior streak form the likely source of Hox activation for anterior-posterior vertebral identity because these are the precursor cells for the paraxial mesoderm. Whereas the primary germ layers are primed to express Hox genes early in gastrulation, Hox gene expression is not activated until the primitive streak in mouse is fully extended during late gastrulation (Forlani, Lawson et al. 2003). Positional information determined by the Hox code is fixed into the PSM as mesodermal cells emerge from the streak (Tam and Beddington 1987). PSM retains its Hox signature identity even when transplanted at an ectopic axial level in the chick embryo.
Hox genes exhibit sequential activation from 3’ → 5’ reflecting their numbered arrangement along the Hox cluster (temporal colinearity). Selective pressure has kept the Hox genes tightly packed, and changes in chromatin structure have an important influence on their ordered expression (Deschamps and van Nes 2005, Iimura and Pourquie 2007). For example, in the PSM Hoxd4 first unpacks, followed in turn by Hoxd8-9, Hox10, and Hoxd11-12. This colinear activation is also reflected in the anterior-posterior location of somites (spatial colinearity). Because the PSM acquires its positional information as cells emerge from the primitive streak (Tam and Beddington 1987), early structures formed during gastrulation are given an anterior identity with 3′ Hox genes as key determinants; progressively later structures express more 5′ Hox genes and acquire a more posterior identity (Krumlauf 1994). Epiblast cells, before migrating through the streak, are somewhat of a mosaic of different Hox signatures (Iimura and Pourquie 2007). Cells of a similar Hox signature ingress through the streak at a certain time, sorting themselves into the metameric lineages in the PSM. The anterior boundary of expression for each gene in the Hox cluster thus moves anteriorly with rostral expansion as posterior tissues are added to the body axis (Figure 3). Dynamic temporal pairing of 3’→5’ Hox activation with the timing of ingression through the anterior streak migration thus lays out the spatial pattern of Hox expression along the trunk (Deschamps and van Nes 2005, Iimura and Pourquie 2007). 5’ Hox overrides more 3’ Hox phenotypes (posterior dominance). Breaking temporal colinearity by disrupting an upstream gene regulatory element in the HoxD cluster (as a model system) produced posterior homeotic transformations coincidentally with an earlier activation of Hoxd genes (Kondo and Duboule 1999).
ATRA is a potent, global activator of Hox gene expression and regional patterning (Dolle, Ruberte et al. 1990, Ruberte, Dolle et al. 1991). Although RAR/RXR liganding may transactivate Hox genes, this effect is not responsible for colinearity (Diez del Corral and Storey 2004). That is known from the observation of a relatively normal pattern of Hox gene activation in Raldh2(−/−) mouse mutants (Niederreither, Subbarayan et al. 1999). On the other hand, perturbations of ATRA signaling have been shown to alter Hox gene expression and induce stage-dependent homeotic transformations of vertebral segments. For example, ATRA excess during gastrulation (GD 7.3) induced posterior homeotic transformations affecting upper vertebral segments (Kessel and Gruss 1991). This can be explained by premature entry of 5’ Hox cells into the streak, resulting in the formation of somites with a more posterior character than normal (Figure 3). The effect was reminiscent of the above-mentioned model where colinearity in the HoxD cluster was broken by depletion of an upstream gene regulatory element (Kondo and Duboule 1999). This implies RARE-derepression of colinear Hox gene activation. It is also reminiscent of findings in vitamin A deficiency (VAD) where fetuses from pregnant VAD rats showed anteriorization of vertebral identity along the entire vertebral column. Shifts in Hox expression domains foreshadowed these transformations, and the window for rescue by exogenous ATRA was late gastrulation (equivalent to GD 7.5 mouse) (Kaiser, Merrill et al. 2003). These findings indicate that a precise distribution of ATRA in PSM is critical for proper regulation of the Hox clock, although ATRA is not obligate to temporal colinearity.
Exogenous ATRA can induce posterior neural tube defects (spina bifida) in the mouse at GD 8.5 although RARγ(−/−) embryos are resistant to this effect (Iulianella, Beckett et al. 1999); however, Raldh2(−/−) mouse embryos exhibit axial shortening due to a block in ATRA synthesis (Niederreither, Subbarayan et al. 1999). Studies in Xenopus showed that RARβ (Rarb2) participates in the control of somite number and size, restriction of the PSM anterior border, among other effects during somitogenesis (Janesick, Tang et al. 2017). This is the RAR subtype most upregulated in response to liganding, and its localization in the trunk somites positions it at the right time and place to respond to ATRA during somitogenesis. In contrast, RARγ2 is the major RAR subtype expressed in the caudal stem cell zone; in the absence of ATRA liganding, RARγ2 is a transcriptional repressor and this state maintains the pool of caudal progenitor cells. When liganded, RARγ2 is a transcriptional activator of caudal gene expression that facilitates somitogenesis but can prematurely terminate body axis extension if uncontrolled. This effect is seen when early Xenopus embryos are treated with the RARγ selective agonist NRX204647 at a concentration of 0.1 μM; in contrast, the inverse-agonist NRX205099 had no effect at that concentration (Janesick, Nguyen et al. 2014). By activating RARγ, the agonist (NRX204647) relieves repression of posterior Hox gene expression and markers of PSM creating posterior truncations. In RAR/RXR complexes, polycomb group (pcG) proteins interact with non-liganded RAR/RXR to recruit nuclear corepressors (NCOR) and silence gene expression. In contrast, trithorax group (trxG) proteins interact with liganded RAR/RXR to recruit nuclear coactivators (NCOA) and activate gene expression (Iimura and Pourquie 2007).
Reductions in somitogenesis and axial length with the RARγ-specific agonist in zebrafish was associated with loss of hoxb13a expression, suggesting RARγ maintains stem/progenitor cells during embryonic development in its nonligated state (Wai, Kawakami et al. 2015). In Xenopus, a switch in RARγ signaling from repressor to activator states could be invoked not only by the RARγ-selective agonist (NRX204647), but also by overexpressing a constitutively active RARγ (VP16-RARγ2) or a dominant-negative nuclear corepressor (c-SMRT, Silencing Mediator of Retinoid and Thyroid hormone receptor) (Janesick, Nguyen et al. 2014). SMRT-dependent repression of RAR is also critical to establish and maintain the somitic Hox code and segmental identity in mice and this involves epigenetic marking of loci (Hong, Fang et al. 2018). Mice deficient in the Polycomb homolog (M33) show homeotic transformations of the axial skeleton, sternal and limb malformations and an aggravation of the skeletal malformations when treated with ATRA on GD 7.5 suggesting M33 plays an epigenetic role in defining access to RAREs in some Hox genes (Core, Bel et al. 1997). Taken together, these findings are consistent with roles for both RARβ and RARγ during somitogenesis: liganded RARγ facilitates expression of RARβ (Janesick, Tang et al. 2017) in the caudal region until required to facilitate body axis cessation when somitogenesis is nearing completion and the progenitor cell pool is exhausted (Olivera-Martinez, Harada et al. 2012). In general, CDX (1/2/4) is a central integrator of FGF/WNT/ATRA signals on Hox gene activation and expression and may, therefore, be a key node in the regulatory network integrating the molecular control over segmentation, posterior axial elongation, and specification of vertebral identity (Deschamps and van Nes 2005).
8. Retinoids in Appendicular Development
The appendicular skeleton (upper and lower extremities in bipeds, forelimb and hindlimb in quadrupeds) is defined in three segments along the proximodistal axis: stylopod (humerus, femur), zeugopod (radius-ulna, tibia-fibula), and autopod (hand, foot). A secondary axis defines anterior-posterior asymmetry (e.g., digits I through V in mouse and humans) and a tertiary axis dorsal-ventral asymmetry. The rudimentary ‘limb-bud’ forms as outcroppings of the flank (forelimb bud, hindlimb bud) composed of surface ectoderm and a mesoderm derived from the lateral plate (LPM). Development and patterning of the appendicular skeleton has been a general paradigm for understanding embryogenesis, and the consequences of physiological (ATRA) and teratological (chemical) disruption of retinoid signaling on limb development has been a subject of much interest and controversy over the decades (Kochhar 1973) (Tickle, Alberts et al. 1982) (Thaller and Eichele 1987) (Lewandoski and Mackem 2009) (Tabin and Wolpert 2007) (Ghyselinck and Duester 2019). The physiological functions of ATRA are reviewed here with a primary focus on mouse limb development but addressing key concepts from experimental embryology in zebrafish, amphibians, and avian species, and for retinoid teratogenicity in rodents, nonhuman primates, and humans.
Forelimb bud initiation:
limb bud induction in the LPM is triggered by a cascade of signaling events involving FGF, WNT, and ATRA. Retinoid signaling is required for initiation of the forelimb bud (but not hindlimb bud) (Zhao, Sirbu et al. 2009, Cunningham, Zhao et al. 2013). For example, the RDH(trex/trex) mouse fetuses lacking the capacity to generate retinaldehyde and subsequently ATRA (as shown by RARE-lacZ reporter transgenes and rescue with exogenous ATRA) display stunted forelimbs and apparently normal hindlimbs, reminiscent of Tyrannosaurus rex (Cunningham, Chatzi et al. 2011). Forelimb development is also disrupted in the Raldh2(−/−) mouse embryo (Niederreither, Subbarayan et al. 1999).
T-box transcription factors Tbx5 and Tbx4 have critically important roles in specifying forelimb and hindlimb identity, respectively through FGF signaling. These are the primary initiators of limb bud outgrowth and the earliest genes expressed in the prospective limb fields. Forelimb expression of Tbx5 in the mouse initiates in the anterior flank at GD 8.5, and Tbx4 mirrors this pattern posteriorly with hindlimb-specific expression at a half-day lag (Naiche and Papaioannou 2003). The failure to initiate forelimb development in association with ATRA deficiency is reflected in loss of Tbx5 and can be alleviated with exogenous ATRA administration to the pregnant dam on GD 8 (Mic, Sirbu et al. 2004). Tbx5 activation is suppressed by FGF8 signals; consequently, Fgf8 expression must be downregulated both in the anterior (cardiac) and posterior (caudal) regions to enable limb initiation. ATRA signaling is a major factor as demonstrated in RALDH2 and RDH10 deficient mouse mutants (Zhao, Sirbu et al. 2009, Cunningham, Zhao et al. 2013). This points to a permissive role for ATRA signaling in conditioning the microenvironment of the prospective limb-bud field for Tbx5 expression through antagonism of FGF8 signaling, similar in concept to craniofacial and axial patterning. Other studies proposed that ATRA might directly activate Tbx5 via a RARE located in intron 2 (Nishimoto, Wilde et al. 2015); however, subsequent enhancer knockout experiments using CRISPR/Cas9 gene editing showed that this RARE is not required for forelimb bud initiation (Cunningham, Lancman et al. 2018). Thus, the most parsimonious model in mice is that ATRA is permissive to forelimb bud initiation by alleviating FGF8 signaling, which then allows another factor (perhaps Wnt) to activate Tbx5 expression.
ATRA is also required for pectoral fin (forelimb homolog) initiation in the zebrafish embryo (Begemann, Schilling et al. 2001). This phenotype is observed in the nof (no-fin) and nls (neckless) mutations in the zebrafish raldh2 locus (Grandel, Lun et al. 2002) (Gibert, Gajewski et al. 2006). A signaling cascade has been proposed where physiological ATRA signals originating in the somitic mesoderm → wnt2b in the intermediate mesoderm → tbx5 in the lateral plate mesoderm → prdm1 in the forelimb bud → FGF10 (Mercader, Fischer et al. 2006). FGF10, in turn, activates Fgf8 expression in the distal ectoderm and thickening that drives polarized outgrowth of the limb-bud. This is consistent with the notion that ATRA antagonizes early axial FGF8 signals that otherwise inhibit the limb field.
Limb patterning:
in addition to mice lacking RALDH2 and RDH10, malformed limbs are observed in mice lacking RARα and RARγ receptors, lacking CYP26B1, or lacking CRABP2 (Cunningham and Duester 2015). These phenotypes reflect the continuing physiological role for ATRA signaling during limb-bud outgrowth and appendicular development; however, the system is complex and data from the literature sometimes contradictory with regards to the involvement in shaping skeletal elements specified along the primary (proximodistal) axis (e.g, stylopod, zeugopod, and autopod) and the secondary (anteroposterior) axis (e.g., number and morphology of the digits). In both cases, polarized outgrowth and patterning is dependent on two major organizing centers of the early limb-bud (Figure 4): the Apical Ectodermal Ridge (AER) distally and the Zone of Polarizing Activity (ZPA) posteriorly (Zakany, Zacchetti et al. 2007).
Figure 4. ATRA signaling during development of the fetal appendicular skeleton.
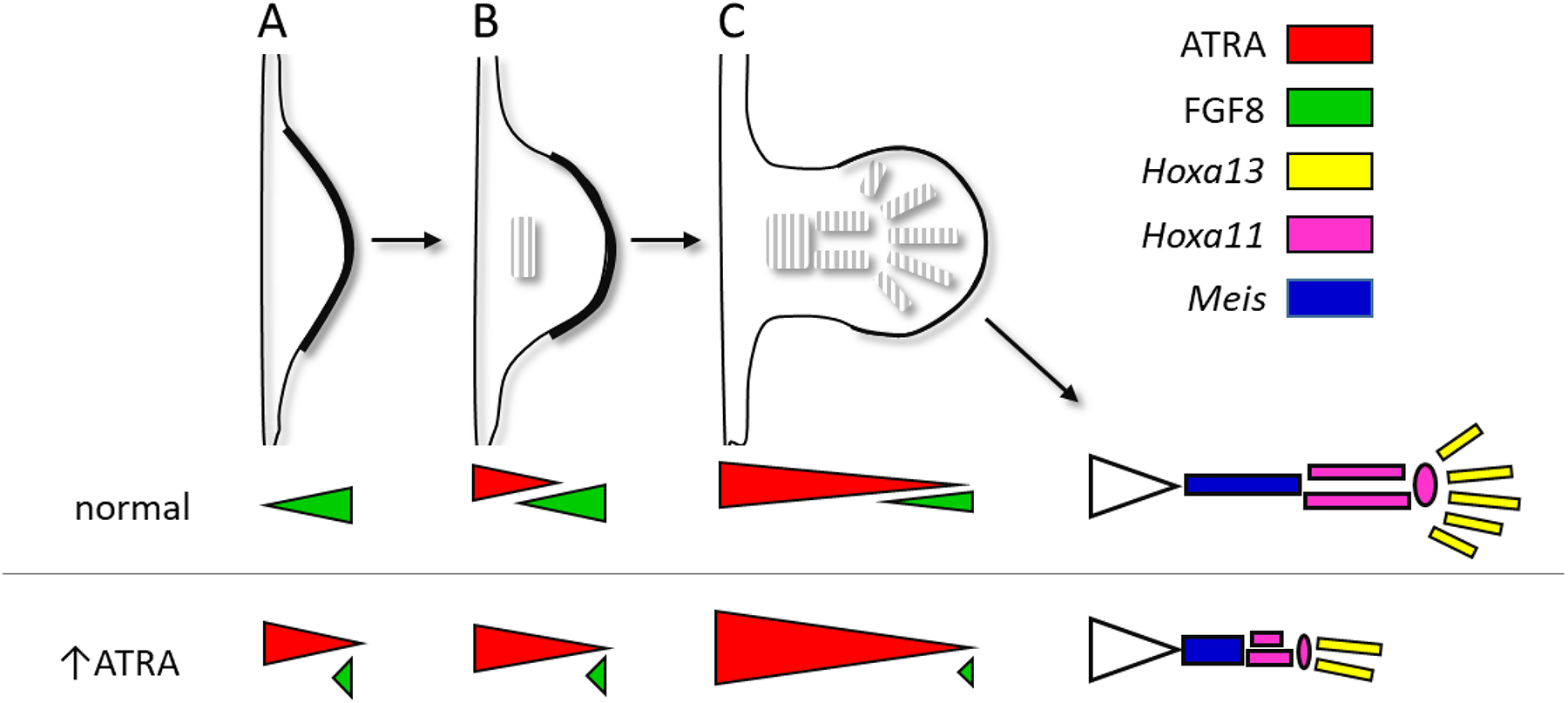
TOP: Mouse forelimb from early outgrowth (A, GD 9.5) to precartilage induction (B, GD10.5) to precartilage pattern (C, GD 11.5); corresponding stages in the hindlimb are delayed by a half day. The precartilage pattern is laid down in proximo-distal fashion for the stylopod (humerus, femur), zeugopod (radius-ulna, tibia-fibula), and autopod (digits of fore- and hind paw). BOTTOM: permissive ATRA signaling on proximo-distal patterning (from Uzkudun, Marcon et al. 2015). ATRA (from RALDH2) enters the proximal limb-bud and is degraded distally by CYP26B1 induced by FGF8; cells leaving the ATRA-free distal mesenchyme have positional values determining regional identity for stylopod (Meis), zeugopod (Hoxa11), and autopod (Hoxa13). Gradients represent two-signal model for ATRA and FGF8 signal inputs; a one-signal model (not shown) was also simulated wherein the time exposed to FGF8 alone determined regional identity.
ATRA synthesized by RALDH2 in the flank enters the limb-bud proximally and, like in the trunk has an opposing distribution to FGF8 signals, here coming distally from the AER (Yashiro, Zhao et al. 2004). Although concentration-dependent effects of ATRA on morphogenesis led to its early characterization as a ‘morphogen’ that can instruct skeletal patterning (Thaller and Eichele 1987), our current understanding indicates the instructional role is perhaps species-specific and that permissive interactions may be more the case for murine development (Riddle, Johnson et al. 1993). Our current understanding again points to a permissive role for ATRA signaling in the mouse via mutual antagonism with FGF8 as is consistent with other organizing centers of the embryo (Lewandoski and Mackem 2009) (Tabin and Wolpert 2007) (Ghyselinck and Duester 2019) (Zuniga 2015).
Proximodistal patterning:
progressive specification of the limb proximodistal segments is based on a timing mechanism in the distal ‘progress zone’. This mesodermal growth zone measures ~300 μm in length and elongates the limb-bud in response to FGF8 elaborated from the AER (Summerbell 1974, Niswander, Tickle et al. 1993, Fallon, Lopez et al. 1994, Mariani, Ahn et al. 2008). Under this model, the fate of mesodermal cells becomes progressively more distal depending on time spent in the progress zone exposed therein to FGF8 (Figure 4). While the timing mechanism is not fully understood (Uzkudun, Marcon et al. 2015), cells falling out of the progress zone condense into precartilage skeletal rudiments. In particular, SOX9 controls the mesenchyme to chondrogenic differentiation (Reinhardt, Gullotta et al. 2019).
In the mouse, early limb-bud outgrowth and regional specification of the skeleton is influenced by ATRA signaling. On one hand, early outgrowth is mediated by FGF signaling from the AER. Target cells in the progress zone progressively condense into proximal (stylopod), intermediate (zeugopod), and distal (autopod) elements of the appendicular skeleton. Localized expression of Cyp26b1 begins in the distal portion of the early limb bud and remains elevated in the progress zone (Yashiro, Zhao et al. 2004). Mice that lack CYP26B1 display proximal deficiencies (meromelia) and missing digits (oligodactyly). These defects in the Cyp26b1-nullizygote are foreshadowed by deficient Sox9 expression in the precartilaginous blastema and excessive cell death, respectively, as early as GD11.5 and were phenocopied in wild-type mice by administration of exogenous ATRA one to three times between GD10.25 and GD 12.25 (Yashiro, Zhao et al. 2004). The extent of molecular proximalization in Cyp26b1-deficient mice was linked to distal expansion of Rarb expression as indicative of ectopic ATRA activity as part of a signaling network with SHH and FGF, but also on CYP26B1-mediated ATRA clearance (Probst, Kraemer et al. 2011).
A one-signal FGF-driven progress zone model for limb proximodistal patterning has been proposed, based on mouse genetic studies, coupled with colinear Hox gene activation. In this model, Hox activation does not require ATRA to specify proximodistal fate but requires ATRA degradation distally to maintain the progress zone and prevent teratogenesis from excess ATRA (Lewandoski and Mackem 2009). ATRA entering the limb-bud from the flank is an indirect ‘proximalizing factor’ of the limb-bud through transactivation of the proximal determinant Meis genes (Meis1, Meis2). This activity earmarks stylopod specification, which is dependent on Hox9 and Hox10 genes in the forelimb and Hox10 genes in the hindlimb, and is counteracted by FGFs from the AER (Mercader, Leonardo et al. 2000, Rosello-Diez, Ros et al. 2011). Like somitogenesis, the antagonistic relationship between ATRA-FGF signals are realized during early limb-bud outgrowth between ATRA (proximalizing factor) and FGF8 (distalizing factor).
In the one-signal model of limb-bud outgrowth continued FGF8 signaling from the AER activates Cyp26b1 expression in the progress zone and therefore drives limb-bud elongation by maintaining distal mesenchyme in a proliferative state. Excessive ATRA signaling would otherwise terminate Fgf8 expression in the progress zone and lead to a precocious involution of the AER before the precartilaginous skeleton has completely formed. Cyp26b1(−/−) mice show abnormal distal expansion of ATRA signaling in both the forelimbs and hindlimbs, with excessive apoptosis and cartilage disruption that correlates with meromelia (limb truncation) and other severe limb deformities reminiscent of retinoid teratogenesis (Yashiro, Zhao et al. 2004). Cyp26b1 is markedly expressed in the AER and distal mesoderm (MacLean, Abu-Abed et al. 2001) and is likely to create an ‘ATRA-free’ progress zone. This reflects a complex signaling module whereby AER-FGF and CYP26B1-ATRA regulate distal progression of limb-bud outgrowth (Probst, Kraemer et al. 2011). Quantitative computer simulation of the ratiometric signaling (Figure 4) show that high ATRA and low FGF8 activate Meis1/2 in the stylopod (humerus, femur); ATRA and FGF4/8 at intermediate levels promote Hoxa11 in the zeugopod (radius-ulna, tibia-fibula); and low ATRA and high FGF4/8 promote Hoxa13 expression in the autopod (hand, foot) (Uzkudun, Marcon et al. 2015). While these findings do not provide evidence that endogenous ATRA signaling is a direct morphogen for proximodistal patterning of the limbs (Duester 2008), they are consistent with a two-signal model observed in the chick whereby ATRA participates in the establishment of proximal fate.
Anteroposterior patterning:
numerous studies have investigated the potential for ATRA to influence anteroposterior patterning of the limb-bud, or so-called ‘polarizing activity’ that determine the numbers, sizes and identities of the digits for example. Some of the most striking findings have followed a line of investigation where carrier beads are soaked with different concentrations of retinoids and then locally applied to various regions in the chick embryonic limb bud, permitting a detailed evaluation of skeletal phenotypes resulting from the timing and position of ATRA-soaked bead application.
Sonic hedgehog (SHH) emanated by the ZPA is the polarizing signal responsible for organizing anteroposterior patterning of the digits (Riddle, Johnson et al. 1993). The ZPA occupies the distal-posterior aspect of the early limb-bud. When ATRA-soaked beads were implanted to the opposite (anterior) margin of the early chick limb-bud, dose-dependent effects were observed such as supernumerary digits (low dose and/or early stage), mirror image digital duplications (medium dose and/or stage), and truncations (high dose and/or late stage) (Summerbell 1983) (Helms, Thaller et al. 1994). The lower dose effects were reminiscent of ectopic ZPA transplantation. When the ZPA region was surgically removed before bead implantation to the anterior margin, ATRA reprogrammed the distal mesenchyme to restore digit formation, but in reverse polarity with respect to anterior-posterior symmetry similar to an ectopic ZPA (Eichele 1989). Beads soaked with synthetic retinoid analogs also caused concentration-dependent duplications when applied to the anterior margin (10- to 100 μg/ml), and truncations at very high concentration (1 mg/ml) (Tamura, Kagechika et al. 1990) or when implanted proximally (Tickle and Crawley 1988). When supernumerary digits were induced at the anterior limb margin by ATRA treatment, their development was preceded by vascular regression in a manner that could be blocked by co-exposure to VEGF, a potent angiogenic factor (Yin and Pacifici 2001). This indicates complex interaction between vascular development and digital specification with regards to ATRA signaling, which implies multiple modes of action on early limb development.
Together, the famous chick bead findings demonstrate the capacity of ATRA signaling to reprogram mesenchyme to an ectopic ZPA. Shh expression is not, however, directly dependent on ATRA in the physiological range (10 μg/ml) (Niswander, Tickle et al. 1994) (Helms, Thaller et al. 1994) (Chen, Dong et al. 1996). But the permissive effect of ATRA on polarizing activity appears to be conserved from fish to mammals (Ogura, Alvarez et al. 1996) (Akimenko and Ekker 1995) and may involve ATRA-regulated Hoxd genes that lie genetically upstream to Shh (Lu, Revelli et al. 1997) (Pickering, Wali et al. 2017).
Interdigital cell death:
ATRA signaling promotes the naturally occurring zones of interdigital cell death that separate the developing digits. Double mouse mutants lacking both the Rarg gene and one or both alleles of Rarb display a severe and fully penetrant interdigital webbing (Dupe, Ghyselinck et al. 1999). Rdh10(trex/trex) mutants also display interdigital webbing, where the hindlimb skeletal pattern was not affected but interdigital cell death was absent at GD 15 (soft tissue syndactyly) (Cunningham, Chatzi et al. 2011). These findings demonstrate that ATRA, although unnecessary for limb patterning is required later for interdigital tissue loss. This is an important point indicating that deficiency in signaling by ATRA could be a cause of (cutaneous, osseous) syndactyly in humans (Dupe, Ghyselinck et al. 1999).
9. Limb teratogenesis
Kochhar in 1973 was among the first to identify teratogenic effects of exogenous ATRA on the mouse limb, invoking dose-dependent phocomelia following a single dose in the range of 1- to 100 mg/kg the pregnant dam (Kochhar 1973). The window of vulnerability in mouse (GD 10–12) coincided with the formation of precartilaginous mesenchymal condensations. ATRA caused dose-dependent phocomelia and digital defects when administered to pregnant mice at 20- to 80 mg/kg on GD 11, where the deficiencies to precartilage presented before overt cartilage differentiation (Kwasigroch, Skalko et al. 1984). Higher teratogenic dosages (e.g, 200 mg/kg) on GD 12 also disrupted joint formation in the hindlimbs (Abu-Hijleh and Padmanabhan 1997). Similar teratological phenotypes have been reported in rats (Yu, Gonzalez et al. 2003) and hamsters (Wiley 1983) indicating generalized susceptibility in rodents. Beyond phocomelia, more severe limb reduction defects/deficiencies have been reported with teratogenic exposures to retinoids at earlier gestational stages. For example, exposure on GD 6 induced ectopic or supernumerary hindlimbs as part of a lower body duplication syndrome (Liao and Collins 2008) and on GD 9 resulted in complete loss of the hindlimbs as part a caudal regression syndrome (Padmanabhan 1998). Discounting broader syndromes where limb malformations are secondary to primary body axis determination, GD 10–12 is the window of vulnerability in mouse for phocomelia and related deformities induced with teratogenic retinoid exposures.
Susceptibility of limb development to retinoid teratogenesis coincides with stages at which limb development is undergoing polarized outgrowth and formation of the precartilaginous skeleton. For example, a fully phocomelic exposure to ATRA in the mouse (100 mg/kg on GD 11) led to the early appearance of excessive cell death in the central prechondrogenic core of the forelimb (Zhou and Kochhar 2004). In fact, expansion of several naturally-occuring zones of physiological cell death in the limb-bud have been observed following ATRA exposure on GD 10 (Bynum 1991). In exposed hamsters (Wiley 1983) a disorganized vasculature was found to encroach on areas of mesenchymal condensation prior to overt differentiation (Wiley 1983). These findings on the cellular basis of retinoid teratogenesis are consistent with disruption of skeletogenesis with or before development of precartilage rudiments of the long bones. It is worth noting that increased bone resorption in the mature skeleton is a well-documented effect of hypervitaminosis A (Conaway, Henning et al. 2013). This effect is related to an imbalance of osteoclast activity in post-menopausal women. Cellular effects of retinoids on bone homeostasis have been observed in neonatal mouse calvarium culture (and other in vitro models), perhaps through RARa-dependent effects on the osteoclast signal RANKL (TNFSF11) that functions in bone and cartilage development. For additional information on ATRA and skeletal homeostasis the reader is referred to excellent reviews by (Conaway, Henning et al. 2013, Henning, Conaway et al. 2015).
Clinical observations with various retinoid congeners have suggested that the major teratogenicity is due to direct effects on the embryo. GD 10 mouse embryos directly exposed to ATRA in whole embryo culture (WEC + 48 h) showed palatal and limb deficiencies, and inhibition of cell proliferation as low as 0.1 μM ATRA (Watanabe and Pratt 1991). When GD 8 mouse embryos were cultured in the presence of isotretinoin (13-cis retinoic acid, also known as Accutane), dysmorphogenesis was first observed in branchial arches I/II at 2 μM followed by the limb-buds at 20 μM (Goulding and Pratt 1986). Isotretinoin, a known human teratogen, causes primarily heart and craniofacial malformations; however, this retinoid congener, as well as a number other natural and synthetic retinoids, do invoke limb dysmorphogenesis in rat WEC +48 h. Differences between in vitro and in utero susceptibilities for the retinoid congeners could, on one hand, reflect maternal or placental effects (Steele, Marlow et al. 1987). On the other hand, an analysis of five retinoids tested in rat WEC showed dysmorphologies resembling the in vivo phenotypes. The NOAECs (no observed adverse effect concentrations) ranged from 0.03 nM to 29.7 μM and correlated well with the teratological LOAELs (lowest observed adverse effect levels) in vivo (Bechter, Terlouw et al. 1992). As such, the teratogenic action of retinoids on the embryo can be largely attributed to direct effects on the embryo and in the absence of maternal factors. Further evidence that the limb-bud itself is a direct target of retinoid teratogenicity comes from in vitro models such as whole organ culture, where precartilage rudiments differentiate into cartilage rudiments that inform organ-level phenotypes, and high-density micromass cultures that enable chondrogenesis to occur de novo from dissociated mesenchymal cells informing tissue-level phenotypes. For example, isotretinoin, ATRA and synthetic retinoids (Ro 10–1670, Ro 11–1430) showed a persistent and dose-dependent inhibition of chondrogenic differentiation in both systems (Zimmermann and Tsambaos 1985).
Limb reduction defects induced by a high teratogenic dose of ATRA (200 mg/kg on GD12.5 mouse) correlated with elevated retinoids within 30 min, peaking at 2 hr then falling sharply by 4 hr (Ward and Morriss-Kay 1995). Analysis of GD 11 mouse limb buds found retinol as the predominant retinoid, although ATRA was enriched over the rest of the embryo for several hours after maternal dosing. The elevation in ATRA content peaked 50-fold over basal (endogenous) levels following a weak teratogenic dose of 10 mg/kg ATRA, and 300-fold after a fully phocomelic dose of 100 mg/kg (Satre and Kochhar 1989). Because teratogenic doses of exogenous ATRA (10- to 100 mg/kg) may result in many-fold higher elevations in ATRA versus endogenous levels, the equivalence of exogenous retinoid teratogenesis to physiological retinoid signaling has been cautioned (Ghyselinck and Duester 2019).
Labeled ATRA was shown to differentially accumulate in regions of the embryo that express cellular retinoic acid binding protein (CRABP), including the developing limb (Hatori, Shigematsu et al. 1991). CRABP function has been implicated in buffering ATRA availability in tissues where ATRA activity is required to be low (Dolle, Ruberte et al. 1990) or where ATRA gradients are important in patterning (Vaessen, Meijers et al. 1990). Indeed, the potency of various retinoid congeners to cause anterior-posterior pattern respecification correlated well with their ability to bind CRABP in both limb development (chick) and regeneration (axolotl) (Maden, Ong et al. 1989). Both CRABP-I and CRABP-II are structurally conserved phylogenetically and subject to cell-specific regulation ontogenetically in the chick limb-bud (Maden, Ong et al. 1988, Maden 1994) and regenerating amphibian limb (Scadding and Maden 1994). CrabpI transcript abundance inversely correlated with tissue-specific susceptibility to retinoid-induced teratogenesis, consistent with a buffering role (Ruberte, Friederich et al. 1992). Due to a RARE sequence the Crabp1 locus is ATRA-inducible (Kleinjan, Dekker et al. 1997). On the other hand, homozygous mice carrying deletions of CrabpI alleles are normal, indicating that CRABP-I does not play a crucial role in ATRA signaling (Gorry, Lufkin et al. 1994). This is in contrast to functional loss of CRABP-II, where CrabpII(−/−) mice show a developmental defect of the forelimb, specifically an additional, postaxial digit at an incidence that varies with genetic background from 10% in outbred CD1 mice to 30% and 50% for inbred strains C57BL/6 and 129Sv, respectively (Fawcett, Pasceri et al. 1995). Aside from this minor defect (polydactyly), mice deficient in CRABP-II (or in both CRABP-I and -II) are otherwise normal. Furthermore, CrabpI(−/−) and CrabpII(−/−) double knockouts are not more sensitive than wild-type embryos to ATRA excess treatment. Thus, CRABP-I and CRABP-II are dispensable to mouse development and none of the functions previously proposed for CRABPs are important enough to account for their evolutionary conservation (Lampron, Rochette-Egly et al. 1995).
The concentration of free ATRA calculated from kinematic coefficients and dissociation constant (Kd) of murine nuclear retinoic acid receptors (RARs) suggests a small dynamic range in the mouse limb-bud, perhaps as low as two-fold with regards to physiological function (Scott, Walter et al. 1994). Endogenous retinoids have been measured in the GD 10.5 mouse embryo before and 3h after maternal exposure to a fully teratogenic dose of ATRA (100 mg/kg) (Horton and Maden 1995). Those results, when compared to endogenous ATRA levels, showed increases of >100-fold in the spinal cord (0.12 → 21.3 μM after treatment); >400-fold in the limb-buds (0.03 → 12.5 μM); and >1000-fold in the forebrain (<0.01 → 10.0 μM). As such, embryos treated to induce malformations experience transitory exposures to massive doses of ATRA. Based on affinity constants RARs would be saturated throughout the limb-bud during both normal and teratological extremes. The more important regulator may be the AER-FGF-CYP26B1 module that maintains an ATRA-free distal zone for FGF-driven outgrowth (noted earlier).
As noted in earlier sections the phenotypic abnormalities observed in compound null mutants of RARs indicates functional redundancy between RARα, RARβ and RARγ (Lohnes, Mark et al. 1995). Although limb defects have not been described in VAD rats, they do appear in RARα/RARγ double null mutant mice (Lohnes, Mark et al. 1994). Those phenotypes include reductions in the scapula and radius (preaxial hemimelia), with weaker sensitivity of the corresponding hindlimb elements, and may reflect the need for ATRA signaling in the controlling the proper amount of limb mesenchyme. In addition, it is noteworthy that the limb phenotypes displayed by RARα/RARγ double null mutant mice and by RARβ/RARγ double null mutant mice altogether recapitulate what is observed in (vitamin A sufficient) fetuses lacking RDH10.
During mouse limb development RARα and RARγ (not RARβ) are uniformly expressed at GD 10 (Dolle, Ruberte et al. 1989). Overexpression of RARα in transgenic animals caused appendicular skeletal defects by blocking chondroblast differentiation of the precartilage anlagen, which could be released with pharmacological antagonists of retinoid signaling (Weston, Rosen et al. 2000). A constitutively active RARα transgene in mice resulted in marked limb defects that recapitulated many features of exogenous retinoid teratogenesis, pinpointing defective chondroblast differentiation as a failure to properly downregulate RARα (Cash, Bock et al. 1997). RARγ transcripts in the developing mouse limb are uniformly distributed through GD 11.5, then found in precartilage condensations (GD 12.5) and cartilages (GD 13.5 onward) (Ruberte, Dolle et al. 1990). An antisense oligodeoxynucleotide to knockdown RARγ function promoted chondrogenesis in limb micromass culture (Motoyama and Eto 1994). These results indicate that RARα and RARγ play a role in the formation of precartilage rudiments and are downregulated during overt chondrogenic differentiation. This is consistent with the teratological findings pinpointing the critical effect of exogenous RA to the stage of overt chondrogenic differentiation in vivo and in vitro.
In contrast, the role of RARβ is more confined to regions of the embryo that are susceptible retinoid teratogenesis. Administration of a fully phocomelic dose of ATRA to pregnant mice on GD 11 induced RARβ transcripts to a greater extent in the limbs versus RARα and RARγ or versus the embryo proper (Harnish, Jiang et al. 1992). Adverse effects on skeletal pattern correlated with the timing of retinoid exposure and the sequence of mesenchymal condensation. Ectopic RARβ promoter activity was detected within 2 h of exposure to ATRA and preceded alterations in the precartilage anlagen (Wood, Ward et al. 1996). Forelimb buds cultured from GD 12.5 mouse embryos in the presence of a RARγ-selective agonist (BMS-189961) showed reduction of chondrogenic progression and increased RAREβ2-lacZ reporter proximally (Galdones and Hales 2008). Together, these findings indicate that RAR-ATRA complexes play a role in position-dependent patterning of the limb skeleton during normal development, where RARβ isoforms block limb mesenchymal cells from expressing their inherent chondrogenic bias (Jiang, Soprano et al. 1995). While these findings support the notion that retinoid-induced RARβ expression plays a unique role in teratogenesis, other findings have shown that induction of RARβ alone is not sufficient to explain the phenotypes, given redundancy in the RAR system (Luo, Pasceri et al. 1995), and that several isoforms of the RARβ gene are independently regulated during limb development (Smith, Kirstein et al. 1995).
Homeobox genes are RAR targets and key regulators of outgrowth and pattern formation during limb development and regeneration, and several studies have shown ATRA-induced dysregulation of Hox gene expression in the developing limb. For example, VAD rat embryos harvested at the 35-somite stage showed reduced expression of Hoxd12 and Hoxd13 (Power, Lancman et al. 1999) and mouse embryos exposed to ATRA at a similar stage showed alterations in Hoxd11 and Hoxd13 expression prior to a delay in the formation of digital rudiments (Wood, Ward et al. 1996). Reciprocal changes in Hoxd13 and RARβ expression have been observed with ATRA-soaked beads applied to chick limb buds, again affecting distal skeletal elements (Hayamizu and Bryant 1994). And finally, the synthetic retinoids TTNPB (RAR agonist) and LG69 (RXR agonist) both induce Hoxb6, Hoxb8 and RARβ in the chick limb-bud (Lu, Eichele et al. 1997). Although normal functions of ATRA cannot be directly inferred from pharmacological studies alone, the evidence from normal and teratological studies implicates ligand bound RAR/RXR as key molecular determinant in both scenarios. These results further support the notion that ATRA-induced RARβ may at least partially account for limb dysmorphogenesis through altering Hox gene expression.
Polycomb-group genes in Drosophila maintain appropriate Hox codes, and mice null for a polycomb-group gene (M33) showed homeotic transformations of the axial skeleton and limb malformations aggravated by exogenous ATRA at GD 7.5 (Core, Bel et al. 1997). These results suggest that M33 defines access to RAREs in the regulatory regions of several Hox genes. Mathematical modeling and simulation show that ATRA forms a mutual antagonistic loop with repressive polycomb group factors in the distal forelimb bud as transcriptional repressors that mediate epigenetic gene silencing by chromatin modification (Yakushiji-Kaminatsui, Kondo et al. 2018).
10. Adverse Outcome Pathway (AOP) framework
Performance-based models that address the regulation, homeostasis, and biological activity of the retinoid signaling pathway will be useful for predictive toxicology based on alternative (non-mammalian) tests. An AOP framework is necessary to organize the relevant data, information and knowledge on molecular initiating events (MIEs) reflecting a disruption in retinoid signaling at critical stages of gestation, and the ensuing cascade of key events (KEs) and their relationships (KERs) leading to adverse outcomes (AOs) of regulatory value in toxicology. Developmental defects in the craniofacial, axial, or appendicular skeletal systems would qualify as they are readily observed in a traditional prenatal developmental toxicology test guideline study (e.g., OECD TG 414).
Both too little and too much vitamin A can have negative consequences on patterning the skeleton. Human VAD is an endemic nutrition problem throughout much of the developing world (West 2003). Recently, the gut microbiome has been identified as a new player in vitamin A metabolism. Commensal and pathogenic bacteria may drastically alter the patterns of vitamin A uptake in the host gut (Iyer and Vaishnava 2019). Genetic errors, synthetic retinoids, or environmental factors are known that can perturb the transport, metabolism, or utilization of endogenous ATRA during critical times in development. Humans and animals may be exposed to compounds in the natural environment that can invoke AOPs for the retinoid system that mimic a state of ATRA deficiency or excess. The putative list of AOPs may be neither complete nor comprehensive but point to the relevant developmental biology from which the mechanistic toxicology can be better understood (Figure 5). Three examples are shown to represent diverse MIEs and specific phenotypes affecting the facial, vertebral, or appendicular skeleton.
Figure 5. Framework and examples of potential AOPs for skeletal dysmorphogenesis linked to disruption of retinoid signaling.
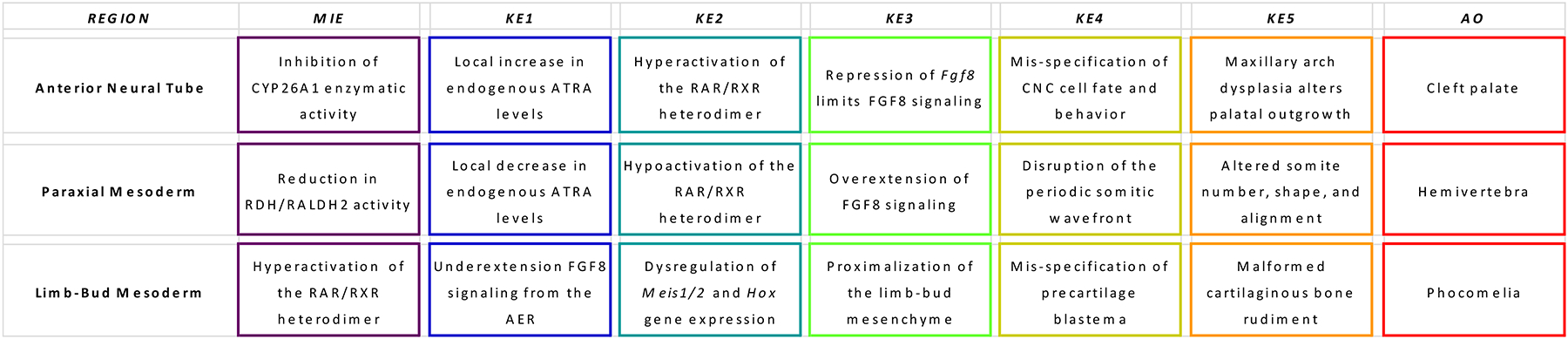
MIE, Molecular Initiating Event; KE, Key Events upstream to downstream; AO, Adverse Outcome. MIEs may include, for example: vitamin A deficiency, chemical inhibitors of RALDH2 or CYP26 enzymatic activity/expression; pharmacological agonists/antagonists of RAR or partner receptor signaling pathways (e.g., RXR/PPARγ); agents that modulate RAR/RXR binding to RARE sites. Subcellular KEs may be reflected in critical imbalances to local ATRA concentration or threshold responses leading to changes in Hox patterning and other molecular determinants of cell lineages. Cellular KEs entail developmental programming of undifferentiated progenitors of body axis determinants, depending on the stage of gestation and region of the embryo affected. Tissue KEs reflect the collective behavior of target cells directed by heterotypic interactions for rudimentary organs. AOs reflect the phenotypes’ resulting from stage and positional alterations in the fetal skeleton.
Subtle disturbances in ATRA homeostasis in vivo dramatically alter fetal morphology. This has been widely demonstrated in genetically engineered mice (Chambon 1996), in animal models of developmental toxicity (Kochhar 2000) and VAD (Kaiser, Merrill et al. 2003), and in human subjects (Lammer, Chen et al. 1985). Table 1 lists estimates for ATRA levels across different developmental systems, indicating the potential ranges of experimental perturbations.
Table 1.
Tissue levels of ATRA reported across different experimental systems
| baseline ATRA (5 somite zebrafish embryo) | < 1 nM | non-morphogenetic | (Shimozono, Iimura et al. 2013) |
| maternal serum (animal study) | 1.7 nM | non-teratogenic | (Daston, Beyer et al. 2014) |
| devTOXqp assay (pluripotent hESC) | 3.0 nM | teratogenic threshold | (Zurlinden, Saili et al. 2020) |
| normal plasma concentration | 5.0 nM | physiological (adult) | (Napoli, Posch et al. 1991) |
| axial gradient (5 somite zebrafish embryo) | 6.0 nM | morphogenetic signal | (Shimozono, Iimura et al. 2013) |
| endodermal differentiation (h-iPSC) | 17 nM | toxicological tipping point | (Saili, Antonijevic et al. 2019) |
| devTOXqp assay (pluripotent h-iPSC) | 19 nM | DevTox potential | (Palmer, Smith et al. 2017) |
| genetic perturbation (mouse) | 30 nM | altered homeostasis | (Helms, Thaller et al. 1994) |
| maternal serum (animal study) | 30 nM | teratogenic potential | (Daston, Beyer et al. 2014) |
| limb-bud (GD 10.5 mouse embryo) | 30 nM | physiological (embryo) | (Horton and Maden 1995) |
| pharmacological kinetics | 1,000 nM | efficacious (therapeutic) | (Helms, Thaller et al. 1994) |
| limb-bud (GD 11 mouse embryo) | 1,500 nM | weakly teratogenic dose | (Satre and Kochhar 1989) |
| limb-bud (GD 10.5 mouse embryo) | 12,500 nM | fully teratogenic dose | (Horton and Maden 1995) |
Whereas VAD animals and RAR gene ‘knockout’ mice indicate that ATRA performs essential functions in skeletal development, different AOP structures will be specific to the stages and sites in the embryo where a presence of endogenous ATRA is critical. One strategy is the intervention in RAR-dependent MIEs at specific developmental stages by means of synthetic retinoids that act as highly effective RAR agonists (e.g., AGN 190121) or antagonists (e.g., AGN 193109). For example, a putative AOP to explain midfacial defects in the mouse linked to functional retinoid deficiency induced with 1 mg/kg AGN 193109 on GD 8.0 (Kochhar, Jiang et al. 1998) would be initiated by RARα/γ antagonism (MIE) whereas antagonism of retinoid function may take a different path to explain the defects observed with the pan-RAR inverse agonist, BMS493 (Mark, Ghyselinck et al. 2006).
Other putative AOPs are needed to explain adverse outcomes linked to non-retinoid disruption of ATRA homeostasis. Citral, an inhibitor of retinol and retinaldehyde dehydrogenases as well as other alcohol and aldehyde dehydrogenases, induced a specific loss of derivatives from the maxillary prominences in the chick embryo that may in part result from disruption of endogenous ATRA synthesis (Shimomura, Kawakami et al. 2015). Ethanol, as a competitive inhibitor of RALDH2, may invoke ATRA deficiency causing phenotypes of the midface in the chick embryo (Kiecker 2016), and in rodent models for human Fetal Alcohol Spectrum Disorder (FASD) (Petrelli, Bendelac et al. 2019), through acetaldehyde formation (Shabtai, Bendelac et al. 2018). Glyphosate-based herbicides increased endogenous ATRA activity in a reporter assay and induced cranial defects in Xenopus embryos observed at the tadpole stage. These defects were ameliorated with RAR antagonists and infer a disruption of CNC development following elevated endogenous ATRA content (Paganelli, Gnazzo et al. 2010); however, it is unclear whether thyroid hormone could also have played a role in this phenomenon. Tributyltin, an antifoulant widely found industrially as a contaminant of dibutyltin in vinyl plastics, binds the RXR and causes retinoid-related craniofacial skeletal deformities in fish embryos (Zhang, Zuo et al. 2012); however, there are deficiencies in this putative AOP because tributyltin has no activity on RARs and the RAR-RXR heterodimer is not permissive to RXR activation.
Inhibition of ATRA degradation has been suggested as a mechanism to explain the dysmorphogenesis of some triazole fungicides on hindbrain segmentation and branchial arch development in cultured rodent embryos that has been hypothesized to result from a local increase of endogenous ATRA (Menegola, Broccia et al. 2006). For example, rat embryos exposed to flusilazole or fluconazole at the 1–3 somite stage displayed ATRA-like dysmorphogenesis of rostral branchial arches (1st, 2nd, 3rd) with concentrations above 6 μM and 60 μM, respectively (Menegola, Broccia et al. 2001). The antifungal properties of azole-derivatives is based on interference with the iron-porphyrin group in cytochrome P450 (CYP51, lanosterol C14 alpha-demethylase) that is required in the synthesis of fungal cell walls; however, this inhibition is not restricted to fungi and also occurs in mammalian CYP450-dependent activities, including CYP26 isozymes that are expressed in the embryo at the sensitive time (Menegola, Broccia et al. 2006). Evidence for a CYP26-mediated degradation effect was shown in cultured rat embryos exposed to fluconazole, where the incidence and severity of branchial arch defects was attenuated with citral (Di Renzo, Broccia et al. 2007). Since the postulated mode of action of azoles is inhibition of CYP enzymatic activity, and some ATRA-responsive patterning genes and proteins (e.g., Msx1, TGFβ1) were down-regulated in the branchial arch (Di Renzo, Corsini et al. 2009, Di Renzo, Rossi et al. 2011), a likely AOP is shown in Figure 5A (Di Renzo, Metruccio et al. 2019).
Congenital vertebral malformations occur in 5 to 10 per 10,000 live births (White and Goldberg 2018). Many are caused by genetic syndromes or intrauterine exposures such as hyperglycemia, carbon monoxide, or antiepileptic drugs. These phenotypes may be classified as a failure of complete vertebral formation (e.g., hemivertebrae), a failure of segmentation, or both. The former is generally attributed to asymmetric vertebral body formation often related to neural tube defects (e.g., spina bifida). Failures of segmentation are characterized by bony fusions between adjacent vertebrae or cranial-caudal border shifts and may also involve changes in ribs and sternum for the associated vertebra. An AOP framework for neural tube and axial defects via modulation of ATRA homeostasis has been outlined as a general mechanism that, when perturbed, may result in manifestations of developmental toxicity that cover a large part of malformations known to occur in humans and experimental animals (Tonk, Pennings et al. 2015). An AOP such as shown in Figure 5B includes key genes in the regulation of retinoid homeostasis, as well as marker genes of neural tube and axial patterning, and tested against existing data of flusilazole exposure in the rat WEC, the zebrafish embryotoxicity test, and the embryonic stem cell test.
Retinoid-induced limb malformations vary in scope and severity with dose and time of exposure. For example, retinoid teratogenicity at early stages of limb-bud growth (GD 9.5 mouse) are primarily restricted to missing digits (ectrodactyly) with varying sensitivities for different inbred mouse strains (Lee, Cantor et al. 2005). A full genome scan localized the susceptibility loci to chromosome 11 (near D11Mit39) syntenic to human Meckel syndrome that includes digital phenotypes. Reports of hindlimb malformations in frogs across North America suggested a likely consequence of retinoid disruption (Degitz, Kosian et al. 2000). Subsequently findings, however, linked the etiology to natural parasites (trematodes) in combination with agricultural runoff (e.g., triazine herbicides, organophosphate insecticides, and synthetic pyrethroids) that decreased the host tadpole’s ability to resist parasitic infection (Kiesecker 2002). As such, the AOP was not initiated by retinoid disruption but in combination with a parallel AOP linked to changes in immunity.
Target cell populations in an AOP will vary based on stage of development, timing and degree of MIE perturbation required to invoke a change. As such, the window of vulnerability for retinoid-induced phocomelia (e.g., GD 10–12) follow later in gestation than for axial defects (e.g., GD 6.5 – 8.5). This temporal factor is further dependent on the type and nature of adverse outcomes. Of importance is the notion that an ATRA-FGF8 antagonistic gradient is a common KE to many potential AOPs. An AOP for phocomelia might look like Figure 5C.
AOP frameworks are an important tool for developmental hazard evaluation because several chemically-distinct classes of developmental toxicants may invoke features of a Fetal Retinoid Syndrome, including anticonvulsants (e.g., valproic acid), some triazole fungicides (e.g., fluconazole), organic metals (e.g., tributyl tin), organophosphates (e.g., aldrin), and flame retardants (e.g., PBDEs). Given the complex relationships noted earlier, a generalized AOP framework for retinoid biology and teratogenesis is highly desirable, but seriously constrained by the complexity of the system and its crosstalk with other morphogenetic signaling pathways and cellular behaviors. Despite this wealth of information only two literature reports refer to an AOP for the retinoid system (Tonk, Pennings et al. 2015, Baker, Boobis et al. 2018). Searching the AOPWiki [https://aopwiki.org/] for ‘retinoic acid’ or ‘retinoid’ returned key events for only 5 AOPs (#ID = 43, 297, 7, 37, 38) at the present time.
11. New Approach Methodologies (NAMs)
Opportunities exist for refining and supplanting current developmental toxicity testing protocols using in vitro data and in silico models in the design and review of revolutionary alternatives to animal testing by experts in the field, alongside their independent validation (Scialli, Daston et al. 2018). New Approach Methodologies (NAMs) is the term adopted as a broadly descriptive reference to any technology, methodology, approach, or combination thereof that can be used to provide information on chemical hazard and risk assessment that avoids the use of intact animals (EPA 2018). NAMs would include methods that evaluate hazard (human health and environmental), exposure, and environmental fate as well as different approaches to integrate NAMs for decision making such as AOPs and integrated approaches to testing and assessment (IATA). Technology platforms include, for example automated in vitro assays for high-throughput screening (HTS) and in silico computational toxicology models to reconstruct complex pathways. For example, 8 in vitro HTS assays for some components in the retinoid system are included in the US ToxCast/Tox21 program (Figure 6).
Figure 6. Case examples for Class Distribution.
Distribution from chemical hits (n=261) having AC50 < 2 μM in one or more of the 8 ToxCast assays (Baker, Boobis et al. 2018). Each assay target is indicated with the number of chemical his registered in the EPA CompTox Chemicals Dashboard (https://comptox.epa.gov/dashboard, last accessed October, 2020). CYP1A1: NVS_ADME_hCYP1A1. RARs: ATG_RARa_TRANS_up, ATG_RARb_TRANS_up, ATG_RARg_TRANS_up. RXRs: ATG_RXRa_TRANS_up, ATG_RXRb_TRANS_up, ATG_RXRg_TRANS_up. DR5: ATG_DR5_CIS_up. As might be expected the DR5 lights up several potential RAR/RXR combinations. First group: vitamin A (retinol) and retinoid ligands (ATRA/RAR, Bexarotene/RXR) references. Second group: Triazole effects on biochemical activity of CYP1A1 as a surrogate for CYP26 isoforms; malformations, vertebral transformations, and caudal regression are linked to CYP26 inhibition (Menegola, Broccia et al. 2001, Kamata, Shiraishi et al. 2008, Tonk, Pennings et al. 2015). Third group: several organochlorine pesticides of a persistent nature have weak RARg-agonist activity and can transactivate retinoid-responsive genes (e.g., CYP26A1) via RARE (Lemaire, Balaguer et al. 2005, Kamata, Shiraishi et al. 2008). Fourth group: several organotin biocides are known to preferentially bind RXRs with nM affinity, but forms a non-permissive RAR/RXR heterodimer (Grun, Watanabe et al. 2006, Brtko and Dvorak 2015).
Briefly, the ToxCast NovaScreen assay (NVS_ADME_hCYP1A1) is a cell-free biochemical assay using a type of enzyme reporter to measure loss-of-signal enzymatic activity (AC50) as it relates to human cytochrome P450, family 1, subfamily A, polypeptide 1 (CYP1A1). Loss of fluorescence intensity signals produced from an NADP-dependent enzymatic reaction involving a fluorogenic substrate for cytochrome P450-linked enzymes substrate, Resorufin Benzyl Ether. Although HTS assays are not available for CYP26a/b/c in ToxCast/Tox21, CYP1A1 effectively degrades ATRA in a cell free biochemical surrogate assay. The Attagene (ATG) assays track changes in transcription factor activity utilizing a library of multiplex reporter transcription unit constructs. In this case, RAR/RXR heterodimers bind to their cognate gene response element, RARE (retinoic acid receptor elements) in this case. Individual transcription units are essentially barcoded to particular receptor. The HepG2 human hepatoma cell line used in this platform retains the potential for Phase I and Phase II metabolic responses to xenobiotics (e.g., expression of various Cytochrome P450s) at levels similar to mature hepatocytes. Finally, DR5 is a cis-regulated assay for the direct repeat element in RARE binding sites and is broadly responsive to chemical changes that increase RARE-dependent transcription. As a cautionary principle, use of these and other HTS data must consider nuances in the methodological, platform, reagent differences, cytotoxicity, and data quality flags. Individual ToxCast assay results provide only one piece of a complex puzzle and must be considered within the larger ToxCast/Tox21 data context in order to advance understanding of potential chemical hazards and their mechanisms (Houck, Judson et al. 2017). Furthermore, there are high-priority deficiencies in the portfolio. For example, assays described in the literature but lacking in ToxCast/Tox21 include dehydrogenases for retinol bioactivation (RDH, RALDH2) (Schindler, Berst et al. 1998, Koppaka, Thompson et al. 2012, Thomas, de Antueno et al. 2016, Attignon, Distel et al. 2017) and oxidoreductases for retinoic acid breakdown (CYP26 a/b/c) (Mulvihill, Kan et al. 2006). As shown earlier, these are critically important for anterior (RALDH2) and posterior (CYP26) development of the fetus and may predict susceptible stages for developmental toxicity. Less complete information is available on disruption of retinoid transport, such as serum transporters (e.g., TTR), molecular transporters (e.g., STRA6), and intracellular binding proteins (e.g., CRABP).
A preliminary analysis of almost 1983 ToxCast chemicals across the 8 assay systems identified 97 having an AC50 < 2 μM in one or more of these assays. Among those on the list were persistent organic pollutants that preferentially activated RARs (e.g., aldrin, dieldrin) and tert-butyl compounds and organotins that preferentially activated RXRs (e.g., butylphenol, tributyltin). These findings are consistent with results from targeted studies in the literature (Lemaire, Balaguer et al. 2005, le Maire, Grimaldi et al. 2009). Other examples include retinoids themselves (ATRA, retinol), anticonvulsants (valproic acid), triazoles (fluconazole), and flame retardants (PBDEs) (Baker, Boobis et al. 2018). In addition, some mitochondrial disrupters displayed activity on DR5 (e.g., strobins, rotenone) that may reflect a dependence of the response on bioenergetics (le Maire, Grimaldi et al. 2009). Note that the chemicals may have activity over multiple types of assays that are not necessarily related to retinoid receptors or signaling.
The complex nature of the retinoid system means that a complete battery of in vitro assays capturing all potential points of retinoid disruption, biological information, and pathway-level crosstalk is unlikely to emerge in the near term. As such, computational modeling will be necessary to build data-driven models for predictive toxicology and quantitative simulation, similar to what has been done for the estrogen signaling pathway (Browne, Judson et al. 2015, Judson, Magpantay et al. 2015, Kleinstreuer, Ceger et al. 2017); (Juberg, Knudsen et al. 2017).
Retinoid signaling-related modeling may prove to be more challenging due to the levels of complexity involved. However, several findings set the stage for promising outcomes in a predictive model for retinoid signaling. (a) RAR was top weighted feature in an early ToxCast (phase I) predictive model for ToxRefDB (Sipes, Martin et al. 2011); (b) retinoic acid ranked #1 in potency across 1065 chemicals tested in a human embryonic stem cell platform (PoD = 10 nM) with up to 84% concordance to animal models for human developmental toxicity (Zurlinden, Saili et al. 2020); (c) RNAseq profile of human iPSC cell differentiation provides a detailed molecular characterization of a toxicological tipping point for retinoid signaling and endodermal toxicity (Point of Departure = 17 nM) (Saili, Antonijevic et al. 2019); and (d) the retinoid signaling pathway also turned up in ToxCast signatures for male reproductive development (Leung, Phuong et al. 2016), cleft palate (Baker, Sipes et al. 2019), and digital paw defects (Ahir, DeWoskin et al. 2019). Translation of exposure-based hazard predictions can utilize in silico prediction tools such as P&G’s DevTox ‘SAR decision tree’ (Wu, Fisher et al. 2013) and translation of exposure-based hazard predictions using in silico prediction tools such as the pregnancy HTTK model and chemotype information (Lumen, Chang et al. 2020). For these models, it is useful to have knowledge of ATRA concentrations/levels in the various systems (Napoli, Posch et al. 1991, Helms, Thaller et al. 1994, Shimozono, Iimura et al. 2013, Daston, Beyer et al. 2014, Palmer, Smith et al. 2017, Saili, Antonijevic et al. 2019, Zurlinden, Saili et al. 2020).
12. Conclusions
This review underscores the importance of ATRA homeostasis to patterning and differentiation of the fetal skeleton. These pathways are complex and connected directly or indirectly to morphogenetic signaling. A critical role of ATRA signaling during gastrulation and early organogenesis influences regional specification and fate of precursor cell populations in the cranial neural crest, paraxial mesoderm, and lateral plate mesoderm. To date, no OECD test guidelines specifically capture the retinoid system for toxicity screening and evaluation. While retinoid disruption may contribute to any number of skeletal endpoints observed in a regulatory guideline developmental toxicity study (e.g., OECD TG 414), related AOP tracks could also explain similar outcomes. This raises the question of whether the ATRA system is a primary target, secondary affiliate, or unrelated to the outcome. Unraveling this complexity will be important for NAMs that may use in vitro data and in silico models for predictive DART. Perturbations evaluated in a prenatal developmental toxicity study conducted for regulatory assessment include a number of adverse outcomes. Xenobiotics can affect multiple molecular pathways so it is not clear how any skeletal defects found will be strictly attributable to or exclusively to an ATRA-signaling-related mechanism. This classic ‘one-to-many problem’ in bioinformatics (e.g., one MIE can diverge into many adverse outcomes; many MIEs may converge onto one adverse outcome) inspires several types of modeling activities for integration and synthesis of retinoid endpoints (including measurement of retinoids and chemical dosimetry).
An AOP framework will be a central resource for informing AOP-based integration of the biology-toxicology and subsequent parameterization of performance-based models for predictive toxicology of the retinoid system and skeletal development. Some information is already available for profiling the retinoid system in vitro (derived from ToxCast/Tox21 datasets) and building performance-based models to predict skeletal dysmorphogenesis in vivo (derived from the literature and ToxRefDB_v2 databases). There is a need to further develop and/or select assays that reflect the known biology of the retinoid signaling system and can be reproduced across laboratories as a fundamental requirement of validation. Such assays should also require corroboration by independent experts.
Future perspectives go beyond what is covered in this review. The strengths and limitations pointed out for predictive toxicology of skeletal development motivate continuing on the track of the retinoid system, in combination with tracks on CNS development, cardiovascular development and other systems. Applying lessons learned from individual developing systems will support future efforts to build an integrated computational model for the ATRA system for applications of NAM data for predictive DART. NAMs can identify potential MIEs that can trigger a series of key events known from the literature to lead to a phenotype from which mechanisms can be inferred. Validation without mammalian test in animal models is in a paradox. Without dynamic simulation, mechanistic inferences from NAMs will remain correlative.
Preface.
All-trans retinoic acid (ATRA) is a conserved signal molecule during morphogenesis, growth and differentiation across diverse organ systems. This review of the literature derives from an annex in Detailed Review Paper (DRP) of the OECD Test Guidelines Programme (Project 4.97) that is intended to support recommendations regarding assay development to determine retinoid system toxicants for developmental and reproductive toxicity. Because mutations of the ATRA system tend to be disruptive of skeletal patterning during early embryogenesis, the review focuses on the embryonic period where skeletal patterning is most vulnerable to disruption of ATRA metabolism and signaling. Literature analysis returned a catalogue of nearly 6,000 publication records broadly annotated for retinoids, embryos, and development and sifted by specific terms in the article’s title, abstract or keywords appropriate to skeletal domains. Specific examples are drawn to support potential Adverse Outcome Pathways (AOPs) that help explain chemical effects on signaling pathways that pattern skeletal development and plausible molecular initiating events (MIEs) linked to critcal effects on ATRA kinematics. Some perspectives on the phylogeny-ontogeny of ATRA signaling are discussed to support it’s conserved role in local cell-cell interactions - the hallmark of multicellular organization. A generalized framework for retinoid teratogenesis supports data from technology platforms for automated high-throughput screening (in vitro) and computational dynamic models (in silico) that address ATRA metabolism, signaling thresholds, genomic responses, and crosstalk with morpho-regulatory processes. Perturbation of several developmental pathways can adversely affect skeletal development. Adverse outcomes evaluated in a prenatal developmental toxicity study conducted for regulatory assessment may include altered growth trajectories, functional and behavioral deficits, structural abnormalities that include both malformations (permanent structural changes that may adversely affect survival, development, or function (sometimes referred to as teratogenicity) and variations (used to indicate a divergence beyond the usual range of structural constitution that may not adversely affect survival or health). From a regulatory developmental toxicity perspective, it is not clear how any skeletal defects found will be strictly attributable to or exclusively to an ATRA-signaling-related mechanism. The reconstruction of toxicity pathways in early embryonic development sets the stage for future perspectives on the potential susceptibility for retinoid system associated modulations on bone health and bone-related disease over the life-course.
Acknowledgements
The authors gratefully acknowledge helpful comments on the DRP draft annex from the following experts: John M. Rogers (US EPA, ORD/CPHEA), Paul C. Brown (US FDA, CDER), Amy C. Nostrandt (US FDA, CDER), Karen L. Hamernik (US EPA, OCSPP), Robert L. Sprando (US FDA, CFSAN), Bruce Blumberg (University of California Irvine, Dept. Developmental and Cell Biology), Shirlee W. Tan (Endocrine Society), Scott M. Belcher (North Carolina State University, Dept. Biological Sciences), Alice Baynes (Institute of Environment, Health and Societies, Brunel University London), Margareta Halin Lejonklou (Swedish Chemicals Agency and Uppsala University), Anne-Lee Gustafson (Swedish Chemicals Agency and Uppsala University), and Manuel Mark (IGBMC, University of Strasbourg, France). A version of this draft is included in an OECD Detailed Review Paper on the Retinoid pathway coordinated by Patience Browne (OECD) and Anne Gourmelon (OECD) with early contributions from Helen Håkansson (Institute of Environmental Medicine, Karolinska Institutet, Sweden).
Footnotes
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- Abe M, Maeda T and Wakisaka S (2008). “Retinoic acid affects craniofacial patterning by changing Fgf8 expression in the pharyngeal ectoderm.” Dev Growth Differ 50(9): 717–729. [DOI] [PubMed] [Google Scholar]
- Abu-Hijleh G and Padmanabhan R (1997). “Retinoic acid-induced abnormal development of hindlimb joints in the mouse.” Eur J Morphol 35(5): 327–336. [DOI] [PubMed] [Google Scholar]
- Ahir B, DeWoskin R, Baker N, Spencer R, Setzer R, Lau C and Knudsen T (2019). “Developmental toxicity Simulated in a dynamic virtual embryo model of early limb-bud outgrowth (in preparation).”
- Akimenko MA, Ekker M (1995). “ Anterior duplication of the Sonic hedgehog expression pattern in the pectoral fin buds of zebrafish treated with retinoic acid” Dev Biol 170 (1): 243–247 [DOI] [PubMed] [Google Scholar]
- Al Tanoury Z, Piskunov A and Rochette-Egly C (2013). “Vitamin A and retinoid signaling: genomic and nongenomic effects.” J Lipid Res 54(7): 1761–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang HL, Deltour L, Hayamizu TF, Zgombic-Knight M and Duester G (1996). “Retinoic acid synthesis in mouse embryos during gastrulation and craniofacial development linked to class IV alcohol dehydrogenase gene expression.” J Biol Chem 271(16): 9526–9534. [DOI] [PubMed] [Google Scholar]
- Attignon EA, Distel E, Le-Grand B, Leblanc AF, Barouki R, de Oliveira E, Aggerbeck M and Blanc EB (2017). “Down-regulation of the expression of alcohol dehydrogenase 4 and CYP2E1 by the combination of alpha-endosulfan and dioxin in HepaRG human cells.” Toxicol In Vitro 45(Pt 3): 309–317. [DOI] [PubMed] [Google Scholar]
- Aulehla A and Pourquie O (2010). “Signaling gradients during paraxial mesoderm development.” Cold Spring Harb Perspect Biol 2(2): a000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N, Boobis A, Burgoon L, Carney E, Currie R, Fritsche E, Knudsen T, Laffont M, Piersma AH, Poole A, Schneider S and Daston G (2018). “Building a developmental toxicity ontology.” Birth Defects Res 110(6): 502–518. [DOI] [PubMed] [Google Scholar]
- Baker N, Knudsen T and Williams A (2017). “Abstract Sifter: a comprehensive front-end system to PubMed.” F1000Res 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N, Sipes N, Franzosa J, Belair DG, Abbott BD, Judson RS and Knudsen TB (2019). “Characterizing cleft palate toxicants using ToxCast data, chemical structure, and the biomedical literature.” Birth Defects Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer JE and Blomhoff R (2005). “A robust characterization of retinoic acid response elements based on a comparison of sites in three species.” The Journal of Steroid Biochemistry and Molecular Biology 96(5): 347–354. [DOI] [PubMed] [Google Scholar]
- Bastien J and Rochette-Egly C (2004). “Nuclear retinoid receptors and the transcription of retinoid-target genes.” Gene 328: 1–16. [DOI] [PubMed] [Google Scholar]
- Bavik C, Ward SJ and Chambon P (1996). “Developmental abnormalities in cultured mouse embryos deprived of retinoic by inhibition of yolk-sac retinol binding protein synthesis.” Proc Natl Acad Sci U S A 93(7): 3110–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechter R, Terlouw GD, Tsuchiya M, Tsuchiya T and Kistler A (1992). “Teratogenicity of arotinoids (retinoids) in the rat whole embryo culture.” Arch Toxicol 66(3): 193–197. [DOI] [PubMed] [Google Scholar]
- Begemann G and Meyer A (2001). “Hindbrain patterning revisited: timing and effects of retinoic acid signalling.” Bioessays 23(11): 981–986. [DOI] [PubMed] [Google Scholar]
- Begemann G, Schilling TF, Rauch GJ, Geisler R and Ingham PW (2001). “The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain.” Development 128(16): 3081–3094. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Aldea D, Oulion S, Subirana L, de Lera AR, Somorjai I and Escriva H (2015). “Evolution of the Role of RA and FGF Signals in the Control of Somitogenesis in Chordates.” PLoS One 10(9): e0136587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaner WS (2001). “Cellular metabolism and actions of 13-cis-retinoic acid.” J Am Acad Dermatol 45(5): S129–135. [DOI] [PubMed] [Google Scholar]
- Blaner WS, Li Y, Brun PJ, Yuen JJ, Lee SA and Clugston RD (2016). “Vitamin A Absorption, Storage and Mobilization.” Subcell Biochem 81: 95–125. [DOI] [PubMed] [Google Scholar]
- Brown JM, Robertson KE, Wedden SE and Tickle C (1997). “Alterations in Msx 1 and Msx 2 expression correlate with inhibition of outgrowth of chick facial primordia induced by retinoic acid.” Anat Embryol (Berl) 195(2): 203–207. [DOI] [PubMed] [Google Scholar]
- Browne P, Judson RS, Casey WM, Kleinstreuer NC and Thomas RS (2015). “Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model.” Environ Sci Technol 49(14): 8804–8814. [DOI] [PubMed] [Google Scholar]
- Brtko J and Dvorak Z (2015). “Triorganotin compounds--ligands for “rexinoid” inducible transcription factors: biological effects.” Toxicol Lett 234(1): 50–58. [DOI] [PubMed] [Google Scholar]
- Bushue N and Wan YJ (2010). “Retinoid pathway and cancer therapeutics.” Adv Drug Deliv Rev 62(13): 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bynum SV (1991). “Morphogenesis of retinoic acid-induced postaxial polydactyly in mice.” Teratology 43(1): 1–9. [DOI] [PubMed] [Google Scholar]
- Cammas L, Romand R, Fraulob V, Mura C and Dolle P (2007). “Expression of the murine retinol dehydrogenase 10 (Rdh10) gene correlates with many sites of retinoid signalling during embryogenesis and organ differentiation.” Dev Dyn 236(10): 2899–2908. [DOI] [PubMed] [Google Scholar]
- Cash DE, Bock CB, Schughart K, Linney E and Underhill TM (1997). “Retinoic acid receptor alpha function in vertebrate limb skeletogenesis: a modulator of chondrogenesis.” J Cell Biol 136(2): 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P (1996). “A decade of molecular biology of retinoic acid receptors.” FASEB J 10(9): 940–954. [PubMed] [Google Scholar]
- Chatzi C, Cunningham TJ and Duester G (2013). “Investigation of retinoic acid function during embryonic brain development using retinaldehyde-rescued Rdh10 knockout mice.” Dev Dyn 242(9): 1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dong D, Kostetskii I, Zile MH (1996). “Hensen’s node from vitamin A-deficient quail embryo induces chick limb bud duplication and retains its normal asymmetric expression of Sonic hedgehog (Shh)” Dev Biol 173 (1): 256–264 [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Garcia E and Schilling TF (2010). “Regulation of facial morphogenesis by endothelin signaling: insights from mice and fish.” Am J Med Genet A 152A(12): 2962–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway HH, Henning P and Lerner UH (2013). “Vitamin a metabolism, action, and role in skeletal homeostasis.” Endocr Rev 34(6): 766–797. [DOI] [PubMed] [Google Scholar]
- Conserva MR, Anelli L, Zagaria A, Specchia G and Albano F (2019). “The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia.” Int J Mol Sci 20(12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core N, Bel S, Gaunt SJ, Aurrand-Lions M, Pearce J, Fisher A and Djabali M (1997). “Altered cellular proliferation and mesoderm patterning in Polycomb-M33-deficient mice.” Development 124(3): 721–729. [DOI] [PubMed] [Google Scholar]
- Cunningham TJ, Brade T, Sandell LL, Lewandoski M, Trainor PA, Colas A, Mercola M and Duester G (2015). “Retinoic Acid Activity in Undifferentiated Neural Progenitors Is Sufficient to Fulfill Its Role in Restricting Fgf8 Expression for Somitogenesis.” PLoS One 10(9): e0137894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Chatzi C, Sandell LL, Trainor PA and Duester G (2011). “Rdh10 mutants deficient in limb field retinoic acid signaling exhibit normal limb patterning but display interdigital webbing.” Dev Dyn 240(5): 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ and Duester G (2015). “Mechanisms of retinoic acid signalling and its roles in organ and limb development.” Nat Rev Mol Cell Biol 16(2): 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Kumar S, Yamaguchi TP and Duester G (2015). “Wnt8a and Wnt3a cooperate in the axial stem cell niche to promote mammalian body axis extension.” Dev Dyn 244(6): 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Lancman JJ, Berenguer M, Dong PDS and Duester G (2018). “Genomic Knockout of Two Presumed Forelimb Tbx5 Enhancers Reveals They Are Nonessential for Limb Development.” Cell Rep 23(11): 3146–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Zhao X, Sandell LL, Evans SM, Trainor PA and Duester G (2013). “Antagonism between retinoic acid and fibroblast growth factor signaling during limb development.” Cell Rep 3(5): 1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daston GP, Beyer BK, Carney EW, Chapin RE, Friedman JM, Piersma AH, Rogers JM and Scialli AR (2014). “Exposure-based validation list for developmental toxicity screening assays.” Birth Defects Res B Dev Reprod Toxicol 101(6): 423–428. [DOI] [PubMed] [Google Scholar]
- Degitz SJ, Kosian PA, Makynen EA, Jensen KM and Ankley GT (2000). “Stage- and species-specific developmental toxicity of all-trans retinoic acid in four native North American ranids and Xenopus laevis.” Toxicol Sci 57(2): 264–274. [DOI] [PubMed] [Google Scholar]
- Dencker L, Gustafson AL, Annerwall E, Busch C and Eriksson U (1991). “Retinoid-binding proteins in craniofacial development.” J Craniofac Genet Dev Biol 11(4): 303–314. [PubMed] [Google Scholar]
- Deschamps J and van Nes J (2005). “Developmental regulation of the Hox genes during axial morphogenesis in the mouse.” Development 132(13): 2931–2942. [DOI] [PubMed] [Google Scholar]
- Di Renzo F, Broccia ML, Giavini E and Menegola E (2007). “Citral, an inhibitor of retinoic acid synthesis, attenuates the frequency and severity of branchial arch abnormalities induced by triazole-derivative fluconazole in rat embryos cultured in vitro.” Reprod Toxicol 24(3–4): 326–332. [DOI] [PubMed] [Google Scholar]
- Di Renzo F, Corsini E, Broccia ML, Marinovich M, Galli CL, Giavini E and Menegola E (2009). “Molecular mechanism of teratogenic effects induced by the fungicide triadimefon: study of the expression of TGF-beta mRNA and TGF-beta and CRABPI proteins during rat in vitro development.” Toxicol Appl Pharmacol 234(1): 107–116. [DOI] [PubMed] [Google Scholar]
- Di Renzo F, Metruccio F, Battistoni M, Moretto A and Menegola E (2019). “Relative potency ranking of azoles altering craniofacial morphogenesis in rats: An in vitro data modelling approach.” Food Chem Toxicol 123: 553–560. [DOI] [PubMed] [Google Scholar]
- Di Renzo F, Rossi F, Prati M, Giavini E and Menegola E (2011). “Early genetic control of craniofacial development is affected by the in vitro exposure of rat embryos to the fungicide triadimefon.” Birth Defects Res B Dev Reprod Toxicol 92(1): 77–81. [DOI] [PubMed] [Google Scholar]
- Diez del Corral R and Storey KG (2004). “Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis.” Bioessays 26(8): 857–869. [DOI] [PubMed] [Google Scholar]
- Dolle P, Ruberte E, Kastner P, Petkovich M, Stoner CM, Gudas LJ and Chambon P (1989). “Differential expression of genes encoding alpha, beta and gamma retinoic acid receptors and CRABP in the developing limbs of the mouse.” Nature 342(6250): 702–705. [DOI] [PubMed] [Google Scholar]
- Dolle P, Ruberte E, Leroy P, Morriss-Kay G and Chambon P (1990). “Retinoic acid receptors and cellular retinoid binding proteins. I. A systematic study of their differential pattern of transcription during mouse organogenesis.” Development 110(4): 1133–1151. [DOI] [PubMed] [Google Scholar]
- Duester G, (2008). “ Retinoic acid synthesis and signaling during early organogenesis.” Cell, 134 (6): 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupe V, Ghyselinck NB, Thomazy V, Nagy L, Davies PJ, Chambon P and Mark M (1999). “Essential roles of retinoic acid signaling in interdigital apoptosis and control of BMP-7 expression in mouse autopods.” Dev Biol 208(1): 30–43. [DOI] [PubMed] [Google Scholar]
- Dupe V and Pellerin I (2009). “Retinoic acid receptors exhibit cell-autonomous functions in cranial neural crest cells.” Dev Dyn 238(10): 2701–2711. [DOI] [PubMed] [Google Scholar]
- Eichele G, (1989). “ Retinoic acid induces a pattern of digits in anterior half wing buds that lack the zone of polarizing activity.” Development 107 (4): 863–867 [DOI] [PubMed] [Google Scholar]
- Escriva H, Holland ND, Gronemeyer H, Laudet V and Holland LZ (2002). “The retinoic acid signaling pathway regulates anterior/posterior patterning in the nerve cord and pharynx of amphioxus, a chordate lacking neural crest.” Development 129(12): 2905–2916. [DOI] [PubMed] [Google Scholar]
- Evans RM and Mangelsdorf DJ (2014). “Nuclear Receptors, RXR, and the Big Bang.” Cell 157(1): 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JF, Lopez A, Ros MA, Savage MP, Olwin BB and Simandl BK (1994). “FGF-2: apical ectodermal ridge growth signal for chick limb development.” Science 264(5155): 104–107. [DOI] [PubMed] [Google Scholar]
- Fawcett D, Pasceri P, Fraser R, Colbert M, Rossant J and Giguere V (1995). “Postaxial polydactyly in forelimbs of CRABP-II mutant mice.” Development 121(3): 671–679. [DOI] [PubMed] [Google Scholar]
- Forlani S, Lawson KA and Deschamps J (2003). “Acquisition of Hox codes during gastrulation and axial elongation in the mouse embryo.” Development 130(16): 3807–3819. [DOI] [PubMed] [Google Scholar]
- Franzosa JA, Bugel SM, Tal TL, La Du JK, Tilton SC, Waters KM and Tanguay RL (2013). “Retinoic acid-dependent regulation of miR-19 expression elicits vertebrate axis defects.” Faseb j 27(12): 4866–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Sato T, Kaneko S, Gotoh O, Fujii-Kuriyama Y, Osawa K, Kato S and Hamada H (1997). “Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos.” Embo j 16(14): 4163–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdones E and Hales BF (2008). “Retinoic acid receptor gamma-induced misregulation of chondrogenesis in the murine limb bud in vitro.” Toxicol Sci 106(1): 223–232. [DOI] [PubMed] [Google Scholar]
- Ghyselinck NB and Duester G (2019). “Retinoic acid signaling pathways.” Development 146(13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibert Y, Gajewski A, Meyer A, Begemann G (2006). “Induction and prepatterning of the zebrafish pectoral fin bud requires axial retinoic acid signaling.” Development 133 (14): 2649–2659 [DOI] [PubMed] [Google Scholar]
- Gorry P, Lufkin T, Dierich A, Rochette-Egly C, Decimo D, Dolle P, Mark M, Durand B and Chambon P (1994). “The cellular retinoic acid binding protein I is dispensable.” Proc Natl Acad Sci U S A 91(19): 9032–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding EH and Pratt RM (1986). “Isotretinoin teratogenicity in mouse whole embryo culture.” J Craniofac Genet Dev Biol 6(2): 99–112. [PubMed] [Google Scholar]
- Grandel H, Lun K, Rauch GJ, Rhinn M, Piotrowski T, Houart C, Sordino P, Kuchler AM, Schulte-Merker S, Geisler R, Holder N, Wilson SW, Brand M (2002). “Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud.” Development 129 (12): 2851–2865 [DOI] [PubMed] [Google Scholar]
- Granstrom G and Kullaa-Mikkonen A (1990). “Experimental craniofacial malformations induced by retinoids and resembling branchial arch syndromes.” Scand J Plast Reconstr Surg Hand Surg 24(1): 3–12. [DOI] [PubMed] [Google Scholar]
- Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T and Blumberg B (2006). “Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates.” Mol Endocrinol 20(9): 2141–2155. [DOI] [PubMed] [Google Scholar]
- Hale F (1935). “The Relation of Vitamin a to Anophthalmos in Pigs.” American Journal of Ophthalmology 18(12): 1087–1093. [Google Scholar]
- Harnish DC, Jiang H, Soprano KJ, Kochhar DM and Soprano DR (1992). “Retinoic acid receptor beta 2 mRNA is elevated by retinoic acid in vivo in susceptible regions of mid-gestation mouse embryos.” Dev Dyn 194(3): 239–246. [DOI] [PubMed] [Google Scholar]
- Hatori A, Shigematsu A, McCormick AM, Willhite CC and Sharma RP (1991). “Temporal distribution of retinoic acid and cellular retinoic acid-binding protein (CRABP) in the fetal hamster.” Exp Mol Pathol 55(1): 38–54. [DOI] [PubMed] [Google Scholar]
- Hayamizu TF and Bryant SV (1994). “Reciprocal changes in Hox D13 and RAR-beta 2 expression in response to retinoic acid in chick limb buds.” Dev Biol 166(1): 123–132. [DOI] [PubMed] [Google Scholar]
- Helms J, Thaller C and Eichele G (1994). “Relationship between retinoic acid and sonic hedgehog, two polarizing signals in the chick wing bud.” Development 120(11): 3267–3274. [DOI] [PubMed] [Google Scholar]
- Henning P, Conaway HH and Lerner UH (2015). “Retinoid receptors in bone and their role in bone remodeling.” Front Endocrinol (Lausanne) 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgreb T, Linhares VL, Menezes DC, Sampaio AC, Yan CY, Cardoso WV, Rosenthal N and Xavier-Neto J (2003). “A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field.” Development 130(22): 5363–5374. [DOI] [PubMed] [Google Scholar]
- Hong SH, Fang S, Lu BC, Nofsinger R, Kawakami Y, Castro GL, Yin Y, Lin C, Yu RT, Downes M, Izpisua Belmonte JC, Shilatifard A and Evans RM (2018). “Corepressor SMRT is required to maintain Hox transcriptional memory during somitogenesis.” Proc Natl Acad Sci U S A 115(41): 10381–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton C and Maden M (1995). “Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo.” Dev Dyn 202(3): 312–323. [DOI] [PubMed] [Google Scholar]
- Houck KA, Judson RS, Knudsen TB, Martin MT, Richard AM, Crofton KM, Simeonov A, Paules RS, Bucher JR and Thomas RS (2017). “Comment on “On the Utility of ToxCast and ToxPi as Methods for Identifying New Obesogens”.” Environ Health Perspect 125(1): A8–A11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubaud A and Pourquie O (2014). “Signalling dynamics in vertebrate segmentation.” Nat Rev Mol Cell Biol 15(11): 709–721. [DOI] [PubMed] [Google Scholar]
- Iimura T, Denans N and Pourquie O (2009). “Establishment of Hox vertebral identities in the embryonic spine precursors.” Curr Top Dev Biol 88: 201–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iimura T and Pourquie O (2007). “Hox genes in time and space during vertebrate body formation.” Dev Growth Differ 49(4): 265–275. [DOI] [PubMed] [Google Scholar]
- Irving DW, Willhite CC and Burk DT (1986). “Morphogenesis of isotretinoin-induced microcephaly and micrognathia studied by scanning electron microscopy.” Teratology 34(2): 141–153. [DOI] [PubMed] [Google Scholar]
- Isoherranen N and Zhong G (2019). “Biochemical and physiological importance of the CYP26 retinoic acid hydroxylases.” Pharmacol Ther: 107400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iulianella A, Beckett B, Petkovich M and Lohnes D (1999). “A molecular basis for retinoic acid-induced axial truncation.” Dev Biol 205(1): 33–48. [DOI] [PubMed] [Google Scholar]
- Iyer N and Vaishnava S (2019). “Vitamin A at the interface of host-commensal-pathogen interactions.” PLoS Pathog 15(6): e1007750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janesick A, Nguyen TT, Aisaki K, Igarashi K, Kitajima S, Chandraratna RA, Kanno J and Blumberg B (2014). “Active repression by RARgamma signaling is required for vertebrate axial elongation.” Development 141(11): 2260–2270. [DOI] [PubMed] [Google Scholar]
- Janesick A, Tang W, Nguyen TTL and Blumberg B (2017). “RARbeta2 is required for vertebrate somitogenesis.” Development 144(11): 1997–2008. [DOI] [PubMed] [Google Scholar]
- Jiang H, Soprano DR, Li SW, Soprano KJ, Penner JD, Gyda M 3rd and Kochhar DM (1995). “Modulation of limb bud chondrogenesis by retinoic acid and retinoic acid receptors.” Int J Dev Biol 39(4): 617–627. [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM and Morriss-Kay GM (2002). “Tissue origins and interactions in the mammalian skull vault.” Dev Biol 241(1): 106–116. [DOI] [PubMed] [Google Scholar]
- Juberg DR, Knudsen TB, Sander M, Beck NB, Faustman EM, Mendrick DL, Fowle JR 3rd, Hartung T, Tice RR, Lemazurier E, Becker RA, Fitzpatrick SC, Daston GP, Harrill A, Hines RN, Keller DA, Lipscomb JC, Watson D, Bahadori T and Crofton KM (2017). “FutureTox III: Bridges for Translation.” Toxicol Sci 155(1): 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RS, Magpantay FM, Chickarmane V, Haskell C, Tania N, Taylor J, Xia M, Huang R, Rotroff DM, Filer DL, Houck KA, Martin MT, Sipes N, Richard AM, Mansouri K, Setzer RW, Knudsen TB, Crofton KM and Thomas RS (2015). “Integrated Model of Chemical Perturbations of a Biological Pathway Using 18 In Vitro High-Throughput Screening Assays for the Estrogen Receptor.” Toxicol Sci 148(1): 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser ME, Merrill RA, Stein AC, Breburda E and Clagett-Dame M (2003). “Vitamin A deficiency in the late gastrula stage rat embryo results in a one to two vertebral anteriorization that extends throughout the axial skeleton.” Dev Biol 257(1): 14–29. [DOI] [PubMed] [Google Scholar]
- Kam RK, Deng Y, Chen Y and Zhao H (2012). “Retinoic acid synthesis and functions in early embryonic development.” Cell Biosci 2(1): 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam RK, Shi W, Chan SO, Chen Y, Xu G, Lau CB, Fung KP, Chan WY and Zhao H (2013). “Dhrs3 protein attenuates retinoic acid signaling and is required for early embryonic patterning.” J Biol Chem 288(44): 31477–31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata R, Shiraishi F, Nishikawa J, Yonemoto J and Shiraishi H (2008). “Screening and detection of the in vitro agonistic activity of xenobiotics on the retinoic acid receptor.” Toxicol In Vitro 22(4): 1050–1061. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Raya A, Raya RM, Rodriguez-Esteban C and Izpisua Belmonte JC (2005). “Retinoic acid signalling links left-right asymmetric patterning and bilaterally symmetric somitogenesis in the zebrafish embryo.” Nature 435(7039): 165–171. [DOI] [PubMed] [Google Scholar]
- Kessel M and Gruss P (1991). “Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid.” Cell 67(1): 89–104. [DOI] [PubMed] [Google Scholar]
- Khatib T, Marini P, Nunna S, Chisholm DR, Whiting A, Redfern C, Greig IR and McCaffery P (2019). “Genomic and non-genomic pathways are both crucial for peak induction of neurite outgrowth by retinoids.” Cell Commun Signal 17(1): 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecker C (2016). “The chick embryo as a model for the effects of prenatal exposure to alcohol on craniofacial development.” Dev Biol 415(2): 314–325. [DOI] [PubMed] [Google Scholar]
- Kiesecker JM (2002). “Synergism between trematode infection and pesticide exposure: a link to amphibian limb deformities in nature?” Proc Natl Acad Sci U S A 99(15): 9900–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, Dekker S, Vaessen MJ and Grosveld F (1997). “Regulation of the CRABP-I gene during mouse embryogenesis.” Mech Dev 67(2): 157–169. [DOI] [PubMed] [Google Scholar]
- Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, Thomas RS, Casey WM, Dix DJ, Allen D, Sakamuru S, Xia M, Huang R and Judson R (2017). “Development and Validation of a Computational Model for Androgen Receptor Activity.” Chem Res Toxicol 30(4): 946–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen TB, Martin MT, Kavlock RJ, Judson RS, Dix DJ and Singh AV (2009). “Profiling the activity of environmental chemicals in prenatal developmental toxicity studies using the U.S. EPA’s ToxRefDB.” Reprod Toxicol 28(2): 209–219. [DOI] [PubMed] [Google Scholar]
- Kochhar DM (1973). “Limb development in mouse embryos. I. Analysis of teratogenic effects of retinoic acid.” Teratology 7(3): 289–295. [DOI] [PubMed] [Google Scholar]
- Kochhar DM (2000). “Teratogenicity of retinoic acid.” Teratology 62(4): 178–180. [DOI] [PubMed] [Google Scholar]
- Kochhar DM, Jiang H, Penner JD, Johnson AT and Chandraratna RA (1998). “The use of a retinoid receptor antagonist in a new model to study vitamin A-dependent developmental events.” Int J Dev Biol 42(4): 601–608. [PubMed] [Google Scholar]
- Kondo T and Duboule D (1999). “Breaking colinearity in the mouse HoxD complex.” Cell 97(3): 407–417. [DOI] [PubMed] [Google Scholar]
- Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD and Vasiliou V (2012). “Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application.” Pharmacol Rev 64(3): 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krezel W, Ruhl R and de Lera AR (2019). “Alternative retinoid X receptor (RXR) ligands.” Mol Cell Endocrinol 491: 110436. [DOI] [PubMed] [Google Scholar]
- Krumlauf R (1994). “Hox genes in vertebrate development.” Cell 78(2): 191–201. [DOI] [PubMed] [Google Scholar]
- Kwasigroch TE, Skalko RG and Church JK (1984). “Mouse limb bud development in submerged culture: quantitative assessment of the effects of in vivo exposure to retinoic acid.” Teratog Carcinog Mutagen 4(3): 311–326. [DOI] [PubMed] [Google Scholar]
- Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, Curry CJ, Fernhoff PM, Grix AW Jr., Lott IT and et al. (1985). “Retinoic acid embryopathy.” N Engl J Med 313(14): 837–841. [DOI] [PubMed] [Google Scholar]
- Lampron C, Rochette-Egly C, Gorry P, Dolle P, Mark M, Lufkin T, LeMeur M and Chambon P (1995). “Mice deficient in cellular retinoic acid binding protein II (CRABPII) or in both CRABPI and CRABPII are essentially normal.” Development 121(2): 539–548. [DOI] [PubMed] [Google Scholar]
- Laue K, Janicke M, Plaster N, Sonntag C and Hammerschmidt M (2008). “Restriction of retinoic acid activity by Cyp26b1 is required for proper timing and patterning of osteogenesis during zebrafish development.” Development 135(22): 3775–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, Balaguer P and Bourguet W (2009). “Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors.” EMBO Rep 10(4): 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, Cantor RM, Abnoosian A, Park E, Yamamoto ML, Hovland DN Jr. and Collins MD (2005). “A gene(s) for all-trans-retinoic acid-induced forelimb defects mapped and confirmed to murine chromosome 11.” Genetics 170(1): 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K and Skromne I (2014). “Retinoic acid regulates size, pattern and alignment of tissues at the head-trunk transition.” Development 141(22): 4375–4384. [DOI] [PubMed] [Google Scholar]
- Lee YJ, McPherron A, Choe S, Sakai Y, Chandraratna RA, Lee SJ and Oh SP (2010). “Growth differentiation factor 11 signaling controls retinoic acid activity for axial vertebral development.” Dev Biol 347(1): 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, Osumi-Yamashita N, Ninomiya Y, Moon CK, Eriksson U and Eto K (1995). “Retinoic acid stage-dependently alters the migration pattern and identity of hindbrain neural crest cells.” Development 121(3): 825–837. [DOI] [PubMed] [Google Scholar]
- Lemaire G, Balaguer P, Michel S and Rahmani R (2005). “Activation of retinoic acid receptor-dependent transcription by organochlorine pesticides.” Toxicol Appl Pharmacol 202(1): 38–49. [DOI] [PubMed] [Google Scholar]
- Leung MC, Phuong J, Baker NC, Sipes NS, Klinefelter GR, Martin MT, McLaurin KW, Setzer RW, Darney SP, Judson RS and Knudsen TB (2016). “Systems Toxicology of Male Reproductive Development: Profiling 774 Chemicals for Molecular Targets and Adverse Outcomes.” Environ Health Perspect 124(7): 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M and Mackem S (2009). “Limb development: the rise and fall of retinoic acid.” Curr Biol 19(14): R558–561. [DOI] [PubMed] [Google Scholar]
- Liao X and Collins MD (2008). “All-trans retinoic acid-induced ectopic limb and caudal structures: murine strain sensitivities and pathogenesis.” Dev Dyn 237(6): 1553–1564. [DOI] [PubMed] [Google Scholar]
- Linville A, Radtke K, Waxman JS, Yelon D and Schilling TF (2009). “Combinatorial roles for zebrafish retinoic acid receptors in the hindbrain, limbs and pharyngeal arches.” Dev Biol 325(1): 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M and Chambon P (1993). “Function of retinoic acid receptor gamma in the mouse.” Cell 73(4): 643–658. [DOI] [PubMed] [Google Scholar]
- Lohnes D, Mark M, Mendelsohn C, Dolle P, Decimo D, LeMeur M, Dierich A, Gorry P and Chambon P (1995). “Developmental roles of the retinoic acid receptors.” J Steroid Biochem Mol Biol 53(1–6): 475–486. [DOI] [PubMed] [Google Scholar]
- Lohnes D, Mark M, Mendelsohn C, Dolle P, Dierich A, Gorry P, Gansmuller A and Chambon P (1994). “Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants.” Development 120(10): 2723–2748. [DOI] [PubMed] [Google Scholar]
- Lorente CA and Miller SA (1978). “The effect of hypervitaminosis A on rat palatal development.” Teratology 18(2): 277–284. [DOI] [PubMed] [Google Scholar]
- Lu HC, Eichele G and Thaller C (1997). “Ligand-bound RXR can mediate retinoid signal transduction during embryogenesis.” Development 124(1): 195–203. [DOI] [PubMed] [Google Scholar]
- Lu HC, Revelli JP, Goering L, Thaller C, Eichele G (1997). “Retinoid signaling is required for the establishment of a ZPA and for the expression of Hoxb-8, a mediator of ZPA formation.” Development 124 (9): 1643–1651 [DOI] [PubMed] [Google Scholar]
- Lumen A, Chang X, Xia M, Zurlinden T, TB K and NC K (2020). “In Vitro Stem-Cell Based Developmental Toxicity Testing and In Vivo Dose Extrapolation. (abstract planned for SOT 2020, manuscript in preparation).”
- Luo J, Pasceri P, Conlon RA, Rossant J and Giguere V (1995). “Mice lacking all isoforms of retinoic acid receptor beta develop normally and are susceptible to the teratogenic effects of retinoic acid.” Mech Dev 53(1): 61–71. [DOI] [PubMed] [Google Scholar]
- Luo Z, Rhie SK and Farnham PJ (2019). “The Enigmatic HOX Genes: Can We Crack Their Code?” Cancers (Basel) 11(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean G, Abu-Abed S, Dolle P, Tahayato A, Chambon P and Petkovich M (2001). “Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development.” Mech Dev 107(1–2): 195–201. [DOI] [PubMed] [Google Scholar]
- Maclean G, Dolle P and Petkovich M (2009). “Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning.” Dev Dyn 238(3): 732–745. [DOI] [PubMed] [Google Scholar]
- Maden M (1994). “Distribution of cellular retinoic acid-binding proteins I and II in the chick embryo and their relationship to teratogenesis.” Teratology 50(4): 294–301. [DOI] [PubMed] [Google Scholar]
- Maden M, Ong DE, Summerbell D and Chytil F (1988). “Spatial distribution of cellular protein binding to retinoic acid in the chick limb bud.” Nature 335(6192): 733–735. [DOI] [PubMed] [Google Scholar]
- Maden M, Ong DE, Summerbell D and Chytil F (1989). “The role of retinoid-binding proteins in the generation of pattern in the developing limb, the regenerating limb and the nervous system.” Development 107 Suppl: 109–119. [DOI] [PubMed] [Google Scholar]
- Mallo M, Vinagre T and Carapuco M (2009). “The road to the vertebral formula.” Int J Dev Biol 53(8–10): 1469–1481. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ (1994). “Vitamin A receptors.” Nutr Rev 52(2 Pt 2): S32–44. [DOI] [PubMed] [Google Scholar]
- Mariani FV, Ahn CP and Martin GR (2008). “Genetic evidence that FGFs have an instructive role in limb proximal-distal patterning.” Nature 453(7193): 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB and Chambon P (2006). “Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis.” Annu Rev Pharmacol Toxicol 46: 451–480. [DOI] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB and Chambon P (2009). “Function of retinoic acid receptors during embryonic development.” Nucl Recept Signal 7: e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB, Kastner P, Dupe V, Wendling O, Krezel W, Mascrez B and Chambon P (1998). “Mesectoderm is a major target of retinoic acid action.” Eur J Oral Sci 106 Suppl 1: 24–31. [DOI] [PubMed] [Google Scholar]
- Marlétaz F, Holland LZ, Laudet V, Schuber M (2006). “Retinoic acid signaling and the evolution of chordates.” Int. J. Biol. Sci 2 (2): 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegola E, Broccia ML, Di Renzo F and Giavini E (2001). “Antifungal triazoles induce malformations in vitro.” Reprod Toxicol 15(4): 421–427. [DOI] [PubMed] [Google Scholar]
- Menegola E, Broccia ML, Di Renzo F and Giavini E (2006). “Postulated pathogenic pathway in triazole fungicide induced dysmorphogenic effects.” Reprod Toxicol 22(2): 186–195. [DOI] [PubMed] [Google Scholar]
- Mercader N, Fischer S, Neumann CJ (2006). “Prdm1 acts downstream of a sequential RA, Wnt and Fgf signaling cascade during zebrafish forelimb induction.” Development 133 (15): 2805–2815 [DOI] [PubMed] [Google Scholar]
- Mercader N, Leonardo E, Piedra ME, Martinez AC, Ros MA, Torres M (2000). “Opposing RA and FGF signals control proximodistal vertebrate limb development through regulation of Meis genes” Development 127 (18): 3961–3970 [DOI] [PubMed] [Google Scholar]
- Mey J (2017). RAR/RXR-mediated signaling. Gene Regulation, Epigenetics and Hormone Signaling. Mandal S. Weinheim, Germany, Wiley‐VCH Verlag GmbH & Co. KGaA. 1. [Google Scholar]
- Mic FA, Sirbu IO, Duester G (2004). “Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation.” J. Biol. Chem 279 (25): 26698–26706 [DOI] [PubMed] [Google Scholar]
- Molotkova N, Molotkov A, Sirbu IO and Duester G (2005). “Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation.” Mech Dev 122(2): 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay G, Ruberte E and Fukiishi Y (1993). “Mammalian neural crest and neural crest derivatives.” Ann Anat 175(6): 501–507. [DOI] [PubMed] [Google Scholar]
- Morriss-Kay GM, Murphy P, Hill RE and Davidson DR (1991). “Effects of retinoic acid excess on expression of Hox-2.9 and Krox-20 and on morphological segmentation in the hindbrain of mouse embryos.” Embo j 10(10): 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama J and Eto K (1994). “Antisense retinoic acid receptor gamma-1 oligonucleotide enhances chondrogenesis of mouse limb mesenchymal cells in vitro.” FEBS Lett 338(3): 319–322. [DOI] [PubMed] [Google Scholar]
- Moutier E, Ye T, Choukrallah MA, Urban S, Osz J, Chatagnon A, Delacroix L, Langer D, Rochel N, Moras D, Benoit G and Davidson I (2012). “Retinoic acid receptors recognize the mouse genome through binding elements with diverse spacing and topology.” J Biol Chem 287(31): 26328–26341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill MJ, Kan JL, Cooke A, Bhagwat S, Beck P, Bittner M, Cesario C, Keane D, Lazarescu V, Nigro A, Nillson C, Panicker B, Smith V, Srebernak M, Sun FL, O’Connor M, Russo S, Fischetti G, Vrkljan M, Winski S, Castelhano AL, Emerson D and Gibson NW (2006). “3-[6-(2-Dimethylamino-1-imidazol-1-yl-butyl)-naphthalen-2-yloxy]-2,2-dimethyl-pro pionic acid as a highly potent and selective retinoic acid metabolic blocking agent.” Bioorg Med Chem Lett 16(10): 2729–2733. [DOI] [PubMed] [Google Scholar]
- Munoz-Sanjuan I, Cooper MK, Beachy PA, Fallon JF and Nathans J (2001). “Expression and regulation of chicken fibroblast growth factor homologous factor (FHF)-4 during craniofacial morphogenesis.” Dev Dyn 220(3): 238–245. [DOI] [PubMed] [Google Scholar]
- Naiche LA and Papaioannou VE (2003). “Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois.” Development 130(12): 2681–2693. [DOI] [PubMed] [Google Scholar]
- Napoli JL, Posch KP, Fiorella PD and Boerman MH (1991). “Physiological occurrence, biosynthesis and metabolism of retinoic acid: evidence for roles of cellular retinol-binding protein (CRBP) and cellular retinoic acid-binding protein (CRABP) in the pathway of retinoic acid homeostasis.” Biomed Pharmacother 45(4–5): 131–143. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Fraulob V, Garnier JM, Chambon P and Dolle P (2002). “Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse.” Mech Dev 110(1–2): 165–171. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Subbarayan V, Dolle P and Chambon P (1999). “Embryonic retinoic acid synthesis is essential for early mouse post-implantation development.” Nat Genet 21(4): 444–448. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Le Roux I, Schuhbaur B, Chambon P and Dolle P (2003). “The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system.” Development 130(11): 2525–2534. [DOI] [PubMed] [Google Scholar]
- Nimmagadda S, Buchtova M, Fu K, Geetha-Loganathan P, Hosseini-Farahabadi S, Trachtenberg AJ, Kuo WP, Vesela I and Richman JM (2015). “Identification and functional analysis of novel facial patterning genes in the duplicated beak chicken embryo.” Dev Biol 407(2): 275–288. [DOI] [PubMed] [Google Scholar]
- Nishimoto S, Wilde SM, Wood S and Logan MP (2015). “RA Acts in a Coherent Feed-Forward Mechanism with Tbx5 to Control Limb Bud Induction and Initiation.” Cell Rep 12(5): 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswander L, Tickle C, Vogel A, Booth I and Martin GR (1993). “FGF-4 replaces the apical ectodermal ridge and directs outgrowth and patterning of the limb.” Cell 75(3): 579–587. [DOI] [PubMed] [Google Scholar]
- Niswander L, Tickle C, Vogel A, Martin G (1994). “ Function of FGF-4 in limb development.” Mol. Reprod. Dev 39 (1): 83–88 discussion 88–89. [DOI] [PubMed] [Google Scholar]
- Noden DM and Trainor PA (2005). “Relations and interactions between cranial mesoderm and neural crest populations.” J Anat 207(5): 575–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, Alvarez IS, Vogel A, Rodriguez C, Evans RM, Izpisua Belmonte JC (1996). “ Evidence that Shh cooperates with a retinoic acid inducible co-factor to establish ZPA-like activity.” Development 122 (2): 537–542 [DOI] [PubMed] [Google Scholar]
- Olivera-Martinez I, Harada H, Halley PA and Storey KG (2012). “Loss of FGF-dependent mesoderm identity and rise of endogenous retinoid signalling determine cessation of body axis elongation.” PLoS Biol 10(10): e1001415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen BR, Reginato AM and Wang W (2000). “Bone development.” Annu Rev Cell Dev Biol 16: 191–220. [DOI] [PubMed] [Google Scholar]
- Padmanabhan R (1998). “Retinoic acid-induced caudal regression syndrome in the mouse fetus.” Reprod Toxicol 12(2): 139–151. [DOI] [PubMed] [Google Scholar]
- Paganelli A, Gnazzo V, Acosta H, Lopez SL and Carrasco AE (2010). “Glyphosate-based herbicides produce teratogenic effects on vertebrates by impairing retinoic acid signaling.” Chem Res Toxicol 23(10): 1586–1595. [DOI] [PubMed] [Google Scholar]
- Palmer JA, Smith AM, Egnash LA, Colwell MR, Donley ELR, Kirchner FR and Burrier RE (2017). “A human induced pluripotent stem cell-based in vitro assay predicts developmental toxicity through a retinoic acid receptor-mediated pathway for a series of related retinoid analogues.” Reprod Toxicol 73: 350–361. [DOI] [PubMed] [Google Scholar]
- Papalopulu N, Clarke JD, Bradley L, Wilkinson D, Krumlauf R and Holder N (1991). “Retinoic acid causes abnormal development and segmental patterning of the anterior hindbrain in Xenopus embryos.” Development 113(4): 1145–1158. [DOI] [PubMed] [Google Scholar]
- Petrelli B, Bendelac L, Hicks GG and Fainsod A (2019). “Insights into retinoic acid deficiency and the induction of craniofacial malformations and microcephaly in fetal alcohol spectrum disorder.” Genesis 57(1): e23278. [DOI] [PubMed] [Google Scholar]
- Pickering J, Wali N, Towers M (2017). “ Transcriptional changes in chick wing bud polarization induced by retinoic acid” Dev. Dyn 246 (9): 682–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power SC, Lancman J and Smith SM (1999). “Retinoic acid is essential for Shh/Hoxd signaling during rat limb outgrowth but not for limb initiation.” Dev Dyn 216(4–5): 469–480. [DOI] [PubMed] [Google Scholar]
- Pratt RM, Goulding EH and Abbott BD (1987). “Retinoic acid inhibits migration of cranial neural crest cells in the cultured mouse embryo.” J Craniofac Genet Dev Biol 7(3): 205–217. [PubMed] [Google Scholar]
- Probst S, Kraemer C, Demougin P, Sheth R, Martin GR, Shiratori H, Hamada H, Iber D, Zeller R and Zuniga A (2011). “SHH propagates distal limb bud development by enhancing CYP26B1-mediated retinoic acid clearance via AER-FGF signalling.” Development 138(10): 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt R, Gullotta F, Nusspaumer G, Unal E, Ivanek R, Zuniga A and Zeller R (2019). “Molecular signatures identify immature mesenchymal progenitors in early mouse limb buds that respond differentially to morphogen signaling.” Development 146(10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M and Dolle P (2012). “Retinoic acid signalling during development.” Development 139(5): 843–858. [DOI] [PubMed] [Google Scholar]
- Rhinn M, Schuhbaur B, Niederreither K and Dolle P (2011). “Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment.” Proc Natl Acad Sci U S A 108(40): 16687–16692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E and Tabin C (1993). “Sonic hedgehog mediates the polarizing activity of the ZPA.” Cell 75(7): 1401–1416. [DOI] [PubMed] [Google Scholar]
- Rosello-Diez A, Ros MA, Ros Torres MAM (2011). “ Diffusible signals, not autonomous mechanisms, determine the main proximodistal limb subdivision.” Science 332 (6033): 1086–1088 [DOI] [PubMed] [Google Scholar]
- Ruberte E, Dolle P, Chambon P and Morriss-Kay G (1991). “Retinoic acid receptors and cellular retinoid binding proteins. II. Their differential pattern of transcription during early morphogenesis in mouse embryos.” Development 111(1): 45–60. [DOI] [PubMed] [Google Scholar]
- Ruberte E, Dolle P, Krust A, Zelent A, Morriss-Kay G and Chambon P (1990). “Specific spatial and temporal distribution of retinoic acid receptor gamma transcripts during mouse embryogenesis.” Development 108(2): 213–222. [DOI] [PubMed] [Google Scholar]
- Ruberte E, Friederich V, Morriss-Kay G and Chambon P (1992). “Differential distribution patterns of CRABP I and CRABP II transcripts during mouse embryogenesis.” Development 115(4): 973–987. [DOI] [PubMed] [Google Scholar]
- Saili K, Antonijevic T, Zurlinden T, Shah I, Deisenroth C and Knudsen T (2019). “Molecular characterization of a toxicological tipping point during human stem cell differentiation.” Reprod Toxicol (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, Rey JP, Ma JX, Staehling-Hampton K and Trainor PA (2007). “RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development.” Genes Dev 21(9): 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satre MA and Kochhar DM (1989). “Elevations in the endogenous levels of the putative morphogen retinoic acid in embryonic mouse limb-buds associated with limb dysmorphogenesis.” Dev Biol 133(2): 529–536. [DOI] [PubMed] [Google Scholar]
- Scadding SR and Maden M (1994). “Retinoic acid gradients during limb regeneration.” Dev Biol 162(2): 608–617. [DOI] [PubMed] [Google Scholar]
- Schindler JF, Berst KB and Plapp BV (1998). “Inhibition of human alcohol dehydrogenases by formamides.” J Med Chem 41(10): 1696–1701. [DOI] [PubMed] [Google Scholar]
- Schuh TJ, Hall BL, Kraft JC, Privalsky ML and Kimelman D (1993). “v-erbA and citral reduce the teratogenic effects of all-trans retinoic acid and retinol, respectively, in Xenopus embryogenesis.” Development 119(3): 785–798. [DOI] [PubMed] [Google Scholar]
- Scialli AR, Daston G, Chen C, Coder PS, Euling SY, Foreman J, Hoberman AM, Hui J, Knudsen T, Makris SL, Morford L, Piersma AH, Stanislaus D and Thompson KE (2018). “Rethinking developmental toxicity testing: Evolution or revolution?” Birth Defects Res 110(10): 840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WJ Jr., Walter R, Tzimas G, Sass JO, Nau H and Collins MD (1994). “Endogenous status of retinoids and their cytosolic binding proteins in limb buds of chick vs mouse embryos.” Dev Biol 165(2): 397–409. [DOI] [PubMed] [Google Scholar]
- Shabtai Y, Bendelac L, Jubran H, Hirschberg J and Fainsod A (2018). “Acetaldehyde inhibits retinoic acid biosynthesis to mediate alcohol teratogenicity.” Sci Rep 8(1): 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T, Kawakami M, Okuda H, Tatsumi K, Morita S, Nochioka K, Kirita T and Wanaka A (2015). “Retinoic acid regulates Lhx8 expression via FGF-8b to the upper jaw development of chick embryo.” J Biosci Bioeng 119(3): 260–266. [DOI] [PubMed] [Google Scholar]
- Shimozono S, Iimura T, Kitaguchi T, Higashijima S and Miyawaki A (2013). “Visualization of an endogenous retinoic acid gradient across embryonic development.” Nature 496(7445): 363–366. [DOI] [PubMed] [Google Scholar]
- Shiotsugu J, Katsuyama Y, Arima K, Baxter A, Koide T, Song J, Chandraratna RA and Blumberg B (2004). “Multiple points of interaction between retinoic acid and FGF signaling during embryonic axis formation.” Development 131(11): 2653–2667. [DOI] [PubMed] [Google Scholar]
- Sipes NS, Martin MT, Reif DM, Kleinstreuer NC, Judson RS, Singh AV, Chandler KJ, Dix DJ, Kavlock RJ and Knudsen TB (2011). “Predictive models of prenatal developmental toxicity from ToxCast high-throughput screening data.” Toxicol Sci 124(1): 109–127. [DOI] [PubMed] [Google Scholar]
- Smith-Thomas L, Lott I and Bronner-Fraser M (1987). “Effects of isotretinoin on the behavior of neural crest cells in vitro.” Dev Biol 123(1): 276–281. [DOI] [PubMed] [Google Scholar]
- Smith SM, Kirstein IJ, Wang ZS, Fallon JF, Kelley J and Bradshaw-Rouse J (1995). “Differential expression of retinoic acid receptor-beta isoforms during chick limb ontogeny.” Dev Dyn 202(1): 54–66. [DOI] [PubMed] [Google Scholar]
- Solecki R, Rauch M, Gall A, Buschmann J, Clark R, Fuchs A, Kan H, Heinrich V, Kellner R, Knudsen TB, Li W, Makris SL, Ooshima Y, Paumgartten F, Piersma AH, Schonfelder G, Oelgeschlager M, Schaefer C, Shiota K, Ulbrich B, Ding X and Chahoud I (2015). “Continuing harmonization of terminology and innovations for methodologies in developmental toxicology: Report of the 8th Berlin Workshop on Developmental Toxicity, 14–16 May 2014.” Reprod Toxicol 57: 140–146. [DOI] [PubMed] [Google Scholar]
- Spoorendonk KM, Peterson-Maduro J, Renn J, Trowe T, Kranenbarg S, Winkler C and Schulte-Merker S (2008). “Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton.” Development 135(22): 3765–3774. [DOI] [PubMed] [Google Scholar]
- Steele CE, Marlow R, Turton J and Hicks RM (1987). “In-vitro teratogenicity of retinoids.” Br J Exp Pathol 68(2): 215–223. [PMC free article] [PubMed] [Google Scholar]
- Strate I, Min TH, Iliev D and Pera EM (2009). “Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system.” Development 136(3): 461–472. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Cook CS and Webster WS (1988). “Teratogens and craniofacial malformations: relationships to cell death.” Development 103 Suppl: 213–231. [DOI] [PubMed] [Google Scholar]
- Summerbell D (1974). “A quantitative analysis of the effect of excision of the AER from the chick limb-bud.” J Embryol Exp Morphol 32(3): 651–660. [PubMed] [Google Scholar]
- Tabin C and Wolpert L (2007). “Rethinking the proximodistal axis of the vertebrate limb in the molecular era.” Genes Dev 21(12): 1433–1442. [DOI] [PubMed] [Google Scholar]
- Taleporos P, Salgo MP and Oster G (1978). “Teratogenic action of bis(dichloroacetyl)diamine on rats: patterns of malformations produced in high incidence at time-limited periods of development.” Teratology 18(1): 5–15. [DOI] [PubMed] [Google Scholar]
- Tam PP and Beddington RS (1987). “The formation of mesodermal tissues in the mouse embryo during gastrulation and early organogenesis.” Development 99(1): 109–126. [DOI] [PubMed] [Google Scholar]
- Tamura K, Kagechika H, Hashimoto Y, Shudo K, Ohsugi K, Ide H (1990). “ Synthetic retinoids, retinobenzoic acids, Am80, Am580 and Ch55 regulate morphogenesis in chick limb bud.” Cell Differ. Development 32 (1): 17–26 [DOI] [PubMed] [Google Scholar]
- Teletin M, Vernet N, Ghyselinck NB and Mark M (2017). “Roles of Retinoic Acid in Germ Cell Differentiation.” Curr Top Dev Biol 125: 191–225. [DOI] [PubMed] [Google Scholar]
- Thaller C and Eichele G (1987). “Identification and spatial distribution of retinoids in the developing chick limb bud.” Nature 327(6123): 625–628. [DOI] [PubMed] [Google Scholar]
- Theunissen PT, Beken S, Beyer BK, Breslin WJ, Cappon GD, Chen CL, Chmielewski G, De Schaepdrijver L, Enright B, Foreman JE, Harrouk W, Hew KW, Hoberman AM, Hui JY, Knudsen TB, Laffan SB, Makris SL, Martin M, McNerney ME, Siezen CL, Stanislaus DJ, Stewart J, Thompson KE, Tornesi B, Van der Laan JW, Weinbauer GF, Wood S and Piersma AH (2016). “Comparison of rat and rabbit embryo-fetal developmental toxicity data for 379 pharmaceuticals: on the nature and severity of developmental effects.” Crit Rev Toxicol 46(10): 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ML, de Antueno R, Coyle KM, Sultan M, Cruickshank BM, Giacomantonio MA, Giacomantonio CA, Duncan R and Marcato P (2016). “Citral reduces breast tumor growth by inhibiting the cancer stem cell marker ALDH1A3.” Mol Oncol 10(9): 1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle C, Alberts B, Wolpert L and Lee J (1982). “Local application of retinoic acid to the limb bond mimics the action of the polarizing region.” Nature 296(5857): 564–566. [DOI] [PubMed] [Google Scholar]
- Tickle C, Crawley A (1988). “The effects of local application of retinoids to different positions along the proximo-distal axis of embryonic chick wings.” Rouxs Arch. Dev. Biol 197 (1): 27–36 [DOI] [PubMed] [Google Scholar]
- Tonk EC, Pennings JL and Piersma AH (2015). “An adverse outcome pathway framework for neural tube and axial defects mediated by modulation of retinoic acid homeostasis.” Reprod Toxicol 55: 104–113. [DOI] [PubMed] [Google Scholar]
- Uehara M, Yashiro K, Mamiya S, Nishino J, Chambon P, Dolle P and Sakai Y (2007). “CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse.” Dev Biol 302(2): 399–411. [DOI] [PubMed] [Google Scholar]
- Uzkudun M, Marcon L, Sharpe J (2015). “Data-driven modelling of a gene regulatory network for cell fate decisions in the growing limb bud.” Mol. Syst. Biol 11 (7): 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US EPA 2018. “Strategic Plan to Promote the Development and Implementation of Alternative Test Methods Within the TSCA Program” U.S. Environmental Protection Agency, Office of Chemical Safety and Pollution Prevention, Washington, DC: (Accessed 22 June 2018). [Google Scholar]
- Vaessen MJ, Meijers JH, Bootsma D and Van Kessel AG (1990). “The cellular retinoic-acid-binding protein is expressed in tissues associated with retinoic-acid-induced malformations.” Development 110(2): 371–378. [DOI] [PubMed] [Google Scholar]
- Vieux-Rochas M, Coen L, Sato T, Kurihara Y, Gitton Y, Barbieri O, Le Blay K, Merlo G, Ekker M, Kurihara H, Janvier P and Levi G (2007). “Molecular dynamics of retinoic acid-induced craniofacial malformations: implications for the origin of gnathostome jaws.” PLoS One 2(6): e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai HA, Kawakami K, Wada H, Muller F, Vernallis AB, Brown G and Johnson WE (2015). “The development and growth of tissues derived from cranial neural crest and primitive mesoderm is dependent on the ligation status of retinoic acid receptor gamma: evidence that retinoic acid receptor gamma functions to maintain stem/progenitor cells in the absence of retinoic acid.” Stem Cells Dev 24(4): 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SJ and Morriss-Kay GM (1995). “Distribution of all-trans-, 13-cis- and 9-cis-retinoic acid to whole rat embryos and maternal serum following oral administration of a teratogenic dose of all-trans-retinoic acid.” Pharmacol Toxicol 76(3): 196–201. [DOI] [PubMed] [Google Scholar]
- Watanabe T and Pratt RM (1991). “Effects of retinoic acid on embryonic development of mice in culture.” Experientia 47(5): 493–497. [DOI] [PubMed] [Google Scholar]
- Webster WS and Ritchie HE (1991). “Teratogenic effects of alcohol and isotretinoin on craniofacial development: an analysis of animal models.” J Craniofac Genet Dev Biol 11(4): 296–302. [PubMed] [Google Scholar]
- Wedden SE (1987). “Epithelial-mesenchymal interactions in the development of chick facial primordia and the target of retinoid action.” Development 99(3): 341–351. [DOI] [PubMed] [Google Scholar]
- Wedden SE, Lewin-Smith MR and Tickle C (1987). “The effects of retinoids on cartilage differentiation in micromass cultures of chick facial primordia and the relationship to a specific facial defect.” Dev Biol 122(1): 78–89. [DOI] [PubMed] [Google Scholar]
- Wedden SE and Tickle C (1986). “Quantitative analysis of the effect of retinoids on facial morphogenesis.” J Craniofac Genet Dev Biol Suppl 2: 169–178. [PubMed] [Google Scholar]
- Wendling O, Ghyselinck NB, Chambon P and Mark M (2001). “Roles of retinoic acid receptors in early embryonic morphogenesis and hindbrain patterning.” Development 128(11): 2031–2038. [DOI] [PubMed] [Google Scholar]
- West KP Jr. (2003). “Vitamin A deficiency disorders in children and women.” Food Nutr Bull 24(4 Suppl): S78–90. [DOI] [PubMed] [Google Scholar]
- Weston AD, Rosen V, Chandraratna RA and Underhill TM (2000). “Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways.” J Cell Biol 148(4): 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JC, Highland M, Kaiser M and Clagett-Dame M (2000). “Vitamin A deficiency results in the dose-dependent acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo.” Dev Biol 220(2): 263–284. [DOI] [PubMed] [Google Scholar]
- White KK and Goldberg MJ (2018). Common neonatal orthopedic conditions. Avery’s Diseases of the Newborn, 10th edition. Gleason E CA and Juul SE, Elsevier, Inc.: 1438–1449. [Google Scholar]
- Wiley MJ (1983). “The pathogenesis of retinoic acid-induced vertebral abnormalities in golden Syrian hamster fetuses.” Teratology 28(3): 341–353. [DOI] [PubMed] [Google Scholar]
- Wiley MJ, Cauwenbergs P and Taylor IM (1983). “Effects of retinoic acid on the development of the facial skeleton in hamsters: early changes involving cranial neural crest cells.” Acta Anat (Basel) 116(2): 180–192. [DOI] [PubMed] [Google Scholar]
- Williams JA, Kondo N, Okabe T, Takeshita N, Pilchak DM, Koyama E, Ochiai T, Jensen D, Chu ML, Kane MA, Napoli JL, Enomoto-Iwamoto M, Ghyselinck N, Chambon P, Pacifici M and Iwamoto M (2009). “Retinoic acid receptors are required for skeletal growth, matrix homeostasis and growth plate function in postnatal mouse.” Dev Biol 328(2): 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M, Burdsal C, Periasamy A, Lewandoski M and Sutherland A (2012). “Mouse primitive streak forms in situ by initiation of epithelial to mesenchymal transition without migration of a cell population.” Dev Dyn 241(2): 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SS, Mear JP, Liang HC, Potter SS, Aronow BJ and Colbert MC (2004). “Large-scale reprogramming of cranial neural crest gene expression by retinoic acid exposure.” Physiol Genomics 19(2): 184–197. [DOI] [PubMed] [Google Scholar]
- Wilson JG, Roth CB and Warkany J (1953). “An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation.” Am J Anat 92(2): 189–217. [DOI] [PubMed] [Google Scholar]
- Wilson V, Olivera-Martinez I and Storey KG (2009). “Stem cells, signals and vertebrate body axis extension.” Development 136(10): 1591–1604. [DOI] [PubMed] [Google Scholar]
- Wise LD, Xue D and Winkelmann CT (2010). “Micro-computed tomographic evaluation of fetal skeletal changes induced by all-trans-retinoic acid in rats and rabbits.” Birth Defects Res B Dev Reprod Toxicol 89(5): 408–417. [DOI] [PubMed] [Google Scholar]
- Wood HB, Ward SJ and Morriss-Kay GM (1996). “Effects of all-trans-retinoic acid on skeletal pattern, 5’HoxD gene expression, and RAR beta 2/beta 4 promoter activity in embryonic mouse limbs.” Dev Genet 19(1): 74–84. [DOI] [PubMed] [Google Scholar]
- Wu S, Fisher J, Naciff J, Laufersweiler M, Lester C, Daston G and Blackburn K (2013). “Framework for identifying chemicals with structural features associated with the potential to act as developmental or reproductive toxicants.” Chem Res Toxicol 26(12): 1840–1861. [DOI] [PubMed] [Google Scholar]
- Yakushiji-Kaminatsui N, Kondo T, Hironaka KI, Sharif J, Endo TA, Nakayama M, Masui O, Koseki Y, Kondo K, Ohara O, Vidal M, Morishita Y and Koseki H (2018). “Variant PRC1 competes with retinoic acid-related signals to repress Meis2 in the mouse distal forelimb bud.” Development 145(19) [DOI] [PubMed] [Google Scholar]
- Yashiro K, Zhao X, Uehara M, Yamashita K, Nishijima M, Nishino J, Saijoh Y, Sakai Y and Hamada H (2004). “Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb.” Dev Cell 6(3): 411–422. [DOI] [PubMed] [Google Scholar]
- Yin M and Pacifici M (2001). “Vascular regression is required for mesenchymal condensation and chondrogenesis in the developing limb.” Dev Dyn 222(3): 522–533. [DOI] [PubMed] [Google Scholar]
- Yip JE, Kokich VG and Shepard TH (1980). “The effect of high doses of retinoic acid on prenatal craniofacial development in Macaca nemestrina.” Teratology 21(1): 29–38. [DOI] [PubMed] [Google Scholar]
- Yu J, Gonzalez S, Martinez L, Diez-Pardo JA and Tovar JA (2003). “Effects of retinoic acid on the neural crest-controlled organs of fetal rats.” Pediatr Surg Int 19(5): 355–358. [DOI] [PubMed] [Google Scholar]
- Yu JK, Meulemans D, McKeown SJ and Bronner-Fraser M (2008). “Insights from the amphioxus genome on the origin of vertebrate neural crest.” Genome Res 18(7): 1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakany J, Zacchetti G and Duboule D (2007). “Interactions between HOXD and Gli3 genes control the limb apical ectodermal ridge via Fgf10.” Dev Biol 306(2): 883–893. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zuo Z, Sun P, Wang H, Yu A and Wang C (2012). “Tributyltin exposure results in craniofacial cartilage defects in rockfish (Sebastiscus marmoratus) embryos.” Mar Environ Res 77: 6–11. [DOI] [PubMed] [Google Scholar]
- Zhang R, Wang Y, Li R and Chen G (2015). “Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism.” Int J Mol Sci 16(6): 14210–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sirbu IO, Mic FA, Molotkova N, Molotkov A, Kumar S and Duester G (2009). “Retinoic acid promotes limb induction through effects on body axis extension but is unnecessary for limb patterning.” Curr Biol 19(12): 1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J and Kochhar DM (2004). “Cellular anomalies underlying retinoid-induced phocomelia.” Reprod Toxicol 19(1): 103–110. [DOI] [PubMed] [Google Scholar]
- Zimmermann B and Tsambaos D (1985). “Retinoids inhibit the differentiation of embryonic-mouse mesenchymal cells in vitro.” Arch Dermatol Res 277(2): 98–104. [DOI] [PubMed] [Google Scholar]
- Zuniga A (2015). “Next generation limb development and evolution: old questions, new perspectives.” Development 142(22): 3810–3820. [DOI] [PubMed] [Google Scholar]
- Zuo H and Wan Y (2017). “Nuclear Receptors in Skeletal Homeostasis.” Curr Top Dev Biol 125: 71–107. [DOI] [PubMed] [Google Scholar]
- Zurlinden T, Saili K, Rush N, Kothiya P, Judson R, Houck K, Hunter E, Baker N, Palmer J, Thomas R and Knudsen T (2020). “Profiling the ToxCast library with a pluripotent human (H9) embryonic stem cell assay for developmental toxicity.” Toxicol Sci 174: 189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]