

Abstract
Cassava (Manihot esculenta Crantz) is a vital carbohydrate source for over 800 million people globally, yet its production in East Africa is severely affected by cassava brown streak disease (CBSD). Genebanks, through ex-situ conservation, play a pivotal role in preserving crop diversity, providing crucial resources for breeding resilient and disease-resistant crops. This study genotyped 234 South American cassava accessions conserved at the CIAT genebank, previously phenotyped for CBSD resistance by an independent group, to perform a genome-wide association analysis (GWAS) to identify genetic variants associated with CBSD resistance. Our GWAS identified 35 single nucleotide polymorphism (SNP) markers distributed across various chromosomes, associated with disease severity or the presence/absence of viral infection. Markers were annotated within or near genes previously identified with functions related to pathogen recognition and immune response activation. Using the SNP candidates, we screened the world’s largest cassava collection for accessions with a higher frequency of favorable genotypes, proposing 35 accessions with potential resistance to CBSD. Our results provide insights into the genetics of CBSD resistance and highlight the importance of genetic resources to equip breeders with the raw materials needed to develop new crop varieties resistant to pests and diseases.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-74161-6.
Keywords: Cassava brown streak disease (CBSD), Genome-wide association study (GWAS), Genetic resources, Disease resistance, Ex-situ conservation, Molecular breeding.
Subject terms: Biotechnology, Genetics, Molecular biology, Plant sciences
Introduction
Cassava (Manihot esculenta Crantz) is the world’s fourth most important staple crop after rice, wheat, and maize, and serves as a vital carbohydrate source for over 800 million people1. In 2021, according to the FAO, global production exceeded 300 million tons, with Africa alone contributing around 60% of its production2. Cassava is Africa’s most important tuberous crop due to its remarkable tolerance to dry environments and infertile soils, thereby serving as a crucial income and nutrition source for small-scale farmers1. However, despite Africa being the major cassava producer, its production is hindered by persistently low yields3. Producing an average 8.5 tons per hectare, yields are low in Africa compared to averages of 22 tons/ha in Asia and 13.2 tons/ha in Latin America2. Low African yields can be attributed to different factors including lack of agronomic interventions, poor stem quality, and distinct pests and diseases, among others3–5.
Climate change has increased the spread of diseases affecting cassava production, threatening food security6,7. Among the diseases that have a significant impact on cassava production, Cassava brown streak disease (CBSD) is one of the major viral diseases causing significant yields losses in East Africa5,8,9. The disease is caused by distinct (+) ssRNA viruses belonging to genus Ipomovirus, family Potyviridae, Cassava brown streak virus (CBSV), and Ugandan cassava brown streak virus (UCBSV)10,11. Since cassava is vegetatively propagated, CBSD can be transmitted through stem cutting, and also by whitefly (Bemisia tabaci)12. The viral infection can cause leaf chlorisis, brown streaks on the stems and root necrosis, rendering the roots inedible5. It has been reported that the disease causes yield losses of up to 70%5 representing the greatest threat to millions of cassava farmers in Central and East Africa13,14.
CBSD-resistant genotypes offer a significant option for effective CBSD control. However, many countries lack CBSD-resistant or tolerant varieties. Cassava breeding is a long-term process (eight years or more), and breeding programs face significant challenges in selecting promising CBSD-resistant materials, given the obstacles posed by virus transmission through whitefly populations and the slow virus infection processes15–17. Despite setbacks, ongoing efforts are identifying sources of resistance genes deploying genome-wide association studies (GWAS). Candidate germplasm is derived from several sources. These include: (i) open-pollinated Tanzanian landraces, wild relatives, and cassava families from Brazil; (ii) populations derived from biparental crosses having contrasting responses to CBSD, with genetically diverse clones derived from the International Institute for Tropical Agriculture (IITA) and the International Center for Tropical Agriculture (CIAT); and (iii) hybridized C1 populations, identifying genomic regions associated with CBSD and linked across different chromosomes9,18–21. Moreover, alternative sources of resistance have been identified in cassava landraces from Colombia, Ecuador and Peru that are conserved in the CIAT genebank22.
Much of the crop diversity of many essential food crops is safeguarded in 1,750 genebanks worldwide23,24. These play a crucial role in developing superior crop varieties that improve yields, climate adaptation, nutrition and resistance to pests and diseases22,25–27. The germplasm collections provide a valuable source of genetic variation that can be used to diversify resistance factors in modern genepools27. Genotyping by sequencing (GBS) has enabled the exploration and characterization of cassava genetic diversity28–30. DArTseq, a GBS platform, offers high-resolution genome-wide genotyping by combining restriction enzyme-based genome complexity reduction method with next-generation sequencing. Efforts to control cassava mosaic disease (CMD), caused by cassava mosaic begomoviruses31 has been achieved through introgression of genes from wild cassava Manihot glaziovii32. Similar cases of using diversity conserved in genebanks to improve resistance to crop pest and diseases have been reported in barley33, wheat34, and bean35.
Colombia, through the International Center for Tropical Agriculture (CIAT), conserves the world’s largest in-vitro collection of cassava and its wild relatives, ensuring the preservation of biodiversity for future generations and supporting global breeding programs by providing access to these genetic resources. This collection consists of 5,577 accessions of the cultivated species and 386 wild relatives belonging to 23 Manihot species. CBSD resistance was found in sixteen accessions from a subset of the cassava core collection after artificial inoculation with CBSV-Mo83 isolates22, highlighting the valuable contribution of germplasm collections as a resource for crop improvement programs.
In the present study, we leveraged the reported phenotypic information by Sheat et al. (2019)22 to perform a genome-wide association with the following objectives: (1) to identify chromosomal regions and genes potentially involved in CBSD resistance within a diverse panel of genebank accessions, (2) to select a set of genebank accessions with potential resistance to CBSD for validation, and (3) to provide an example of how genomics, data integration and statistics can support and facilitate informed access to genetic resources conserved in national or international genebanks, and while also providing insights into the molecular mechanisms underlying disease resistance.
Results
Cassava accessions used in this study and source of phenotypic data
With the aim of conducting a trait association analysis, we utilized 234 germplasm accessions conserved at CIAT in Palmira, Colombia. These accessions had previously been evaluated for CBSD resistance by Sheat et al. in 2019, who artificially inoculated cassava plantlets with the most pathogenic virus isolate (CBSV-Mo83), through auxiliary bud grafting22. CBSD symptoms severity was assessed by Sheat et al. (2019) using a five-point scale36, where 0 indicates no symptoms on leaves and stems, and 5 indicates severe symptoms, including wilting, followed by plant death. Additionally, virus presence in each sample was determined using quantitative reverse transcription PCR (qRT-PCR) by the same independent research team22.
These 234 CBSD phenotyped accessions were collected or donated to the CIAT genebank from 20 countries, primarily from South America, with the majority coming from Colombia, followed by Brazil, Peru and Venezuela. All accessions are landraces, except for six that are improved lines from Brazil, Nigeria, and Thailand. Sheat et al. (2019) reported 16 accessions with no symptoms on leaves and stems, 27 accessions on scale 2, 56 accessions on scale 3, 98 on scale 4 and 37 on scale 522. Among the 16 on scale 1, only seven accessions demonstrated high resistance against CBSV-Mo83 isolate, showing an absence of viral infection when confirmed by qRT-PCR (COL40, COL2182, COL144, ECU41, PER221, PER556, and PER333)22. All these accessions were genotyped for SNP markers, and the phenotypic data were extracted from Sheat et al. (2019) (Supplementary Table S1).
High density genome‑wide DArTseq markers and selection of SNP markers
Genotyping generated 121,405 SNP markers distributed over the cassava genome. Approximately 87% (105,826) of the SNP markers were aligned to the eighteen cassava chromosomes, and about 1.7% (2,078) were mapped to different scaffolds of the cassava reference genome v6.137. A total of 11% (13,501 SNPs) were not mapped to the reference genome. The highest number of markers mapped onto chromosome 1 (Chr1) (1,594), while the lowest number mapped onto Chr14 (984 SNPs) (Fig. 1A). The proportion of marker missing values across loci and individuals ranged from 0.92 to 0 and from 0.31 to 0.01, respectively. Minor allele frequency (MAF) values ranged from 0.002 to 0.500, with an average value of 0.162. Average marker count (AvgMarkerCount) and marker reproducibility (two marker-quality parameters) ranged from 2.2 to 184 and from 0.90 to 1, respectively (Supplementary Table S2). To assess the population structure, a subset of 17,989 high-quality markers was selected based on four criteria described in the Methods section. For the association analysis, we further refined this subset by selecting only the markers that mapped onto the eighteen chromosomes of the cassava reference genome v6.137, resulting in a total of 16,452 SNP markers (Fig. 1B).
Fig. 1.
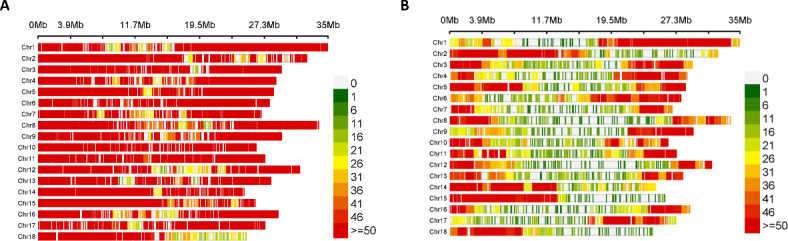
Genome-wide distribution of SNP markers showing the density of SNPs within a 1 Mb window size. The distribution of the markers across the eighteen chromosomes of the cassava reference genome v6.1 is shown in (A) before filtering, and (B) after filtering.
Genome-wide association study
The population structure of the GWAS panel was estimated using three different approaches: (i) principal component analysis (PCA); (ii) admixture analysis, using sparse non-negative matrix factorization (snmf), and (iii) agglomerative hierarchical clustering. In the PCA, the first eight principal components (PCs) accounted for 22.6% of the genetic variation observed in the data (Fig. 2A). Specifically, the first PC accounted for 5.2%, the second for 3.4%, the third for 3.3%, and the remaining five PCs accounted for 10.7% collectively. When examining the population structure based on the country of origin, three major groups of accessions were observed (Fig. 2B). One group primarily consisted of accessions from Colombia, Peru, and Venezuela (G1), while a second group was predominantly composed of accessions from Brazil and Paraguay (G2). Additionally, a third group comprised accessions from Brazil, along with some from Colombia and Venezuela (G3). In terms of population structure based on CBSD symptom severity phenotype, most accessions showed no clear separation for scales 2 to 5. However, all 16 accessions were categorized within scale 1 (showing no symptoms on leaves and stems) and corresponded to group 1 (G1 in Fig. 2C). Consequently, when considering the presence (1) or absence (0) of the virus detected by qRT-PCR, the seven accessions that remained uninfected by the virus and were considered resistant by Sheat et al. (2019)22 were all clustered in G1 (Fig. 2D). In the admixture analysis with snmf, the entropy criterion suggested a higher number of genetic clusters in comparison to the PCA, with tentative 8 genetic clusters in the data (Supplementary Fig. S1). Moreover, the hierarchical clustering analysis detected the major three clusters of the PCA, as well as the eight clusters detected by the admixture analysis (Fig. 3A,B). However, the accessions’ assignment in each cluster were mostly consistent with the clusters detected by the admixture analysis (K = 3 and K = 8). The linkage disequilibrium (LD) decay pattern across the 18 chromosomes was consistent, showing a rapid decay as the distance between markers increased, with an average genome-wide LD of 0.021 (Supplementary Fig. S2). A total of three principal components were utilized for the association analyses, that accounted for 11,9% of the genetic variation observed to correct for population stratification effects and minimize false positives.
Fig. 2.
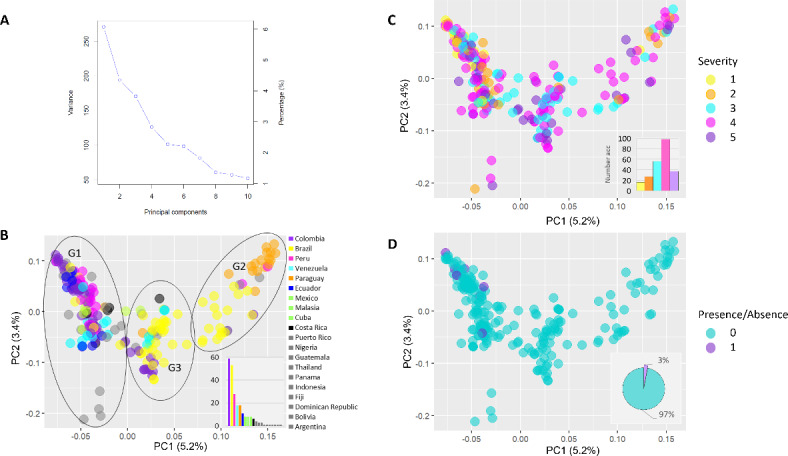
Plots of the two principal components (PCs) showing the distribution of accessions in PC1 and PC2. (A) depicts the variance explained by the first ten principal components and their respective percentages. In (B), colors represent the country of origin of each accession, with three major groups shown within circles (G1, G2, and G3). (C) indicates colors corresponding to the five-point scale of CBSD symptom severity phenotype as determined by Sheat et al. (2019)22. Similarly, colors in (D) represent the presence or absence of the CBSV-Mo83 virus isolate as reported by the same authors.
Fig. 3.
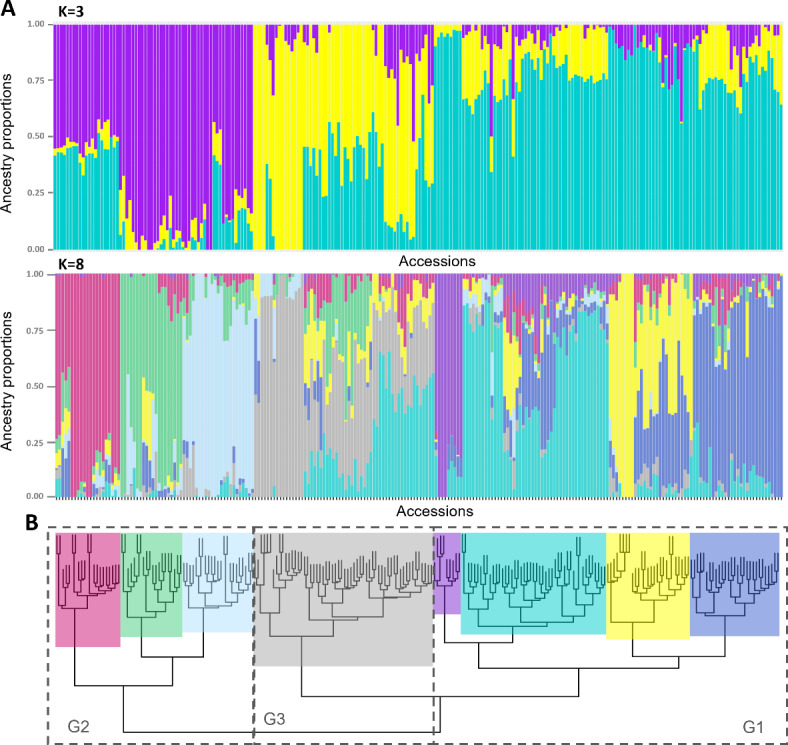
Population structure and clustering analyses of the studied samples. (A) Bar plots showing the ancestry coefficients from K = 3 and K = 8 based on the admixture analysis. (B) Clusters detected by hierarchical clustering analysis using an Identity-by-State distance matrix and the ‘Ward.2D’ clustering method. Three and eight clusters are displayed with dashed lines or colors, respectively.
Three statistical models were used for the GWAS: the Mixed Linear Model (MLM), Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway (BLINK), and Fixed and random model Circulating Probability Unification (FarmCPU)38–41. These models identified 35 significant associations across multiple chromosomes (Table 1), including five linked to CBSD severity on chromosomes 3, 7, 11 and 15, and thirty associated with CBSV infection on multiple chromosomes (Table 1; Fig. 4A,B). The Quantile-Quantile (QQ) plot confirmed statistical significance, with most observed -log10(p-values) following the expected distribution, except for significant associations (Fig. 4C,D). Among the five markers linked to CBSD severity, FarmCPU identified all five, one was identified by all three models, and two by both BLINK and FarmCPU (Supplementary Fig. S3). These SNPs explained 0 to 8.4% of phenotypic variation (PVE), with AlleleIDs 7118148:40-G/T on chromosomes 7 and 13856723:54-G/C on chromosomes 11 accounting for 8.4% and 8%, respectively (Table 1). The functional annotation analysis using the cassava reference genome v6.1 revealed that the five SNPs were located within or near to encoding genes. The AlleleID 7118148:40-G/T on Chr7 was a variant within a non-coding region upstream of the Manes.07G118400 gene, which encodes glycerol-3-phosphate acyltransferase 5 (GPAT). The AlleleID 13856723:54-G/C on Chr11, was annotated as a synonymous variant within a coding region in the Manes.11G122600 gene, which encodes to a RHOMBOID-like protein 13 (Table 1 and Supplementary Table S3).
Table 1.
List of significant markers identified from the GWAS analyses using the CBSD symptom severity and presence/absence of CBSV phenotype, as determined by Sheat et al. (2019)22.
| Phenotype | Chr | AlleleID | Ref/Alt | Chr_pos | Gene related | Variant annotation | Model | PVE(%) | p-value | Bonferroni correction |
|---|---|---|---|---|---|---|---|---|---|---|
| CBSD severity | 3 | 7112046-16-A/C | A/C | 26,416,990 | Manes.03G173800 | splice_acceptor_variant | FarmCPU | 0.0 | 2.90E–06 | 4.76E–02 |
| 7122503-44-G/T | G/T | 27,997,604 | Manes.03G197900 | 5_prime_UTR_variant | FarmCPU | 0.0 | 6.15E–08 | 1.01E–03 | ||
| 7 | 7118148-40-G/T | G/T | 24,643,336 | Manes.07G118400 | upstream_transcript_variant | BLINK | 8.4 | 1.33E–07 | 2.19E–03 | |
| FarmCPU | 0.0 | 1.52E–06 | 2.49E–02 | |||||||
| 11 | 13856723-54-G/C | G/C | 23,187,439 | Manes.11G122600 | synonymous_variant | BLINK | 8.0 | 2.26E–08 | 3.72E–04 | |
| FarmCPU | 0.0 | 2.54E–06 | 4.19E–02 | |||||||
| 15 | 7119826-59-T/C | T/C | 22,387,295 | Manes.15G185900 | 5_prime_UTR_variant | FarmCPU | 5.9 | 1.81E–08 | 2.98E–04 | |
| Presence/ absence virus | 1 | 7116322-33-A/G | A/G | 26,393,776 | Manes.01G156200 | intron_variant | MLM | 1.2 | 2.46E–06 | 4.04E–02 |
| BLINK | 11.2 | 2.12E–08 | 3.48E–04 | |||||||
| FarmCPU | 2.9 | 1.53E–08 | 2.52E–04 | |||||||
| 13855347-8-A/T | A/T | 32,704,262 | Manes.01G250600 | 3_prime_UTR_variant | BLINK | 1.5 | 6.21E–20 | 1.02E–15 | ||
| 2 | 20486307-57-T/G | T/G | 1,707,248 | Manes.02G021400 | 3_prime_UTR_variant | MLM | 28.2 | 5.27E–08 | 8.67E–04 | |
| BLINK | 24.9 | 2.52E–35 | 4.15E–31 | |||||||
| 7118726-36-A/G | A/G | 1,906,690 | Manes.02G023900 | downstream_transcript_variant | MLM | 0.6 | 2.81E–06 | 4.62E–02 | ||
| BLINK | 0.0 | 8.79E–07 | 1.45E–02 | |||||||
| FarmCPU | 4.1 | 1.42E–06 | 2.33E–02 | |||||||
| 7113688-40-G/A | G/A | 9,304,881 | Manes.02G127300 | upstream_transcript_variant | BLINK | 0.0 | 2.79E–06 | 4.59E–02 | ||
| 20485616-18-G/A | G/A | 9,617,457 | Manes.02G131400 | 5_prime_UTR_variant | BLINK | 2.9 | 1.45E–13 | 2.38E–09 | ||
| 20500103-66-C/T | C/T | 16,005,821 | Manes.02G194400 | intron_variant | MLM | 0.0 | 1.13E–08 | 1.87E–04 | ||
| 20490170-25-A/T | A/T | 17,064,412 | Manes.02G200000 | splice_acceptor_variant | BLINK | 1.4 | 1.53E–06 | 2.52E–02 | ||
| FarmCPU | 1.4 | 1.02E–09 | 1.68E–05 | |||||||
| 4 | 13861704-29-C/A | C/A | 17,592,579 | Manes.04G064300 | intron_variant | FarmCPU | 0.0 | 3.33E–15 | 5.47E–11 | |
| 5 | 20496717-36-A/G | A/G | 11,465,069 | Manes.05G116000 | intron_variant | BLINK | 0.2 | 3.49E–07 | 5.74E–03 | |
| 20497481-41-C/T | C/T | 19,957,250 | Manes.05G140500 | upstream_transcript_variant | FarmCPU | 0.0 | 5.81E–07 | 9.56E–03 | ||
| 6 | 7132461-6-C/A | C/A | 26,120,228 | Manes.06G159700 | synonymous_variant | MLM | 0.1 | 2.22E–06 | 3.65E–02 | |
| 7 | 20504508-6-T/C | T/C | 23,142,139 | unknown | intergenic_variant | BLINK | 8.1 | 4.79E–09 | 7.88E–05 | |
| FarmCPU | 7.1 | 1.59E–07 | 2.62E–03 | |||||||
| 8 | 7112869-8-C/G | C/G | 32,642,954 | Manes.08G165800 | missense_variant | FarmCPU | 6.7 | 2.18E–06 | 3.58E–02 | |
| 9 | 7148270-42-A/C | A/C | 26,108,329 | Manes.09G143200 | missense_variant | MLM | 33.3 | 9.06E–08 | 1.49E–03 | |
| BLINK | 20.1 | 2.36E–14 | 3.89E–10 | |||||||
| 11 | 20473300-48-A/C | A/C | 17,561,191 | Manes.11G100900 | synonymous_variant | BLINK | 0.6 | 1.59E–21 | 2.62E–17 | |
| 7125725-8-A/C | A/C | 25,090,966 | Manes.11G138500 | intron_variant | MLM | 8.8 | 1.25E–06 | 2.05E–02 | ||
| BLINK | 6.7 | 2.81E–06 | 4.63E–02 | |||||||
| 7116418-22-C/A | C/A | 26,470,720 | Manes.11G154300 | intron_variant | FarmCPU | 2.7 | 1.97E–09 | 3.24E–05 | ||
| 12 | 20488092-21-C/G | C/G | 9,548,450 | Manes.12G081300 | 3_prime_UTR_variant | MLM | 0.0 | 3.04E–06 | 5.00E–02 | |
| 13 | 7147375-42-A/C | A/C | 24,876,235 | Manes.13G121100 | intron_variant | BLINK | 2.4 | 6.52E–19 | 1.07E–14 | |
| FarmCPU | 0.0 | 2.46E–08 | 4.05E–04 | |||||||
| 14 | 20497806-14-T/G | T/G | 8,260,281 | Manes.14G102400 | upstream_transcript_variant | MLM | 0.0 | 2.06E–07 | 3.39E–03 | |
| 15 | 20473618-24-G/C | G/C | 2,128,122 | Manes.15G027800 | intron_variant | BLINK | 3.2 | 4.07E–11 | 6.69E–07 | |
| FarmCPU | 0.0 | 2.24E–12 | 3.69E–08 | |||||||
| 7126319-64-T/C | T/C | 9,954,369 | Manes.15G130900 | missense_variant | MLM | 0.2 | 2.45E–06 | 4.03E–02 | ||
| 20491807-17-C/A | C/A | 11,631,786 | Manes.15G148600 | splice_acceptor_variant | BLINK | 3.0 | 4.01E–12 | 6.59E–08 | ||
| 16 | 7107809-32-T/C | T/C | 24,021,740 | Manes.16G083000 | exonic_splice_region_variant | MLM | 0.2 | 3.43E–08 | 5.64E–04 | |
| FarmCPU | 0.0 | 2.09E–07 | 3.44E–03 | |||||||
| 20481832-32-G/C | G/C | 27,000,390 | Manes.16G115300 | splice_acceptor_variant | BLINK | 1.2 | 7.98E–17 | 1.31E–12 | ||
| 7112183-41-G/A | G/A | 28,218,989 | Manes.16G132500 | intron_variant | MLM | 5.7 | 5.71E–07 | 9.40E–03 | ||
| BLINK | 1.5 | 1.01E–07 | 1.66E–03 | |||||||
| 17 | 13856211-29-G/A | G/A | 18,683,421 | Manes.17G049300 | exonic_splice_region_variant | MLM | 0.0 | 1.93E–07 | 3.17E–03 | |
| 7124234-46-A/T | A/T | 24,150,948 | Manes.17G100500 | intron_variant | BLINK | 3.3 | 2.64E–08 | 4.35E–04 | ||
| FarmCPU | 0.0 | 9.98E–10 | 1.64E–05 | |||||||
| 18 | 13857824-33-C/G | C/G | 23,405,196 | Manes.18G144800 | synonymous_variant | BLINK | 0.6 | 1.79E–09 | 2.94E–05 |
Fig. 4.

Genome-wide association studies and statistical model results. (A) Manhattan plots for the CBSD severity phenotype using MLM, BLINK and FarmCPU models and, (B) for the presence/absence of CBSV infection phenotype. QQ plots for (C) the CBSD severity phenotype and, (D) for the presence/absence of CBSV infection phenotype with the three statistical models used. In Manhattan plots dashed lines represent False discovery rate thresholds and continuous lines Bonferroni threshold, respectively.
Among the 30 significant associations with presence/absence of CBSV infection phenotype (Supplementary Table S1), 12 were identified by FarmCPU, 13 by MLM, and 19 by BLINK on chromosomes 1, 2, 4, 5, 6, 7, 8, 9, 11, 12, 13, 14, 15, 16, 17, and 18 (Table 1; Fig. 4B). The QQ plot revealed that BLINK’s observed -log10(p-values) exhibited a more pronounced deviation from the expected distribution in contrast to MLM and FarmCPU (Fig. 4D). Comparing the significant associations across the three models, 12 markers were identified by more than one model, including two markers identified by all three: 7116322:33-A/G on Chr1 and 7118726:36-A/G on Chr2 (Table 1 and Supplementary Fig. S3). Phenotypic variation across the 30 significant markers ranged from 0 to 33% with an average of 4.6%, varying by model (Table 1). Five markers showed percentages of PVE above 6%: 7116322-33-A/G on Chr1, 20486307-57-T/G on Chr2, 20504508-6-T/C on Chr7, 7148270-42-A/C on Chr9 and 7125725-8-A/C on Chr11. Four of these were annotated within or near the genes Manes.01G156200, Manes.02G021400, Manes.09G143200 and Manes.11G138500. These genes are known to encode Protein phosphatase 2 C family protein, RING/U-box superfamily protein; NB-ARC domain-containing disease resistance protein and hydrolase family protein / HAD-superfamily protein, respectively (Table 1 and Supplementary Table S3). The markers, 20486307-57-T/G on Chr2 and 7148270-42-A/C on Chr9 showed the highest phenotypic variation, averaging 26.5 and 26.7%, respectively, across the MLM and BLINK models.
Another twenty-five associations were located within or close to genes known to encode for: guanosine triphosphate (GTP) binding protein beta 1 (3.3% phenotypic variation explained); photosystem I subunit D-2 (3.2%); DDOST_48_kDa_subunit (3%); Rab5-interacting family protein (2.9%); Galactose oxidase/kelch repeat superfamily protein (1.5%); P-loop containing nucleoside triphosphate hydrolase (1.4%); proline extensin-like receptor kinase 1 (PERK) (1.2%); peroxidase superfamily protein (0.6%); and O-methyltransferase family protein (0.6%), among others listed in Table 1. Along all 35 significant variants the most common were intron variants (10); 3’ or 5’ untranslated region (UTR) variants (6); upstream or downstream transcript variants (5); synonymous variants (4); splice acceptor variants (4), missense variants (3); exonic splice region variants (2), and one intergenic variant (Table 1 and Supplementary Table S3). The missense variant found on Chr9 (which explains the highest phenotypic variation (33%) in a NB-ARC domain-containing disease resistance protein) causes an amino acid change at the N-terminal of the protein (Table 1). Specifically, it changes the amino acid at position 24 from glutamic acid to alanine.
Identification of cassava accessions within the genebank exhibiting potential resistance
Four markers were selected to help identify potential unexplored CBSD resistance among cassava accessions within the largest cassava genebank collection. Among these markers, the two associations for CBSD severity phenotype (7118148-40-G/T on Chr7 and 13856723-54-G/C on Chr11), and two associations for the presence/absence of the virus phenotype (20486307-57-T/G on Chr2 and 7148270-42-A/C on Chr9), were selected. The selection of these markers was based on two criteria (i) their ability to explain a high percentage of the phenotypic variation and, (ii) their potential to define the favorable genotype based on the phenotype distribution. The favorable genotype conferring resistance to CBSD symptoms severity and absence of viral infection was determined using the phenotypic distribution for each genotype (Supplementary Fig. S4). The genotype frequency for markers associated with low disease severity was 0.201 and 0.056 for 7118148-40-G/T and 13856723-54-G/C, respectively. Moreover, the genotype frequency for markers associated with absence of viral infection was even lower, at 0.009 and 0.004, for 20486307-57-T/G and 7148270-42-A/C, respectively (Supplementary Table S4). We assessed the presence of favorable genotypes for these four markers within a panel of 5,302 cassava accessions conserved at the genebank in Palmira, Colombia, at CIAT. None of the accessions displayed all four favorable genotypes simultaneously, and a total of 4,008 accessions did not display any of the four favorable genotypes. Among those accessions that harbor favorable genotypes, two accessions (COL40 and COL40A) harbored three of the favorable genotypes, while 61 accessions harbored two, and 1,230 accessions harbored at least one. From those accessions that harbored at least one genotype, a total of 1,199 accessions harbored favorable genotypes for either of the two associations for CBSD severity phenotype (7118148-40-G/T on Chr7 and 13856723-54-G/C on Chr11), while the other 31 accessions harbored favorable genotypes for either of the two associations for the presence/absence of the virus phenotype (20486307-57-T/G on Chr2 and 7148270-42-A/C on Chr9).
We selected 63 accessions harboring 3 or 2 favorable genotypes, along with the 31 accessions with a favorable genotype for 20486307-57-T/G on Chr2 and 7148270-42-A/C on Chr9, resulting in a total of 94 accessions. After, assessing the genetic redundancy within these accessions, a total of 41 groups were identified, with 26 consisting of a single accession and 15 consisting of groups of accessions ranging from 2 to 24 (Supplementary Table S5). Among the group of 94 accessions, the following six COL2182, COL40, ECU41, PER315, PER353, and PER556 were previously classified at scale 1 for CBSD severity, indicating no symptoms on leaves and stems22. Additionally, four of these six were reported to be free of virus infection following virus inoculation by Sheat et al. (2019)22 (Supplementary Table S1). After selecting one accession per MLG/group and removing those groups of accessions clustering with the six accessions already evaluated by Sheat et al. (2019)22, were proposed a set of 35 genetically distant accessions with potential resistance to CBSD (Table 2). From these sets, most accessions are landraces (33) from: Colombia (16), followed by Peru (6), Ecuador (5), Venezuela (3), Brazil (3), Guatemala (1) and Puerto Rico (1) (Table 2; Fig. 5).
Table 2.
List of accession with potential resistance to CBSD.
| Accession name | Country of Origin | Biological status | Doi |
|---|---|---|---|
| BRA1170 | BRA | Landrace | 10.18730/PCP0Y |
| BRA1A | BRA | Landrace | 10.18730/P9697 |
| BRA947 | BRA | Landrace | 10.18730/P90A1 |
| CG1118-118 | COL | Breeding line | 10.18730/PDBVT |
| COL1033 | COL | Landrace | 10.18730/PCZTB |
| COL1035B | COL | Landrace | 10.18730/PC654 |
| COL1134 | COL | Landrace | 10.18730/PBN8J |
| COL1313 | COL | Landrace | 10.18730/PDV6U |
| COL2010 | COL | Landrace | 10.18730/P8WP~ |
| COL2017 | COL | Landrace | 10.18730/PCH5Q |
| COL2074 | COL | Landrace | 10.18730/P9Q1S |
| COL2524 | COL | Landrace | 10.18730/PCW11 |
| COL493 | COL | Landrace | 10.18730/P9T6F |
| COL636 | COL | Landrace | 10.18730/PAN4T |
| COL690 | COL | Landrace | 10.18730/PDW3W |
| COL776 | COL | Landrace | 10.18730/P8G30 |
| COL785 | COL | Landrace | 10.18730/P8YRS |
| COL985 | COL | Landrace | 10.18730/PA54* |
| ECU137 | ECU | Landrace | 10.18730/P8S9= |
| ECU150 | ECU | Landrace | 10.18730/P8S4Y |
| ECU166 | ECU | Landrace | 10.18730/PB962 |
| ECU181 | ECU | Landrace | 10.18730/PCTGT |
| ECU4 | ECU | Landrace | 10.18730/PB9PJ |
| GUA8 | GTM | Landrace | 10.18730/PB0BF |
| PER337 | PER | Landrace | 10.18730/PACTK |
| PER382 | PER | Landrace | 10.18730/PACNE |
| PER412 | PER | Landrace | 10.18730/P9HR4 |
| PER467 | PER | Landrace | 10.18730/PACE7 |
| PER471 | PER | Landrace | 10.18730/P9HF* |
| PER480 | PER | Landrace | 10.18730/PBNS= |
| PTR37 | PRI | Landrace | 10.18730/P8NT= |
| SG702-13 | COL | Breeding line | 10.18730/PBS3Y |
| VEN168 | VEN | Landrace | 10.18730/PCPD6 |
| VEN277 | VEN | Landrace | 10.18730/PBW9N |
| VEN47 | VEN | Landrace | 10.18730/PABEC |
Fig. 5.
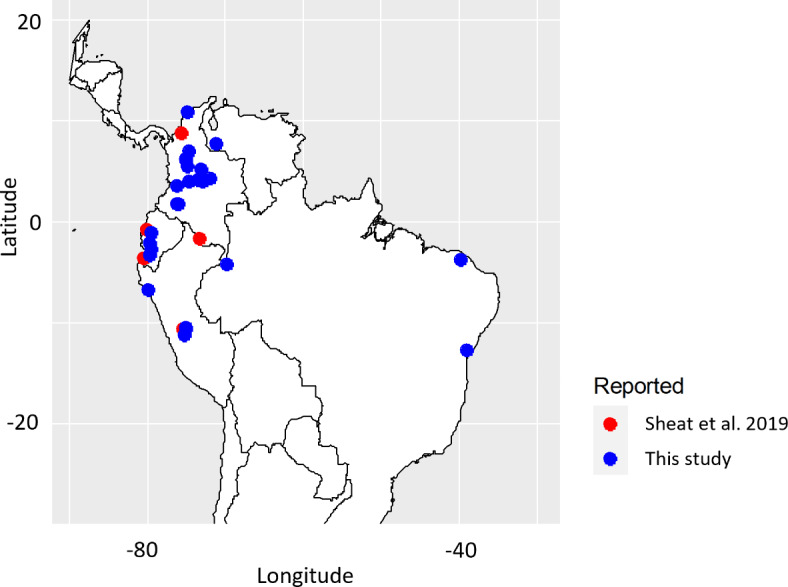
A map displaying the thirty-five accessions out of the 41 accessions selected that have coordinates of collection sites, proposed with potential resistance to CBSD. Those reported in this study are shown in blue. Accessions reported as resistant by Sheat et al. 2019 are presented in red.
Discussion
Despite continuous efforts over recent decades to identify sources of resistance to CBSD, the disease continues to cause significant losses for cassava farmers in Africa. To identify chromosomal regions and genes potentially involved in CBSD resistance, we used a set of 234 accessions from the world’s largest cassava collection and conducted a GWAS using available phenotypic data reported by an independent research team in 201922. Sheat et al. (2019) assessed these accessions for CBSD severity symptoms and virus presence by qRT-PCR after artificially inoculating the plants with a severe isolate of CBSV from Mozambique (CBSV-Mo83)22. A total of fifteen accessions remained free of symptoms on leaves and stems, and seven of these either exhibited necrosis symptoms on roots and tested negative for virus infection. Moreover, two accessions (COL40 and COL2182) remained uninfected when inoculated with other viral isolates from Kenya and Tanzania (UCBSV-Ke125 and CBSV-Tan70)22. We genotyped these 234 accessions using DArTseq technology41, which provides a cost-effective method for high-density genome coverage across the eighteen chromosomes of the cassava genome. This technology has been previously used for cassava diversity studies, characterization, and trait selection28–30,42,43. More than 80% of the SNPs markers were able to be mapped to the reference genome37. We applied a series of marker-parameter filters to select a subset of high-quality markers for assessing population structure and performing association analysis.
Our population structure analysis detected three major clusters and eight sub-clusters, which could be differentiated based on PCA, admixture and hierarchical clustering analysis. The three clusters correspond to the origin of the accessions: one group (G1) mostly composed of countries from the western part of South America (including Colombia, Peru, and Venezuela); a second group (G2) from the eastern part (mainly Brazil and Paraguay), and a third cluster (G3) including Brazil, Colombia, Venezuela, and other countries. A study by Perez et al. (2023) reports similar clustering, but with less groups44. Perez et al. (2023) studied the genetic diversity of a subset of 481 accessions from the genebank collection, identifying two major genepools and seven genetic subgroups that co-localize with different eco-geographic regions44. The two major genepools were defined as ‘North’ & ‘Northwest’ of Amazon River basin (ARB) and ‘South’ & ‘Southeast’ of ARB. The former genepool included four genetic subgroups, while the latter included two. Interestingly, of the 481 accessions used in the Perez et al. (2023) study, 114 are included in our study. Moreover, three major clusters were observed in a previous study when assessing the genetic redundancy of 5,302 accessions of the cultivated cassava collection45. Assessing the population structure using the phenotypic data revealed that all sixteen resistant accessions reported by Sheat et al. (2019)22 clustered in G1, suggesting that the resistance arises in varieties from the western part of South America. Furthermore, the population structure of the three clusters found within the 234 accessions resembled that of the three major clusters obtained in a pervious study with 5,302 accessions, and the major clusters obtained45. We therefore opted to use three principal components to account for the population structure in the GWAS. This approach helps to avoid false positives or spurious associations39.
Our GWAS identified 35 SNP associations for the two phenotypes evaluated. Among the five associations for CBSD symptom severity, two located on chromosomes 7 and 11, at positions 24,643,336 and 23,187,439, respectively, explaining 8.4% and 8.0% of the phenotypic variation. The associated SNP on chromosome 7 was annotated as an upstream transcript variant in the gene encoding glycerol-3-phosphate acyltransferase 5 (GPAT5). In other plant species, the acyltransferase GPAT5 is required for the synthesis of suberin in the seed coat and root46. Although widespread across various tissues at specific locations during plant growth, suberin is also synthesized in response to stress and wounds, providing a barrier to pathogens47,48. The associated SNP on chromosome 11 was annotated as a synonymous variant in the gene encoding the Rhomboid-like protein 13 (RBL13). Rhomboid-like proteins (RBLs) are intramembrane proteases with a variety of regulatory roles in cells49. While the precise role of RBL13 remains unclear, it has been observed that a Rhomboid-like protease gene originating from an interspecies translocation, including from sugar beet provides resistance to cyst nematodes50.
Among the 30 associations identified with the phenotype of presence/absence of CBSV infection, two were commonly identified by all models used (MLM, BLINK and FarmCPU) on Chr 1 and 2, and 10 were commonly associated by at least two models. Of these, two SNPs: 20486307-57-T/G on Chr 2 and 7148270-42-A/C on Chr 9, showed the highest phenotypic variation, averaging 26.5% and 26.7%, respectively, across the models. The SNP 20486307-57-T/G was annotated as a 3’ untranslated region (UTR) variant at the gene encoding a RING/U-box superfamily protein. Really Interesting New Gene (RING) finger proteins are characterized by containing 40–60 residues and are believed to function as E3 ubiquitin ligase51. Numerous RING-finger proteins have been found in plants, playing important roles in plant growth, stress resistance, and signal transduction51,52. The RING-finger domain serves as a protein–protein interaction domain, essential for catalyzing the E3 ligase activity of these proteins51,53. Recent studies have demonstrated that RING-finger proteins play a role in biotic stress responses across numerous species54,55. The SNP 7148270-42-A/C on Chr 9 was annotated as a missense variant in a gene encoding an NB-ARC domain-containing disease-resistance protein. This domain is present in many resistance proteins involved in pathogen recognition and the subsequent activation of the innate immune response56. The associated SNP causes an amino acid change at the N-terminal part of the protein, at position 24, from glutamic acid to alanine. Glutamic acid has a longer side chain with a carboxyl group (-COOH) at the end, making it a negatively charged, polar amino acid, often involved in active or binding sites of enzymes due to its ability to donate and accept protons57. Alanine has a shorter side chain consisting of a single methyl group (-CH3), making it a non-polar, hydrophobic amino acid, often involved in maintaining the hydrophobic core of proteins and typically found in regions that do not require specific reactivity58.
Despite the advances in genomics, few studies have investigated chromosomal regions and genes potentially involved in CBSD resistance compared to other crops. These studies have identified associations with root necrosis and foliar symptoms on chromosomes 1, 2, 4, 5, 6, 11, 12, 13, 15, 17 and 189,18,20,21. Most of these studies have used bi-parental crosses between African landraces, breeding lines, or materials such as Kiroba and Namikonga, closely related to the cassava wild relative Manihot glaziovii. These have introgressed regions across different chromosomes18,19. Nzuki et al. (2017) identified eleven significant QTLs using an F1 cross developed between the Tanzanian landrace, Kiroba, and a breeding line, AR37-8018. The research has found two QTLs associated with CBSD root necrosis only (Chr 5 and 12), seven associated with foliar symptoms only (Chr 4, 6, 17 and 18), and others on Chr 11 and 15 associated with both CBSD foliar and root necrosis symptoms18. Moreover, they identified introgression segments of Manihot glaziovii on chromosomes 17 and 18 overlapping with the QTL associated with foliar symptoms18. Using a bi-parental cross of Namikonga and Albert (two Tanzanian farmer varieties), Masumba et al. (2017), identified three QTLs linked to CBSD resistance in Namikonga on Chr 2, 11 and 1819. A total of twenty-seven genes were identified on Chr 2, including two leucine-rich repeat (LRR) proteins and a signal recognition particle19. Later, Kayondo et al. (2018), using GWAS found two regions associated to CBSD, one on Chr 4 which co-localizes with a Manihot glaziovii introgression segment, and one on Chr 11, containing a cluster of NBS-LRR genes20. Another recent study identified QTLs on chromosomes 1, 6, 13, and 18 in genomic selection population21. All these previous studies used disease severity symptoms to identify trait-marker associations for CBSD resistance.
In our study we used a panel of genetically diverse germplasm conserved in an international cassava genebank, originating from 20 countries, mostly landraces from South America. We also used disease severity symptoms found in other studies and a second binary “phenotype” of presence/absence of viral infection that to our knowledge has not been previously used for GWAS to identify association for CBSV resistance. Comparing our findings with previous studies, we found associations on previously reported chromosomes: one on each of chromosomes 4, 6, 12, 13, and 18; two on chromosomes 1, 5 and 17; four on chromosome 11 and 15; and six on chromosome 2. The locus on chromosome 18 has already been reported by Kayondo et al. (2018)20. We identified new associations on previously unreported chromosomes, including one SNP on chromosomes 8, 9 and 14, two on chromosome 3 and 7, both separated within each other by 1.5 Mb. The two SNPs on chromosome 7 were identified with different phenotypes and explain around 8.0% of the phenotypic variation, and 3 SNPs on chromosome 16. Among all these, seven new associations were related to the binary phenotype of presence/absence of viral infection on chromosomes 7, 8, 9, 14 and 16. Interestingly, no associations were commonly identified between the two studied phenotypes, suggesting different mechanisms involved in virus replication and symptom expression. Binary traits are often used in human GWAS to assess disease presence or absence, and have been widely applied in plant GWAS, typically using the MLM model59,60. Assessing presence or absence of CBSV infection, as reported by Sheat et al. (2019)22, can help to identify resistance mechanisms.
Germplasm collections serve as a reservoir of genetic variation, which can be utilized to diversify resistance factors in modern crop varieties. Colombia, through CIAT, conserves the world’s largest in-vitro collection of cassava. By using four selected markers identified in this study, (20486307-57-T/G on Chr2, 7118148-40-G/T on Chr7, 7148270-42-A/C on Chr9 and 13856723-54-G/C on Chr11) and the genotypic data of 5,302 cultivated cassava accessions of CIAT, we predict potential resistance to CBSD on 35 genetically distant accessions originally from seven countries in South America for validation tests. These markers were selected based on their ability to explain a high percentage of the phenotypic variation and their potential to define a favorable genotype based on the two-phenotype distribution (CBSD symptoms severity and CBSV infection). Other markers represented challenges in defining a favorable genotype and were therefore not considered. The proposed candidate accessions can accelerate the development of improve crop varieties, and the identified markers can support markers-assisted selection in breeding programs.
Our study demonstrates how genomic resources derived from genebank collections, integration of data from previous studies from genebanks users, and statistical analysis can support and facilitate informed access to and use of genetic resources, particularly in large national or international collections. Additionally, it provides insights into the molecular mechanisms underlying disease resistance.
Methods
Germplasm and phenotypic data used in this study
A subset of 234 accessions from the 629 of the CIAT cassava core collection, conserved in Palmira, Colombia, was used in this study. The accessions originate from 20 countries, primarily in South America, including Colombia (59), Brazil (53), and Peru (28), followed by Venezuela (18), Paraguay (18), Ecuador (11), and other countries such as Argentina, Bolivia, Costa Rica, Cuba, the Dominican Republic, Guatemala, Mexico, Panama and Puerto Rico. Fourteen accessions originated from countries outside South America, namely Fiji, Indonesia, Malaysia, Nigeria, and Thailand. This set of accessions was selected based on available CBSD resistance phenotypic data reported by Sheat el. al. (2019) in supplementary materials22. Sheat et al. (2019) reported the severity of CBSD symptoms and the presence or absence of viral infection for the 234 accessions. This information was obtained after artificial inoculation with the pathogenic virus isolate (CBSV-Mo83) through auxiliary bud grafting22. The severity of CBSD symptoms was categorized on a five-point scale: no symptoms on leaves and stems (1); very mild stem symptoms, inconspicuous symptoms on leaves only (2); moderate symptoms on leaves only (3); severe symptoms on leaves and stems (4), and wilting, followed by plant death (5). Additionally, virus infection data consisted of two categories: presence (1) or absence (0)22. The accessions names, origins, biological status, severity scales of CBSD, and presence or absence of viral infection are summarized in Supplementary Table S1.
DNA extraction and genotyping
Leaf tissue was collected from in-vitro conserved plantlets and stored at -20 °C. Subsequently, samples were lyophilized for 3 days at -50 °C and 0.002 mbar. Approximately 10 mg of lyophilized leaf tissue was used for DNA extraction, as previously described by Carvajal-Yepes et al. (2024)45. In brief, samples were homogenized and lysed with Cetyltrimethylammonium bromide (CTAB) extraction buffers61. After vortex mixing and incubation at 65 °C for 30 min, the samples were mixed and incubated with an equal volume of chloroform: Isoamyl alcohol 24:1 (Sigma-Aldrich, USA). Following centrifugation, the aqueous phase was collected in another tube and mixed with equal volume of cold isopropanol, followed by 1 h of incubation at -20 °C. Upon removal of the supernatant, the formed pellets were washed with 80% cold ethanol. Subsequently, the pellets were air-dried and resuspended in TE buffer (pH 8.0, Alpha Teknova, USA) with 40 µg of RNase (QIAGEN, Germany), and incubated for 30 min at 37 °C. The extracted DNA was stored at -20 °C. The concentration and purity of DNA was estimated by calculating the absorbance at 260/280 nm, while the integrity of DNA was assessed by electrophoresis using 0.8% agarose gel in 0.5X TBE stained with GelRed (2 µl/100 ml of gel).
A total of 50ul of genomic DNA, with a concentration of 50ng/µl, was shipped to Diversity Array Technology in Canberra, Australia, for genotyping by sequencing using DArTseq™ technology, which involves a combination of MseI and PstI restriction enzymes. The prepared DNA libraries were sequenced following the protocol described by Nadeem et al. (2018)62. SNP calling was performed with DS14 software (Diversity Arrays Technology P/L).
Filter applied to select high-quality SNP markers
To assess the population structure, a subset of 17,989 high-quality markers was selected based on the following four criteria: (i) missing values (≤ 0.2); (ii) minor allele frequency (MAF ≥ 0.005); (iii) average marker count (≥ 5 and ≤ 75), which represents the average number of sequence-tag copies of a marker (calculated by averaging the mean number of seq-tag copies on the two SNP alleles), and (iv) reproducibility (≥ 0.99). The latter is an estimate calculated by DArTseq, which assesses the proportion of technical replicate assay pairs for which the calls of a given marker were consistent. To conduct the association analysis, we selected markers that mapped to the eighteen chromosomes of the cassava reference genome v6.1, obtaining a total of 16,452 SNP markers34.
Genetic diversity and population structure
Population structure analysis was conducted using the snpgdsPCA function from the SNPRelate package v1.6.463, in the R program v4.2.264 to compute eigenvectors and eigenvalues for principal component analysis (PCA). Additionally, a pairwise genetic distance matrix (1-IBS, identity by state) was calculated using 17,989 SNP markers. The distance matrix was used for agglomerative clustering with the minimum variance clustering method (Ward.D2)65, employing the hclust function in the stats R package v4.2.265. The find.clusters function in the adegenet R package v2.1.10 was used to identify clusters using K-means and the Bayesian Information Criterion (BIC)66. Furthermore, the best estimation of K ancestral populations was determined using the snfm function of LEA R package v3.2.067. For analysis, the SNPs were transformed into genlight objects and converted into STRUCTURE input files with the gl2structure function of the dartR R package v2.9.768. The structure-formatted files were then converted into the geno format using the struc2geno function of LEA. Visual representations, including bar plots of admixture coefficients and cross-entropy values plots across different K values, were generated using the ggplot2 R package v3.3.369.
Genome-wide association analysis
The association analyses were conducted using 234 cassava accessions and 16,452 SNP markers, as well as two types of phenotypic data: (i) CBSD symptom severity, and (ii) presence or absence of cassava brown streak virus (CBSV), as determined by Sheat et al. (2019)22. The SNP markers were filtered to select those with missing values below 0.2, MAF above 0.005, average marker count (AvgMarkerCount) between 5 and 75 and reproducibility above 0.99. The AvgMarkerCount parameter represents the average number of sequence-tag copies of a marker and is calculated by averaging the mean number of sequence-tag copies of the two SNP alleles. Reproducibility assesses the proportion of technical replicate assay pairs for which the calls of a given marker were consistent. This parameter is estimated by DArT P/L. Three statistical models were implemented in Genomic Association and Prediction Integrated Tool version 3 (GAPIT3)70. The models used include MLM, BLINK and FarmCPU. This analysis employed the Bonferroni correction method with a significant threshold set at 0.05. The associations are visualized in Manhattan plots with values above 5.5 -log10(p-value). Linkage disequilibrium (LD) was estimated using correlation coefficients (r²) between pairs of loci on each chromosome, based on 16,451 SNPs with a minor allele frequency above 0.05, using the gl.report.ld.map function from the dartR package v2.9.768. Results were visualized with the ggplot2 package v3.4.3.
Functional annotation
The functional annotation was conducted using the Next-Generation Sequencing Experience Platform (NGSEP) software v4.3.171 to determine whether the significant markers are located within or near genes, regulatory regions, or functional elements, and to assess the potential functional consequences of genetic variants. The VCF annotate option, from NGSEP, was used with default settings: 1000 bp offset upstream and a 300 bp offset downstream, with a splice donor/acceptor offset of 2, a splice region intron offset of 10, and a splice region exon offset of 2. The cassava reference genome v6.137 was utilized for this analysis. Subsequently, the identified associated genes and proteins were reviewed for their function using Pfam 202172.
Selection of a subset of potential CBSD-Resistant accessions
Favorable genotypes for the four selected markers (7118148-40-G/T, 13856723-54-G/C, 20486307-57-T/G and 7148270-42-A/C) were done based on the phenotype distribution of CBSD severity and the presence (1) or absence (0) of the viral infection as reported by Sheat et al., (2019). The four markers were reviewed in a panel of 5,307 cassava cultivated accessions for which DArTseq genotypic data is available (Supplementary Table S6). Accessions harboring from 3 to 2 favorable genotypes were selected, as well as accessions with a favorable genotype for either of these two markers 20486307-57-T/G and 7148270-42-A/C. To identify genetic distinctness and redundancy within the selection of accessions we implemented the procedure reported by Carvajal-Yepes et al. (2024) and the dataset of 5,307 accessions and 7,180 SNP markers45. In brief, we calculated Identity-By-State (IBS) distances using the 1-IBS function in PLINK v1.073, and collapsed multilocus genotypes (MLGs) within accessions using a genetic distance threshold of 0.015 to identify a set of unique accessions45.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was generously funded by the CGIAR Genebank Initiative and the Global Diversity Crop Trust. We give special thanks to Vincent Johnson, consultant to the Alliance of Bioversity International and CIAT Science Writing Service, for English and copy editing of this manuscript, Miguel Correa for suggesting alternative approaches for data analysis, Monica Velez, Ericson Aranzales and Norma Manrique for their outstanding collaboration in providing plant material and preparing samples.
Author contributions
J.A.O. conceived the experiment(s), conducted all analyses, and supported writing of the original manuscript, M.C.Y. conceived and designed the study and wrote the original manuscript, D.C.L. supported and guided analyses of the results, G.W. and P.W. gave editorial input into the manuscript and contributed with the discussion. All authors reviewed the manuscript.
Data availability
The genotypic data that support the findings of this study have been deposited in Dataverse in the following link: 10.7910/DVN/H4PDE5.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Adebayo, W. G. Cassava production in Africa: A panel analysis of the drivers and trends. Heliyon. 9 (2023). [DOI] [PMC free article] [PubMed]
- 2.FAOSTAT. https://www.fao.org/faostat/en/#home
- 3.Nyirakanani, C. et al. Farmer and field survey in cassava-growing districts of Rwanda reveals key factors associated with cassava brown streak disease incidence and cassava productivity. Front. Sustain. Food Syst. 5, 699655 (2021). [Google Scholar]
- 4.Chikoti, P. C., Mulenga, R. M., Tembo, M. & Sseruwagi, P. Cassava mosaic disease: A review of a threat to cassava production in Zambia. J. Plant. Pathol. 101, 467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chikoti, P. C. & Tembo, M. Expansion and impact of cassava brown streak and cassava mosaic diseases in Africa: A review. Front. Sustain. Food Syst. 6, 1076364 (2022). [Google Scholar]
- 6.Newton, A. C., Johnson, S. N. & Gregory, P. J. Implications of climate change for diseases, crop yields and food security. Euphytica. 179, 3–18 (2011). [Google Scholar]
- 7.Kriticos, D. J. et al. Improving climate suitability for Bemisia tabaci in East Africa is correlated with increased prevalence of whiteflies and cassava diseases. Sci. Rep. 10, 1–17 (2020). [DOI] [PMC free article] [PubMed]
- 8.Hillocks, R. & Thresh, M. Cassava mosaic and cassava brown streak virus diseases in Africa. (2000).
- 9.Kawuki, R. S. et al. Eleven years of breeding efforts to combat cassava brown streak disease. Breed. Sci. 66, 560–571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter, S. et al. Analysis of cassava brown streak viruses reveals the presence of distinct virus species causing cassava brown streak disease in East Africa. J. Gen. Virol. 91, 1365–1372 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Mbanzibwa, D. R. et al. Genetically distinct strains of Cassava brown streak virus in the Lake Victoria Basin and the Indian Ocean coastal area of East Africa. Arch. Virol. 154, 353–359 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Maruthi, M. N. et al. Transmission of Cassava brown streak virus by Bemisia tabaci (Gennadius). J. Phytopathol. 153, 307–312 (2005). [Google Scholar]
- 13.Hillocks Illocks, R. J., Aya, R., Mtunda Tunda, M. D., Kiozia Iozia, H. & K. & Effects of brown streak virus disease on yield and quality of cassava in Tanzania. J. Phytopathol. 149, 389–394 (2001). [Google Scholar]
- 14.Ndyetabula, I. L. et al. Analysis of interactions between Cassava Brown Streak Disease Symptom types facilitates the determination of varietal responses and yield losses. Plant. Dis. 100, 1388–1396 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Ano, C. U. et al. Cassava Brown Streak Disease Response and Association with agronomic traits in Elite Nigerian cassava cultivars. Front. Plant. Sci. 12 (2021). [DOI] [PMC free article] [PubMed]
- 16.Mukiibi, D. R. et al. Resistance of advanced cassava breeding clones to infection by major viruses in Uganda. Crop Prot. 115, 104–112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rey, C. & Vanderschuren, H. Cassava Mosaic and Brown Streak diseases: Current perspectives and beyond. Annu. Rev. Virol. 4, 429–452 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Nzuki, I. et al. QTL mapping for Pest and Disease Resistance in Cassava and coincidence of some QTL with introgression regions derived from Manihot glaziovii. Front. Plant. Sci. 8, (2017). [DOI] [PMC free article] [PubMed]
- 19.Masumba, E. A. et al. QTL associated with resistance to cassava brown streak and cassava mosaic diseases in a bi-parental cross of two Tanzanian farmer varieties, Namikonga and Albert. Theor. Appl. Genet. 130, 2069–2090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kayondo, S. I. et al. Genome-wide association mapping and genomic prediction for CBSD resistance in Manihot esculenta. Sci. Rep. 8, (2018). [DOI] [PMC free article] [PubMed]
- 21.Nandudu, L., Kawuki, R., Ogbonna, A., Kanaabi, M. & Jannink, J. L. Genetic dissection of cassava brown streak disease in a genomic selection population. Front. Plant. Sci. 13 (2023). [DOI] [PMC free article] [PubMed]
- 22.Sheat, S., Fuerholzner, B., Stein, B. & Winter, S. Resistance against Cassava Brown Streak viruses from Africa in Cassava Germplasm from South America. Front. Plant. Sci. 10 (2019). [DOI] [PMC free article] [PubMed]
- 23.Engels, J. M. M. Plant genetic resources management and conservation strategies: Problems and progress. Acta Hortic. 623, 179–191 (2003). [Google Scholar]
- 24.FAO. The Second Report on the State of the World’s Plant (Rome, 2010).
- 25.Westengen, O. T., Skarbø, K., Mulesa, T. H. & Berg, T. Access to genes: Linkages between genebanks and farmers’ seed systems. Food Secur. 10, 9–25 (2018). [Google Scholar]
- 26.Innes, N. L. Gene banks and their contribution to the breeding of disease resistant cultivars. Euphytica. 63, 23–31 (1992). [Google Scholar]
- 27.Dinglasan, E., Periyannan, S. & Hickey, L. T. Harnessing adult-plant resistance genes to deploy durable disease resistance in crops. Essays Biochem. 66, 571 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson, M. E. et al. Collection and characterization of cassava germplasm in Comoros. Genet. Resour. Crop Evol. 71, 341–361 (2024). [Google Scholar]
- 29.Busconi, M. et al. Validation of SNP markers for diversity analysis, quality control, and trait selection in a biofortified cassava population. Plants. 13, 2328 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adu, B. G. et al. High-density DArT-based SilicoDArT and SNP markers for genetic diversity and population structure studies in cassava (Manihot esculenta Crantz). PLoS One. 16, (2021). [DOI] [PMC free article] [PubMed]
- 31.Zhou, X. et al. Evidence that DNA-A of a geminivirus associated with severe cassava mosaic disease in Uganda has arisen by interspecific recombination. J. Gen. Virol. 78(Pt 8), 2101–2111 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Fondong, V. N. The search for resistance to Cassava Mosaic geminiviruses: How much we have accomplished, and what lies ahead. Front. Plant. Sci. 8, (2017). [DOI] [PMC free article] [PubMed]
- 33.Thauvin, J. N. et al. Genome-wide association study for resistance to rhynchosporium in a diverse collection of spring barley germplasm. Agronomy. 12, 782 (2022). [Google Scholar]
- 34.Riaz, A. et al. Mining Vavilov’s treasure chest of wheat diversity for adult plant resistance to Puccinia Triticina. Plant. Dis. 101, 317–323 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Nkhata, W. et al. Genome-wide association analysis of bean fly resistance and agro-morphological traits in common bean. PLoS One 16, (2021). [DOI] [PMC free article] [PubMed]
- 36.Rwegasira, G. M. & Rey, C. M. E. Response of selected cassava varieties to the incidence and severity of Cassava Brown Streak Disease in Tanzania. J. Agric. Sci. 4, (2012).
- 37.Bredeson, J. V. et al. Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nat. Biotechnol. 34, 562–570 (2016). [DOI] [PubMed]
- 38.Staff, T. P. G. Correction: Iterative usage of fixed and Random Effect Models for Powerful and efficient genome-wide Association studies. PLoS Genet. 12, e1005957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Segura, V. et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830 (2012). [DOI] [PMC free article] [PubMed]
- 41.Kilian, A. et al. Diversity arrays technology: A generic genome profiling technology on open platforms. Methods Mol. Biol. 888, 67–89 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Adu, B. G. et al. Whole genome SNPs and phenotypic characterization of cassava (Manihot esculenta Crantz) germplasm in the semi-deciduous forest ecology of Ghana. Ecol. Genet. Genom. 17, 100068 (2020). [Google Scholar]
- 43.Pierre, N. et al. Genetic diversity of local and introduced cassava germplasm in Burundi using DArTseq molecular analyses. bioRxiv. 10.1101/2021.08.09.455732 (2021). [DOI] [PMC free article] [PubMed]
- 44.Perez-Fon, L. et al. Integrated genetic and metabolic characterization of latin American cassava (Manihot esculenta) germplasm. Plant. Physiol. 192, 2672–2686 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carvajal-Yepes, M. et al. Identifying genetically redundant accessions in the world’s largest cassava collection. Front. Plant. Sci. 14, (2024). [DOI] [PMC free article] [PubMed]
- 46.Belsson, F., Li, Y., Bonaventura, G., Pollard, M. & Ohlrogge, J. B. The acyltransferase GPAT5 is required for the synthesis of suberin in seed coat and root of Arabidopsis. Plant. Cell. 19, 351–368 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dean, B. B. & Kolattukudy, P. E. Synthesis of Suberin during wound-healing in Jade leaves, Tomato Fruit, and Bean pods. Plant. Physiol. 58, 411–416 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lulai, E. C. & Corsini, D. L. Differential deposition of suberin phenolic and aliphatic domains and their roles in resistance to infection during potato tuber (Solanum tuberosum L.) Wound-healing. Physiol. Mol. Plant. Pathol. 53, 209–222 (1998). [Google Scholar]
- 49.Lavell, A. et al. Proteins associated with the Arabidopsis thaliana plastid rhomboid-like protein RBL10. Plant. J. 108, 1332–1345 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar, A. et al. A rhomboid-like protease gene from an interspecies translocation confers resistance to cyst nematodes. New Phytol. 231, 801–813 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Borden, K. L. B. RING domains: Master builders of molecular scaffolds? J. Mol. Biol. 295, 1103–1112 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Li, W., He, M., Wang, J. & Wang, Y. P. Zinc finger protein (ZFP) in plants-a review. Plant. Omics (2013).
- 53.Metzger, M. B., Pruneda, J. N., Klevit, R. E. & Weissman, A. M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta. 1843, 47–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marino, D. et al. Arabidopsis ubiquitin ligase MIEL1 mediates degradation of the transcription factor MYB30 weakening plant defence. Nat. Commun. 4, (2013). [DOI] [PubMed]
- 55.Hong, J. K., Choi, H. W., Hwang, I. S. & Hwang, B. K. Role of a novel pathogen-induced pepper C3-H-C4 type RING-finger protein gene, CaRFPI, in disease susceptibility and osmotic stress tolerance. Plant. Mol. Biol. 63, 571–588 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Van Ooijen, G. et al. Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J. Exp. Bot. 59, 1383–1397 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Guzzo, A. V. The influence of amino acid sequence on protein structure. Biophys. J. 5, 809–822 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kubyshkin, V. & Budisa, N. The Alanine World Model for the development of the amino acid repertoire in protein biosynthesis. Int. J. Mol. Sci. 20, 5507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Monnot, S. et al. Deciphering the genetic architecture of plant virus resistance by gwas, state of the art and potential advances. Cells. 10, 3080 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burton, P. R. et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447, 661 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dellaporta, S. L., Wood, J. & Hicks, J. B. A plant DNA minipreparation: Version II. Plant. Mol. Biol. Rep. 1, 19–21 (1983). [Google Scholar]
- 62.Nadeem, M. A. et al. Characterization of genetic diversity in Turkish common bean gene pool using phenotypic and whole-genome DArTseq-generated silicoDArT marker information. PLoS One. 13, (2018). [DOI] [PMC free article] [PubMed]
- 63.Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 28, 3326–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.R Core Team. R: A Language and Environment for Statistical ## Computing. R Foundation for Statistical Computing. (2021). https://www.R-project.org/, Vienna, Austria.
- 65.Murtagh, F. & Legendre, P. Ward’s hierarchical agglomerative clustering method: Which Algorithms Implement Ward’s Criterion? J. Classif. 31, 274–295 (2014). [Google Scholar]
- 66.Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 11, (2010). [DOI] [PMC free article] [PubMed]
- 67.Frichot, E. & François, O. L. E. A. An R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929 (2015). [Google Scholar]
- 68.Mijangos, J. L., Gruber, B., Berry, O., Pacioni, C. & Georges, A. dartR v2: An accessible genetic analysis platform for conservation, ecology and agriculture. Methods Ecol. Evol. 13, 2150–2158 (2022). [Google Scholar]
- 69.Wickham, H. ggplot2.(2016). 10.1007/978-3-319-24277-4
- 70.Lipka, A. E. et al. GAPIT: Genome association and prediction integrated tool. Bioinformatics. 28, 2397–2399 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Gonzalez-García, L. N., Lozano-Arce, D., Londoño, J. P., Guyot, R. & Duitama, J. Efficient homology-based annotation of transposable elements using minimizers. Appl. Plant. Sci. 11, e11520 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genotypic data that support the findings of this study have been deposited in Dataverse in the following link: 10.7910/DVN/H4PDE5.