

Abstract
Amegakaryocytic thrombocytopenia is a rare haematologic disorder characterised by a profound reduction in platelet production due to the near absence of megakaryocytes, leading to severe thrombocytopenia. This report elucidates the diagnostic and therapeutic challenges encountered in managing such rare haematological conditions alongside significant cardiovascular disease. We detail the case of a patient who presented with chest pain and was diagnosed with non-ST elevation myocardial infarction (NSTEMI). Subsequent investigations revealed severe thrombocytopenia and underlying triple vessel disease, complicating immediate surgical intervention. Initial management strategies aimed at treating presumed immune thrombocytopenia proved ineffective. A definitive diagnosis of amegakaryocytic thrombocytopenia was established following a bone marrow biopsy. Despite treatment adjustments, including the administration of thrombopoietin agonists and immunosuppression, platelet counts improved but did not reach levels safe for coronary artery bypass grafting. This case underscores the importance of diagnosis and treatment of systemic disorders prior treatment of cardiac disease. It also demonstrates the importance of interdisciplinary cooperation in the treatment of a complex patient case.
LEARNING POINTS
Amegakaryocytic thrombocytopenia is a rare cause of thrombocytopenia that can co-occur with cardiac conditions.
The diagnosis of amegakaryocytic thrombocytopenia can be difficult as it can be mistaken for immune mediated thrombocytopenia, but distinction is critical as treatments differ.
Modern patient care frequently requires collaboration between different subspecialities.
Keywords: NSTEMI, thrombocytopenia, amegakaryocytic thrombocytopenia, triple vessel disease, CABG
CASE PRESENTATION
A 55-year-old male presented to the emergency department with a month-long history of intermittent, left-sided, sub-sternal, non-pleuritic, burning chest pain that had worsened on the day of admission. This prompted an emergency department visit. The patient reported a history of low platelets, recent epistaxis and easy bruising, but denied experiencing fevers, chills, shortness of breath, palpitations, syncope, unintentional weight loss, rash or haematuria. Physical examination revealed no acute distress, clear lungs, a regular heart rate and rhythm without murmurs, and noticeable bruising on the chest and bilateral legs. His medical history included minimal interactions with healthcare providers and previously diagnosed hypertension.
Laboratory findings indicated severe thrombocytopenia with a platelet count of 11,000/μl, a haematocrit of 35%, an elevated high-sensitivity troponin T initially at 155 ng/l peaking at 289 ng/l and a brain natriuretic peptide level of 2,247 pg/ml. Electrocardiography showed normal sinus rhythm with frequent premature ventricular contractions and inferolateral T-wave inversions (Fig. 1). Echocardiography revealed left ventricular systolic dysfunction with an ejection fraction of 36.2% and grade 1 diastolic dysfunction (Fig. 2). A coronary angiogram showed diffuse coronary disease, with significant stenoses and chronic total occlusions in multiple arteries. The diagnosis was triple vessel disease; however, coronary artery bypass grafting (CABG) was deferred due to the high haemorrhagic and mortality risks associated with his thrombocytopenia.
Figure 1.

ECG on patient’s initial presentation to the hospital.
Figure 2.
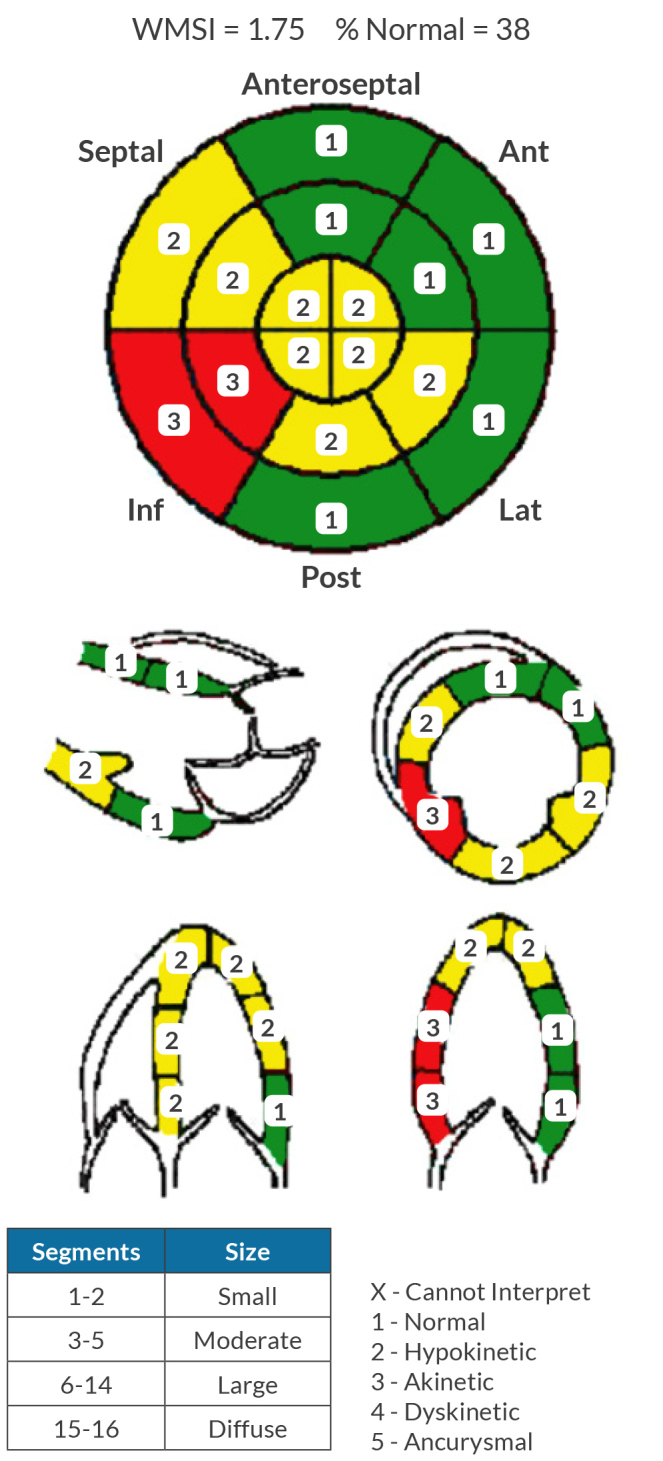
Initial transthoracic echocardiogram report.
The patient was treated with atorvastatin, aspirin, metoprolol, losartan, isosorbide mononitrate and PRN nitroglycerin. Initial therapies for presumed immune thrombocytopenia with glucocorticoids, IVIG and rituximab proved ineffective. After an 11-day hospital stay, he was discharged for outpatient haematological follow-up. A bone marrow biopsy revealed minimal megakaryocytes on CD-61 staining and normal trilineage haematopoiesis, confirming a diagnosis of amegakaryocytic thrombocytopenia (AMT). AMT is a rare diagnosis with congenital and acquired forms. Our patient’s positive antinuclear antibody with elevated inflammatory markers such as erythrocyte sedimentation rate, C-reactive protein and anti-DNA suggest an autoimmune aetiology, such as the acquired form of AMT. The exact prevalence of acquired AMT is unknown due to the primary source of information being case reports and small case series. Treatment with cyclosporine and eltrombopag was initiated to stimulate thrombopoietin production.
Seven months post-discharge, a repeat echocardiogram on guideline-directed medical therapy, including angiotensin receptor/neprilysin inhibitor, spironolactone, metoprolol succinate, acetylsalicylic acid, atorvastatin and isosorbide mononitrate, demonstrated an improvement in ejection fraction to 51–55% and showed concentric remodelling (Fig. 3). The patient’s platelet count improved to approximately 50,000 and he is now pending cardiothoracic surgery evaluation for potential CABG.
Figure 3.
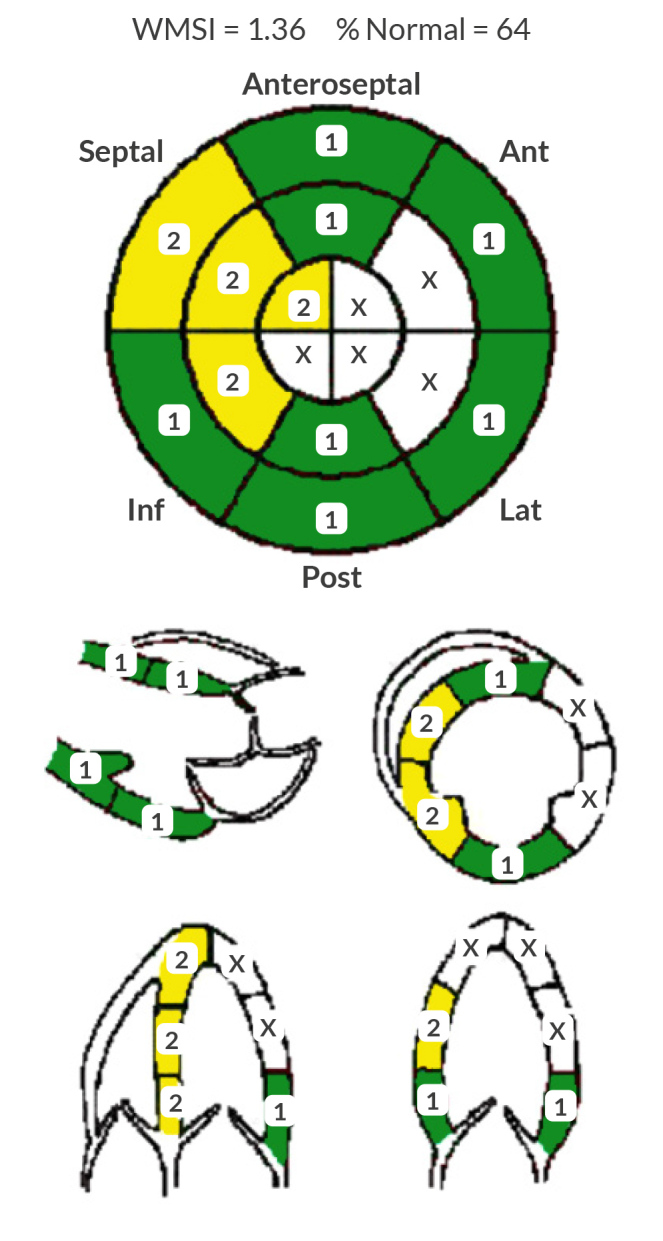
Follow-up transthoracic echocardiogram report 7 months after discharge.
DISCUSSION
The care of the complex cardiac patient can pose a significant challenge. With the increasing specialisation of medical providers increased collaboration across departments is needed in the current era of medical treatment. Our patient is a prime example of this, having both significant cardiovascular disease and haematologic disease that was complicated by diagnostic ambiguity.
The first consideration was which reperfusion strategy would best benefit the patient. The 2021 ACC/AHA guidelines recommend CABG over percutaneous coronary intervention (PCI) for patients with significant left coronary artery disease and multivessel disease, as CABG generally offers better outcomes[1]. However, the presence of thrombocytopenia substantially increases the risk of severe bleeding, mortality and other major adverse events post-CABG[2]. While PCI is less invasive, it is contraindicated in cases of significant thrombocytopenia because standard post-PCI therapy requires dual antiplatelet therapy (DAPT), which poses a bleeding risk that could extend from one month to one year based on individual patient factors[1]. Current literature lacks data on the tolerance of DAPT in thrombocytopenic patients[3], rendering it unsuitable for our patient until resolution of his thrombocytopenia, initially presumed to be caused by immune thrombocytopenia (ITP).
As both reperfusion strategies were delayed by thrombocytopenia attention was turned to the diagnosis and treatment of this condition. ITP is diagnosed by exclusion, and a bone marrow biopsy is generally not indicated unless the patient fails to respond to glucocorticoids or IVIG[4]. Both therapies made little difference in our patient’s thrombocytopenia, and other potential aetiologies for their thrombocytopenia were explored. AMT, a rare haematological disorder characterised by a significantly reduced number of megakaryocytes in the bone marrow, was subsequently diagnosed. AMT typically involves abnormalities in the thrombopoietin receptor pathway, which is crucial for megakaryocyte proliferation and differentiation[5]. Unlike ITP, which is managed with therapies aimed at reducing platelet destruction, AMT requires stimulation of megakaryocyte production through agents such as thrombopoietin agonists or even bone marrow transplantation in severe cases. The diagnosis of AMT is confirmed via bone marrow biopsy, where immunohistochemical staining for CD61 reveals a marked reduction in megakaryocyte count[6]. AMT can be acquired or inherited. Congenital cases are rare and generally present in childhood with severe thrombocytopenia and bleeding complications. Congenital cases are usually due to an acquired mutation in the MPL gene[7], which encodes thrombopoietin receptor, a critical component in the maintenance of adequate platelet production. Acquired cases can be triggered by autoimmune disease or viral illness but many cases are idiopathic, our patient’s positive antinuclear antibody and elevated inflammatory markers such as erythrocyte sedimentation rate, C-reactive protein and anti-DNA suggest an autoimmune aetiology, which further supports the use of immunosuppressive and immunomodulatory treatments[8].
In our review of similar patient presentations, we have only found one case in which a patient was described as having coronary artery disease and AMT[9]. Unlike in our case, PCI was considered optimal or equivalent to CABG. Treatment consisted of PCI with platelet transfusions and DAPT treatment for one week, followed by monotherapy with clopidogrel. With platelets maintained between 15k and 30k, the patient had no cardiac complaints at a three-month follow-up. Without precise data, monotherapy with clopidogrel must be carefully considered in patients with thrombocytopenia, while weighing the increased risk of bleeding and stent thrombosis against the risk of an impending cardiac event. This case in addition to ours highlights the medical complexity of patients with multiple major disease processes occurring simultaneously.
This case highlights the complexity of diagnosing and managing thrombocytopenia in patients needing coronary revascularisation, underscoring the necessity for tailored therapeutic strategies based on a precise diagnosis.
CONCLUSION
This case illustrates the importance of multidisciplinary collaboration in a patient with concomitant cardiac and haematologic conditions. As additional specialisations of cardiology such as cardio oncology emerge, it will be more important than ever to construct a patient’s multidisciplinary team to best fit their condition. In this case it was the collaboration between cardiology and haematology that effectively bridged our patient to his needed surgical intervention and optimisation of their cardiovascular and haematologic outcomes.
Footnotes
Conflicts of Interests: The Authors declare that there are no competing interests.
Patient Consent: The patient provided written consent for this publication. Patient information was redacted from this report for patient privacy.
REFERENCES
- 1.Lawton JS, Tamis-Holland JE, Bangalore S, Bates ER, Beckie TM, Bischoff JM, et al. 2021 ACC/AHA/SCAI Guideline for Coronary Artery Revascularization: Executive summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e7–17. doi: 10.1161/CIR.0000000000001039. Erratum in: Circulation 2022; 145:e771. [DOI] [PubMed] [Google Scholar]
- 2.Nammas W, Dalén M, Rosato S, Gherli R, Reichart D, Gatti G, et al. Impact of preoperative thrombocytopenia on the outcome after coronary artery bypass grafting. Platelets. 2019;30:480–486. doi: 10.1080/09537104.2018.1466389. [DOI] [PubMed] [Google Scholar]
- 3.Yusuf SW, Iliescu C, Bathina JD, Daher IN, Durand JB. Antiplatelet therapy and percutaneous coronary intervention in patients with acute coronary syndrome and thrombocytopenia. Tex Heart Inst J. 2010;37:336340. [PMC free article] [PubMed] [Google Scholar]
- 4.Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Jr, Crowther MA American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190–4207. doi: 10.1182/blood-2010-08-302984. [DOI] [PubMed] [Google Scholar]
- 5.Tirthani E, Said MS, De Jesus O. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024. Jan, Amegakaryocytic thrombocytopenia. [PubMed] [Google Scholar]
- 6.Levy I, Laor R, Jiries N, Bejar J, Polliack A, Tadmor T. Amegakaryocytic thrombocytopenia and subsequent aplastic anemia associated with apparent Epstein-Barr virus infection. Acta Haematol. 2018;139:7–11. doi: 10.1159/000484595. [DOI] [PubMed] [Google Scholar]
- 7.Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer. 2011;57:199–203. doi: 10.1002/pbc.22927. [DOI] [PubMed] [Google Scholar]
- 8.Dragani M, Andreani G, Familiari U, Marci V, Rege-Cambrin G. Pure red cell aplasia and amegakaryocytic thrombocytopenia in thymoma: the uncharted territory. Clin Case Rep. 2020;8:598–601. doi: 10.1002/ccr3.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed AO, Abdelghani M, Abu-Tineh M, Ibrahim F, Bakr M, Rahhal A, et al. ACS in a patient with amegakaryocytic thrombocytopenia: a management dilemma. Circulation. 2022;146 doi: 10.1161/circ.146.suppl_1.9440. [DOI] [Google Scholar]