

Abstract
Many strategies have been developed to produce high levels of biologically active recombinant proteins in plants for biopharmaceutical purposes. However, the production of an active form of human iduronate-2-sulfatase (hIDS) for the treatment of Hunter syndrome by enzyme replacement therapy (ERT) is challenging due to the requirement for cotranslational modification by a formylglycine-producing enzyme encoded by sulfatase modifying factor 1 (hSUMF1) at the Cys84 residue, which converts it to C(alpha)-formylglycine. In this study, we have shown that hIDS can be highly expressed in N. benthamiana by using different constructs. Among them, BiP-GB1-L-dCBD1-2L-8xHis-L-6xHis-3L-EK-hIDS-HDEL (GB1-CBD1-hIDS) showed a high expression level when transiently co-expressed with the turnip crinkle virus gene silencing suppressor P38 and GB1-fused human calreticulin (GB1-CRT1) as a folding enhancer. The hSUMF1 was co-expressed with hIDS for cotranslational modification. The full-length recombinant proteins were purified using Ni2+-NTA affinity resin followed by enterokinase treatment to obtain tag-free hIDS. The N-terminal fragment was removed using microcrystalline cellulose (MCC) beads. The purified active form of hIDS can successfully cleave the sulfate group from an artificial substrate, 4-nitrocatechol sulfate, at a similar level to commercial hIDS expressed in animal cells. These results suggest that plants could be a promising platform for the production of recombinant hIDS.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-73778-x.
Keywords: Nicotiana benthamiana, Recombinant protein production, Iduronate-2-sulfatase, Sulfatase modifying factor 1, Lysosomal storage disease, Enzyme replacement therapy
Subject terms: Plant biotechnology, Molecular engineering in plants
orcid.org/0000-0002-1388-1367.
Mucopolysaccharidosis II (MPS II), also known as Hunter syndrome (OMIM 309900), is a rare X-linked recessive lysosomal storage disease (LSD) caused by mutations in the lysosomal enzyme gene encoding iduronate-2-sulfatase (I2D or IDS, EC 3.1.6.13) located on chromosome Xq28 1. This defect leads to the accumulation of glycosaminoglycans (GAGs) due to the impaired degradation by the deficient IDS enzyme, disrupting cellular metabolism and causing organ dysfunction. Excessive amounts of chondroitin sulfate B (dermatan sulfate) and heparitin sulfate (heparan sulfate) are excreted in the urine2.
MPS II presents as a multisystem disorder with two main clinical phenotypes: severe and attenuated. Severe MPS II patients typically present with intellectual disability, coarse facial features, short stature, skeletal deformities, joint stiffness, retinal degeneration, chronic diarrhea, progressive hearing loss and communicating hydrocephalus3. In contrast, attenuated MPS II patients have similar somatic symptoms but a slower rate of progression and no central nervous system involvement. Life expectancy and disease progression differ significantly between these two phenotypes. Patients with severe MPS II often succumb to the disease in their second decade, whereas some patients with the attenuated form survive into their fifth or sixth decade4.
Currently, ERT is the primary treatment for MPS II patients, although additional treatments such as stem cell transplantation using bone marrow or peripheral blood haematopoietic stem cells are also available5. Two types of recombinant enzymes, idursulfase (Elaprase®) and idursulfase-beta (Hunterase®), are commonly used for ERT. Idursulfase is produced in a human cell line, while idursulfase-beta is produced in Chinese hamster ovary (CHO) cells6. Idursulfase administration by intravenous injection has shown improvements in several parameters, including the 6-minute walk test, forced vital capacity, liver and spleen volumes, joint range of motion and urinary GAG levels7. Similarly, treatment with idursulfase-beta has been shown to improve clinical symptoms in MPS II patients8.
Despite the success of these enzyme therapies, a major drawback is their high cost, partly due to their production in human cell lines and CHO cells8. Consequently, there is a growing demand for alternative production systems with lower costs. Recent studies have explored the production of these lysosomal enzymes in various microorganisms such as Pichia pastoris and Escherichia coli6. However, these microorganisms have certain limitations, including the absence of post-translational modifications, endotoxin contamination, protein misfolding and aggregation9.
As an alternative approach, plants have recently attracted considerable interest as production hosts for recombinant enzymes10. In particular, taliglucerase alfa, used to treat Gaucher disease, has been successfully produced in carrot cells and has received FDA approval in the USA. In this case, a suspension culture system containing transgenic carrot cells was used to produce the enzyme11. Similarly, a culture cell system utilizing BY-2 cells has been used to produce pegunigalsidase alfa for potential use in the treatment of Fabry disease12. These approaches offer promising solutions to the cost issues associated with current ERT.
In addition to the suspension cell culture systems, whole plants, particularly N. benthamiana, have emerged as a preferred host platform due to potential advantages such as rapid production of recombinant proteins, low maintenance costs, easy scalability and no risk of contamination with human pathogens13. Various methods have been developed to harness the potential of plants for recombinant protein production, with the expression vector standing out as a critical component, capable of yielding up to 800 mg/kg fresh weight of leaves when used for transient expression in N. benthamiana14.
In this study, our goal was to achieve high levels of active hIDS production in N. benthamiana. Since the target gene can be transiently expressed in leaf cells by Agrobacterium-mediated plant transformation15, we expected rapid production of recombinant proteins within a week. To achieve this, we designed recombinant genes with different domains for endoplasmic reticulum (ER) targeting, translation enhancement, protein accumulation in the ER and protein purification, which were then tested for their ability to facilitate high-level expression of hIDS. Co-expression of human calreticulin (CRT1) was also used to enhance hIDS expression. In addition, we developed a method to produce tag-free hIDS through a series of steps including Ni2+-NTA affinity chromatography, enterokinase cleavage and microcrystalline cellulose (MCC) bead-based removal of the N-terminal fusion fragment. Finally, we investigated whether hIDS expressed in the ER of N. benthamiana leaf cells could be converted to its active form by co-expression of hSUMF1 encoding the formylglycine producing enzyme. Our results provide evidence that tag-free active hIDS can be produced at high levels in N. benthamiana.
Results
Design of chimeric constructs for expression of hIDS and hSUMF1 and their expression in N. benthamiana
In designing the recombinant constructs, we prioritized three key aspects: achieving high expression levels, facilitating efficient purification, and enabling tag-free production of the target protein. To enhance expression, we chose the endoplasmic reticulum (ER) as the expression site for hIDS, based on previous findings demonstrating about the stability of foreign proteins in the ER16. To ensure ER targeting, we used the leader sequence of Arabidopsis BiP1 (immunoglobulin binding protein) to target the protein to the ER (Fig. 1A). In addition, we incorporated an ER retention motif, HDEL, at the C-terminus of the recombinant protein to ensure its retention within the ER. For efficient solubilization and expression, we introduced the multifunctional B1 domain of streptococcal protein G (GB1). The GB1 domain is highly soluble and has been shown to improve the solubility of fusion proteins in E. coli17. In plants, the GB1 domain increased expression levels of recombinant proteins in N. benthamiana18. We have positioned GB1 at the C-terminus of the BiP leader sequence. Notably, the GB1 domain also facilitates direct detection using HRP-conjugated secondary antibodies without the need for a primary antibody during Western blot analysis19. We also considered the inclusion of the M domain, which contains several N-glycosylation sites known to increase protein expression levels. Studies have shown that the M domain can increase expression levels 6 to 7-fold in N. benthamiana20. We inserted the M domain adjacent to the BiP leader sequence for further enhancement of recombinant protein expression (Fig. 1A).
Fig. 1.

Expression of recombinant hIDS and hSUMF1 in Nicotiana benthamiana by Agrobacterium-mediated infiltration. (A) Schematic diagram of the constructs used for the expression of recombinant hIDS and CRT1. Two different recombinant hIDS constructs were generated using different domains as indicated in the schematic diagram. In addition, hSUMF1 and GB1-CRT1 recombinant constructs were generated for this study as indicated. LB, Left Border; 35 S, CaMV 35 S promoter; 35 S T, CaMV 35 S terminator; TCV-CP, P38 gene silencing suppressor; MacT, MacT promoter; BiP, leader sequence of Arabidopsis BiP1; GB1, B1 domain of streptococcal protein G; L, Linkers; M, a small region (A231 to D290) from extracellular domain of protein tyrosine phosphatase receptor type C (CD45); dCBD1, two copies of cellulose binding domain 1; CBM3, family 3 cellulose binding module-3; His, polyhistidine residues; EK, enterokinase cleavage site; HDEL, ER retention motif; RD29B T, RD29B terminator; HSP T, HSP terminator; 3PR T, 3PR terminator consisting of 35 S terminator with PINII terminator and RB7 matrix attachment sequence; RB, Right Border. (B) Expression level analysis of GB1-CBD1-hIDS and GB1-CBM3-hIDS constructs upon co-expression with GB1-CRT1. The indicated constructs were co-infiltrated into leaf tissues of N. benthamiana. Total protein extracts (5 µg) from the leaf tissues harvested at the indicated dpi were separated by SDS-PAGE and stained with Coomassie brilliant blue (left panel) or analyzed by Western blotting using secondary HRP-conjugated anti-IgG antibody (right panel) (Sec IgG) and western blotting image was spliced to show effective comparison between two constructs by drawing a vertical line (full image is provided in the Supplementary Figure S2A). The arrows indicate the positions of recombinant proteins. Lane M, protein size standard; lane NT, non-transformed leaves; lanes 3–5, 3, 5 and 7 dpi of GB1-CBD1-hIDS, respectively; lanes 6–8, 3, 5 and 7 dpi of TPE of GB1-CBM3-hIDS, respectively and *, non-specific band. (C) Expression of hSUMF1 recombinant construct. The hSUMF1 construct was introduced into leaf tissues of N. benthamiana. Total protein extracts (8 µg) from leaf tissues collected at 3, 5 and 7 dpi were separated by SDS-PAGE and stained with Coomassie brilliant blue or analyzed by Western blotting using anti-HA antibody. Lane M, protein size standard; lane NT, non‐transformed leaves; lane 3–5, hSUMF1 co-expressed with GB1-CRT1 and *, non-specific band.
To purify the recombinant proteins, we incorporated affinity tags into the constructs. To facilitate purification using Ni2+-NTA resins, we chose to use a His tag. We incorporated either two His tags, one with 8 histidine residues and another with 6 histidine residues, or a single His tag with 6 histidine residues before the enterokinase cleavage site (Fig. 1A). To achieve tag-free production of hIDS, we aimed to remove all fusion domains by proteolytic cleavage. Enterokinase was chosen for this purpose due to its ability to specifically cleave without leaving residues after the cleavage site21. We inserted an enterokinase cleavage site, DDDDK, at the N-terminus of hIDS. In addition, we included either family 3 cellulose binding module-3 (CBM3) or two copies of a cellulose binding domain 1 (dCBD1) as affinity tags to facilitate removal of the N-terminal domains after cleavage with enterokinase using low-cost MCC beads (Fig. 1A)22. To increase the flexibility between these domains in the recombinant proteins, we inserted single or multiple copies of the GGGGS linker (L) between them. As a result, the final construct had a structure of BiP-GB1-L-dCBD1-2xL-8xHis-L-6xHis-3xL-EK-hIDS-HDEL (GB1-CBD1-hIDS) or BiP-GB1-M-3xL-CBM3-L-6xHis-3xL-EK-hIDS-HDEL (GB1-CBM3-hIDS). For the expression of these recombinant hIDS constructs, we used an expression vector containing the MacT promoter23, a 21 bp immediate 5’UTR known for its high translational efficiency24, RD29B23, and 3PR terminator25. The 21 bp immediate 5’ UTR has been shown to have several times higher translation efficiency in different plant species26. For the expression of hSUMF1, we designed a relatively simple construct consisting of only the BiP leader sequence and an HA epitope at the N- and C-termini, respectively (Fig. 1A).
To enhance the expression of the target genes, we also investigated the incorporation of two additional genes as secondary helper genes. One of these genes encodes the turnip crinkle virus (TCV) gene silencing suppressor P3827 and the other is the homo sapiens calreticulin 1 (CRT1) gene28. The P38 gene was introduced into the same expression vector (Fig. 1A). Although this study did not directly investigate the effect of P38 on hIDS expression, previous studies described that P38 increases the expression level of various proteins by suppressing the gene silencing effect29. For CRT, we generated a fusion construct BiP-GB1-CRT1 (GB1-CRT1) to facilitate high-level expression of CRT1 (Fig. 1A)30.
Transient expression of recombinant hIDS and hSUMF1 in N. benthamiana
The expression level of hIDS was examined by preparing several constructs with different domains and ER retention signal (Supplementary Figure S1A). As a control for the basal expression level of hIDS, constructs with or without the C-terminal HDEL motif, the ER retention signal, were prepared. The hIDS construct without HDEL showed an almost undetectable level of expression. The expression levels were gradually increased by fusing GB1 at the N-terminus of hIDS (Supplementary Figure S1B). Based on the expression levels of different constructs of hIDS, two different constructs were selected for further studies: GB1-CBD1-hIDS and GB1-CBM3-hIDS (Fig. 1A).
These constructs, together with GB1-CRT1, were introduced into Agrobacterium for expression in N. benthamiana leaf tissues. Leaf tissues harvested at 3, 5 or 7 dpi were used for the preparation of total protein extracts. Evaluation of the hIDS and GB1-CRT1 recombinant constructs was performed by Western blot analysis and Coomassie brilliant blue (CBB) staining of SDS-PAGE gels. The image of same Western blotting membrane with same exposure was spliced to show an effective comparison between GB1-CBD1-hIDS and GB1-CBM3-hIDS constructs by drawing a vertical separation line (Fig. 1B and Supplementary Figure S2A). In leaf tissue infiltrated with GB1-CBD1-hIDS and GB1-CRT1, two bands were observed at positions of 110 and 65 kDa, corresponding to the expected sizes based on the calculated molecular masses of GB1-CBD1-hIDS and GB1-CRT1, respectively. The presence of the GB1 domain in these recombinant proteins allowed the simultaneous detection of both hIDS and CRT1 proteins by HRP-conjugated secondary anti-IgG (Sec IgG) during Western blot analysis19. Similarly, two bands at 120 and 65 kDa were detected in leaf tissue infiltrated with GB1-CBM3-hIDS and GB1-CRT1 constructs. Additional minor bands observed in the Western blot image were also detected in non-transformed plant extracts, indicating their non-specific nature. These results suggest that the recombinant hIDS proteins can be expressed without significant degradation. However, the expression levels of GB1-CBD1-hIDS and GB1-CBM3-hIDS were different. GB1-CBD1-hIDS showed high expression at 3 dpi, which was maintained at similar levels up to 7 dpi. Quantification based on image signal intensity showed the highest expression level at 5 dpi, which was 1.32-fold higher than at 3 dpi and 7 dpi. In contrast, GB1-CBM3-hIDS showed its highest expression at 5 dpi, followed by a decrease at 3 and 7 dpi. Furthermore, GB1-CBD1-hIDS was expressed at higher levels than GB1-CBM3-hIDS at all three time points (Supplementary Figure S2B). Therefore, GB1-CBD1-hIDS expressed at 5 dpi was selected for the subsequent analysis.
It is well known that hIDS is converted to its active form by cotranslational modification of Cys84 to C-alpha-formylglycine by the formylglycine-generating enzyme encoded by hSUMF131. We therefore started by investigating the expression of hSUMF1 in N. benthamiana. The hSUMF1 construct was introduced into Agrobacterium and the Agrobacterium suspension was then used to infiltrate leaf tissue of N. benthamiana. The expression of hSUMF1 was assessed at 3, 5 and 7 dpi by Western blot analysis using an anti-HA antibody. As shown in Fig. 1C, hSUMF1 was detected at the expected position of 45 kDa and its expression remained stable at all time points examined. Next, we co-expressed GB1-CBD1-hIDS and hSUMF1 by co-infiltration of Agrobacteria carrying GB1-CBD1-hIDS, hSUMF1 and GB1-CRT1. The ratio of Agrobacteria carrying GB1-CBD1-hIDS and hSUMF1 was adjusted to either 1:1 or 1:4. Total protein extracts from leaf tissues collected at 5 dpi were separated by SDS-PAGE and analyzed by Western blotting using an anti-HA antibody or Coomassie brilliant blue (CBB) staining. GB1-CBD1-hIDS, GB1-CRT1 and hSUMF1 were detected at positions of 110 kDa, 65 kDa and 45 kDa, respectively (Fig. 2). Notably, the expression level of GB1-CBD1-hIDS and hSUMF1 remained similar at both 1:1 and 1:4 ratios, indicating that the expression of GB1-CBD1-hIDS is not affected by the co-expression of hSUMF1.
Fig. 2.

Co-expression of recombinant hIDS and hSUMF1 in Nicotiana benthamiana. The GB1-CBD1-hIDS and hSUMF1 constructs were co-infiltrated at ratio of 1:1 or 1:4. Total protein extracts (8 µg) from leaf tissues collected at 3, 5 and 7 dpi were separated by SDS-PAGE and stained with CBB (left panel), or analyzed by Western blotting using anti-HA antibody (right panel). Lane M, protein size standard; lane NT, non-transformed leaves; lane 3–5, GB1-CBD1-hIDS co-expressed hSUMF1 with a ratio of 1:1; lane 6–8, GB1-CBD1-hIDS co-expressed hSUMF1 with a ratio of 1:4.
Production of tag-free hIDS from plants
After co-expression of GB1-CBD1-hIDS with hSUMF1, we proceeded for the purification of recombinant active form of GB1-CBD1-hIDS (GB1-CBD1-hIDS-su). Since GB1-CBD1-hIDS-su was His-tagged, we used Ni2+-NTA affinity chromatography as the first purification step. Using 10 volumes (v/w) of extraction buffer for 20 g of leaf tissue harvested at 5 dpi, total protein extracts were applied to a column containing Ni2+-NTA resin. The flow-through (FT1) was then reapplied to the resin to maximize protein binding. The column was then washed twice with elution buffer containing 10 mM imidazole to remove loosely bound non-specific proteins from the resin. Finally, GB1-CBD1-hIDS-su was eluted using elution buffer supplemented with 500 mM imidazole. Throughout the purification process, fractions from different steps were collected and analyzed by SDS-PAGE followed by Coomassie brilliant blue (CBB) staining and Western blotting using an HRP-conjugated anti-mouse IgG secondary antibody (Fig. 3A). Predominantly GB1-CBD1-hIDS-su at 110 kDa was detected in the elution fraction (E). In addition, a small amount of GB1-CBD1-hIDS-su was observed in the flow-through fractions (FT1 and FT2) and the first wash (W1), but not in the W2 fraction (Fig. 3A).
Fig. 3.

Purification for active form hIDS-su from total protein extracts. (A) Purification of GB1-CBD1-hIDS-su. Total protein extracts (TPE) from infiltrated leaves were purified using Ni2+-NTA resin. Proteins in the fractions from the purification process were separated by SDS-PAGE followed by CBB staining (left panel) and Western blot analysis (right panel). In the CBB staining, Lane M, protein size standard; lane NT, non-transformed leaves; lane 3, total protein extract of hIDS; lane 4–5, flow though samples; lane 6–7, washing-off solutions; lane 8, eluents; lane 9, proteins bound to Ni2+-NTA after elution using 500 mM imidazole. (B) Elution profile of Ni2+-NTA purified GB1-CBD1-hIDS-su on size exclusion column. Purified proteins using Ni2+-NTA affinity column chromatography were loaded to the size exclusion column and fractions were collected. (C) Analysis of fractions from size exclusion chromatography of GB1-CBD1-hIDS-su. The indicated fractions (40–53) were separated by SDS-PAGE, followed by CBB staining. (D) Quantification of purified GB1-CBD1-hIDS-su. Fractions from 40 to 53 of size exclusion chromatography for GB1-CBD1-hIDS-su were combined and concentrated to 4 mL. 1, 2, 4 or 8 µL of purified GB1-CBD1-hIDS-su were separated by SDS-PAGE along with human serum albumin (HSA, 0.25 to 2.0 µg) as reference. The concentration was estimated by comparing HSA.
Size exclusion chromatography was used to further purify GB1-CBD1-hIDS-su and to remove minor contaminants from the Ni2+-NTA affinity resin eluent. The eluent was first concentrated by centrifugal force using a Centricon device and then loaded onto the size exclusion column. Fractions were then collected, focusing on the main peak (40 to 53), and analyzed for protein purity by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining and Western blotting using an HRP-conjugated secondary antibody. Our analysis confirmed that the fractions within the main peak (40 to 53) contained predominantly GB1-CBD1-hIDS-su with minimal impurities (Fig. 3B and C). Therefore, the fractions from 40 to 53 were pooled and further concentrated to 4 mL using Centricon. Protein concentration was determined using the Bradford method by comparing the intensity of the CBB-stained GB1-CBD1-hIDS-su band with a reference protein, human serum albumin (HSA), at different concentrations. The final protein concentration of GB1-CBD1-hIDS-su was estimated to be approximately 515 ng/µL (Fig. 3D).
Following the same purification protocol, we purified GB1-CBD1-hIDS expressed without co-expression of hSUMF1 (Supplementary Figure S4A). The total yield of GB1-CBD1-hIDS in 4 mL was approximately 345 ng/µL, as determined by the Bradford method and by comparing the band intensity of CBB-stained hIDS with that of human serum albumin (HSA) at different levels (Supplementary Figure S4B). The purified GB1-CBD1-hIDS or GB1-CBD1-hIDS-su had concentrations of 0.69 and 1.03 mg/g FW, respectively, indicating a high yield of recombinant hIDS fusion proteins. In addition, the plants remain in good condition after overexpression of hIDS in N. benthamiana and no visible necrosis was observed in the infiltrated leaves after Agrobacterium infiltration (Supplementary Figure S3A and S3B).
We aimed to produce tag-free hIDS-su from purified GB1-CBD1-hIDS-su. In the GB1-CBD1-hIDS construct, we incorporated an enterokinase site at the N-terminus of hIDS. Purified GB1-CBD1-hIDS-su was incubated with enterokinase to release tag-free hIDS-su in vitro. The 110 kDa protein band largely disappeared and was replaced by a new band at 70 kDa for tag-free hIDS-su together with a band at 40 kDa corresponding to GB1-CBD1, indicating that enterokinase specifically cleaved GB1-CBD1-hIDS-su with high efficiency (Fig. 4A).
Fig. 4.

Production and purification of tag-free hIDS via enterokinase cleavage. (A) Proteolytic cleavage of purified GB1-CBD1-hIDS-su using enterokinase. Purified GB1-CBD1-hIDS-su (2.5 mg) was incubated with enterokinase (50 units) at 25oC for 16 h. An aliquot of incubated samples together with other controls was separated by SDS-PAGE followed by CBB staining. Lane M, protein size standard; lane NT, non-transformed leaves; lane 3, GB1-CBD1-hIDS-su purified using Ni2+-NTA resins; lane 4, enterokinase-treated GB1-CBD1-hIDS-su; lane 5, MCC bead-bound proteins; lane 6, supernatant contains only hIDS-su protein. (B) Quantification of tag-free hIDS-su. The incubated mixture from A was incubated with MCC beads and the supernatant was collected to recover the tag-free hIDS-su. The supernatant was concentrated using Centricon to 4 mL of final volume. The indicated volumes of hIDS-su were separated by SDS-PAGE together the indicated amount of human serum albumin (HSA), followed by staining with CBB.
MCC beads were then used to purify tag-free hIDS-su from the enterokinase digestion mixture. The N-terminal fragment containing CBD1 showed a high binding affinity to MCC beads. Therefore, the cleavage reaction mixture was incubated with MCC beads and the supernatant containing the tag-free hIDS-su was collected. This approach allowed us to remove the N-terminal fragment as well as any uncleaved small amounts of full-length GB1-CBD1-hIDS-su (Fig. 4A). Similarly, we produced tag-free hIDS expressed alone (Supplementary Figure S5A). Tag-free hIDS-su and hIDS were quantified using the Bradford method by comparing the band intensity of CBB-stained hIDS-su and hIDS with that of HSA at different concentrations (Fig. 4B and Supplementary Figure S5B). The final concentrations of hIDS and hIDS-su were approximately 185 and 365 ng/µL, corresponding to 0.37 and 0.73 mg/g FW, respectively. These results indicate that we were able to produce tag-free hIDS in high yield from N. benthamiana with a purity of > 95% in both cases.
Co-expressing hIDS with hSUMF1 leads to a high level of activity
We evaluated the activity of hIDS produced in N. benthamiana (Supplementary Figure S6A). Enzyme activity was determined by measuring the amount of p-nitrocatechol released from the substrate p-nitrocatechol sulfate. First, a standard curve was constructed using p-nitrocatechol at various concentrations such as 3.125, 6.25, 12.5, 25, 50, 100, 200 and 400 nmol (Supplementary Figure S6B)32. The specific activity of plant-produced hIDS and hIDS-su was measured together with commercial hIDS expressed in HEK293 (Cat. No. 10337-H08H, Sino Biological, USA) as a positive control and assay buffer only and assay buffer with p-nitrocatechol sulfate salt as a negative control. Plant-produced hIDS exhibited an activity of 1.01 ± 0.01 nmol/min/µg, whereas hIDS-su co-expressed with hSUMF1 at a ratio of 1:1 and 1:4 exhibited activities of 2.76 ± 0.23 and 8.10 ± 0.06 nmol/min/µg, respectively, indicating that hIDS can be activated by co-expressed hSUMF1 in plants in a manner dependent on the expression level of hSUMF1. The positive and negative controls showed activities of approximately 9.78 ± 0.09 and 0.27 nmol/min/µg, respectively (Fig. 5).
Fig. 5.

Enzymatic activity of tag-free hIDS produced in N. benthamiana. Purified tag-free hIDS were incubated with an artificial substrate p-nitrocatechol sulfate dipotassium salt (4-PNCS) at 37oC for 24 h. Blank, assay buffer alone; hIDS/HEH293, commercial hIDS produced in HEK293 used as positive control; hIDS, hIDS produced without hSUMF1; hIDS-su(1:1 and 1:4), hIDS produced with co-expression of hSUMF1 at ratios of 1:1 and 1:4, respectively. Error bars indicate the mean ± standard deviation (n = 5). Statistical data analysis was performed by Student’s t test (GraphPad Prism 6) and asterisks indicate significant differences (***p < 0.0001).
Plant-produced hIDS-su shows functional stability
We further characterized plant produced hIDS-su under different conditions. We used hIDS-su produced under the condition of co-infiltration of GB1-CBD1-hIDS and hSUMF1 in a ratio of 1:4. Enzymatic activity was measured at different pH and temperature conditions4. The highest activity was observed at pH 5.0, followed by a 50% and 83% decrease in activity at pH 3.0 and 9.0, respectively (Fig. 6A). The effect of pH on the enzymatic activity of plant-produced hIDS was similar to that of lysosomal hIDS33.
Fig. 6.

The effect of pHs and temperatures on the enzymatic activity of plant-produced hIDS-su. (A) Effect of pHs on the activity of hIDS-su. Purified hIDS-su was incubated at different pH values for 24 h of reaction before measuring enzymatic activity. The preincubated samples were used for enzymatic activity assay at pH 5.0 ± 0.2. (B) Effect of temperature on the activity of hIDS-su. Purified hIDS-su was incubated at various temperatures such as 4oC, 25oC, 37oC, 45oC, and 60oC for 24 h before reaction and the preincubated proteins were used for enzymatic activity measurement at 37oC. The activities at the standard conditions (pH 5.0 and 37oC) at time 24 h were taken as 100%. The activities at various pH values and temperatures were reported as relative activity to those at the standard conditions. The data were presented as mean ± standard deviation (n = 5).
Regarding the thermal stability of the enzyme, it exhibited the highest activity at 37°C, followed by 45°C and 60°C (Fig. 6B). In addition, plant-produced hIDS showed much lower enzymatic activity at lower temperatures such as 25°C and 4°C, similar to that produced in animal cells34, confirming that plant-produced hIDS has similar biophysical properties to that produced in animal cells.
hSUMF1 modifies cysteine residue at active site of hIDS to formylglycine at high efficiency in plants
We investigated whether the cysteine residue at the active site of hIDS was modified to formylglycine by co-expression of hSUMF1. Purified hIDS expressed alone or together with hSUMF1 was analyzed by LC-MS/MS. After trypsin digestion, the 84th position cysteine or formylglycine residue was included in the 28-residue peptides. They were then analyzed by Total Ion Chromatogram (TIC), Extracted Ion Chromatogram (XIC) and MS Spectrum35. The results showed that the 28-residue peptide produced by hIDS was detected at the position of 1024.14 Da. In contrast, the 28-residue peptide from hIDS-su(1:4) was detected at both 1024.14 Da and 1000.17 Da with an intensity of 203 and 687, respectively (Supplementary Figure S7A-E). The ratio between modified and unmodified at the 84th cysteine residue was 3.38. These results indicate that hIDS was modified to the active form with 77% efficiency when co-expressed with hSUMF1.
Discussion
In this study, we developed a strategy to produce the active form of hIDS in a cost-effective manner. Recently, N. benthamiana has been widely used for rapid and large-scale expression of recombinant proteins via Agrobacterium-mediated infiltration36. In addition, transgenic plants with stable expression of recombinant genes can provide even more cost-effective means of producing recombinant proteins, although this approach requires a longer period of time to establish transgenic lines37. Recently, many viral vectors have been developed to achieve high levels of recombinant protein expression in plants, with yields ranging from 0.5 to 5 g/kg fresh leaf biomass or more than 50% of total soluble proteins38. In addition, the viral vector gene is not integrated into the plant genome. This approach holds promise for the economic production of a wide range of recombinant proteins, including hIDS, paving the way for potential therapeutic applications.
We implemented several strategies to express hIDS at high levels in N. benthamiana and to obtain tag-free hIDS from hIDS fusion proteins. First, we aimed to reduce the cost of hIDS production by using N. benthamiana as an expression host and expressing the recombinant hIDS fusion gene via Agrobacterium-mediated transient expression. In addition, we inserted a 21 bp translation-enhancing sequence in the immediate 5’ UTR to increase translation efficiency26. We also used the GB1 domain at the C-terminus of the BiP leader sequence to increase the expression level and solubility of the protein19. The GB1 domain has also been shown to increase the solubility of aggregation-prone recombinant proteins in E. coli39,40. The expression level achieved for GB1-CBD1-hIDS was 2 g/kg fresh leaf weight, which is considered high. Previous studies have shown that model proteins such as GFP can be obtained at levels of 3–5 g/kg fresh weight when expressed in the cytosol using high expression vectors such as magnICON® and geminiviral vectors41. However, expression levels of true targets such as IgG are typically lower than GFP. In this study, using a traditional binary vector with translation-enhancing domains, we achieved expression levels close to 2 g/kg fresh weight. Further increases in expression levels would be desirable to further reduction of production costs.
In designing the recombinant construct for hIDS, an important consideration was the purification strategy from total protein extracts. Several approaches can be used to purify proteins produced in plants, similar to other systems such as animal cells and E. coli. One approach is to secrete proteins into the apoplast when plant cells are grown in medium42, while another is to purify proteins from total cellular extracts of whole plants43. Our approach was to use affinity tag-based purification. Therefore, we included His tags to facilitate protein purification from total protein extracts. In addition, we included another affinity tag, double CBD1 or CBM3 domains, which have a high binding affinity to MCC beads. The CBD1 or CBM3 tag was designed to remove the N-terminal fragment released after cleavage with enterokinase. Indeed, we were able to purify GB1-CBD1-hIDS-su from total protein extracts to a high degree of purity using Ni2+-NTA resin, although not to complete purity. A final consideration was the production of tag-free hIDS. We incorporated a proteolytic cleavage site for enterokinase at the N-terminus of hIDS-su. Enterokinase is known to cleave on the C-terminal side of the lysine residue at the cleavage site, DDDDK44. Thus, the GB1-CBD1-hIDS-su can be proteolytically cleaved into two fragments, yielding the N-terminal domain with all the fusion domains and the C-terminal domain with only hIDS. For efficient cleavage by enterokinase, we have inserted His tags and 3 copies of the GGGGS linker at the N-terminus of the DDDDK site to allow easy access to the cleavage site by enterokinase. Indeed, after treatment with enterokinase, GB1-CBD1-hIDS-su was cleaved with an efficiency of over 90%.
A crucial aspect of hIDS production is ensuring that the active form of the enzyme is produced in plants. Like other members of the sulfatase family, hIDS undergoes a common post-translational modification in which a Cys or Ser residue on the active enzyme core is converted to formylglycine (FGly)45. Specifically, in the case of hIDS, the cysteine at position 84 is cotranslationally modified to FGly within the endoplasmic reticulum (ER) to produce active enzyme46. The enzyme responsible for this conversion is hSUMF1, which converts the 84th residual cysteine in the hIDS active site to FGly. The process of SUMF1-mediated FGly modification of hIDS occurs in two steps: first, hSUMF1 oxidizes the cysteine residue to a sulfenic acid intermediate, and then it transfers a formyl group from its own active site to the sulfenic acid intermediate to form FGly. Previous studies have shown that when different types of sulfatases are expressed in prokaryotes, about 60% carry the FGly residue at their active site, while the remaining 40% contain the original amino acid residue encoded by the gene sequence47. Plant cells lack enzymes capable of converting the cysteine residue to FGly. One approach to overcome this is to co-express hSUMF1 with hIDS in plant cells. Transient expression in N. benthamiana allows multiple genes to be expressed simultaneously, either by co-infiltrating two different types of Agrobacteria or by using a binary vector carrying multiple target genes. Indeed, when hIDS was expressed alone, it showed no significant activity with the artificial substrate 4-nitrocatechol sulfate. However, when hSUMF1 was co-expressed with GB1-CBD1-hIDS at a ratio of GB1-CBD1-hIDS to hSUMF1 of 1:1 and 1:4, the activity of hIDS increased 3-fold and 8-fold, respectively, compared to that without hSUMF1 co-expression. This result suggests that the level of hSUMF1 co-expressed with hIDS is critical for the full conversion of hIDS to the active form. In addition, biochemical analysis of hIDS co-expressed with hSUMF1 showed that the modification of Cys84 to FGly84 occurred with 77% efficiency in N. benthamiana.
It is important to consider the biophysical similarity of plant hIDS to its counterparts in animal cells. To investigate this, we examined the activity of plant-produced hIDS in terms of pH optimum and heat stability. Remarkably, plant-produced hIDS exhibited similar behavior to that produced in animal cells, indicating comparable biophysical properties. This similarity was observed in terms of pH optimum and temperature stability, further confirming that plant-produced hIDS is biophysically equivalent to its animal cell-derived counterpart48.
Since we have fused the HDEL motif as an ER retention signal to the C-terminus of hIDS, it was expected that hIDS localizes to the ER. Thus, the N-glycosylation pattern of hIDS should be high mannose form. In animal cells, proteins with the high mannose form of N-glycan can be taken up by the mannose-6-phosphate (M6P) receptor that recognizes the mannose residues of N-glycan. Therefore, the high mannose form of N-glycan of hIDS is crucial for its uptake in mammalian cells and enzyme replacement therapy. This strategy has been successfully employed with taliglucerase alfa (Elelyso®)11,49.
In conclusion, this study represents a significant milestone as it demonstrates, to the best of our knowledge, the successful production of highly active hIDS in plants by co-expression with hSUMF1. Moving forward, further research is warranted to validate our initial findings regarding the production of active hIDS in N. benthamiana. In addition, it is important to assess the safety and efficacy of recombinant hIDS produced in plants, paving the way for potential therapeutic applications.
Materials and methods
Plasmid construction
The Homo sapiens iduronate-2-sulfatase (hIDS), transcript variant 1, mRNA proprotein sequence (GenBank Accession No. NM_000202.8, Region 245–1819) contains 525 amino acid residues that were codon-optimized for N. benthamiana. The hIDS gene was chemically synthesized with several additional molecular features. These include an XmaI restriction endonuclease site, a purification tag consisting of 6 histidine residues and an enterokinase cleavage site (Asp-Asp-Asp-Asp-Lys▼) at the 5’ end. In addition, an endoplasmic reticulum (ER) retention motif, HDEL, and an XhoI restriction endonuclease site at the 3’ end of the sequence were incorporated (provided by Gene Universal, USA). Similarly, the Homo sapiens sulfatase modifying factor 1 (hSUMF1) transcript variant 1, mRNA proprotein sequence (GenBank Accession No. NM_182760.4, Region 187–1209) included 341 amino acids that were codon-optimized for N. benthamiana and chemically synthesized with specific features. These include a BamHI restriction endonuclease site at the 5’ end, an HA tag for epitope recognition, an ER retention signal motif (His-Asp-Glu-Leu or HDEL) and an XhoI restriction endonuclease site at the 3’ end (Gene Universal, USA).
To generate a chimeric construct containing CBD1 (GenBank Accession No. EU179903.1) mentioned as pTEX1L-BiP-GB1-L-dCBD1-2L-8xHis-L-6xHis-3L-EK-hIDS-HDEL (GB1-CBD1-hIDS), we used a PCR-based approach. First, the BiP-GB1 (BiP, GenBank Accession No. D89341.1; GB1 domain, PDB Accession No. 3GB1_A) and double CBD1 (dCBD1) fragments were amplified by PCR using the XbaI_BiP forward primer/BamHI_GB1 reverse primer and BamHI_dCBD1 forward primer/XmaI_dCBD1 reverse primer sets, respectively (Supplementary Table S1). These two PCR fragments were fused by overlapping PCR to yield the BiP-GB1-L-dCBD1-2L-8xHis-L fragment, and the resulting PCR products were digested with XbaI and XmaI restriction endonucleases. In addition, the chemically synthesized 6xHis-3L-EK-hIDS-HDEL fragment was digested with XmaI and XhoI restriction endonucleases. These two digested DNA fragments were ligated to a modified binary pCAMBIA-1300 vector (GenBank Accession No. AF234296.1), mentioned as pTEX1L-BiP-HA-mCor1-LysM-His-HDELvector50 digested with XbaI and XhoI restriction endonucleases to generate the GB1-CBD1-hIDS construct.
To generate another chimeric construct containing M domain (GenBank Accession No. AY539692.1) and CBM3 (GenBank Accession No. HQ232851.1) indicated as pTEX1L-BiP-GB1-M-3L-CBM3-L-6xHis-3L-EK-hIDS-HDEL (GB1-CBM3-hIDS) was generated by PCR using BamHI_MP forward primer/MP reverse primer and MP_CBM3 forward primer/XmaI_CBM3 reverse primer sets with MSC-GcCAα3as a template36. These two PCR products were fused by overlap PCR to generate M-3L-CBM3-L. The M-3L-CBM3-L fragment was digested with BamHI and XmaI restriction endonucleases and used to replace dCBD1 of the GB1-CBD1-hIDS construct to generate the final GB1-CBM3-hIDS construct.
A few additional constructs of the hIDS gene (Supplementary Figure S1A) such as pTEX1L-BiP-hIDS (hIDS-x), pTEX1L-BiP-hIDS-HDEL (hIDS-H), pTEX1L-BiP-L-dCBD1-2L-8xHis-L-6xHis-3L-EK-hIDS-HDEL (CBD1-hIDS) were generated by PCR using multiple primers (Supplementary Table S1).
The synthesized hSUMF1 gene was digested with BamHI and XhoI restriction endonucleases and ligated into the pTEX1L-BiP-HA-mCor1-LysM-His-HDEL vector digested with the same restriction endonucleases50 to generate pTEX1L-BiP-hSUMF1-HA-HDEL (hSUMF1).
To generate pTEX1L-BiP-GB1-CRT-HA-HDEL (GB1-CRT1) construct containing CRT1 (GenBank Accession No. NM_004343.4), at first the pTEX1L-BiP-CRT-HA-HDEL construct was digested with BamHI and XhoI restriction endonucleases and then replaced with the GB1-CBD1-hIDS construct digested with the same restriction endonucleases.
To ensure the accuracy of the DNA sequences for all expression constructs, a thorough confirmation was performed by DNA sequencing performed by SolGent Analysis Services (Korea).
Plant material and growth
Model wild-type Nicotiana benthamiana plants were obtained following the implementation of relevant institutional, national, and international laws and regulations. The seeds were kindly provided by Professor Doil Choi at the Department of Plant Science, Seoul National University (Seoul, Korea). The Cellular Systems Biology lab’s greenhouse at Pohang University of Science and Technology (Pohang, Korea) was utilized to grow the plants. The plants were grown in soil composed of 30 g zeolite, 130 g perlite, 705.3 g coco-peat, 130 g peat moss, and 0.1 g wetting agent. The specific plant growth conditions were a 14 h light/10 h dark cycle, with a light intensity of 130–150 µEm−2sec−1 at 26 ± 2 °C and relative humidity of 40–60% for 4–6 weeks. Four-week-old plants were selected for Agrobacterium-mediated syringe infiltration, while six-week-old plants were selected for Agrobacterium-mediated vacuum infiltration. These methods were used for the transient expression of recombinant proteins according to the protocols described by Razzak et al36.
Expression of hIDS and hSUMF1 in N. benthamiana by Agrobacterium-mediated infiltration
The expression constructs, including both the target and modifying genes, were transformed into Agrobacterium tumefaciens (GV3101, Ti pMP90) using a MicroPulser electroporator (Cat. No. 165–2100, Bio-Rad, USA) at 1.8 kV with a 1-mm gap electroporation cuvette51. Colony PCR was then performed to identify positive colonies, which were then cultured in 5 mL LB media supplemented with kanamycin (50 µg/mL) and rifampicin (50 µg/mL).
The overnight Agrobacterium cultures (5 mL) were grown at 28oC and transferred to 50 mL LB media for syringe infiltration or 400 mL LB media for vacuum infiltration into N. benthamiana leaves. After a further 16 h of growth, bacterial cells were harvested by centrifugation at 5,000 xg for 15 min at 25°C. The resulting pellets were resuspended in infiltration buffer (10 mM MES, pH 5.6 and 10 mM MgSO4)52. The cell density was adjusted to 0.6 OD600 by adding additional infiltration buffer to the suspension.
To optimize target gene expression, Agrobacterium carrying the target gene and GB1-CRT1 were co-infiltrated in a 5:1 ratio. For the co-expression of GB1-CBD1-hIDS and hSUMF1, three Agrobacterium cell suspensions, each containing GB1-CBD1-hIDS, hSUMF1 or GB1-CRT1, were mixed at two different ratios (5:5:2 and 5:20:2) to achieve final ratios of 1:1 and 1:4 for GB1-CBD1-hIDS and hSUMF1, respectively. In all cases, the final Agrobacterium suspension was adjusted to 0.6 OD600. Acetosyringone (200 µM), a chemical inducer of virulence gene expression in Agrobacterium, was added to the Agrobacterium suspension and incubated for 2 h at room temperature in the dark prior to infiltration53,54.
In the small-scale expression analysis, four-week-old N. benthamiana plants were used for syringe infiltration. The Agrobacterium-containing infiltration medium was gently injected into the abaxial surface of the first three healthy leaves from the top of each plant. This was achieved by gently pressing with a 1 mL plastic syringe without a needle until the entire leaf surface became wet55.
For the large-scale production and purification of recombinant hIDS, vacuum infiltration was used on six-week-old N. benthamiana plants. The plants were inverted and submerged in a vessel containing the Agrobacterium suspension, and a vacuum pressure of 50 to 400 mbar was applied for 2 min. The vacuum was then slowly released to allow the medium to be absorbed by all the leaves. The plants were then transferred to the greenhouse for further growth56.
Protein extraction and analysis
Leaves were harvested at 3, 5 and 7 days post infiltration (dpi) and immediately stored at -80°C prior to extraction. Leaf tissue (100 mg) was pulverized under liquid nitrogen using a mortar and pestle. The resulting leaf powder was homogenized with 500 µL of protein extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20, 2 mM DTT and 2 mM PMSF). The extraction mixture was vortexed vigorously for 2 min and then sonicated for 2 min with 4 pulses of 10 s each followed by a 5 s interval. After sonication, the extraction mixture was centrifuged at 16,000 xg for 15 min at 4°C. The resulting supernatant and pellet fractions were collected separately to assess the expression levels of the target proteins.
For analysis, 5 µg (35 µL) of total protein extracts (TPE) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by Western blotting. HRP-conjugated sheep anti-mouse IgG secondary antibody (1:5,000) (Cat. No. A90-146 A, Fortis Life Sciences, USA) was used for the detection of hIDS. The expression levels of leaf tissues harvested at 3, 5 and 7 dpi were compared between GB1-CBD1-hIDS and GB1-CBM3-hIDS constructs using ImageJ2 software (NIH, USA) and the results were plotted using GraphPad Prism 6 (Dotmatics, USA).
For hSUMF1 expression analysis, Western blotting analysis was performed using anti-HA antibody (1:5,000) as the primary antibody (Cat. No. H3663, Sigma, USA) and HRP-conjugated anti-mouse IgG (1:5000) as the secondary antibody. In addition, the expression levels of GB1-CBD1-hIDS and hSUMF1 were assessed by Western blotting using anti-HA (1:5000) as the primary antibody, followed by incubation with an HRP-conjugated anti-mouse antibody (1:5,000). Chemiluminescence signals were captured using a LAS 4000 image capture system (Fujifilm Corporation, Japan) by exposing the membranes to light for 10 s under dark conditions. In addition, the SDS-PAGE gels and membranes were stained with Coomassie brilliant blue R-250 for visualization.
Purification of recombinant proteins using Ni2+-NTA resin and size exclusion chromatography
Recombinant hIDS with a polyhistidine tag (6xhis) was purified by Ni2+-NTA affinity chromatography (Qiagen, Valencia, CA) according to the manufacturer’s instructions. First, fresh leaf tissue (20 g) from GB1-CBD1-hIDS co-expressed with hSUMF1 was crushed and homogenized with 10 volumes (w/v) of protein extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20, and 2 mM PMSF). The 200 mL extraction mixture was supplemented with 0.3% activated charcoal and 0.3% polyvinylpyrrolidone, followed by incubation at 4oC for 30 min. After centrifugation (3 times at 16,000 xg at 4oC for 15 min), the supernatant containing total protein extract (TPE) was collected by passing through 22–25 μm rayon-polyester with an acrylic binder Miracloth (Millipore, Merck, USA). The final TPE was filtered through a 0.45 μm syringe filter membrane (GVS filter technology, USA). Imidazole was then added to a final concentration of 2 mM. The TPE was then applied to Ni2+-NTA affinity column equilibrated with 10 mL protein extraction buffer. At the end of the flow, the beads were washed twice with 50 mL PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4) containing 10 mM imidazole. The recombinant hIDS was eluted with 20 mL PBS containing 500 mM imidazole. The elution volume was reduced to 4 mL and the elution buffer was exchanged with PBS using a 10-kDa cutoff membrane filter (Amicon® Ultra-0.5 device, Millipore, Merck, USA).
The purified GB1-CBD1-hIDS was further purified by size exclusion chromatography (Hi Load® 16/600 Superdex® 200 prep grade, Cytiva, Sigma-Aldrich, Merck, USA). Two column volumes were washed with filtered (0.2 μm pore, 47 mm circles, Whatman Nylon Membrane, Cytiva, USA) double-distilled water and PBS, respectively. 1 mL of sample was injected after centrifugation (16,000 xg at 4oC for 15 min) and filtration (0.2 μm pore, Whatman GD/X syringe filters, Merck, USA). All eluted fractions were collected in 96 fraction collection tubes over 1.5 h. The major peak fractions such as 40 to 53, 53 to 80, and 80 to 95 were collected separately and then concentrated using a 10-kDa cutoff membrane filter (Amicon® Ultra-0.5 device, Millipore, Merck, USA). The final concentrated sample was analyzed by Western blotting and CBB staining to confirm the purity of GB1-CBD1-hIDS co-expressed with hSUMF1 (designated as GB1-CBD1-hIDS-su). The final volume of purified protein was 4 mL after SEC steps. Following the same purification protocol, we purified GB1-CBD1-hIDS expressed without co-expression of hSUMF1. The concentration of GB1-CBD1-hIDS-su and GB1-CBD1-hIDS was determined by the band intensity of CBB staining and the Bradford method. Purified GB1-CBD1-hIDS and GB1-CBD1-hIDS-su were mixed with glycerol to 30% of the final concentration and aliquots of protein samples were stored at -80oC for later use.
Proteolytic cleavage by enterokinase
To cleave full-length recombinant hIDS with enterokinase, purified recombinant GB1-CBD1-hIDS-su or GB1-CBD1-hIDS (1 mg/mL) was mixed with two volumes of enterokinase reaction buffer (20 mM Tris-HCl at pH 8. 0, 50 mM NaCl, and 2 mM CaCl2) and then incubated with 10 µL (50 U) enterokinase light chain (Cat. No. P8070, New England Biolabs, USA) at 25oC for 16 h under gentle rotation at 150 rpm according to the manufacturer’s instructions. Enterokinase proteolytically cleaves after the lysine residue at its cleavage site Asp-Asp-Asp-Asp-Lys▼ located at the N-terminus of hIDS-su or hIDS. The N-terminal fragment containing GB1-CBD1 was removed using 50 μm microcrystalline cellulose (MCC) beads (Cat. No. S5504, Sigma, USA) after incubation for 2 h at 4oC, as dCBD1 binds to MCC beads22. After centrifugation at 16,000 xg for 15 min at 4oC, the supernatant containing tag-free hIDS-su or hIDS was collected. Enterokinase-digested proteins were analyzed by Western blotting and CBB staining. Protein quantification for tag-free hIDS-su or hIDS was performed by CBB staining and Bradford protein assay (Bio-Rad, USA).
Enzyme activity assay
Enzyme activity was assessed spectrophotometrically using the artificial substrate 4-nitrocatechol sulfate dipotassium salt (4-PNCS) (Cat. No. N-7251, Sigma, USA)57. Purified recombinant protein samples (5 µg) and control samples were diluted in 250 µL reaction buffer (50 mM sodium acetate, pH 5.0, and 100 mM NaCl). 2 mM substrate solution was prepared in the same reaction buffer. Then, 250 µL of protein sample was mixed with an equal volume of 250 µL substrate solution and incubated at 37°C for 24 h for the enzymatic reaction. The reaction was stopped by adding 500 µL of reaction stop solution (200 mM NaOH, pH 13.3). Enzyme activity was measured in 5 replicates at 200 µL/well in the 96-well maxi-binding plate (Cat. No. 32296, SPL Life Sciences, Korea). The assay included recombinant hIDS produced in HEK293 cells (Cat. No. 10337-H08H, Sino Biological, USA) as a positive control and assay buffer alone as a negative control. A standard curve was generated using p-nitrocatechol (PNC) (Cat. No. N15553, Sigma, USA) at different concentrations (nmol) to quantify the specific enzyme activity of recombinant hIDS. Spectrophotometric absorbance was measured at 510 nm in endpoint mode using a spectrophotometer (SpectraMax ABS, Molecular Devices, USA). Enzyme activity was calculated according to the following formula:
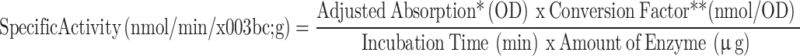 |
*Adjusted for substrate blank, **Derived using calibration standard p-nitrocatechol (PNC) (Catalog # N15553, Sigma, USA).
Characteristics of recombinant hIDS
The effect of pH on hIDS activity was investigated using reaction buffers with different pH values ranging from 3.0 to 9.0. In addition, to evaluate the temperature stability, the enzyme activity was measured at different temperatures, including 4, 25, 37, 45, and 60oC. The activity at different pH and temperature conditions was expressed as relative activity (%) to that at pH 5.0 or temperature 37oC using the following formula:
 |
Cotranslational modification analysis
Cotranslational modification of the active site cysteine residue to formylglycine by hSUMF1 was analyzed for plant-produced purified hIDS and hIDS-su(1:4) as inactive and active forms of hIDS, respectively. Protein samples were digested with 100 µg/µL of trypsin (Roche, Cat. No. 11418025001) in 50 mM Tris-HCl for 17 h at 37oC to isolate 28-residue peptides containing the cotranslational modification residue with Cys84 in inactive form (SPNIDQLASHSLLFQNAFAQQAVCAPSR) and FGly84 at position 84 in active form (SPNIDQLASHSLLFQNAFAQQAVG*APSR). The modification was identified by liquid chromatography tandem mass spectrometry (LC-MS/MS) at Gyeonggido Business & Science Accelerator (Korea). The LC method was performed with Agilent 1290 Infinity II chromatography and AB SCIEX Q-TOF 5600+ mass spectrometry using Acquity UPLC Peptide C18, 1.7 μm, 2.1 × 150 mm column at 50oC with a flow rate of 0.3 mL/min. The injected sample volume was 20 µL and the solvent was DW (0.1% FOA):ACN (0.1% FOA). The MS method was implemented using the positive ([M + H]+) detection ion mode with a scan range of TOF MS 50 ~ 2000 Da. Peak View, BPV flex and Protein Pilot software were used for peptide mapping. The relative proportion between the FGly-form peptide and the Cys-form peptides at position 84 was determined using the peak areas from the extracted ion chromatogram (XIC) data.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants (2022H1D3A2A0209352712 and RS-2023-00235511) from NRF of the Ministry of Science and ICT, and a grant from Nature Biotech Research Program (2023) of Gyeongbuk, Republic of Korea.
Author contributions
I.H. conceived the project, I.H. and M.H.S. designed the experiments, M.H.S. conducted most of the experiments, T.M. contributed technical assistance, M.E. aided protein purification, H.H.N. and A.Y. performed activity assays, M.H.S. and I.H. interpreted results and wrote the manuscript.
Data availability
All data generated or analysed in this study are included in this published article and supplementary information. Raw datasets in the current study and any additional questions can be addressed to the corresponding author(s).
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fukuhara, Y. et al. A cDNA analysis disclosed the discordance of genotype-phenotype correlation in a patient with attenuated MPS II and a 76-base deletion in the gene for iduronate-2-sulfatase. Mol. Genet. Metab. Rep.25, 100692. 10.1016/j.ymgmr.2020.100692 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whiteman, D. A. & Kimura, A. Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): the past, the present and the future. Drug Des. Devel Ther.11, 2467–2480. 10.2147/dddt.S139601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stapleton, M. et al. Newborn screening for mucopolysaccharidoses: measurement of glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep.22, 100563. 10.1016/j.ymgmr.2019.100563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pimentel, N. et al. Production and characterization of a human lysosomal recombinant iduronate-2‐sulfatase produced in Pichia pastoris. Biotechnol. Appl. Biochem.65, 655–664 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Pinto et al. Precision medicine for lysosomal disorders. Biomolecules. 10, 1110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomatsu, S. et al. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des. Dev. Therapy, 1937–1953 (2015). [DOI] [PMC free article] [PubMed]
- 7.Bradley, L. A., Haddow, H. R. & Palomaki, G. E. Treatment of mucopolysaccharidosis type II (Hunter syndrome): results from a systematic evidence review. Genet. Med.19, 1187–1201 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Cho, S. Y., Sohn, Y. B. & Jin, D. K. An overview of Korean patients with mucopolysaccharidosis and collaboration through the Asia Pacific MPS network. Intractable Rare Dis. Res.3, 79–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrer-Miralles, N., Domingo-Espín, J., Corchero, J. L., Vázquez, E. & Villaverde, A. Microbial factories for recombinant pharmaceuticals. Microb. Cell. Fact.8, 1–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schillberg, S. & Finnern, R. Plant molecular farming for the production of valuable proteins–critical evaluation of achievements and future challenges. J. Plant. Physiol.258, 153359 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Tekoah, Y. et al. Large-scale production of pharmaceutical proteins in plant cell culture—the protalix experience. Plant. Biotechnol. J.13, 1199–1208 (2015). [DOI] [PubMed] [Google Scholar]
- 12.van der Veen, S. J., Hollak, C. E., van Kuilenburg, A. B. & Langeveld, M. Developments in the treatment of fabry disease. J. Inherit. Metab. Dis.43, 908–921 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wani, K. I. & Aftab, T. Plant Molecular Farming: Applications and new Directions (Springer Nature, 2022).
- 14.Soni, A. P., Lee, J., Shin, K., Koiwa, H. & Hwang, I. Production of recombinant active human TGFβ1 in nicotiana Benthamiana. Front. Plant. Sci.13, 922694 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bally, J. et al. The rise and rise of Nicotiana benthamiana: a plant for all reasons. Ann. Rev. Phytopathol.56, 405–426 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Wandelt, C. I. et al. Vicilin with carboxy-terminal KDEL is retained in the endoplasmic reticulum and accumulates to high levels in the leaves of transgenic plants. Plant. J.2, 181–192. 10.1046/j.1365-313X.1992.t01-41-00999.x (1992). [DOI] [PubMed] [Google Scholar]
- 17.Hammarström, M., Hellgren, N., van den Berg, S., Berglund, H. & Härd, T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci.11, 313–321. 10.1110/ps.22102 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xueming Zheng, X. W. X. F. D. D. & Feihu, W. Expression and purification of human epidermal growth factor (hEGF) fused with GB1. Biotechnol. Biotechnol. Equip.30, 813–818. 10.1080/13102818.2016.1166984 (2016). [Google Scholar]
- 19.Song, S. J., Diao, H. P., Moon, B., Yun, A. & Hwang, I. The B1 domain of streptococcal protein G serves as a multi-functional tag for recombinant protein production in plants. Front. Plant. Sci.13, 878677 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang, H., Park, Y., Lee, Y., Yoo, Y. J. & Hwang, I. Fusion of a highly N-glycosylated polypeptide increases the expression of ER-localized proteins in plants. Sci. Rep.8, 4612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shahravan, S. H., Qu, X., Chan, I. S. & Shin, J. A. Enhancing the specificity of the enterokinase cleavage reaction to promote efficient cleavage of a fusion tag. Protein Express Purif.59, 314–319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islam, M. R. et al. Cost-effective production of tag‐less recombinant protein in Nicotiana Benthamiana. Plant. Biotechnol. J.17, 1094–1105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song, S. J. et al. SARS-CoV-2 spike trimer vaccine expressed in Nicotiana benthamiana adjuvanted with Alum elicits protective immune responses in mice. Plant. Biotechnol. J.20, 2298–2312. 10.1111/pbi.13908 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, Y. et al. The immediate upstream region of the 5’-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res.42, 485–498. 10.1093/nar/gkt864 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yun, A., Kang, J., Lee, J., Song, S. J. & Hwang, I. Design of an artificial transcriptional system for production of high levels of recombinant proteins in tobacco (Nicotiana Benthamiana). Front. Plant. Sci.14, 1138089 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, Y. et al. The immediate upstream region of the 5′-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res.42, 485–498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu, F., Ren, T. & Morris, T. J. The coat protein of turnip crinkle virus suppresses posttranscriptional gene silencing at an early initiation step. J. Virol.77, 511–522 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia, X. Y., He, L. H., Jing, R. L. & Li, R. Z. Calreticulin: conserved protein and diverse functions in plants. Physiol. Plant.136, 127–138 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Thomas, C. L., Leh, V., Lederer, C. & Maule, A. J. Turnip crinkle virus coat protein mediates suppression of RNA silencing in nicotiana Benthamiana. Virology. 306, 33–41. 10.1016/S0042-6822(02)00018-1 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Margolin, E. et al. Co-expression of human calreticulin significantly improves the production of HIV gp140 and other viral glycoproteins in plants. Plant. Biotechnol. J.18, 2109–2117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dierks, T. et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human Cα-formylglycine generating enzyme. Cell. 113, 435–444 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Wiegmann, E. M. et al. Arylsulfatase K, a novel lysosomal sulfatase. J. Biol. Chem.288, 30019–30028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strovel, E. T., Cusmano-Ozog, K., Wood, T., Yu, C. & Committee, A. L. Q. A. Measurement of lysosomal enzyme activities: a technical standard of the American College of medical genetics and genomics (ACMG). Genet. Med.24, 769–783 (2022). [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez-López, A. et al. Recombinant human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) produced in the methylotrophic yeast Pichia pastoris. Sci. Rep.6, 29329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mashima, R., Ohira, M., Okuyama, T. & Tatsumi, A. Quantification of the enzyme activities of iduronate-2-sulfatase, N-acetylgalactosamine-6-sulfatase and N-acetylgalactosamine-4-sulfatase using liquid chromatography-tandem mass spectrometry. Mol. Genet. Metab. Rep.14, 36–40. 10.1016/j.ymgmr.2017.12.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Razzak, M. A., Lee, D. W., Lee, J. & Hwang, I. Overexpression and purification of Gracilariopsis Chorda carbonic anhydrase (GcCAα3) in Nicotiana Benthamiana, and its immobilization and use in CO2 hydration reactions. Front. Plant. Sci.11, 563721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters, J. & Stoger, E. Transgenic crops for the production of recombinant vaccines and anti-microbial antibodies. Hum. Vacc. 7, 367–374 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Gleba, Y., Klimyuk, V. & Marillonnet, S. Viral vectors for the expression of proteins in plants. Curr. Opin. Biotechnol.18, 134–141 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Peti, W. & Page, R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Express Purif.51, 1–10 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Sugase, K., Landes, M. A., Wright, P. E. & Martinez-Yamout, M. Overexpression of post-translationally modified peptides in Escherichia coli by co-expression with modifying enzymes. Protein Express Purif.57, 108–115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen, Q., He, J., Phoolcharoen, W. & Mason, H. S. Geminiviral vectors based on bean yellow dwarf virus for production of vaccine antigens and monoclonal antibodies in plants. Hum. Vaccines. 7, 331–338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang, X., Chung, K. P., Lin, W. & Jiang, L. Protein secretion in plants: conventional and unconventional pathways and new techniques. J. Exp. Bot.69, 21–37 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Łojewska, E., Kowalczyk, T., Olejniczak, S. & Sakowicz, T. Extraction and purification methods in downstream processing of plant-based recombinant proteins. Protein Express Purif.120, 110–117 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Yuan, L. D. & Hua, Z. C. Expression, purification, and characterization of a biologically active bovine enterokinase catalytic subunit in Escherichia coli. Protein Express Purif.25, 300–304 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Poutou-Piñales, R. A. et al. Human sulfatase transiently and functionally active expressed in E. Coli K12. Electron. J. Biotechnol.13, 5–6 (2010). [Google Scholar]
- 46.Dierks, T. et al. Posttranslational formation of formylglycine in prokaryotic sulfatases by modification of either cysteine or serine. J. Biol. Chem.273, 25560–25564 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Dierks, T. et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 121, 541–552 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Recksiek, M., Selmer, T., Dierks, T., Schmidt, B. & von Figura, K. Sulfatases, trapping of the sulfated enzyme intermediate by substituting the active site formylglycine. J. Biol. Chem.273, 6096–6103 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Grabowski, G. A., Golembo, M. & Shaaltiel, Y. Taliglucerase alfa: an enzyme replacement therapy using plant cell expression technology. Mol. Genet. Metab.112, 1–8. 10.1016/j.ymgme.2014.02.011 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Song, S. J. et al. Plant-based, adjuvant‐free, potent multivalent vaccines for avian influenza virus via Lactococcus surface display. J. Integr. Plant. Biol.63, 1505–1520 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Shamloul, M., Trusa, J., Mett, V. & Yusibov, V. Optimization and utilization of Agrobacterium-mediated transient protein production in Nicotiana. JoVE, e51204 (2014). [DOI] [PMC free article] [PubMed]
- 52.Nosaki, S., Hoshikawa, K., Ezura, H. & Miura, K. Transient protein expression systems in plants and their applications. Plant. Biotechnol.38, 297–304 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wydro, M., Kozubek, E. & Lehmann, P. Optimization of transient Agrobacterium-mediated gene expression system in leaves of Nicotiana Benthamiana. Acta Biochim. Pol.53, 289–298 (2006). [PubMed] [Google Scholar]
- 54.Zheng, L. et al. An improved and efficient method of Agrobacterium syringe infiltration for transient transformation and its application in the elucidation of gene function in poplar. BMC Plant. Biol.21, 1–19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leuzinger, K. et al. Efficient agroinfiltration of plants for high-level transient expression of recombinant proteins. JoVE, e50521 (2013). [DOI] [PMC free article] [PubMed]
- 56.Chen, Q. et al. Agroinfiltration as an effective and scalable strategy of gene delivery for production of pharmaceutical proteins. Adv. Tech. Biol. Med.10.4172/atbm.1000103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Strovel, E. T., Cusmano-Ozog, K., Wood, T. & Yu, C. Measurement of lysosomal enzyme activities: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Medi. 24, 769–783. 10.1016/j.gim.2021.12.013 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed in this study are included in this published article and supplementary information. Raw datasets in the current study and any additional questions can be addressed to the corresponding author(s).