

Abstract
This report describes the development and characterization of a comprehensive collection of CHO cell glycosylation mutants with significant potential for advancing glycobiology and biotechnology. EPO-Fc and trastuzumab, two model molecules, were produced using these mutants to assess the effects of mutated glycogenes, and LC-MS/MS analysis was employed to quantitatively analyse their N-glycans. EPO-Fc exhibited exclusively homogeneous Man9 glycans only when nearly all α-mannosidases in the genome were inactivated, except lysosomal MAN2B1. Some mutants lacking GnT-I activity produce mostly Man5 N-glycans, while their O-glycan and glycolipid profiles can differ due to other mutations in the cell. GnT-II deficiency prevents GnT-V from adding GlcNAc to the core N-glycan, resulting in branches attaching solely to the α1,3-linked mannose, leaving the α1,6-linked mannose free. The mutant-produced antibody’s single-branched glycan contains more sialic acid than the dual-branched glycans produced in CHO-K1 cells. Trastuzumab produced in these mutants provided insights into how Fc N-glycans impact the antibody’s interaction with FcγR1 and FcγR2a, FcγR3a, and their influence on antibody-dependent cellular cytotoxicity (ADCC). In the study of Fc glycans in Fc-FcγR1 and FcγR2a interactions, we observed a consistent glycan-related impact on binding to both receptors, indicating a common interaction mechanism between Fc glycans and both FcγRI and FcγRIIa. CHO mutants produced trimeric gp120 demonstrated distinct reactivity with multiple broadly neutralizing anti-HIV antibodies, confirming the involvement of gp120 glycans in interactions with specific broadly neutralizing antibodies. Finally, one of the mutants produced human β-glucocerebrosidase with uniform Man5 N-glycans, showcasing its potential for glycoengineered production and enhancement in therapeutic efficacy.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-73722-z.
Keywords: CHO cell glycosylation mutants, Genome editing, LC-MS analysis of N-glycans, Fc gamma receptors (FcγRs), Fc N-glycans, Recombinant gp120, Recombinant human β-glucocerebrosidase
Subject terms: Carbohydrates, Glycobiology
Introduction
Protein glycosylation is a common posttranslational modification that significantly impacts protein folding, stability and activity1,2. Altering glycan structures in biotherapeutics can dramatically affect their efficacy3. In antibody therapeutics, removing core fucose from the N-glycan boosts affinity for FcγRIIIa on NK cells, enhancing ADCC activity4. Cerezyme, a recombinant human β-glucocerebrosidase, for treating Gaucher’s disease, requires N-glycan modification to expose core mannose residues. These residues are recognized by mannose receptors and taken up by macrophages where its substrate Glc-Cer is accumulated5,6.
HIV’s envelope glycoprotein (Env) gp120 is critical for entering CD4 + cells. It is heavily glycosylated, predominantly with immature high mannose type glycans, with only a small portion converted to complex glycans. Research indicates that many broadly neutralizing anti-HIV antibodies (bnAbs) target glycan or glycan-peptide combinations on gp1207–10. This highlights the importance of generating gp120 with precise glycan attachments to advance our understanding of HIV infection and immune response.
Chinese hamster ovary (CHO) cells are widely used for biotherapeutic production due to their human-like glycosylation. However, the complex nature of protein glycosylation in CHO cells often results in substantial heterogeneity in recombinant therapeutics. Isolating CHO cell glycosylation mutants using cytotoxic lectins enabled the production of glycoproteins with reduced glycan heterogeneity11. Advances in genome editing have enhanced the ability to create CHO cell glycosylation mutants for producing precise glycan structures.
This report describes the development and analysis of a panel of CHO cell glycosylation mutants, obtained through cytotoxic lectin selection and genome editing. Each mutant’s glycosylation capabilities were assessed with two model proteins: erythropoietin-Fc fusion (EPO-Fc) and the IgG1 antibody trastuzumab. HILIC-UPLC-QTOF mass spectrometry was utilized to quantify the abundance of each glycan. Unlike the commonly used MALDI-TOF method, this method offers novel insights into the glycosylation abilities of specific glycogenes through quantitative comparisons. We investigated how different Fc glycans affect antibody interactions with various Fcγ receptors and their impact on ADCC activation. Additionally, we studied how gp120 N-glycans affect interactions with broadly neutralizing anti-gp120 antibodies, using trimeric gp120 produced in CHO cell mutants. Lastly, with one of our mutants, we successfully produced human β-glucocerebrosidase with consistent Man5 glycans.
Results
CHO cells with mutated nucleotide sugar transporters
Nucleotide sugar transporters transfer nucleotide sugars from the nucleus or cytosol to the ER or Golgi apparatus, where they serve as substrates for glycosylation12. Using the cytotoxic lectin Maackia amurensis agglutinin (MAA), were isolated two mutants, CHO-gmt1 (CHO-glycosylation mutant 1) and CHO-gmt2. The previously reported CHO MAR-11 cells belong to the CHO-gmt1 group of mutants13. CHO-gmt2 lacks a functional UDP-galactose transporter14. Using genome editing, CHO-gmt3 and CHO-gmt5 were created by inactivating the GDP-fucose transporter gene in CHO-K1 and CHO-gmt1 cells15,16.
In this study, we used HILIC-UPLC-QTOF-MS (LC-MS)16 to analyse N-glycans from two model molecules, EPO-Fc and trastuzumab, produced in different CHO mutants17. LC-MS surpasses commonly used MALDI-TOF18,19 by providing comprehensive insights into both the composition and quantification of individual glycans within glycan mixtures. When ambiguity in glycan characterisation arose, we combined LC-MS with MS2 fragmentation and/or exoglycosidase treatment. EPO-Fc and trastuzumab were produced in diverse CHO-gmt lines, with corresponding gene mutations listed in Table 1, while Supplementary Table 1 provides a list of their corresponding gene mutations. Supplementary Fig. 1 and Supplementary Fig. 2 display glycan profiles for EPO-Fc and trastuzumab from various CHO-gmt lines, along with quantitative LC-MS analysis of different glycans. A combination of Oxford and Consortium for Functional Glycomics nomenclature has been used to annotate individual glycan structures20,21. In this report, the alphanumeric notation conveys specific glycan structural information. The letters represent the monosaccharide type: F for fucose, A for Antennary GlcNAc, B for Bisecting GlcNAc, M for Mannose, G for Galactose, and S for Sialic acid. The number that comes after the alphabet indicates the number of monosaccharides present. The number in the curved brackets ( ) indicates the linkage. The number in the square brackets [ ] indicates the arm the monosaccharide is on. This system differs from the common biotech usage. In biotech, G0F stands for glycan with zero galactose with one fucose, while G0 represents zero galactose without fucose. Using this system, they are named as F(6)A2 and A2, respectively.
Table 1.
CHO glycosylation mutants and the corresponding inactivated genes.
| Cell line | Mutations |
|---|---|
| CHO-K1 | |
| CHO-gmt1 | SLC35A1 |
| CHO-gmt2 | SLC35A2 |
| CHO-gmt3 | SLC35C1 |
| CHO-gmt4 | MGAT1 |
| CHO-gmt5 | SLC35A1, SLC35C1 |
| CHO-gmt6 | SLC35A1, MGAT1 |
| CHO-gmt7 | SLC35A2, MGAT1 |
| CHO-gmt8 | SLC35A1, SLC35C1, MGAT1 |
| CHO-gmt9 | SLC35A2, SLC35C1 |
| CHO-gmt10 | SLC35A1, SLC35A2 |
| CHO-gmt11 | SLC35A1, SLC35A2, MGAT1 |
| CHO-gmt12 | SLC35C1, MGAT1 |
| CHO-gmt13 | SLC35A2, MGAT2 |
| CHO-gmt14 | SLC35A2, SLC35C1, MGAT2 |
| CHO-gmt15 | SLC35A2, MGAT4A, MGAT4B, MGAT5 |
| CHO-gmt17 | SLC35C1, MGAT1, MAN2A1 |
| CHO-gmt17x | SLC35C1, MGAT1, MAN2A1, MAN2A2 |
| CHO-gmt18 | MAN1A1, MAN1A2, MAN1C1, MANEA |
| CHO-gmt19 | MAN1A1, MAN1A2, MAN1C1, MAN1B1 |
| CHO-gmt20 | MAN1A1, MAN1A2, MAN1C1, MANEA, MAN1B1 |
| CHO-gmt21 | MGAT2 |
| CHO-gmt22 | SLC35C1, MGAT2 |
| CHO-gmt26 | MAN1A1, MAN1A2, MAN1C1, MANEA, MAN1B1, EDEM1, EDEM2, EDEM3 |
| CHO-gmt29 | MAN1A1, MAN1A2, MAN1C1, MANEA, MAN1B1, EDEM1, EDEM2, EDEM3, MAN2B1 |
| CHO-gmt30 | MAN2A1, MAN2A2, MAN1A1, MAN1A2, MAN1C1, MANEA, EDEM1, EDEM2, EDEM3 |
| CHO-gmt32 | MAN2A1, MAN2A2, MAN1A1, MAN1A2, MAN1C1, MANEA, MAN1B1, EDEM1, EDEM2, EDEM3 |
| CHO-gmt35 | MAN2A1, MAN2A2, MAN1A1, MAN1A2, MAN1C1, MANEA, MAN1B1, EDEM1, EDEM2, EDEM3, MAN2B1 |
Mutated genes in CHO cell glycosylation mutants discussed in the report. Mutated genes in CHO cell glycosylation mutants are represented by gene symbols in the table, while the corresponding common names are listed below.
SLC35A1: CMP sialic acid transporter.
SLC35A2: UDP galactose transporter.
SLC35C1: GDP fucose transporter.
MGAT1: GnTI / α-1,3-mannosyl-glycoprotein 2-β-N-acetylglucosaminyltransferase.
MGAT2: GnTII / α-1,6-mannosyl-glycoprotein 2-β-N-acetylglucosaminyltransferase.
MGAT4A: GnTIVa / α-1,3-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase A.
MGAT4B: GnTIVb / α-1,3-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase B.
MGAT5: GnTV / α-1,6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase.
MAN2A1: α-mannosidase II.
MAN2A2: α-mannosidase II X.
MAN1A1: α-mannosidase IA.
MAN1A2: α-mannosidase IB.
MAN1C1: α-mannosidase IC.
MANEA: endo α-mannosidase I.
MAN1B1: ER α-mannosidase I.
EDEM1: ER degradation-enhancing α-mannosidase-like protein 1.
EDEM2: ER degradation-enhancing α-mannosidase-like protein 2.
EDEM3: ER degradation-enhancing α-mannosidase-like protein 3.
MAN2B1: lysosomal α-mannosidase.
CHO-K1 cells efficiently produce highly branched, core-fucosylated, and highly sialylated N-glycans on EPO-Fc (Supplementary Fig. 1a). The Asn297 in the Fc of EPO-Fc was mutated to Ala, limiting N-glycans to the EPO segment. In contrast, CHO-K1-produced trastuzumab mainly exhibits core-fucosylated bi-antennary complex glycans with variable galactose content and minimal sialic acid (Supplementary Fig. 2a). As anticipated, N-glycans from CHO-gmt1-produced EPO-Fc and trastuzumab are devoid of sialic acid, as shown in Supplementary Fig. 1b and Supplementary Fig. 2b.
The EPO rescue assay identified an MAA-resistant CHO cell line carrying an inactive UDP-galactose transporter gene (Slc35a2) (Supplementary Fig. 3). Sequencing the Slc35a2 cDNA revealed a 1-nucleotide insertion, inducing a gene frameshift (Supplementary Table 1). Co-transfecting CHO cells with other glycogenes showed no effect on EPO glycosylation, despite confirmed gene expression through RT-qPCR22. CHO cells with a dysfunctional UDP-galactose transporter are called CHO-gmt2 cells. EPO-Fc from CHO-gmt2 cells mostly lacked galactose (Supplementary Fig. 1c). Likewise, trastuzumab N-glycan in CHO-gmt2 cells were predominantly fucosylated and lacked galactose (Supplementary Fig. 2c).
The Slc35c1 gene, encoding for the GDP-fucose transporter, was inactivated in CHO-K1 cells through ZFNs, TALENs and CRISPR-Cas9, creating CHO-gmt3 cells16. EPO-Fc and trastuzumab N-glycans from CHO-gmt3 cells lacked core fucose (Supplementary Fig. 1d, Supplementary Fig. 2d). CHO-gmt5 lines arose from ZFN-mediated Slc35c1 gene inactivation in CHO-gmt1 cells15, producing N-glycans devoid of sialic acid and fucose (Supplementary Fig. 1f, Supplementary Fig. 2f). Additionally, CHO-gmt9 cells were generated by ZFN-mediated Slc35c1 gene inactivation in CHO-gmt2 cells, leading to fucose-free and galactose-free N-glycans on EPO-Fc and trastuzumab (Supplementary Fig. 1j, Supplementary Fig. 2j). For clarity regarding the creation of these CHO mutants, Supplementary Fig. 4 provides a comprehensive roadmap.
CHO cells with dysfunctional GnT-I gene
Ricinus communis agglutinin-I (RCA-I) was used to isolate CHO-gmt4 cells. Intriguingly, only CHO cells with inactivated N-acetylglucosaminyltransferase I (GnT-I) survived RCA-I treatment21,23. The EPO rescue assay revealed that over 200 surviving clones all lacked functional MGAT1 gene, which encodes GnT-I. cDNA sequencing confirmed GnT-I mutations in all RCA-I-resistant clones. N-glycans in CHO-gmt4-produced EPO-Fc consisted primarily of fucosylated and non-fucosylated Man5 glycans, as well as paucimannose structures such as Man4, Man3, and their fucosylated derivatives (Supplementary Fig. 1e). In contrast, trastuzumab N-glycan in CHO-gmt4 were mostly fucose-free Man5 glycans, with limited Man4 and Man3 glycans (Supplementary Fig. 2e). These findings suggest that the N-glycans on EPO-Fc are more prone to further processing, such as fucosylation, compared to IgG antibodies.
CHO cells with dysfunctional nucleotide sugar transporters and GnT-I
Compared to CHO-K1 cells, CHO-gmt1, CHO-gmt2 and CHO-gmt5 cells showed increased RCA-I sensitivity. We isolated RCA-I-resistant cells from each mutant line, naming them CHO-gmt6, CHO-gmt7, and CHO-gmt8. EPO rescue assay and cDNA sequencing confirmed mutated GnT-I gene in all mutants (Supplementary Table 1; Supplementary Fig. 3). Thus, CHO-gmt6 had dysfunctional SLC35A1 and MGAT1, CHO-gmt7 lacked SLC35A2 and MGAT1 activities, and CHO-gmt8 lacked functional SLC35A1, SLC35C1, and MGAT1 (Supplementary Table 1). Because of MGAT1 impairment, EPO-Fc N-glycans from CHO-gmt4, CHO-gmt6, CHO-gmt7, and CHO-gmt8 cells shared comparable glycoprofiles, mainly consisting of Man5 glycan (Supplementary Fig. 1e, g, h, i). A benefit of these mutants is their identical N-glycan production, while O-glycans and glycolipids can vary. Trastuzumab N-glycans from CHO-gmt4, CHO-gmt6, CHO-gmt7, and CHO-gmt8 cells exhibited mainly fucose-free Man5 glycans (Supplementary Fig. 2e, g, h, i). Using CRISPR-Cas9, CHO-gmt10 and CHO-gmt11 cells were generated by Slc35A1 gene disruption in CHO-gmt2 and CHO-gmt7 cells. Supplementary Table 2 outlines the targeted sites by ZFNs and CRISPR-Cas9 in this study. In CHO-gmt10 cells, EPO-Fc N-glycans are mostly fucosylated galactose-free (Supplementary Fig. 1k), whereas trastuzumab N-glycans are mainly G0F and G0 glycans (Supplementary Fig. 2k). In CHO-gmt11 cells, the N-glycan profile resembles that of CHO-gmt4 cells in both EPO-Fc and trastuzumab, as MGAT1 activity is impaired (Supplementary Fig. 1l; Supplementary Fig. 2l).
CHO cell mutants that produce homogenous fucose-free Man5 N-glycans
Since CHO-gmt4 produces fucosylated Man5 glycans on EPO-Fc, we treated CHO-gmt3 cells with RCA-I, resulting in CHO-gmt12 cells. EPO-Fc from CHO-gmt12 featured fucose-free Man5 and Man4 glycans (Supplementary Fig. 1m), while trastuzumab from CHO-gmt12 exhibited mainly fucose-free Man5 glycans (Supplementary Fig. 2m).
The Man2a1 gene, encoding α-mannosidase II, in CHO-gmt12 was disrupted with CRISPR-Cas9, resulting in CHO-gmt17 cells. Analysis of EPO-Fc from CHO-gmt17 showed a predominance (90.68%) of Man5 structures (Supplementary Fig. 1q), with minor amounts (2.8%) of Man4 and Man3 glycans. Trastuzumab from CHO-gmt17 contained nearly exclusive Man5 glycans (Supplementary Fig. 2q). Subsequently, CHO-gmt17x was created by CRISPR-Cas9 targeting Man2a2 in CHO-gmt17 cells. MAN2A2, or α-mannosidase IIx, plays a role in removing mannose residues from Man5 glycans24. The majority of glycans on EPO-Fc from CHO-gmt17x were mainly fucose-free Man5 (95.38%) (Supplementary Fig. 1r), whereas CHO-gmt17x-produced trastuzumab consisted almost entirely of Man5 glycans (Supplementary Fig. 2r).
CHO cell mutants that produce N-glycans with reduced branches
CHO-gmt15 was created using CRISPR-Cas9 to inactivate Mgat4a, Mgat4b, and Mgat5 genes in CHO-gmt2 cells. These genes introduce extra GlcNAc branches to bi-antennary complex N-glycans, resulting the formation of tri- and tetra-antennary complex N-glycans. Analysis of N-glycans in CHO-gmt15 cells revealed predominant fucosylated bi-antennary GlcNAc-terminated N-glycans in both EPO-Fc and trastuzumab. Additionally, fucosylated mono-antennary GlcNAc-terminated N-glycans were observed in EPO-Fc (Supplementary Fig. 1p, Supplementary Fig. 2p). Small amounts of mono-galactosylated N-glycans (along with sialylation) were identified, consistent with findings in other CHO-gmt2-derived cell lines.
CHO-gmt13 and CHO-gmt14 were generated by CRISPR-Cas9 targeting of the Mgat2 gene in CHO-gmt2 and CHO-gmt9 cells, respectively. MGAT2 adds the second GlcNAc residue to the core N-glycan at the α1,6-linked mannose, forming a β1,2 linkage. N-glycans on the EPO-Fc from CHO-gmt13 cells are shown in Supplementary Fig. 1n. Lack of MGAT2 leaves the α1,6-linked mannose of the core N-glycan unmodified. In CHO-gmt13 cells, the two GlcNAc residues are both linked to the α1,3-linked mannose arm, deviating from the typical bi-antennary structure yet maintaining the same molecular mass. N-glycans on trastuzumab from CHO-gmt13 cells exhibited primarily fucosylated core structure with a single GlcNAc residue (Supplementary Fig. 2n). The N-glycan profiles of EPO-Fc and trastuzumab from CHO-gmt14 cells resemble those from CHO-gmt13 cells, with the exception that they lack fucose (Supplementary Fig. 1o, Supplementary Fig. 2o). Among all mutants, trastuzumab from CHO-gmt14 cells carries the smallest N-glycan, consisting of just six sugar residues.
Similarly, CHO-gmt21 and CHO-gmt22 resulted from CRISPR-Cas9 targeting Mgat2 in CHO-K1 and CHO-gmt3 cells. Analysing N-glycan structures of CHO-gmt21-produced EPO-Fc revealed exclusive attachment of the branches to the α1,3-linked mannose of the core structure, with the α1,6-linked mannose arm remaining unchanged (Supplementary Fig. 1v). Trastuzumab from CHO-gmt21 cells had more sialic acid in its mono-antennary than the CHO-K1-produced antibody (Supplementary Fig. 2v). N-glycans of EPO-Fc and trastuzumab from CHO-gmt22 cells closely resemble those from CHO-gmt21 cells, except for lacking fucose (Supplementary Fig. 1w, Supplementary Fig. 2w). Complex N-glycans on EPO-Fc in CHO-gmt13, CHO-gmt14, CHO-gmt21, and CHO-gmt22 all have unmodified α1,6-linked mannose arms, confirmed by mannosidase treatment. Supplementary Fig. 5 shows the mannosidase treatment of CHO-gmt22-produced N-glycans, confirming the presence of unblocked mannose residue. Mannosidase treatments yield consistent results for EPO-Fc from CHO-gmt13, CHO-gmt14 and CHO-gmt21 (data not shown). This confirms that GnT-II inactivity prevents GnT-V from adding GlcNAc to the α1,6-linked mannose arm in EPO-Fc. Interestingly, CHO-gmt21 and CHO-gmt22-produced trastuzumab exhibited higher sialylated single arm N-glycans than CHO-K1 cells and CHO-gmt3 cells, possibly due to enhanced sialyltransferase accessibility on mono-antennary N-glycans in IgG antibodies.
Generation of CHO cell mutants that produce homogenous Man9 N-glycans
As per current understanding, MAN1B1 (ER α-mannosidase I) generates Man8 isomer B, while MANEA (Golgi endo-α-mannosidase) generates Man8 isomer A. Subsequently, MAN1A1, MAN1A2, and MAN1C1, namely Golgi α1–2 mannosidase IA, IB and IC, trim Man8 to Man5. Following the addition of a GlcNAc residue to the α1-3Man arm of the Man5 glycan by GnT-I, the α1–3 and α1-6-linked mannose residues on the α1-6Man arm are removed by α-mannosidase II and α-mannosidase IIx (MAN2A1, MAN2A2)25,26. The Man5 (Man5GlcNAc2) glycan plays a vital role as an intermediate in the N-glycosylation pathway. Mutants unable to reach this stage are incapable of producing complex or hybrid type N-glycans.
Using CRISPR-Cas9, three mutants were created: CHO-gmt18 with Man1a1, Man1a2, Man1c1, and Manea gene disruptions (Man1b1 unchanged), CHO-gmt19 with Man1a1, Man1a2, Man1c1 and Man1b1 inactivations (Manea gene functional), and CHO-gmt20 with all 5 genes (Man1a1, Man1a2, Man1c1, Man1b1 and Manea) knocked out. Figure 1 shows the creation of CHO-gmt35 cells producing consistent Man9 N-glycans. The left and right columns in Fig. 1, display N-glycan profiles of EPO-Fc and trastuzumab in various CHO mutants. Although CRISPR/Cas9 disrupted these a-mannosidases in CHO cells, the presence of complex N-glycans in EPO-Fc indicate that trimming of mannose residues to Man5 glycans was not entirely halted. Glycans linked to EPO-Fc and trastuzumab from wild-type CHO-K1 cells are shown as controls in Fig. 1a, b, respectively. EPO-Fc glycans from CHO-gmt18, CHO-gmt19, and CHO-gmt20 are shown in Fig. 1c, e, g, respectively. Clearly, EPO-Fc from these mutants exhibit significantly higher levels of high mannose type glycans (CHO-gmt18, 51%; CHO-gmt19, 65.1%; CHO-gmt20, 75.2%) compared to CHO-K1 cells (0.73%). EPO-Fc from all three mutants contained Man6, Man7 and Man8 glycans, while Man9 glycans are only present in CHO-gmt19 and CHO-gmt20 cells. Supplementary Fig. 1s, t, u show detailed glycan profiles from these three mutants. In contrast, CHO-gmt18, CHO-gmt19, and CHO-gmt20 produced Man6, Man7, and Man8 glycans on trastuzumab without complex glycans. Man9 glycans were exclusively found on trastuzumab produced in CHO-gmt19 and CHO-gmt20 cells as shown in Fig. 1d, f, h. Supplementary Fig. 2s, t, u provides a closer look at the specific glycans attached to trastuzumab in these three mutants. These findings suggest that Man1b1 knocking out may be more effective than Manea in preventing mannose trimming. Notably, trastuzumab from all three mutants lacked complex N-glycans, indicating reduced efficiency of mannosidases trimming on antibody glycans, possibly due to structural constraints in the Fc region.
Fig. 1.

Creating CHO cell mutants for Man9 N-glycan production. EPO-Fc and trastuzumab were stably expressed in diverse CHO cells lines, purified, and their N-glycan composition was analysed using LC-MS. Glycan composition results for EPO-Fc (left) and trastuzumab (right). (a, b) CHO-K1; (c, d) CHO-gmt18; (e, f) CHO-gmt19; (g, h) CHO-gmt20; (i, j) CHO-gmt26; (k, l) CHO-gmt29; (m, n) CHO-gmt32; (o, p) CHO-gmt35. The Y-axis shows the fluorescence intensity.
These findings highlight the essential roles of the five mannosidases in normal mannose trimming. Yet, upon their inactivation, other α-mannosidases likely engage in this process. For instance, ER degradation-enhancing α-mannosidase–like protein 1, 2, and 3 (EDEM1, EDEM2, EDEM3) are recognized α-mannosidase responsible for removing mannose residues from misfolded glycoproteins27. CHO-gmt26 cells were created using CRISPR-Cas9 targeting of Edem1, Edem2, and Edem3 genes in CHO-gmt20 cells. When comparing the EPO-Fc between CHO-gmt20 and CHO-gmt26, the overall high-mannose glycan population remained similar (75.2% vs. 74.1%). However, the Man9 content in EPO-Fc from CHO-gmt26 rose from 15.2 to 28.2% (Fig. 1g, i, Supplementary Fig. 1u, x). Man9 N-glycan levels on trastuzumab from CHO-gmt26 significantly exceeded those from CHO-gmt20 cells (Fig. 1h, j, Supplementary Fig. 2u, x). This indicates that EDEMs possess α1,2-mannosidase activities for correctly folded glycoproteins and antibodies. The precise site of EDEM-mediated mannose trimming in the ER or Golgi remains unclear.
MAN2B1, a class II α-mannosidases, is believed to reside in lysosomes and plays a role in breaking down N-glycans released during glycoprotein turnover28. This enzyme exhibits broad specificity, capable of cleaving all a-mannosidic linkages29. MAN2A1 and MAN2A2, Golgi-resident class II α-mannosidases, are crucial for converting GlcNAcMan5GlcNAc2 to GlcNAcMan3GlcNAc2. They achieve this by removing α1,3- and α1,6-linked mannose residues from the α1,6-Man arm of the core structure24. The gene knockout in the creation of CHO-gmt17x cells, as previously discussed, strongly supports their suggested functions. Three more mutants were generated in CHO-gmt26 cells using CRISPR-Cas9: CHO-gmt29 targeted Man2b1, CHO-gmt32 targeted Man2a1 and Man2a2, and CHO-gmt35 targeted Man2b1, Man2a1, and Man2a2.
Compared to N-glycans of EPO-Fc and trastuzumab from CHO-gmt26 cells, knocking out Man2b1 alone (CHO-gmt29) had no obvious impact on mannose trimming (Fig. 1k and Supplementary Fig. 1y; Fig. 1l and Supplementary Fig. 2y). When both Man2a1 and Man2a2 were inactivated (CHO-gmt32), high-mannose glycan levels of PEO-Fc significantly rose from 74.06 to 100%, predominantly Man9 glycans at 97.94% (Fig. 1m; Supplementary Fig. 1z). Trastuzumab N-glycans from CHO-gmt32 cells entirely shifted to Man9 glycans (Fig. 1n and Supplementary Fig. 2z). Thus, MAN2A1 and MAN2A2 play a clear role in mannose trimming when other a-mannosidases are absent. Comparing EPO-Fc N-glycans in CHO-gmt35 and CHO-gmt32 cells, Man8 glycans decreased from 2.06% (CHO-gmt32) to 1.33% (CHO-gmt35) (Fig. 1o; Supplementary Fig. 1aa). The amount of Man9 from trastuzumab produced in CHO-gmt32 and CHO-gmt35 remained consistent at nearly 100% (Fig. 1p and Supplementary Fig. 2aa). These results indicate that all other mannosidases can contribute to mannose trimming, except for MAN2B1.
Impact of fc glycans on Fc-FcγRIIIa interactions
The Fc region of IgG activates diverse cellular responses by interacting with Fcγ receptors (FcγRs)30,31. Glycosylation on Asn297 of IgG1 is crucial for Fc conformation and FcγR binding32,33. Key FcγRs include FcγRI (CD64), FcγRIIa-c (CD32a-c), and FcγRIIIa-b (CD16a-b)34,35. We examined the influence of various glycans on Fc-FcγRI, IIa, and IIIa interactions using trastuzumab derived from CHO cell glycosylation mutants.
The Fc-FcγRIIIa interaction is vital for antibody dependent cellular cytotoxicity (ADCC). Removing core fucose from the Fc N-glycan greatly enhances this interaction4. To study trastuzumab’s interaction with FcγRIIIa, we produced the extracellular domain of human FcγRIIIa with a His-tag in CHO cells. Glycoengineered trastuzumab was immobilized on the Surface Plasmon Resonance (SPR) sensor chip, and the trastuzumab-FcγRIIIa interaction was evaluated with BiaCore. The binding affinities (KD) determined by BiaCore are summarised in Supplementary Tables 3 and illustrated in Fig. 2a. Additionally, we performed ELISA to compare antibody and FcγRIIIa binding. The His-tagged FcγRIIIa extracellular domain was incubated on nickel-coated plates, and antibody-FcγRIIIa binding was quantified through a standard ELISA (Fig. 2b). Both methods showed consistent trends, with decreasing KD values from BiaCore corresponding to increased antibody binding observed in ELISA (Fig. 2a, b). These findings confirm ELISA’s reliability in assessing Fc-FcγRIIIa interactions under these conditions. Removing core fucose significantly increased Fc-FcγRIIIa affinity. Mutants without core fucose (CHO-gmt3, CHO-gmt5 and CHO-gmt9) showed a substantial increase in affinity for FcγRIIIa. Our SPR data indicated that the enhanced affinity of the afucosylated antibody for FcγRIIIa is primarily due to a slower off rate. This is evident in the substantially lower KD values: 69 nM, 46 nM and 80 nM for trastuzumab produced in CHO-gmt3, CHO-gmt5, and CHO-gmt9, respectively, compared to the 560 nM KD for CHO-K1-produced trastuzumab (Supplementary Table 3).
Fig. 2.

Impact of Fc glycan on Fc-FcγR interactions. (a) Trastuzumab’s interaction with FcγRIIIa was analysed with BiaCore, and the KD values are shown graphically, with detailed KD data summarised in Supplementary Table 3. (b) Trastuzumab’s interaction with FcγRIIIa was evaluated using standard ELISA. (c) The interaction between trastuzumab and FcγRI was determined with a standard ELISA. (d) Trastuzumab’s interaction with FcγRIIa was also determined using a standard ELISA.
Trastuzumab from CHO-gmt4 and CHO-gmt8 cells, lacking GnT-I activity, displayed afucosylated Man3-5 glycans (Supplementary Fig. 2e, i). This led to increased FcγRIIIa affinity, with KD values of 129 nM and 200 nM for trastuzumab from CHO-gmt4 and CHO-gmt8, respectively (Fig. 2a). Similar results were observed with other high mannose glycans, such as CHO-gmt18, 19, 35, as indicated by BiaCore and ELISA analyses (Fig. 2a, b). However, the effect was not as significant as in CHO-gmt3, which produces afucosylated complex biantennary N-glycans. Trastuzumab from CHO-gmt4, 8, 18, 19, 35 all lack core-fucose, potentially enhancing binding, but the larger high mannose glycan might not fit as effectively into the interaction pocket as the afucosylated complex biantennary N-glycans4.
Interestingly, compared to trastuzumab from CHO-gmt2 and CHO-K1 cells, the single-armed N-glycans on trastuzumab produced by CHO-gmt13 and CHO-gmt21 cells decreased FcγRIIIa affinity to 780 nM and 799 nM, respectively, regardless of galactose on the glycan (Fig. 2a). This decrease in FcγRIIIa affinity was confirmed by the ELISA (Fig. 2b). The smaller single-armed glycan may hinder effective interaction with FcγRIIIa. This is evident in afucosylated single-arms glycoforms from CHO-gmt22 and CHO-gmt23 cells, which did not achieve the same affinity as CHO-gmt3 (Fig. 2a, b).
Impact of GalT1 and GnT-III overexpression on fc glycan structures and FcγRIIIa interaction
Co-expression of b-1,4-galactosyltransferase 1 (B4GalT1) in CHO-K1 and CHO-gmt3 cells led to a significant increase in galactosylation levels of the antibody, from 51 to 77% and from 27 to 80%, respectively (Supplementary Fig. 6a, b). This increase in galactosylation was accompanied by an improvement in affinity with FcγRIIIa, with KD values decreasing from 560 nM to 257 nM for CHO-K1 cells and from 69 nM to 53 nM for CHO-gmt3 cells, respectively (Supplementary Table 3, Fig. 2a). These findings were confirmed by the ELISA results (Fig. 2b). Our observations are consistent with a previous study showing improved FcγRIIIa affinity through increased galactosylation36.
Overexpressing GnT-III in CHO-K1, CHO-gmt2, and CHO-gmt9 cells significantly raised bisecting GlcNAc levels from 0% to 87–97% (Supplementary Fig. 6c, d, e). To differentiate between bisecting GlcNAc and tri-antennary glycans with identical molecular mass, LC-MS/MS analysis, similar to a previous method, was used37. Bisecting GlcNAc presence was verified through MS/MS analysis, as demonstrated in Supplementary Fig. 7. Bisecting GlcNAc presence appeared to have no impact on fucosylation in trastuzumab produced in CHO-K1 and CHO-gmt2 cells (Supplementary Fig. 2a, c; Supplementary Fig. 6c, d). However, it significantly reduced fucosylation in CHO-gmt13 (from 93 to 74%) and CHO-gmt21 cells (from 92 to 70%) (Supplementary Fig. 2n, v; Supplementary Fig. 6g, i). GnT-III overexpressing and introducing bisecting GlcNAc in CHO-K1, CHO-gmt2, CHO-gmt9, CHO-gmt13, and CHO-gmt21 cells all enhanced their FcγRIIIa binding affinity (Fig. 2a, Supplementary Table 3).
Impact of fc glycan structures on Fc-FcγRI and FcγIIa interactions
To explore Fc glycans on Fc-FcγRI and FcγIIa interactions, we assessed interaction affinities between FcγRI and FcγRIIa extracellular domains and trastuzumab glycosylation variants using ELISA. As shown in Fig. 2c, d, glycans on trastuzumab clearly impacts its interaction with FcγRI and FcγRIIa, though somewhat less pronounced than with FcγRIIIa. In Fig. 2c, trastuzumab glycovariants are ordered by their FcγRI affinity, high to low. The same arrangement demonstrates a corresponding decrease in FcγRIIa affinity (Fig. 2d). This similarity implies a consistent glycan-related impact on the binding to both receptors. For example, GnT-III overexpression in CHO-gmt13 increased its FcγRIIIa binding (Fig. 2a, b), it also enhanced Fc affinity for both FcγRI and FcγRIIa (Fig. 2c, d). Overexpressing GalT1 in CHO-K1 and CHO-gmt3, moderately raised binding affinity for both FcγRI and FcγRIIa (Fig. 2c, d). Thus, GalT1 overexpression enhances Fc binding affinity for all three receptors, consistent with a previous study38. Results suggest that core fucose has no impact on Fc-FcγRI and Fc-FcγRIIa interactions, highlighting a distinction in the interaction between Fc glycan and FcγRIIIa compared to FcγRI and FcγRIIa. Previous studies suggested the significant role of Fc glycan in antibody-FcγRI interaction39. Our results not only validated their findings but also indicated a similar interaction mode between Fc glycan and FcγRI/FcγRIIa.
Influence of fc glycans on trastuzumab-induced ADCC in breast cancer cell lines
Trastuzumab targets overexpressed HER2 antigen in breast cancer cells. To assess Fc glycan’s impact on ADCC, we used the ADCC Reporter Bioassay kit to study trastuzumab glycovariant cytotoxicity in breast cancer cells. Five human breast cancer cell lines (SK-BR-3, HCC1954, MDA-MB-231, MDA-MB-453, MDA-MB-468) were included in this study. HER2 levels in these cell lines were assessed via FACS using commercial Herceptin (Fig. 3a). The cell lines exhibited diverse HER2 expression. MDA-MB-468, a triple-negative breast cancer cell line, served as a Her2 expression-negative control. Results indicate MDA-MB-231 had the lowest Her2 expression, while SK-BR-3 displayed the highest Her2 expression.
Fig. 3.
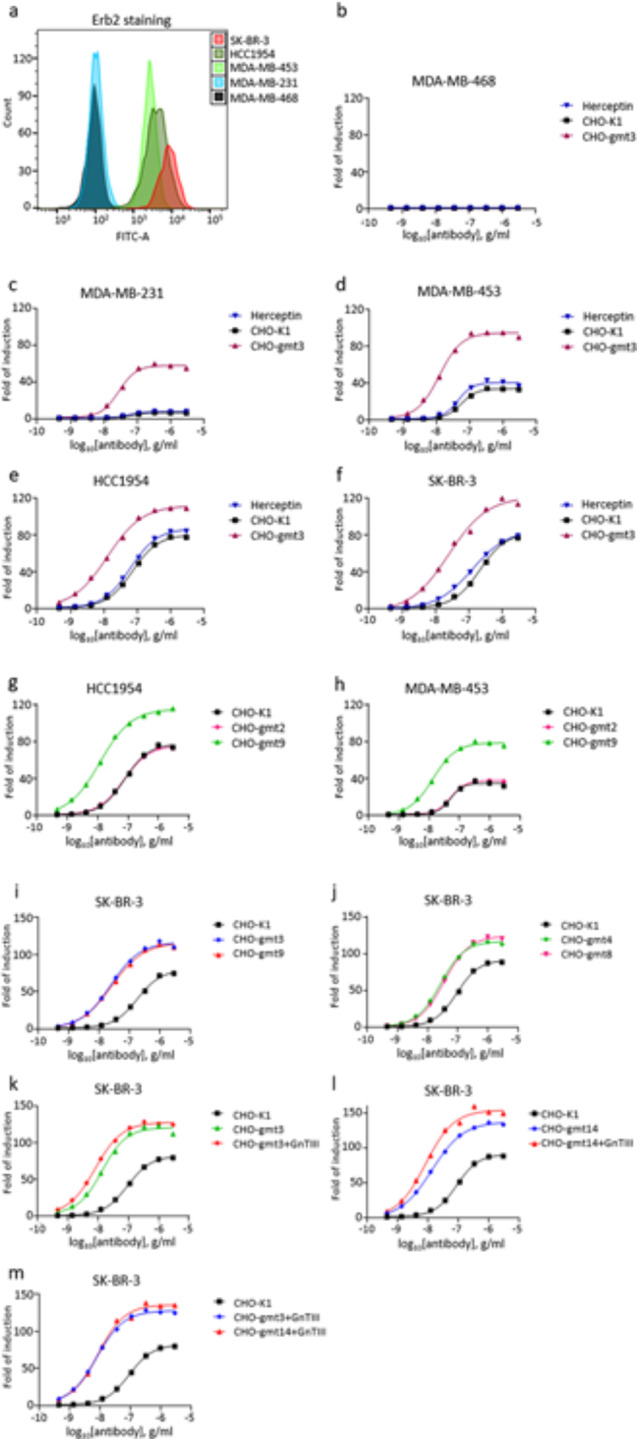
In vitro ADCC analysis of glycoengineered trastuzumab on breast cancer lines. (a) FACS analysis of Her2 expression levels using commercial Herceptin in MDA-MB-468, MDA-MB-231, MDA-MB-453, HCC1954, and SK-BR-3 breast cancer cell lines. (b-f) Comparing the cell-based ADCC activity of trastuzumab from CHO-K1 and CHO-gmt3 cells with the commercial drug Herceptin. (g-h) Comparing ADCC activity of trastuzumab from CHO-K1, CHO-gmt2 and CHO-gmt9 on HCC1954 and MDA-MB-453. (i-m) Comparison of cell based ADCC activity of trastuzumab produced from CHO-K1 and various CHO-gmts in SK-BR-3.
Figure 3b, c, d, e, f show ADCC Reporter Bioassay results for trastuzumab from CHO-K1 and CHO-gmt3 cells, and Herceptin, tested on five breast cancer cell lines. Among the cell lines MB-231, MDA-MB-453, HCC1954, and SK-BR-3, afucosylated trastuzumab from CHO-gmt3 showed significantly stronger response than trastuzumab from CHO-K1 and Herceptin. Her2-negative MDA-MB-468 cells did not exhibit ADCC induction with any antibodies (Fig. 3b). A notable observation was the inverse corelation between Her2 levels and antibody-mediated cell killing response. Compared to Herceptin and CHO-K1-derived trastuzumab, CHO-gmt3-produced trastuzumab exhibited significantly greater ADCC signaling induction: around 7- fold in MDA-MB-231 cells, 2.5-fold in MDA-MB-453 cells, and 1.5-fold in HCC1954 and SK-BR-3 cells (Fig. 3b, c, d, e, f). Previous studies showed that afucosylation of IgG1 antibodies significantly enhances ADCC activity for low antigen density, compared to normally glycosylated antibodies40. Thus, investigating afucosylated trastuzumab or rituximab in patients with low HER2 or CD20 levels could be promising. HCC1954 and MDA-MB-453 cell lines exhibited similar Her2 expression, with HCC1954 showing slightly elevated levels. Consequently, the afucosylated antibodies (CHO-gmt9) induced stronger ADCC response in HCC1954 (Fig. 3g) compared to MDA-MB-453 cells (Fig. 3h). Conversely, the fucosylated antibodies (CHO-gmt2) were comparable to unmodified CHO-K1 (Fig. 3g, h). Moreover, SK-BR-3 cells, selected for its highest Her2 expression, served as the target cells for testing a panel of trastuzumab glycovariants. Trastuzumab from CHO-gmt3 (afucosylated and galactosylated) and CHO-gmt9 (predominately afucosylated and agalactosylated) triggered nearly identical ADCC levels, showing a 3.8-fold increase in maximum ADCC compared to CHO-K1-produced antibody (Fig. 3i). Therefore, CHO-gmt9 might excel as a host for producing targeted anticancer antibodies, enhancing ADCC via consistent N-glycan profiles and minimizing batch variations (Supplementary Fig. 2j).
We assessed the effect of high mannose, bisecting GlcNAc, and mono-antennary complex structures on ADCC using the highly responsive SK-BR-3 target cells (Fig. 3j, k, l, m). Trastuzumab from CHO-gmt4 and CHO-gmt8 cells, dominated by fucose-free Man5 N-glycans (Supplementary Fig. 2e, i), showed a 1.3-fold rise in maximum ADCC response versus CHO-K1 cell-produced trastuzumab (Fig. 3j). The introduction of 68% bisecting glycans into trastuzumab by expressing GnT-III in CHO-gmt3 cells (Supplementary Fig. 6e) led to a slight increase in maximum ADCC compared to trastuzumab produced in CHO-gmt3 cells (Fig. 3k). Trastuzumab from CHO-gmt14 cells, with galactose-free and fucose-free single-arm glycans, exhibited a 1.6-fold higher maximum ADCC than CHO-K1-produced trastuzumab (Fig. 3l). These findings (Fig. 3k, l) align closely with the Biacore dinging data (Fig. 2a). Furthermore, in CHO-gmt14 cells, GnT-III overexpression led to a 1.8-fold increase in ADCC compared to CHO-K1-produced trastuzumab (Fig. 3l). Trastuzumab derived from CHO-gmt3 and CHO-gmt14 cells, co-expressing GnT-III, showed the highest ADCC activity among all tested glycovariants (Fig. 3m). Trastuzumab from CHO-gmt14 with overexpressed GnT-III elicited the highest level of ADCC signalling in SK-BR-3 cells.
Glycoengineered trimeric HIV-1 envelope glycoprotein (env) for studying glycan specificity of bnAbs against HIV
Most broadly neutralizing antibodies (bnAbs) against HIV target the trimeric HIV Env glycoprotein, consisting of gp120 and gp41. This HIV Env trimer is shielded by dense glycans, often impacting bnAb-antigen interactions41. BG505 SOSIP.664 gp140 is a recombinant soluble trimeric protein, closely mimicking the native membrane bound Env spike of HIV BG505 strain42. Like the native Env, BG505 SOSIP.664 gp140 has occupied glycosylation sites with premature high mannose type N-glycans9.
We produced His-tagged BG505 SOSIP.664 gp140 (SOSIP Env) in CHO-K1, CHO-gmt4 and CHO-gmt35 cells. Following purification with Ni++-column, N-glycans were released with PNGase F and analysed using LC-MS. Figure 4a shows the acquired glycan profiles. SOSIP Env protein from CHO-K1 cells (SOSIP Env-K1) primarily features high mannose-type glycans, with some small complex glycan peaks. Man9 stands out as the most abundant type of high mannose glycans. The SOSIP Env protein derived from CHO-gmt4 cells (SOSIP Env-Gmt4) lacks complex N-glycans due to the absence of GnT-I activity. Notably, it exhibits a distinct high mannose-type glycan distribution, with Man5 being the predominant form, in contrast to CHO-K1 cells. Further investigation is needed to determine if the absence of complex glycans enhances the activity of a-mannosidases. Nonetheless, the aim of producing a complex N-glycan-free SOSIP Env protein was accomplished. SOSIP Env-Gmt35 protein produced in CHO-gmt35 cells (SOSIP Env-Gmt35) exclusively featured Man9 glycans (Fig. 4a). Supplementary Fig. 8 displays the detailed N-glycan distribution of SOSIP Env produced by these three cell lines.
Fig. 4.

Effect of HIV gp140 glycans on interactions with anti-HIV bnAbs. (a) Comparison of glycosylation patterns of BG505 SOSIP.664 trimers produced in CHO-K1, CHO-gmt4 and CHO-gmt35 cells. Broadly neutralizing anti-HIV antibodies binding of these BG505 SOSIP.664 trimers were studied by ELISA. The Y-axis shows the relative fluorescence intensity, with the highest peak as 100%. (b) 2G12, (c) PG9, (d) PG16, (e) PGT121, (f) PGT128.
We produced five well-studied bnAbs targeting gp120 using CHO-K1 cells: 2G12, PGT121, PGT128, PG9, and PG16. Supplementary Table 4 contains amino acid sequences of these five antibodies. 2G12 binds the terminal dimannose structure of Man9 glycans via an unusual heavy chain domain exchange43. PG9 penetrate the glycan shield through its long CDR3 region, fitting precisely between two N-glycans to access the protein backbone on the V1/V2 region44. PG16, a close relative of PG9, shares many binding characteristics with PG9, including binding to the same Asn160 glycan. However, PG16 distinguishes itself from PG9 by also binding to a sialylated complex type structure in the second N-glycan45. PGT121 did not bind Man9 N-glycan on glycan arrays but did bind complex-type N-glycan on glycan arrays and in crystal structures. However, PGT121 recognized high-mannose-only HIV envelopes, whether isolated or in virions46,47. Like PG9, PGT128 interacts with two high-mannose type N-glycans (Man9 and Man5), and the V3 loop region upon binding to an engineered gp120 outer domain48.
To assess how gp120 glycans affect binding affinities of five bnAbs, we conducted ELISA on SOSIP Env produced in CHO-K1, CHO-gmt4, and CHO-gmt35 cells. As expected, 2G12 consistently bound to all three proteins: SOSIP Env-K1, SOSIP Env-Gmt4, and SOSIP Env-Gmt35. This suggests that the Man9 glycans recognised by 2G12 remain unaffected in CHO-gmt4-produced SOSIP Env, despite a notable reduction in total Man9 content in CHO-gmt4-produced SOSIP Env (Fig. 4b). PG9 exhibited comparable affinity for both SOSIP Env-K1 and SOSIP Env-Gmt4, suggesting its binding is not dependent on complex glycans. However, its binding to SOSIP Env-Gmt35 significantly declined, suggesting a potential interference due to the presence of a large Man9 glycan (Fig. 4c). PG16 failed to bind SOSIP Env-Gmt35, likely due to the hindrance from a large Man9 glycan. Its affinity was lower than PG9 when binding to SOSIP Env-CHO-K1 and SOSIP Env-CHO-Gmt4. Interestingly, PG16 bound more strongly to SOSIP Env-CHO-Gmt4 than to SOSIP Env-CHO-K1 (Fig. 4d). Whether a Man5 glycan binds the antibody better requires further investigation. Since PG16 is believed to interact with sialic acid on complex glycans, the removal of complex glycans in SOSIP Env-CHO-Gmt4 should reduce the interaction45. The smaller high mannose glycans in SOSIP Env-CHO-Gmt4 may contribute to the enhanced binding, though this connection needs further investigation. PGT121 displayed comparable binding to SOSIP Env from three cell lines, suggesting that complex N-glycans are not crucial for PGT121 to bind gp120 (Fig. 4e). Hence, high-mannose N-glycan is crucial for PGT121’s binding to gp120, despite earlier suggestions of potential interaction with complex glycans46. PGT128, targeting the terminal dimannose of Man9 N-glycan, showed similar binding profiles to all three SOSIP Env proteins (Fig. 4f). These results strongly indicate that HIV Env produced in these mutants can be valuable tools for probing glycans’ role in bnAbs’ recognition. Significant potential exists for expanding this study, including mutants like CHO-gmt1 and CHO-gmt2.
Production of β-glucocerebrosidase (GCase) with uniform Man5 glycans using CHO-gmt17x cells
Recombinant GCase production in CHO-gmt4 cells eliminates the need for enzymatic treatment to obtain mannose-terminated N-glycans. We created stable GCase-producing CHO-gmt4 transfectants and isolated a high-producing clone. The purified GCase is primarily consists of Man5, alongside fucosylated Man5, Man4, and fucosylated Man4 glycans. Similar to generating CHO-gmt17 cells, we knocked out the GDP-fucose transporter and a-mannosidase II genes in GCase-producing CHO-gmt4 cells. We then knocked out the a-mannosidase IIx gene in CHO-gmt17 cells, resulting in GCase-producing CHO-gmt17x cells (Fig. 5a). GCase protein possesses five putative N-glycosylation sequons, with the first four sites confirmed to be glycosylated49. Our quantitative analysis reveals that glycans derived from CHO-gmt4 cells display the least homogeneity, comprising 76.78% of Man5, 13.51% of Man4, 6.23% of F(6)Man5, and 3.48% F(6)Man4. Glycans from CHO-17 cells comprise 87.31% Man5 and 12.69% Man4, whereas glycans from CHO-17x cells exhibit exceptional homogeneity with 95.34% Man5 and 4.66% Man. To elucidate the glycan distribution at each glycosylation sites, we performed a site-specific glycosylation analysis50,51, and the results are shown in Fig. 5b. Analysis of the results confirmed the non-glycosylation at Asn501. The exclusive presence of Man5 N-glycans at Asn58 in all three cell lines is intriguing, indicating limited N-glycan processing at this site. This may be due to restricted access to modification enzymes, possibly because of structural constraints, despite its proximity to the N-terminus and the absence of apparent obstruction from other protein regions52. In contrast, glycans at the remaining three glycosylation sites underwent further processing, including fucose addition and mannose residue removal. Glycan diversity decreases as more genes are inactivation in CHO-17 and CHO-gmt17x cells.
Fig. 5.

Producing b-glucocerebrosidase with uniform Man5 glycans. (a) Recombinant b-glucocerebrosidase (GCase) from CHO-gmt4, CHO-gmt17 and CHO-17x was purified and analysed by LC-MS. The Y-axis shows the relative fluorescence intensity, with the highest peak as 100%. (b) Quantitative site-specific glycan analysis of recombinant GCase from CHO-gmt4, CHO-gmt17 and CHO-gmt17x cells. Glycosylation sites are indicated, with the Y-axis showing glycan composition percentage. Glycan species are color-coded.
Discussion
We developed and characterized a panel of CHO glycosylation mutant cell lines capable of producing glycans with diverse structures. EPO-Fc and trastuzumab were produced in each mutant, and the attached N-glycans were quantitatively analysed using LC-MS. Our study with EPO-Fc and trastuzumab revealed that achieving homogenous Man9 N-glycan production requires the inactivation of all the a-mannosidases in the genome, except lysosomal MAN2B1. CHO-gmt4, CHO-gmt6, CHO-gmt7, and CHO-gmt11 cells all lack GnT-I activity and produce predominantly Man5 N-glycans, their O-glycan and glycolipid profiles can be different. We showed that lack of GnT-II activity prevents GnT-V from adding GlcNAc to the core structure, causing branches to attach exclusively to the α1,3-linked mannose, leaving the α1,6-linked mannose free. The antibody’s mono-antennary glycan has a greater sialic acid content than bi-antennary glycans produced in CHO-K1 cells. With trastuzumab as a model IgG1 antibody, we systematically analysed how Fc N-glycans affect interactions between the Fc region and three FcγRs. Adding bisecting GlcNAc with GnT-III enhanced trastuzumab’s affinity to FcγRIIIa in all tested mutants. Using trastuzumab glycovariants, we observed Fc glycans influencing the interaction between Fc and FcγRI/FcγRIIa, revealing a shared interaction mechanism for both FcγRI and FcγRIIa. The trastuzumab glycovariants showed a clear correlation between their binding affinity to FcγRIIIa and their in vitro ADCC cell-killing efficacy. We produced a soluble trimeric HIV Env gp140 protein based on the sequence of the HIV BG505 strain using CHO-K1, CHO-gmt4, and CHO-gmt35 cells. The glycan dependency of five bnAbs against HIV spike protein was confirmed using these trimeric gp140 proteins. These results strongly suggest that HIV Env produced in these mutants can be valuable tools for studying the role of glycans in bnAbs’ recognition. Future studies could include additional mutants like CHO-gmt1 and CHO-gmt2.
In most of these mutants, multiple genes have been inactivated. Traditional mouse gene knockout methods are inadequate for achieving this, as inactivating many of these glycogenes would be lethal to the animal. However, at the cellular level, these mutations do not significantly impact cell growth. All mutants have been adapted to serum-free suspension cultures and stably transfected for antibody and glycoprotein production.
The use of EPO-Fc and an IgG1 antibody as model molecules for assessing the effect of each mutation on N-glycans proved highly successful. The three N-glycans attached to EPO are readily accessible for glycan modifications in the Golgi, resulting in a mixture of highly processed N-glycans. The Fc N-glycans, located between the two CH2 domains, encounter structural constraints that limit the activity of glycosidases and glycotransferases. Consequently, the Fc glycans exhibit less complexity compared to those of EPO-Fc. The two model molecules complement each other, creating an excellent system for assessing the influence of various glycogenes. This was particularly evident during the development of Man9-producing mutants.
LC-MS was used for quantitative analysis of glycan structure produced by each mutant, highlighting its clear advantage over the commonly used MALDI-TOF method. This advantage was particularly notable in cells that overexpressed galactosyltransferase, as well as during the development of CHO-gmt17x and CHO-gmt35 cells.
The production of a wide range of homogeneous Fc glycans, biologics incorporating both Man5 and Man9, glycoengineered gp140 specifically designed for studying bnAbs, all clearly highlights their vast potential for driving advancements in glycobiology and biotechnology.
Methods
Cell cultures
Adherent CHO-K1 cells and CHO glycosylation mutant cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen/Gibco) supplemented with 10% fetal bovine serum (FBS) (Invitrogen/Gibco, New Zealand), at 37 °C with 5% CO2. Suspension CHO-K1 cells and glycosylation mutant cells were cultured in a chemically defined, serum-free 50/50 medium, comprising a 1:1 ratio mixture of HyClone PF-CHO Multi-powder system (GE Healthcare) and CD CHO medium (Thermo Fisher Scientific). The cells were incubated in a 37 °C, 8% CO2 shaking incubator. The 50/50 medium was supplemented with 0.05% Pluronic F-68 acid (Thermo Fisher Scientific) and 6 mM L-glutamine (Thermo Fisher Scientific).
Cell lines
The Chinese hamster ovary-K1 (CHO-K1) cell lines used in this work were obtained from two different sources: CHO-K1 (ATCC) was purchased from the American Type Culture Collection (ATCC), while CHO-K1 (Mich) was obtained from Dr. Donald K. MacCallum at the University of Michigan in Ann Arbor. Human breast cancer cell lines were obtained from ATCC (ATCC) and cultured according to their respective recommended protocols.
Generation of CHO glycosylation mutants (CHO-gmts)
CHO-gmt1 cells were isolated from CHO-K1 cells via Maackia amurensis agglutinin (MAA) treatment as described earlier13. CHO-gmt2 cells were similarly isolated through MAA selection, while CHO-gmt3 cells were generated from CHO-K1 cells using different genome editing techniques16. CHO-gmt4 cells were isolated from CHO-K1 cells using RCA-I selection23. CHO-gmt5 cells were created by ZFN-mediated knockout of the Slc35c1 gene in CHO-gmt1 cells15. CHO-gmt6, CHO-gmt7, and CHO-gmt8 cells were isolated from CHO-gmt1, CHO-gmt2, and CHO-gmt5 cells through RCA-I selection, following the method described earlier23.
To generate additional CHO glycosylation mutants, we used CRISPR-Cas9 technology to knock out various glycosyltransferases, sugar nucleotide transporters, and mannosidases in different CHO cell lines. Potential CRISPR-Cas9 target sites in each gene were identified using the website http://staff.biosustain.dtu.dk/laeb/crispy/.53 We selected target sequences near the translational start site and with minimal predicted off-target sites. CRISPR-Cas9 constructs were designed using the GeneArt CRISPR Nuclease Vector with OFP Reporter Kit (Thermo Fisher Scientific), following the manufacturer’s protocol. The target sequences are provided in Supplementary Table 1.
FACS analysis and cell sorting
Two days after transfection with CRISPR-Cas9 constructs, CHO cells expressing orange fluorescent protein (OFP) were individually sorted into 96-well plates using FACSAriaIII (Becton Dickinson). Briefly, the adherent cells were detached from propagation flasks using Trypsin-EDTA (0.25%) (Thermo Fisher Scientific), centrifuged at 200 × g for 5 min, and washed twice with cold DPBS. The cells were then resuspended in serum-supplemented growth medium and prepared for sorting.
FACS analyses were also performed using FITC-conjugated MAA and Concanavalin A (Con A) lectins to evaluate mannosidases knockout mutants during the development of CHO-gmt35 cells. MAA is a lectin with a specific affinity for α2,3-linked sialic acid to galactose residues in complex N-glycans54,55, and it binds to sialic acid-containing N-glycans on CHO cells. Con A specifically recognise a-mannose residues on cell surface. In wild-type CHO-K1 cells, FITC-MAA exhibits the strongest binding, while FITC-Con A displays the lowest binding. In mannosidases knockout cells, as complex glycans decrease and high mannose type glycans increase on CHO cells, the binding patterns of the two lectins reversed. These lectins served as indicators for identifying Man9-producing CHO mutants, eliminating the need for N-glycan structure analysis to evaluate the results. For instance, during the creation of CHO-gmt26 from CHO-gmt20 cells achieved by knocking out EDEM1, EDEM2, and EDEM3, FITC-Con A exhibited maximal staining and MAA-FITC displayed minimal staining only when all three genes were inactivated. The lectin staining procedure was conducted at 4 °C, unless otherwise stated. Briefly, 1 × 106 cells were centrifuged at 200 × g for 5 min and subsequently washed twice with cold DPBS. Next, the cells were incubated in blocking solution (1% (w/v) BSA in DPBS) for 30 min. This was followed by incubation in 8 µg/ml FITC-MAA, or FITC-Con A, in the blocking solution. The treated cells were washed twice with cold DPBS, and then resuspended in serum-supplemented growth medium and subjected to FACS sorting.
Analyses of genomic DNA and cDNAs in CHO glycosylation mutants
After treatment with various reagents like lectins or CRISPR-Cas9 constructs, single cells were isolated, cultured to confluence, and genomic DNA was extracted from the clones using QuickExtract DNA Extraction Solution (Epicentre). Next, we PCR-amplified and sequenced the targeted genomic regions to detect mutations, with the sequencing primers for various detailed in Supplementary Table 2. PCR amplicons were cloned using the Zero Blunt TOPO PCR Cloning Kit for Sequencing (Thermo Fisher Scientific) and then sequenced to confirm mutation presence.
Mutant cell lines isolated with cytotoxic lectins like MAA and RCA-I were first analysed using the EPO rescue assay to identify the mutated gene23. Briefly, mutant CHO cells are transfected with constructs expressing human EPO and a glycogene. If the glycogene complements the mutated gene, EPO will undergo full glycosylation, which is clearly visualized on an isoelectric focusing (IEF) gel. After identifying the mutated gene, we isolate mRNA and sequence the open reading frame to pinpoint the specific mutation. In brief, total RNA was extracted with the RNeasy Mini Kit (Qiagen). Then, we synthesized cDNA and amplified the target gene using gene-specific primers and the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Thermo Fisher Scientific). The amplicons were then sequenced to identify mutations. Genome editing-generated mutations were determined using the same method, and a summary of all mutations is available in Supplementary Table 2. Each gene mutation is compared to the corresponding sequence in wild-type CHO-K1 cells. For genes with multiple alleles identified, each allele’s mutation is listed. Cell lines from the same parental cell line with shared mutation(s) are grouped together in Supplementary Table 2.
Glutamine synthetase (GS) knockout from CHO cell
The GS gene was knocked out in CHO-K1 and CHO-gmt cells using TALEN-mediated gene editing as previously described56.
T7E1 mismatch assay
Genomic DNA from CHO cells transfected with ZFNs, CRISPR-Cas9, or TALEN was extracted using the DNeasy Blood & Tissue Kit (Qiagen) 48-72-hours post-transfection for T7E1 mismatch assay, as described earlier16.
Expression constructs
The human erythropoietin (EPO) open reading frame (ORF) was cloned into the pcDNA3.1 expression vector (Thermo Fisher Scientific). The DNA sequence encoding EPO-Fc fusion protein was constructed as previously described23. An IRES sequence was used to link the GS selection marker downstream of the ORFs.
The heavy chain and the light chain of trastuzumab, with optimized signal peptides as described earlier57 were cloned into the pcDNA3.1 vector, in a tricistronic configuration as previously described58. The light chain and heavy chain sequences of bnAbs 2G12, PG9, PG16, PGT121, and PGT128 (Supplementary Table 4) were also cloned into pcDNA3.1 vector, using GS as the selection marker in the same tricistronic configuration.
The ectodomains of FcγRI (NM_000566.3, residue 1-292), FcγRIIa (NM_001136219.1, residue 1-217), and FcγRIIIa (NM_000569, residue 1-208) were cloned into the pcDNA3.1 vector with a C-terminal 6x His tag. GSR324C served as the selection marker, fused downstream of expressed proteins in a bicistronic configuration using an IRES sequence56.
The coding sequence of 6x His-tagged BG505 SOSIP.664 gp140 Env (SOSIP Env) was cloned into the pcDNA 3.1 vector driven the CMV promoter. GS served as the selection marker, fused with an IRES sequence in a bicistronic arrangement. The Chinese hamster furin protease (NM_001243986.1) was cloned into the same vector under the SV40 promoter using the following primers: Forward primer: CAACACCCGGGGCCACCATGGAGCTGAGGCCCTGGTTGCTATGGGTGG, Reverse primer: GGTTGCCCGGGTCAAAGGGCGCTCTGGTCTTTGATAAAGGC. The furin sequence was then linked to the Neomycin selection marker using an IRES sequence.
Transfection of adherent CHO cells
Adherent CHO cells were seeded into 6-well plates at a density of 3 × 105 cells/ml one day prior to transfection. On the day of transfection, cells were transfected using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific) following the manufacturer’s protocol. Briefly, Lipofectamine LTX was diluted in Opti-MEM (Thermo Fisher Scientific), and DNA along with Plus reagent were diluted separately in Opti-MEM. The suspensions were then combined, mixed, and added onto the cells.
Adaptation of adherent CHO cells to serum-free suspension culture
Adherent CHO cells were gradually transitioned to serum-free suspension culture by serially diluting the serum content in the growth medium over several passages with the appropriate suspension medium ratios. At 2.5% serum, the cell lines were transferred into shake flasks and cultured in incubator shaker until they were fully adapted to serum-free media.
Stable transfection of suspension cells
Suspension CHO cells were electroporated using the SG Cell line 4D-Nucleofector X Kit (LONZA) in the 4D-Nucleofector System (LONZA), following the manufacturer’s protocol. Briefly, 1 × 107 cells were transfected with 5 µg of DNA in 100 µl Nucleofector Solution SG. Two days after transfection, the medium was switched to glutamine-free 50/50 medium for selection. After the recovery period, the medium was supplemented with 25 µM L-methionine sulfoximine (MSX) (Merck) to selectively culture cells with the GS selection marker. Subsequently, cells were allowed to recover and grow until reaching confluence. Cells with the attenuated GS selection marker (GSR324C) underwent one round of glutamine-free selection without MSX.
To produce HIV SOSIP Env, the construct was electroporated into CHO-K1, CHO-gmt4 and CHO-gmt35 cells at a ratio of 10 µg plasmid DNA per ten million cells, using the SG Cell line 4D-Nucleofector X Kit from LONZA. Selection was performed using glutamine-free 50/50 media supplemented with 600 µg/ml Geneticin (Thermo Fisher Scientific).
Isoelectric focusing (IEF) analysis of EPO
Immobilised pH gradient (IPG) IEF was performed following our previously published protocol13.
Large-scale production of recombinant proteins and antibodies
Recombinant EPO-Fc, trastuzumab, 2G12, PGT121, PGT128, PG9, PG16, and BG505 SOSIP.664 gp140 were produced in large-scale in different CHO cell lines. After stable transfection with constructs encoding the recombinant proteins, large-scale cultures of 100 to 240 ml were completed. Cultures were grown for approximately 6–7 days. When cell viability was still above 90%, the supernatants were collected, filtered using Nalgene Rapid-Flow filter units with 0.2 μm PES membrane (Thermo Fisher Scientific), and stored at 4 °C for purification.
Protein A purification of antibodies and EPO-Fc
EPO-Fc purification utilized HiTrap Protein A 5 ml column (GE Healthcare, Chicago, IL), while trastuzumab and bnAbs purification employed HiTrap MabSelect SuRe 5 ml column (GE Healthcare), with an AKTA purifier (GE Healthcare). On the purification day, supernatants from large-scale culture were equilibrated with 0.5 volume of 20 mM phosphate buffer (pH 7.2). the column was pre-equilibrated with the equilibration buffer, and then the supernatant was injected into the column. Subsequently, the column was washed with equilibration buffer to remove unbound proteins. EPO-Fc purification used 100 mM glycine pH 3.0 for elution, while trastuzumab and bnAbs purification used 100 mM acetic acid pH 3.5 for elution. Fractions with the highest UV280 readings were pooled, and the pH was adjusted to pH 6.6 with 1 M Trizma. Protein quantification was performed using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific).
Immobilized metal affinity chromatography for purification of recombinant his-tagged proteins
The supernatant containing His-tagged FcγRs and SOSIP Env were purified using His HiTrap HP column (GE Healthcare Life Sciences) with the ÄKTA purification system (GE Healthcare Life Sciences). Briefly, the column was equilibrated with 20 mM sodium phosphate buffer pH 7.4 containing 500 mM NaCl and 20 mM imidazole. Then, the pre-filtered supernatant containing recombinant His-tagged protein was loaded onto the column. The bound column was washed with 20 mM sodium phosphate buffer pH 7.4 containing 500 mM NaCl and 40 mM imidazole. The proteins were eluted by increasing the imidazole concentration to 200 mM. The FcγRs were buffer exchanged with 20 mM sodium phosphate pH 7 buffer, and the concentration was determined using NanoDrop 2000.
Purification of BG505 SOSIP.664 trimer
Approximately 200 ml of harvested supernatant containing BG505 SOSIP.664 trimer was diluted 1:1 with binding buffer (20mM sodium phosphate, 0.5 M sodium chloride, 20mM imidazole pH 7.4). Samples were loaded onto a pre-equilibrated HisTrap HP 1 ml column (Cytiva) at a flow rate of 0.8 ml/min on an AKTA purifier UPC10. Columns were washed with wash buffer (20mM sodium phosphate, 0.5 M sodium chloride, 40mM imidazole pH 7.4) for 20 column volume (CV) at 0.8 ml/min. Gradient elution was performed from 40 mM to 200 mM imidazole in buffer (20mM sodium phosphate, 0.5 M sodium chloride) over 30 CV at 1 ml/min, with 2 ml fractions collected. Selected fractions were then pooled and buffer-exchanged to pH 8.2 using 20 mM Tris buffer with 75 mM sodium chloride.
Biacore analysis
The binding affinities and kinetics between the glycoengineered antibodies and FcγRIIIa ectodomain were measured using a Biacore T200 instrument. The antibody was immobilized on a CM5 sensor chip (GE Life Sciences) using standard amine coupling techniques, and all SPR measurements were performed at 25 °C. Flow line 1 served as a blank for all sensor chips, while all experimental runs were conducted using a binding buffer containing 20 mM 3-morpholinopropane-1-sulfonic acid (MOPS), 100 mM sodium chloride, pH 7.4. Purified FcγRIIIa at different concentrations was diluted in the running buffer (20 mM MOPS, 100 mM sodium chloride, pH 7.4) and injected over all 4 flow cells at a flow rate of 20 µl/min for 1a duration of 10 min. The dissociation phase continued for 40 min, and surface regeneration was not required as the response rate returned to baseline at the end of each cycle. The FcγRIIIa analyte concentration was serially diluted two-fold from 2000 nM to 15.625 nM. Sensorgrams were analysed using Biacore T200 Evaluation Software, and all sensorgrams were generated by subtracting the blank injection response from the analyte injected flow cell responses. Equilibrium fitting was done to obtain the dissociation constant (KD) using the maximum response unit (Rmax) from each measured concentration range. Association rates (kon) and dissociation rates (koff) were calculated using a 1:1 binding model fitting. kon was determined by measuring the slope of the line fitted to the observed association rates, while koff was determined by averaging the observed dissociation rates.
ADCC reporter bioassay
Trastuzumab’s ADCC activity was assessed using the ADCC Reporter Bioassay (Promega, Fitchburg, WI) by following the manufacturer’s protocol. In the assay, a panel of breast cancer cell lines (MDA-MB-468, MDA-MB-231, MDA-MB-453, HCC1954, and SK-BR-3) was used as target cells instead of the WIL-S cells provided in the kit. Target cells were detached from propagation flasks using Accutase solution (Merck, Germany) and seeded in 96-well plates at a density of 1.25 × 104 cells / 100 µl medium / well, approximately 20–24 h before the assay. This ensured an effector cell to target cells ratio (E: T ratio) of 6:1. After incubating the ADCC Bioassay Effector Cells, luminescence was measured using the Tecan Infinite 200 microplate reader (Tecan Trading AG, Switzerland). The results were analysed, and data graphed as Fold of Induction ((induced – background) / (control – background)) versus Log10 [antibody] and fitted to a 4PL curve using GraphPad Prism software (GraphPad Software). Each experiment was triplicated, yielding consistent results. results with similar trends were obtained. One was shown in the figure.
ELISA analysis to determine the binding affinity of trastuzumab to different FcγRs
A nickel coated plate (Pierce) was incubated with either 0.5 µg/ml of His-tagged FcγRI or His-tagged FcγRIIa, or 0.2 µg/well of His-tagged FcγIIIa for 2 h at room temperature. The plate was then washed three times with PBST (PBS/0.05% Tween-20). Glycoengineered trastuzumab samples were added to each well and incubated for 2-hour at 37 °C. Unbound antibodies were then removed by washing the wells three times with PBST. The detection antibody (Goat POD-anti human IgG (H + L), Jackson ImmunoResearch) was added and incubated at room temperature for 2 h, followed by washing. Substrate solution (1 step ultra TMB-ELISA, Thermo Fisher Scientific) was added to each well, and the reaction was stopped with 2 M sulfuric acid. Absorbance at 450 nm was then measured with SoftMax Pro software and a VersaMax ELISA Microplate reader. Each experiment was triplicated, and the mean reading was recorded.
ELISA to assess the binding affinities between the SOSIP Env and the bnAbs
Purified His-tagged SOSIP Env from CHO-K1, CHO-gmt4 and CHO-gmt35 were coated onto Nunc MaxiSorp 96-well plate at 2 mg/ml (50 ml/well) in 1x PBS for 2 h at room temperature. Unbound SOSIP Env proteins were washed off with TBS-0.05% Tween-20 (TBST), followed by overnight blocking with 3% BSA/TBST at 4 °C or 1-hour room temperature block. After washing off unbound BSA with TBST, Protein A purified bnAbs from CHO-K1 stable pools were added at 4-fold dilutions (starting at 10 µg/ml) and incubated for 1.5 h at room temperature. Unbound bnAbs were removed with three TBST washes. Bound bnAbs were detected using Horse radish peroxidase-conjugated rabbit anti-human IgG (H + L) (Jackson ImmunoResearch) in 1% BSA/TBST at 1:5000 for 1.5 h at room temperature, followed by five TBST washes before adding the substrate solution (1-step Ultra TMB-ELISA Substrate solution, Thermo Fisher Scientific). The colorimetric reaction was stopped with 0.2 M sulfuric acid, and the plates were read at 450 nm using Tecan Pro with Infinite 7.1 software. Each experiment was triplicated, and the mean reading was recorded.
Glycan analysis by HILIC-UPLC-QTOF
N-glycans were released from 15 µg of intact purified protein by PNGase F treatment followed by fluorescent labelling and HILIC clean up using Glycoworks RapiFluor-MS (RFMS) N-glycan kit (Waters Corporation, Milford, MA, US) according to the manufacturer’s protocol. The fluorescent labelled N-glycans were then dried down and reconstituted sequentially in 9 µL water, 10 µL dimethyl formamide (DMF), and 21 µL LC-MS grade acetonitrile (ACN) (> 99.9% purity, Fisher Chemical, Waltham, MA, US) (total of 40 µL).
Ten microliters (10 µL) of the reconstituted glycans were injected onto a Waters LC-MS/MS system which consists of a H-class Acquity UPLC system connected to a Xevo G2-S quadrupole-time of flight (QTOF) mass spectrometer, controlled by Waters UNIFI Biopharmaceutical software (version 1.8, Waters Corporation). Separation was performed across a Waters Acquity UPLC glycan BEH amide column (2.1 mm x 150 mm, 1.7 μm) using a gradient of buffer A (50 mM ammonium formate (pH 4.4)) and buffer B (100% ACN) with the following multi-step gradient; 75–51% B for 40 min at 0.4 mL/min, 51% B to 0% B in 1.5 min, 0% B for 3 min while flow rate is adjusted down from 0.4 to 0.2 mL/min, 0–75% B in 3.6 min at 0.2 mL/min, 75% B for 4.5 min at 0.2 mL/min, and 75% B for another 7.4 min while flow rate was increased from 0.2 back to 0.4 mL/min. Glycan signal was detected at excitation wavelength of 265 nm and emission wavelength of 425 nm. The retention time of each chromatographic peak was converted into a glucose unit (GU) by fitting into a calibration curve established by an external standard of RFMS-labelled dextran ladder (Waters Corporation, Milford, MA, US) using a cubic spline polynomial. The GU value of each chromatographic peak was search against the RFMS database (Waters Corporation) for assignment of possible N-glycan structures in the UNIFI software. Mass spectrum data was also used to identify the correct structure based on accurate mass confirmation (5 ppm error). Full MS scan was acquired in positive mode in the range of m/z 100–2000 using a capillary voltage of 2750 V, cone voltage at 15 V, source temperature at 120 °C, desolvation gas flow at 800 L/H, desolvation temperature at 300 ˚C and scan time of 1 s. Glu-fibrinopeptide (m/z 785.8421) was introduced as a lock spray mass to calibrate the mass accuracy of the instrument during the run.
MSMS was performed from the mass range of m/z 150 to 2000 at a scan time of 0.5 s for charge states 1 to 3 and ion intensity of 2000. Ramp collision energy was set to 10 eV to 30 eV.
Most peaks contained 1 N-glycan and this case LC fluorescent peak area was used to quantitate using the UNIFI software.
Mannosidase digestion
RFMS labelled samples were dried down and reconstituted in a mixture of glycobuffer 4, zinc and α1–2,3,6 mannosidase (New England Biolabs) and incubated for 72 h at 37 °C to ensure complete digestion. HILIC clean up using Glycoworks HILIC µElution plate (Waters Corporation) was performed following digestion.
Site-specific analysis of glucocerebrosidase N-glycopeptides by LC-MS/MS
Sample preparation for N-glycopeptide analysis was performed using the filter-aided sample preparation (FASP) method adapted from Wiśniewski et al. In brief, 30 µg of the protein was first denatured and reduced using 4 M urea and dithiothreitol (DTT) at 56 °C for 30 min. The protein was then transferred to a 10 K MWCO filter (Pall Corporation) and centrifuged at 14,000 g for 10 min at room temperature to remove the urea and DTT followed by addition of iodoacetamide for alkylation in the dark for 30 min. After 30 min, the iodoacetamide was removed by centrifugation and the protein which remained on filter was washed thrice with 50 mM ammonium bicarbonate. Trypsin (sequencing grade from Promega, Madison, WI, US) was then added in 1:20 ratio (enzyme/protein) and incubated at 37 ˚C for 16 h. The digested products were collected subsequently as flow-through from the filter by centrifugation. The peptides were dried down and reconstituted in 0.1% formic acid (Merck, Darmstadt, Germany) for analysis on LC-MS/MS.
The treated sample was injected and analyzed by nanospray LC-MS/MS on an Orbitrap Fusion Tribrid (Thermo Fisher Scientific, Waltham, MA, US) with an Easy-Spray source coupled to a nano LC system (Ultimate 3000 RSLCnano, Thermo Fisher Scientific) with a C18 trap column, (Acclaim PepMap100 C18, 5 μm, 5 mm x 300 μm, Thermo Fisher Scientific). Glycopeptides were enriched onto the trap column using a loading solvent of 0.1% trifluoroacetic acid (TFA) (Merck, Darmstadt, Germany) at a flow rate of 15 µL/min for 5 min before being eluted and separated on a 50 μm x 15 cm Easy-Spray C18, 2 μm nano column (Thermo Fisher Scientific) at a flow rate of 300 nL/min using the following multi-step gradient; 4% B for 5.5 min, 4 to 95% B in 49.5 min, 95% B for 7 min, 95–4% B in 2 min, 4% B for 15 min. Solvent A is 0.1% formic acid and solvent B is 0.1% formic acid in ACN.
Full FT-MS scan was acquired in the range of m/z 400–3000, with the source voltage set at 2000 V, ion transfer tube temperature at 300 °C, resolution at 120 K, AGC target of 4 × 105 and maximum injection time of 50 ms. MS/MS experiment was carried out using HCD with the following parameters: top speed 5 s, charge state 2–7, dynamic exclusion at 15 s with mass tolerance of ± 10 ppm, intensity greater the 5 × 104, isolation window of 1.6 m/z, collision energy at 20% and 40%, resolution of 30 K, AGC target of 1 × 105 and maximum injection time of 100 ms.
N-glycopeptide data analysis
Identification and quantification of glycopeptides and their glycosylation sites were performed using Byonic (Protein Metrics Inc., ver. 3.1.0) integrated into Proteome Discoverer (v2.2.0.388, Thermo Fisher Scientific) as well as manual inspection of the raw mass spectral data using the software Xcalibur (ver. 4.1.50, Thermo Fisher Scientific). The following parameters settings were used with the enzyme for sample digestion set to trypsin (full), two maximum missed cleavages, precursor mass tolerance at 15 ppm, fragment mass tolerance at 20 ppm, common modifications (3 per peptide) were dynamic methionine oxidation, asparagine and glutamine deamidation and static cysteine carbamidomethylation, rare modifications were set to one N-glycan per peptide and searched against an in-house created N-glycan library database comprising of 20 glycans with 1% false discovery rate.
Glycopeptides were identified by searching their MSMS fragmentation. In particular, the presence of oxonium ions (m/z 204.0867, 366.1396), peptide sequence (b- and y-ions fragmentation), elongation of the oligosaccharide as well as, the complete intact mass of the glycopeptides was checked manually for complete agreement.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank all current and former lab members for their invaluable discussions and support throughout this project. This project was partially funded by the grant from the A*STAR BMRC Strategic Positioning Fund (BMRC SPF) for “GylcoSing, A Centre for Glycobiotechnology and Glycomics in Singapore.” We thank the BiaCore lab at Nanyang Technological University for conducting the antibody-FcγR interaction analysis.
Author contributions
R.H. and K.F.C. generated and analysed the CHO glycosylation mutants, while also producing and purifying EPO-Fc and the antibodies in these mutants. R.H. conducted ADCC experiments using breast cancer cell lines. P.C.L. prepared materials and conducted ELISA experiments for antibody-FcγR interaction. P.C.L. produced anti-HIV bnAbs, P.C.L. and K.F.C. produced and purified His-tagged BG505 SOSIP.664 gp140 (SOSIP Env), as well as performed ELISA analysis with the bnAbs. W.S. prepared martials for Biacore analysis and the materials and ELISA experiments for antibody-FcγR interaction. Y.L.T. produced and purified recombinant β-glucocerebrosidase (GCase). C.W and S.J.T. structure analysis of glycans. T.N.K. and I.W. glycan data analysis. Z.S. conceived, planed, and wrote the manuscript for this project.
Data availability
All data supporting the findings of this study are available within the paper and its supplementary information. Requests for resources and reagents should be directed to Zhiwei Song (song_zhiwei@bti.a-star.edu; or songzw2017@gmail.com).
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Li, H. & d’Anjou, M. Pharmacological significance of glycosylation in therapeutic proteins. Curr. Opin. Biotechnol.20, 678–684 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Solá, R. J. & Griebenow, K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharm. Sci.98, 1223–1245 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solá, R. J. & Griebenow, K. Glycosylation of therapeutic proteins: An effective strategy to optimize efficacy. BioDrugs24, 9–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira, N. A., Chan, K. F., Lin, P. C. & Song, Z. The less-is-more in Therapeutic Antibodies: Afucosylated anti-cancer Antibodies with Enhanced antibody-dependent Cellular Cytotoxicity 5, 693–711 (Taylor & Francis, 2018). [DOI] [PMC free article] [PubMed]
- 5.Friedman, B. et al. A comparison of the pharmacological properties of carbohydrate remodeled recombinant and placental-derived β-glucocerebrosidase: Implications for clinical efficacy in treatment of Gaucher disease. Blood93, 2807–2816 (1999). [PubMed] [Google Scholar]
- 6.Grabowski, G. A. et al. Enzyme therapy in type 1 gaucher disease: Comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann. Intern. Med.122, 33–39 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Horiya, S., MacPherson, I. S. & Krauss, I. J. Recent strategies targeting HIV glycans in vaccine design. Nat. Chem. Biol.10, 990–999 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doores, K. J. The HIV glycan shield as a target for broadly neutralizing antibodies. FEBS J.282, 4679–4691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritchard, L. K. et al. Structural constraints determine the glycosylation of HIV-1 envelope trimers. Cell. Rep.11, 1604–1613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Struwe, W. B. et al. Site-specific glycosylation of virion-derived HIV-1 Env is mimicked by a soluble trimeric immunogen. Cell. Rep.24, 1958–1966. e1955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patnaik, S. K. & Stanley, P. Lectin-resistant CHO glycosylation mutants. Methods Enzymol.416, 159–182 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Song, Z. Roles of the nucleotide sugar transporters (SLC35 family) in health and disease. Mol. Aspects Med.34, 590–600 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Lim, S. F., Lee, M. M., Zhang, P. & Song, Z. The golgi CMP-sialic acid transporter: A new CHO mutant provides functional insights. Glycobiology18, 851–860 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang, P., Chan, K. F., Haryadi, R., Bardor, M. & Song, Z. CHO Glycosylation Mutants as Potential host Cells to Produce Therapeutic Proteins with Enhanced Efficacy, 63–87 (Future Trends in Biotechnology, 2013). [DOI] [PubMed]
- 15.Zhang, P. et al. Identification of functional elements of the GDP-fucose transporter SLC35C1 using a novel Chinese hamster ovary mutant. Glycobiology22, 897–911 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Chan, K. F. et al. Inactivation of GDP-fucose transporter gene (Slc35c1) in CHO cells by ZFNs, TALENs and CRISPR‐Cas9 for production of fucose‐free antibodies. Biotechnol. J.11, 399–414 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Lauber, M. A. et al. Rapid preparation of released N-glycans for HILIC analysis using a labeling reagent that facilitates sensitive fluorescence and ESI-MS detection. Anal. Chem.87, 5401–5409 (2015). [DOI] [PubMed] [Google Scholar]
- 18.North, S. J. et al. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol. Chem.285, 5759–5775 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang, Z. et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat. Biotechnol.33, 842–844 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Harvey, D. J. et al. Proposal for a standard system for drawing structural diagrams of N-and O‐linked carbohydrates and related compounds. Proteomics9, 3796–3801 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Varki, A. et al. Symbol nomenclature for glycan representation. Proteomics9, 5398–5399 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, P. et al. A functional analysis of N-glycosylation-related genes on sialylation of recombinant erythropoietin in six commonly used mammalian cell lines. Metab. Eng.12, 526–536 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Goh, J. S. et al. RCA-I-resistant CHO mutant cells have dysfunctional GnT I and expression of normal GnT I in these mutants enhances sialylation of recombinant erythropoietin. Metab. Eng.12, 360–368 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Misago, M. et al. Molecular cloning and expression of cDNAs encoding human alpha-mannosidase II and a previously unrecognized alpha-mannosidase IIx isozyme. Proc. Natl. Acad. Sci.92, 11766–11770 (1995). [DOI] [PMC free article] [PubMed]
- 25.Lal, A. et al. Substrate specificities of recombinant murine golgi α1, 2-mannosidases IA and IB and comparison with endoplasmic reticulum and Golgi processing α1, 2-mannosidases. Glycobiology8, 981–995 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Ikeda, Y., Ihara, H., Tsukamoto, H., Gu, J. & Taniguchi, N. Mannosyl (beta-1, 4-)-glycoprotein beta-1, 4-N-acetylglucosaminyltransferase (MGAT3); β1, 4-NAcetylglucosaminyltransferase III (GnT-III, GlcNAcT-III). In Handbook of Glycosyltransferases and Related Genes, Second Edition, 209–222 (Springer Japan, 2014).
- 27.Xu, C. & Ng, D. T. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol.16, 742–752 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Malm, D. & Nilssen, Ø. Alpha-mannosidosis. Orphanet J. Rare Dis.3, 1–10 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao, Y. F., Lal, A. & Moremen, K. W. Cloning, expression, purification, and characterization of the human broad specificity lysosomal acid α-mannosidase. J. Biol. Chem.271, 28348–28358 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Hulett, M. D. & Hogarth, P. M. Molecular basis of fc receptor function. Adv. Immunol.57, 1–127 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Lu, J. et al. Structural recognition and functional activation of FcγR by innate pentraxins. Nature456, 989–992 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feige, M. J. et al. Structure of the murine unglycosylated IgG1 fc fragment. J. Mol. Biol.391, 599–608 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Radaev, S. & Sun, P. D. Recognition of IgG by Fcγ receptor: The role of fc glycosylation and the binding of peptide inhibitors. J. Biol. Chem.276, 16478–16483 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Nimmerjahn, F. & Ravetch, J. V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol.8, 34–47 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Caaveiro, J. M., Kiyoshi, M. & Tsumoto, K. Structural analysis of Fc/FcγR complexes: A blueprint for antibody design. Immunol. Rev.268, 201–221 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Subedi, G. P. & Barb, A. W. The Immunoglobulin G1 N-glycan Composition Affects Binding to each low Affinity Fc γ Receptor, 8, 1512–1524 (Taylor & Francis, 2016). [DOI] [PMC free article] [PubMed]
- 37.Kayili, H. M. Identification of bisecting N-glycans in tandem mass spectra using a procainamide labeling approach for in-depth N-glycan profiling of biological samples. Int. J. Mass Spectrom.457, 116412 (2020). [Google Scholar]
- 38.Thomann, M. et al. In vitro glycoengineering of IgG1 and its effect on fc receptor binding and ADCC activity. PLoS ONE10, e0134949 (2015). [DOI] [PMC free article] [PubMed]
- 39.Lu, J. et al. Structure of FcγRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc. Natl. Acad. Sci.112, 833–838. (2015). [DOI] [PMC free article] [PubMed]
- 40.Niwa, R. et al. Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin. Cancer Res.11, 2327–2336 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Haji-Ghassemi, O., Blackler, R. J., Young, M., Evans, S. V. & N., and Antibody recognition of carbohydrate epitopes. Glycobiology25, 920–952 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Sanders, R. W. et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP. 664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog.9, e1003618 (2013). [DOI] [PMC free article] [PubMed]
- 43.Calarese, D. A. et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science300, 2065–2071 (2003). [DOI] [PubMed] [Google Scholar]
- 44.McLellan, J. S. et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature480, 336–343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pancera, M. et al. Structural basis for diverse N-glycan recognition by HIV-1–neutralizing V1–V2–directed antibody PG16. Nat. Struct. Mol. Biol.20, 804–813 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mouquet, H. et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci.109, E3268-E3277 (2012). [DOI] [PMC free article] [PubMed]
- 47.Julien, J. P. et al. Broadly neutralizing antibody PGT121 allosterically modulates CD4 binding via recognition of the HIV-1 gp120 V3 base and multiple surrounding glycans. PLoS Pathog.9, e1003342 (2013). [DOI] [PMC free article] [PubMed]
- 48.Pejchal, R. et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science334, 1097–1103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berg-Fussman, A., Grace, M., Ioannou, Y. & Grabowski, G. Human acid beta-glucosidase. N-glycosylation site occupancy and the effect of glycosylation on enzymatic activity. J. Biol. Chem.268, 14861–14866 (1993). [PubMed] [Google Scholar]
- 50.Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Canis, K., Garénaux, E. & Boe, J. F. Site-Specific N-glycosylation Analysis of Recombinant Proteins by LC/MS E. Mass Spectrometry of Glycoproteins: Methods and Protocols, 133–154 (2021). [DOI] [PubMed]
- 52.Dvir, H. et al. X-ray structure of human acid‐β‐glucosidase, the defective enzyme in Gaucher disease. EMBO Rep.4, 704–709 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ronda, C. et al. Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol. Bioeng.111, 1604–1616 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knibbs, R. N., Goldstein, I. J., Ratcliffe, R. M. & Shibuya, N. Characterization of the carbohydrate binding specificity of the leukoagglutinating lectin from Maackia amurensis. Comparison with other sialic acid-specific lectins. J. Biol. Chem.266, 83–88 (1991). [PubMed] [Google Scholar]
- 55.Wang, W. C. & Cummings, R. D. The immobilized leukoagglutinin from the seeds of Maackia amurensis binds with high affinity to complex-type asn-linked oligosaccharides containing terminal sialic acid-linked alpha-2,3 to penultimate galactose residues. J. Biol. Chem.263, 4576–4585 (1988). [PubMed] [Google Scholar]
- 56.Lin, P. C. et al. Attenuated Glutamine Synthetase as a Selection Marker in CHO Cells to Efficiently Isolate Highly Productive Stable Cells for the Production of Antibodies and Other Biologics, 5, 965–976 (Taylor & Francis, 2019). [DOI] [PMC free article] [PubMed]
- 57.Haryadi, R. et al. Optimization of heavy chain and light chain signal peptides for high level expression of therapeutic antibodies in CHO cells. PLoS ONE10, e0116878. 10.1371/journal.pone.0116878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ho, S. C. et al. IRES-mediated tricistronic vectors for enhancing generation of high monoclonal antibody expressing CHO cell lines. J. Biotechnol.157, 130–139. 10.1016/j.jbiotec.2011.09.023 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and its supplementary information. Requests for resources and reagents should be directed to Zhiwei Song (song_zhiwei@bti.a-star.edu; or songzw2017@gmail.com).