

Abstract
BACKGROUND:
Lung transplantation remains the sole curative option for patients with idiopathic pulmonary fibrosis (IPF), but donor organs remain scarce, and many eligible patients die before transplant. Tools to optimize the timing of transplant referrals are urgently needed.
METHODS:
Least absolute shrinkage and selection operator was applied to clinical and proteomic data generated as part of a prospective cohort study of interstitial lung disease (ILD) to derive clinical, proteomic, and multidimensional logit models of near-term death or lung transplant within 18 months of blood draw. Model-fitted values were dichotomized at the point of maximal sensitivity and specificity, and decision curve analysis was used to select the best-performing classifier. We then applied this classifier to independent IPF and non-IPF ILD cohorts to determine test performance characteristics. Cohorts were restricted to patients aged ≤72 years with body mass index 18 to 32 to increase the likelihood of transplant eligibility.
RESULTS:
IPF derivation, IPF validation, and non-IPF ILD validation cohorts consisted of 314, 105, and 295 patients, respectively. A multidimensional model comprising 2 clinical variables and 20 proteins outperformed stand-alone clinical and proteomic models. Following dichotomization, the multidimensional classifier predicted near-term outcome with 70% sensitivity and 92% specificity in the IPF validation cohort and 70% sensitivity and 80% specificity in the non-IPF ILD validation cohort.
CONCLUSIONS:
A multidimensional classifier of near-term outcomes accurately discriminated this end-point with good test performance across independent IPF and non-IPF ILD cohorts. These findings support refinement and prospective validation of this classifier in transplant-eligible individuals.
Keywords: idiopathic pulmonary fibrosis, interstitial lung disease, proteomics, survival, biomarker
Progressive pulmonary fibrosis is a common manifestation of interstitial lung disease (ILD) that results in irreversible parenchymal scarring, declining lung function, and poor survival.1,2 Idiopathic pulmonary fibrosis (IPF) is considered the prototypical progressive pulmonary fibrosis, but a subset of those with non-IPF forms of ILD also develop this condition, leading to similarly poor outcomes.3 While antifibrotic therapy is now approved for the treatment of IPF and progressive non-IPF ILD, currently available drugs slow, rather than stop or reverse fibrosis.4,5 Lung transplantation is an additional treatment option for a subset of patients with IPF, but donor organs are scarce, and many patients die before a transplant evaluation is completed or after being listed for transplant.6,7 While expert guidance has been published by the International Society for Heart and Lung Transplantation (ISHLT), the optimal timing of transplant referral for those with progressive pulmonary fibrosis remains unclear.8 Moreover, some patients develop and quickly succumb to rapidly progressive disease,9 missing the opportunity to undergo transplant evaluation. To optimize the timing of lung transplant referral, tools to accurately identify those most likely to benefit from lung transplant evaluation are urgently needed.
Clinical prediction models effectively discriminate mortality risk in patients with IPF10–12 and non-IPF ILDs.13 However, these models fail to predict near-term progression,14 suggesting that they are a better measure of disease severity than biological activity. Moreover, clinical models rely heavily on age, leaving it unclear how well they would perform in younger individuals, who are more likely to be considered for lung transplant. A host of blood-based prognostic biomarkers have been proposed for patients with progressive pulmonary fibrosis, including peripheral blood leukocyte telomere length, a composite gene expression-based signature, many stand-alone proteins, and composite proteomic signatures.15–23 While these measures remain promising, none have been widely adopted for clinical use. This reality stems in part from uncertainly around biomarker test performance and the way biomarker classification would change clinical management.
In this investigation, we leveraged clinical and high-throughput proteomic data to develop an actionable biomarker to support lung transplant referral in patients with IPF. Using machine learning, we derived clinical, proteomic, and multidimensional prediction models of a composite end-point of death or transplant within 18 months of blood draw. We then dichotomized fitted values for the best-performing model and tested this binary classifier in an independent IPF cohort to determine test performance characteristics. To assess generalizability to other ILD subtypes, we then applied this classifier to an independent non-IPF ILD cohort.
Methods
Cohorts
Consecutive patients with IPF and non-IPF ILD who underwent proteomic profiling as part of the PRECISIONS study24 were eligible for inclusion. The PRECISIONS molecular cohort consists of individuals with IPF and non-IPF ILD participating in prospective registries and biorepositories administered by the Pulmonary Fibrosis Foundation (PFF) Patient Registry (March 2016-June 2018), University of Virginia (September 2018-November 2021), University of California-Davis (July 2016-April 2021), and University of Chicago (March 2007-July 2017). To enrich the cohort with patients potentially eligible for lung transplant, those aged > 72 or with body mass index (BMI) < 18 or > 32 were excluded. Those with missing survival and BMI data were also excluded, as were those without forced vital capacity (FVC) and diffusion capacity of the lung for carbon monoxide (DLCO) performed within 12 months of blood draw to ensure each model could be directly compared in the same population.
Study-specific protocols were approved at UC-Davis (protocol #875917), UChicago (protocol #13-1180), and UVA (protocol #20937). Patients with IPF from the PPF Patient Registry25 comprised the IPF discovery cohort, while those with IPF from the Universities of Virginia, California-Davis, and Chicago comprised the IPF validation cohort. A pooled cohort of patients with non-IPF ILD from all 4 registries comprised the non-IPF ILD validation cohort. PFF Patient Registry participants from the University of Chicago and University of Virginia were excluded to ensure no overlap between cohorts. The University of California-Davis did not contribute patients to the PFF Patient Registry.
Proteomic platform and data
Proteomic data were generated using the Olink (Uppsala, Sweden) Explore 3072 platform, which provides semiquantitative measures for 2921 analytes. This and other Olink platforms use proximity extension assays to quantify proteins, which provide excellent sensitivity and specificity for low-abundance proteins.26,27 Intensity normalization is used to aggregate data across plates. A value is returned for all proteins, with those falling below the lower limit of detection imputed and flagged. Normalized protein data were log2 transformed and modeled continuously.
Statistical analysis
Continuous measures are presented as means with standard deviations and compared using a student’s t-test. Categorical measures are presented as count and percentage and compared using a chi-square test. Three separate logistic regression models were fit in the IPF derivation cohort with clinical, proteomic, and multidimensional (clinical and proteomic) data to identify variables predictive of a composite dichotomous end-point of death or transplant within 18 months of blood draw. Variable selection was performed using the least absolute shrinkage and selection operator with 10-fold cross validation used for parameter tuning.28 Clinical variables available for selection included age, sex, race, BMI, smoking history, percent predicted FVC, and percent predicted DLCO. Proteins included all 2921 generated using the Olink Explore platform. A multidimensional model was derived using all available clinical and proteomic data.
Fitted values for each model were dichotomized at the point of maximal sensitivity and specificity using Youden’s index29 and the best-performing classifier was identified using receiver operator curve and decision curve analysis.30,31 We then applied the best-performing classifier to the IPF validation cohort and pooled non-IPF ILD validation cohort to determine test performance characteristics and generalizability.
Eighteen-month transplant-free survival was plotted for classification groups using the Kaplan-Meier estimator. Univariate and multivariate Cox proportional hazards regression models were used to estimate outcome risk for those with a high-risk classification. The proportional hazards assumption was checked and satisfied for all models. All statistical analyses were performed using Stata (StataCorp. 2022. Release 17. College Station, TX). Statistical significance was set a p < 0.05.
Results
Of 937, 368 and 623 eligible patients from the IPF derivation, IPF validation, and non-IPF ILD validation cohorts, respectively, 314, 105, and 295 met inclusion and exclusion criteria (Figure S1). Baseline characteristics at the time of blood draw for each cohort are shown in Table 1. IPF cohorts were similar in age, sex, race, and smoking history. Those in the derivation cohort had lower percent predicted FVC and DLCO and higher antifibrotic use when compared to the IPF validation cohort. As expected, the non-IPF ILD cohort was younger, with fewer males, whites, smokers, and outcomes than the IPF cohorts. Baseline immunosuppressant use was higher in the non-IPF validation cohort. Near-term outcome occurred in 25.5% (n = 80), 28.6% (n = 30), and 18.3% (n = 54) of patients in the IPF derivation, IPF validation, and non-IPF ILD validation cohorts, respectively. Lung transplant accounted for 35% to 49% of these near-term outcomes depending on the cohort, supporting transplant eligibility for a large minority of patients satisfying this outcome. When comparing 18-month transplant-free survival between cohorts (Figure S2), those with non-IPF ILD displayed slightly favorable survival when compared to the IPF cohorts (plogrank = 0.03), while there was no difference in transplant-free survival between IPF cohorts (plogrank = 0.59).
Table 1.
Baseline Characteristics and Outcomes for Derivation and Validation Cohorts
| Characteristic | IPF derivation cohort (n = 314) |
IPF validation cohort (n = 105) |
p-valuea | Non-IPF ILD validation cohort (n = 295) |
|---|---|---|---|---|
| Age, mean (± SD) | 65.5 (6.3) | 66.2 (4.5) | 0.29 | 59.5 (10.6) |
| Male sex, n (%) | 235 (74.8) | 73 (69.5) | 0.31 | 130 (44.1) |
| White race, n (%) | 293 (93.3) | 94 (89.5) | 0.21 | 226 (76.6) |
| Ever smoker, n (%) | 194 (61.8) | 64 (61.0) | 0.91 | 145 (49.2) |
| FVC %, mean (± SD) | 66.1 (16.1) | 71.6 (17.9) | 0.003 | 66.4 (19.5) |
| DLCO %, mean (± SD) | 41.3 (16.4) | 51.4 (18.3) | < 0.001 | 45.4 (18.6) |
| Antifibrotic treated, n (%) | 262 (83.4) | 72 (68.6) | 0.002 | 31 (10.5) |
| Immunosuppressant treated, n (%) | 38 (12.1) | 2 (1.9) | < 0.001 | 112 (38.0) |
| Outcomes | ||||
| Death or transplant | 80 (25.5) | 30 (28.6) | 0.42 | 54 (18.3) |
| Death | 41 (13.1) | 19 (18.1) | 35 (11.9) | |
| Lung transplant | 39 (12.4) | 11 (10.5) | 19 (6.4) |
Abbreviations: DLCO, diffusion capacity of the lung for carbon monoxide; FVC, forced vital capacity; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis.
Antifibrotic = nintedanib or pirfenidone.
Immunosuppressant = prednisone, mycophenolate mofetil, azathioprine, cyclophosphamide, or rituximab.
Comparing IPF derivation and validation cohorts.
When applying least absolute shrinkage and selection operator to the IPF derivation cohort, 5 clinical variables were selected for the clinical prediction model, 16 proteins for the proteomic prediction model, and 2 clinical variables and 20 proteins for the multidimensional prediction model (Table S1). After dichotomization of fitted values for each model according to Youden’s index, 115 (36.6%), 112 (35.7%), and 91 (29.0%) patients were classified as high-risk for near-term outcome. In comparative analysis, the multidimensional model and binary classifier provided the highest net benefit over a wide range of threshold probabilities when compared to the clinical and proteomic models and classifiers (Figure 1). Individual measures of test performance corroborated these findings (Table 2). The multidimensional classifier was advanced for further testing, as this provided the highest specificity (88%) and positive predictive value (68%) without sacrificing sensitivity or negative predictive value relative to the other 2 classifiers. Based on likelihood ratios for this multidimensional classifier, a high-risk classification would increase the pretest probability of a near-term outcome (based on observed prevalence) from 26% to approximately 70% (Figure 2). A low-risk classification would reduce the pretest probability from 26% to approximately 7% (Figure 2).
Figure 1.
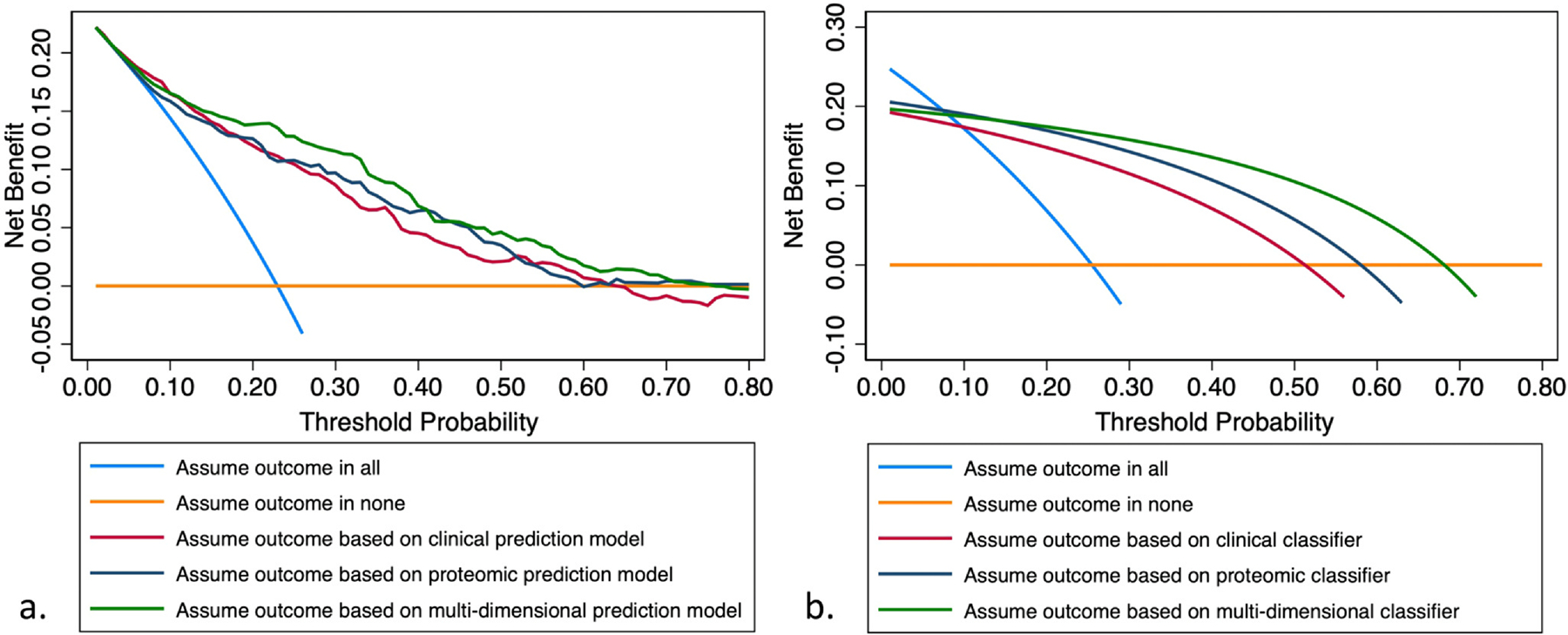
Decision curve analysis comparing clinical, proteomic, and multidimensional models (a) and binary classifiers (b) of near-term outcome.
Table 2.
Comparison of Test Performance Characteristics for Clinical, Proteomic, and Multidimensional Classifiers of 18-Month Death or Transplant in the IPF Derivation Cohort (n = 314)
| Classification | Clinical classifier | Proteomic classifier | Multidimensional classifier | |||
|---|---|---|---|---|---|---|
| Outcome (+) | Outcome (−) | Outcome (+) | Outcome (−) | Outcome (+) | Outcome (−) | |
| High risk | 61 | 54 | 65 | 47 | 62 | 29 |
| Low risk | 19 | 180 | 15 | 187 | 18 | 205 |
| Sensitivity | 0.76 | 0.81 | 0.78 | |||
| Specificity | 0.77 | 0.80 | 0.88 | |||
| (+) Predictive value | 0.53 | 0.58 | 0.68 | |||
| (−) Predictive value | 0.90 | 0.93 | 0.92 | |||
| (+) Likelihood ratio | 3.30 | 4.05 | 6.25 | |||
| (−) Likelihood ratio | 0.31 | 0.23 | 0.26 | |||
Abbreviations: IPF, idiopathic pulmonary fibrosis.
Figure 2.
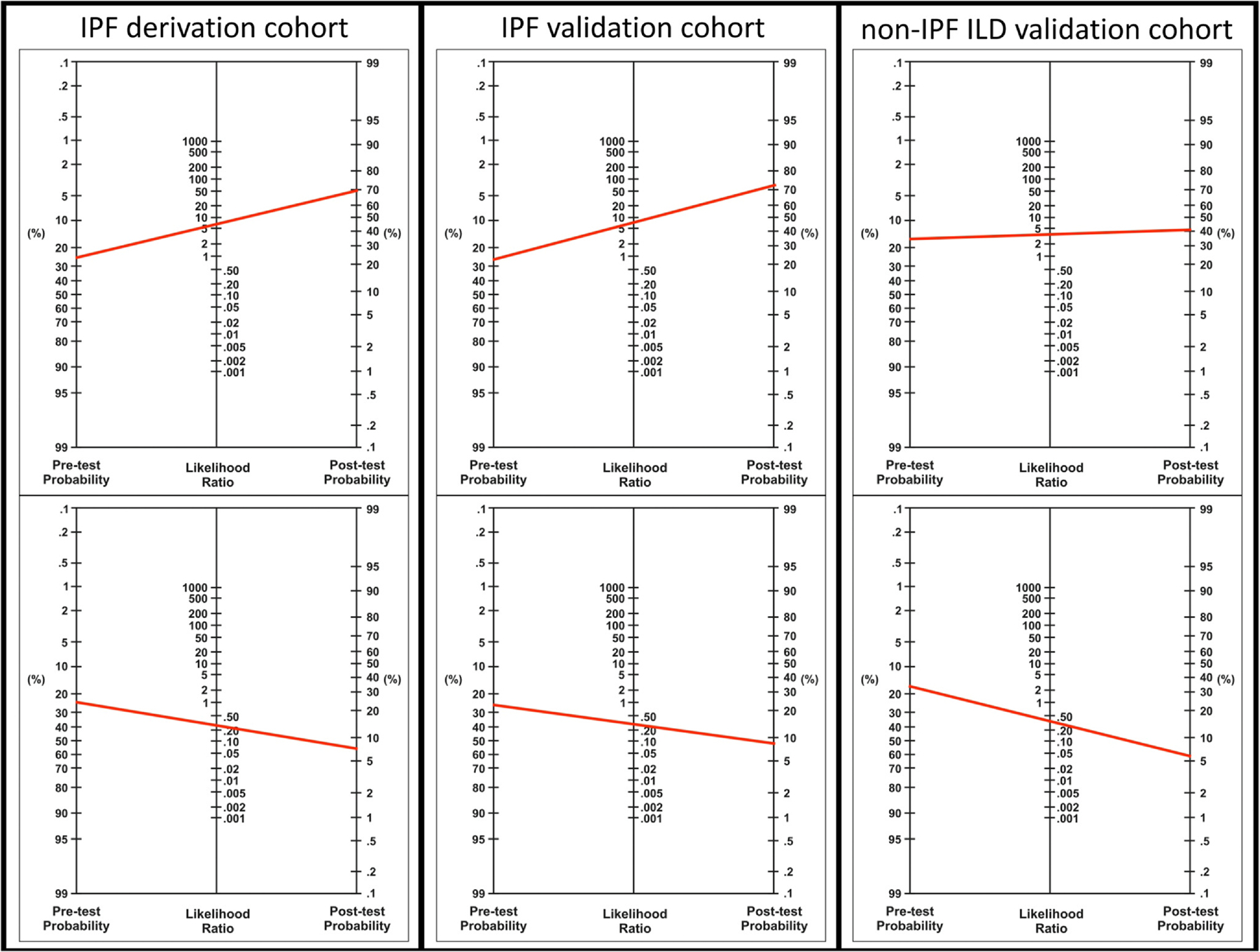
Nomograms for change in pre- and post-test probability based on likelihood ratio tests for IPF derivation, IPF validation, and non-IPF ILD validation cohorts. Pretest probabilities established using the observed incidence of 18-month death or transplant. ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis.
When applied to the IPF validation cohort, the classifier had lower sensitivity (70%), but higher specificity (92%), producing high positive (78%), and negative (88%) predictive values (Table 3). Using the observed 18-month cumulative incidence of death or transplant (29%) to establish pretest probability, likelihood ratios for this cohort suggested that a high-risk classification would increase the pretest probability of 18-month death or transplant from 29% to just over 70% (Figure 2). A low-risk classification would reduce the pretest probability from 29% to just less than 10% (Figure 2). After combining IPF derivation and validation cohorts, test performance was consistent across different thresholds of age (Table S3), baseline lung function (Tables S4 and S5), antifibrotic use (Table S6), and for death and transplant modeled as separate events (Table S7).
Table 3.
Multidimensional Classifier Test Performance Characteristics for Predicting 18-Month Death or Transplant When Applied to Independent IPF and Non-IPF ILD Validation Cohorts
| Classification | IPF validation cohort (n = 105) | Non-IPF ILD validation cohort (n = 295) | ||
|---|---|---|---|---|
| Outcome (+) | Outcome (−) | Outcome (+) | Outcome (−) | |
| High risk | 21 | 6 | 38 | 47 |
| Low risk | 9 | 69 | 16 | 194 |
| Sensitivity | 0.70 | 0.70 | ||
| Specificity | 0.92 | 0.80 | ||
| (+) Predictive value | 0.78 | 0.45 | ||
| (−) Predictive value | 0.88 | 0.92 | ||
| (+) Likelihood ratio | 8.75 | 3.61 | ||
| (−) Likelihood ratio | 0.33 | 0.37 | ||
Abbreviations: ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis.
When applied in the non-IPF ILD validation cohort, sensitivity was 70% and specificity 80%. The negative predictive value remained high (92%), but the positive predictive value dropped significantly (45%), owing to a lower outcome prevalence in this cohort (Table 3). Likelihood ratios for this cohort suggested that a high-risk classification would increase the pretest probability of near-term outcome (based on observed prevalence) from 18% to approximately 40% (Figure 2), again using observed 18-month incidence of death or transplant to establish the pretest probability. A low-risk classification would reduce the pretest probability from 18% to approximately 6% (Figure 2). When exploring test performance measures in key non-IPF ILD subgroups, the negative predictive value was high across cohorts, but the positive predictive value was low for those with connective tissue disease associated ILD and fibrotic hypersensitivity pneumonitis (Table S2). Test performance in those with non-IPF idiopathic interstitial pneumonia was similar to what was observed in the IPF cohorts.
When plotting transplant-free survival by multidimensional classification, those with a high-risk classification showed significantly worse survival than those with a low-risk classification (Figure 3). High-risk classification status was associated with significantly increased outcome risk in the IPF derivation (hazard ratio (HR) 13.39; 95% confidence interval (CI) 6.05–29.65; p < 0.0001), IPF validation (HR 14.02; 95% CI 8.25–23.8; p < 0.0001), and non-IPF ILD validation cohorts (HR 7.56; 95% CI 4.21–13.58; p < 0.0001) in unadjusted analysis. Adjustment for age, sex, race, smoking history, BMI, baseline immunosuppressant exposure, and baseline antifibrotic exposure did not impact these measures of association (Table S8). Lung function was not included to avoid collinearity, as percent predicted FVC and DLCO were both included in the multidimensional classifier.
Figure 3.
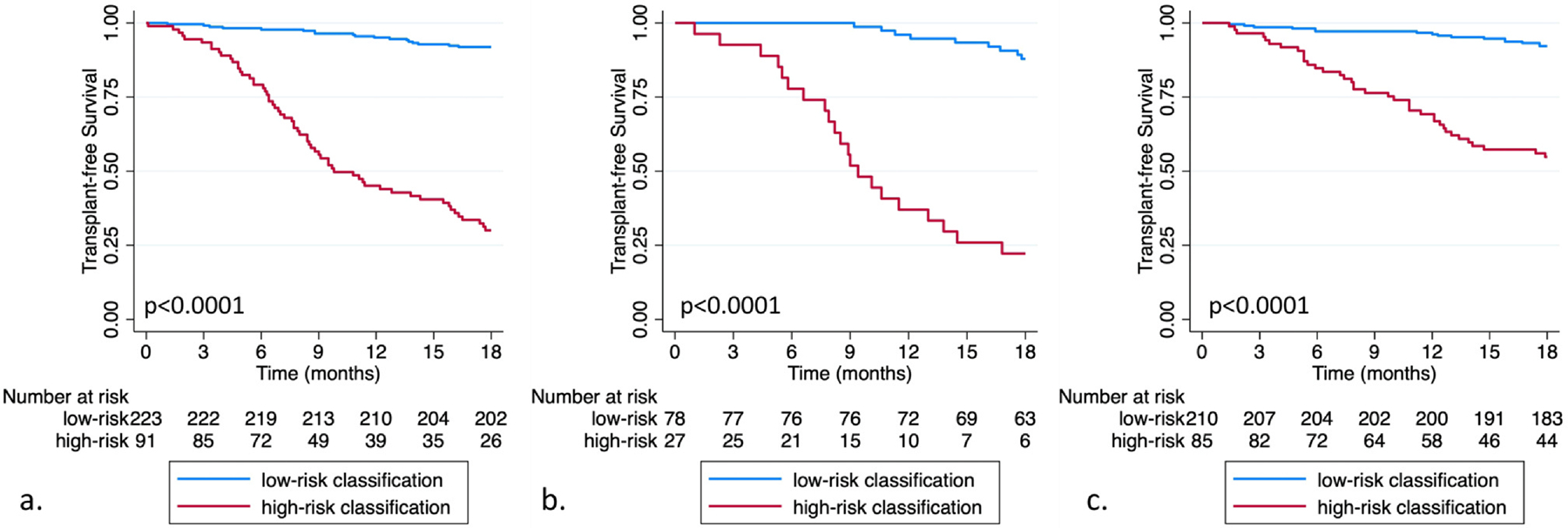
Eighteen-month transplant-free survival for idiopathic pulmonary fibrosis (IPF) derivation (a) IPF validation (b) and non-IPF interstitial lung disease validation cohorts (c) stratified by multidimensional biomarker classification.
Discussion
In this investigation, we present a novel multidimensional classifier of near-term outcome in patients with IPF. This classifier provided good test performance across independent IPF and non-IPF ILD cohorts and produced a positive likelihood ratio of greater than 6 across independent IPF cohorts, suggesting clinically actionable results. Test performance was less compelling when this classifier was applied to those with non-IPF ILD. Beyond measures of test performance, this multidimensional classifier strongly discriminated outcomes across cohorts, producing higher hazard ratios than most categorical biomarkers reported to date. Prospective validation of these findings in transplant-eligible individuals with IPF could help optimize the timing of lung transplant referral and inform future ISHLT guidelines based on classifier risk stratification.
These findings build upon a wealth of clinical and molecular biomarker data published in ILD to date. Clinical features, including age, sex, lung function, and several composite indices that include these measures, have been shown to strongly predict outcomes in patients with IPF and other fibrotic ILDs.10–13 Our findings corroborate the importance of lung function, which was among the variables of highest importance selected by our multidimensional model. Among molecular biomarkers, those most widely validated to date include peripheral blood leukocyte telomere length, a composite 52-gene signature, stand-alone proteins including KL6, matrix metalloproteinase 7, keratin 19 and its breakdown product CYFRA 21–1, neoepitopes, and several recently described composite proteomic signatures.15–23 However, while the survival association observed for these biomarkers is clearly reproducible, none have proven to be clinically useful. This reality stems from several key features required of a biomarker.
First, a biomarker must provide actionable results.32 Because antifibrotic therapy is recommended for most patients with IPF upon diagnosis,33 no biomarkers published to date have been shown to alter this management strategy. While one could argue that high-risk molecular classification could be used to enrich clinical trial cohorts, few trials employ death as a primary end-point, leaving it unclear whether those with a high-risk classification are any more likely to experience a change in FVC, the current gold standard for establishing therapeutic efficacy.34 We attempted to overcome this limitation by selecting an IPF population for whom management may differ based on classifier status. The ISHLT consensus currently advises early referral for all patients diagnosed with IPF, citing unpredictable disease trajectory as its rationale.8 Multidimensional classification would allow for more nuanced timing based on prognosis. In finding that nearly 70% to 80% of those with a high-risk classification will die or need a transplant within 18 months of blood draw, our results suggest that this classifier could justify immediate lung transplant referral and expedited work-up for high-risk patients, even with baseline lung function higher than ISHLT recommended referral thresholds. Conversely, we found that < 10% of patients with a low-risk classification died or required transplant over the next 18 months, which could help inform the pace of work-up, decision to list, and timing of listing for those with a low-risk classification.
Key to establishing actionable results for a biomarker is intuitive categorization and transparent reporting of test performance characteristics.32 While continuous biomarker measures often explain a larger proportion of outcome risk than categorical measures, the latter is typically needed for interpretation and clinical adoption. Importantly, measures of association (e.g., risk ratios, odds ratios, and hazard ratios) do not necessarily equate to meaningful measures of test performance (e.g., sensitivity, specificity, predictive values, and likelihood ratios). Underscoring this point is our observation that, while a high-risk classification was associated with > 7-fold increase in outcome risk in those with non-IPF ILD, our multidimensional classifier did not change post-test probability in a clinically meaningful manner. Conversely in IPF cohorts, a high-risk classification corresponded with a high positive likelihood ratio and meaningfully increased post-test probability of 18-month death or transplant, highlighting its potential to prompt early referral.
Augmentation or outperforming easily acquired clinical data is another key biomarker attribute. Clinical prediction models have been shown to discriminate IPF and non-IPF ILD survival with high accuracy across diverse cohorts and settings.10,11,13,14 As such, outperforming these easily estimated models is a challenge. For this reason, we fitted a stand-alone clinical prediction model for comparison. While we found that the stand-alone proteomic model did outperform the clinical model, a multidimensional model that capitalized on both lung function and 20 proteins outperformed both. Decision curve analysis, an efficient approach to comparing models and allows for a comparison of models over a range of outcome probabilities,30,31 corroborated these findings.
While our findings support refinement and prospective validation of this promising biomarker in patients with IPF, point estimates generated using semiquantitative data have limited external validity due to batch effects and normalization protocols.35,36 Commercially available, targeted quantitative multiplex assays are emerging and have the potential to advance this and other composite biomarkers toward clinical implementation.37 Our findings do not support the immediate advancement of this tool for prospective validation in those with non-IPF ILD, as the modest positive and negative likelihood ratios were un-likely to alter post-test probability in a clinically meaningful way. Subgroup analysis suggested that the reduction in test performance was isolated to those with connective tissue disease and fibrotic hypersensitivity pneumonitis. Inflammatory pathobiology may have contributed to these findings and prior work by members of our group suggested that inflammatory proteins may play a more prominent role in non-IPF ILD progression when compared to IPF.22,23 These findings and others showing poor outcomes among those with non-IPF ILD suggest dedicated investigation in this population is urgently needed.
Our study has several important limitations. First, while we attempted to restrict the cohort to individuals that were potentially eligible for lung transplant, it remains unclear what proportion were referred and how many were deemed ineligible due to unmeasured comorbid conditions. Nearly 50% of patients in the derivation cohort who met the primary end-point did so by undergoing lung transplants, suggesting that most were likely eligible. Sensitivity analysis suggested that the multidimensional classifier performed similarly irrespective of the end-point modeled, increasing confidence that those who died were molecularly similar to those who successfully underwent transplant. Next, clinical prediction models have been developed to predict death in patients listed for lung transplant.38,39 Our data did not allow for direct comparison to these validated models but suggests that a molecular-based approach could further augment current models. Next, our use of a dichotomous outcome fails to account for those who were censored before reaching the end of the follow-up period; however, few patients were censored (n = 26/714 patients) before 18 months. Next, our cohort was pre-dominantly white, leaving it unclear whether these findings would extend to a more racially diverse population. Finally, semiquantitative proteomic data do not always highly correlate with quantitative measures of the same protein,40 as we recently found a strong correlation for nearly 70% of quantified proteins, but a weak correlation for over 30%.23 For this reason, and those outlined above, prospective validation using quantitative methodology is needed.
Conclusion
Recent advances in proteomic technology have allowed for the identification of novel biomarkers of clinically relevant end-points in patients with IPF and non-IPF ILD. Beyond the biological importance of these biomarkers, a number have a high potential to improve prognostication and risk stratification. Here, we present a composite biomarker tool that showed good test performance for discriminating near-term outcomes among patients with IPF across independent cohorts. Prospective validation of these findings in transplant-eligible patients with IPF would help guide clinical decision-making and optimize the timing of lung transplant referral for this population.
Supplementary Material
Acknowledgments
We thank all patients who participated in the Pulmonary Fibrosis Foundation (PFF) Patient Registry. We also thank the investigators, clinical research coordinators, and other staff at participating PFF Care Centers for providing clinical data, the PFF, which established and has maintained the Registry since 2016, and lastly, the many generous donors.
Role of the Funding Source
This study was funded by the NHLBI, which had no role in the design or conduct of the study. All authors had full access to all data, and the corresponding author had final responsibility for the decision to submit for publication.
Disclosure statement
Dr Kim reports grants from the National Institutes of Health, Pulmonary Fibrosis Foundation, and Chest foundation. Dr Adegunsoye reports grants from the National Institutes of Health and personal fees from Genentech, Inogen, Medscape, Abbvie, PatientMpower, and Boehringer Ingelheim. Dr Strek reports grants from the National Institutes of Health, Pulmonary Fibrosis Foundation, and Boehringer Ingelheim, and personal fees for DSMB work from Fibrogen and Brisol Myers Squibb. Dr Dilling reports personal fees from Boehringer Ingelheim. Dr Flaherty reports personal fees from Up To Date, Roche/Genentech, Bellerophon, Respivant, Shionogi, DevPro, AstraZeneca, Pure Health, Horizon, FibroGen, Sun Pharmaceuticals, Pliant, United Therapeutics, Arrowhead, Lupin, Polarean, Pure Tech, CSL Behring, Daewoong, Dispersol, Immunet, NeRRe Therapeutics, Insilico, Vicore, Glaxo Simth Kline and Merck. Dr Martinez reports grants from the National Institutes of Health and personal fees from Afferent/Merck, Boehringer Ingelheim, Biogen, Bristol Myers Squibb, DevPro, Nitto, Patara/Respivant, ProMedior/Roche. Dr Noth reports grants from the National Institutes of Health and Veracyte and personal fees from Boehringer Ingelheim and Sanofi. Dr Oldham reports grants from the National Institutes of Health and personal fees from Boehringer Ingelheim, Roche, Genentech, Endeavor BioMedicines, Lupin Pharmaceuticals AmMax Bio, and Novartis. Drs Pugashetti, Combs, Ma, Linderholm, Chen, and Whelan report no conflicts.
Financial support
NHLBI—T32HL007749 (JVP), K23HL150301 (JSK), K23HL146942 (AA), UH3HL145266 (FJM, IN) R01HL169166 (JMO).
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.healun.2024.03.018.
Data sharing statement
Individual-level data will be made available on BioLINCC within 12 months of publication.
References
- 1.Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J 2020;55:2000085. 10.1183/13993003.00085-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cottin V, Hirani NA, Hotchkin DL, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev 2018;27:180076. 10.1183/16000617.0076-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pugashetti JV, Adegunsoye A, Wu Z, et al. Validation of proposed criteria for progressive pulmonary fibrosis. Am J Respir Crit Care Med 2023;207:69–76. 10.1164/rccm.202201-0124OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083–92. 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 5.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071–82. 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 6.Laporta Hernandez R, Aguilar Perez M, Lázaro Carrasco MT, Ussetti Gil P. Lung transplantation in idiopathic pulmonary fibrosis. (In eng). Med Sci (Basel) 2018;6. 10.3390/medsci6030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapnadak SG, Raghu G. Lung transplantation for interstitial lung disease. (In eng). Eur Respir Rev 2021;30:210017. 10.1183/16000617.0017-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leard LE, Holm AM, Valapour M, et al. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. (In eng). J Heart Lung Transplant 2021;40:1349–79. 10.1016/j.healun.2021.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40. 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 10.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012;156:684–91. 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 11.Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 2003;167:962–9. 10.1164/rccm.2111053. [DOI] [PubMed] [Google Scholar]
- 12.Chandel A, Pastre J, Valery S, et al. Derivation and validation of a simple multidimensional index incorporating exercise capacity parameters for survival prediction in idiopathic pulmonary fibrosis. Thorax 2023;78:368–75. 10.1136/thoraxjnl-2021-218440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest 2014;145:723–8. 10.1378/chest.13-1474. [DOI] [PubMed] [Google Scholar]
- 14.Ley B, Bradford WZ, Vittinghoff E, et al. Predictors of mortality poorly predict common measures of disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;194:711–8. 10.1164/rccm.201508-1546OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herazo-Maya JD, Sun J, Molyneaux PL, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med 2017;5:857–68. 10.1016/S2213-2600(17)30349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maher TM, Oballa E, Simpson JK, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med 2017;5:946–55. 10.1016/S2213-2600(17)30430-7. [DOI] [PubMed] [Google Scholar]
- 17.Maher TM, Stowasser S, Nishioka Y, et al. Biomarkers of extra-cellular matrix turnover in patients with idiopathic pulmonary fibrosis given nintedanib (INMARK study): a randomised, placebo-controlled study. Lancet Respir Med 2019;7:771–9. 10.1016/S2213-2600(19)30255-3. [DOI] [PubMed] [Google Scholar]
- 18.Stuart BD, Lee JS, Kozlitina J, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med 2014;2:557–65. 10.1016/S2213-2600(14)70124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molyneaux PL, Fahy WA, Byrne AJ, et al. CYFRA 21–1 predicts progression in idiopathic pulmonary fibrosis: a prospective long-itudinal analysis of the PROFILE cohort. Am J Respir Crit Care Med 2022;205:1440–8. 10.1164/rccm.202107-1769OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowman WS, Echt GA, Oldham JM. Biomarkers in progressive fibrosing interstitial lung disease: optimizing diagnosis, prognosis, and treatment response. Front Med (Lausanne) 2021;8:680997. 10.3389/fmed.2021.680997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clynick B, Corte TJ, Jo HE, et al. Biomarker signatures for progressive idiopathic pulmonary fibrosis. (In eng). Eur Respir J 2022;59:2101181. 10.1183/13993003.01181-2021. [DOI] [PubMed] [Google Scholar]
- 22.Bowman WS, Newton CA, Linderholm AL, et al. Proteomic biomarkers of progressive fibrosing interstitial lung disease: a multicentre cohort analysis. Lancet Respir Med 2022;10:593–602. 10.1016/S2213-2600(21)00503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oldham JM, Huang Y, Bose S, et al. Proteomic biomarkers of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2023. 10.1164/rccm.202301-0117OC. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Podolanczuk AJ, Kim JS, Cooper CB, et al. Design and rationale for the prospective treatment efficacy in IPF using genotype for NAC selection (PRECISIONS) clinical trial. (In eng). BMC Pulm Med 2022;22:475. 10.1186/s12890-022-02281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang BR, Edwards R, Freiheit EA, et al. The pulmonary fibrosis foundation patient registry. rationale, design, and methods. (In eng). Ann Am Thorac Soc 2020;17:1620–8. 10.1513/AnnalsATS.202001-035SD. [DOI] [PubMed] [Google Scholar]
- 26.Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 2014;9:e95192. 10.1371/journal.pone.0095192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundberg M, Eriksson A, Tran B, et al. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res 2011;39:e102. 10.1093/nar/gkr424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranstam J, Cook JA. LASSO regression. 1348–1348 Br J Surg 2018;105. 10.1002/bjs.10895. [DOI] [Google Scholar]
- 29.Youden WJ. Index for rating diagnostic tests. Cancer 1950;3:32–5. . [DOI] [PubMed] [Google Scholar]
- 30.Sadatsafavi M, Adibi A, Puhan M, et al. Moving beyond AUC: decision curve analysis for quantifying net benefit of risk prediction models. Eur Respir J 2021;58:63. 10.1183/13993003.01186-2021. [DOI] [PubMed] [Google Scholar]
- 31.Vickers AJ, van Calster B, Steyerberg EW. A simple, step-by-step guide to interpreting decision curve analysis. (In eng). Diagn Progn Res 2019;3:18. 10.1186/s41512-019-0064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pletcher MJ, Pignone M. Evaluating the clinical utility of a biomarker: a review of methods for estimating health impact. (In eng). Circulation 2011;123:1116–24. 10.1161/circulationaha.110.943860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. (In eng). Am J Respir Crit Care Med 2022;205:e18–47. 10.1164/rccm.202202-0399ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paterniti MO, Bi Y, Rekić D, et al. Acute exacerbation and decline in forced vital capacity are associated with increased mortality in idiopathic pulmonary fibrosis. (In eng). Ann Am Thorac Soc 2017;14: 1395–402. 10.1513/AnnalsATS.201606-458OC. [DOI] [PubMed] [Google Scholar]
- 35.Sivakumar P, Ammar R, Thompson JR, et al. Integrated plasma proteomics and lung transcriptomics reveal novel biomarkers in idiopathic pulmonary fibrosis. Respir Res 2021;22:273. 10.1186/s12931-021-01860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakhin AV, Tarantul VZ, Gening LV. Aptamers: problems, solutions and prospects. Acta Nat 2013;5:34–43〈https://www.ncbi.nlm.nih.gov/pubmed/24455181〉. [PMC free article] [PubMed] [Google Scholar]
- 37.Olink Flex. Olink proteomics. Accessed 10 April, 2024. Available at: 〈https://olink.com/content/uploads/2023/03/white-paper-ensuring-quality-with-flexibility-olink-flex-validation-verification-1.pdf〉.
- 38.Egan TM, Bennett LE, Garrity ER, et al. Predictors of death on the UNOS lung transplant waiting list: results of a multivariate analysis. (In eng). J Heart Lung Transplant 2001;20:242. 10.1016/s1053-2498(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 39.Chan EY, Goodarzi A, Graviss EA, et al. Still valid: reassessing a lung transplant recipient risk of death model. J Heart Lung Transplant 2021;40(Suppl 4):S159–60. 10.1016/j.healun.2021.01.478. [DOI] [Google Scholar]
- 40.Raffield LM, Dang H, Pratte KA, et al. Comparison of proteomic assessment methods in multiple cohort studies. (In eng). Proteomics 2020;20:e1900278. 10.1002/pmic.201900278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual-level data will be made available on BioLINCC within 12 months of publication.