

Key Points
-
•
PV with or without rituximab was tested in relapsed/refractory CLL, including after progression during treatment with a covalent BTKi.
-
•
These fixed-duration treatments were safe and showed promising efficacy in both covalent BTK-naïve and BTK-exposed patients.
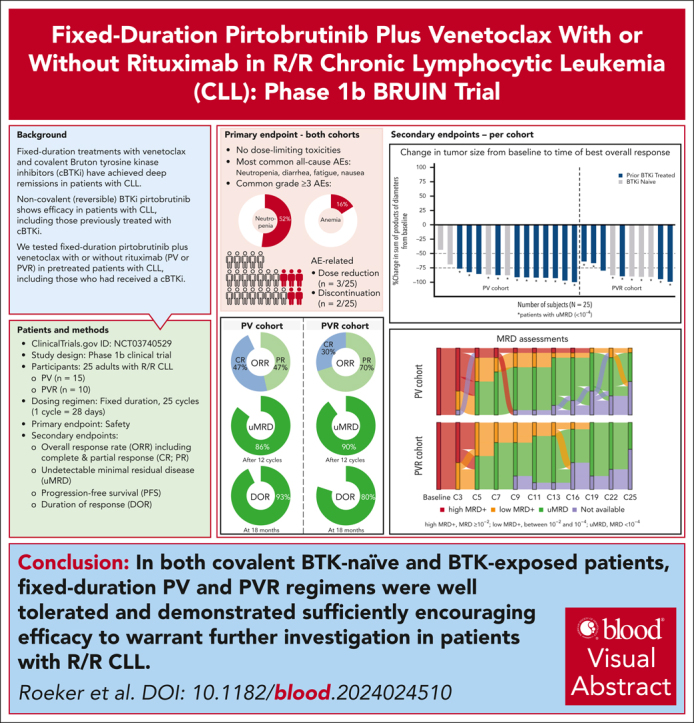
Visual Abstract
Abstract
Pirtobrutinib is a highly selective, noncovalent (reversible) Bruton tyrosine kinase inhibitor (BTKi). Patients with relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL) were treated with fixed-duration pirtobrutinib plus venetoclax (PV) or pirtobrutinib plus venetoclax and rituximab (PVR) in this phase 1b trial. Prior covalent BTKi therapy was allowed, but not prior treatment with venetoclax. Patients were assigned to receive PV (n = 15) or PVR (n = 10) for 25 cycles. Most patients (68%) had received prior covalent BTKi therapy. At the data cutoff date, the median time on study was 27.0 months for PV and 23.3 months for PVR. Overall response rates were 93.3% (95% confidence interval [CI], 68.1-99.8) for PV and 100% (95% CI, 69.2-100.0) for PVR, with 10 complete responses (PV: 7; PVR: 3). After 12 cycles of treatment, 85.7% (95% CI, 57.2-98.2) of PV and 90.0% (95% CI, 55.5-99.7) of PVR patients achieved undetectable minimal residual disease (<10-4) in peripheral blood. Progression-free survival at 18 months was 92.9% (95% CI, 59.1-99.0) for PV patients and 80.0% (95% CI, 40.9-94.6) for PVR patients. No dose-limiting toxicities were observed during the 5-week assessment period. The most common grade ≥3 adverse events (AEs) for all patients included neutropenia (52%) and anemia (16%). AEs led to dose reduction in 3 patients and discontinuation in 2. In conclusion, fixed-duration PV or PVR was well tolerated and had promising efficacy in patients with R/R CLL, including patients previously treated with a covalent BTKi. This trial was registered at www.clinicaltrials.gov as #NCT03740529.
Roeker and colleagues report the results of a phase 1b trial of the noncovalent Bruton tyrosine kinase inhibitor pirtobrutinib plus venetoclax with rituximab (PVR) or without rituximab (PV) for relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL). Therapy was well tolerated, and responses were excellent, with overall responses over 90% (93% for PV and 100% for PVR); 90% of patients treated with PVR achieved undetectable minimal residual disease in the peripheral blood. Both regimens have promising efficacy for R/R CLL.
Introduction
Chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL) are chronic malignancies characterized by clonal proliferation of mature B cells. For many years, chemotherapy in combination with an anti-CD20 monoclonal antibody was the cornerstone of early treatment options for patients with CLL/SLL. However, standard treatments now mainly consist of targeted therapies, including the covalent Bruton tyrosine kinase (BTK) inhibitors ibrutinib, acalabrutinib, and zanubrutinib,1, 2, 3 and the B-cell lymphoma 2 (BCL-2) inhibitor.4, 5, 6 Although monotherapies with targeted agents have improved the outcomes of patients with CLL/SLL, combination strategies have emerged to address some of the limitations of single-agent therapy.7
Covalent BTK inhibitors as single agents have been administered until toxicity or disease progression.8 Despite improvements in survival, the covalent BTK inhibitors alone rarely achieve a complete response (CR) or undetectable minimal residual disease (uMRD), thus necessitating their continuous administration to maintain efficacy.9 A major drawback of continuous dosing has been the potential for cumulative adverse events (AEs) that may negatively impact quality of life and the ability to adhere to treatment, and thereby may compromise the durability of disease control and outcomes for patients.10,11 Up to 40% of patients discontinue ibrutinib, primarily because of drug-related toxicity.12 Importantly, although both acalabrutinib and zanubrutinib have demonstrated lower incidences of grade ≥3 AEs than ibrutinib (acalabrutinib: 68.8% vs 74.9%; zanubrutinib: 67.3% vs 70.4%, respectively), ∼15% of patients still discontinued acalabrutinib or zanubrutinib because of drug-related AEs. These AEs occurred during a median treatment duration of ∼38 months for acalabrutinib and ∼28 months for zanubrutinib.2,13
Another challenge associated with continuous treatment with a covalent BTK inhibitor remains acquired treatment resistance and consequent disease progression,14,15 whereas fixed-duration regimens may be less likely to generate such resistance mutations.16 In addition, surveys of adults with CLL indicate that often patients prefer fixed-duration treatment regimens over continuous dosing.17, 18, 19 Clinical trials of venetoclax combined with an anti-CD20 antibody demonstrated that a fixed-duration treatment strategy was a viable option.4,5 Furthermore, venetoclax combined with covalent BTK inhibitors in doublet fixed-duration regimens has also shown promising efficacy in both phase 2 and 3 trials.20, 21, 22, 23, 24 More recently, fixed-duration triplet regimens combining venetoclax, a covalent BTK inhibitor, and an anti-CD20 antibody have demonstrated promising outcomes; however, these triplet combinations have not been proven to be superior to doublet combinations.25, 26, 27 Additionally, the AE profiles of these regimens are an important consideration that may limit their use. Cardiac events occurred more commonly among patients treated with the combination of venetoclax and ibrutinib, likely due to ibrutinib, than among patients treated with chemoimmunotherapy.21,23,28 Cytopenias were less common with venetoclax and ibrutinib than with chemoimmunotherapy, and infection rates varied across studies and were either comparable or slightly higher with venetoclax and ibrutinib than with chemoimmunotherapy.21,29 In contrast, infection rates were higher with ibrutinib, venetoclax, and obinutuzumab than with venetoclax and an anti-CD20 antibody.27 Single-arm studies of triplet combinations have demonstrated a risk of cytopenias, in addition to AEs reported with monotherapy, such as headache, fatigue, bruising, nausea, diarrhea, infusion-related reactions, hypertension, arthralgias, and infection.30
Pirtobrutinib is a noncovalent (reversible) BTK inhibitor that demonstrates low nanomolar potency against both wild-type and C481-mutant BTK, the most common mutation associated with resistance to covalent BTK inhibitors.31 Pirtobrutinib monotherapy is administered continuously until toxicity or disease progression. In the BRUIN phase 1/2 clinical trial, 247 patients with CLL/SLL who had previously received a covalent BTK inhibitor were treated with continuous pirtobrutinib monotherapy and had an overall response rate (ORR) of 82.2%.32 Treatment with pirtobrutinib monotherapy was associated with mainly low-grade AEs. The most common grade ≥3 AEs were infections (28.1%) and neutropenia (26.8%). The incidences of grade ≥3 atrial fibrillation (1.3%), major hemorrhage (2.2%), and hypertension (3.5%) were low. AEs were manageable, leading to low dose reduction and treatment discontinuation rates (4.7% and 2.8%, respectively). This favorable safety profile is consistent with pirtobrutinib’s high degree of BTK selectivity and minimal off-target inhibition.33 On the basis of these findings, pirtobrutinib monotherapy was approved in 2023 by the US Food and Drug Administration for the treatment of adults with CLL/SLL who have received at least 2 prior lines of systemic therapy, including a BTK inhibitor and a BCL-2 inhibitor.34,35
The observed safety profile and clinical activity of pirtobrutinib monotherapy makes pirtobrutinib an attractive candidate to combine with venetoclax and anti-CD20 antibodies. In this phase 1b clinical trial, we evaluated the safety and efficacy of fixed-duration pirtobrutinib in combination with either venetoclax (PV) or venetoclax and rituximab (PVR) in patients with previously treated CLL/SLL, including those who had received a covalent BTK inhibitor.
Methods
Study design and treatment
The primary objective of the phase 1b portion of the BRUIN trial was to evaluate the safety of fixed-duration pirtobrutinib plus venetoclax, with or without rituximab, in adults with relapsed or refractory (R/R) CLL/SLL. The first 6 patients were enrolled in each cohort according to a 3 + 3 design to determine the safety of combination treatments. Patients received treatment for 25 cycles or until disease progression, unacceptable toxicity, or withdrawal. Each treatment cycle was 28 days. Briefly, starting in cycle 1, oral pirtobrutinib was administered daily at the approved dose of 200 mg.36 Venetoclax and rituximab delivery were each based on the labeled CLL dose.37,38 To minimize the risk of tumor lysis syndrome (TLS), the first treatment cycle consisted of pirtobrutinib (with or without rituximab) to debulk the tumor before initiation of venetoclax. Venetoclax was started in cycle 2 at 20 mg per day and escalated weekly over 5 weeks to the target dose of 400 mg per day, as per the standard ramp-up schedule. Rituximab was administrated intravenously at 375 mg/m2 for cycle 1 and then at 500 mg/m2 for cycles 2 to 6. Both pirtobrutinib and venetoclax were given in combination through cycle 25, for a total of 24 cycles. After treatment discontinuation, patients were followed up approximately every 3 months for up to 2 years.
The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guideline, the principles of the Declaration of Helsinki, and all other applicable regulatory requirements. The protocol was approved by institutional review boards or independent ethics committees of all participating institutions. All patients provided written informed consent. This study was registered with ClinicalTrials.gov (NCT03740529).
Patient eligibility
Adults (≥18 years) were eligible for this study if they had R/R CLL/SLL and if venetoclax or venetoclax plus rituximab would be an appropriate treatment. Prior anti-CD20 antibody and/or covalent BTK inhibitor therapy was allowed; however, prior BCL-2 inhibitor use was not permitted. Other eligibility criteria included adequate hematologic status within 7 days of starting study treatment; Eastern Cooperative Oncology Group status from 0 to 2; and adequate hepatic and renal function. Patients were excluded if they had received a hematopoietic stem cell transplant or chimeric antigen receptor T-cell therapy within 60 days before starting study treatment. Patients were also excluded if they had known central nervous system involvement, significant cardiovascular disease, history of a major bleeding event with a prior BTK inhibitor, or current prescription for warfarin. Patients currently receiving anticoagulation therapy with warfarin were excluded, but other anticoagulants or antiplatelets were allowed. Baseline mutations in TP53, BTK, and PLCG2 were assessed using targeted next-generation sequencing panels (NeoGenomics Laboratories, Inc, Fort Myers, FL). The presence of TP53 mutation had a limit of detection of 5% variant allele frequency.
Safety assessment
The primary end point was safety as assessed by the frequency and severity of AEs. AEs were graded according to the Common Terminology Criteria for Adverse Events, version 5.0. AEs were coded using the Medical Dictionary for Regulatory Activities, version 24.0. The dose-limiting toxicity (DLT) assessment period occurred from the first administration of all agents in the combination regimen through the first 5 weeks of treatment (ie, cycle 2 day 1 to cycle 3 day 8). DLT was defined as any grade ≥3 nonhematologic or hematologic toxicity (with exceptions as defined in the supplemental Methods, available on the Blood website) or any-grade toxicity resulting in discontinuation, dose reduction, or administration of <75% of the planned doses. For a combination to be considered safe for further study, a DLT rate <33% was required (ie, 0 of 3 or 1 of 6 patients). A safety review committee determined whether additional patients or doses should be evaluated on the basis of the DLT rate in the first 6 enrolled patients receiving combination treatment. If the safety review committee deemed the initial dose safe for the first 6 enrolled patients, additional patients could be enrolled to each dosing combination at this dose to further investigate tolerability, efficacy, and pharmacokinetics (PK). Patients were first enrolled to receive PV to assess the safety of dual combination treatment. After PV was deemed safe in the first 3 patients who completed the DLT assessment period, patients were then enrolled to receive PVR. The final enrolled number of patients who received PV (n = 15) or PVR (n = 10) was considered sufficient to gain preliminary assessment of the primary end point of safety.
Tolerability of pirtobrutinib was assessed by relative dose intensity and calculated by dividing the actual dose intensity (milligram per day) by the planned dose intensity (milligram per day), until treatment discontinuation.
Efficacy assessment
Secondary efficacy end points included ORR, best overall response, duration of response, progression-free survival (PFS), overall survival (OS), and uMRD rate. All response end points were assessed by investigators according to criteria from the 2018 International Workshop on Chronic Lymphocytic Leukemia (iwCLL).39 ORR was defined as the proportion of patients with a CR or partial response (PR). Response assessments were performed by computed tomography every 2 cycles for the first year, every 3 cycles for the second year, and every 6 months thereafter. Hematologic or radiologic CRs were confirmed by bone marrow biopsy. Baseline molecular characteristics were assessed centrally. Blood was collected to assess MRD at screening, every 2 cycles from cycle 3 to cycle 12, and then every 3 cycles from cycle 13 to cycle 25. uMRD in peripheral blood was defined as <1 CLL cell per 10 000 nucleated cells (10–4 sensitivity) via clonoSEQ assay (Adaptive Biotechnologies, Seattle, WA).40 The PK of pirtobrutinib and venetoclax were evaluated for the 2 combination treatments. Blood samples were collected serially for PK analyses on cycle 1 day 8, cycle 2 day 8, cycle 3 day 8, and cycle 4 day 1.
Statistical analysis
The data cutoff date for safety and efficacy analyses was 5 May 2023. Safety and efficacy were assessed in all patients who received at least 1 dose of the study drug. Descriptive statistics were used to summarize the findings. ORR was calculated with a 2-sided 95% confidence interval (CI) based on the exact binomial distribution. Distributions of time-to-event end points were estimated using the Kaplan-Meier method. SAS (version 9.4; SAS Institute, Cary, NC) was used for statistical analyses.
The data cutoff date for PK analyses was 5 February 2023. PK analysis of pirtobrutinib and venetoclax was performed using noncompartmental methods in Phoenix version 8.3.5 (Certara, Princeton, NJ). Using Phoenix, the following PK parameters were calculated from plasma concentrations if appropriate: maximum drug concentration (Cmax), time to maximum plasma concentration (Tmax), area under the concentration vs time curve from time 0 to t (AUC0–t), AUC from time 0 extrapolated to infinity (AUC0–∞), clearance or apparent oral clearance, volume of distribution or apparent volume of distribution, and terminal elimination half-life. Descriptive statistics were used to summarize PK data.
Results
Study population and disposition
Between August 2020 and July 2021, 25 patients with CLL in the United States and Australia were enrolled to receive treatment with PV (n = 15) or PVR (n = 10) (Table 1). No patients with SLL were enrolled. The median age for all patients was 66 years (range, 39-78). Most patients were male (PV: 87%; PVR: 70%). The median prior lines of therapy for all patients were 2 (range, 1-4). All patients had an Eastern Cooperative Oncology Group functional performance status of either 0 (56%) or 1 (44%). Most patients had received previous treatment with anti-CD20 antibody therapy (72%), covalent BTK inhibitor therapy (68%), and chemotherapy (56%). Among the 17 patients who received a previous covalent BTK inhibitor, 12 (71%) had discontinued treatment of the prior BTK inhibitor because of progressive disease and the other 5 (29%) because of toxicity. At baseline, high-risk cytogenetic and molecular features among patients with available pretreatment peripheral blood samples for central assessment identified BTK C481 mutation in 9 of 23 (39%), PLCG2 mutation in 1 of 23 (4%), 17p deletion in 4 of 20 (20%), TP53 mutation in 6 of 23 (26%), both 17p deletion and/or TP53 mutation in 7 of 19 (37%), 11q deletion in 8 of 20 (40%), and unmutated IGHV in 16 of 20 (80%) patients (Table 1).
Table 1.
Patient characteristics at baseline
| PV (n = 15) | PVR (n = 10) | Total (N = 25) | |
|---|---|---|---|
| Age in y, median (range) | 66 (49-77) | 69 (39-78) | 66 (39-78) |
| Sex, n (%) | |||
| Female | 2 (13) | 3 (30) | 5 (20) |
| Male | 13 (87) | 7 (70) | 20 (80) |
| ECOG PS | |||
| 0 | 7 (47) | 7 (70) | 14 (56) |
| 1 | 8 (53) | 3 (30) | 11 (44) |
| Mutation status, n (%)∗ | |||
| BTK C481 mutant | 6 (40) | 3 (38) | 9 (39) |
| BTK C481 wild type | 9 (60) | 5 (63) | 14 (61) |
| PLCG2 mutation status, n (%)† | |||
| Yes | 1 (7) | 0 (0) | 1 (4) |
| No | 14 (93) | 8 (100) | 22 (96) |
| del(17p) by FISH, n (%)‡ | |||
| Yes | 1 (9) | 3 (33) | 4 (20) |
| No | 10 (91) | 6 (67) | 16 (80) |
| TP53 mutation status, n (%)§ | |||
| Mutated | 3 (20) | 3 (38) | 6 (26) |
| Unmutated | 12 (80) | 5 (63) | 17 (74) |
| del(17p) and/or TP53 mutation status, n (%)|| | |||
| Yes | 3 (27) | 4 (50) | 7 (37) |
| No | 8 (73) | 4 (50) | 12 (63) |
| del(11q) by FISH, n (%)¶ | |||
| Yes | 4 (36) | 4 (44) | 8 (40) |
| No | 7 (63) | 5 (56) | 12 (60) |
| IGHV mutation status, n (%)# | |||
| Mutated | 3 (27) | 1 (11) | 4 (20) |
| Unmutated | 8 (73) | 8 (89) | 16 (80) |
| Prior lines of systemic therapy, median (range) | |||
| All therapies | 1 (1-2) | 2 (1-4) | 2 (1-4) |
| BTKi therapies | 1 (0-1) | 1 (0-2) | 1 (0-2) |
| Prior therapies, n (%) | |||
| Anti-CD20 antibody∗∗ | 11 (73) | 7 (70) | 18 (72) |
| BTKi | 11 (73) | 6 (60) | 17 (68) |
| Chemotherapy | 8 (53) | 6 (60) | 14 (56) |
| PI3K agent | 1 (7) | 2 (20) | 3 (12) |
| Reasons for BTKi discontinuation, n (%)†† | |||
| Progressive disease | 8 (73) | 4 (67) | 12 (71) |
| Toxicity | 3 (27) | 2 (33) | 5 (29) |
Total percentage might be different than the sum of the individual components because of rounding. Molecular features were analyzed by a central research laboratory. Percentages were calculated for patients with available data.
BTKi, Bruton tyrosine kinase inhibitor; ECOG PS, Eastern Cooperative Oncology Group Performance Status; FISH, fluorescence in situ hybridization; IGHV, immunoglobulin heavy variable; PI3K, phosphatidylinositol 3-kinase.
2 patients had missing data (PV: 0; PVR: 2).
2 patients had missing data (PV: 0; PVR: 2).
5 patients had missing data (PV: 4; PVR: 1).
2 patients had missing data (PV: 0; PVR: 2).
6 patients had missing data (PV: 4; PVR: 2).
5 patients had missing data (PV: 4; PVR: 1).
5 patients had missing data (PV: 4; PVR: 1).
Includes both patients who had anti-CD20 as monotherapy and as combination therapy.
Calculated as the percentage of patients who received prior BTK inhibitor. If >1 reason was noted for discontinuation, disease progression took priority.
At the time of the data cutoff, median time on treatment was 23.0 months (interquartile range [IQR], 22.0-23.3) for all patients, 22.8 (IQR, 20.5-23.1) months for PV, and 23.0 (IQR, 21.9-23.3) months for PVR. The median time on study was 25.2 months (IQR, 23.3-28.3) for all patients, 27.0 (IQR, 24.7-32.1) months for PV, and 23.3 (IQR, 21.9-24.2) months for PVR. As of the data cutoff, treatment was ongoing (>21 months) in 3 patients (PV: 1; PVR: 2) and had been discontinued in 22 patients (PV: 14; PVR: 8). Of these 22 patients, 14 (PV: 9; PVR: 5) completed all 24 cycles of combination therapy. Treatment was discontinued early in 8 patients (PV: 5; PVR: 3) because of progressive disease (PV: 2), AEs (PVR: 2), death due to AE that was considered unrelated to treatment by the investigator (PVR: 1), or other reasons (PV: 3) (supplemental Table 1). Three additional deaths occurred, 1 after discontinuing treatment for progressive disease, 1 after discontinuing treatment for AE, and 1 after the completion of the planned 25 cycles of therapy.
Safety
No DLTs were observed in either treatment combination during the per-protocol 5-week observation period. The incidence of DLTs during 8 weeks of combination treatment was also reviewed in the first 6 patients treated with PV or PVR, and no DLTs were observed. Two cases of grade ≥3 neutrophil count decrease (PV: 1; PVR: 1) were observed during this 8-week period, which did not meet the DLT criteria. All treatment-emergent AEs within 12 months after the initial dose of venetoclax were retrospectively analyzed for the first 6 patients in each cohort for further safety validation. Seven AEs that would have been classified as DLTs were observed among 3 patients. One patient experienced grade 5 COVID-19 pneumonia; 1 patient experienced grade 3 cerebral hemorrhage, diabetic ketoacidosis, and grade 5 cardiac failure; and 1 patient experienced grade 3 back pain, bone pain, and noncardiac chest pain. All patients were receiving PVR, and only the grade 3 cerebral hemorrhage was deemed related to treatment. The median relative dose intensity for all patients was 98.4% for pirtobrutinib, 98.4% for venetoclax, and 100% for rituximab. The most common AEs and AEs of special interest (ie, those previously associated with BTK inhibitors) are shown in Table 2., Table 3., respectively. The most common all-cause AEs were nausea (60%), fatigue (53%), neutropenia (47%), and diarrhea (47%) for patients receiving PV, and neutropenia (70%), diarrhea (60%), fatigue (50%), and nausea (40%) for patients receiving PVR. Neutropenia was the most common all-cause grade ≥3 AE (PV: 47%; PVR: 60%). All-cause grade ≥3 AEs of special interest included hypertension (PV: 7%; PVR: 10%), bleeding (PV: 0%; PVR: 10%), and serious infections (PV: 27%; PVR: 40%). No atrial arrhythmias were observed. With the exception of neutropenia, treatment-related grade ≥3 AEs were uncommon. One PV patient developed grade 4 TLS during dose escalation to 400-mg venetoclax (supplemental Results). Symptoms resolved after intravenous fluids and temporary dialysis for refractory hyperkalemia, and the patient achieved the planned venetoclax target dose of 400 mg daily by cycle 3 day 1 and completed all 24 cycles of combination therapy at this dose.
Table 2.
AEs in 15 patients treated with PV
| All-cause AEs (≥25%), % |
Treatment-related AEs, % |
|||
|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| AE | ||||
| Neutropenia∗ | 7 (46.7) | 7 (46.7) | 7 (46.7) | 7 (46.7) |
| Nausea | 9 (60.0) | 0 (0.0) | 7 (46.7) | 0 (0.0) |
| Fatigue | 8 (53.3) | 0 (0.0) | 5 (33.3) | 0 (0.0) |
| Diarrhea | 7 (46.7) | 2 (13.3) | 4 (26.7) | 2 (13.3) |
| Hypophosphatemia | 5 (33.3) | 0 (0.0) | 2 (13.3) | 0 (0.0) |
| Constipation | 4 (26.7) | 0 (0.0) | 3 (20.0) | 0 (0.0) |
| Cough | 4 (26.7) | 0 (0.0) | 1 (6.7) | 0 (0.0) |
| Platelet count decreased | 4 (26.7) | 1 (6.7) | 4 (26.7) | 1 (6.7) |
| Vomiting | 4 (26.7) | 0 (0.0) | 2 (13.3) | 0 (0.0) |
| AEs of interest† | ||||
| Infections‡ | 12 (80.0) | 4 (26.7) | 7 (46.7) | 3 (20.0) |
| Arthralgia | 4 (26.7) | 0 (0.0) | 2 (13.3) | 0 (0.0) |
| Bruising§ | 3 (20.0) | 0 (0.0) | 3 (20.0) | 0 (0.0) |
| Hypertension | 3 (20.0) | 1 (6.7) | 2 (13.3) | 0 (0.0) |
| Rash|| | 1 (6.7) | 0 (0.0) | 1 (6.7) | 0 (0.0) |
| Hemorrhage¶ | 1 (6.7) | 0 (0.0) | 1 (6.7) | 0 (0.0) |
| Atrial fibrillation/flutter# | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Aggregate of neutropenia and neutrophil count decrease.
AEs of interest are those that were previously associated with covalent BTK inhibitors regardless of occurrence rate.
Aggregate of all preferred terms indicating infection and including COVID-19. Infection-related grade 3 to 4 (no grade 5) AEs included COVID-19 pneumonia, pneumonia, oral candidiasis, sepsis, soft tissue infection, and abscess.
Aggregate of contusion, petechiae, ecchymosis, and increased tendency to bruise.
Aggregate of all preferred terms including rash.
Aggregate of all preferred terms including hemorrhage or hematoma.
Aggregate of atrial fibrillation and atrial flutter.
Table 3.
AEs in 10 patients treated with PVR
| All-cause AEs (≥25%), % |
Treatment-related AEs, % |
|||
|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| AE | ||||
| Neutropenia∗ | 7 (70.0) | 6 (60.0) | 7 (70.0) | 6 (60.0) |
| Diarrhea | 6 (60.0) | 0 (0.0) | 6 (60.0) | 0 (0.0) |
| Fatigue | 5 (50.0) | 1 (10.0) | 2 (20.0) | 0 (0.0) |
| Nausea | 4 (40.0) | 0 (0.0) | 4 (40.0) | 0 (0.0) |
| Constipation | 4 (40.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) |
| Infusion-related reaction | 4 (40.0) | 2 (20.0) | 4 (40.0) | 2 (20.0) |
| Anemia | 3 (30.0) | 1 (10.0) | 2 (20.0) | 1 (10.0) |
| Back pain | 3 (30.0) | 1 (10.0) | 1 (10.0) | 0 (0.0) |
| Chills | 3 (30.0) | 0 (0.0) | 3 (30.0) | 0 (0.0) |
| Dyspnea | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Cough | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dry mouth | 3 (30.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) |
| AEs of interest† | ||||
| Infections‡ | 9 (90.0) | 4 (40.0) | 2 (20.0) | 1 (10.0) |
| Bruising§ | 2 (20.0) | 0 (0.0) | 2 (20.0) | 0 (0.0) |
| Rash|| | 3 (30.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) |
| Arthralgia | 4 (40.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) |
| Hemorrhage¶ | 2 (20.0) | 1 (10.0) | 1 (10.0) | 1 (10.0) |
| Hypertension | 1 (10.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) |
| Atrial fibrillation/flutter# | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Aggregate of neutropenia and neutrophil count decrease.
AEs of interest are those that were previously associated with covalent BTK inhibitors regardless of occurrence rate.
Aggregate of all preferred terms indicating infection and including COVID-19. Infection-related grade 3-4 (no grade 5) AEs included COVID-19, COVID-19 pneumonia, urinary tract infection, cellulitis, and cystitis.
Aggregate of contusion, petechiae, ecchymosis, and increased tendency to bruise.
Aggregate of all preferred terms including rash.
Aggregate of all preferred terms including hemorrhage or hematoma.
Aggregate of atrial fibrillation and atrial flutter.
Treatment-related dose reductions occurred in 3 patients (PV:1; PVR: 2). The dose of pirtobrutinib for the patient in the PV group was reduced because of diarrhea and platelet count decrease. Doses of both pirtobrutinib and venetoclax were reduced in 2 patients in the PVR group because of platelet count decrease in 1 case and neutropenia in the other. Neutropenia also led to dose interruptions in 7 patients (PV: 5; PVR: 2) and was treated with growth-factor support in 10 patients (PV: 4; PVR: 6). Treatment was discontinued early in 2 patients because of treatment-related neutropenia or urinary tract infection (PV: 0; PVR: 2) (supplemental Table 1). No treatment-related discontinuations or deaths were observed among patients receiving PV.
Efficacy
All 25 patients were evaluable for efficacy, and the ORR was 96.0% (95% CI, 79.6-99.9), with 10 CRs and 14 PRs (Table 4). ORR was 93.3% (95% CI, 68.1-99.8) for the 15 patients receiving PV, and 100% (95% CI, 69.2-100.0) for the 10 patients receiving PVR. CR rates were 46.7% (95% CI, 21.3-73.4) for patients receiving PV and 30.0% (95% CI, 6.7-61.3) for patients receiving PVR. Overall, 17 patients had previously been treated with a covalent BTK inhibitor, and 8 were naïve to BTK inhibitor therapy. Among the patients with prior exposure to a BTK inhibitor, 8 achieved CR and 9 PR, and among those who were BTK inhibitor–naïve, 2 achieved CR, 5 PR, and 1 stable disease. Nine patients had a BTK C481s mutation, with 4 achieving CR and 5 PR. The 1 patient with a PLCG2 mutation achieved CR. Overall median time to best response was 2.4 months (IQR, 1.9-14.3), corresponding to the time of the first disease assessment. Tumor size of target lesions decreased by at least 50% from baseline for 96% of patients and by at least 75% from baseline for 84% of patients (Figure 1). The decrease in tumor size by at least 75% was similar among the 17 patients previously treated with a BTK inhibitor (88%) and the 8 patients who were BTK inhibitor–naïve (75%) (Figure 1). Median duration of follow-up for response was 20.3 (IQR, 18.4-21.2) months for all patients, 20.3 (IQR, 18.4-23.9) months for PV patients, and 20.3 (IQR, 18.9-20.3) months for PVR patients. Duration of response estimates at 18 months were 87.5% (95% CI, 66.1-95.8) for all patients, 92.9% (95% CI, 59.1-99.0) for PV patients, and 80.0% (95% CI, 40.9-94.6) for PVR patients.
Table 4.
Best overall response of pirtobrutinib-based combination treatments
| PV (n = 15) |
PVR (n = 10) |
Total (N = 25) |
|
|---|---|---|---|
| 15 | 10 | 25 | |
| ORR, % (95% CI) | 93.3 (68.1-99.8) | 100 (69.2-100) | 96.0 (79.6-99.9) |
| Best response, n (%)∗ | |||
| CR | 7 (46.7) | 3 (30.0) | 10 (40.0) |
| PR | 7 (46.7) | 7 (70.0) | 14 (56.0) |
| SD | 1 (6.7) | 0 | 1 (4.0) |
| PD | 0 | 0 | 0 |
| Median time to best response, mo (IQR) | 3.9 (1.9- 14.7) | 1.9 (1.8- 10.7) | 2.4 (1.9-14.3) |
The data cutoff date was 5 May 2023.
NE, not evaluable; PD, progressive disease; SD, stable disease.
ORR is the number of patients with a best response of CR or PR divided by the total number of patients. Response status per iwCLL 2018 as assessed by the investigator.
Figure 1.

Change in tumor size from baseline to the time of best overall response. Tumor size was calculated as the sum of product diameters of the target lesions. Asterisks (∗) indicate patients who achieved uMRD (<10–4).
Median duration of follow-up for PFS was 22.1 (IQR, 20.1-23.0) months for all patients, 22.1 (IQR, 20.1-25.7) months for PV, and 22.1 (IQR, 20.7-22.1) months for PVR. Median PFS and OS were not reached for either the doublet or triplet combination. PFS and OS estimates at 24 months for all 25 patients were 79.5% (95% CI, 52.0-92.3) and 87.5% (95% CI, 66.1-95.8), respectively. PFS and OS estimates at 24 months for PV were 79.6% (95% CI, 37.1-94.9) and 92.9% (95% CI, 59.1-99.0), respectively (Figure 2A,C). The PFS estimate at 24 months for PVR patients was not evaluable, whereas the PFS estimate at 18 months was 80.0% (95% CI, 40.9-94.6), and the OS estimate at 24 months was 80.0% (95% CI, 40.9-94.6) (Figure 2B,D).
Figure 2.

PFS and OS rates. Kaplan-Meier curves for PFS (A-B) and OS (C-D) for patients treated with pirtobrutinib-based combination therapies. Tick marks indicate censored data.
MRD
Twenty-four patients were evaluable for MRD assessment in peripheral blood (PV: 14; PVR: 10). Median time to first uMRD, assessed at a sensitivity threshold of 10–4, was 4.3 months for patients receiving PV (n = 14) and 3.7 months for patients receiving PVR (n = 10). The uMRD rate after completion of cycle 12 was 70.8% for the 24 evaluable patients, including 67% of patients receiving PV and 70% of patients receiving PVR (Figure 3). During treatment, uMRD was achieved by 85.7% (95% CI, 57.2-98.2) of patients receiving PV and 90.0% (95% CI, 55.5-99.7) of patients receiving PVR (Figure 3). Among patients who achieved uMRD, all except 1 sustained their uMRD status during subsequent MRD assessments (Figure 3). To evaluate the depth of MRD response, the uMRD rate was also assessed at sensitivity thresholds of 10–5 and 10–6. At the 10–5 threshold, uMRD was achieved by 79.2% (95% CI, 57.8-92.9) of all patients, 78.6% (95% CI, 49.2-95.3) of PV patients, and 80.0% (95% CI, 44.4-97.5) of PVR patients (Figure 3). At the 10–6 threshold, uMRD was achieved by 62.5% (95% CI, 40.6-81.2) of all patients, 57.1% (95% CI, 28.9-82.3) of PV patients, and 70.0% (95% CI, 34.8-93.3) of PVR patients (Figure 3).
Figure 3.

Depth of MRD response in peripheral blood. MRD was assessed by clonoSEQ assay (Adaptive Biotechnologies, Seattle, WA) at screening and starting with cycle 3 day 1. (A-B) Sankey plots showing changes in MRD status at a sensitivity of <1 CLL cell per 10 000 nucleated cells (<10–4). (C-D) Line graphs showing MRD response for individual patients at a sensitivity of <1 CLL cell per 1 000 000 nucleated cells (<10–6). MRD was assessed in 24 evaluable patients during 25 cycles of therapy. MRD data for 1 patient in the PV cohort are not shown because calibration did not identify a suitable dominant DNA sequence for MRD tracking. The study protocol required a lead-in cycle of pirtobrutinib monotherapy followed by 24 cycles of combination therapy with venetoclax, for a total of 25 cycles. C, cycle; D, day.
Twelve patients (PV: 6; PVR: 6) who achieved uMRD (10–4) were classified as partial responders, either solely because of residual lymphadenopathy (n = 8) or because of pending bone marrow biopsy for CR confirmation (n = 4). Of these 12 patients, 7 achieved deep levels of uMRD (10–6), including 3 with residual lymphadenopathy and 4 patients who met the iwCLL criteria for CR but were awaiting confirmation of normal bone marrow. With the limited number of patients, there was no clear association between uMRD and timing or depth of response.
PK
For those receiving PV, 260 pirtobrutinib samples and 120 venetoclax samples from 14 patients were included in the PK analysis. For those receiving PVR, 198 pirtobrutinib samples and 98 venetoclax samples from 10 patients were included in the PK analysis. Pirtobrutinib and venetoclax PK were similar in both combination regimens, suggesting that addition of rituximab did not affect the PK of either pirtobrutinib or venetoclax (Figure 4; supplemental Tables 2-3). There was no apparent drug–drug interaction between pirtobrutinib and venetoclax.
Figure 4.

Pirtobrutinib and venetoclax concentrations in patients treated with pirtobrutinib and venetoclax with or without rituximab. For pirtobrutinib concentrations, the open circles of cycle 1 day 8 represent pirtobrutinib alone, and closed symbols from cycle 2 and later represent pirtobrutinib with venetoclax.
Discussion
Fixed-duration combination treatments with covalent BTK inhibitors and venetoclax have achieved deep remissions and long-term disease control in patients with CLL/SLL; however, the real-world utility of these regimens has been limited. Moreover, in the modern era, many patients receive covalent BTK inhibitors as frontline continuous therapy, making their use in combination regimens significantly less relevant in the relapsed setting. In this phase 1b trial, the noncovalent (reversible) BTK inhibitor pirtobrutinib demonstrated promising activity when combined with venetoclax, with or without rituximab, in patients with R/R CLL. Among all patients treated with PV or PVR, the ORR was 96.0%, including 40% of patients who achieved a CR, and the PFS at 24 months was 79.5%. Notably, these clinical responses were observed in patients who were previously treated with covalent BTK inhibitors (68%), most of whom had developed covalent Bruton tyrosine kinase inhibitor resistance (71%). In addition, the uMRD (<10–4) rate in peripheral blood after 12 cycles as measured by next-generation sequencing was 70.8% for patients receiving PV or PVR. Moreover, uMRD (<10–4) was achieved in 87.5% of patients at some time during the trial. Early treatment discontinuation due to disease progression was rare, occurring in 2 patients receiving PV. In addition, PK analyses indicated that there were no apparent drug–drug interactions between pirtobrutinib and venetoclax. Both combination therapies had PK exposures comparable to those of pirtobrutinib and venetoclax monotherapies.34,37 Taken together, these findings demonstrate that pirtobrutinib combined with venetoclax, with or without rituximab, has promising efficacy in patients with R/R CLL, including those who were previously treated with a covalent BTK inhibitor.
Fixed-duration pirtobrutinib-based combination therapies were also well tolerated. Observed AEs were consistent with those seen with pirtobrutinib, venetoclax, and rituximab monotherapy. Among the 22 patients who had discontinued study treatment at the time of data cutoff, early treatment discontinuation due to AEs was relatively uncommon (9%). Moreover, the tolerability of pirtobrutinib-based combination treatments led to high rates of patient adherence to treatment as evidenced by relative dose intensities for all study drugs of ≥98%. Collectively, these data suggest that pirtobrutinib combined with venetoclax with or without rituximab was well tolerated in patients with R/R CLL.
Achieving uMRD, defined as <1 CLL cell per 10 000 leukocytes, during or after fixed-duration treatment has been shown to predict more favorable PFS and OS.39,41 However, comparing uMRD results of clinical trials of different fixed-duration combination treatments can be challenging. Sample source (peripheral blood or bone marrow), time of assessment, depth of assessment, and assay technique can influence the estimated rate of MRD. In this phase 1b trial, MRD was assessed using next-generation sequencing (clonoSEQ assay) at 3 thresholds (10–4, 10–5, and 10–6) in peripheral blood at various times during treatment. During treatment, uMRD (10–4) was achieved by 85.7% of patients receiving PV and 90.0% of patients receiving PVR. Many clinical trials have shown that venetoclax combined with a covalent BTK inhibitor can achieve high rates of uMRD and induce deep remissions that are maintained after stopping treatment.20,22, 23, 24,29,42,43 Unlike patients in these trials, however, most patients in this phase 1b trial had been previously treated with a covalent BTK inhibitor, suggesting that pirtobrutinib-based combination treatments can achieve deep remissions in relapsed CLL patients even after treatment failure on a covalent BTK inhibitor. Whether MRD can be used to identify patients who would benefit from shorter or longer durations of fixed therapy remains an ongoing research question.
These results may compare relatively favorably to results from the phase 3 MURANO trial.5 In MURANO, fixed-duration venetoclax and rituximab were given to previously treated patients with R/R CLL. After patients had completed 12 cycles of treatment, uMRD (<10–4) was 60% in peripheral blood. For PV or PVR, the peripheral blood uMRD (<10–4) rate was 70.8% after 12 cycles of treatment. A notable distinction between the MURANO study and this trial lies in the patient population. In MURANO, very few patients had prior exposure to a covalent BTK inhibitor. In contrast, most patients in this phase 1b trial (68%; 17/25) had previous exposure to a covalent BTK inhibitor.
The CR rate (40%) observed in this phase 1b study was not concordant with the uMRD rate (87.5%). Four patients who achieved uMRD (10–4) were classified as partial responders awaiting bone marrow biopsy confirmation of CR. These patients met all other iwCLL criteria for CR. Moreover, all 4 patients achieved deep uMRD at the 10–6 threshold. However, if morphologically normal bone marrow has not been confirmed, the best available response per the iwCLL criteria is PR. It is therefore possible that the absence of bone marrow assessment in these 4 patients may have led to an underestimation of the CR rate in this phase 1b study. In addition, 8 patients were classified as partial responders solely because of residual lymphadenopathy, and it is typical of most recent studies in CLL that the CR rate is lower than the rate of uMRD, usually because of small residual lymphadenopathy.
This study has important limitations. Our sample size was too small to meaningfully compare the relative tolerability and efficacy of PV and PVR regimens with each other or other treatments. Important differences between these combination regimens may include the mode of administration (eg, intravenous for PVR vs oral for PV) and safety profile (eg, infusion-related reactions with PVR or possibly more TLS with PV due to less debulking). These treatment attributes may be important when selecting an optimal treatment for a patient. Several differences in baseline characteristics between treatment groups, including functional status, molecular features, and prior lines of therapy, may have influenced apparent differences in safety profiles and outcomes. Longer follow-up and large sample size are needed to establish the safety of fixed-duration treatment and better estimate the durability of responses. Additionally, the 5-week observation period for DLTs only included 1 week of full-dose venetoclax. Additional retrospective analysis of the 28-day combination therapy period including full-dose venetoclax and analysis of the 12-month full-dose venetoclax treatment period further support the safety of the current dosage based on the 3 + 3 decision rule.
In summary, both fixed-duration PV and PVR were well tolerated and demonstrated sufficiently promising efficacy to warrant further investigation in patients with R/R CLL. The BRUIN CLL-322 phase 3 trial comparing fixed-duration PVR with venetoclax and rituximab in previously treated CLL is ongoing (ClinicalTrials.gov identifier: NCT04965493), and other pirtobrutinib-based combinations are underway in patients with previously untreated CLL (ClinicalTrials.gov identifiers: NCT05536349, NCT05677919). In addition, the investigator-initiated phase 3 CLL-18 trial, which will use an MRD-guided approach to evaluate pirtobrutinib in combination with venetoclax vs obinutuzimab in combination with venetoclax, is being planned.
Conflict-of-interest disclosure: L.E.R. has served as a consultant for AbbVie, Ascentage, AstraZeneca, BeiGene, Janssen, Loxo Oncology, Pharmacyclics, Pfizer, and TG Therapeutics and as a continuing medical education speaker for DAVA, Curio, Medscape, and PeerView; holds minority ownership interest in Abbott Laboratories; received travel support from Loxo Oncology; and has received research funding (paid to the institution) from Adaptive Biotechnologies, AstraZeneca, Genentech, AbbVie, Pfizer, Loxo Oncology, Aptose Biosciences, Dren Bio, and Qilu Puget Sound Biotherapeutics. J.A.W. reports personal fees from Loxo Oncology during the conduct of the study; personal fees from Janssen, Pharmacyclics, AstraZeneca, AbbVie, BeiGene, Genentech, Merck, and Newave; and grants from Pharmacyclics, Schrodinger, and AbbVie, outside the submitted work. C.Y.C. reports personal fees from Loxo Oncology during the conduct of the study; grants and personal fees from Roche, AbbVie, and Bristol Myers Squibb; and personal fees from Menarini, Kite, Janssen, Gilead, Genmab, BeiGene, and AstraZeneca outside the submitted work. C.C.C. reports personal fees and honoraria and payment to institution for clinical trial from Loxo Oncology, during the conduct of the study; personal fees from AbbVie, Allogene, AstraZeneca, BeiGene, Genentech, Janssen, MEI Pharma, MingSight, Octapharma, and TG Therapeutics; and payment to institution for clinical trial from AbbVie outside the submitted work. N.N.S. reports personal fees from Loxo Oncology, during the conduct of the study; personal fees and research support and consultancy from Miltenyi Biotec; and personal fees from Incyte, Celgene, Kite, and Verastem, outside the submitted work. M.R.P. reports consultancy, board of director’s membership, or advisory roles for Janssen, EMD Serono, Pfizer, Pharmacyclics, Bayer, Genentech, and Loxo Oncology, during the conduct of the study. N.L. received research funding from Loxo Oncology, Juno, Oncternal, Verastem, TG Therapeutics, MingSight, and Octapharma, and has been in a consulting role for AbbVie, AstraZeneca, BeiGene, Genentech, Celgene, Gilead, Janssen, and Pharmacyclics. D.E.T., B.N., X.Z., N.M., and S.C.M. report full-time employment with Loxo Oncology during the study. C.W. and S.C. report full-time employment with Eli Lilly and Company during the conduct of the study. J.R.B. has served as a consultant for AbbVie, Acerta/AstraZeneca, Alloplex Biotherapeutics, BeiGene, Galapagos NV, Genentech/Roche, Grifols Worldwide Operations, InnoCare Pharma Inc, iOnctura, Kite, Loxo/Lilly, Merck, Numab Therapeutics, Pfizer, and Pharmacyclics, and received research funding from BeiGene, Gilead, iOnctura, Loxo/Lilly, MEI Pharma, and TG Therapeutics. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the patients, their families, and the trial teams at the participating centers. They thank Bastien Nguyen from Loxo Oncology at Lilly for creating the minimal residual disease figures and Louisa Ludwig-Begall from Evidera for creating the visual abstract.
The BRUIN trial is supported by funding from Loxo Oncology, a wholly-owned subsidiary of Eli Lilly and Company. Institutional expenses incurred during the study were also supported in part by the National Institutes of Health, National Cancer Institute Cancer Center support grant P30CA008748. Medical writing support was provided by Michael Franklin from Evidera, which was funded by Eli Lilly and Company and conducted in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3). J.A.W. is a clinical scholar of the Leukemia & Lymphoma Society.
Authorship
Contribution: D.E.T. conceptualized and designed the study; D.E.T., B.N., C.W., X.Z., S.C., D.L., D.R., N.M., and S.C.M. verified and interpreted acquired study data and conducted the analysis; C.W. and X.Z. conducted statistical analyses; L.E.R., J.A.W., C.Y.C., C.C.C., N.N.S., W.G.W., M.R.P., N.L., and J.R.B. acquired and interpreted the study data; and all authors had access to and verified the data, vouch for the completeness and accuracy of the data and adherence to the protocol, agreed to the content of the manuscript and submission, had access to primary clinical trial data, and critically revised the manuscript and approved the final version.
Footnotes
Eli Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available upon request 6 months after the indication studied has been approved in the United States and the European Union and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Barr PM, Owen C, Robak T, et al. Up to 8-year follow-up from RESONATE-2: first-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022;6(11):3440–3450. doi: 10.1182/bloodadvances.2021006434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrd JC, Hillmen P, Ghia P, et al. Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: results of the first randomized phase III trial. J Clin Oncol. 2021;39(31):3441–3452. doi: 10.1200/JCO.21.01210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hillmen P, Brown JR, Eichhorst BF, et al. ALPINE: zanubrutinib versus ibrutinib in relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma. Future Oncol. 2020;16(10):517–523. doi: 10.2217/fon-2019-0844. [DOI] [PubMed] [Google Scholar]
- 4.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225–2236. doi: 10.1056/NEJMoa1815281. [DOI] [PubMed] [Google Scholar]
- 5.Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107–1120. doi: 10.1056/NEJMoa1713976. [DOI] [PubMed] [Google Scholar]
- 6.Seymour JF, Kipps TJ, Eichhorst BF, et al. Enduring undetectable MRD and updated outcomes in relapsed/refractory CLL after fixed-duration venetoclax-rituximab. Blood. 2022;140(8):839–850. doi: 10.1182/blood.2021015014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung C, Umoru G, Abboud K, Hobaugh E. Sequencing and combination of current small-molecule inhibitors for chronic lymphocytic leukemia: where is the evidence? Eur J Haematol. 2023;111(1):15–28. doi: 10.1111/ejh.13973. [DOI] [PubMed] [Google Scholar]
- 8.Shadman M. Diagnosis and treatment of chronic lymphocytic leukemia: a review. JAMA. 2023;329(11):918–932. doi: 10.1001/jama.2023.1946. [DOI] [PubMed] [Google Scholar]
- 9.Eyre TA, Hori S, Munir T. Treatment strategies for a rapidly evolving landscape in chronic lymphocytic leukemia management. Hematol Oncol. 2022;40(2):129–159. doi: 10.1002/hon.2943. [DOI] [PubMed] [Google Scholar]
- 10.Thompson MC, Mato AR. Treatment of chronic lymphocytic leukemia after discontinuation of Bruton's tyrosine kinase inhibitors. Hematol Oncol Clin North Am. 2021;35(4):793–806. doi: 10.1016/j.hoc.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Gaballa S, Pinilla-Ibarz J. BTK inhibitors in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2021;16(5):422–432. doi: 10.1007/s11899-021-00645-1. [DOI] [PubMed] [Google Scholar]
- 12.Mato AR, Nabhan C, Thompson MC, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874–879. doi: 10.3324/haematol.2017.182907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JR, Eichhorst B, Hillmen P, et al. Zanubrutinib or ibrutinib in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2023;388(4):319–332. doi: 10.1056/NEJMoa2211582. [DOI] [PubMed] [Google Scholar]
- 14.Gu D, Tang H, Wu J, Li J, Miao Y. Targeting Bruton tyrosine kinase using non-covalent inhibitors in B cell malignancies. J Hematol Oncol. 2021;14(1):40. doi: 10.1186/s13045-021-01049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghia P, Wierda WG, Barr PM, et al. Relapse after first-line fixed duration ibrutinib + venetoclax: high response rates to ibrutinib retreatment and absence of BTK mutations in patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) with up to 5 years of follow-up in the phase 2 captivate study [abstract] Blood. 2023;142(suppl 1):633. [Google Scholar]
- 17.Ravelo A, Myers K, Brumble R, et al. Understanding patient preferences for chronic lymphocytic leukemia treatments [abstract] Blood. 2022;140(suppl 1):10803–10805. [Google Scholar]
- 18.Sportoletti P, Laurenti L, Chiarenza A, et al. Patients' preferences for chronic lymphocytic leukemia treatment: the CHOICE study. Hematol Oncol. 2024;42(1):e3216. doi: 10.1002/hon.3216. [DOI] [PubMed] [Google Scholar]
- 19.Ravelo A, Myers K, Ervin C, et al. Patient preferences for fixed versus treat-to-progression therapies in chronic lymphocytic leukemia [abstract] Blood. 2023;142(suppl 1):3706. [Google Scholar]
- 20.Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380(22):2095–2103. doi: 10.1056/NEJMoa1900574. [DOI] [PubMed] [Google Scholar]
- 21.Munir T, Cairns DA, Bloor A, et al. Chronic lymphocytic leukemia therapy guided by measurable residual disease. N Engl J Med. 2024;390(4):326–337. doi: 10.1056/NEJMoa2310063. [DOI] [PubMed] [Google Scholar]
- 22.Munir T, Moreno C, Owen C, et al. Impact of minimal residual disease on progression-free survival outcomes after fixed-duration ibrutinib-venetoclax versus chlorambucil-obinutuzumab in the GLOW study. J Clin Oncol. 2023;41(21):3689–3699. doi: 10.1200/JCO.22.02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niemann CU, Munir T, Moreno C, et al. Fixed-duration ibrutinib-venetoclax versus chlorambucil-obinutuzumab in previously untreated chronic lymphocytic leukaemia (GLOW): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023;24(12):1423–1433. doi: 10.1016/S1470-2045(23)00452-7. [DOI] [PubMed] [Google Scholar]
- 24.Tam CS, Allan JN, Siddiqi T, et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: primary analysis of the CAPTIVATE FD cohort. Blood. 2022;139(22):3278–3289. doi: 10.1182/blood.2021014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cramer P, Fürstenau M, Robrecht S, et al. Obinutuzumab, acalabrutinib, and venetoclax, after an optional debulking with bendamustine in relapsed or refractory chronic lymphocytic leukaemia (CLL2-BAAG): a multicentre, open-label, phase 2 trial. Lancet Haematol. 2022;9(10):e745–e755. doi: 10.1016/S2352-3026(22)00211-3. [DOI] [PubMed] [Google Scholar]
- 26.Huber H, Tausch E, Schneider C, et al. Final analysis of the CLL2-GIVe trial: obinutuzumab, ibrutinib, and venetoclax for untreated CLL with del(17p)/TP53mut. Blood. 2023;142(11):961–972. doi: 10.1182/blood.2023020013. [DOI] [PubMed] [Google Scholar]
- 27.Eichhorst B, Niemann CU, Kater AP, et al. First-line venetoclax combinations in chronic lymphocytic leukemia. N Engl J Med. 2023;388(19):1739–1754. doi: 10.1056/NEJMoa2213093. [DOI] [PubMed] [Google Scholar]
- 28.Hillmen P, Pitchford A, Bloor A, et al. Ibrutinib and rituximab versus fludarabine, cyclophosphamide, and rituximab for patients with previously untreated chronic lymphocytic leukaemia (FLAIR): interim analysis of a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023;24(5):535–552. doi: 10.1016/S1470-2045(23)00144-4. [DOI] [PubMed] [Google Scholar]
- 29.Kater AP, Owen C, Moreno C, et al. Fixed-duration ibrutinib-venetoclax in patients with chronic lymphocytic leukemia and comorbidities. NEJM Evid. 2022;1(7) doi: 10.1056/EVIDoa2200006. [DOI] [PubMed] [Google Scholar]
- 30.Ryan CE, Lampson BL, Tyekucheva S, et al. Updated results from a multicenter, phase 2 study of acalabrutinib, venetoclax, obinutuzumab (AVO) in a population of previously untreated patients with CLL enriched for high-risk disease [abstract] Blood. 2022;140(suppl 1):837–838. [Google Scholar]
- 31.Gomez EB, Ebata K, Randeria HS, et al. Pirtobrutinib preclinical characterization: a highly selective, non-covalent (reversible) BTK inhibitor. Blood J. 2023;142(1):62–72. doi: 10.1182/blood.2022018674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mato AR, Woyach JA, Brown JR, et al. Pirtobrutinib after a covalent BTK inhibitor in chronic lymphocytic leukemia. N Engl J Med. 2023;389(1):33–44. doi: 10.1056/NEJMoa2300696. [DOI] [PubMed] [Google Scholar]
- 33.Jensen JL, Mato AR, Pena C, Roeker LE, Coombs CC. The potential of pirtobrutinib in multiple B-cell malignancies. Ther Adv Hematol. 2022;13 doi: 10.1177/20406207221101697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.US Food and Drug Administration Jaypirca (pirtobrutinib). Prescribing information. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216059Orig1s000Corrected_lbl.pdf
- 35.FDA grants accelerated approval to pirtobrutinib for chronic lymphocytic leukemia and small lymphocytic lymphoma. 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pirtobrutinib-chronic-lymphocytic-leukemia-and-small-lymphocytic
- 36.US Food and Drug Administration Jaypirca (pirtobrutinib). Prescribing information. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216059s001lbl.pdf
- 37.US Food and Drug Administration Venclexta (venetoclax). Prescribing information. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf
- 38.US Food and Drug Administration Rituxan (rituximab). Prescribing information. 2012. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/103705s5367s5388lbl.pdf
- 39.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760. doi: 10.1182/blood-2017-09-806398. [DOI] [PubMed] [Google Scholar]
- 40.Ching T, Duncan ME, Newman-Eerkes T, et al. Analytical evaluation of the clonoSEQ Assay for establishing measurable (minimal) residual disease in acute lymphoblastic leukemia, chronic lymphocytic leukemia, and multiple myeloma. BMC Cancer. 2020;20(1):1–15. doi: 10.1186/s12885-020-07077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wierda WG, Rawstron A, Cymbalista F, et al. Measurable residual disease in chronic lymphocytic leukemia: expert review and consensus recommendations. Leukemia. 2021;35(11):3059–3072. doi: 10.1038/s41375-021-01241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillmen P, Rawstron AC, Brock K, et al. Ibrutinib plus venetoclax in relapsed/refractory chronic lymphocytic leukemia: the CLARITY study. J Clin Oncol. 2019;37(30):2722–2729. doi: 10.1200/JCO.19.00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niemann CU, Levin MD, Dubois J, et al. Venetoclax and ibrutinib for patients with relapsed/refractory chronic lymphocytic leukemia. Blood. 2021;137(8):1117–1120. doi: 10.1182/blood.2020008608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.