

Abstract
For mitogen-activated protein kinases (MAPKs) a shallow surface—distinct from the substrate binding pocket—called the D(ocking)-groove governs partner protein binding. Screening of broad range of Michael acceptor compounds identified a double-activated, sterically crowded cyclohexenone moiety as a promising scaffold. We show that compounds bearing this structurally complex chiral warhead are able to target the conserved MAPK D-groove cysteine via reversible covalent modification and interfere with the protein-protein interactions of MAPKs. The electronic and steric properties of the Michael acceptor can be tailored via different substitution patterns. The inversion of the chiral center of the warhead can reroute chemical bond formation with the targeted cysteine towards the neighboring, but less nucleophilic histidine. Compounds bind to the shallow MAPK D-groove with low micromolar affinity in vitro and perturb MAPK signaling networks in the cell. This class of chiral, cyclic and enhanced 3D shaped Michael acceptor scaffolds offers an alternative to conventional ATP-competitive drugs modulating MAPK signaling pathways.
Subject terms: Kinases, Mechanism of action, X-ray crystallography
Póti et al developed cyclic, tuneable and chiral warheads against the protein-protein interaction surface of mitogen-activated protein kinases, which form a reversible covalent bond with nucleophilic surface residues to modulate cellular signaling networks.
Introduction
Cysteine, because of its nucleophilic thiol group, can be covalently modified by various electrophiles, especially by Michael acceptors1. This covalent targeting option has become the basis of many contemporary drug discovery programs, particularly in the kinase field. More specifically, blocking protein function through the covalent modification of key cysteine residues next to the catalytic site has been used to inhibit protein kinases involved in cellular signaling2–5. During the past decades several approved and experimental drugs having an acrylamide warhead, an easily installed Michael acceptor, have been developed targeting specific noncatalytic cysteines6.
Although low-reactivity Michael acceptors are wittingly used for covalent inhibitor developments, their nonselective reactivity may lead to adverse (off-target) effects and inhibitor depletion in the cellular environment by millimolar amounts of free nucleophiles (e.g., glutathione/GSH)7. Thus, there is a need to broaden the current catalog of suitable Michael acceptors, by using chemoproteomic platforms and/or rational design8–11.
Mitogen-activated protein kinases (MAPKs) play crucial roles in cellular physiology and altered activation of MAPKs underlies many diseases (e.g., cancer or inflammation)12–14. Distinct concepts and practical solutions for the modulation of MAPK signaling pathways are in great demand because acquired somatic mutations in currently used upstream targets (e.g., RAS, RAF, MEK for the ERK pathway) and/or compensatory changes at network level often cause resilience against treatments15.
Most kinase inhibitors bind in the deep nucleotide binding crevice between the N-terminal and C-terminal lobes of protein kinases. These ATP competitive drugs need to bind with high affinity (low nanomolar) as they compete against the millimolar concentration of ATP in the cell. As a complementary approach to blocking kinase activity, cellular signaling may also be inhibited by blocking the protein-protein interactions (PPI) of kinases. MAPKs have a dedicated shallow PPI surface which is used by activators and substrates for binding16–18. Protein partners all bind to this MAPK D(ocking)-groove by short linear motifs located in their unstructured regions19. The D-groove has a conserved cysteine (Cys161 in ERK2, Cys162 in p38α, and Cys163 in JNK1) and is comprised of a negatively charged area (referred to as the CD groove) and three small hydrophobic pockets20. We posited that blocking the binding of bona fide signaling clients at this shallow groove provides an alternative to classical ATP competitive kinase inhibitors. However, targeting shallow PPI pockets is normally challenging, because the modest positive enthalpic contribution is canceled out by a large negative entropic cost. One solution is to exploit covalent bond formation between the inhibitor and the target to balance the high entropic cost and use this as an anchor to search for compatible moieties that may further increase the enthalpic contribution.
In this study, we show that the MAPK D-groove cysteine is amenable to Michael addition by an intrapeptide acrylamide warhead. Moreover, a biochemical, “electrophile first” in vitro screen identified a structurally complex, chiral and enhanced 3D shaped Michael acceptor that covalently bound to the D-groove cysteine located next to the hydrophobic pockets. Intriguingly, MAPK binding was found to be reversible and had low micromolar affinity with some of the hit compounds. Reversible warheads provide an alternative to the practice of using an irreversible electrophile (e.g., acrylamide), particularly if one considers that unwanted cross-reactions in the cell are likely to be more devastating if happened through irreversible covalent bond formation. The reversible covalent bond provided an appropriate anchor interaction, thus binding affinity and specificity could be modulated by further synthetic elaboration of the warhead. Moreover, the cyclic and chiral nature of the warhead scaffold offers further advantage compared to open-chain (acyclic) acryl- or cyanoacryl-based designs: the local, and inherently chiral environment of specific nucleophiles on the protein surface can be better exploited in creating specific inhibitors, for example by varying the configuration of substituent groups in the cyclic warhead scaffold.
Results
The ERK2 docking groove contains a surface cysteine amenable to Michael addition
The short linear motifs from partner proteins may bind in the MAPK docking groove in two different N-to-C directions: there are classical or reverse D-motif containing peptides (revD)20. The binding mode of peptides will be governed by the arrangement of positively charged (θ) and hydrophobic amino acids (φU, φL, φA, φB) and by the spacing between these (Fig. 1a). Peptides normally do not use all hydrophobic pockets but hydrophobic amino acids contacting φL and φA pockets are common. Crystal structures of MAPKs in complex with natural, SynthD, and SynthRevD peptides showed that the D-groove cysteine falls next to the main-chain atoms between φL and φA residues. These amino acids are one or two residues apart, and we hypothesized that the nucleophilic thiol of cysteine 161 (ERK2), which is universally conserved in all MAPKs, may be covalently modified by an electrophilic group through Michael addition, as this had been shown for some noncatalytic cysteines in kinases located next to the ATP binding pocket5,21. Our first-generation MAP kinase docking groove inhibitors were peptide-based, in which an intrachain acrylamide warhead was used as a Michael acceptor to selectively target the MAPK D-groove cysteine via an irreversible covalent adduct. The crystal structure of ERK2 in complex with an artificial revD peptide (SynthRevDcov) revealed the structure of this complex (Fig. 1b and Supplementary Table 1) and further characterization showed that targeting Cys161 by the intrachain warhead requires a high-affinity binder context and precise geometry between the beta carbon of the warhead and the sulfur atom of the D-groove cysteine. Formation of the covalent bond is a slow process, with a half-time of approximately 30 min under physiological conditions for a nanomolar binder (Supplementary Figs. 1–3).
Fig. 1. Covalent binding of sterically crowded cyclohexenon compounds in the MAPK D-groove is resilient to GSH.

a Topography of the MAPK D-groove. The negatively charged aspartate residues (D318 and D321) are colored red, Cys161 is colored yellow. The panel on the left shows the crystal structure of the ERK2-SynthRevD protein-peptide complex (PDB ID: 4FMQ)20. The inset shows the position of the D-peptide (in black), the nucleotide (in orange) and the catalytic region (D149; in red). b Crystal structure of ERK2 with an acrylamide containing artificial peptide (SynthRevDCOV; PDB ID: (8PSR). The panel shows the Fo-Fc omit map (2ϭ) calculated with the final model but without the peptide-cysteine adduct. The inset shows the region between φL and φA for the free (SynthRevD, black) or the Michael acceptor (intrachain acrylamide) containing covalently bound peptide (SynthRevDCOV, blue). c Gallery of the Michael acceptor warheads represented in the compound collection (Group 1-7) with the structure of one of the hit compounds from Group 3 (1). Capacity of the small molecule to block ERK2 binding to a fluorescently labeled D-peptide probe was monitored in a quantitative binding assay. Panels below show competitive binding curves with 1 in the absence or presence of 1 mM GSH. Kiapp is a proxy for the MAPK binding affinity of unlabeled compounds. (FB: Fraction Bound; error indicates the parameter estimation error based on the least square method, n = 3). d Binding of GSH to a cyclohexenone-based warhead. 2-biotin was immobilized on the SPR streptavidin chip by biotin capture and GSH was injected over the chip surface at different concentrations. The panel shows the equilibrium binding data (with red diamonds) fitted to a 1:1 stoichiometric binding model (in black line). The expected RUmax at the applied capture level is ~15, the error of the determined KD and the RUmax shows the parameter estimation error based on the least square method. The panel below shows the results of a kinetic binding experiment with 1 mM GSH injected over the chip with lower 2-biotin capture level (RUmax is ~3.5; result of one representative experiment). Based on this measurement k_chem(off) is 0.07 s−1 using the 1:1 reversible binding model. The association rate falls outside the reliable measurement range of the instrument (>1000 M−1s−1), but if Kchem is 1 mM, based on the equilibrium measurement, then k_chem(on) is ~70 M−1s−1. e The scheme of reversible inhibitor (I) binding to a cysteine thiol on the surface of a protein (P). The equilibrium constants based on this 2-step scheme for the formation of I-Pcov complex as well as the corresponding kinetic rates are shown on the left. The two formulas below represent the overall KD expressed in different forms for the I (inhibitor) + P (protein) reaction where I is a reversible covalent inhibitor. The formula on the right can be obtained from the classical definition of KD shown on the left with the equilibrium concentrations of the different species in steady-state. A division of the latter by [I:P] leads to the following formula: ([P] * [I] / [I:P]) / (1 + ([I-Pcov] / [I:P])), where ([P] * [I]) / [I:P] equals to KD´ and [I-Pcov] / [I:P] is equal to Kchem´ by definition, giving the final equation KD = KD´ / (1 + (1/Kchem´)) that can be used to describe the binding of a reversible covalent inhibitor to a protein surface thiol. Kchem´ is characteristic for the activity of the inhibitor to form the reversible covalent bond with the target thiol, while KD´ is characteristic for noncovalent contact formation of the inhibitor on a protein surface surrounding the thiol of the target cysteine. Protein and reversible covalent inhibitor binding can be described by an overall KD. In the presence of free thiols in solution (e.g., GSH) the inhibitor equipped with a double-activated, sterically crowded cyclohexenone-based warhead will “prefer” the protein surface cysteine because of additional (noncovalent) contacts (see panel on the right on top; Kchem refers to the intrinsic activity of the warhead with a free thiol such as GSH). Source data are provided as a Source Data file.
Many biologically active natural products, including various terpenoids such as oridonin (a natural diterpenoid widely used in Chinese herbal medicines), contain electrophilic Michael acceptor fragments22,23. Other antitumor or anti-inflammatory herbal compounds such as withaferin A (from Withania somnifera) or zerumbone (from ginger, Zingiber zerumbet) contain cyclic α,β-unsaturated ketone moieties and react with nucleophilic residues (e.g., cysteines) of proteins (vimentin, NFκB or Keap1, HuR, respectively). Accordingly, the reactive functionalities are often embedded in a chiral and complex molecular environment, which is strikingly different from the acrylamide appendage used in contemporary drug developments. Inspired by these natural products and learning that the MAPK D-groove cysteine can be specifically targeted by Michael addition under some conditions, we continued to identify small molecules that may replace artificial peptides described above.
Screening with Michael acceptor containing building blocks
Structural characterization of MAPK−D-peptide complexes revealed that the D-groove cysteine is surrounded by hydrophobic pockets and natural peptides often use hydrophobic side chains to contact and fill these small pockets. Therefore, to identify small molecules that can target the MAPK D-groove cysteine, we constructed a library of compounds containing different Michael acceptor warheads endowed with small hydrophobic moieties. Our primary aim was to outreach this study toward structurally more complex, chiral and terpenoid-like electrophiles. Thus, we selected our previously developed synthetic building blocks used in various total syntheses24,25. Owing to their distinct structural features, these electrophiles have an enhanced steric crowding around the reactive center which was envisioned to evoke a “frustrated” state, namely thiol-adduct formation might be compromised both thermodynamically and kinetically. Accordingly, higher selectivity in the cysteinome and lower reactivity toward GSH were expected. In addition to the compounds used as intrachain acrylamide warheads (Group 1) described earlier, the collection contained 75 compounds representing six types of Michael acceptors: 2) cyclohexenone, 3) terpenoid-like, sterically crowded and chiral cyclohexenone, 4) chalcone, 5) cyanoacryl, 6) sterically crowded cyanoacryl and 7) nitroalkene; and each group contained molecules where substituent groups (R1–R17) varied (Supplementary Table 2). Since the chemical environment of the α,β-unsaturated carbonyl affects the electronic properties of the warhead, the reactivity of the beta carbon in the different groups varies, moreover in “frustrated” compounds (Group 3 and 6) reactivity is also affected by steric hindrance (Fig. 1c).
The collection was used to identify molecules capable of interfering with the binding of fluorescently labeled D-motif peptides to ERK2, p38α or JNK1 (Supplementary Fig. 4). Group 3 (“frustrated” cyclohexenone) and Group 7 (nitroalkene) compounds showed interference with reporter D-motif peptide binding. Group 7 compounds were found ineffective when the same screen was repeated under high concentration of GSH (10 mM), therefore they were not pursued further, while Group 3 molecules retained most of their inhibitory effect and were further investigated.
MAPK D-groove binding of double-activated, sterically crowded cyclohexenone compounds
A quantitative follow-up assay with compound 1 demonstrated that binding to ERK2 (Ki ~7 µM) is unaffected in the presence of 1 mM GSH (see Fig. 1c). In order to gain insights into the apparent “tolerance” of sterically crowded cyclohexenone compounds to GSH, thiol reactivity of a test compound containing a biotin tag (2-biotin) was characterized by surface plasmon resonance (SPR) measurements. The equilibrium KD (or Kchem) was ~1 mM, moreover a kinetic binding experiment suggested reversible adduct formation (Fig. 1d). Binding of 3, a congener derivative, to a simple thiol compound (beta-mercaptoethanol; BME) was also analyzed by isothermal calorimetry (ITC) and Kchem was also found to be low millimolar (~2 mM) in this different analysis (Supplementary Fig. 5). The 3R-GSH adduct (3 R is the R stereoisomer of the racemic 3) could also be detected by mass spectrometry and the ratio of free 3 R and its covalent adduct did not increase between measurements taken right after components were added together (~5 min) or after some incubation time (2 h), suggesting that the on-rate is indeed fast at the examined millimolar GSH concentration (Supplementary Fig. 6a). The 3R-BME adduct was also detected in NMR experiments, moreover, the result of a jump-dilution experiment suggested that the dissociation of the covalent adduct is indeed fast: the adduct completely dissociated during the lag time of the measurement (~2.5 min). This is consistent with the result of the SPR analysis with GSH and 2-biotin (koff = 0.07 s−1) based on which the half-life of the thiol adduct is calculated to be ~ 10 s (t1/2 = ln2/koff) (Supplementary Fig. 6b).
In contrast to GSH, the (apparent) binding affinity of 1 or 3 to MAPKs is low micromolar (Table 1). Preferential binding, namely low micromolar versus millimolar, towards the MAPK D-groove cysteine could be explained by the latter thiol being in a more complex environment where noncovalent binding (I:P) at the protein surface could indirectly facilitate and contribute to the formation of the covalent adduct (I-Pcov) and also “protect” from GSH thiols. Higher affinity thus emerges from a combination of chemical reactivity on the target (Kchem′) and noncovalent contacts (KD′) (Fig. 1e). The overall KD of a reversible covalent inhibitor can be calculated: KD = KD′/1 + (1/Kchem′), and this formula shows that millimolar noncovalent binding affinity (KD′) could be efficiently increased by a similar Kchem′ value (mM), because the KD would drop to micromolar for a reversible covalent inhibitor using both noncovalent and covalent mechanisms. Naturally, the Kchem determined for the off-target GSH thiol is likely different from Kchem′, because the latter could be greatly affected by the target thiol’s local chemical and topological environment on the protein surface, nevertheless a Kchem′= 10 mM (covalent) would decrease the KD compared to KD′ (noncovalent) by ~100-fold and Kchem′= 100 mM would decrease it by ~10-fold. Based on the scheme above the formation of the I-Pcov complex was assumed to follow a 2-step process. This provides a pragmatic solution for a complex problem with some inherent limitations related to parameter calculation underlying the noncovalent versus covalent components. Undoubtedly, the concrete mechanistic details related to covalent bond formation and thiol elimination related to k3 (or kchem_on) and k4 (or kchem_off) kinetic rates, respectively, for example are not directly addressed. Fortunately, the steady-state KD′ and Kchem′, which are important parameters for reversible inhibitor design, could be experimentally directly addressed or approximated (see later).
Table 1.
Summary of apparent binding affinities of Group 3 (double-activated, sterically crowded cyclohexenone) compounds
| ERK2 Kiapp/µM | p38 Kiapp/µM | JNK1 Kiapp/µM | logP | ||
|---|---|---|---|---|---|
| 1 | 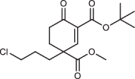 |
10.8 ± 1.6 | 1.2 ± 0.0 | 47.6 ± 10.5 | 2.796 |
| 3 | 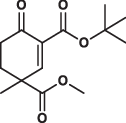 |
16.0 ± 1.2 | 1.6 ± 0.2 | 7.4 ± 1.3 | 1.797 |
| 4 | 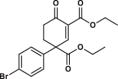 |
101.7 ± 7.6 | 2.5 ± 0.3 | 8.1 ± 3.2 | 3.102 |
| 5 | 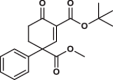 |
11.3 ± 0.5 | 1.2 ± 0.1 | 18.8 ± 3.9 | 2.728 |
| 6 | 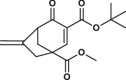 |
12.7 ± 1.2 | 0.8 ± 0.1 | 2.1 ± 0.2 | 1.963 |
| 7 | 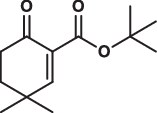 |
31.2 ± 1.3 | 19.0 ± 1.6 | 110.3 ± 14.5 | 2.644 |
| 8 | 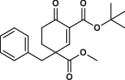 |
4.3 ± 0.2 | 0.5 ± 0.1 | 4.8 ± 1.1 | 3.020 |
| 9 | 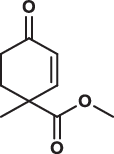 |
> 200 | > 200 | > 200 | 1.085 |
| 10 | 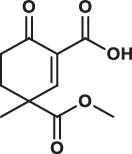 |
> 200 | > 200 | > 200 | 0.540 |
| 11 | 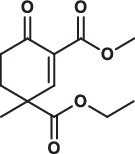 |
172.0 ± 30.4 | 8.5 ± 0.7 | > 200 | 1.018 |
| 12 | 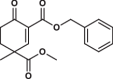 |
16.2 ± 1.0 | 5.4 ± 0.4 | 4.0 ± 0.0 | 2.198 |
| 13 | 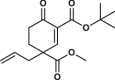 |
15.7 ± 1.0 | 2.7 ± 0.2 | 2.9 ± 0.7 | 2.353 |
| 14 | 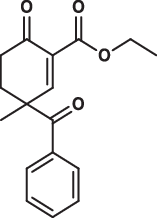 |
144.3 ± 22.3 | 10.9 ± 0.5 | 35.3 ± 5.4 | 2.728 |
| 15 | 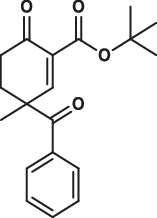 |
10.9 ± 1.2 | 2.2 ± 0.2 | 14.5 ± 2.7 | 3.507 |
| 16 | 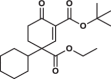 |
26.5 ± 2.0 | 4.0 ± 0.5 | 47.0 ± 8.3 | 3.747 |
| 17 | 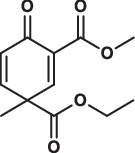 |
> 200 | > 200 | > 200 | 0.794 |
Data show the mean ± parameter error estimates from weighted least square method, see Supplementary Fig. 7; logP are calculated values.
MAPKs have an overall similar D-groove topography, but they display some sequence variation around the D-groove20. In agreement with this, compounds showed varying degree of selectivity towards the three MAPKs tested (e.g., 4: ~40-fold difference for p38 versus ERK2, 5: > 10-fold for p38 versus JNK1, and 6: > 5-fold for JNK1 versus ERK2). While all compounds bound to p38α with the highest affinity (~0.5–10 µM), most molecules–depending on the substituent groups (R5, R6 and R7)–also bound to ERK2 and JNK1 with medium micromolar (2–10 µM) affinity (Table 1 and Supplementary Fig. 7). Stronger binding to p38α may be explained by the more flexible overall structure of p38α26: the docking groove of this MAPK accommodates an optimal cysteine-warhead adduct conformation easier and/or noncovalent contacts mediated by the directing moiety at C2 can contribute more to overall binding energy.
After establishing that Group 3 molecules were promising hit molecules, we characterized ERK2 binding of these cyclic Michael acceptors and the contribution of different substituent groups to ERK2 binding was examined (Fig. 2a). This structure-activity relationship (SAR) analysis established the importance of the electron withdrawing ester group in C4 next to the electrophilic centrum C3, and it also showed that bulky hydrophobic groups (e.g., tert-butyl or benzyl) attached to C2 or C4 increase binding affinity. In agreement with this, binding affinity (namely, the capacity of the compounds to compete with docking peptide binding in the D-peptide displacement assay) showed moderate correlation with the logP value (lipophilicity) of compounds (Pearson’s coefficients for ERK2, p38α, and JNK1 were −0.678, −0.590, −0.672). Replacing ERK2 D-groove cysteine with alanine greatly decreased binding ( ~ 40-fold) as expected (Fig. 2b). Moreover, specific targeting of the D-groove cysteine (C162) in p38α was confirmed by a similar experiment. This MAPK contains another cysteine next to the groove (C119). 8 R bound with decreased affinity to the C162A mutant but binding was unaffected to the C119V mutant, indicating that the compound specifically targets the D-groove cysteine (C162) (Supplementary Fig. 8). In contrast to the slow and irreversible intrachain acrylamide adduct formation observed between artificial peptides and ERK2, covalent adduct formation of cyclic Michael acceptors (tested with 8 R) is fast and reversible (Fig. 2c, d).
Fig. 2. Structure-activity relationship (SAR) analysis of cyclohexenone containing compounds binding to ERK2.

a Summary of competitive fluorescence polarization-based protein-peptide assays (see Supplementary Fig. 7). The numbering of the carbon atoms of the cyclohexenone ring (1–6) is indicated on 3 in the middle. (Kiapp values show the mean with parameter estimation error based on the least square method, n = 3). b Mutating Cys161 to alanine greatly decreases the apparent binding affinity of 8. Panels on the left show the direct binding titration with protein (WT or C161A) and reporter D-peptide (fluorescently labeled), panels on the right show the results of the competition experiments. (Kiapp values show the mean with parameter estimation error based on the least square method, n = 3). c Reversibility of ERK2-cyclohexenone adduct formation. Panels show the intact mass before (left) and after dialysis (right) for ERK2-8R. 5 µM ERK2 was mixed with 50 µM 8R and analyzed by LC-MS, then the sample was dialyzed in buffer (20 mM Tris pH = 8, 200 mM NaCl, 2 mM TCEP) overnight and analyzed by LC-MS again (n = 1). Notice that the ratio of the ERK2 intact mass after the dialysis increases and the ratio of the adduct decreases in the deconvoluted mass spectrum, indicating that 8R adduct formation is reversible (Mwt(8S) = 344). 8R or 8S refers to the (R) and (S) stereosimers of 8, respectively, and all other stereoisomers of a given compound is similarly labeled with this simpler labeling scheme henceforth. (FP: fluorescence polarization in artificial units; FB: fraction bound). d Real-time monitoring of ERK2-8R binding by surface plasmon resonance (SPR). ERK2 was covalently linked to the SPR chip surface (CM-5) by amine-coupling. 8R was injected over this chip surface at 50 µM concentration (expected RUmax is ~45; one representative experiment is shown out of three). e Crystal structure of ERK2-8S and -8R complexes (PDB ID: (8PSW and 8PSY, respectively). Middle panels display Fo-Fc simulated annealing omit maps shown at 2ϭ for the covalent adduct. (ERK2 is shown in cartoon, the Cys161-small molecule adduct and the main chain of 159–163 are shown in sticks; these panel show the two structures in the same view). The panel to the right shows an overlay of the ERK2-8S complex (see panel on the left) with the ERK2-SynthrevDCOV (PDB ID: 8PSR) structure demonstrating that the cyclohexenone core and the different substituent groups of the small molecule occupy the same pockets (φL, φA, φB) used by the hydrophobic amino acids of the D-peptide. Source data are provided as a Source Data file.
We used X-ray crystallography to determine the structure of ERK2-8R and -8S complexes (see Supplementary Table 1). Although 8R and 8S differ only in their configuration at C4, both molecules could bind to Cys161 covalently and their cyclohexenone moiety with its pendant substituents occupy the hydrophobic pockets (φA, φL and φB) (Fig. 2e). The binding pose of the cyclohexenone moiety was confirmed by testing a derivative of 3 (3’R with a 5-methyl group at C5). The 5-methyl group in 3’R would clash with the surface and 3’R indeed bound to ERK2 as well as to other MAPKs with less affinity (Supplementary Fig. 9). The cyclic structure of the cysteine targeting covalent warhead seems to be a key feature, since similar molecules 39 or 40 with an acrylester or cyanoacrylester, which are open-chain (acyclic) Michael acceptor warheads, were far less efficient in binding to the MAPK D-groove (Supplementary Fig. 10).
In contrast to broadly applied acrylester/acrylamide warheads that form irreversible adducts with thiol groups, the covalent adduct formation by these cyclohexenone compounds is reversible. The reversible Michael adduct formation tendency can be attributed to both electronic (as in the case of cyanoacrylates) and counteracting steric crowding effects. The ester group adjacent to C2 has an explicitly strong electron withdrawing effect making the unsaturated beta (C3) carbon atom more reactive towards the sulfhydryl group of the cysteine, and replacement with an amide group or other less electron withdrawing group in the C2 position is expected to lower the reactivity in the Michael reaction due to its mitigated electron withdrawing capacity. While amide (18–23) or oxazole (24) bearing compounds also formed a reversible covalent bond with ERK2 similarly to ester-containing warheads, these analogs of the ester compounds (e.g., 18 vs 3, 20 vs 12) or other variants (19, 21–24) bound less efficiently to all MAPKs (Table 2; Supplementary Figs. 11 and 12). The Michael acceptor moiety can also be embedded into a 5-membered cyclopentenone ring (25–27). Unexpectedly, the electronically fine-tuned cyclopentenone derivative 27 bound to JNK1 the best (see Table 2), while all other tested inhibitors bound p38α with the highest affinity. These examples demonstrate that the nature of the electron withdrawing group at C2, other pendant substituents at C4 and C5 positions or the size of the ring have a significant impact on the capacity of these cyclic scaffolds to target the MAPK docking groove cysteine. The capacity for reversible thiol targeting and micromolar binding affinity to MAPKs make the chiral cyclohexenone/cyclopentenone compounds a promising set of hit molecules as a starting point for further optimization.
Table 2.
Summary of apparent binding affinities of compounds with different electron withdrawing groups at C2 or with a cyclopentenone scaffold
| ERK2 Kiapp/µM | p38 Kiapp/µM | JNK1 Kiapp/µM | logP | ||
|---|---|---|---|---|---|
| 18 | 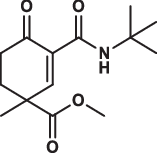 |
> 200 | 74.7 ± 14.0 | > 200 | 1.370 |
| 19 | 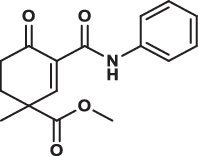 |
> 200 | 19.3 ± 3.1 | > 200 | 2.094 |
| 20 | 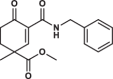 |
32.8 ± 2.7 | 42.2 ± 2.9 | 97.4 ± 24.1 | 1.771 |
| 21 | 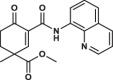 |
71.4 ± 9.2 | 38.9 ± 6.2 | > 200 | 2.642 |
| 22 | 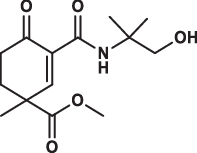 |
> 200 | 53.0 ± 11.1 | > 200 | 0.342 |
| 23 | 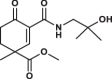 |
> 200 | 55.9 ± 6.5 | > 200 | 0.342 |
| 24 | 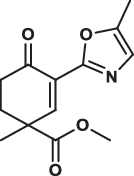 |
42.7 ± 3.6 | 21.2 ± 2.8 | 112.2 ± 32.7 | 1.909 |
| 25 | 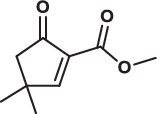 |
135.7 ± 14.1 | 28.0 ± 2.4 | > 200 | 1.085 |
| 26 | 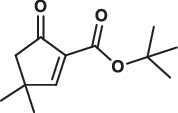 |
168.7 ± 8.7 | 27.4 ± 3.3 | > 200 | 2.254 |
| 27 | 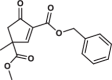 |
37.2 ± 4.0 | 9.4 ± 2.1 | 2.1 ± 0.3 | 1.808 |
Data show the mean ± parameter error estimates from weighted least square method, see Supplementary Fig. 12; logP are calculated values.
Characterization of enantiomerically enriched cyclohexenone compounds
Most of the tested cyclohexenone compounds contain a quaternary asymmetric center at C4 and molecules were originally synthesized as racemic mixtures. To test whether the stereoisomers have different affinity, we produced enantiomerically enriched compounds by stereoselective synthesis24. Most of the different stereoisomers bound to MAPKs with modest difference in binding affinity (<3-fold), however, the stereoisomers of bicyclic 6 containing two chirality centers (6S,S and 6R,R) bound with ~10-fold difference (Table 3 and Supplementary Fig. 13). To elucidate how stereochemistry and the pendant substituents at key positions affect the binding mode, we determined the crystal structure of three different enantiomeric pairs bound to ERK2: ERK2-8R/8S (see earlier), ERK2-12R/12S, and ERK2-3R/3S (Fig. 3a, Supplementary Fig. 14 and see Supplementary Table 1). High-resolution structural data (~1.6–2.0 Å) on these complexes revealed that the configuration of the emerging C3 stereogenic center can greatly vary. Note that in addition to the existing stereogenic center at C4, new chirality centers at C3 and even at C2 (in the oxo tautomer) will be created upon covalent adduct formation. The absolute configuration will depend on the directing substituent group in C2 (e.g., tert-butyl or benzyl) as well as on the original configuration (e.g., S or R) and/or substituent group size (methyl or benzyl next to carboxymethyl) at C4. Moreover, the apparent binding affinity is also affected by the substituent group at C5. The 5-methyl derivative of 3R (3’R) bound more weakly to ERK2 (and to the other MAPKs, too; see Supplementary Fig. 9) because this somewhat more crowded cyclohexenone derivative would likely be less compatible with binding pocket topography around the cysteine-warhead adduct. Structural comparisons between apoERK2, ERK2–D-peptide and ERK2-cyclic warhead complexes show that in the latter case the hydrophobic pocket area of the D-groove widens to accommodate the cyclohexenone ring at φA. For the 3R/3S enantiomeric pair, the binding mode is fundamentally different and is affected by the configuration of the stereogenic center at C4: 3R forms a cysteine-adduct, while 3S forms a mixture (~50–50%) of cysteine(161)- and histidine(125)-adducts in the crystal structure. This unexpected behavior of 3S could only be explained by its different chiral configuration which is likely not optimal in the chiral environment around Cys161, but it is a better fit for the nearby His125, which unexpectedly is also a suitable nucleophile for the Michael acceptor of the cyclic warhead.
Table 3.
Summary of apparent binding affinities of C4 stereoisomers
| ERK2 Kiapp/µM | p38 Kiapp/µM | JNK1 Kiapp/µM | ||
|---|---|---|---|---|
| 3R | 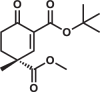 |
12.3 ± 0.4 | 1.7 ± 0.1 | 23.6 ± 1.5 |
| 3S | 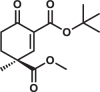 |
4.9 ± 0.2 | 8.3 ± 0.7 | 6.5 ± 0.6 |
| 8R | 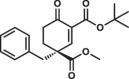 |
5.9 ± 0.4 | 0.8 ± 0.1 | 1.2 ± 0.3 |
| 8S | 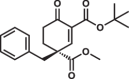 |
6.3 ± 0.3 | 0.8 ± 0.1 | 1.8 ± 0.4 |
| 12R | 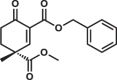 |
10.1 ± 1.0 | 4.8 ± 0.4 | 27.6 ± 3.9 |
| 12S | 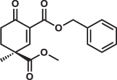 |
24.7 ± 1.5 | 5.5 ± 0.6 | 15.3 ± 2.4 |
| 6S,S | 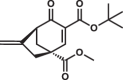 |
66.7 ± 5.1 | 6.2 ± 0.4 | 8.0 ± 0.7 |
| 6R,R | 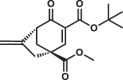 |
8.4 ± 0.7 | 0.6 ± 0.0 | 1.0 ± 0.1 |
Data show the mean ± parameter error estimates from weighted least square method, see Supplementary Fig. 13.
Fig. 3. Structural comparison of the ERK2 covalent adducts with different cyclohexenone containing compounds.

a Panels show the crystallographic complexes of ERK2-8S or -8R, ERK2-12R or -12S, and ERK2-3R or -3S (PDB ID: 8PSW, 8PSY, 8PT0, 8PT1, 8PST, 8PT5, respectively). The absolute configuration at C4 is R or S; 8S/8 R, 12 R/12S and 3 R/3S are enantiomeric pairs but new asymmetric centers form upon adduct formation at C3 and C2, too. The cysteine adduct in the ERK2-12R complex has two alternative C-S covalent bond conformations (see Supplementary Fig. 14). The ERK2 D-groove is shown with surface representation from the same view for 8S/8 R, 12 R/12S and 3 R (and the atoms of Cys161 on the surface are colored yellow). Note that, the crystal structure of the ERK2-3S complex is shown in a view 180° rotated along the Z axis (perpendicular to the plane of the panel) compared to the other surface panels. This latter complex has a cysteine (yellow) as well as a histidine adduct (salmon) (and the D-groove is shown in surface representation apart from the amino acid adducts, which are shown in sticks). Naturally, one molecule can form only one of the two adducts, and the final structural model fitted best with the crystallographic data if the two alternative adducts were equally present. (The conformation of the intact cysteine or the histidine side chain is shown in black.). b 6 R,R forms a reversible covalent adduct with the imidazole side chain of histidine in the MAPK D-groove (see Supplementary Fig. 14). The crystal structure of the ERK2-6R,R complex (PDB ID: 8PT3) with the Fo-Fc simulated annealing omit map shown at 2ϭ for the histidine adduct is shown in the middle panel. The panel on the right shows the MAPK docking groove in surface representation highlighting the different hydrophobic pockets (φA, φB, φL) and the polar CD groove.
The two enantiomers of bridged bicyclic 6, namely (6S,S and 6R,R) showed the greatest difference in their apparent binding affinity for all three MAPKs tested. Because of the extra bridge between C4 and C6, the warhead is more rigid and extended perpendicular to the cyclohexenone ring. Importantly, the binding of this more complex three-dimensional, bulkier warhead proved to be more sensitive to the configuration at C4 as the R,R stereoisomer bound 8-10-fold better compared to the S,S stereoisomer (see Table 3). The crystal structure of the ERK2-6R,R complex revealed an unexpected binding mode of this C4-C6 bridged compound: instead of the cysteine(161) thiol, the compound formed the reversible covalent adduct with the imidazole side chain of histidine(125) and the cysteine thiol remained intact (Fig. 3b). Independently from histidine adduct formation in the MAPK D-groove suggested by X-ray crystallography, covalent adduct formation of 6R,R with histidine (i.e. N-acetyl-L-histidine) in solution could also be confirmed by mass spectrometry (Supplementary Fig. 15).
The impact of covalent bond formation on compound binding affinity/energetics
It is noteworthy that synthetic molecules <300 Da in size can bind to the hydrophobic pocket area of the MAPK D-groove and compete with natural peptide binding, albeit the latter are naturally larger (>1000–2000 Da) and achieve similar affinities by making extensive contacts throughout the entire D-groove (CD-groove + hydrophobic pockets). The relatively high binding affinity of double-activated, sterically crowded cyclohexenone/pentenone compounds are likely due to a combination of two reversible mechanisms and emerges both from noncovalent and covalent binding. We used the peptide displacement assay to assess the contribution of these two distinct mechanisms. First, we demonstrate that the fluorescence polarization signal, reporting on the level of complex formation between the MAPK and the fluorescently labeled reporter peptide, does not depend on the incubation time in the presence of the small molecule inhibitor at any of the used concentrations (Fig. 4a). This suggests that cyclohexenone/pentenone compounds bind in the MAPK D-groove with fast dynamics and the peptide displacement assay could be used to report on steady-state binding affinity. (Technically, this assay determines apparent Ki values for the small compounds, which may be used as a proxy for their KD. Note that in the peptide displacement assay it is practical to assume a one-step binding model, thus the Ki determined based on the noncovalent binding competition equation must be referred to as apparent Ki, Kiapp. For competitors whose binding do not include a reversible covalent mechanism it is correct to refer to this value simply as Ki.) Next, we synthesized the saturated derivatives of 6R,R and 3R (6”R,R or 3”R) which did not contain the reactive olefin moieity required for covalent binding and assessed their binding to MAPKs (see Fig. 4a). Although the overall structure of the cyclohexane ring in 6”R,R or 3”R may not be the same as in the original unsaturated compounds, their lack of competition in this assay suggests that the covalent contribution plays a major role in binding and it may greatly increase binding affinity as expected.
Fig. 4. Contribution of the covalent mechanism to binding.

a Binding measurements with the original or saturated versions of 6R,R and 3R. 6R,R was tested in the competitive peptide displacement assay for the importance of the incubation time with three MAPKs: the measurements were taken right after setting up the binding reaction ( ~ 5 min; first row) or after 1 h (second row). Note that the competition binding profiles are unchanged. Lower panels show the results of similar experiments but using the saturated form of two cyclohexenone based compounds (6”R,R and 3”R). Note that these latter compounds could not compete well with reporter D-peptide binding even up to 1 mM concentration, suggesting that the C2 = C3 double bound is very important for binding as expected. (Kiapp values show the mean with parameter estimation error based on the least square method, n = 3; FB: fraction bound.). b Binding energy contribution of noncovalent and covalent mechanism in the binding of 8 to ERK2. The upper panels show the results of the peptide displacement assay using a weak binding reporter peptide (pepMK2) (FP: fluorescence polarization, FB: fraction bound; n = 3). Left panel: direct titration experiment with the ERK2 double mutant lacking both D-groove nucleophiles (C161A/H125L mutant); right panel: competitive titration with 8. Summary of the energy contribution of the noncovalent and covalent binding mechanisms is shown below. The values for ERK2 WT (Kiiapp ~4 μM) and for the single ERK2 C161A mutant (Kiapp ~160 μM) are taken from the experiment shown on Fig. 2b. The Kiapp value measured with the ERK2 C161A/H125L double mutant shows the noncovalent contribution (gray bar). Based on the scheme introduced on Fig. 1e, the Kchem´ value for ERK2 WT and 8 binding can be calculated. (FP: fluorescence polarization in arbitrary units; FB: fraction bound.). c 1H NMR spectra of the 3R-BME and 6 R,R–His-Test adducts. The left panel shows the structure of 3 R and of its thiol-adduct diastereomers (enol forms). Panels below show the 1H NMR spectra of the initial sample 3R (14.1 mM) and after mixing it with 1.4 molar equivalent BME and collected after 90 min. (The HC proton was in an overlapping position.) The panel on the right shows the structure of the 6R,R–His-Test covalent adduct (enol form) with the corresponding spectra for the His-Test sample alone (5 mM), the initial 6R,R sample (17.2 mM), and after mixing the latter with 2.6 molar equivalent His-Test (collected after 60 min). 1H NMR spectra were recorded in PBS (D2O; pH ~7.2) with 75% DMSO. The major stereoisomer of the imidazole adduct is R ( > 95 %). The 6.02 ppm protons from the reference compound was used as the internal standard (IS) important to be able to calculate Kchem(His) (n = 1; see Supplementary Note 1). d The structures of compounds analyzed by electronic circular dichroism (ECD) measurements with their Kchem(thiol) value determined based on BME titration experiments (colored parts of the molecules highlight important differences; a tert-butyl ester or amide in 3R vs 18R, a bridged skeleton in 6R,R, and anilide in 19’S). An example of this measurement for the 6R,R + BME reaction is shown on the panels right of the dashed vertical line (n = 1). These show the ECD spectrum of 6R,R (black) in PBS (pH ~7.4) compared with those produced by addition of different equivalents of BME (from 2 to 32 equivalents) and the inset figure shows the enlarged 275–410 nm wavelength range with 0–250 equivalents of BME; the concentration of 6R,R was 250 μM) and the right panel shows the change of the ECD signal of 6 R,R (∆ε) in the function of increasing concentration of BME monitored at 341 nm. Curve fitting produced the value of 409 ± 68 (M−1) for the equilibrium constant, corresponding to Kchem(thiol) value of ~2.5 mM for this compound (see Supplementary Note 2). Source data are provided as a Source Data file.
Earlier we showed that the C161A mutant of ERK bound to 8 with greatly reduced affinity compared to wild-type (WT) ( ~ 160 μM vs 4 μM, respectively; see Fig. 2b). In principle, this finding allows a quantitative assessment on the importance of the C-S(Cys) covalent bond based on the ΔΔG = R*T*ln(Kiapp(WT) – Kiapp(C161A)) equation. However, the adjacent histidine (H125) may potentially also contribute to the covalent mechanism as it was observed in the crystal structure of some of the MAPK adducts (see Fig. 3; note that the crystal structures of 8 with ERK2 displayed only the Cys-adduct, however, the structures only document the energetically most favorable adduct if the energy is not greatly different between the two alternative adducts). Therefore, we also tested if the His-adduct could also play a role in the binding of 8 to ERK2. To this end, we mutated His125 to a leucine in addition to the C161A mutation and measured the binding of 8 to this ERK2 double mutant and determined its Kiapp to be weak (420 μM) as expected. In the peptide displacement assay we had to use a much weaker reporter D-peptide to be able to quantitatively address this weaker interaction (Kiapp > 200 μM): CF-labeled pepMK2 binds to ERK2 with only medium micromolar affinity (KD ~ 50 μM) in contrast to the stronger CF-labeled RHDF1 peptide (KD ~ 2 μM) used in the earlier measurements. In brief, these experiments gave a quantitative view on the energy landscape of C-S(Cys) and/or C-N(His) adduct formation versus noncovalent binding of 8 to ERK2 (provided that Cys/His do not contribute to noncovalent binding)(Fig. 4b). The analysis confirms that noncovalent binding (KD′ ~ 0.4 mM; ΔG = −4.7 kcal/mol) indeed represents a major contribution in overall binding (KD ~ 0.004 mM; ΔG = −7.3 kcal/mol), while the covalent contribution (Kchem′ ~ 10 mM; ΔG = −2.7 kcal/mol) is smaller (where for 8 this mostly comes from the Cys-adduct). Interestingly, Kchem′ in the MAPK D-groove is greater, meaning less covalent contribution, than for a model thiol determined for a similar compound (Kchem(GSH) ~ 1 mM; see Fig. 1d), highlighting that adduct formation in the MAPK D-groove is less optimal compared to that of with a free thiol. This latter is an important finding in understanding how cyclohexenone/pentenone-based warhead containing compounds achieve selectivity for different on-target nucleophiles (e.g., located in the MAPK D-groove) and make preferential adducts with these compared to chemically more favorable adducts with free thiols (e.g., GSH or other off-target nucleophiles): the noncovalent contribution of the compound compensates for the relative loss in intrinsic chemical reactivity on the target protein’s surface in a specific fashion, moreover, as a collateral advantage the koff will decrease due to the bimodal (noncovalent+covalent) mechanism (see later).
Next, we focused on the determination of the intrinsic reactivity of 3R and 6R,R towards a free thiol (GSH) and imidazole group (namely histidine in His-Test; N-acetyl-L-histidine methyl ester). NMR measurements clearly indicated the formation of the respective covalent adducts (Fig. 4c). Unfortunately, NMR titration measurements could not be used to determine Kchem(thiol) for the 3R + BME reaction since 3R had to be used in >10 mM concentration to be able to record good quality 1H NMR spectra and addition of any amounts of BME titrant indicated that the titration mix was already at saturation regarding adduct formation; and this finding was in agreement with the result of the ITC measurement where the Kchem of the 3 + BME reaction was determined to be 2 mM, see supplementary Fig. 5). Fortunately, a similar experiment designed to obtain the intrinsic reactivity of this compound for histidine was successful: the Kchem(His) of the 6R,R+His-Test reaction was ~ 166 mM. The NMR analysis was useful to demonstrate the formation of the thiol and imidazole adducts but required relatively high amounts of sample and the spectra had to be recorded in DMSO:PBS (D2O, pH ~7.2) (3:1) solvent, therefore, to be able to analyze a greater diversity of compounds under more physiological conditions (in 1 x PBS, pH ~ 7.4), an alternative analytic tool was needed. Finally, we came up with the idea of exploiting the chirality of our compounds and used more sensitive electronic circular dichroism (EDC) measurements. With this method in hand we could gain further insight into the intrinsic chemical thiol reactivity of electronically differently tuned warheads. To this end, we examined the reactivity of 3R (with tert-butyl ester, as reference,) 18R (with tert-butyl amide, with less electron-withdrawing potential), 19’S (with aniline amide, presumably a more reactive electrophile compared to 18R), and 6R,R (with a bridged structure, with more limited accessibility to the reactive C3 carbon) with BME (Fig. 4d). To follow the conjugate addition of BME to optically active cyclohexenone Michael acceptors, the ECD spectra of Michael acceptors were recorded and titrations with different equivalents of BME were monitored with ECD measurements. In the conjugate addition, the conjugated carbon-carbon double bond of the Michael acceptor is saturated, which eliminates the previously existing conjugation with the carbonyl groups and introduced two new chirality centers. This modification in the chromophore and the two new chirality centers are expected to induce significant changes in the ECD spectra, which is suitable to monitor and characterize the conjugate addition of BME. This comparative analysis showed that the bridged skeleton in 6R,R was similarly reactive as the reference 3R compound (Kchem ~ 1.5 mM), while 18R with an aliphatic amide was substantially weaker ( ~ 10 mM) as expected, but the anilide 19’S was as reactive as the ester compound (3R). Briefly, this data shows that thiol adduct formation of cyclohexenone warheads can be fine-tuned by changing the susbtituents at the carboxylic moiety. Naturally, the Kchem values determined in these measurements provide the chemical driving force for covalent adduct formation on the protein target (Kchem’), however, the latter may greatly differ from the former depending on the local chemical and/or sterical environment of the nucleophile located on the protein surface. For example, the high millimolar Kchem(His) value of 6R,R is presumably lowered in the ERK2 D-groove (Kchem’ < Kchem) since the ERK2-6R,R crystallographic complex clearly showed a preference for the His-adduct versus the Cys-adduct (see Fig. 3b).
In summary, these experiments showed that the warhead’s cyclic structure in combination with some additional noncovalent, likely hydrophobic, component is the major contributor to overall MAPK D-groove binding and it seems to be a prerequisite for the “specific” covalent mechanism; where the latter may provide different amounts of covalent contribution, depending on the interplay between the warhead and the chiral protein surface with its distinct chemical properties, compared to a free thiol/imidazole reaction.
High-affinity and MAPK-specific binding of a peptide-warhead chimera
The crystal structures showed that the cyclohexenone moiety fills up the φA and φB pockets but the charged CD groove remains unoccupied (Fig. 5a). This provided a great opportunity to extend the previously identified molecules towards that direction. In order to produce compounds that fill this polar pocket, one of the steric modulating groups at C4 in 3S was transformed into a propargyl ester group (28S), which was then linked to a peptide through an N-terminal azide using CuACC click chemistry (28S-pepMNK1_C). Importantly, the terminal acetylenic group orthogonally reacted in a Cu(I) catalyzed click reaction and the highly reactive electrophilic Michael acceptor center remained intact. In silico docking showed that the triazolyl group may direct the moieties towards the CD groove by fitting into φL. Indeed, when the CD-groove binding half of the natural MNK1 peptide was clicked to 28S the apparent binding affinity greatly increased (p38α; Kiapp ~ 3 nM), the koff decreased, and this effect was MAPK-specific (Supplementary Fig. 16). These findings, supported by independent ITC measurements (Supplementary Fig. 17), demonstrated that the cyclohexenone core could be easily decorated further to increase binding affinity and to enhance MAPK specificity.
Fig. 5. Extension of the cyclohexenone scaffold towards the CD-groove by click chemistry.

a Crystal structure of the ERK2-pepMNK1 complex (PDB ID: 2Y9Q top) and the model of the in silico docked (hypothetical) 28S-methyl-triazole molecule (bottom) are shown on the left. The CD-groove binding C-terminal half of the MNK1 peptide with N-terminal azide (N3-pepMNK1_C) was connected to 28S by copper(I)-catalyzed azide-alkyne cycloaddition. Results of SPR experiments with a p38α sensor chip are shown below. Sensorgrams show the binding of pepMNK1 or the 28S-peptide chimera injected under saturating conditions, in 20 µM or 30 nM concentration, respectively (n = 1). Arrows indicate the start of the association or the dissociation phase. (RU: response units). b Binding of 28S-pepMNK1_C to wild-type (WT) or D-groove nucleophile mutant versions of p38α (C162A mutated in the D-groove cysteine, H126L mutated at an adjacent nucleophile position (H126) or C162A/H126L double mutant). The peptide chimera was labeled with carboxyfluorescein (CF) to be able to determine the KD directly. Note that elimination of the thiol had a bigger impact than that of the imidazole ring, suggesting that cysteine is a more important nucleophile in this peptide chimera context. (Kiapp values show the mean with parameter estimation error based on the least square method, n = 3; FP: fluorescence polarization in arbitrary units.). c Kinetic peptide displacement experiments. Wild-type (WT) or C162A mutant version of p38α was mixed with 50 nM 28S-pepMNK1_C_CF to obtain ~80–90% complex formation and incubated for half an h (78 nM or 625 nM, respectively). A great excess of unlabeled pepMNK1 (40 μM; Ki ~ 0.2 μM) was added and the dissociation of the p38α/28S-pepMNK1_C_CF complex was monitored in time. (Data are presented as mean values and error bars show SD, albeit smaller than the symbols of the data points; n = 3, parallel independent measurements; FP: fluorescence polarization in arbitrary units.). Source data are provided as a Source Data file.
To facilitate the characterization of different MAPK docking groove binding compounds and to assess their capacity to interfere with MAPK D-groove mediated signaling events, we developed a quantitative in vitro kinase assay based on the concept of phosphorylation-assisted luciferase complementation (PhALC)27. PhALC assay results with D-SENSOR in the presence of 10 mM GSH showed lower IC50 values for the composite inhibitors compared to that of their fragments (Supplementary Fig. 18). The D-groove binding peptide-warhead chimera provided an opportunity to measure the contribution of the C-S covalent bond to binding affinity and kinetics in a high-affinity, noncovalent binding context (since the peptide region makes extensive contacts in the CD-groove section of the MAPK D-groove and mediates micromolar binding on its own; which is a distinct context compared to small molecules equipped only with a small additional hydrophobic moiety such as a tert-butyl or phenyl group). To this end, we measured the binding affinity of the 28S-pepMNK1_C chimera with wild-type p38α (WT) and the Cys162Ala mutant (C162A) using a C-terminally labeled chimera peptide (28S-pepMNK1_C_CF; CF: carboxyfluorescein) (Fig. 5b). These experiments showed that the chimera peptide binds ~10-fold stronger to WT compared to the C162A mutant, moreover, results of independent ITC experiments also showed a good agreement with this finding (see Supplementary Fig. 17). Similarly to ERK2, a histidine (H126) may also serve as an alternative nucleophile in p38α, however, mutation of this latter residue to leucine only marginally affected binding affinity, and in agreement with this, the C162A/H126L double mutant did not show further decreased affinity compared to the C162A mutant. Results of these experiments highlight an important contribution of the reversible thiol adduct in overall binding affinity as expected. (Note that the decreased binding affinity of p38αC162A is not due to a weaker noncovalent binding mechanism since this mutant bound a CF-labeled reporter peptide with the same binding affinity compared to wild-type, see Supplementary Fig. 8). Based on these measurements, the contribution of the warhead moiety in this bivalent chimera context towards the overall KD is even more modest compared to that of 8 (Kchem′ ~100 mM vs 10 mM; see Fig. 4b). The reversible covalent mechanism, however, may play a major role in improving the kinetic aspect of the composite chimera as an inhibitor by decreasing the koff (see Fig. 5a, b). To this end, we carried out an experiment that directly addressed this “kinetic” contribution. Intact (WT) or C162A version of p38α were mixed with the fluorescently labeled peptide chimera to achieve ~80–90% complex formation and then unlabeled pepMNK1 (Ki ~0.2 μM) was added in great excess (40 μM) and dissociation of the MAPK–labeled peptide chimera complex was monitored in time in the peptide displacement assay (Fig. 5c). These experiments showed that in this “competitive environment” the peptide chimera lasted far longer if the MAPK D-groove cysteine were intact (t1/2 ~ 9 min; calculated with an exponential decay equation), compared to the C162A mutant where the complex was found to be fully dissociated already in the first time point (the lag time of the measurement is about 30 s). Despite that this half-life increase is not high in this artificial peptide chimera context, it still highlights an important aspect of the reversible covalent mechanism: it makes the protein-inhibitor complex longer-lived even under conditions where there are high amounts of noncovalent (natural) binders/competitors present. This is a very useful capacity from an inhibitor designer’s point of view, however the trade-off is that the kon may somewhat also decrease. Intuitively, and also supported by the results of experiments with intrachain acrylamide containing peptides (see Fig. 1b and Supplementary Fig. 1-3), formation of the covalent adduct has more restricted stereochemical requirements compared to simple noncovalent binding. This makes the activation energy barrier landscape far more complex in the context of a composite bivalent inhibitor compared to simpler warhead binding. In spite of these complications emerging from the more complex nature of bivalent binding, results with the peptide-warhead chimera demonstrate that the double-activated, sterically crowded cyclohexenone/pentenone scaffold may provide a practical solution to increase the binding affinity, specificity, and residence time of more complex compounds containing extra functionalities contacting additional surface pockets on MAPKs.
Extension of the D-groove binding cyclohexenone warhead scaffold with different moieties
Despite their promising binding capacity, peptide containing conjugates will likely have only limited use as inhibitors in cells, therefore the original cyclohexenone scaffold was decorated further to produce more complex but still compact small molecules. 6R,R contained a direct extension at C4 where this carbon was linked to C6 creating a more complex C4-C6 bridged warhead. Moreover, the propargyl ester group at C4 (in 28S and 28 R) could be extended by using orthogonal azide-alkyne cycloaddition or Sonogashira coupling to add distinct functionalities directly to the warhead moiety, and some of these extensions led to molecules that had comparable in vitro binding affinity to that of the simpler designs (Table 4 and Supplementary Fig. 19). The effect of these molecules on MAPK-based signaling networks in the cell is expected to be complex because they will compete with dozens, possibly hundreds of MAPK D-groove mediated interactions including binding to activator kinases, substrates, scaffolds, and/or phosphatases (i.e. partner proteins), where these latter naturally also compete with each other and their affinity vary from submicromolar (~0.1 μM) to medium micromolar ( ~ 10–20 μM)19,28. In addition, the extra moieties may potentially modify the effects of the warhead core, as these could make additional contacts and become important for more selective blockage of specific binary MAPK-partner protein binding events, which, however, would only be apparent in cells. First, we tested the effect of D-groove binding compounds on MAP2K-MAPK binding in live HEK293T cells using the NanoBiT PPI assay (MAPK kinases or MAP2Ks are the natural activators of MAPKs and binary binding between them depends on intact docking)19,20,29 (Fig. 6a). Experiments with 28S and 32S showed that extending the propargyl group of 28S with an extra moiety could indeed increase the inhibitory effect, for example 32S blocked MKK7-JNK1 better than 28S. Moreover, 6R,R seemed to have better inhibitory capacity on MAP2K-MAPK binding compared to its enantiomer (6S,S). The effect of these molecules on MKK6-p38α binding was analyzed further and we found that 6R,R efficiently blocked this interaction already 15 min after adding to cells, indicating that its cellular uptake and its target engagement is fast, while 6S,S did not affect this interaction in agreement with its weaker binding to p38α (Supplementary Fig. 20). Next, the impact of 32S and 32 R, which are enantiomers with N,N-dimethylaniline extension at C4, was tested on binary binding between MKK1 and ERK2, ERK2 and RSK1, and ERK2 and MKP3, which all depend on intact D-groove mediated binding (MKK1 is one of the upstream activator kinases of ERK2; RSK1 is one of the downstream substrates of ERK2; MKP3 is one of the known MAPK phosphatases)20. Results of these cell-based tests suggested that MAPK-binding potency does not only depend on the nature of the extended moiety but also on additional structural features set by the configuration at C4. This finding was corroborated by another experiment carried out in HeLa cells where ERK and RSK phosphorylation were monitored by quantitative western blots upon EGF stimulation. This experiment showed that 32S (10 μM) is very efficient in blocking MKK1-mediated ERK phosphorylation and this stereoisomer showed somewhat stronger effects compared to 32R (Supplementary Fig. 21).
Table 4.
Summary of apparent binding affinities for different C4-extended cyclohexenone compounds
| ERK2 Kiapp/µM | p38 Kiapp/µM | JNK1 Kiapp/µM | logP | ||
|---|---|---|---|---|---|
| 28S | 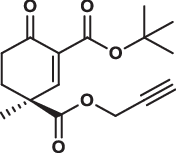 |
1.0 ± 0.1 | 0.8 ± 0.6 | 3.4 ± 0.5 | 1.800 |
| 28R | 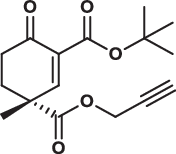 |
3.8 ± 0.2 | 3.5 ± 0.2 | 17.0 ± 2.0 | 1.800 |
| 29S | 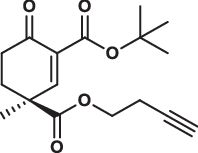 |
1.4 ± 0.2 | 1.1 ± 0.1 | 2.4 ± 0.3 | 2.190 |
| 30S | 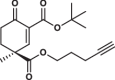 |
0.8 ± 0.1 | 0.9 ± 0.2 | 1.7 ± 0.4 | 2.580 |
| 31S | 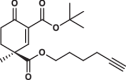 |
1.0 ± 0.1 | 1.3 ± 0.0 | 1.6 ± 0.4 | 2.970 |
| 32S | 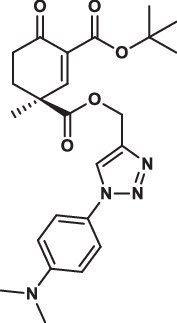 |
2.3 ± 0.1 | 3.5 ± 0.9 | 2.4 ± 0.8 | 3.014 |
| 32R | 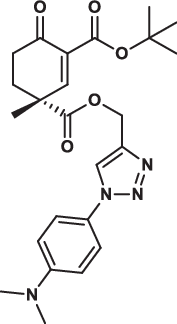 |
15.7 ± 2.9 | 12.8 ± 3.0 | 13.6 ± 6.1 | 3.014 |
| 33S | 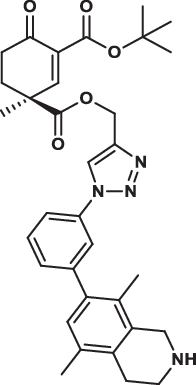 |
4.4 ± 0.7 | 2.8 ± 0.7 | 8.8 ± 2.6 | 4.877 |
| 34S | 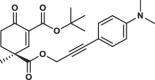 |
-* | 3.1 ± 0.5 | 1.3 ± 0.6 | 3.285 |
| 34R | 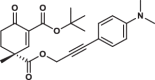 |
-* | 3.9 ± 0.8 | 2.4 ± 0.9 | 3.285 |
| 35S | 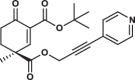 |
1.0 ± 0.0 | 1.5 ± 0.3 | 2.7 ± 0.6 | 2.614 |
| 35R | 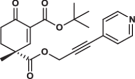 |
3.8 ± 0.2 | 1.8 ± 0.7 | 2.9 ± 1.1 | 2.614 |
Data show the mean ± parameter error estimates from weighted least square method, see Supplementary Fig. 19; logP are calculated values. (* indicates only partial inhibition, suggesting weak binding).
Fig. 6. Characterization of compounds with extended cyclohexenone scaffolds in cell-based tests.

a Results with the NanoBiT luciferase fragment-complementation assay. The panel on the left shows the scheme of the dynamic, luciferase complementation-based protein-protein interaction (PPI) assay (NanoBiT)29. The assay was done in live HEK293T cells using 10 μM inhibitor concentration and compounds were added 1.5-hours before the measurements (“-”: control with 0.05 % DMSO). Top panels show the effect of 28S, 32S, 6S,S and 6R,R on MAP2K-MAPK binding. MKK7, MKK1, and MKK6 are the cognate upstream activator kinases (MAP2Ks) of JNK1, ERK2, and p38α, respectively. Note that 32S is an extended version of 28S with the same configuration at C4, while 6S,S and 6R,R are stereoisomers containing two stereogenic centers (at C4 and C6; shown with arrows) with opposite configurations. MKK7_D0, MKK1_D0 and MKK6_D0 are MAP2K constructs that do not contain their D-groove binding docking motifs – located in their disordered N-terminal region – and were used to show that binding is indeed D-motif/docking dependent. Lower panels show the effect of 32S and 32R, which are stereoisomers with the same extension at C4, on MKK1-ERK2, ERK2-RSK1, and ERK2-MKP3 binary binding. Data are presented as mean values and error bars show SD (n = 3, upper panels, or n = 4, lower panels; luminescence for the latter is shown in arbitrary units; parallel independent measurements). (*p < 0.05, **p < 0.01, ***p < 0.001; two-sided, unpaired t-test). b Effect of inhibitors on AP-1 transcription factor promoter activity. The scheme shows that PMA treatment activates protein kinase C (PKC) which ultimately leads to the phosphorylation (P) of AP-1 transcription factor dimers promoting AP-1 mediated gene expression. The process, in addition to c-Jun phosphorylation by JNK, is also positively affected by phosphorylation of c-Jun dimerization partners (ATF and c-Fos). This latter is promoted by p38 and/or ERK1/2. In the Reporter AP-1 – HEK293 Recombinant Cell Line PMA stimulation increases the transcription of the reporter luciferase gene (Luc). AP-1 promoter driven transcription of the luciferase reporter gene was monitored by measuring luminescence after 6 h in unstimulated cells (-) or cells stimulated with phorbol 12-myristate 13-acetate ( + PMA). Inhibitors were co-administered with PMA. The first panel shows that p38i and ERKi – two ATP competitive inhibitors, SB202190 and SCH772984, respectively, used in 1 μM concentration – indeed attenuate AP-1 mediated transcription as expected. D-groove binding inhibitors (6 R,R, 32S or 32 R) were then used in 1, 3, or 10 μM concentrations in another experiment. 6R,R shows significant inhibition at 3 μM concentration and above, while 32S and 32R, which are enantiomeric pairs with extended moieties at C4, appear to be stronger inhibitors in this MAPK dependent cell-based test. 32S performs significantly better than 32R at all three concentrations tested. The next panel at the bottom shows the results of another experiment with 34S and 35S, containing N,N-dimethylaniline or pyridine moiety, respectively (but C4 was extended by the same chemical solution, i.e. Sonogoshira coupling). 28S, 34S, and 35S are all S stereoisomers at C4 but they contain different moieties (propargyl ester, N,N-dimethylaniline or pyridine) or different linkers for C4 extension (1,4-substituated 1,2,3-triazole: nonlinear or arylalkyne: linear). The last panel shows that extending 28S with an appropriate extra moiety such as in 35S greatly increases the potency of the compound in this MAPK-dependent assay. 35S exerts a greater inhibitory effect at each concentration tested below 10 μM. Data are presented as mean values and error bars show SD (n = 3, parallel independent measurements). (*p < 0.05, **p < 0.01, ***p < 0.001; two-sided, unpaired t-test). Source data are provided as a Source Data file.
In further experiments MAPK-mediated transcription from the AP-1 promoter was monitored by using the AP-1 Reporter – HEK293 cell line containing an AP-1 promoter+luciferase gene cassette stably integrated into the genome. This cellular system gives a luminescence-based output and reports on MAPK activities within the cell where ERK, p38 and JNK activation and AP-1 factor phosphorylation all contribute to the final output, in which the action of the MAPKs is synergistic30. PMA-induced activation of AP-1 promoter mediated transcription, a biological output mediated by MAPK phosphorylated AP-1 dimers comprised of endogenous c-Jun, ATF and c-Fos transcription factors, was indeed abrogated not only by JNK inhibitors31 but also by ATP-competitive ERK or p38 inhibitors. Experiments with D-groove binding inhibitors demonstrated that the inhibitory capacity of the compounds was greatly affected by the nature of the extension (N,N-dimethylaniline or pyridine; compare 34S vs 35S), stereochemistry (compare 32S vs 32 R), and the chemical link (1,4-substituated 1,2,3-triazole: nonlinear or arylalkyne: linear; compare 32S vs 34S; see Table 4) (Fig. 6b). These latter all seem to affect the capacity of the more complex molecules to contact additional surface residues and/or to interfere with contacts required for partner protein binding. From a functional standpoint, differential impact on activator kinase (e.g., MKK1), deactivating phosphatase (e.g., MKP3), and/or substrate (e.g., RSK) binding will likely have different effects on MAPK controlled signaling events in the cell.
To address the selectivity of one of the MAPK D-groove binding compounds, 28S was further tested in experiments on potential off-targets: 1) ribosomal S6 kinase (RSK), which is a downstream component of ERK signaling pathways and had been targeted via its ATP-pocket cysteine by a covalent warhead32, 2) an ATP-pocket cysteine in ERK2 (C166) formally shown to be covalently targeted by hypothemycin (a natural polyketide)33, and 3) cyclin-dependent kinase 7 (CDK7)34, which is a close relative of MAPKs. These proteins contain an accessible and sensitive cysteine that had formally been targeted by covalent inhibitors, however the model compound (28S) used in 10 µM concentration left off-target cysteine related kinase activity intact, while it efficiently blocked MAPK D-groove cysteine dependent functionality as expected (Supplementary Fig. 22). In summary, these results demonstrate that the double-activated, sterically crowded cyclohexenone/pentenone scaffold can be easily and orthogonally elaborated further toward compounds that modulate MAPK signaling network activity by influencing MAPK D-groove mediated binding.
Discussion
There have been several attempts to find small molecules capable of binding in the MAPK D-groove and thus to block MAPK signaling35–38. Interestingly, the D-groove of all MAPKs (e.g., ERK, p38 and JNK) contain a conserved surface cysteine and covalent modification of this residue may directly interfere with MAPK-partner protein binding. Indeed, it has been shown that a compound, which had originally been identified as a JNK D-groove binder, covalently bound to this cysteine in ERK239. Here, we show that all MAPKs contain a D-groove cysteine that can be targeted by Michael acceptor-based warheads.
Despite the historic successes of covalent drugs, found mainly through serendipitous discoveries in the past, there was skepticism against these compounds due to their potential cross-reactivity with other proteins. However, after reevaluating their risks and benefits, there is a recent resurgence of covalent drugs approved for the clinic40,41. Furthermore, safety concerns may also be mitigated by the development of warheads whose reactivity is fine-tuned so that they could form the cysteine adduct only on the desired target, and/or off-target reactions are mitigated by the reversible nature of the covalent bond forming warhead42.
The highly reducing environment in the cell would first appear to limit the usability of thiol Michael addition reactions in drug design (equivalent of 1–10 mM reduced glutathione, GSH)43. However, the reactivity of α,β-unsaturated carbonyl containing electrophiles with thiols can be fine-tuned and these Michael acceptors may mediate the formation of irreversible or reversible covalent bonds with cysteines in proteins11. First, we showed that a weakly reactive acrylamide (intrapeptide) warhead may form an irreversible covalent bond with the D-groove cysteine on MAPKs provided that it is properly positioned in the context of a high-affinity binding D-groove targeting peptide. This finding prompted us to assemble a Michael acceptor molecular library comprised of building blocks originally used for the synthesis of terpenoid natural products. Gratifyingly, some of the compounds blocked D-motif peptide binding, and we later focused on the sterically crowded (frustrated), double-activated cyclohexenone scaffold because such compounds were functional in the presence of millimolar amounts of reduced glutathione (GSH). SAR analysis suggested that electron withdrawing (e.g., ester) groups as substituents at C2 and C4 of the cyclohexenone ring together with a hydrophobic moiety linked to C2 was critical for low micromolar binding. Such a multi-functionalized cyclohexenone scaffold seemed to have the appropriate reactivity towards the MAPK D-groove cysteine but its hydrophobic substituent group (e.g., tert-butyl or benzyl) was also needed to direct the electrophile into the hydrophobic pocket next to the D-groove cysteine. Because of enhanced steric crowding, “frustration”, next to the reactive center of the Michael acceptor and the presence of additional electron withdrawing groups, covalent bond formation with cysteine residue is reversible and is more selective. We posit that these two structural elements are key factors so that the electrophilic warhead would be capable of finding the right target protein with a specific cysteine in a complex reducing environment such as in the inside of the cell.
Unexpectedly, we found that in addition to cysteine thiol, some of the cyclic warheads could shift the nucleophile preference and target the imidazole side chain of histidine. Histidine covalent adduct formation is far more exotic compared to cysteine targeting44,45. The imidazole side chain of a histidine residue located in the MAPK D-groove, next to the cysteine thiol, is an alternative nucleophile for reversible covalent adduct formation. Nucleophile choice for Michael addition appears to be governed by an interplay between protein surface topography and the chirality of the cyclic warhead structure.
The reversible nature of the thiol targeting warhead provides several advantages over irreversible warheads for biological tool design and potentially for drug development: resilience against GSH, tunable reactivity and residence time. The metabolic properties of some of the compounds with different cyclohexenone scaffolds were examined in a preliminary pharmacokinetic study31 (using rat hepatocyte culture and blood plasma). Hepatic clearance of 3 was found to be intermediate but plasma stability due to high esterase activity was low, since this compound contained esters at C2 and C4. Gratifyingly, experiments with other more complex compounds showed that steric congestion at C4 makes the compounds much more stable in plasma. Fortunately, one of the great advantages of the presented double-activated cyclohexenone/pentenone scaffolds is the relative ease of their synthetic elaboration, and this is a great asset in engineering their metabolic properties in the future.
The electronic properties and thus the reactivity of the Michael acceptor of the cyclic warhead architecture can be tailored via a modular synthetic approach allowing the straightforward incorporation of different substituent groups. Additionally, organocatalytic synthetic approach allows efficient control over the stereochemistry of substituent groups at key positions next to C3. Variation of pendant substituents in C2 and/or C4 could be exploited in directing different moieties towards different pockets near the targeted cysteine to increase binding strength or to affect other binding properties, namely kinetics and specificity. The cyclic warhead structure seems to allow more selective chemical bond formation with cysteines and/or with other nucleophilic side chains (e.g., histidine) located in a specific chiral environment. Thus, a special substrate-directed stereocontrol is emerging as reflected by the stereogenic centers formed upon addition of cysteine and histidine moieties. In stark contrast, stereospecific synthetic control is not yet realized with open-chain warhead scaffolds and is expected to be far less straightforward (cyclic vs. acyclic stereocontrol).
Extension of the warhead scaffold towards the charged CD-groove increased binding affinity and the residence time of the inhibitor. This finding suggests that the cyclohexenone warhead can indeed be used as an anchor to develop MAPK inhibitors that target this key PPI surface. This was substantiated by results of cell-based tests in which larger small molecules—stereospecifically extended versions of the original cyclohexenone scaffold—displayed distinct capacities in affecting MAPK based cellular processes, such as MAPK controlled transcription, by interfering with MAPK-based binary binding to activators, substrates and/or phosphatases. We showed that D-groove binding compounds can selectively target the D-groove cysteine in a MAPK (see Supplementary Fig. 8), moreover, other compounds with different (ATP-competitive) directing groups with the same covalent warheads did not target the D-groove cysteine as expected31. Unfortunately, proteome-wide selectivity of sterically crowded, double-activated cyclohexenone/pentenone containing compounds cannot be easily tested because their reversible covalent binding poses a challenge for classical chemoproteomics. There is a need to develop an equilibrium binding based approach allowing unbiased off-target identification for reversible covalent binders.
Our initial motivation was the development of D-groove binding compounds but the reversible covalent thiol targeting capacity of the identified double-activated, sterically crowded (“frustrated”) cyclohexenone warhead prompted us to investigate if it may also be used to target other cysteines apart from the D-groove cysteine. To this end, we examined the possibility of replacing the widely used acrylamide (open-chain) warhead with our cyclohexenone/pentenone warheads in an ATP-competitive JNK inhibitor where a cysteine located next to the ATP-pocket of JNK was the nucleophilic target31. We found that the cyclic warhead scaffold can be successfully used in this different system too, suggesting a broader application for cyclic enone-based warheads.
Methods
Protein expression and peptide synthesis
MAPKs were expressed in E. coli and purified as described earlier20. Activated human ERK2, p38α, and JNK1 (used in the PhALC assay) for the biochemical assays were produced by co-expressing the MAPKs with constitutively active GST-tagged MAP2Ks in E. coli with bicistronic plasmids. Double phosphorylation of the activation loop of MAPKs was confirmed in western blots with anti-phospho MAPK-specific antibody and/or mass spectrometry. MAPKs were expressed with an N-terminal His6-tag that was cleaved off by the TEV protease after purification with Ni-NTA affinity resin, and then samples were further purified using ion-exchange chromatography (MonoQ, GE Healthcare). Dephosphorylated MAPKs were produced with GST-tagged λ-phage phosphatase and purified similarly (and were used for FP measurements and ERK2 for crystallization). Peptides from MAPK partner proteins (MKK6, RHDF1) and SynthD and SynthRevD peptides were described earlier19,20. These peptides bind to ERK2 and/or p38α. Based on the JNK interacting region of JIP1, a high-affinity JNK binding peptide was developed based on evolutionary sequence conservation information (evJIP1) observed across JIP1 vertebrate orthologs (from fish to human). Unlike other known weak JNK binding D-motifs, this peptide bound to JNK1 with submicromolar affinity20. Mutant versions of MAPKs were generated by using chemically synthesized DNA oligonucleotides with the two-step PCR-based mutagenesis protocol, and mutated proteins were expressed/purified as described above for wild-type proteins (Supplementary Table 3).
Peptides were chemically synthesized using solid phase peptide synthesis (on Rink Amid resin, PS3 peptide synthesizer, Protein Technologies) with Fmoc/tBu strategy and purified by RP-HPLC using a Jupiter 300 Å C18 column (Phenomenex). The quality of the peptides was verified by HPLC-MS (Shimadzu LCMS-2020).
Protein and peptide binding assays, library screen
Fluorescence polarization (FP) based protein-peptide and protein-protein binding experiments were carried out as earlier described20. Briefly, known MAPK docking groove binding peptides were N-terminally labeled by carboxyfluorescein (RHDF1: SLQRKKPPWLKLDIPS, evJIP1: PPRRPKRPTSLDLPSTPSL). All measurements with ERK2 or p38α were done with the RHDF1 peptide, while the evJIP1 peptide was used with JNK1. An exception to this was when the binding affinity of 8 was determined with ERK2 C161A/H125L where the pepMK2 reporter peptide (CF-IKIKKIEDASNPLLLKRRKK) was used. 50-100 nM labeled reporter peptide was mixed with MAPKs in a concentration to achieve ~50-80% complex formation. The unlabeled competitor (peptide or small molecule) was added in increasing amounts and the FP signal was measured in a Cytation 3 (BioTek Instruments) fluorescence plate reader in black 384 well plates in 20 µL volume. The binding buffer contained the following: 20 mM Tris pH = 8.0, 100 mM NaCl, 0.05 % Brij35, which was supplemented with 1 or 10 mM GSH when indicated. The Ki for each competitor was determined by fitting the data to a competition binding equation46. Titration experiments were carried out in triplicates, and the average FP signal was used for fitting the data in Origin 2018 (OriginLab, USA). The small molecule library screen was carried out similarly, but the competitor concentration was set to 100 µM. In the direct binding experiments or in the peptide displacement competitive assay setup, FP measurements were taken, unless otherwise indicated, after ~30 min incubation time.
SPR measurements
For surface plasmon resonance (SPR) measurements ERK2 was captured on a Biacore CM5 sensor chip by amine coupling using a Biacore S200 instrument (GE Healthcare). The SPR running buffer was the following: 1X PBS supplemented with 0.05% Tween 20, 0.2 mM TCEP, 5% DMSO. For p38α measurements, the protein was expressed with an N-terminal His10-tag and an NTA sensor chip was used for capturing the protein, and the SPR running buffer was the following: 10 mM Tris pH = 8.0, 300 mM NaCl, 0.2 mM TCEP, 0.05% Tween 20. For SPR measurements where the small molecule was the ligand (with a biotin tag), the molecules were immobilized on a Biacore CAP sensor chip, and the SPR running buffer was the following: 10 mM HEPES pH = 7.4, 150 mM NaCl, 0.05% Tween 20. All SPR measurements were done using a Biacore S200 instrument (GE Healthcare) at room temperature and included double referencing.
ITC measurements
p38α was injected from the syringe ( ~ 0.3 mL) into the measuring cell (1.4 mL) using a Microcal VP-ITC instrument (Malvern). The concentration of p38α in the syringe was 170 μM, 500 μM, 818 μM, 100 μM for pepMNK1 (19 μM), 28S (60 μM), N3-pepMNK1_C (50 μM), and 28S-pepMNK1_C (10 μM) measurements, respectively. For the measurement with the p38α C162A mutant the protein concentration in the syringe was 180 μM and the concentration of the peptide-chimera in the measuring cell was 30 μM. The buffer in the measuring cell was: 10 mM HEPES pH = 7.4, 150 mM NaCl, 10 mM TCEP (+1% DMSO in the case of 28S); bacterially expressed, purified recombinant p38α was dialyzed into the same buffer before injected into the measuring cell in 15 μL aliquots. The binding isotherms were fit to a “one set of sites” binding model using the Origin 2018 extension of the VP-ITC analysis software.
Mass spectrometric analysis
The molecular weights of MAPKs and their conjugates were identified using a Triple TOF 5600+ hybrid Quadrupole-TOF LC/MS/MS system (Sciex, USA) equipped with a DuoSpray IonSource coupled with a Perkin Elmer Series 200 micro LC system. Data acquisition and processing were performed using Analyst TF software version 1.7.1 (Sciex, USA). Chromatographic separation was achieved by Thermo Beta Basic C8 (50 mm × 2,1 mm, 3 µm, 150 Å) HPLC column. The sample was eluted in gradient elution mode using solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in ACN). The initial condition was 10% B followed by a linear gradient to 55% B by 12 min, to 95% B by 3 min, 15 to 17.5 min 95% B was retained; and from 17.5 to 18 min back to initial condition with 10% eluent B and retained from 18 to 20 min. Flow rate was set to 0.4 ml/min. The column temperature was 40 °C and the injection volume was 5 µl. UV-VIS spectrometer was used at 254 nm wavelength. Nitrogen was used as the nebulizer gas (GS1), heater gas (GS2), and curtain gas with the optimum values set at 30, 30 and 35 (arbitrary units), respectively. Data were acquired in positive electrospray mode in the mass range of m/z = 300 to 2500, with 1 s accumulation time. The source temperature was 350 °C and the spray voltage was set to 5500 V. Declustering potential value was set to 80 V. Peak View SoftwareTM V.2.2 (Sciex, USA) was used for deconvoluting the raw electrospray data to obtain the neutral molecular masses.
The off-target thiol or histidine adducts form in a dynamic fashion at physiological pH (~7.4) based on earlier experiments, but unexpectedly we were able to detect the GSH- or histidine-small molecule adducts with 3R and 6R,R by LC-MS. We posit that the reason for this is due to the highly acidic conditions of the LC separation step (0.1% formic acid, pH ~ 2.7) under which adduct dissociation is greatly slowed down. The molecules (50 μM) were mixed with 10 mM GSH or N-acetyl-L-histidine in PBS (pH ~7.4) and subjected to LC-MS analysis immediately (lag time ~ 5 min) or after 1 h of incubation. The LC-MS procedure was the following. Chromatographic separation was performed on a Kinetex F5 (100 × 3 mm, 3 µm, 100 Å) HPLC column (Phenomenex, USA). Gradient elution was applied by using water containing 0.1% formic acid (eluent A) and acetonitrile containing 0.1% formic acid (eluent B). The initial composition of solvents was 5% of eluent B. A linear gradient was applied by 4.5 min to reach 95% of eluent B. This was held for 1 min and the initial solvent composition was set back by 0.3 min followed by a 2.2 min equilibration part. The flow rate was 0.6 mL/min. 2 µL of samples were injected. The column temperature was set to 40 °C. Data were acquired in positive electrospray mode. The mass range was set to m/z 100–1500 with an accumulation time of 0.5 s. Source conditions were: spray voltage: 5000 V, nebulizer gas (GS1), drying gas (GS2) and curtain gas (CUR) values were set to 40, 40 and 45 arbitrary unit, respectively. The declustering potential (DP) was set to 80 V and source temperature was set to 450 °C. The resolution of the instrument was above 25000 over the entire mass range.
NMR experiments on covalent adduct formation
NMR experiments were conducted on a Varian NMR System spectrometer operating at 600 MHz. Off-target adduct formation was analyzed through the reaction between the respective Michael acceptor (3R or 6R,R) and the corresponding model compound (BME or His-Test) by 1H NMR. His-Test was synthesized from N-acetyl-L-histidine, referred to as Ac-His and used in the LC-MS experiment earlier, to neutralize its carboxyl group: N-acetyl-L-histidine methyl ester. His-Test was much better suited to carry out the 1H NMR experiments because the pH could be kept constant during titration experiments. All reagents were dissolved and the experiments were carried out in PBS (D2O, pH ~7.2) with 75% DMSO. Details of the measurements and the assignment of the spectra are described in Supplementary Note 1.
Electronic circular dichroism (ECD) analysis of Michael adduct formation with BME
ECD measurements were carried out with a J-815 spectropolarimeter (Jasco). Raw spectra were corrected with the background ECD spectra and normalized for the concentration of the substrate and cell length. 0.025 M stock solutions of the substrate and BME were prepared in acetonitrile and PBS, respectively. The individual samples for ECD measurements were prepared by dilution of the substrate stock solution by using varying amounts of the BME stock solution and the PBS buffer. The final substrate concentration was set to 0.25 mM for the aliphatic substrate 3R, 6R,R and 18R and 0.05 mM for substrate 19’S containing an aromatic moiety. The acetonitrile content of the samples was 1% in each case. The background ECD samples for the processing were similarly prepared using only acetonitrile instead of the substrate stock solution. Further details of the measurements and the quantitative analysis to obtain the Keq (M−1) of the reaction, from which Kchem was calculated by taking the inverse of this value, is described in Supplementary Note 2.
X-ray structure determination
ERK2 (15 mg/ml) was mixed with the SynthRevD-36_opt peptide or small molecules in a 1:1.5 stoichiometric ratio in the presence of 2 mM AMP-PNP. Complexes of ERK2 with peptides and small molecules were crystallized in a custom PEG Screen containing 6 different molecular weight PEGs under four different pH conditions (5.5, 6.5, 7.5 and 8.5). Diffraction quality crystals grew in 10–30% PEG1000, PEG3350, PEG6000 or PEG8000 in pH = 5.5, 7.5 or 8.5. The crystallization conditions for the crystals from which the final crystallographic data was collected were the following: ERK2-SynthRevD-36_opt: 20% PEG8000, 0.1 M Na-citrate pH = 5.5; ERK2-8S: 30% PEG1000, 0.1 M HEPES pH = 7.5; ERK2-8R: 30% PEG1000, 0.1 M Na-citrate pH = 5.5; ERK2-3R: 20% PEG8000, 0.1 M Na-citrate pH = 5.5; ERK2-3S: 10% PEG8000, 0.1 M Na-citrate pH = 5.5; ERK2-12R: 30% PEG3350, 0.1 M Tris pH = 8.5; ERK2-12S: 20% PEG8000, 0.1 M HEPES pH = 7.5; ERK2-6R,R: 10% PEG8000, 0.1 M Tris pH = 8.5.
Crystals were frozen in mother liquor supplemented with 20-25% glycerol. Crystallographic data were collected at ESRF beamline ID23-1 at a wavelength of 1 Å or at the EMBL PETRA III beamline P14 (ERK2- 6R,R and ERK2-3S; wavelength: 0.9763 Å). The crystal structures of all ERK2 complexes were solved by molecular replacement using the ERK2 structure (from PDB ID: 4NIF) as the search model with PHASER47. The crystallographic model was refined using PHENIX48. Building of the cysteine adducts and the unnatural residue was done using JLigand 1.0.4049.
Computational methods
The logP value of the compounds were calculated using the RDKit Descriptor Calculation Node in Knime (https://www.knime.com/rdkit). Docking of the hypothetical 28S-methyl-triazole compound was done by using the flexible side-chain method in AutoDock4 to predict the binding mode of the covalent complex. The docking simulations were conducted using the default settings of the Lamarckian Genetic Algorithm. The ranking of ligand docking conformations was based on a semi-empirical force field-derived scoring function50.
In vitro kinase assay (PhALC) and in vitro IC50 determination
The Phosphorylation-Assisted Luciferase Complementation assay (PhALC) was used to measure MAPK activity in vitro for fast and cost-effective identification/characterization of any compound blocking MAPK activity27. The principle of this assay is the following: the general MAPK phosphorylation target motif (S/TP) is positioned C-terminal from a MAPK binding D-motif and this SENSOR construct is fused with the small fragment of the luciferase enzyme (NanoBiT small subunit, deep sea shrimp, Oplophorus glacilirostris, Promega). In the Recognition Construct (RC), the Pin1 WW domain binding specifically to the phosphorylated MAPK target motif is fused with the large fragment of the luciferase enzyme (NanoBiT large subunit)29. Upon SENSOR phosphorylation the RC will bind the SENSOR and triggers the assembly of the luciferase enzyme which will produce photons as it turns over its substrate (coelenterazine). Both constructs are produced with an N-terminal maltose binding protein (MBP) and a C-terminal histidine tag for high-yield bacterial expression and for simple affinity-resin purification. The two purified constructs (SENSOR and RC) were mixed with activated MAPKs, the reaction was started by injecting ATP into the reaction mix containing the luciferase enzyme substrate coelenterazine, and the luminescence signal was monitored over time. The PhALC assay was used as a semi-high throughput, microplate compatible biochemical assay to obtain IC50 values of any compounds blocking MAPK activity. In more details: The PhALC constructs (SENSOR and RC) were expressed in E. coli using a modified pET expression vector (pET-MBP) and standard bacterial expression conditions in LB where the expression of proteins was induced by the addition of 0.2 mM IPTG at 25 °C for 4 h. Proteins were purified on Ni-NTA resin which was followed by another affinity purification step using maltose resin. RC and SENSOR were typically used in 1 µM concentrations with 1–10 nM double-phosphorylated MAPKs produced and purified as described earlier19. The coelenterazine concentration was 200 µM and the reaction was started by adding 0.1 mM ATP. The kinase assay buffer contained 20 mM HEPES pH = 7.4, 100 mM NaCl, 0.05 % IGEPAL, 5 mM MgCl2. The luminescence signal was monitored up to 30 min and the slope at the linear range (typically up to 5 min) was calculated based on linear regression. The p38 and ERK SENSOR contained the CNK3 (LKKEKSAILDLYIPPP) or MEF2A D-motif (SRKPDLRVVIPP), and the JNK sensor had the more JNK-specific pepPDE4B motif (GDGISRPTTLPLTTLP)19,20. SENSORs contained the same MAPK phosphorylation target sequence compatible with WW domain binding: VPRTPVS. This sequence motif was positioned C-terminal from the D-motif in D-SENSORs, separated by a flexible linker (HMGSGSSGGSSGSGSVD). For IC50 determination the competitor was added in increasing concentrations, incubated ~30 min in the reaction mix before starting the luminescence measurements and the normalized slope of the luminescence signal was fitted to the dose-response equation in Origin2018 (OriginLab, USA).
Cell culture
For MAPK-activated and AP-1 promoter mediated transcription measurements, 20,000 Reporter AP-1–HEK293 cells (BPS Bioscience, #60405) were seeded into a 96-well plate in 100 µl DMEM (Gibco) supplemented with 10% FBS. After cells became adherent (confluency: 80%) the inhibitors were added in Opti-MEM (Gibco) and following 2 h of incubation cells were stimulated with 6 ng/ml PMA solution for 6 h. The luminescence signal was read out using a BioTek Cytation 3 microplate reader after adding freshly prepared Steadylite Plus solution (PerkinElmer) according to the manufacturer’s protocol.
For EGF stimulation measurements, 25,000 HeLa cells (ATCC, CCL-2) were seeded into 48-well plates and were grown till 80% confluence (typically 24 hrs) in 200 µl DMEM supplemented with 10% FBS and were serum-starved (0% FBS) for 16 h before pretreatment with inhibitors (10 μM) for 1.5 h. Cells were stimulated by 100 ng/mL EGF for the indicated time, the media was removed, and cells were lysed in 70 µl 1X SDS-PAGE loading buffer, 10 µl was loaded onto 4–20% Mini-PROTEAN TGX Precast gels (Bio-Rad) or 5–15% Tris-glycine SDS-PAGE gels, and gels were blotted to nitrocellulose membrane. The DMSO control contained 0.05% DMSO, an equal amount to inhibitor treated cells. Western-blot results were analyzed using Odyssey CLx imaging system (LI-COR) and fluorescently labeled secondary antibodies: IRDye 680RD (Goat anti-mouse IgG; LI-COR #925-68070; 1:10000) or IRDye 800CW (Goat anti-rabbit IgG, Li-Cor #926-32211; 1:5000). The primary antibodies were the following: anti-p44/42 MAPK (ERK1/2) (L34F12) Mouse mAb (Cell Signaling #4696; 1:3000 dilution; referred to as ERK antibody), anti-phospho-p44/42 MAPK (Thr202/Tyr204) Rabbit Ab (Cell Signaling #9101; 1:3000 dilution; referred to as ppERK), anti-phospho-p90RSK (S380) (D3H11) Rabbit mAb (Cell Signaling 11989; 1:2000 dilution; referred to as pRSK), anti-α-tubulin (Sigma #T6199; 1:10000 dilution, referred to as TUB).
NanoBiT protein-protein interaction assay
For luciferase complementation NanoBiT assays (Promega), HEK293T cells (ATCC, CRL-3216) were transfected with LgBiT and SmBiT containing plasmids using Lipofectamine 3000 in Opti-MEM (Gibco). cDNAs of human proteins were sub-cloned into LgBiT and SmBiT expression vectors: ERK2, p38α and JNK1 were expressed as LgBiT fusions and partner protein constructs had SmBiT fusion tags (see Supplementary Table 3). Cells were serum-starved overnight in DMEM (Gibco), and luciferase activity was measured in 96-well white plates in a luminescence plate reader (Cytation 3, BioTek) at 37 °C after the addition of 20 μM Coelenterazine h (301-10 hCTZ; Prolumen Ltd, USA) at the maximum level of luminescence (3-8 min). The MKK1_D0, MKK6_D0 and MKK7_D0 constructs lacked the first N-terminal 31, 19 or 62 residues, respectively. For longer real-time NanoBiT signal measurements, cells were handled similarly but the Nano-Glo Live Cell Assay System (with the furimazine luciferase substrate; Promega, #N2011) was used according to the manufacturer’s protocol to improve the stability of the luminescence signal over time.
Characterization and synthesis of small molecules
All reactions were carried out using oven-dried glassware and anhydrous solvents unless noted otherwise. Flash silica chromatography was performed on silica gel (ZEOprep 60 25-40 µm, ZEOCHEM) with the indicated eluents. Reverse phase chromatography was performed using a gradient method on a Gemini® 5 µm C18 110 Å column (H2O:MeCN = 95:5 (0.1 % HCOOH) to 100% MeCN (0.1 % HCOOH)). Thin-layer chromatography was performed on silica plates (Kieselgel 60 F254 Merck). Compounds were visualized by UV (254 nm) or KMnO4 or p-anisaldehyde staining. Chemical shifts are referenced to the residual solvent signals (CHCl3: δ = 7.26 ppm for 1H, δ = 77.0 ppm for 13C). Data are reported as follows: chemical shifts (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dt = doublet of triplets, dq = doublet of quartets, dqd = doublet of quartet of doublets, td = triplet of doublets, tt = triplet of triplets, tdt = triplet of doublet of triplets), coupling constants (Hz), and integration. All spectra were recorded with the standard spectrometer pulse sequences and settings using a Varian 300 MHz, 500 MHz, or 600 MHz spectrometer. The enantiomeric excess of the products was determined on a chiral stationary phase HPLC (Daicel Chiralpak ID 250 × 4.6 mm, 5 µm column) using Hexane:iPrOH = 9:1 as eluent. All starting materials were purchased from Aldrich, TCI, or Fluorochem and used without further purification unless stated otherwise. Anhydrous THF and toluene were distilled from sodium/benzophenone, while anhydrous DMF and DMSO were kept under Ar on 4 Å molecular sieves. The synthetic procedures for compounds 1–35, their derivatives, unnatural amino acids (36–38; used to make some artificial peptides), 39–40 (open-chain acryl- or cyanoacrylester compounds), and His-Test are described in Supplementary Note 3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
We are thankful to Zoltán Dobi, Kristóf Hegedüs, Máté Gyöngyösi and Péter Angyal for contributing compounds to the Michael acceptor containing molecular library, to Máté Répási for assistance in synthesis, and to beamline scientists at ESRF for their support at data collection, and to Isabel Bento (EMBL-Hamburg) for some of the X-ray data collection. T. K. thanks István Fábián for the helpful discussion on the determination of the equilibrium constants by ECD. A.R. is grateful to the EU-OPENSCREEN ERIC (www.eu-openscreen.eu) for stimulating conferences, meetings and logistical support. This work was supported by the National Research Development and Innovation Office (NKFIH) grants (KKP 126963 awarded to A.R.), VEKOP-2.3.3-15-2016-00011, and by the Hungarian Academy of Sciences (KEP-10/2019).
Author contributions
Á.L.P., D.B., T.S., and A.R. conceived of the project idea, made intellectual contributions, designed experiments, analyzed and interpreted the data, and wrote the manuscript. D.B., K.O., O.E. and SZ. V. synthesized compounds, and S.M. produced peptide-compound chimaeras. Á.L.P. crystallized all protein-small molecule complexes. Á.L.P. and K.P. developed the PhALC assay and characterized the compounds by biochemical measurements. A.A. carried out all cell-based assays, and K.A. performed all SPR measurements. G.T. carried out the NMR measurements required for the analysis of the off-target adducts, while S.B.K. and T.K. performed the ECD measurements and analysis. G.G. and P.S. collected X-ray data and determined the crystal structures. T.I. and K.N. carried out MS measurements.
Peer review
Peer review information
Nature Communications thanks Thomas Brett, Matthias Wymann and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
Crystal structures of ERK2-SynthRevDCOV, -8R, -8S, -12R, -12S, -3R, -3S, and -6R,R are deposited in the Protein Data Bank with accession codes 8PSR, 8PSW, 8PSY, 8PT0, 8PT1, 8PST, 8PT5 and 8PT3, respectively. The following X-ray structures are available from the PDB: 4NIF, 4FMQ and 2Y9Q. Source data are provided in this paper.
Competing interests
A.R., T.S., Á.L.P., D.B., A.A., P.S., K.A., O.E., T.I., and S.V. are inventors in a patent application pending approval (PCT/HU2023/050079) on the use of cyclic designer scaffolds for the covalent targeting of proteins via Michael addition. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ádám Levente Póti, Dániel Bálint.
Change history
10/28/2024
In this article, compounds 34S and 34R in Table 4 did not display properly. The original article has been corrected.
Contributor Information
Tibor Soós, Email: soos.tibor@ttk.hu.
Attila Reményi, Email: remenyi.attila@ttk.hu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52574-1.
References
- 1.Gehringer, M. & Laufer, S. A. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J. Medicinal Chem.62, 5673–5724 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Liu, Q. et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol.20, 146–159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller, R. M. & Taunton, J. Targeting protein kinases with selective and semipromiscuous covalent inhibitors. Methods Enzymol.548, 93–116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao, Z., Liu, Q., Bliven, S., Xie, L. & Bourne, P. E. Determining cysteines available for covalent inhibition across the human kinome. J. Med. Chem.60, 2879–2889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaikuad, A., Koch, P., Laufer, S. A. & Knapp, S. The cysteinome of protein kinases as a target in drug development. Angew. Chem. - Int. Ed.57, 4372–4385 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Jackson, P. A., Widen, J. C., Harki, D. A. & Brummond, K. M. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem.60, 839–885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Resnick, E. et al. Rapid covalent-probe discovery by electrophile-fragment screening. J. Am. Chem. Soc.141, 8951–8968 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Backus, K. M. et al. Proteome-wide covalent ligand discovery in native biological systems. Nature534, 570–574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grossman, E. A. et al. Covalent ligand discovery against druggable hotspots targeted by anti-cancer natural products. Cell Chem. Biol.24, 1368–1376.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berdan, C. A. et al. Parthenolide covalently targets and inhibits focal adhesion kinase in breast cancer cells. Cell Chem. Biol.26, 1027–1035.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnan, S. et al. Design of reversible, cysteine-targeted michael acceptors guided by kinetic and computational analysis. J. Am. Chem. Soc.136, 12624–12630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavoie, H., Gagnon, J. & Therrien, M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol.21, 607–632 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Canovas, B. & Nebreda, A. R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol.22, 346–366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeke, A., Misheva, M., Reményi, A. & Bogoyevitch, M. A. JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev.80, 793–835 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smalley, I. & Smalley, K. S. M. ERK inhibition: a new front in the war against MAPK pathway-driven cancers? Cancer Discov.8, 140–142 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Tanoue, T., Maeda, R., Adachi, M. & Nishida, E. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J.20, 466–479 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang, C. I., Xu, B. E., Akella, R., Cobb, M. H. & Goldsmith, E. J. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell9, 1241–1249 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Sammons, R. M., Ghose, R., Tsai, K. Y. & Dalby, K. N. Targeting ERK beyond the boundaries of the kinase active site in melanoma. Mol. Carcinog.58, 1551–1570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeke, A. et al. Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol. Syst. Biol.11, 837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garai, A. et al. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal.5, ra74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, T. et al. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol.19, 140–154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding, Y. et al. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem.122, 102–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gersch, M., Kreuzer, J. & Sieber, S. A. Electrophilic natural products and their biological targets. Nat. Prod. Rep.29, 659–682 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Berkes, B. et al. Expedient and diastereodivergent assembly of terpenoid decalin subunits having quaternary stereocenters through organocatalytic Robinson annulation of nazarov reagent. Chemistry22, 18101–18106 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Martin, G., Angyal, P., Egyed, O., Varga, S. & Soós, T. Total syntheses of dihydroindole aspidosperma alkaloids: reductive interrupted fischer indolization followed by redox diversification. Org. Lett.22, 4675–4679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar, G. S. et al. Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc. Natl Acad. Sci. Usa.115, 4655–4660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Póti, Á. L. et al. Phosphorylation-assisted luciferase complementation assay designed to monitor kinase activity and kinase-domain-mediated protein-protein binding. Int. J. Mol. Sci.24, 14854 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanoue, T., Adachi, M., Moriguchi, T. & Nishida, E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol.2, 110–116 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol.11, 400–408 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Whitmarsh, A. J. & Davis, R. J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. (Berl.).74, 589–607 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Bálint, D. et al. Reversible covalent c-Jun N-terminal kinase (JNK) inhibitors targeting a specific cysteine by precision-guided Michael-acceptor warheads. Nat. Commun. 10.1038/s41467-024-52573-2. [DOI] [PMC free article] [PubMed]
- 32.Cohen, M. S., Zhang, C., Shokat, K. M. & Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science308, 1318–1321 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rastelli, G., Rosenfeld, R., Reid, R. & Santi, D. V. Molecular modeling and crystal structure of ERK2-hypothemycin complexes. J. Struct. Biol.164, 18–23 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Kwiatkowski, N. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature511, 616–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hancock, C. N. et al. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. J. Med. Chem.48, 4586–4595 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Stebbins, J. L. et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl Acad. Sci. Usa.105, 16809–16813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller, C. J., Muftuoglu, Y. & Turk, B. E. A high throughput assay to identify substrate-selective inhibitors of the ERK protein kinases. Biochem. Pharmacol.142, 39–45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sammons, R. M. et al. A novel class of common docking domain inhibitors that prevent ERK2 activation and substrate phosphorylation. ACS Chem. Biol.14, 1183–1194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaoud, T. S. et al. Modulating multi-functional ERK complexes by covalent targeting of a recruitment site in vivo. Nat. Commun. 10, 5232 (2019). [DOI] [PMC free article] [PubMed]
- 40.Singh, J., Petter, R. C., Baillie, T. A. & Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov.10, 307–317 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Boike, L., Henning, N. J. & Nomura, D. K. Advances in covalent drug discovery. Nat. Rev. Drug Discov.21, 881–898 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradshaw, J. M. et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol.11, 525–531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem.263, 17205–17208 (1988). [PubMed] [Google Scholar]
- 44.Jakob, C. G. et al. Novel modes of inhibition of wild-type isocitrate dehydrogenase 1 (IDH1): direct covalent modification of His315. J. Med. Chem.61, 6647–6657 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Joshi, P. N. & Rai, V. Single-site labeling of histidine in proteins, on-demand reversibility, and traceless metal-free protein purification. Chem. Commun. (Camb.).55, 1100–1103 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Wang, Z. X. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett.360, 111–114 (1995). [DOI] [PubMed] [Google Scholar]
- 47.McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr.40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams, P. D. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. Biol. Crystallogr.66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lebedev, A. A. et al. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D. Biol. Crystallogr.68, 431–440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bianco, G., Forli, S., Goodsell, D. S. & Olson, A. J. Covalent docking using autodock: two-point attractor and flexible side chain methods. Protein Sci.25, 295–301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystal structures of ERK2-SynthRevDCOV, -8R, -8S, -12R, -12S, -3R, -3S, and -6R,R are deposited in the Protein Data Bank with accession codes 8PSR, 8PSW, 8PSY, 8PT0, 8PT1, 8PST, 8PT5 and 8PT3, respectively. The following X-ray structures are available from the PDB: 4NIF, 4FMQ and 2Y9Q. Source data are provided in this paper.