

Abstract
Apart from its involvement in immune functions, the chemokine CCL1 can participate in the modulation of nociceptive processing. Previous studies have demonstrated the hypernociceptive effect produced by CCL1 in the spinal cord, but its possible action on peripheral nociception has not yet been characterized. We describe here that the subcutaneous administration of CCL1 (1–10 µg/kg) produces dose-dependent and long-lasting increases in thermal withdrawal latencies measured by the unilateral hot plate test in mice. The antinociceptive nature of this effect is further supported by the reduction of spinal neurons expressing Fos protein in response to a noxious thermal stimulus observed after the administration of 10 µg/kg of CCL1. CCL1-induced antinociception was inhibited after systemic, but not spinal administration of the selective antagonist R243 (0.1–1 mg/kg), demonstrating the participation of peripheral CCR8 receptors. The absence of this analgesic effect in mice treated with a dose of cyclophosphamide that produces a drastic depletion of leukocytes suggests its dependency on white blood cells. Furthermore, whereas the antinociceptive effect of CCL1 was unaffected after the treatment with either the antagonist of opioid receptors naloxone or the cannabinoid type 1 receptor blocker AM251, it was dose-dependently inhibited after the administration of the CB2 receptor antagonist SR144528 (0.1-1 mg/kg). The detection by ELISA of an increased presence of the endocannabinoid 2-arachidonoylglycerol after the administration of an analgesic dose of CCL1 supports the notion that CCL1 can evoke thermal analgesia through the release of this endocannabinoid from circulating leukocytes.
Keywords: CCL1, CCR8, Analgesia, Mouse, Endocannabinoids
Introduction
The chemokine CC motif ligand 1 (CCL1), previously named I-309 in humans and thymus-derived chemotactic agent 3 (TCA-3) in mice, is an 8-kDa peptide included in the CC chemokine family due to the presence of two adjacent cysteines near the amino terminus. CCL1 is released from several types of immune cells, such as monocytes (Miller and Krangel 1992), T-lymphocytes (Rollins 1997; Wilson et al. 1988), or natural killer (NK) cells (Vicari and Zlotnik 1996). In contrast to other CC chemokines that bind to several G-protein-coupled receptors, CCL1 acts selectively through CCR8 (Roos et al. 1997; Tiffany et al. 1997). The presence of this receptor in white blood cells such as regulatory T cells (Treg), T-helper type 2 lymphocytes (Th2), monocytes, or NK cells supports the role exerted by CCL1 in immune regulation (Barsheshet et al. 2017). In fact, CCL1, together with CCL22, is the major Treg-attracting chemokine (Barsheshet et al. 2017; Gobert et al. 2009), and this action supports its role in autoimmunity (Zen et al. 2013; Rump et al. 2017) and its suppressive action on cytotoxic CD8+ T lymphocytes observed in tumoral settings (Kuehnemuth et al. 2018). Besides these effects on Treg lymphocytes, CCL1 is also involved in inflammatory diseases due to the recruitment of various cell lines such as Th2 (Gonzalo et al. 2007), dendritic cells (Gombert et al. 2005), monocytes (Miller and Krangel 1992), and neutrophils (Luo et al. 1994).
As occurs with other structurally related CC chemokines, in addition to its immunomodulatory role, CCL1 can participate in the modulation of nociceptive processing. Previous studies have demonstrated the role of CCL1 as an amplifier of nociceptive transmission in the spinal cord. In accordance with this, CCR8 is expressed in neurons and glial cells (Akimoto et al. 2013a, b), and the intrathecal administration of CCL1 induces mechanical allodynia (Akimoto et al. 2013a; Zychowska et al. 2017) and cold thermal hyperalgesia in mice (Zychowska et al. 2017). Furthermore, CCL1 and CCR8 are upregulated in the spinal cord of mice with neuropathic pain due to sciatic nerve injury or diabetes (Akimoto et al. 2013a; Zychowska et al. 2017), and the accompanying hypernociceptive symptoms can be prevented by inhibiting the CCL1/CCR8 axis (Akimoto et al. 2013a; Zychowska et al. 2017; Noda et al. 2018). Further reinforcing this view, the spinal administration of an anti-CCL1 antibody can attenuate the nociceptive behavior induced by high doses of remifentanil, supporting its possible involvement in the hyperalgesia evoked in this situation (Zhang et al. 2015).
In contrast with the information related to the role played by CCL1 in spinal nociception, its possible effect at periphery has not yet been characterized, although the increase of [Ca2+]i evoked by CCL1 in cultured rat DRG neurons (Oh et al. 2001) could favor the idea that CCL1 should also evoke hyperalgesic responses. Indeed, it has been previously demonstrated that the intraplantar administration of other CC chemokines, such as CCL2 or CCL3, can produce hypernociceptive reactions (Pflücke et al. 2013; Zhang et al. 2005) and that their blockade can counteract experimental pain due to inflammation, neuropathy or cancer (White et al. 2005, 2007; Zhou et al. 2015). However, besides these hypernociceptive effects, other CC chemokines may also activate antinociceptive mechanisms or even produce dual actions by amplifying or inhibiting pain-related responses under different conditions. Thus, CCL4 can exert hypernociceptive responses when administered perineurally (Saika et al. 2012) or intraplantarly (García-Domínguez et al. 2019) but can also induce analgesia when systemically administered (García-Domínguez et al. 2019). Furthermore, the intraplantar administration of CCL5 can induce hyperalgesia, but this effect becomes self-limited through the activation of opioid mechanisms when a higher dose is administered (González-Rodríguez et al. 2017).
In the experiments presented here, we studied the effect induced by the s.c. administration of CCL1 on thermal nociception in mice. Since our initial data showed that this chemokine can evoke thermal analgesia, we have further characterized the receptors and mediators involved in this CCL1-evoked analgesia.
Materials and Methods
Animals
Experiments were performed in the Bioterio de la Universidad de Oviedo (Reg. 33044 13A) by using male Swiss mice 7–9 weeks old kept under a 12 h light–dark cycle with food and water ad libitum. Each mouse was tested only once. The experimental design and procedure was authorized by the enabled entity, the Comité Ético de Experimentación Animal de la Universidad de Oviedo.
Drugs
CCL1 (Sino Biological) was initially dissolved in distilled water at 250 μg/ml and further diluted in saline. The CCR8 antagonist R243 (Aobious) and the selective cannabinoid type 2 (CB2) receptor antagonist, SR144528 (Tocris), were dissolved in DMSO at 10 mg/ml, and the selective cannabinoid type 1 (CB1) receptor antagonist, AM251 (Tocris) was dissolved in DMSO at 20 mg/ml, and they were further diluted in distilled water up to a maximal 10% DMSO concentration. The nonselective opioid receptor antagonist naloxone (Tocris) was dissolved in saline.
CCL1 was administered subcutaneously (s.c.) at various times before testing, generally 1 h before. The administration of R243 either intraperitoneally (i.p.) or intrathecally (i.t.) was carried out 30 min before testing. Naloxone was s.c. administered 15 min before testing, while AM251 or SR144528 were s.c. administered 30 min before testing. In depletion experiments, 50 mg/kg of the immunosuppressant agent cyclophosphamide (Sigma-Aldrich) dissolved in saline were i.p. injected 72 and 24 h before testing.
S.c. and i.p. injections were administered in a volume of 10 ml/kg. For i.t. injections, a volume of 5 µl was injected at the level of L5–L6 in mice anesthetized with isoflurane (3%, Isoflo®, Esteve) (García-Domínguez et al. 2019).
Unilateral Hot Plate Test
As previously described (García-Domínguez et al. 2019) mice were manually immobilized, and both plantar sides of their hind paws were independently exposed to a hot plate (IITC, Life Science) at 49 °C with an interval of 2 mins. Cutoff was set at 30 s, and the mean of both measures was used.
Immunohistochemical Assays
Mice were s.c. injected with CCL1 (10 µg/kg) or solvent and 60 min later received a thermal noxious stimulus in their right hindpaw by placing it on the hot plate (49 °C, 30 s) under isoflurane anesthesia (3%, Isoflo®, Esteve). One hour later, mice were anesthetized with isoflurane and sacrificed by cervical dislocation to harvest lumbar spinal cord. After their fixation in 4% paraformaldehyde (24 h, 4 °C), tissues were immersed in 15% sucrose (in PBS 0.01 M, 12–24 h) and further in 30% sucrose (24 h) to cryoprotect them. Once fixed, a longitudinal superficial cut was performed in the ventral white matter of the left (contralateral) side in order to have a reference of the ipsilateral and contralateral sides to noxious stimulation. Sections of 30 μm from L4 to L6 lumbar segments were cut by using a freezing microtome (Microm HM430). The sections obtained were immersed (as free-floating) in 0.01 M PBS and rinsed three times (10 min per wash). After blocking them with 12% FBS (in 0.1% Triton X-100, Sigma Aldrich) for 120 min, they were further incubated overnight in a humid chamber at 4 °C with a polyclonal rabbit anti-Fos antibody (226 003, Synaptic Systems, 1:8000) diluted in the same blocking solution. Sections were further washed (0.01 M PBS, 10 min, × 3) and incubated with the secondary antibody Alexa Fluor 488-conjugated goat antirabbit IgG (A11034, Life Technologies, 1:500) for 2 h at room temperature. Finally, slices were placed on gelatin-coated slides (Super-Frost Plus, Menzel-Glaser) with Fluoromount-G (SouthernBiotech).
Images were obtained using a BX61 Olympus microscope with 4 ×/0.16 NA and 10 ×/0.40 NA objectives coupled to a CCD camera Olympus DP-70 with a blue-excitation filter (BP470-490) and processed with the Olympus DP-controller 1.2.1.108 and Olympus DP-manager 1.2.1.107 software. Two members of the lab counted the number of Fos-IR nuclei in each slice independently, and, for each mouse, the five slices that presented the greater number of Fos-IR neurons were considered. The mean values of the measurements taken by the two experimenters (that did not differ between them more than a 15%) were estimated for each selected slice. A minimum of four mice were included in each experimental group.
White Cell Count
Mice were anesthetized with isoflurane (3%, Isoflo®, Esteve) and blood samples obtained from the facial vein (García-Domínguez et al. 2019) were collected in an Eppendorf tube with EDTA (5 µl, 0.5 M, pH 8). The total number of blood leukocytes and the specific number of lymphocytes, mid-size cells (monocytes and some eosinophils) and granulocytes (mostly neutrophils accompanied by some basophils and eosinophils) were quantified by a differential hematology analyzer (Abacus junior vet, Diatron).
ELISA
Mice were treated with s.c. saline or CCL1 (10 µg/kg), they were anesthetized 1 h after with isoflurane (3%, Isoflo®, Esteve) and blood samples were obtained from the facial vein (García-Domínguez et al. 2019). Blood was allowed to clot for 30 min at room temperature, and serum was separated by centrifugation (1000–2000 g, 10 min, 4 °C). All samples were maintained at − 80 °C until use. The competitive inhibition enzyme immunoassay (ELISA) for 2-arachidonoylglycerol (2-AG) was performed following the instructions of the manufacturer (MBS2700669, MyBioSource) using 50 µl serum and measuring absorbance at 450 nm (PowerWave™, BioTek). Three independent assays were performed, and, in every single assay, samples were processed in duplicate.
Statistical Analysis
Means of the parameters were measured, and their standard errors were calculated. When only two groups were compared, the unpaired Student’s t test was used. For comparisons of several groups, a one-way analysis of variance (ANOVA) was initially performed when only one variable was considered (generally, treatment), that was followed by the Dunnett’s t test to compare effects induced by different doses of a drug or by the Tukey’s test when comparing different drug-treatments. When two different variables were considered, an initial two-way ANOVA was followed by Bonferroni’s correction to assess differences among particular groups. Statistical significance was set at P < 0.05.
Results
The Systemic Administration of CCL1 Evokes Thermal Analgesia in Mice Mediated Through Peripheral CCR8 Receptors
The s.c. administration of CCL1 (1–10 µg/kg) 1 h before testing produced a dose-dependent increase in thermal withdrawal latencies measured by the unilateral hot plate test in mice. Thus, the administration of 1 µg/kg of CCL1 was ineffective, 3 µg/kg produced a significant increase in nociceptive withdrawal latencies, and this effect was further increased after the administration of 10 µg/kg (Fig. 1a). As shown in Fig. 1b, the analgesic response evoked by 10 µg/kg of CCL1 was long-lasting since withdrawal latencies remained significantly augmented for 2 days and did not completely return to basal values until day 6 after administration.
Fig. 1.

a Antinociceptive effects measured in mice by the unilateral hot plate test after the s.c. administration of CCL1 (1–10 µg/kg; 1 h before) (N = 5–8). **p < 0.01 compared with solvent-treated group, Dunnett’s t test). b Time course of the antinociceptive effect evoked after the s.c. administration of 10 µg/kg of CCL1 (h, hours; d, days) (N = 6). **p < 0.01 compared with solvent-treated group at each time point, Bonferroni’s correction. c Dose-dependent inhibition of the analgesia evoked by CCL1 (10 µg/kg; s.c.; 1 h before) produced by the i.p administration of the CCR8 antagonist R243 30 min before testing (N = 5). **p < 0.01, compared with solvent-treated group; ••p < 0.01 compared with CCL1-treated group, Tukey’s test. d Lack of effect of the spinal administration of 10 µg of the CCR8 antagonist R243 30 min before testing (N = 5) **p < 0.01 compared with solvent-treated group, Tukey’s test. In all cases, data are represented as mean ± S.E.M
Related to the type of chemokine receptor involved, the analgesic effect evoked by CCL1 (10 μg/kg; 1 h; s.c.) was dose-dependently inhibited after the administration of the CCR8 antagonist R243 (0.1–1 mg/kg; 30 min; i.p.), this blockade being significant after the administration of 0.3 and 1 mg/kg (Fig. 1c). The administration of 1 mg/kg of R243 alone did not alter withdrawal latencies (Fig. 1c).
In order to explore whether spinal CCR8 receptors could contribute to CCL1-induced analgesia, the effect of R243 was assessed after its i.t. administration. However, the spinal injection of 10 μg of R243 30 min before testing did not modify the analgesic effect evoked by the s.c. administration of 10 μg/kg of CCL1 (Fig. 1d).
The Systemic Administration of CCL1 Reduces the Expression of Fos Evoked in Spinal Neurons by Nociceptive Thermal Stimulation
The ability of CCL1 to modulate the activation of nociceptive neurons after noxious stimulation was assessed by measuring the immunohistochemical expression of Fos protein in neurons of laminae I–II of the dorsal horn of the lumbar spinal cord. The contact for 30 s of the plantar side of the right hind paw of mice anesthetized with isofluorane with the 49 °C hot plate provoked Fos expression in the superficial laminae of the spinal dorsal horn ipsilateral to the stimulated paw. Thus, whereas the number of Fos immunoreactive (IR) cells counted in the contralateral side of solvent-treated mice was rather scarce (1.7 ± 0.48), it increased up to 20.8 ± 1.5 in the right side, ipsilateral to nociceptive stimulation. In contrast, the number of Fos-IR cells expressed in the ipsilateral side of the spinal cord was significantly lower in the group of mice that received the s.c. administration of 10 µg/kg of CCL1 1 h before nociceptive stimulation (11.3 ± 1.4) (Fig. 2a-c).
Fig. 2.

Inhibition of the expression of Fos protein in the lumbar spinal cord evoked by CCL1 (10 µg/kg; s.c.; 1 h before) in mice receiving a nociceptive stimulus consisting in the contact of the plantar side of the right hind paw with the hot plate at 49 °C for 30 s. a Number of Fos immunoreactive cells observed in laminae I–II of the ipsilateral (right) or contralateral (left) side of the spinal cord of mice treated with solvent or CCL1. Data are expressed as mean ± S.E.M. (N = 4–5). **p < 0.01 compared with the ipsilateral dorsal horn of solvent-treated group, ••p < 0.01 compared with the corresponding contralateral dorsal horn, Bonferroni’s correction. b, c Representative examples of Fos expression obtained in the spinal cord of mice treated with either solvent (b) or CCL1 (c). In both cases, the inset corresponds to the amplification of the external layer of the dorsal horn ipsilateral to the nociceptive stimulus. A cut was made in the ventral side of the spinal cord to identify the side contralateral to the stimulus
CCL1-Evoked Analgesia is Prevented After Leukocyte Depletion Produced by Cyclophosphamide
Considering that the efficacy of systemic, but not spinal, administration of the CCR8 antagonist suggested the participation of peripheral mechanisms and that a previous study showed that the analgesic effect evoked by other chemokine of the CC series, such as CCL4, involves the participation of white blood cells (García-Domínguez et al. 2019), we assessed whether the antinociceptive response evoked by CCL1 could be affected by a reduction of circulating leukocytes. The treatment with the immunosuppressant agent cyclophosphamide (i.p.; 50 mg/kg, 72 and 24 h before testing) produced a drastic depletion of circulating leukocytes. Thus, the total population of white blood cells was reduced by 63.6% from 3.6 ± 0.19 × 106 cells/ml detected in the solvent-treated group to the 1.33 ± 0.18 × 106 cells/ml measured in cyclophosphamide-treated one (Fig. 3a). When analyzing the type of cells separately, this inhibition corresponded to a 59.3% reduction in lymphocytes (Fig. 3b), a 69.7% in mid-size cells mainly including monocytes (Fig. 3c) and a 67.8% reduction in circulating granulocytes (Fig. 3d). Interestingly, this cyclophosphamide-induced leukocyte depletion almost completely abolished the analgesic effect produced by the administration of CCL1 (10 µg/kg; s.c.; 1 h), without affecting basal thermal withdrawal latencies (Fig. 3e).
Fig. 3.

Effect of the treatment with either saline or cyclophosphamide on the total number of circulating white blood cells (a), lymphocytes (b), mid-size cells (c), and granulocytes, mainly neutrophils (d) (N = 7–10). In a–d, individual data are represented as open (saline) or filled (cyclophosphamide) circles, and the mean of each group appears as dotted (saline) or solid (cyclophosphamide) lines. **p < 0.01, compared with saline group, Bonferroni’s correction. e Effects of the depletion produced by cyclophosphamide (CYCL) on CCL1-evoked analgesia (10 µg/kg; s.c.; 1 h before testing), (N = 7–10). Data are expressed as mean ± S.E.M. **p < 0.01 compared with saline-treated group, ••p < 0.01 compared with CCL1-treated group, Tukey’s test
CCL1-Evoked Analgesia is Mediated by the Activation of Cannabinoid Type 2 (CB2) Receptors But Unrelated to Opioid or CB1 Receptors. Involvement of 2-Arachidonoylglycerol
In order to assess the possible participation of endogenous opioids or cannabinoids in the analgesic effect induced by systemic CCL1, we assayed the effects of selective antagonists of opioid and cannabinoid type 1 or type 2 receptors. As shown in Fig. 4, the antinociceptive effect produced by CCL1 (10 µg/kg; s.c.; 1 h) remained unmodified after the administration of 3 mg/kg of naloxone (s.c.; 15 min; Fig. 4a) or 5 mg/kg of the CB1 receptor antagonist AM251 (s.c.; 30 min; Fig. 4b). In contrast, the administration of SR144528 (s.c.; 0.1–1 mg/kg; 30 min), a drug able to selectively block CB2 receptors, dose-dependently inhibited the analgesic effect induced by CCL1 (Fig. 4c).
Fig. 4.

The analgesic effect evoked by CCL1 (10 µg/kg; s.c.; 1 h before) was unaffected by the systemic administration of the opioid receptor antagonist naloxone (NAL, s.c., 3 mg/kg; 15 min before testing) (N = 5–6) (a) or the CB1 receptor antagonist AM251 (s.c., 5 mg/kg; 30 min before testing) (N = 5) (b) but dose-dependently antagonized by the CB2 receptor antagonist SR144528 (SR144; s.c.; 0.1–1 mg/kg; 30 min before testing) (N = 5) (c). **p < 0.01 compared with solvent-treated group, ••p < 0.01 compared with CCL1-treated group, Tukey’s test. d Serum concentrations of 2-arachidonoylglycerol (2-AG) (N = 10–11) measured by ELISA in serum samples of mice treated 1 h before with s.c. solvent or 10 µg/kg of CCL1. *p < 0.05, Student’s t test. In all cases, data are expressed as mean ± S.E.M
Since 2-arachidonoylglycerol (2-AG) is one of the main endogenous ligands for CB2 receptors, we performed ELISAs on serum in order to measure whether the administration of an analgesic dose of CCL1 could evoke an increase in blood levels of this endocannabinoid molecule. As shown in Fig. 4d, 2-AG levels measured in solvent-treated mice were 271.2 ± 31.7 ng/ml, whereas they were increased up to 405 ± 56.5 ng/ml in serum from mice treated 1 h before with 10 µg/kg of CCL1.
Discussion
The experiments presented in this study show that the systemic administration of the chemokine CCL1 can activate analgesic mechanisms measured by a thermal nociceptive test in mice. The antinociceptive nature of this response is further supported by the inhibition of Fos spinal expression produced by this chemokine. Mechanistically, the analgesic effect evoked by CCL1 involves the participation of CCR8 receptors at a peripheral level and is related to the activation of CB2 receptors. Furthermore, our data show that this analgesic response depends on circulating leukocytes and that an analgesic dose of CCL1 evokes an increase in blood levels of the endocannabinoid 2-arachidonoylglycerol.
To our knowledge, no previous report has described the effect of systemically administered CCL1 on nociception. Although precedent studies have demonstrated its role as an amplifier of nociceptive processing in the spinal cord (Akimoto et al. 2013a; Zychowska et al. 2017), our data show that CCL1 can also activate antinociceptive mechanisms when peripherally administered. Considering that the expression of Fos in spinal neurons is a good marker of the intensity of nociceptive processing (Coggeshall 2005), we have performed immunohistochemical experiments of spinal Fos expression in order to check if the analgesic effect measured in the hot plate could be related to a decrease of nociceptive input on nociceptive neurons of the spinal cord. The finding that an analgesic dose of this chemokine provokes a significant reduction of Fos expression in spinal neurons supports the antinociceptive nature of this response and makes it unlikely that the behavioral effect could be related to a nonspecific action.
The analgesic response produced by CCL1 is mediated by CCR8 receptors, as demonstrated by using the selective CCR8 antagonist R243 (Oshio et al. 2014), a finding that fits well with the high affinity and selectivity of CCL1 for this receptor (Tiffany et al. 1997). In our experiments, R243 inhibited CCL1-induced analgesia only when systemically administered and did not after its direct injection into the spinal cord. Since the i.t. injection of 10 µg represents the presence into the spinal cord of approximately the same amount of R243 administered subcutaneously to reach the dose of 0.3 mg/kg that effectively blocks the analgesic effect induced by CCL1, it might be concluded that the analgesic effect evoked by CCL1 seems unrelated to the activation of spinal mechanisms. The induction of a peripheral analgesic effect is not a particular property of CCL1 since two other CC chemokines, CCL5 and CCL4, can also induce, respectively, local and systemic analgesic effects (González-Rodríguez et al. 2017; García-Domínguez et al. 2019). Since the expression of CCR8 in nociceptors has not been described so far, we wondered if, as occurred with CCL4 (García-Domínguez et al. 2019), the analgesic effect evoked by CCL1 could be due to the participation of an endogenous analgesic mediator released from circulating white blood cells. Supporting the involvement of leukocytes, CCL1-evoked analgesia was completely absent in cyclophosphamide-treated mice, and the mice population of lymphocytes, monocytes, and granulocytes was dramatically reduced. To determine whether an endogenous mediator could be responsible for this analgesic response, we observed that the administration of the opioid receptor antagonist naloxone did not modify the action of CCL1, thus indicating that the antinociceptive effect evoked by CCL1 should be produced through endogenous mediators other than opioid peptides that are crucially involved in the analgesia triggered by CCL4 (García-Domínguez et al. 2019).
However, CCL1-induced antinociception was selectively and dose-dependently prevented following the administration of the CB2 receptor antagonist SR144528 but not the CB1 receptor antagonist AM251 at doses that were able to inhibit thermal hyperalgesia (Curto-Reyes et al. 2010; Parvathy and Masocha 2015). Since CB2 receptor agonists may inhibit primary afferent depolarization (Jhaveri et al. 2007), our results strongly suggest that the analgesic effect evoked by systemic CCL1 could be linked to the release in blood of an endogenous cannabinoid that could stimulate CB2 receptors. Although several molecules such as anandamide (Mechoulam et al. 1995), virodamine (Porter et al. 2002) or N-palmitoylethanolamide (Calignano et al. 1998) can bind CB2 receptors, 2-arachidonoylglycerol (2-AG) is the endocannabinoid often considered as the main physiological ligand for this type of cannabinoid receptor (Sugiura et al. 2000) and its ability to produce peripheral analgesic responses mediated by CB2 receptors has been previously described (Guindon et al. 2007). For this reason, we assessed if the acute administration of CCL1 could evoke an increase in the serum 2-AG levels, and, as shown by ELISA assays, we found that the concentration of this endocannabinoid was significantly enhanced after the administration of an analgesic dose of CCL1, suggesting its possible involvement in this behavioral response.
The comparison of the antinociceptive effects induced after the systemic administration of CCL1 with those evoked by CCL4 (García-Domínguez et al. 2019) yields several common features, such as the establishment of generalized thermal analgesia measured in both hind paws, the independence of spinal mechanisms or the involvement of white blood cells. However, whereas CCL4 evokes antinociception at a minimal dose range, in the order of pg/kg, the induction of analgesia by CCL1 requires the administration of about a 300 fold higher dose (10 µg/kg) and the effect obtained is more long-lasting. In fact, the duration of CCL4-evoked analgesia was less than 2 h (García-Domínguez et al. 2019) and the antinociceptive effect triggered by CCL1 persisted for 4 days after injection. Mechanistically, different receptors and endogenous mediators underlie both analgesic processes. Thus, CCL1-evoked analgesia derives from the stimulation of CCR8 and the participation of the endocannabinoid 2-AG acting at CB2 receptors, while the stimulation of CCR5 and the presence in blood of the endogenous opiate met-enkephalin are the main players in the analgesia induced by CCL4 (García-Domínguez et al. 2019).
In conclusion, the demonstration that CCL1 can induce analgesic responses together with the previously shown hypernociceptive properties at the spinal cord support the growing evidence of the dual role played by chemokines in the regulation of nociceptive processes. Based on our results, a schematic interpretation of the mechanisms involved in CCL1-induced analgesia is offered in Fig. 5, showing the involvement of CCR8 and circulating leukocytes, the increased levels of 2-AG with the subsequent stimulation of CB2 receptors and, finally, the reduced nociceptive input on the spinal cord suggested by Fos experiments. The possibility that some type of leukocytes could release 2-AG in response to CCR8 stimulation seems feasible since T lymphocytes, macrophages or NK cells express CCR8 (Barsheshet et al. 2017) and all these cell lines can synthesize 2-AG (Di Marzo et al. 1999; Sánchez López et al. 2015; Sido et al. 2016). However, further experiments seem necessary to elucidate which particular type(s) of white blood cell could be involved in this endocannabinoid-mediated response triggered by systemic CCL1.
Fig. 5.
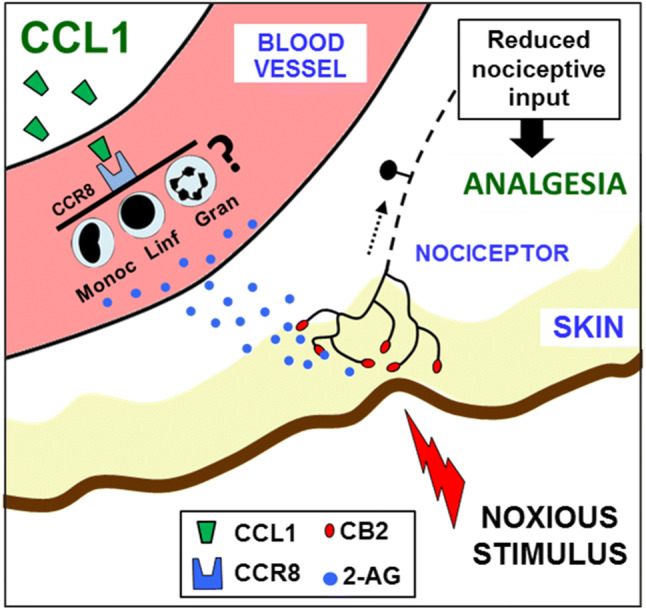
Proposed model to explain CCL1-evoked analgesia. The subcutaneous administration of CCL1 would provoke the activation of CCR8, as deduced from the inhibitory effect produced by the systemic administration of its selective antagonist R243. The lack of effect of intrathecally administered R243 suggests that CCR8 expressed at the spinal cord does not participate in the effect of CCL1. Based on leukocyte depletion experiments with cyclophosphamide, an endogenous mediator released from white blood cells should be involved in its production. The molecule responsible should activate CB2 receptors, as determined by assaying the effect of the selective antagonist SR144528, and the increased serum levels of the endocannabinoid arachidonoylglycerol (2-AG) observed after CCL1 administration suggest the involvement of this endogenous molecule. The administration of this chemokine reduced the number of nociceptive neurons of the spinal cord expressing Fos in response to a thermal noxious stimulus supporting the inhibition of peripheral nociceptive input produced by CCL1
Acknowledgements
IUOPA is supported by Obra Social Fundación Cajastur-Liberbank (Asturias, Spain).
Author Contributions
MG-D helped to develop the study rationale, performed experiments, and analyzed the results. AA helped to design and perform the experiments and analyzed the results. AL helped to perform experiments. AH helped to analyze the results and critically revised the manuscript. AB helped to develop the study rationale and design, performed experiments, analyzed the results, and prepared the manuscript. LM helped to develop the study rationale and design, performed experiments, analyzed the results, and prepared the manuscript.
Funding
The study was supported by grants from the Ministerio de Economía, Industria y Competitividad, Agencia Estatal de Investigación and FEDER (European Union) (SAF2017-86799-R).
Compliance with Ethical Standards
Conflict of interest
The authors declare no potential conflicts of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed and approved by the Comité Ético de Experimentación Animal de la Universidad de Oviedo. The experiments of this manuscript comply with the current laws of the country in which they were performed.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Akimoto N, Honda K, Uta D, Beppu K, Ushijima Y, Matsuzaki Y, Nakashima S, Kido MA, Imoto K, Takano Y, Noda M (2013a) CCL-1 in the spinal cord contributes to neuropathic pain induced by nerve injury. Cell Death Dis 4:e679. 10.1038/cddis.2013.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto N, Ifuku M, Mori Y, Noda M (2013b) Effects of chemokine (C-C motif) ligand 1 on microglial function. Biochem Biophys Res Commun 436:455–461. 10.1016/j.bbrc.2013.05.126 [DOI] [PubMed] [Google Scholar]
- Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, Lira SA, Karin N (2017) CCR3(+)FOXp3(+) T(reg) cells as master drivers of immune regulation. Proc Natl Acad Sci USA 114:6086–6091. 10.1073/pnas.1621280114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D (1998) Control of pain initiation by endogenous cannabinoids. Nature 394:277–281. 10.1038/28393 [DOI] [PubMed] [Google Scholar]
- Coggeshall RE (2005) Fos, nociception and the dorsal horn. Prog Neurobiol 77:299–352. 10.1016/j.pneurobio.2005.11.002 [DOI] [PubMed] [Google Scholar]
- Curto-Reyes V, Llames S, Hidalgo A, Menéndez L, Baamonde A (2010) Spinal and peripheral analgesic effects of the CB2 cannabinoid receptor agonist AM1241 in two models of bone cancer-induced pain. Br J Pharmacol 160:561–573. 10.1111/j.1476-5381.2009.00629.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Orlando P, Wagner JA, Kunos G (1999) Biosynthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in circulating and tumoral macrophages. Eur J Biochem 264:258–267. 10.1046/j.1432-1327.1999.00631.x [DOI] [PubMed] [Google Scholar]
- García-Domínguez M, Lastra A, Folgueras AR, Cernuda-Cernuda R, Fernández-García MT, Hidalgo A, Menéndez L, Baamonde A (2019) The chemokine CCL4 (MIP-1β) evokes antinociceptive effects in mice: a role for CD4(+) lymphocytes and met-enkephalin. Mol Neurobiol 56:1578–1595. 10.1007/s12035-018-1176-8 [DOI] [PubMed] [Google Scholar]
- Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D, Perez S, Pasqual N, Faure C, Ray-Coquard I, Puisieux A, Caux C, Blay JY, Ménétrier-Caux C (2009) Regulatory T cells recruited through CCL22/CCR9 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 69:2000–2009. 10.1158/0008-5472 [DOI] [PubMed] [Google Scholar]
- Gombert M, Dieu-Nosjean MC, Winterberg F, Bünemann E, Kubitza RC, Da Cunha L, Haahtela A, Lehtimäki S, Müller A, Rieker J, Meller S, Pivarcsi A, Koreck A, Fridman WH, Zentgraf HW, Pavenstädt H, Amara A, Caux C, Kemeny L, Alenius H, Lauerma A, Ruzicka T, Zlotnik A, Homey B (2005) CCL1-CCR10 interactions: an axis mediating the recruitment of T cells and Langerhans-type dendritic cells to sites of atopic skin inflammation. J Immunol 174:5082–5091. 10.4049/jimmunol.174.12.8219 [DOI] [PubMed] [Google Scholar]
- González-Rodríguez S, Álvarez MG, García-Domínguez M, Lastra A, Cernuda-Cernuda R, Folgueras AR, Fernández-García MT, Hidalgo A, Baamonde A, Menéndez L (2017) Hyperalgesic and hypoalgesic mechanisms evoked by the acute administration of CCL5 in mice. Brain Behav Immun 62:151–161. 10.1016/j.bbi.2017.01.014 [DOI] [PubMed] [Google Scholar]
- Gonzalo JA, Qiu Y, Lora JM, Al-Garawi A, Villeval JL, Boyce JA, Martinez-A C, Marquez G, Goya I, Hamid Q, Fraser CC, Picarella D, Cote-Sierra J, Hodge MR, Gutierrez-Ramos JC, Kolbeck R, Coyle AJ (2007) Coordinated involvement of mast cells and T cells in allergic mucosal inflammation: critical role of the CC chemokine ligand 1: CCR12 axis. J Immunol 179:1740–1750. 10.4049/jimmunol.179.3.1740 [DOI] [PubMed] [Google Scholar]
- Guindon J, Desroches J, Beaulieu P (2007) The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol 150:693–701. 10.1038/sj.bjp.0706990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Sagar DR, Elmes SJR, Kendall DA, Chapman V (2007) Cannabinoid CB2 receptor-mediated anti-nociception in models of acute and chronic pain. Mol Neurobiol 36:26–35. 10.1007/s12035-007-8007-7 [DOI] [PubMed] [Google Scholar]
- Kuehnemuth B, Piseddu I, Wiedemann GM, Lauseker M, Kuhn C, Hofmann S, Schmoeckel E, Endres S, Mayr D, Jeschke U, Anz D (2018) CCL1 is a major regulatory T cell attracting factor in human breast cancer. BMC Cancer 18:1278. 10.1186/s12885-018-5117-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Laning J, Devi S, Mak J, Schall TJ, Dorf ME (1994) Biologic activities of the murine β-chemokine TCA3. J Immunol 153:4616–4624 [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kamin-ski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90. 10.1016/0006-2952(95)00109-D [DOI] [PubMed] [Google Scholar]
- Miller MD, Krangel MS (1992) The human cytokine I-309 is a monocyte chemoattractant. Proc Natl Acad Sci USA 89:2950–2954. 10.1073/pnas.89.7.2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Tomonaga D, Kitazono K, Yoshioka Y, Liu J, Rousseau JP, Kinkead R, Ruff MR, Pert CB (2018) Neuropathic pain inhibitor, RAP-103, is a potent inhibitor of microglial CCL1/CCR19. Neurochem Int 119:184–189. 10.1016/j.neuint.2017.12.005 [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ (2001) Chemokines and glycoprotein 120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci 21:5027–5035. 10.1523/JNEUROSCI.21-14-05027.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshio T, Kawashima R, Kawamura YI, Hagiwara T, Mizutani N, Okada T, Otsubo T, Inagaki-Ohara K, Matsukawa A, Haga T, Kakuta S, Iwakura Y, Hosokawa S, Dohi T (2014) Chemokine receptor CCR21 is required for lipopolysaccharide-triggered cytokine production in mouse peritoneal macrophages. PLoS ONE 9:e94445. 10.1371/journal.pone.0094445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathy SS, Masocha W (2015) Coadministration of indomethacin and minocycline attenuates established paclitaxel-induced neuropathic thermal hyperalgesia: Involvement of cannabinoid CB1 receptors. Sci Rep 5:10541. 10.1038/srep10541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflücke D, Hackel D, Mousa SA, Partheil A, Neumann A, Brack A, Rittner HL (2013) The molecular link between C–C-chemokine ligand 2-induced leukocyte recruitment and hyperalgesia. J Pain 14:897–910. 10.1016/j.jpain.2013.02.012 [DOI] [PubMed] [Google Scholar]
- Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, Felder CC (2002) Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther 301:1020–1024. 10.1124/jpet.301.3.1020 [DOI] [PubMed] [Google Scholar]
- Roos RS, Loetscher M, Legler DF, Clark-Lewis I, Baggiolini M, Moser B (1997) Identification of CCR25, the receptor for the human CC chemokine I-309. J Biol Chem 272:17251–17254. 10.1074/jbc.272.28.17251 [DOI] [PubMed] [Google Scholar]
- Rollins BJ (1997) Chemokines. Blood 90:909–928 [PubMed] [Google Scholar]
- Rump L, Mattey DL, Kehoe O, Middleton J (2017) An initial investigation into endothelial CC chemokine expression in the human rheumatoid synovium. Cytokine 97:133–140. 10.1016/j.cyto.2017.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saika F, Kiguchi N, Kobayashi Y, Fukazawa Y, Kishioka S (2012) CC-chemokine ligand 4/macrophage inflammatory protein-1β participates in the induction of neuropathic pain after peripheral nerve injury. Eur J Pain 16:1271–1280. 10.1002/j.1532-2149.2012.00146.x [DOI] [PubMed] [Google Scholar]
- Sánchez López AJ, Román-Vega L, Ramil Tojeiro E, Giuffrida A, García-Merino A (2015) Regulation of cannabinoid receptor gene expression and endocannabinoid levels in lymphocyte subsets by interferon-β: a longitudinal study in multiple sclerosis patients. Clin Exp Immunol 179:119–127. 10.1111/cei.12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sido JM, Nagarkatti PS, Nagarkatti M (2016) Production of endocannabinoids by activated T cells and B cells modulates inflammation associated with delayed-type hypersensitivity. Eur J Immunol 46:1472–1479. 10.1002/eji.201546181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T, Suhara Y, Takayama H, Waku K (2000) Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem 275:605–612. 10.1074/jbc.275.1.605 [DOI] [PubMed] [Google Scholar]
- Tiffany HL, Lautens LL, Gao JL, Pease J, Locati M, Combadiere C, Modi W, Bonner TI, Murphy PM (1997) Identification of CCR31: a human monocyte and thymus receptor for the CC chemokine I-309. J Exp Med 186:165–170. 10.1084/jem.186.1.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicari AP, Zlotnik A (1996) Mouse NK1.1+ T cells: a new family of T cells. Immunol Today 17:71–76. 10.1016/0167-5699(96)80582-2 [DOI] [PubMed] [Google Scholar]
- White FA, Bhangoo SK, Miller RJ (2005) Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov 4:834–844. 10.1038/nrd1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ (2007) Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci USA 104:20151–20158. 10.1073/pnas.0709250104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SD, Burd PR, Billings PR, Martin CA, Dorf ME (1988) The expression and regulation of a potential lymphokine gene (TCA3) in CD4 and CD8 T cell clones. J Immunol 141:1563–1570 [PubMed] [Google Scholar]
- Zen Y, Liberal R, Nakanuma Y, Heaton N, Portmann B (2013) Possible involvement of CCL1-CCR36 interaction in lymphocytic recruitment in IgG4-related sclerosing cholangitis. J Hepatol 59:1059–1064. 10.1016/j.jhep.2013.06.016 [DOI] [PubMed] [Google Scholar]
- Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ (2005) A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci USA 102:4536–4541. 10.1073/pnas.0406030102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LL, Shu RC, Li Nan, Wang ZF, Wang CY, Wang HY, Yu YH, Wang G-L (2015) Involvement of CCL1/CCR38 in spinal cord dorsal horn in remifentanil-induced hyperalgesia in rats. J Anesth Perioper Med 2:53–60. 10.1097/AJP.0000000000000319 [Google Scholar]
- Zhou YQ, Gao HY, Guan XH, Yuan X, Fang GG, Chen Y, Ye DW (2015) Chemokines and their receptors: potential therapeutic targets for bone cancer pain. Curr Pharm Des 21:5029–5033. 10.2174/1381612821666150831141931 [DOI] [PubMed] [Google Scholar]
- Zychowska M, Rojewska E, Piotrowska A, Kreiner G, Nalepa I, Mika J (2017) Spinal CCL1/CCR8 signaling interplay as a potential therapeutic target - Evidence from a mouse diabetic neuropathy model. Int Immunopharmacol 52:261–271. 10.1016/j.intimp.2017.09.021 [DOI] [PubMed] [Google Scholar]