

Abstract
Background
Understanding the resistance mechanisms of tumor is crucial for advancing cancer therapies. The prospective MATCH-R trial (NCT02517892), led by Gustave Roussy, aimed to characterize resistance mechanisms to cancer treatments through molecular analysis of fresh tumor biopsies. This report presents the genomic data analysis of the MATCH-R study conducted from 2015 to 2022 and focuses on targeted therapies.
Methods
The study included resistant metastatic patients (pts) who accepted an image-guided tumor biopsy. After evaluation of tumor content (TC) in frozen tissue biopsies, targeted NGS (10 < TC < 30%) or Whole Exome Sequencing and RNA sequencing (TC > 30%) were performed before and/or after the anticancer therapy.
Patient-derived xenografts (PDX) were established by implanting tumor fragments into NOD scid gamma mice and amplified up to five passages.
Results
A total of 1,120 biopsies were collected from 857 pts with the most frequent tumor types being lung (38.8%), digestive (16.3%) and prostate (14.1%) cancer. Molecular targetable driver were identified in 30.9% (n = 265/857) of the patients, with EGFR (41.5%), FGFR2/3 (15.5%), ALK (11.7%), BRAF (6.8%), and KRAS (5.7%) being the most common altered genes. Furthermore, 66.0% (n = 175/265) had a biopsy at progression on targeted therapy. Among resistant cases, 41.1% (n = 72/175) had no identified molecular mechanism, 32.0% (n = 56/175) showed on-target resistance, and 25.1% (n = 44/175) exhibited a by-pass resistance mechanism. Molecular profiling of the 44 patients with by-pass resistance identified 51 variants, with KRAS (13.7%), PIK3CA (11.8%), PTEN (11.8%), NF2 (7.8%), AKT1 (5.9%), and NF1 (5.9%) being the most altered genes. Treatment was tailored for 45% of the patients with a resistance mechanism identified leading to an 11 months median extension of clinical benefit.
A total of 341 biopsies were implanted in mice, successfully establishing 136 PDX models achieving a 39.9% success rate. PDX models are available for EGFR (n = 31), FGFR2/3 (n = 26), KRAS (n = 18), ALK (n = 16), BRAF (n = 6) and NTRK (n = 2) driven cancers. These models closely recapitulate the biology of the original tumors in term of molecular alterations and pharmacological status, and served as valuable models to validate overcoming treatment strategies.
Conclusion
The MATCH-R study highlights the feasibility of on purpose image guided tumor biopsies and PDX establishment to characterize resistance mechanisms and guide personalized therapies to improve outcomes in pre-treated metastatic patients.
Graphical Abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s12943-024-02134-4.
Keywords: Cancer, Metastatic, Resistance, PDX, Targeted therapy, Biopsy
Introduction
In recent years, significant technological advancements in molecular biology techniques and the development of patient-derived models have led to exponential progress in cancer therapy. Alongside classic chemotherapy, a wide range of new drugs including tyrosine kinase inhibitors (TKI), immune checkpoint inhibitors, endocrine therapy, or DNA repair and epigenetic modulators, are being used in monotherapy or in combinations.
Despite these promising developments, the identification of specific and reliable biomarkers capable of predicting treatment response remains a significant challenge. Certain indicators, such as PD-L1 status [1], tumor mutational burden [2], and mismatch repair deficiency [3], offer partial predictive value for responses to immune checkpoint inhibitors. Additionally, the presence of specific targetable alterations, occurring in genes such as ALK, BRAF, EGFR, MET, NTRK, RET and ROS1, serves as a reliable predictor of response to kinase inhibitors. Nevertheless, responses to these inhibitors, while often prolonged, eventually give way to disease progression due to acquired resistance mechanisms. Presently, some of these multidimensional resistance mechanisms have been elucidated, paving the way for more tailored therapeutic approaches. As novel cancer treatments continue to be developed and investigated in clinical trials, it is essential to devise research strategies to systematically investigate new resistance mechanisms to cancer therapies.
The lack of relevant and aggressive models is a major obstacle for the development of new therapeutics. In this context, in vitro models are not the most suitable due to the impossibility to reflect physiological proprieties, particularly in terms of pharmacokinetics. Conversely, patient-derived xenografts (PDX), consisting of fresh patients’ tumor tissue grafted into immunocompromised mice, have shown their ability to recapitulate the main characteristics of their originated tumors. PDX maintain cancer stromal components and architecture as well as cellular heterogeneity [4–7] and are therefore candidates of choice for personalized medicine [8–11]. PDX models are not only essential to deepen our understanding of acquired resistance mechanisms to therapies, but also to identify and validate overcoming strategies by testing novel molecules or combination strategies [12–14].
The prospective MATCH-R study (NCT02517892) [15] was specifically designed to discover acquired resistance mechanisms in patients with solid tumors treated with innovative therapies and to build a PDX preclinical platform allowing the development of a wide collection of unique models crucial for the development of new therapeutic strategies. We analyzed in the present study the output of this large repeated biopsy clinical trial for identification of molecular mechanisms of resistance as well as PDX relevancy with a special focus on the targeted therapy cohort of patients.
Materials and methods
Study design and procedure
The MATCH-R clinical trial (NCT02517892) was a monocentric, prospective study conducted between 2015 and 2022 and led by Gustave Roussy. The trial’s primary objective was to investigate how tumors evolve in patients in response to various anticancer treatments such as chemotherapy, endocrine therapy, monoclonal antibodies, tyrosine kinase inhibitors, and immunotherapy. Additionally, the trial aimed to identify new mechanisms of resistance to anticancer agents and develop PDX to model these resistances in vivo. The endpoints of the trial included genotyping the molecular alterations associated with resistance and comparing the molecular profiles of patients before treatment and at the progression of anticancer therapies potentially leading to treatment tailoring. Each individual genomic report was reviewed and discussed within a multidisciplinary tumor board to evaluate the presence of actionable therapeutics targets, based on ESCAT classification. The key inclusion criteria included the following: (1) Patients affiliated to a social security regimen; (2) Patients scheduled to receive anticancer agents or currently receiving anticancer agents; (3) Tumor lesion accessible to core biopsies; (4) Patients who were fully informed, able to comply with the protocol and who signed the informed consent. This protocol was approved by the CPP (“Comité de protection des personnes”) and by the ANSM (“Agence nationale de sécurité du médicament et des produits de santé”) and adhered to the principles in the Guideline for Good Clinical Practice and the Declaration of Helsinki. Written informed consent was obtained from all patients included in this study.
Molecular profiling
After signed consent, the patient underwent a biopsy or a tumor resection stored as formalin-fixed paraffin-embedded (FFPE) and fresh frozen (FF) samples. From FF samples, a molecular profiling analysis was conducted for samples with a tumor content exceeding 10%, as evaluated by a senior pathologist on a hematoxylin and eosin slide. Tumor DNA and RNA were extracted using the AllPrep DNA/RNA Mini Kit (Qiagen), and germline DNA was extracted using the Maxwell® RSC Blood DNA Kit (Promega, Charbonnières-Les-Bains, France).
When the tumor content ranged from 10 to 30%, a targeted next-generation sequencing (NGS) analysis was performed using the customized MOSC4 panel (Ion AmpliSeq custom design, ThermoFisher Scientific, Illkirch, France). This panel was specifically designed to encompass 82 genes associated with cancer. For the final patients included in the trial, the Oncomine Comprehensive Assay v3 (ThermoFisher Scientific, Illkirch, France) was utilized, which covers 161 genes and allow for the detection of single nucleotide variation, copy number variations and gene fusions. The bioinformatics analysis of the sequencing data was carried out using TorrentSuite software and variantCaller (ThermoFisher Scientific, Illkirch, France). The median coverage depth on retained variants is over 700 reads, offering a sensitivity down to 5% of variant allelic frequency.
For biopsies with a tumor content exceeding 30%, whole exome sequencing (WES) and RNA sequencing (RNAseq) were conducted. These sequencing methods utilized different capture kits to adapt to technological advancements, such as SureSelect Clinical Research Exome V2 and V6 kits from Agilent Technologies followed by Exome 2.0 kit from Twist Biosciences. These high throughput analyses were applied to the frozen patient tumors and the PDX.
The pathogenicity of the variants was determined using the ACMG variant classification [16].
The OncoKB classification was used to determine whether an alteration was actionable [17, 18].
Development of patients-derived xenografts (PDX)
All animal procedures and studies were performed in accordance with the approved guidelines for animal experimentation by the ethics committee at University Paris Saclay (CEEA 26, Project 2014_055_2790) following the regulations set by the European Union. The animals were housed in pathogen-free conditions and provided unlimited food and water access. To establish the tumor models, fresh tumor fragments were implanted beneath the renal capsule [19] of 6 to 8-week-old male NOD scid gamma (NSG) mice obtained from Charles River Laboratories within 1 to 12 h after the patient biopsy. Subsequently, the xenografts were serially propagated subcutaneously from one mouse to another for up to five passages. In the frame of a collaboration with XenTech company, selective pressure was applied to selected PDX using the inhibitor to which the patient had developed resistance to validate its pharmacological status. All PDX generated are listed with their molecular and clinical associated data on the following web page https://pdx.gustaveroussy.fr (Supplementary File 1).
In vivo drug response studies
PDXs were subcutaneously grafted (1 graft per mouse) into either nude or NSG mice. The grafts were allowed to grow until the tumor volume reached a size ranging of 80–200 mm3. Subsequently the mice were randomly assigned to either the treatment groups or the vehicle group. To monitor the progression of the tumors, their volume was measured twice weekly using calipers throughout the treatment period.
Statistical analyses
R statistical software was used; Sankey diagram was created using the function sankeyNetwork available in the networkD3 package (version 0.4) and the violin plot was created using the ggstatsplot function (version 0.9.5).
Oncoprint was drawn using the cBioPortal online tool [20, 21].
Data availability
All the WES/RNAseq raw data files from patient’s samples and PDX models included in this study are deposited at the European Genome–phenome Archive (EGA) using the accession code EGAD50000000697 (Supplementary Table 1). Access to this shared dataset is controlled by the institutional Data Access Committee, and requests for access can be sent to the corresponding author. Further information about EGA can be found at https://ega-archive.org/. Any additional information required to reanalyze the data reported in this article is available upon request by filling out the data request form for Gustave Roussy clinical trials at https://redcap.link/DataRequestClinicalTrialsGustaveRoussy. The steering committee and the sponsor will review the requests on a case-by-case basis. In case of approval, a specific agreement between the sponsor and the researcher may be required for data transfer.
Results
Study flow and patient population
The study flow is illustrated in Fig. 1A. From January 2015 to December 2022, a total of 1,120 biopsies were performed on 857 patients, with 220 patients (25.7%) undergoing a second biopsy, 38 (4.4%) a third biopsy, 4 (0.5%) a fourth, and one patient having a fifth biopsy. The repeated biopsies were mainly performed due to treatment progression, insufficient material, or unsuccessful tumor sequencing.
Fig. 1.

Global patient population. A. Study flow chart. B. Oncoprint depicting 1230 pathogenic alterations identified in the initial biopsy of 563 patients, all of whom exhibited at least 1 pathogenic alteration. C. Frequency of the highest OncoKb alteration in each first biopsy
The median age of the 857 patients was 63.3 years (interquartile range: 54.7–70.5). The majority of the patients were male, accounting for 56.0% (n = 480/857) of the cohort. The five most prevalent cancer types within this cohort were lung cancers (38.9%; n = 333/857), digestive cancers (16.3%; n = 140/857), prostate cancers (14.1%; n = 121/857), genito-urinary cancers outside of the prostate (10.0%; n = 86/857) and gynecological cancers (5.8%; n = 50/857). In terms of treatment regimen, 30.9% (n = 265/857) of the patients received targeted therapy, 19.3% (n = 165/857) received immunotherapy, 14.1% (n = 121/857) received endocrine therapy [22, 23] and 35.7% (306/857) received another type of treatment (such as conventional cytotoxic chemotherapy) (Table 1 and Supplementary Table 1).
Table 1.
Characteristics of the patients included in the MATCH-R trial
| Patients with at least one biopsy (n = 857) | ||
|---|---|---|
| Age at inclusion | Mean (SD) | 61.8 (12.2) |
| Median (IQR) | 63.3 (54.7—70.5) | |
| Min—Max | 22—93 | |
| Gender | Male | 480 (56.0%) |
| Female | 377 (44.0%) | |
| Primary tumor type | Lung | 333 (38.9%) |
| Digestive | 140 (16.3%) | |
| Prostate | 121 (14.1%) | |
| Urothelial | 86 (10.0%) | |
| Gynecological | 50 (5.8%) | |
| Other | 34 (4.0%) | |
| Hematological | 28 (3.3%) | |
| Head and neck | 21 (2.5%) | |
| Sarcoma | 20 (2.3%) | |
| Breast | 16 (1.9%) | |
| Skin | 8 (0.9%) | |
| Type of treatment received | Targeted therapy | 265 (30.9%) |
| Immunotherapy | 165 (19.3%) | |
| Endocrine therapy | 121 (14.1%) | |
| Other | 306 (35.7%) |
Genomic landscape of the MATCH-R patients
Overall, out of 857 patients, molecular analysis was not feasible for 254 (29.6%) due to low tumor content (TC) (< 10%) or insufficient DNA and/or RNA. A total of 812 biopsies with adequate TC (> 10%) were collected from these patients, and molecular profiling was conducted on this subset. Specifically, 520 (64.0%) biopsies underwent both WES and RNAseq, 164 (20.2%) were analyzed using WES only, 106 (13.1%) using a targeted NGS panel due to low TC (ranging from 10 to 30%), and 22 (2.7%) using RNAseq only. Additionally, 341 (41.4%) of these biopsies were implanted in mice, resulting in 136 (16.5%) PDX models generated (Fig. 1A).
Focusing on the 661 molecular profiling performed on the first biopsy, at the time of inclusion in the clinical trial, at least one pathogenic alteration was identified for 563 (84.3%) of these biopsies. In total, 1384 pathogenic alterations were identified, 1294 (93.5%) single nucleotide variations, 64 (4.6%) focal copy number alterations, and 26 (1.9%) fusion transcripts. Among the genes mutated in at least 10 patients in this pan-cancer cohort, the five most affected were TP53 (63%), KRAS (21%), EGFR (10%), PIK3CA (10%) and ARID1A (9%) (Fig. 1B).
Furthermore, at least one actionable alteration was identified in 357 (53.4%) out of the 661 first biopsies performed. When considering the OncoKB actionability scale, 25% of these alterations were classified as level 1, 4% as level 2, 6% as level 3 and 27% as level 4 (Fig. 1C).
Targeted therapy cohort and resistance mechanisms
Among the 857 patients who underwent a biopsy, 265 (30.9%) were known to harbor a molecular cancer driver either through molecular profiling conducted as part of the protocol or through standard molecular diagnosis. The five most common oncogenic driver alterations were found in the genes EGFR, FGFR2/3, ALK, BRAF and KRAS (Fig. 2A). EGFR was the most prevalent gene, identified in 41.5% (n = 110/265) of the patients. All 110 EGFR mutations were identified in lung cancer patients. An FGFR2/3 alteration was detected in 15.5% (n = 41/265) of the patients. Among the 41 cases with FGFR2/3 alterations, the majority, 51.2% (n = 21/41), originated from digestive cancer, followed by urothelial cancer 34.1% (n = 14/41), gynecological cancers 7.3% (n = 3/41), and 2 from other types of cancer. ALK alterations were observed in 11.7% (n = 31/265) of the patients. The large majority, 90.3% (n = 28/31), of ALK alterations were identified in lung cancer patients, while one case each came from digestive cancer, sarcoma, and thyroid cancer. BRAF alterations were detected in 6.8% (n = 18/265) of the patients. The majority of these, 55.6% (n = 10/18), were found in lung cancer cases, followed by digestive cancers 22.2% (n = 4/18), other cancers 16.7%, (n = 3/18), and 1 case in urological cancer. Finally, KRAS mutations were identified in 5.7% (n = 15/265) of the patients. Among these KRAS mutations, the largest proportion 53.3% (n = 8/15) originated from digestive cancers, followed by lung cancer 33.3% (n = 5/15), and gynecological cancers 13.3% (n = 2/15).
Fig. 2.

Targeted therapy cohort. A. Sankey diagram depicting the tumor type of origin of the 265 patients harboring a molecular driver and the 594 patients without any molecular driver identified. B. Flow chart representing the resistance mechanisms identified in the 265 patients harboring a molecular driver. C. Pie chart depicting the by-pass resistance mechanisms identified in the 265 patients harboring a molecular driver
Subsequent repeated biopsies were used to investigate acquired resistance mechanisms. Out of 265 patients, 175 (66.0%) underwent additional molecular profiling on biopsies collected after developing resistance to targeted therapies. The resistance mechanism remained unidentified for 72 patients (41.1%). Among the patients with identified resistance mechanisms, 56 (32.0%) experienced an on-target resistance mechanism, 44 (25.1%) had a by-pass resistance mechanism, and 3 patients had histological transformation explaining the resistance to the targeted therapy (Fig. 2B). The molecular profiles of the 44 patients with a by-pass resistance mechanism revealed the presence of 51 alterations. The most frequently altered genes among these patients were KRAS (13.7%, n = 7/51), PIK3CA (11.8%, n = 6/51), PTEN (11.8%, n = 6/51), NF2 (7.8%, n = 4/51), AKT1 (5.9%, n = 3/51), and NF1 (5.9%, n = 3/51) (Fig. 2C).
EGFR
Among the 110 EGFR-mutated patients treated with a TKI, a molecular profile was performed at the time of progression in 70.0% (n = 77/110) of them.
The disease progression was experienced after first- or second-generation TKI for 24 patients (31.2%). In terms of baseline oncogenic driver, 14 patients (58.3%) harbored a deletion in exon 19, 8 patients (33.3%) carried a p.(Leu858Arg) mutation, 1 patient had both p.(Leu858Arg) and p.(Thr790Met) mutations, and 1 had a p.(Gly719Ala) mutation.
Regarding the mechanisms of resistance observed in patients treated with first- or second-generation TKI, 14 patients (58.3%) demonstrated an on-target resistance mechanism. As expected, the predominant mechanism was the presence of the p.(Thr790Met) mutation, which was found in 13 patients (92.9%). Additionally, 1 other on-target alterations was identified: p.(Arg776Cys). For 10 patients (41.7%), the resistance mechanisms were not identified. Of note, no by-pass resistance mechanisms were identified in these patients (Fig. 3A).
Fig. 3.

Resistance mechanisms in EGFR, FGFR2/3 and ALK driven patients. A & B. Sunburst charts representing the EGFR cohorts. The innermost layer depicts the baseline molecular driver and the identified resistance mechanism are presented in the outermost layer. C & D. Sunburst charts illustrating the FGFR2/3 cohorts. The innermost layer depicts the baseline molecular driver and the identified resistance mechanism are presented in the outermost layer. E. Sunburst chart representing the ALK cohort. The innermost layer depicts the baseline molecular driver and the identified resistance mechanism are presented in the outermost layer
Out of the 77 EGFR-mutated patient who received an EGFR TKI treatment and for whom a biopsy at progression was available, 53 (68.8%) experienced disease progression while being treated with the third-generation TKI osimertinib. Among these patients, the most prevalent oncogenic driver observed at baseline was the combination of deletion in exon 19 and p.(Thr790Met) mutation, which was present in 27 patients (50.9%). Other oncogenic drivers included: deletion in exon 19 alone, observed in 10 patients (18.9%), p.(Leu858Arg) mutation observed in 5 patients (9.4%), combination of p.(Leu858Arg) and p.(Thr790Met) mutations observed in 6 patients (11.3%) and finally other drivers such as the combination of deletion in exon 19 and EGFR amplification (n = 2), the combination of p.(Glu709Ala) and p.(Gly719Ala) mutations, the combination of p.(Ser768Ile) and p.(Leu858Arg) mutations, and the presence of p.(Leu747Ser) mutation, were observed in 5 patients (9.4%).
A mechanism of resistance to third-generation EGFR TKI was identified for 32 patients (60.4%) who were treated with osimertinib. For 20 patients (37.7%), a by-pass mechanism was observed involving 25 genes. The most common by-pass mechanisms were PTEN with 4 mutations, KRAS 3 mutations, PIK3CA 3 mutations, RET 2 mutations, FGFR3 2 fusions and MET with 2 amplifications. An on-target resistance mechanism was observed for 11 patients (20.8%). The most frequent mutation observed was EGFR:p.(Cys797Ser), which was seen in 9 patients (81.8%). The EGFR:p.(Cys797Gly) mutation was observed in 1 patients and p.(Val843Ile) in 1 patient. A histological transformation from non-small cell lung cancer to small cell lung cancer was observed in 1 patient. Finally, for 20 (37.7%) patients, no molecular mechanisms explaining the resistance were observed (Fig. 3B).
FGFR2/3
Among the 41 patients with FGFR2/3 alterations, a molecular profiling performed at progression in the MATCH-R clinical trial was available for 32 of them (78.0%).
The baseline oncogenic driver was a FGFR2 fusion for 15 of these patients (46.9%) mainly involving cholangiocarcinoma patients. The most frequent fusion partner of FGFR2 was BICC1 observed in 3 patients, followed by STRN4 (n = 2/21) in 2 patients. Other FGFR2 partners included CCSER2, DIP2C, ERC1, FAM160B1, INA, MITF, PAWR, PXN, and WDHD1. Regarding FGFR2 mutations, FGFR2:p.(Cys382Arg) was identified in two patients (Fig. 3C). Additionally, the baseline oncogenic driver alteration was a FGFR3 mutation for 15 patients (46.9%), mainly suffering from urothelial cancer, with 11 patients (34.4%) presenting with a FGFR3:p.(Ser249Cys) mutation, 3 patients (9.4%) harboring a FGFR3:p.(Tyr373Cys) and 1 patient with a FGFR3:p.(Arg248Cys) mutation (Fig. 3D).
In terms of resistance mechanisms to FGFR2 or FGFR3 inhibitors (erdafitinib, futibatinib or pemigatinib), an on-target mechanism was identified in 15 patients (46.9%). These on-target mechanisms were monoclonal for 11 of these patients (68.8%, n = 11/16). By-pass mechanisms were observed in 7 patients (21.9%) and occurred in different genes such as EGFR, NF1, PIK3CA, NF2 or MSH6. Lastly, no resistance mechanism was identified for 10 patients (31.3%).
ALK (Fig. 3E)
A molecular profiling performed in the context of the MATCH-R clinical trial was available for 21 out of the 31 patients (67.7%) known to harbor an ALK molecular driver. Among these patients, ALK systematically involved fusion events, with EML4 emerging as the predominant fusion partner, observed in 11 patients (52.4%). Additionally, there were unique fusion occurrences involving CLTC and STRN in individual patients. The fusion partner of ALK remained unidentified for 8 patients (38.1%).
Regarding the resistance, an on-target mechanism was identified in 9 patients (42.9%) which was a unique mutation for 6 of them (66.7%). A by-pass resistance mechanism was identified in 2 patients involving NF2 in both cases. One histological transformation from non-small cell lung cancer to small cell lung cancer was also identified. Finally, the resistance mechanism to the ALK inhibitor remained unidentified for 9 patients (42.9%).
Treatment orientation based on molecular profiling
Overall, out of the 100 patients who received a targeted therapy and developed either an on-target or a by-pass resistance mechanism, treatment adaptation based on molecular profiling was implemented for 45 (45.0%) of them. Among the 56 patients with an on-target resistance mechanism, a new line of treatment was proposed for 34 of them (60.7%). Similarly, out of the 44 patients with a by-pass resistance mechanism, a new line of treatment was proposed for 11 patients (25.0%).
The best response observed to the new line of treatment could be assessed for 29 (64.4%) patients. Among these patients, 11 (37.9%) had a stable disease, 11 (37.9%) had a partial response, 6 (20.7%) had a progressive disease. Additionally, one patient achieved a complete response.
The median duration of treatment was 11 months (ranging from 0 to 101 months) and the main reason for stopping the adapted treatment was progressive disease, identified in 23 out of 29 patients (79.3%) overall.
Among the 29 patients who had an evaluable response to the new line of treatment, 20 patients (69.0%), were diagnosed with an EGFR-driven cancer. Fourteen (70.0%) of them were initially treated by a first- or a second-generation EGFR TKI and all developed an on-target resistance involving the EGFR:p.(Thr790Met) mutation. Switching to a third-generation EGFR TKI appeared to be effective in overcoming this identified resistance mechanism. The median duration of treatment with this new line was 18 months, with some patients still on osimertinib for 78, 87, and even 101 months. On the other hand, resistance to third-generation TKI primarily involved by-pass resistance mechanisms (in 5 out of 6 patients) occurring in HER2, MET, KRAS, ALK and NF1. The treatment duration of the new line of treatment was shorter when compared to resistance to first or second generation TKIs with a median duration of 2 months.
For the four ALK-driven patients who had an evaluable clinical response to the adapted treatment, the median duration of treatment was 15 months and for the three FGFR2/3 patients the median was 11 months.
However, a few patients with on-target resistance mutations did not respond to next-generation therapies specifically designed to target those alterations. For example, patients MR396 and MR475 experienced rapid disease progression (1 and 6 months, respectively) despite treatment with osimertinib targeting the EGFR:p.(Thr790Met) mutation. Similarly, patient MR517 who harbored the ROS1:p.(Gly2032Arg) resistance mutation to lorlatinib, failed to respond to reprotectinib, with progression occurring within 2 months. Patient MR439, treated with brigatinib for the ALK:p.(Gly1269Ala) mutation showed disease progression within only a few days. Conversely, some patients demonstrated prolonged responses with single-agent therapies instead of combination, targeting exclusively the resistance mechanism. For instance, patient MR12 responded to erlotinib for 43 months after developing the EGFR:p.(Leu861Arg) mutation, which emerged following progression on a first-line MET inhibitor (Table 2).
Table 2.
Treatment orientation based on molecular profiling
| ID | Gene | Molecular driver | Last treatment | Resistance mechanism | Treatment adapted | Best response | Cause of end of treatment | Duration (m) |
|---|---|---|---|---|---|---|---|---|
| 448 | ALK | EML4::ALK | Alectinib | ALK G1202R + ALK T1151M | Brigatinib | PR | PD | 10 |
| 1001 | ALK | ALK Fusion Unknown partner | Alectinib | ALK G1202R | Lorlatinib | PR | PD | 28 |
| 1188 | ALK | EML4::ALK | Alectinib | ALK V1180L | Lorlatinib | PR | PD | 20 |
| 439 | ALK | EML4::ALK | Lorlatinib | ALK G1269A | Brigatinib | PD | PD | 0 |
| 157 | EGFR | del19 | Afatinib | EGFR T790M | Osimertinib + Necitumumab | PR | PD | 11 |
| 376 | EGFR | del19 | Afatinib | EGFR T790M | Osimertinib | PR | PD | 26 |
| 16 | EGFR | del19 | Erlotinib | EGFR T790M + PTEN MUT | Osimertinib | PR | Ongoing | 101 |
| 129 | EGFR | del19 | Erlotinib | EGFR T790M | Osimertinib | CR | Ongoing | 87 |
| 222 | EGFR | del19 | Erlotinib | EGFR T790M | Osimertinib | PR | Ongoing | 78 |
| 296 | EGFR | L858R | Erlotinib | EGFR T790M | Osimertinib | PR | PD | 18 |
| 387 | EGFR | del19 | Erlotinib | EGFR T790M + EGFR V769M | Osimertinib | SD | PD | 13 |
| 396 | EGFR | del19 | Erlotinib | EGFR T790M | Osimertinib | PD | PD | 1 |
| 436 | EGFR | L858R | Erlotinib | EGFR R776C | Osimertinib | SD | PD | 18 |
| 445 | EGFR | del19 | Erlotinib | EGFR T790M | Osimertinib | SD | PD | 16 |
| 475 | EGFR | del19 | Erlotinib | EGFR T790M | Osimertinib | PD | PD | 6 |
| 8 | EGFR | L858R | Gefitinib | EGFR T790M | Osimertinib | SD | Ongoing | 46 |
| 22 | EGFR | del19 | Gefitinib | EGFR T790M | Osimertinib | PR | PD | 58 |
| 303 | EGFR | del19 | Gefitinib | EGFR T790M | Osimertinib | PR | PD | 12 |
| 1 | EGFR | del19 + T790M | Osimertinib | HER2 AMP | Paclitaxel + Trastuzumab | SD | Toxicity | 2 |
| 7 | EGFR | L858R + T790M | Osimertinib | MET AMP | TED11449 (MET inhibitor) | SD | PD | 1 |
| 69 | EGFR | del19 + T790M | Osimertinib | KRAS MUT + MET AMP | MET inhibitor + Erlotinib | SD | PD | 2 |
| 240 | EGFR | del19 + T790M | Osimertinib | STRN::ALK | Brigatinib | SD | PD | 5 |
| 258 | EGFR | L858R + T790M | Osimertinib | EGFR V843I | Afatinib | SD | lost at follow up | 6 |
| 330 | EGFR | del19 + T790M | Osimertinib | NF1 MUT | ERK inhibitor + BRAF inhibitor | PD | PD | 0 |
| 248 | FGFR | Y373C | Erdafitinib | MSH6 MUT | ICOS + PD1 inhibitor | PR | PD | 24 |
| 345 | FGFR | FGFR2::BICC1 | Futibatinib | polyclonal FGFR2 MUT + TSC1 MUT | Everolimus | SD | PD | 10 |
| 422 | FGFR | FGFR2::PXN | Pemigatinib | polyclonal FGFR MUT + PIK3CA MUT | Everolimus | SD | PD | 11 |
| 12 | MET | splice exon 14 | Crizotinib | EGFR L861R | Erlotinib | PD | PD | 43 |
| 517 | ROS1 | CD74::ROS1 | Lorlatinib | ROS1 G2032R | Repotrectinib | PD | PD | 2 |
PDX establishment
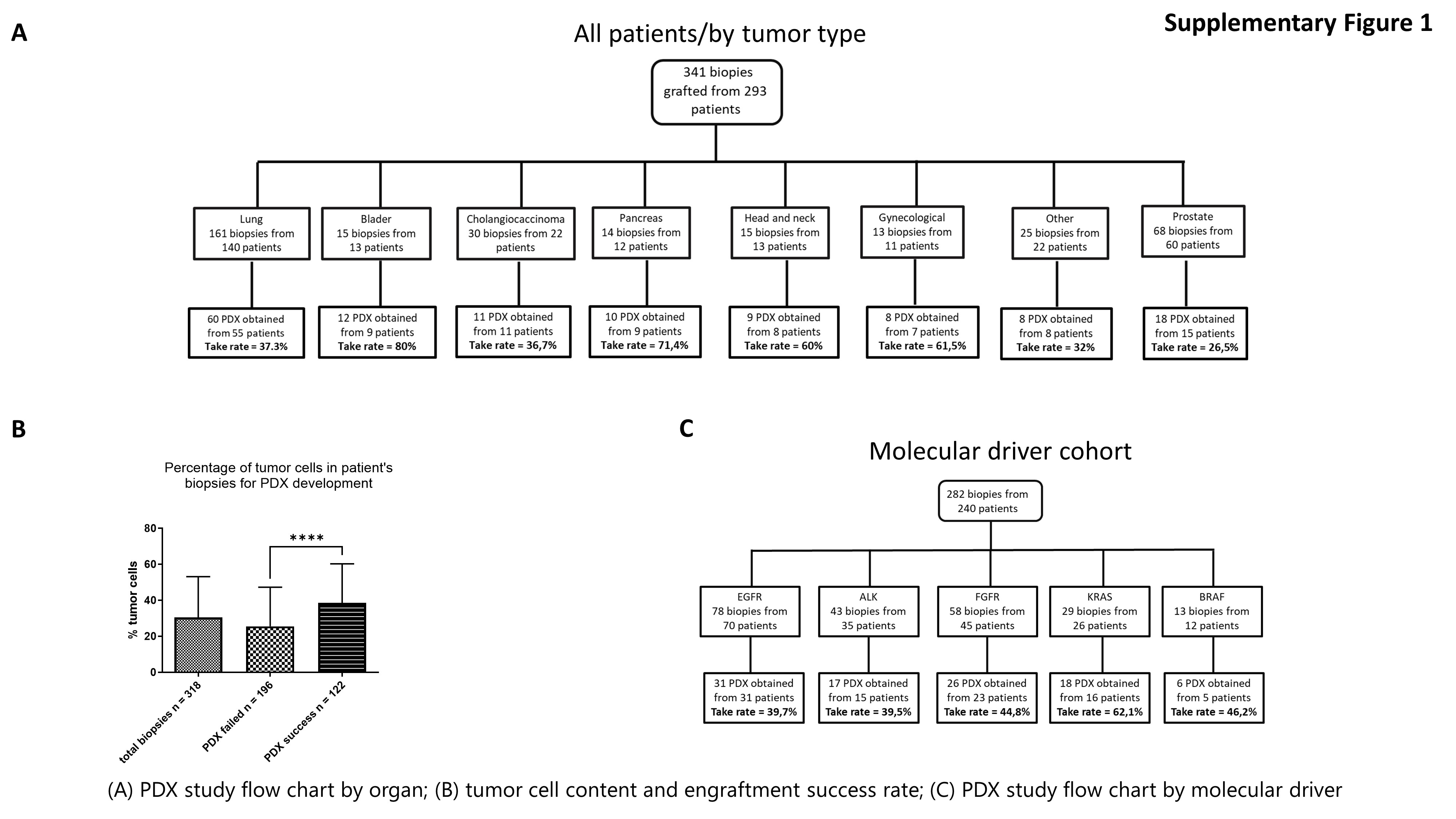
Out of the 341 biopsies grafted in mice, 136 patients-derived xenograft (PDX) models were successfully established, reaching an overall success rate of 39.9% (n = 136/341) (Fig. 4) (https://pdx.gustaveroussy.fr). The success rates of tumor engraftment varied by pathology, with bladder cancer achieving an 80.0% (n = 12/15) success rate, followed by pancreas at 71.4% (n = 10/14), gynecological at 61.5% (n = 8/13), head and neck at 60.0% (n = 9/15), non-small cell lung cancer at 37.3% (n = 60/161), cholangiocarcinoma at 36.7% (n = 11/30), and prostate at 26.5% (n = 18/68) (Supplementary Fig. 1A). Notably, the success rate of engraftment appeared to be positively associated with the tumor cell content of the other biopsies collected simultaneously for sequencing (Supplementary Fig. 1B).
Fig. 4.

PDX models available. Illustration of the PDX developed based on the molecular driver and the targeted therapy that led to acquired resistance
Regarding the targeted therapy cohort, 31 PDX models were available from EGFR patients, 26 from FGFR2/3 patients (14 with FGFR2 fusion, eight with FGFR3:p.(Ser249Cys), three with FGFR3:p.(Tyr373Cys), and one with FGFR2:p.(Arg248Cys)), 16 from ALK patients, 15 from KRAS patients, and 6 from BRAF patients. The global take rate of the PDX models from tumors harboring an oncogenic molecular driver was 44.3%, with variations among the different drivers: KRAS (62.1%), BRAF (46.2%), FGFR2/3 (44.8%), EGFR (39.7%) and ALK (39.5%) (Supplementary Fig. 1C).
PDX with resistance to targeted therapies available
The high value of the PDX models generated lies in the fact that most of them originate from tumors collected at the time of acquired resistance in patients. After first- second-generation EGFR inhibitors, we generated four PDX models with on-target resistance mechanisms, including three models carrying an EGFR:p.(Thr790Met) mutation, and one model with an EGFR:p.(Arg776Cys) mutation. Additionally, among the 25 models established post-osimertinib, resistant models harbored on-target resistance, four had an EGFR:p.(Cys797Ser) mutation, and one an EGFR:p.(Cys797Gly) mutation. In contrast to the models obtained after first- second-generation inhibitors, we acquired PDX models that developed by-pass pathway resistance to osimertinib. Three showed a PIK3CA mutation PIK3CA:p.(Asn545Lys) or PIK3CA:p.(Arg108His), two had a fusion FGFR3::TACC3 or STRN::ALK), two displayed a MET amplification, and one exhibited a NRAS mutation NRAS:p.(Gln61Leu).
In terms of resistant models to FGFR2/3 TKI, PDX models were available for nine patients with a FGFR2 fusion, five with a FGFR3:p.(Ser249Cys) mutation, two harboring a FGFR3:p(Tyr373Cys) mutation, and one each harboring FGFR2:p.(Cys382Arg) and FGFR3:p.(Arg248Cys) mutations. Among these models, seven harbored on-target resistance mutations FGFR2:p.(Asn549Lys), FGFR2:p.(Asp650His), FGFR2:p.(Val565Phe), FGFR2:p.(Val564Leu), FGFR3:p.(Val754Met), four were established from patients presenting by-pass resistance mechanisms (PTEN, EGFR, NF1, PIK3CA), two PDX models from the same patient with an MSI profile presented multiple by-pass resistance mechanisms, and three models had unknown resistance mechanisms.
Resistant PDX models to ALK/ROS1 inhibitors, were available for eight patients with an EML4::ALK fusion, one patient with a CD74::ROS1 fusion, and one patient harboring a STRN::ALK fusion. Among these models, five harbored a single on-target resistance mutation ALK:p.(Gly1202Arg), ALK:p.(Val1180Leu), ALK:p.(Phe1174Leu), or ALK:p.(Leu1196Met), and one PDX model carried two on-target resistance mutations ALK:p.(Gly1202Arg) + ALK:p.(Thr1151Met). Moreover, two PDX models were established from patients presenting by-pass resistance mechanisms, NF2 skipping of exon 10 + NF2:p.(Lys543Asn) and NF1:p.(Lys1385Arg) and four models had unknown resistance mechanisms.
In addition to the previously mentioned results related to EGFR, FGFR2/3, and ALK, our team has successfully developed numerous PDX models that exhibit acquired resistance to targeted therapies (Fig. 4). As the development of KRAS inhibitors targeting the KRAS:p.(Gly12Cys) and KRAS:p.(Gly12Asp) mutations has more recently become a new challenge in the clinic in our KRAS cohort, we successfully developed 15 PDX models, including nine with the KRAS:p.(Gly12Cys) mutation and six with the KRAS:p.(Gly12Asp) mutation. Among these PDX models, four came from patients who acquired resistance to KRAS inhibitors. For two PDX models, by-pass resistance mechanisms were identified, involving a PIK3CA:p.(Glu545Lys) mutation and multiple gene amplifications. Furthermore, we have successfully established eight PDX models that exhibit resistance to various targeted therapies, including BET inhibitors, everolimus, EED inhibitors, NOTCH1 inhibitors, and HER2 inhibitors. Importantly, our research efforts have resulted in the development of PDX models derived from patients who have experienced resistance to PARP inhibitors such as olaparib, niraparib or rucaparib, ATR inhibitors such as ceralasertib, as well as to MAPK pathway inhibitors like vemurafenib, dabrafenib, or trametinib, and to NTRK inhibitors.
PDX closely reproduce patient’s tumor
As expected, the PDX models closely resembled the genetic tumors found in patients. In this study, when comparing pathogenic alterations, the mean concordance between the PDX models and the patients’ tumors reached 75.3% overall. When looking into details, the mean concordance was 100% for NTRK and HER2 models. The mean concordance was also notably high for ALK (90.0%), KRAS (82.0%), EGFR (81.0%), FGFR2/3 (81.0%), and BRAF models (72.0%) as depicted by Supplementary Fig. 2 and Supplementary Table 2.
For 40% (n = 55/136) of PDX models, in the frame of a collaboration with XenTech company, selective pressure was applied to the mice using the inhibitor to which the patient had developed resistance to validate its pharmacological status. Globally 84% (n = 46/55) of the PDX reproduced the pharmacological status of the patient’s tumor.
For example, PDX models MR7 and MR441-R, which exhibit resistance mechanisms involving MET amplification and the acquisition of EGFR:p.(Cys797Ser) respectively, both demonstrate resistance to osimertinib treatment (Fig. 5A). Within the PDX models of ALK-positive patients, PDX model MR619 displayed resistance to alectinib, consistent with the patient's clinical resistance profile marked by the acquisition of a secondary mutation, ALK:p.(Gly1202Arg) (Fig. 5B). Another PDX model, MR448-RE, exhibited lorlatinib resistance and retained the compound mutations, ALK:p.(Gly1202Arg) and ALK:p.(Thr1151Met) (Fig. 5B).
Fig. 5.

Pharmacological status of PDX models. In vivo pharmacological evaluation was performed by measuring the tumor volume in vehicle-treated and TKI-treated mice for: A. Two EGFR-resistant PDX models (MR7 and MR441) treated with 25 mg/kg of Osimertinib and vehicle. B. Two ALK-resistant PDX models (MR619, from cholangiocarcinoma, and MR448re, from lung) treated with 50 mg/kg of Alectinib and 20 mg/kg of Lorlatinib, respectively. C. Two FGFR3 bladder PDX models from the same patient: one from a biopsy in a stable disease site (MR86-SD) and one from a resistant disease site (MR86-PD), treated with 15 mg/kg of erdafitinib and vehicle. D. Two FGFR2-resistant cholangiocarcinoma PDX models (MR313 and MR369) treated with 1 mg/kg of Pemigatinib or vehicle
MR86-PD PDX model, treated with erdafitinib, revealed a PIK3CA mutation PIK3CA:p.(Glu545Lys) upon resistance analysis. This indicated a by-pass pathway resistance mechanism, confirmed by subsequent treatment response in the PDX model, not observed in PDX models obtained from stable disease sites (Fig. 5C). Patients MR313 and MR369 developed pemigatinib resistance due to secondary mutations FGFR2:p.(Asp650His) and FGFR2:p.(Val565Leu), respectively, with corresponding PDX models exhibiting pemigatinib resistance (Fig. 5D).
Overcoming strategies investigated in PDX models
The use of PDX models serves as a valuable and complementary approach in enhancing our understanding of resistance mechanisms and enables us to evaluate the efficacy of therapeutic combinations to overcome such resistance. Notably, we have conducted successful validations in four instances, demonstrating that combination therapy can effectively overcome resistance:
-
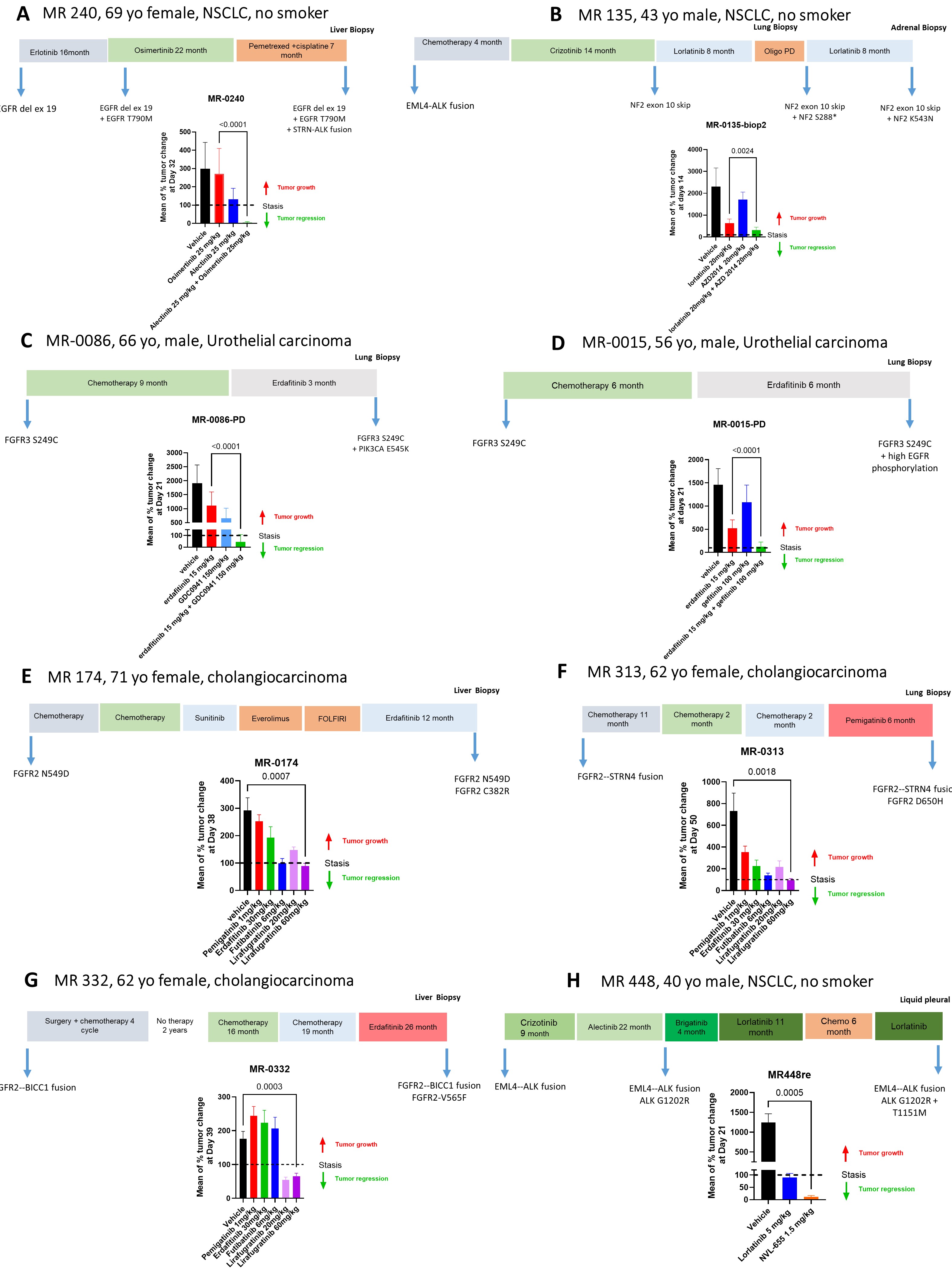
Case 1: MR240 (Supplementary Fig. 3A).
After 22 months of osimertinib treatment, patient MR240 developed STRN::ALK fusion-induced resistance. To explore alternatives, we tested alectinib and osimertinib combination efficacy using PDX models, revealing a robust response [14].
-
Case 2: MR135 (Supplementary Fig. 3B).
Double-hit alteration involving NF2 caused lorlatinib resistance in patient MR135. Investigating resistance mechanisms, we found that combining mTOR inhibitor vistusertib with lorlatinib in PDX models effectively overcame the resistance [13].
-
Case 3 and 4: MR15 and MR86 (Supplementary Fig. 3C and 3D).
Identifying by-pass pathways during erdafitinib treatment, MR15 model exhibited increased EGFR phosphorylation, while MR86 had a PIK3CA:p.(Glu545Lys) mutation. Targeting these abnormalities, we combined erdafitinib with gefitinib for MR15 and with PIK3CA inhibitor pictilisib for MR86, proving the efficacy of these tailored therapeutic combinations in both cases [12].
PDX models have also proven invaluable for assessing the efficacy of next-generation TKIs in overcoming resistance associated with secondary kinase domain mutations from previous generations. For instance, we recently demonstrated that the irreversible FGFR2-selective inhibitor lirafugratinib exhibited significant potency against PDXs established from biopsies of cholangiocarcinoma patients. These biopsies were collected at the onset of acquired resistance to reversible FGFR inhibitors and contained resistant secondary mutations, specifically FGFR2:p.(Asn549Asp) (Supplementary Fig. 3E), FGFR2:p.(Asp650His) (Supplementary Fig. 3F) and FGFR2:p.(Asn549Asp) (Supplementary Fig. 3G) [24].
Similarly, the latest generation ALK TKI, NVL-655, demonstrated remarkable activity against MR448re PDX that was established following resistance to lorlatinib due to compound ALK mutations ALK:p.(Gly1202Arg) + ALK:p.(Thr1151Met) (Supplementary Fig. 3H) [25].
Discussion
Advances in molecular profiling of tumors have revolutionized cancer treatment by allowing personalized treatment based on the unique characteristics of each patient's cancer. Several prospective studies have explored the feasibility of using real-time molecular findings to guide individualized treatment approaches. For instance, the BATTLE trial was the first biomarker-based study that required biopsies and demonstrated the feasibility of selecting personalized treatment based on molecular biomarkers, resulting in a disease control rate of 46% in chemoresistant NSCLC patients after 8 weeks [26]. In the MOSCATO-01 trial, heavily pre-treated patients achieved a clinical benefit to targeted therapies in 33% of cases, with treatment assignments based on tumor molecular profiling [27]. The WINTHER trial revealed a disease control rate of 26.2% when DNA sequencing or RNA expression was used to determine therapy in patients with colon, head and neck, and lung cancers [28].
The MATCH-R study broadens the scope by providing insights into acquired resistance mechanisms across diverse cancer types but also by adjusting the treatment strategy based on repeated molecular profiling of tumors once resistance occurs. By utilizing high-throughput sequencing techniques like WES and RNAseq, the study successfully identified a molecular resistance mechanism in 57.1% (n = 100/175) of patients undergoing targeted therapies. These results highlight the importance in clinical practice of performing re-biopsy in patients at each radiological progression when feasible. The strength of the MATCH-R clinical trial lies in the integration of these molecular analyses with comprehensive discussions held during molecular tumor board meetings, allowing for informed consideration of treatment options. Consequently, a new line of biology guided treatment was proposed for 45.0% (n = 45/100) of patients with progressive disease. This percentage is comparable to the 49% observed in the I-PREDICT study, which explored the feasibility of selecting a customized multidrug regimen upon the identification of multiple molecular alterations through DNA sequencing [29]. However, in some cases, despite the new line of treatment being specifically designed to target the identified resistance mechanism, no or short clinical response was observed. This lack of response may be due to the presence of subclonal populations that were missed by the molecular analysis of the tissue sample, likely as a result of tumor heterogeneity [30], or to alternative resistance mechanisms like epigenetic modifications not detectable by standard genetic analysis [31].
Looking into details of the resistance mechanisms identified, our data suggests that 25.1% (n = 44/175) of patients treated with targeted therapies will develop a by-pass resistance mechanism. One of the challenges in clinical practice is the development of new therapeutic strategies to effectively treat these patients. One suggested approach is to combine drugs that are already used in monotherapy. Combinations of targeted therapies have already demonstrated significant success, such as the combination of a BRAF inhibitor and MEK inhibitor in BRAF:p.(Val600Glu) mutated melanoma [32]. The data obtained from our PDX models indicate the potential efficacy of combining a therapy targeting the original oncogenic driver with another therapy targeting the by-pass mechanism. Previous studies [12–14, 33–36] have also demonstrated the utility of PDX models in testing these combinations. However, the challenge in clinical practice will be to manage the toxicities encountered and determine synergistic doses with minimal side effects between the compounds. PDX models can assist in pharmacokinetic/pharmacodynamics studies to validate the minimum doses required to achieve a tumor effect. Our PDX models have shown a mean similarity of 75.3% in terms of pathogenic driver, aligning closely with the 75.0% observed in another comprehensive pan cancer study focused specifically on PDX models [9]. Our extensive collection of PDX models will serve as a valuable preclinical tool for identifying crucial mechanisms that drive acquired resistance to current targeted therapies. Moreover, they offer opportunities for developing innovative strategies to overcome or preempt treatment failure. Currently, other therapeutic strategies, such as bi-specific antibodies, will enable us to simultaneously target the original oncogenic driver and the by-pass resistance mechanism. For instance, the use of amivantamab, a bispecific antibody targeting EGFR and MET [37], offers a promising approach to address this new clinical need.
One limitation of this study is that biopsy samples were not obtainable for 230 patients. However, an alternative approach could be the utilization of NGS on circulating tumor DNA (ctDNA). This method has already demonstrated clinical utility in capturing actionable alterations [38] and identifying resistance mechanisms to targeted therapies. A recent study reported a resistance mechanism in 34% of patients treated with targeted therapy using ctDNA-based NGS [39].
Another limitation of our study is the selection of patients for inclusion but also for PDX development that were impacted by investigator’s interest and the representation of tumor types does not therefore recapitulate the general cancer patient population. Similarly, the availability of therapies impacted on the number of driver genes represented in this study. For instance, the low percentage of KRAS mutations in our cohort relies on the unavailability of KRAS inhibitor at the time of patient inclusions (starting in 2015) and a rate closer to real incidence would be obtained if the study would be conducted now that several KRAS inhibitors are in clinical trials.
Finally, a deep characterization of the heterogeneity of cancer cell populations as well as microenvironment changes over the treatment courses were not feasible in MATCH-R using bulk sequencing of patient samples. Gustave Roussy recently implemented the UNLOCK program that will go a step forward into the longitudinal evolution of cancer during innovative treatments by performing single cell sequencing and spatial transcriptomics to better integrate these aspects in acquired resistance.
The MATCH-R study has facilitated advancements in the era of personalized medicine for multiple reasons. Firstly, it has served as an invaluable resource for scientists and clinicians by providing relevant samples from patients who have developed acquired resistance. For instance, specific studies have focused on resistance to androgen receptor inhibitors in prostate cancer [22, 23], and a novel predictive biomarker of immune-checkpoint blockade resistance called LIF has been discovered [40]. Additionally, the study has shed light on new resistance mechanisms for osimertinib [14, 41], lorlatinib [13], and dabrafenib-trametinib [42] in lung cancer patients with EGFR, ALK, and BRAF mutations, as well as for FGFR2/3 TKIs in urothelial cancer or cholangiocarcinoma patients with FGFR2/3 alterations [12], [24].
Secondly, MATCH-R now represents a substantial repository of high-throughput sequencing data. It includes 679 WES and 544 RNAseq data from patient biopsies, along with clinical information. Additionally, there are 94 WES and 77 RNAseq data available from matching patient-derived xenograft (PDX) models, all accessible through the EGA for further analysis (EGAD50000000697).
Furthermore, MATCH-R has been crucial in guiding participating patients towards molecularly targeted treatments. It has allowed them to benefit from tailored treatments after developing acquired resistance, thanks to the molecular profiling of their tumors. This personalized approach has significantly improved patient outcomes.
Lastly, the MATCH-R study has generated an almost unprecedented collection of molecularly characterized PDX models. This collection holds immense potential for future research and advancement in the field of personalized medicine.
Overall, the MATCH-R study has been a step forward in personalized medicine, providing invaluable samples, extensive sequencing data, tailored treatments, and a remarkable collection of PDX models.
Supplementary Information
{kind=link}
Additional file 1: Supplementary Figure 1: (A) PDX study flow chart by organ; (B) tumor cell content and engraftment success rate; (C) PDX study flow chart by molecular driver.
{kind=link}
Additional file 2: Supplementary Figure 2: Violin plot illustrating the molecular comparison of the variant pathogen between PDX models and their associated patients.
{kind=link}
Additional file 3: Supplementary Figure 3: Validation of therapeutic strategies in PDX models. Graphical representation of patients' treatment course and mean percent tumor volume change of single agents and combined treatments in resistant PDX models for: A. EGFR PDX (MR240) was treated with 25 mg/kg of osimertinib, 25 mg/kg of alectinib, either in monotherapy or in combination. Original publication in [14]. B. ALK PDX (MR135-Biop2) was treated with 20 mg/kg of lorlatinib and 20 mg/kg of an mTOR inhibitor (vistusertib), either in monotherapy or in combination. Original publication in [13]. C & D. Two FGFR3-positive PDX models, MR86-PD and MR15-PD, were treated with erdafitinib at 15 mg/kg. MR86-PD was also treated with a PIK3CA inhibitor (pictilisib) at 150 mg/kg, and MR15-PD was treated with gefitinib at 100 mg/kg, either in combination or as monotherapy. Original publication in [12]. E& F & G. PDXs from cholangiocarcinoma patients resistant to reversible FGFR inhibitors with secondary mutations treated by the indicated doses of FGFR inhibitors including lirafugratinib. Original publication in [24]. H. MR448re PDX resistance to lorlatinib treated with the ALK TKI, NVL-655. Re-analysis of data previously published in [25] included here for comparison only. For full data and information, please refer to the original publication in [25].
Additional file 4: Supplementary Table 1: List of the 857 successfully biopsied patients with their histology, molecular driver and availability of WES and RNAseq data.
Additional file 5: Supplementary Table 2: List of mutations identified in PDX and matching tumor.
Additional file 6: Supplementary File 1: Web page referencing available PDX models.
Acknowledgements
We thank XenTech company for performing pharmacological evaluation of PDX models in the frame of a collaboration. We thank Alexandre Halimi and Emilie Natali for processing the patient samples for tumor cell content and nucleic acid extractions.
Authors’ contribution
D.V., L.B., K.B., J.F.A., L.F. contributed to writing the main manuscript and preparing the figures. F.F., A.H., M.A., F.B.D., A.G., D.P., F.A., F.B., S.P., J.C.S., B.B., Y.L. were responsible for patient recruitment. J.C.S., F.A., B.B., Y.L., L.F., K.A.O. conceptualized the study. D.V., L.L., N.P.M. conducted the molecular analyses. L.T. was responsible for biopsy collection. L.B., C.B., A.D.S., F.B. developed the PDX models. C.N., M.N.C. gathered the clinical data. S.N., G.J.C. performed the bioinformatic analyses. S.M handled the statistical analysis. J.Y.S. reviewed the H&E histological slides. All authors reviewed and approved the manuscript.
Funding
The work of L. Friboulet is supported by an ERC Consolidator Grant (agreement number 101044047). The work of D. Vasseur is supported by Institut Servier and by Philippe Foundation.
Availability of data and materials
All PDX generated are listed with their molecular and clinical associated data on the following web page https://pdx.gustaveroussy.fr. All the WES/RNAseq raw data files from patient’s samples and PDX models included in this study are deposited at the European Genome–phenome Archive (EGA) using the accession code EGAD50000000697 (Supplementary Table 1). Access to this shared dataset is controlled by the institutional Data Access Committee, and requests for access can be sent to the corresponding author. Further information about EGA can be found at https://ega-archive.org/. Any additional information required to reanalyze the data reported in this article is available upon request by filling out the data request form for Gustave Roussy clinical trials at https://redcap.link/DataRequestClinicalTrialsGustaveRoussy. The steering committee and the sponsor will review the requests on a case-by-case basis. In case of approval, a specific agreement between the sponsor and the researcher may be required for data transfer.
Declarations
Ethics approval and consent to participate
The MATCH-R clinical trial (NCT02517892) was a monocentric, prospective study conducted between 2015 and 2022 and led by Gustave Roussy. This protocol was approved by the CPP (“Comité de protection des personnes”) and by the ANSM (“Agence nationale de sécurité du médicament et des produits de santé”) and adhered to the principles in the Guideline for Good Clinical Practice and the Declaration of Helsinki. Each individual genomic report was reviewed and discussed within a multidisciplinary internal review board. All animal procedures and studies were performed in accordance with the approved guidelines for animal experimentation by the ethics committee at University Paris Saclay (CEEA 26, Project 2014_055_2790) following the regulations set by the European Union. The animals were housed in pathogen-free conditions and provided unlimited food and water access.
Patients able to comply with the protocol were fully informed and signed an informed consent. Written informed consent was obtained from all patients included in this study.
Consent for publication
Not applicable.
Competing interests
D. Vasseur: speaker for Roche and Astra Zeneca JC. Soria: Full time employee of Amgen. Share-holder of Amgen. LF: Research funding from Debiopharm, Incyte, Relay Therapeutics, Sanofi and Nuvalent. F. Facchinetti : personal fees from BeiGene outside the submitted work. Y. Loriot : personal fees, nonfinancial support, and other support from Janssen during the conduct of the study, as well as personal fees, nonfinancial support, and other support from MSD, Pfizer, Merck KGaA, Astellas, Gilead, Bristol Myers Squibb, and Roche, nonfinancial support and other support from Incyte, other support from Exelixis, and personal fees and other support from Taiho outside the submitted work. S. Michiels : personal fees from DSMB or scientific committee study membership for Kedrion, Biophytis, Servier, IQVIA, Yuhan, and Roche outside the submitted work. L. Tselikas : personal fees from Incyte during the conduct of the study, as well as personal fees from GE Healthcare, Quantum Surgical, and Boston Scientific outside the submitted work. B. Besse : other support from AbbVie, BioNTech SE, Bristol Myers Squibb, Chugai Pharmaceutical, CureVac AG, Daiichi Sankyo, F. Hoffmann-La Roche Ltd, PharmaMar, Regeneron, Sanofi-Aventis, and Turning Point Therapeutics, other support from AbbVie, Eli Lilly and Company, Ellipses Pharma Ltd, F.Hoffmann-La Roche Ltd, Genmab, Immunocore, Janssen, MSD, Ose Immunotherapeutics, Owkin, and Taiho oncology, other support from AstraZeneca, BeiGene, Genmab A/S, GlaxoSmithKline, Janssen, MSD, Ose Immunotherapeutics, PharmaMar, Roche-Genentech, Sanofi, and Takeda, and other support from AbbVie, AstraZeneca, Chugai Pharmaceutical, Daichii Sankyo, Hedera Dx, Janssen, MSD, Roche, Sanofi-Aventis, and Springer Healthcare Ltd during the conduct of the study. F. André : grants from AstraZeneca, Guardant Health, Novartis, Owkin, Pfizer, Eli Lilly and Company, Roche, and Daiichi Sankyo, other support from Boston Pharmaceutics, Gilead, and Servier, and grants and other support from N-Power medicine outside the submitted work. A. Hollebecque : personal fees and nonfinancial support from Amgen, personal fees from Basilea, Bristol Myers and Squibb, Servier, Relay Therapeutics, Taiho, MSD, Seagen, grants and personal fees from Incyte, and nonfinancial support from Pierre Fabre outside the submitted work, as well as reports being a PI of the TransThera (Tinengotinib) phase III trial. A. Gazzah: travel accommodations, congress registration expenses from Boehringer Ingelheim, Novartis, Pfizer, and Roche. Consultant/Expert role for Novartis. Principal/sub-Investigator of Clinical Trials for Aduro Biotech, Agios Pharmaceuticals, Amgen, Argen-X Bvba, Arno Therapeutics, Astex Pharmaceuticals, Astra Zeneca, Aveo, Bayer Healthcare Ag, Bbb Technologies Bv, Beigene, Bioalliance Pharma, Biontech Ag, Blueprint Medicines, Boehringer Ingelheim, Bristol Myers Squibb, Ca, Celgene Corporation, Chugai Pharmaceutical Co., Clovis Oncology, Daiichi Sankyo, Debiopharm S.A., Eisai, Exelixis, Forma, Gamamabs, Genentech, Inc., Gilead Sciences, Inc., GlaxoSmithKline, Glenmark Pharmaceuticals, H3 Biomedicine, Inc., Hoffmann La Roche Ag, Incyte Corporation, Innate Pharma, Iris Servier, Janssen, Kura Oncology, Kyowa Kirin Pharm, Lilly, Loxo Oncology, Lytix Biopharma As, Medimmune, Menarini Ricerche, Merck Sharp & Dohme Chibret, Merrimack Pharmaceuticals, Merus, Millennium Pharmaceuticals, Nanobiotix, Nektar Therapeutics, Novartis Pharma, Octimet Oncology Nv, Oncoethix, Oncomed, Oncopeptides, Onyx Therapeutics, Orion Pharma, Oryzon Genomics, Pfizer, Pharma Mar, Pierre Fabre, Rigontec Gmbh, Roche, Sanofi Aventis, Sierra Oncology, Taiho Pharma, Tesaro, Inc., Tioma Therapeutics, Inc., Xencor. Research Grants from AstraZeneca, BMS, Boehringer Ingelheim, Janssen Cilag, Merck, Novartis, Pfizer, Roche, Sanofi. Non-financial support (drug supplied) from AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Johnson & Johnson, Lilly, Medimmune, Merck, NH TherAGuiX, Pfizer, Roche. D. Planchard: Consulting, advisory role or lectures: AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi Sankyo, Eli Lilly, Merck, MedImmune, Novartis, Pfizer, prIME Oncology, Peer CME, Roche. Honoraria: AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Eli Lilly, Merck, Novartis, Pfizer, prIME Oncology, Peer CME, Roche. Clinical trials research as principal or co-investigator (Institutional financial interests): AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, Pfizer, Roche, Medimmune, Sanofi Aventis, Taiho Pharma, Novocure, and Daiichi Sanky. Travel, Accommodations, Expenses: AstraZeneca, Roche, Novartis, prIME Oncology, Pfizer. M. Aldea: Expenses from Sandoz. No disclosures were reported by the other authors.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Damien Vasseur, Ludovic Bigot and Kristi Beshiri contributed equally to this work.
Contributor Information
Yohann Loriot, Email: yohann.loriot@gustaveroussy.fr.
Luc Friboulet, Email: luc.friboulet@gustaveroussy.fr.
References
- 1.Baroz AR, Mambetsariev I, Fricke J, Pharaon R, Tan T, Kidambi T, et al. Elevated Eosinophil Count Following Pembrolizumab Treatment for Non-Small Cell Lung Cancer. Cureus. 2021;13(7):e16266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jardim DL, Goodman A, de Melo GD, Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39(2):154–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee V, Murphy A, Le DT, Diaz LA. Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. Oncologist. 2016;21(10):1200–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goéré D, Mariani P, et al. Characterization of a Large Panel of Patient-Derived Tumor Xenografts Representing the Clinical Heterogeneity of Human Colorectal Cancer. Clin Cancer Res. 2012;18(19):5314–28. [DOI] [PubMed] [Google Scholar]
- 5.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–25. [DOI] [PubMed] [Google Scholar]
- 6.Katsiampoura A, Raghav K, Jiang ZQ, Menter DG, Varkaris A, Morelli MP, et al. Modeling of Patient-Derived Xenografts in Colorectal Cancer. Mol Cancer Ther. 2017;16(7):1435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guenot D, Guérin E, Aguillon-Romain S, Pencreach E, Schneider A, Neuville A, et al. Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J Pathol. 2006;208(5):643–52. [DOI] [PubMed] [Google Scholar]
- 8.Dudová Z, Conte N, Mason J, Stuchlík D, Peša R, Halmagyi C, et al. The EurOPDX Data Portal: an open platform for patient-derived cancer xenograft data sharing and visualization. BMC Genomics. 2022;23(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun H, Cao S, Mashl RJ, Mo CK, Zaccaria S, Wendl MC, et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment. Nat Commun. 2021;12(1):5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28(10):2595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cocco E, de Stanchina E. Patient-Derived-Xenografts in Mice: A Preclinical Platform for Cancer Research. Cold Spring Harb Perspect Med. 2024;14(7):a041381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Facchinetti F, Hollebecque A, Braye F, Vasseur D, Pradat Y, Bahleda R, et al. Resistance to Selective FGFR Inhibitors in FGFR-Driven Urothelial Cancer. Cancer Discov. 2023;13(9):1998–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Recondo G, Mezquita L, Facchinetti F, Planchard D, Gazzah A, Bigot L, et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin Cancer Res. 2020;26(1):242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Facchinetti F, Braye F, Yurchenko AA, Bigot L, Ponce S, et al. Single-cell DNA-seq depicts clonal evolution of multiple driver alterations in osimertinib-resistant patients. Ann Oncol. 2022;33(4):434–44. [DOI] [PubMed] [Google Scholar]
- 15.Recondo G, Mahjoubi L, Maillard A, Loriot Y, Bigot L, Facchinetti F, et al. Feasibility and first reports of the MATCH-R repeated biopsy trial at Gustave Roussy. NPJ Precis Oncol. 2020;4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suehnholz SP, Nissan MH, Zhang H, Kundra R, Nandakumar S, Lu C, et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024;14(1):49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017;1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Wang JX, Xue H, Lin D, Dong X, Gout PW, et al. Subrenal capsule grafting technology in human cancer modeling and translational cancer research. Differentiation. 2016;91(4–5):15–9. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bigot L, Sabio J, Poiraudeau L, Annereau M, Menssouri N, Helissey C, et al. Development of Novel Models of Aggressive Variants of Castration-resistant Prostate Cancer. European Urology Oncology. 2023;S2588931123002262. [DOI] [PubMed]
- 23.Menssouri N, Poiraudeau L, Helissey C, Bigot L, Sabio J, Ibrahim T, et al. Genomic Profiling of Metastatic Castration-Resistant Prostate Cancer Samples Resistant to Androgen Receptor Pathway Inhibitors. Clin Cancer Res. 2023;29(21):4504–17. [DOI] [PubMed] [Google Scholar]
- 24.Facchinetti F, Loriot Y, Braye F, Vasseur D, Bahleda R, Bigot L, et al. Understanding and Overcoming Resistance to Selective FGFR inhibitors Across FGFR2-Driven Malignancies. Clin Cancer Res. 2024. [DOI] [PMC free article] [PubMed]
- 25.Lin JJ, Horan JC, Tangpeerachaikul A, Swalduz A, Valdivia A, Johnson ML, et al. NVL-655 Is a Selective and Brain-Penetrant Inhibitor of Diverse ALK-Mutant Oncoproteins, Including Lorlatinib-Resistant Compound Mutations. Cancer Discov. 2024;OF1–20. [DOI] [PMC free article] [PubMed]
- 26.Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Tsao A, et al. The BATTLE Trial: Personalizing Therapy for Lung Cancer. Cancer Discov. 2011;1(1):44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massard C, Michiels S, Ferté C, Le Deley MC, Lacroix L, Hollebecque A, et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017;7(6):586–95. [DOI] [PubMed] [Google Scholar]
- 28.Rodon J, Soria JC, Berger R, Miller WH, Rubin E, Kugel A, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25(5):751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. 2016;13(6):335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wajapeyee N, Gupta R. Epigenetic Alterations and Mechanisms That Drive Resistance to Targeted Cancer Therapies. Cancer Res. 2021;81(22):5589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. 2012;367(18):1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Wu W, Cai C, Zhang H, Shen H, Han Y. Patient-derived xenograft models in cancer therapy: technologies and applications. Sig Transduct Target Ther. 2023;8(1):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med. 2019;25(9):1422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 2018;24(7):961–7. [DOI] [PubMed] [Google Scholar]
- 36.Yaeger R, Mezzadra R, Sinopoli J, Bian Y, Marasco M, Kaplun E, et al. Molecular Characterization of Acquired Resistance to KRASG12C-EGFR Inhibition in Colorectal Cancer. Cancer Discov. 2023;13(1):41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neijssen J, Cardoso RMF, Chevalier KM, Wiegman L, Valerius T, Anderson GM, et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J Biol Chem. 2021;296:100641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bayle A, Peyraud F, Belcaid L, Brunet M, Aldea M, Clodion R, et al. Liquid versus tissue biopsy for detecting actionable alterations according to the ESMO Scale for Clinical Actionability of molecular Targets in patients with advanced cancer: a study from the French National Center for Precision Medicine (PRISM). Ann Oncol. 2022;33(12):1328–31. [DOI] [PubMed] [Google Scholar]
- 39.Bayle A, Belcaid L, Palmieri LJ, Teysonneau D, Cousin S, Spalato-Ceruso M, et al. Circulating tumor DNA landscape and prognostic impact of acquired resistance to targeted therapies in cancer patients: a national center for precision medicine (PRISM) study. Mol Cancer. 2023;22(1):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loriot Y, Marabelle A, Guégan JP, Danlos FX, Besse B, Chaput N, et al. Plasma proteomics identifies leukemia inhibitory factor (LIF) as a novel predictive biomarker of immune-checkpoint blockade resistance. Ann Oncol. 2021;32(11):1381–90. [DOI] [PubMed] [Google Scholar]
- 41.Enrico D, Lacroix L, Chen J, Rouleau E, Scoazec JY, Loriot Y, et al. Oncogenic Fusions May Be Frequently Present at Resistance of EGFR Tyrosine Kinase Inhibitors in Patients With NSCLC: A Brief Report. JTO Clin Res Rep. 2020;1(2):100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Facchinetti F, Lacroix L, Mezquita L, Scoazec JY, Loriot Y, Tselikas L, et al. Molecular mechanisms of resistance to BRAF and MEK inhibitors in BRAFV600E non-small cell lung cancer. Eur J Cancer. 2020;132:211–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figure 1: (A) PDX study flow chart by organ; (B) tumor cell content and engraftment success rate; (C) PDX study flow chart by molecular driver.
Additional file 2: Supplementary Figure 2: Violin plot illustrating the molecular comparison of the variant pathogen between PDX models and their associated patients.
Additional file 3: Supplementary Figure 3: Validation of therapeutic strategies in PDX models. Graphical representation of patients' treatment course and mean percent tumor volume change of single agents and combined treatments in resistant PDX models for: A. EGFR PDX (MR240) was treated with 25 mg/kg of osimertinib, 25 mg/kg of alectinib, either in monotherapy or in combination. Original publication in [14]. B. ALK PDX (MR135-Biop2) was treated with 20 mg/kg of lorlatinib and 20 mg/kg of an mTOR inhibitor (vistusertib), either in monotherapy or in combination. Original publication in [13]. C & D. Two FGFR3-positive PDX models, MR86-PD and MR15-PD, were treated with erdafitinib at 15 mg/kg. MR86-PD was also treated with a PIK3CA inhibitor (pictilisib) at 150 mg/kg, and MR15-PD was treated with gefitinib at 100 mg/kg, either in combination or as monotherapy. Original publication in [12]. E& F & G. PDXs from cholangiocarcinoma patients resistant to reversible FGFR inhibitors with secondary mutations treated by the indicated doses of FGFR inhibitors including lirafugratinib. Original publication in [24]. H. MR448re PDX resistance to lorlatinib treated with the ALK TKI, NVL-655. Re-analysis of data previously published in [25] included here for comparison only. For full data and information, please refer to the original publication in [25].
Additional file 4: Supplementary Table 1: List of the 857 successfully biopsied patients with their histology, molecular driver and availability of WES and RNAseq data.
Additional file 5: Supplementary Table 2: List of mutations identified in PDX and matching tumor.
Additional file 6: Supplementary File 1: Web page referencing available PDX models.
Data Availability Statement
All the WES/RNAseq raw data files from patient’s samples and PDX models included in this study are deposited at the European Genome–phenome Archive (EGA) using the accession code EGAD50000000697 (Supplementary Table 1). Access to this shared dataset is controlled by the institutional Data Access Committee, and requests for access can be sent to the corresponding author. Further information about EGA can be found at https://ega-archive.org/. Any additional information required to reanalyze the data reported in this article is available upon request by filling out the data request form for Gustave Roussy clinical trials at https://redcap.link/DataRequestClinicalTrialsGustaveRoussy. The steering committee and the sponsor will review the requests on a case-by-case basis. In case of approval, a specific agreement between the sponsor and the researcher may be required for data transfer.
All PDX generated are listed with their molecular and clinical associated data on the following web page https://pdx.gustaveroussy.fr. All the WES/RNAseq raw data files from patient’s samples and PDX models included in this study are deposited at the European Genome–phenome Archive (EGA) using the accession code EGAD50000000697 (Supplementary Table 1). Access to this shared dataset is controlled by the institutional Data Access Committee, and requests for access can be sent to the corresponding author. Further information about EGA can be found at https://ega-archive.org/. Any additional information required to reanalyze the data reported in this article is available upon request by filling out the data request form for Gustave Roussy clinical trials at https://redcap.link/DataRequestClinicalTrialsGustaveRoussy. The steering committee and the sponsor will review the requests on a case-by-case basis. In case of approval, a specific agreement between the sponsor and the researcher may be required for data transfer.