

Abstract
In this study, we measured the frequency of revertants of a cytopathic strain of the duck hepatitis B virus that bears a single nucleotide substitution in the pre-S envelope protein open reading frame, resulting in the amino acid substitution G133E. Cytopathic virus mixed with known amounts of a genetically marked wild-type virus was injected into ducklings. Virus outgrowth was accompanied by a coselection of wild-type and spontaneous revertants during recovery of the ducklings from the acute liver injury caused by death of the G133E-infected cells. The frequency of individual revertants in the selected noncytopathic virus population was estimated by determining the ratio of each revertant to the wild-type virus. Spontaneous revertants were found to be present at frequencies of 1 × 10−5 to 6 × 10−5 per G133E genome inoculated. A mathematical model was used to estimate that the mutation rate was 0.8 × 10−5 to 4.5 × 10−5 per nucleotide per generation.
Duck hepatitis B virus (DHBV) belongs to the family Hepadnaviridae, a small group of enveloped viruses which cause persistent liver infections and replicate their DNA genomes through reverse transcription of an RNA intermediate (26). Because of the involvement of reverse transcription, the replication of the viral genome is assumed to be error prone and to give rise to a high degree of genetic variation. In humans, HBV can cause acute and chronic liver disease, and genetic variation is believed to be related to the clinical outcome of hepadnavirus infection (for a review, see reference 8). In recent years, it has become more fully appreciated how the presence of a complex mixture of variants in the virus population within a single host, the quasispecies, may enable rapid adaptation to changing environments (5), in particular to selection forces such as antiviral drugs (1), vaccines (4), immunomodulatory substances (20), and other changes in the host's immune response, leading to the emergence of various resistance and escape mutants (25).
Major mechanisms contributing to the complexity of the viral quasispecies are the generation of errors during viral genome replication, environmentally induced mutations, and recombination. Of these, errors in replication are thought to constitute the greatest source for generating variants. The frequency of replication errors in hepadnaviruses is a function of the fidelities of three polymerization reactions which occur during the life cycle in two different compartments. In the nucleus, covalently closed circular DNA (cccDNA)-templated positive-strand RNA synthesis is carried out by cellular RNA polymerase (RNA pregenome synthesis), and in the viral core particles, RNA-templated negative-strand DNA synthesis and DNA-templated positive-strand DNA synthesis are both carried out by reverse transcriptase. The relative contributions of the various enzymes involved in different polymerization steps to the overall mutation rate of the virus are not known.
In spite of the reported widespread occurrence of genetic variation in hepadnavirus infection, little is known about the natural mutation rate of HBV, and most estimates are extrapolations from that of RNA viruses and retroviruses, which have been more intensively studied (6, 9, 19, 23). Experimental approaches used to measure variation in hepadnaviruses have generally focused on estimating the variation that has occurred within infected individuals over a known period of time (2, 3, 7, 10, 22). Two difficulties in interpreting such measurements are the lack of knowledge about selective pressures that may have influenced the frequency of any particular variant in the virus population and the history of the dynamic state of virus replication during the interval considered. Since the frequency of any unselected variant in the population will reflect the number of generations of virus replication that have occurred, and fitness selection depends on the dynamics of the infection (29), it is difficult to relate measurements of mutant frequency to the mutation rate per generation.
In this study, we determined the frequency of mutations at several individual sites in the DHBV genome by assaying the occurrence of spontaneous reversion of a single-base-change cytopathic variant of DHBV (14, 15, 17) in an experimental infection of young ducklings. Because revertants could be coselected in parallel with a known amount of wild-type (WT) virus (16), we were able to measure the frequency of revertants relative to the added WT genomes. We used reasonable assumptions to construct a mathematical model for relating the frequency of revertants to the mutation rate per generation.
MATERIALS AND METHODS
Plasmids.
The DHBV type 16 (DHBV-16) WT genome was a head-to-tail-dimer construct derived from that sequenced by Mandart et al. (18) and cloned into pSP65. The G133E mutant viral genome carried a single-amino-acid codon change from glycine to glutamic acid at residue 133 (G133E) in the large envelope protein (14) produced by site-directed mutagenesis of a guanylate residue at nucleotide (nt) 1198 to adenylate (G1198A). In addition, this mutant genome carried a silent nucleotide substitution of cytidylate at nt position 1178 to adenylate (C1178A), resulting in the destruction of a SmaI site present in the WT genome. The presence or absence of the SmaI site was used to determine the virus variant in the duckling serum. Neither nucleotide substitution altered the DHBV polymerase open reading frame, which overlaps the pre-S region.
Cell culture and transfection.
WT and mutant viruses were produced by transfection of the chicken hepatoma cell line LMH (13) with respective DHBV DNA as previously described (27). Supernatants from the transfected LMH cells were collected daily from day 3 to day 10 posttransfection, clarified by low-speed centrifugation, and stored at 4°C before use. The titer of virus of the supernatants was determined by Southern blot hybridization with a 32P-labeled riboprobe following selective DNA extraction from enveloped virus particles (14). Hybridization was quantitated by phosphorimaging (Molecular Dynamics, Sunnyvale, Calif.). Virus used for injection was concentrated by precipitation with 10% (wt/vol) polyethylene glycol 8000 (Sigma, St. Louis, Mo.) (15).
Animals and infections.
One-day-old White Pekin ducklings obtained from Metzer Farms (Redland, Calif.) were used in experiment 1, and ducklings from Privett Hatchery (Portales, N. Mex.) were used in experiment 2. Ducklings testing negative for DHBV DNA by dot hybridization were infected by intravenous injection of a volume of 300 μl of a suspension of DHBV on day 4 posthatching. All birds were fed an unrestricted diet and received humane care throughout the study in accordance with guidelines issued by the National Institutes of Health.
In experiment 1, six groups of four ducklings each were infected with inocula of G133E at a dose of 108 viral genomes per bird either alone or combined with WT DHBV added in ratios of 10−2 to 10−6. One group of three ducklings was infected with WT virus only, at a dose of 108. Three birds that served as uninfected controls received no inoculum. In experiment 2, ducklings were infected with a mixture of 107 genomes of WT DHBV combined with 107 genomes of G133E, E133G, D129E, G133K, Q135R, Q135H, or T140I. The birds were monitored for weight change (experiment 1) and viremia (experiments 1 and 2) for up to 25 days after infection in experiment 1 and up to 11 days after infection in experiment 2. DHBV DNA in the serum was detected by dot hybridization using a 32P-labeled riboprobe specific for the detection of the viral minus strand (24) and quantified by phosphorimaging.
PCR amplification, determination of virus variants, and cloning of DHBV DNA from PCR products.
Amplification was performed on DNA derived from the equivalent of 2.5 μl of serum by mixing serum samples with an equal amount of 10 mM Tris-HCl (pH 7.4)–1 mM EDTA and incubating the mixture at 96°C for 10 min (21). The entire pre-S region was amplified by using sense primer P1 and reverse primer P2. A restriction site for ApaI (New England BioLabs, Beverly, Mass.) was introduced into the 5′ end of P1 (underlined below). In addition, this primer was biotinylated. Sequences of the primers were as indicated: P1 5′-CGCGGGGCCCAAGAGCATTTCCTA, nt 584 to 603, sense; P2, 5′-CCGATTAGGCCAGCTAGTAT, nt 1324 to 1305, reverse. A 741-bp fragment was produced from the viral DNA template by using 2.5 U of Taq DNA polymerase (Promega, Madison, Wis.) for 35 cycles of 15 s at 95°C, 15 s at 55°C, and 30 s at 72°C in a GeneAmp PCR System 9700 (Perkin-Elmer Cetus, Norwalk, Conn.).
The PCR product from the WT DHBV contained a single SmaI site yielding two digestion products, one of 592 and one of 149 bps, however, this restriction site was absent in PCR products derived from the mutant G133E genome. A pGEM-7Zf(+) vector (600 ng) (Promega), carrying a single SmaI site in its sequence, was introduced in the digestion reaction with SmaI as an internal control for complete digestion.
For cloning, PCR amplification products from serum were digested with the restriction enzymes ApaI and KpnI (both from New England BioLabs) and ligated into an appropriately cut pGEM-7Zf(+) vector (Promega) by using a rapid DNA ligation kit (Roche Molecular Biochemicals, Indianapolis, Ind.). Recombinant plasmids were cloned into DH5α bacterial cells (GIBCO/BRL, Gaithersburg, Md.).
DNA sequencing of PCR-amplified products and cloned DHBV DNA.
For direct sequencing, the biotinylated PCR products were adsorbed to streptavidin-coated beads (Dynabeads M-280; Dynal A.S., Oslo, Norway) and washed according to the manufacturer's instructions with the help of a magnetic particle concentrator (Dynal catalogue no. 120.04; Dynal A.S.). The nonbiotinylated minus-strand products were released from the beads by denaturation in 0.1 N NaOH, and the biotinylated plus-strand products were sequenced on the beads with 5 pmol of P2 and 35S-dATP using the T7 Sequenase version 2.0 dGTP reagent kit (Amersham Life Science, Cleveland, Ohio).
The sequence of cloned PCR products was determined by using primers P3 (5′-TTGGCCTGCTGGGGCGGGAA, nt 992 to 1011, sense), P4 (5′-GGTGGTTTCCGGTGGTCTTT, nt 1136 to 1117, reverse), M13reverse (Stratagene, La Jolla, Calif.), and M13(−20)forward (Stratagene).
Cloning of PCR-derived DNA into replication-competent genomes.
Six candidate revertant mutations identified by sequencing of PCR amplification products of the pre-S region were cloned into the DHBV-16 WT genome. PCR products amplified with P1 and P2 (see above) were purified by phenol extraction, precipitated with ethanol, and digested with the restriction enzymes PflF1 and XhoI or KpnI (all from New England BioLabs) respectively. Subsequently, they were subjected to low-melting-point agarose electrophoresis (GIBCO/BRL) and visualized by ethidium bromide staining. The bands of interest were cut out under low-wave UV illumination and, after heating to 65°C, directly used for ligation into the DHBV-16 WT genome construct which had been digested with the appropriate restriction enzymes to remove the corresponding WT pre-S sequences. Ligation was performed with T4 DNA ligase (GIBCO/BRL) overnight at 14°C. DH5α bacterial cells (GIBCO/BRL) were used for cloning.
RESULTS
The purpose of this study was to determine the spontaneous mutation rate during an experimental infection of young ducklings with the avian hepadnavirus DHBV. We used the reversion rate of a cytopathic variant of DHBV as a measure of mutation rate. Noncytopathic revertants are easily detected, since we previously showed that they are rapidly selected in vivo (16). In addition, we sought to identify the sites of the various mutations in the DHBV genome that cause reversion of the cytopathic phenotype.
Strategy and rationale of the experiment.
Previous studies have shown that infection of ducklings with a variant of DHBV of the viral large envelope (pre-S) domain, namely, G133E, was cytopathic for the birds, leading to severe acute but transient noninflammatory liver injury (17). Recovery from acute liver disease was shown to be accompanied by the appearance of noncytopathic spontaneously arising revertant viruses (16). At the molecular level, hepatocellular injury was preceded by elevated levels of DHBV cccDNA in the hepatocytes, and enhanced cytoplasmic staining for viral capsid proteins was observed within the cells. A single amino acid change of glycine to glutamic acid at position 133 (G133E) in the pre-S domain of DHBV was directly responsible for these phenomena, as the production of new viral cccDNA in hepatocytes is regulated by the viral pre-S protein (27). It is believed that WT pre-S protein directs capsids containing relaxed circular DNA, the precursor of cccDNA, into a pathway for assembly of virus particles, which are secreted from the infected cells, excluding their utilization for cccDNA synthesis in the nuclei, possibly through direct interaction with the capsid protein of the virus itself (14).
In previous work, we showed that authentic WT DHBV added to the G133E inoculum could be detected among the population of noncytopathic viruses that grew out during recovery from acute liver injury (16). Hence, WT virus, when added in known amounts to the G133E inoculum, could be used as an internal reference for measuring the frequency of spontaneous mutants that behave like WT virus during outgrowth. Noncytopathic mutants were expected to carry a same-site mutation or various second-site suppressor mutations in the pre-S envelope gene and could be identified by sequencing. We assumed that the frequency at which any such mutant arises reflects the mutation rate at that nucleotide.
The amount of each spontaneous revertant in the final noncytopathic virus population was compared with the amount of reference WT virus to determine approximately how frequently that revertant was generated and replicated compared to parental virus during outgrowth of the parent virus in vivo. The frequency of each revertant was calculated as the ratio of that revertant to WT virus in the final noncytopathic virus population, multiplied by the ratio of WT virus to G133E virus in the inoculum. Because multiple rounds of infection occur in vivo, and the reversion frequency is a cumulative measure of the revertants present in the inoculum plus those generated in each round of infection, the reversion frequency is dependent on the amount of virus replication during spread of infection and replacement (see Appendix).
Viremia and weight gain after mixed infection.
In experiment 1, a total of 24 ducklings (six groups consisting of four birds each) were infected at 4 days of age with inocula of 108 viral genomes of G133E, either alone (group 1) or mixed with WT DHBV at a frequency of 10−2 to 10−6 WT genomes per G133E genome (groups 2 to 6, respectively). One group of three ducklings was infected with 108 genomes of WT virus only (group 7). Three birds that served as uninfected controls received no inoculum (group 8). Ducklings were bled and weighed daily from day 3 to day 14 and at 21 and 25 days postinfection. The presence of viral DNA in the serum was monitored by PCR, and the respective viral DNA titers were determined by dot hybridization and phosphorimaging.
The amount of DHBV DNA (mean log molecules per milliliter) detected in serial bleeds collected over the course of experiment 1 from all groups of birds is shown in Fig. 1. Viremia achieved an early maximum already by day 6 postinfection in WT virus-infected birds (group 7), whereas virus levels in G133E-infected birds (group 1) or mixed-infection groups (groups 2 to 6) peaked at slightly later times. Viremia in the serum declined or fluctuated over time after its peak in all groups of ducklings. Virus was not detected in any birds from the uninfected control group 8.
FIG. 1.

Viremias in eight groups of ducklings. Viremias were determined by dot hybridization and phosphorimaging. The minimum viremia detectable by this method was approximately 2 × 106 genomes per ml. The mean log value for all positive samples within a group for each time point was plotted.
The mean body weights for all eight groups of ducklings are shown in Fig. 2. As previously reported, infection with G133E resulted in a retardation of the normal rapid growth. Coinfection with WT virus caused a moderation of this effect, seemingly in proportion to the amount of WT virus included in the inoculum. This moderating effect may be due to protection of hepatocytes infected with WT virus from damage caused by G133E. The transient retardation of growth in G133E-infected ducklings reflects the previously demonstrated hepatocytopathic effect of the mutant virus, and the resumption of normal weight gain is due to the replacement of the cytopathic virus population with noncytopathic viruses (16).
FIG. 2.

Body weights in eight groups of ducklings. All birds in each group were weighed at the indicated times, and the mean body weight at each time point was plotted.
Outgrowth of WT and putative revertant viruses in mixed infections with G133E and WT virus.
To see whether recovery from liver disease reflected by the resumption of normal growth was primarily due to outgrowth of WT virus or to spontaneous revertants in mixed-virus-infected birds (groups 2 to 6), we carried out PCR amplifications of the DHBV region which carries the genetic tag and G133E mutation and assayed for the presence (WT virus population) or absence (G133E and/or spontaneous revertant population) of the SmaI site at position 1178. As seen in Fig. 3, the PCR products obtained from the serum of ducklings from groups 5 and 6 lacked any detectable SmaI cleavage. These virus populations therefore consisted predominantly of spontaneous revertants and G133E virus with no detected WT internal standard. In contrast, in serum samples from birds infected with larger amounts of WT internal standard added to the inoculum (groups 2, 3, and 4), SmaI-digestible viral DNA appeared during the course of the experiment in amounts that were directly related to the presence of WT virus in the inoculum. These results suggested that, by 21 days postinfection, coincident with recovery from acute liver injury, the major virus population found in the serum of ducklings shifted from the G133E genotype to the WT genotype (group 2) mixed with spontaneous revertant genotypes (groups 3 to 6). Accordingly, the frequency of all spontaneous revertants would be approximately equal to the proportion of WT reference virus in the inoculum that produced an equal mixture of WT and revertant virus in the ducklings at 21 days. (After melting and reannealing during the final cycles of PCR, only 25% of double-stranded products would contain SmaI sites in both strands and be digested with SmaI.) In this experiment, the inoculum most closely producing such a mixture was that containing WT virus at a frequency of 10−4.
FIG. 3.

PCR assay of the emergence of WT virus in ducklings with mixed infections. Serum samples from individual infected ducks, represented in separate lanes, were amplified by PCR. The products were each mixed with 600 ng of pGEM-7Zf(+) DNA and digested with SmaI to detect the appearance of WT internal reference genomes. The upper band (C) is the control added to each reaction mixture to monitor for complete digestion. The double bands represent the undigested PCR product of G133E-derived genomes (G) and the digested product of WT internal reference genomes (W).
To confirm the presence of potential spontaneous revertants and WT internal reference genomes in the serum of different animals, we directly sequenced the PCR amplification products obtained from birds of groups 2, 4, and 6 by day 21 postinfection. The results obtained from one animal in each of the respective groups are shown in Fig. 4. Consistent with the data shown in Fig. 3, no WT internal reference was present in the direct sequence from a bird of group 6, as indicated by the absence of a WT-specific band (C) at the SmaI site (CCCGGG). In groups 2 and 4, both the WT-specific band (C) and the G133E-specific band (G) could be seen in the mass sequence, with the G band being enriched in the sequence from group 4 birds. No putative spontaneous revertants, indicated by minor bands marked with arrows, were detectable in the direct sequence of the PCR amplification product in the bird from group 2 (the nucleic acid sequence at position 1198 of the pre-S region showed either WT GGA for glycine or cytopathic mutant GAA for glutamate). A population of various potential spontaneous revertants was visible in the direct sequences from birds of groups 4 and 6.
FIG. 4.

Direct sequence of PCR products derived from representative birds in groups 2, 4, and 6. Sequencing reactions were performed on biotinylated PCR products as described in the text. The sequence ladder runs from bottom to top in the plus-strand direction. The positions of the SmaI site present in the WT internal reference and codon 133 are indicated. Arrows mark sites of potential reverting mutations. aa, amino acid.
Nature and location of mutations in the pre-S region of spontaneous revertant viruses.
In order to resolve the individual spontaneous revertant sequences in G133E-infected ducklings, we cloned and sequenced the C-terminal region of the pre-S domain obtained from PCR products of four mixed-virus-infected birds from group 4 at various time points postinfection. In one bird (no. 440), pre-S sequences were analyzed at multiple times during infection. In a second bird (no. 438), the only viremic serum sample available was from 5 days postinfection, and all subsequent samples were negative by PCR. A total of six libraries of cloned pre-S sequences were prepared, and up to 31 separate sequences were determined for each library. As listed in Table 1, a spectrum of amino acid changes and corresponding codon changes was observed in the libraries, with the libraries from 3 and 5 days postinfection showing little heterogeneity, while libraries prepared from later time points contained various amounts of the internal reference WT virus and low amounts of the cytopathic virus G133E. The remaining sequences in the libraries from 14 and 21 days postinfection contained the G133E-specific genetic tag accompanied by changes at one residue (86 of 93) or occasionally a second residue (14 of 86). In cases where mutations occurred at two sites, one of these mutations was usually a unique example (one exception, E133K + Q135R) and was considered to be possibly introduced by PCR. This interpretation is consistent with the observed frequency of single mutations in the WT internal reference genomes sequenced (1 of 17), which is not significantly different.
TABLE 1.
Pre-S genotypes of viruses from group 4 infected ducks
| Bird No. | Days p.i.b | Viral genotypea | Frequency | Codon change |
|---|---|---|---|---|
| 440 | 3 | G133E | 24/24 | None |
| 440 | 14 | G133E | 1/31 | None |
| WT | 1/31 | None | ||
| E133G | 1/31 | GAA→GGA | ||
| E133K | 18/31 | GAA→AAA | ||
| E133K + S136S | 1/31 | GAA→AAA + TCT→TCC | ||
| E133K + Q121R | 1/31 | GAA→AAA + CAG→CGG | ||
| E133K + K123R | 1/31 | GAA→AAA + AAG→AGG | ||
| E133K + G164S | 1/31 | GAA→AAA + GGT→AGT | ||
| E133K + Q135R | 1/31 | GAA→AAA + CAG→CGG | ||
| Q135H | 1/31 | CAG→CAC | ||
| Q135H | 1/31 | CAG→CAT | ||
| Q135H + Q145R | 1/31 | CAG→CAT + CAG→CGG | ||
| P130L + P151Q | 1/31 | CCA→CTA + CCA→CAA | ||
| V150V | 1/31 | GTG→GTA | ||
| 440 | 21 | G133E | 2/17 | None |
| WT | 4/17 | None | ||
| E133G | 2/17 | GAA→GGA | ||
| E133K | 1/17 | GAA→AAA | ||
| D129E | 3/17 | GAT→GAG | ||
| D129E | 2/17 | GAT→GAA | ||
| Q135H | 2/17 | CAG→CAT | ||
| Q135R | 1/17 | CAG→CGG | ||
| 437 | 14 | G133E | 4/31 | None |
| WT | 5/31 | None | ||
| E133G | 7/31 | GAA→CGA | ||
| E133K | 6/31 | GAA→AAA | ||
| D129E | 3/31 | GAT→GAG | ||
| D129E + K161E | 1/31 | GAT→GAG + AAA→GAA | ||
| D129E + T140A | 1/31 | GAT→GAA + ACT→GCT | ||
| Q135R | 2/31 | CAG→CGG | ||
| T140I + S136S | 1/31 | ACT→ATT + TCT→TCA | ||
| P119L | 1/31 | CCT→CTT | ||
| 438 | 5 | G133E | 23/31 | None |
| WT | 0/31 | None | ||
| E133K | 7/31 | GAA→AAA | ||
| H141H | 1/31 | CAT→CAC | ||
| 439 | 21 | G133E | 0/31 | None |
| WT | 6/31 | None | ||
| WT + K154E | 1/31 | AAA→GAA | ||
| E133G | 1/31 | GAA→GGA | ||
| E133K | 12/31 | GAA→AAA | ||
| E133K + A149S | 1/31 | GAA→AAA + GCG→TCG | ||
| E133K + T155T | 1/31 | GAA→AAA + ACT→ACC | ||
| D129E | 1/31 | GAT→GAG | ||
| D129E | 1/31 | GAT→GAA | ||
| Q135R + L137H | 1/31 | CAG→CGG + CTC→CAC | ||
| T140I | 1/31 | ACT→ATT | ||
| Q135H | 2/31 | CAG→CAT | ||
| Q135H | 2/31 | CAG→CAC | ||
| Q135H + D128G | 1/31 | CAG→CAC + GAC→GGC |
All revertants with substitutions at positions other than 133 retain the G133E mutation.
p.i., postinfection.
A total of eight mutations were found in more than one library, and seven of these were considered to be candidate noncytopathic revertants. Of these, the codon 133 mutant, E133G, representing a true reversion to WT, was observed in samples analyzed from three different birds. Major second-codon mutations resulted in the substitutions D129E, Q135R, Q135H, and T140I. Two of these substitutions (D129E and Q135H) were generated by alternative codon changes (Table 1). The remaining commonly found mutation was the substitution E133K. This mutation, previously shown to retain a cytopathic phenotype, was always present in the population of revertants, especially during the earlier times, and decreased in frequency at later times (compare 14 and 21 days, bird 440). Because this mutant is cytopathic, shows a unique dynamics of appearance, and is unable to compete with true revertants (14; data not shown), we chose to exclude it from the analysis of revertant frequencies. We do not understand the behavior associated with the emergence of this mutant.
Functional tests of candidate pre-S mutants.
To test whether the growth rates of the candidate spontaneous revertants allowed them to expand in parallel with the WT internal reference, we determined their ability to compete with the WT virus in a mixed infection of ducklings. Pre-S regions containing the four observed codon changes resulting from the six mutations observed in more than one library were substituted into a WT genome and transfected into LMH cells. To verify the absence of additional mutations, the whole pre-S region between the sites used for cloning was sequenced in each construct. Virus stocks of revertants were prepared, and ducklings were inoculated together with WT virus in a 1:1 ratio (experiment 2). To estimate the ability of the cytopathic virus to compete with WT, G133E was also included among the combinations of mixed infections. The region of viral DNA containing the genetic tag was amplified from the earliest virus-positive serum sample from each bird, and the PCR products were subjected to SmaI digestion and agarose gel electrophoresis. The data obtained are shown in Fig. 5.
FIG. 5.

PCR assay for the genotype of virus in ducks with mixed infections. The first viremic serum sample from each group of ducklings infected with WT plus the indicated candidate revertant was amplified by PCR, and the products were mixed with pGEM DNA and digested with SmaI exactly as described for Fig. 3. The positions of the control (C), the G133E-specific (G), and the WT-specific (W) bands are indicated.
Although the cytopathic G133E virus had been previously shown to produce transiently higher levels of virus than did WT in a primary duck hepatocyte infection, its presence was not detected in the viremic serum samples when competed with WT. This result indicates that the cytopathic virus sustained a growth disadvantage during spread of infection in the liver in spite of its transiently elevated virus release. E133G virus, the same-site revertant, maintained a 1:1 ratio to WT virus during outgrowth in vivo as expected. The same was true for all second-site revertants tested in different birds, with the possible exception that the T140I second-site mutant was slightly decreased below WT after outgrowth. These results, to a first approximation, justify the use of the internal reference WT virus to estimate the revertant frequencies at the various sites detected, since the WT and revertant populations expand in parallel. The data also provide indirect evidence that the putative revertants were probably noncytopathic since the cytopathic phenotype of G133E (Fig. 5) and of E133K (data not shown) apparently caused a reduction in its rate of spread during infection in vivo.
Frequency of revertants.
For estimating the frequencies of the various spontaneous revertants, the ratios of seven spontaneous revertants which occurred most often in the viral population to the WT internal standard were calculated by dividing the total number of respective revertant clones observed by the number of WT clones observed in the libraries. The frequency of WT clones in this viral population was defined as 10−4 per G133E genome, since, in group 4, WT virus had been added to the inoculum at a frequency of 10−4. As seen in Table 2, the relative mutation frequencies for these specific sites in the DHBV genome varied from 1 × 10−5 to 6 × 10−5, with the same-site reversion being present at the highest frequency.
TABLE 2.
Relative frequencies of spontaneous revertants in group 4 ducklings
| Genotype | Amino acid change | No. of clones | Calculated frequency (10−4)a |
|---|---|---|---|
| Internal WT reference | |||
| 1198G | 133G | 17 | 1.0 |
| Revertants | |||
| A1198G | E133G | 11 | 0.6 |
| T1187G | D129E | 8 | 0.5 |
| T1187A | D129E | 4 | 0.2 |
| A1204G | Q135R | 5 | 0.3 |
| G1205T | Q135H | 6 | 0.4 |
| G1205C | Q135H | 4 | 0.2 |
| C1219T | T140I | 2 | 0.1 |
The ratio of spontaneous revertants to WT internal reference (10−4).
Types of mutations observed.
The mutations observed in the seven authentic revertants included three transitions and four transversions. In contrast, the remaining 16 sporadic mutations observed consisted of 12 transitions and 4 transversions. Whether the different ratios of transitions to tranversions in the two groups of mutants are significant and reflect two different mechanisms for generating mutations is not known (e.g., errors in replication versus PCR-generated errors). No deletions were detected, and this is not surprising because of the high degree of selection for function in this system.
DISCUSSION
In the previous studies cited, we demonstrated that infection of young ducklings with a cytopathic hepadnavirus, G133E, resulted in acute noninflammatory liver injury, followed within 2 to 3 weeks by recovery and restoration of normal liver histology. Recovery was attributed to the emergence of spontaneous noncytopathic revertants, which replaced the cytopathic virus in the liver. Thus, liver injury likely reflected the process of virus replacement, with G133E-infected cells dying and being replaced by revertant-infected cells. Infection with the cytopathic virus could therefore be considered to be transient, involving a limited amount of virus replication before elimination from the liver.
In this study, we determined how frequently revertant viruses were produced during the transient G133E replication that occurred in this system. The frequency of revertant production may be taken as a crude measure of the rate at which hepadnaviruses generate variants by spontaneous random mutation. To measure the frequency of reversion, we utilized a genetically marked WT virus as an internal reference. Since true revertants, by definition, would be indistinguishable from the WT reference in their growth and subsequent selection in the duckling, the ratio of such revertants to the WT reference could be used to calculate the total number of revertants generated.
For example, in an inoculum containing 108 G133E virus produced from transfected plasmid DNA, and a reversion rate at a single site of 10−5 per G133E genome synthesized, the corresponding revertant population would be 103 virus particles. After addition to the inoculum of 104 WT virus as the internal reference and injection into ducklings, each infecting viral genome would be converted to cccDNA and begin to produce progeny cccDNA by intracellular amplification and extracellular spread of infection. The increase in cccDNA derived from the input revertant genomes would exactly parallel the increase in cccDNA from the WT reference virus; however, revertant genomes would continue to be generated de novo as a by-product of the replication of G133E cccDNA. These new cohorts of revertants will be converted to cccDNA and replicate, also expanding in parallel with the WT internal reference population. As a result, as all three virus populations expand in vivo, and new cohorts of revertants are generated from the parental G133E, the ratio of revertant virus to WT virus will increase over time. As the proportion of replicating G133E genomes diminishes due to death of infected cells or overgrowth of WT and revertant cccDNA, new cohorts of revertants contributed by G133E replication become less significant, and eventually the ratio of revertants and WT virus stabilizes. The final ratio is a measure of the sum of the revertants present in the inoculum plus those generated in vivo and the total WT reference in the inoculum. Since the ratio of WT reference virus to G133E in the inoculum is known, the amount of each revertant per inoculated G133E genome accumulated over multiple generations required for spread of infection throughout the liver can be inferred. In our experiments, the frequency of revertants determined in this manner was between 1 × 10−5 and 6 × 10−5 per G133E genome inoculated.
The relationship between the frequency of revertants that we measured and the mutation rate, i.e., the probability of mutation per nucleotide per generation, is complex because we have measured the frequency of specific mutations over an unknown number of generations of G133E DNA replication. In a mathematical model presented in the Appendix, we have estimated that the final frequency of each specific revertant in our experiment is probably between 4- and 23-fold higher than the reversion rate at that site. For the same-site revertant, E133G, which grows at the same rate as does WT virus, the final revertant frequency was 6 × 10−5, implying that the rate of the mutation E133G was between 0.26 × 10−5 and 1.5 × 10−5 per generation. Accordingly, the error rate for all three possible substitutions at this position would be 0.8 × 10−5 to 4.5 × 10−5 errors per genome synthesized, or one error per 7 to 42 viral genomes synthesized. All specific second-site revertants were found at somewhat lower frequencies (Table 2), suggesting that these revertants may have replicated at a slightly reduced rate compared with that of the true WT revertant (E133G) and the reference virus, even though a reduced replication rate was not detected in the competition experiment. In support of this notion, the revertant T140I, which was present at the lowest frequency, appeared to undergo a weak negative selection in mixed infection with WT. In addition, error frequencies may differ at different positions and for specific substitutions (6, 9).
In this limited survey, substitutions at three sites in the vicinity of residue 133, namely, amino acids 129, 135, and 140, were able to revert the G133E cytopathic phenotype, presumably by restoring the normal function of the pre-S protein in virus release and cccDNA regulation. This result does not necessarily mean that the region inclusive of amino acids 129 to 140 is part of a functional site of interaction of the pre-S protein with its ligand, e.g., mature capsids. It is not known how the G133E mutation affects pre-S function in virus production and regulation of cccDNA synthesis. While the mutation may directly alter a functional site on the protein, it is also possible that the formation of a domain of the protein that contains the functional site is affected by the mutation, or that the G133E mutation causes a partial folding defect resulting in reduced amounts of properly folded protein. Each such defect could potentially be suppressed by substitution of amino acids neighboring the original mutation.
Interestingly, the glutamate at position 133 of the pre-S open reading frame, known to be cytopathic in the context of DHBV-16, is naturally present in most DHBVs of Chinese or Australian origin without apparent cytopathicity (28). These viruses all carry a lysine at position 135 of the pre-S protein and not a glutamine, as does DHBV-16. The selection for revertants with Q135R or Q135H as second-site mutations might therefore be anticipated if certain basic amino acid side chains at this position can suppress the phenotype resulting from a glutamate at position 133. Although the subsititution Q135K can theoretically be generated by a single nucleotide substitution (CAG→AAG), this change would introduce a stop codon (TCA→TAA) in the overlapping P open reading frame and would be inconsistent with viability. While P gene mutations may be generated during the reverse transcription step, genomes bearing these mutations were unlikely to be propagated during expansion of the revertant pool.
We do not understand the early emergence of the mutant E133K from the population of cytopathic viruses and its later decline seen for bird 440 (Table 1). We previously showed that this mutant is unable to compete effectively with WT virus in a mixed infection and that infection of ducklings with the E133K virus produces an acute liver injury similar to that seen with G133E (16). It is possible that initially this mutant had some undefined advantage over WT revertants that was confined to early stages of infection of the duckling and that the cytopathic phenotype caused rapid selection against it during later stages. Alternatively, the mutant may be generated by an extraordinarily high mutation frequency at position 1199. We have not adequately tested either of these possibilities.
ACKNOWLEDGMENTS
We thank Wengang Yang and Francis Lim for valuable suggestions and advice during the course of these experiments, Wengang Yang for inoculating ducklings in experiment 1, and William S. Mason, Fox Chase Cancer Center, for helpful discussions and critical reading of the manuscript.
This work was supported by grants CA42542 and CA84017 from the National Cancer Institute.
Appendix
In the experiments described in this paper, we infected ducklings with inocula containing three types of viruses: a cytopathic virus, G133E (G); a genetically marked WT reference virus (W); and spontaneous revertants of G133E (R) produced as a result of errors in replication. Here we examine how the spontaneous revertants are predicted to accumulate relative to the WT reference virus as the population of the G133E parent expands and is displaced by WT and revertant virus in the liver.
The model. Since the spontaneous revertants were produced in a transfection of plasmid G133E DNA, we assume that they arose in a single cycle of transcription and DNA synthesis and therefore that any particular revertant R1 was present in the G-containing inoculum in an amount R1(0) = G(0) × m1, where m1 is the probability of formation of a particular revertant per G133E genome synthesized and R1(0) and G(0) represent the fraction of liver cells infected by R1 and G, respectively, at t = 0. The complete collection of possible revertants at t = 0, R(0), can be represented by R(0) = R1(0) + R2(0) +…+ Rn(0) = G(0) × m, where m = m1 + m2 +… mn. During the spread of infection, assuming that m << 1, and neglecting back mutations, the increase in the three virus populations is described by the following set of equations:
 |
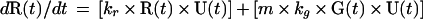 |
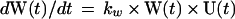 |
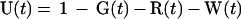 |
where U(t) is the fraction of the liver that is uninfected; kg, kr, and kw are the rate constants for synthesis of G, R, and W, respectively; and kd is the rate constant for loss of G by death of G133E-infected cells. In this model, the following assumptions are made. (i) Expansion of virus into the uninfected liver proceeds according to first-order kinetics. (ii) Death of G133E-infected cells proceeds according to first-order kinetics. (iii) Loss of G133E by cell death does not result in loss of W or R. (iv) Death of G133E-infected hepatocytes results in the immediate production of new hepatocytes susceptible to infection by any of the three viruses. (v) The size of the liver does not change. While none of these assumptions is strictly true, deviation of the model from the actual conditions does not substantially change the final outcome with respect to how the revertant-to-WT ratio changes over time. The reason for this is that the majority of the increase in the revertant-to-WT ratio occurs during the expansion of the G133E population into the uninfected liver, when the three virus populations are growing independently and there is an excess of uninfected cells.
Initial values. The values for G(t), R(t), and W(t) were calculated using numerical integration to visualize the expansion of the three virus populations in the liver and the replacement of G133E and to determine the final R/W ratio. In these calculations, we assumed that the inoculum of 108 viral genomes resulted in infection of 1:1,000 of the liver, i.e., G(0) = 10−3. The reversion rate, m, was arbitrarily set equal to 10−5 per genome, so that R(0) = 10−8. W(0) was set equal to R(0). Therefore, the ratio R(t)/W(t) indicates the increase in the frequency of revertants over the reversion rate due to the accumulation of revertants through multiple generations. The values for the various rate constants were chosen with the following rationale. Since the experiment in Fig. 5 shows that, in the viremia resulting from inoculation of ducklings with a 1:1 mixture of WT and G133E, G133E was not detected, we conclude that the net rate of virus spread of G133E when U ∼ 1 is less than that of WT virus. Therefore, (kg − kd) < kw. It is known that G133E-infected primary duck hepatocytes transiently produce cccDNA and virus at higher levels than do WT-infected cells (15) and therefore kg > kw. We set the rate constant for WT cccDNA synthesis equal to 1.6 day−1 (11, 12) and varied kg at between 1.6 and 8.0 day−1. Calculations were run with values of kd adjusted so that the net growth rate of G133E was between 0.5 and 0.9 times the WT growth rate. Values for G(t), R(t), and W(t) were determined over a period of 25 days. In all runs using the criteria listed above, the R(t)/W(t) ratio stabilized within 25 days. The results of a run using values of kg and kg − kd in the midrange of the values tested are shown in Fig. A1A, and a plot of the R(t)/W(t) ratio is shown in Fig. A1B.
FIG. A1.

Simulated growth of three virus populations in the liver of an infected duckling. (A) Numerical calculations of the fraction of liver infected with G133E (G), G133E revertant (R), and WT viruses (W). Initial conditions were m = 10−5 revertants/genome synthesized, G(0) = 10−3, R(0) = 10−8 [R(0) = m × G(0)], W(0) = 10−8, kw = 1.6 day−1, kr = kw, kg = 3.2 day−1, and kd = 1.92 day−1 [(kg − kd)/kw = 0.8]. Calculations were run over a simulated time of 25 days. (B) R/W ratio during the simulation in panel A.
A series of calculations varying kg and kg − kd as indicated above was performed, the final ratio R/W at 25 days was determined, and the values are plotted in Fig. A2. It can be seen that the increase in the ratio of revertant to the WT internal reference occurring after maximum expansion of the G133E population is directly dependent on both the relative growth rate of G133E, kg, and the net growth rate of G133E, kg − kd. The dependence on kg is due to the increased rate of revertant production. The dependence on the net growth rate, kg − kd, is due to the prolonged ability of G133E to compete with preexisting WT and revertants during the G133E expansion and thereby to continue to add significant cohorts of revertants to the replicating revertant pool. Moreover, these results also apply to the increase in ratio of each individual revertant to WT since the outcome of the model is relatively independent of the actual mutation rate and all revertants grow in parallel.
FIG. A2.

Accumulation of revertants as a function of the growth properties of G133E. Calculations similar to those shown for Fig. A1 were performed using the indicated growth rate constant (kg) and the net growth rate constant (kg − kd) for G133E. The rate constants were normalized to that of WT (kw). The ratio of revertant to WT virus (R/W) at 25 days postinfection is plotted. Since R(0)/W(0) = 1, the ratio R/W represents the excess revertant accumulation due to growth of G through multiple generations.
In the experiments, we measured the ratio of a specific revertant, E133G, to WT to be approximately 0.6 after replacement of G133E in the liver. To calculate limits on the inferred ratio of this revertant to WT in the inoculum, we may refer to Fig. A2, which indicates that the increase in the ratio over that in the inoculum would lie in the range between approximately 4- and 23-fold (3.97 to 22.8). Accordingly, we estimate the ratio in the inoculum to have been between 0.026 and 0.15. Since WT reference virus in the inoculum was present at a frequency of 10−4, the E133G revertant would have been present at a frequency of 0.26 × 10−5 to 1.5 × 10−5. We take this to be the specific reversion rate per viral genome synthesized. Because this reversion corresponds to a specific nucleotide substitution, the total error rate per nucleotide per genome would be three times the specific error rate or 0.8 × 10−5 to 4.5 × 10−5 per nucleotide per genome synthesized.
REFERENCES
- 1.Allen M I, Deslauriers M, Andrews C W, Tipples G A, Walters K A, Tyrrell D L, Brown N, Condreay L D. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Hepatology. 1998;27:1670–1677. doi: 10.1002/hep.510270628. [DOI] [PubMed] [Google Scholar]
- 2.Argentini C, La Sorsa V, Bruni R, D'Ugo E, Giuseppetti R, Rapicetta M. Hepadnavirus evolution and molecular strategy of adaptation in a new host. J Gen Virol. 1999;80:617–626. doi: 10.1099/0022-1317-80-3-617. [DOI] [PubMed] [Google Scholar]
- 3.Blackberg J, Kidd-Ljunggren K. Occult hepatitis B virus after acute self-limited infection persisting for 30 years without sequence variation. J Hepatol. 2000;33:992–997. doi: 10.1016/s0168-8278(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 4.Carman W F. The clinical significance of surface antigen variants of hepatitis B virus. J Viral Hepatitis. 1997;4:11–20. doi: 10.1111/j.1365-2893.1997.tb00155.x. [DOI] [PubMed] [Google Scholar]
- 5.Domingo E, Escarmis C, Menendez-Arias L, Holland J J. Viral quasispecies and fitness variations. In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. San Diego, Calif: Academic Press, Inc.; 1999. pp. 141–161. [Google Scholar]
- 6.Durbin R K, Stollar V. Sequence analysis of the E2 gene of a hyperglycosylated, host restricted mutant of Sindbis virus and estimation of mutation rate from frequency of revertants. Virology. 1986;154:135–143. doi: 10.1016/0042-6822(86)90436-8. [DOI] [PubMed] [Google Scholar]
- 7.Girones R, Miller R H. Mutation rate of the hepadnavirus genome. Virology. 1989;170:595–597. doi: 10.1016/0042-6822(89)90455-8. [DOI] [PubMed] [Google Scholar]
- 8.Gunther S, Fischer L, Pult I, Sterneck M, Will H. Naturally occurring variants of hepatitis B virus. Adv Virus Res. 1999;52:25–137. doi: 10.1016/s0065-3527(08)60298-5. [DOI] [PubMed] [Google Scholar]
- 9.Hahn C S, Rice C M, Strauss E G, Lenches E M, Strauss J H. Sindbis virus ts103 has a mutation in glycoprotein E2 that leads to defective assembly of virions. J Virol. 1989;63:3459–3465. doi: 10.1128/jvi.63.8.3459-3465.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hannoun C, Horal P, Lindh M. Long-term mutation rates in the hepatitis B virus genome. J Gen Virol. 2000;81:75–83. doi: 10.1099/0022-1317-81-1-75. [DOI] [PubMed] [Google Scholar]
- 11.Jilbert A R, Freiman J S, Burrell C J, Holmes M, Gowans E J, Rowland R, Hall P, Cossart Y E. Virus-liver cell interactions in duck hepatitis B virus infection. A study of virus dissemination within the liver. Gastroenterology. 1988;95:1375–1382. doi: 10.1016/0016-5085(88)90375-7. [DOI] [PubMed] [Google Scholar]
- 12.Jilbert A R, Miller D S, Scougall C A, Turnbull H, Burrell C J. Kinetics of duck hepatitis B virus infection following low dose virus inoculation: one virus DNA genome is infectious in neonatal ducks. Virology. 1996;226:338–345. doi: 10.1006/viro.1996.0661. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi T, Nomura K, Hirayama Y, Kitagawa T. Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Res. 1987;47:4460–4464. [PubMed] [Google Scholar]
- 14.Lenhoff R J, Summers J. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J Virol. 1994;68:4565–4571. doi: 10.1128/jvi.68.7.4565-4571.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenhoff R J, Summers J. Construction of avian hepadnavirus variants with enhanced replication and cytopathicity in primary hepatocytes. J Virol. 1994;68:5706–5713. doi: 10.1128/jvi.68.9.5706-5713.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenhoff R J, Luscombe C A, Summers J. Competition in vivo between a cytopathic variant and a wild-type duck hepatitis B virus. Virology. 1998;251:85–95. doi: 10.1006/viro.1998.9394. [DOI] [PubMed] [Google Scholar]
- 17.Lenhoff R J, Luscombe C A, Summers J. Acute liver injury following infection with a cytopathic strain of duck hepatitis B virus. Hepatology. 1999;29:563–571. doi: 10.1002/hep.510290236. [DOI] [PubMed] [Google Scholar]
- 18.Mandart E, Kay A, Galibert F. Nucleotide sequence of a cloned duck hepatitis B virus genome: comparison with woodchuck and human hepatitis B virus sequences. J Virol. 1984;49:782–792. doi: 10.1128/jvi.49.3.782-792.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mansky L M, Temin H M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mutimer D. Hepatitis B virus antiviral drug resistance: from the laboratory to the patient. Antivir Ther. 1998;3:243–246. [PubMed] [Google Scholar]
- 21.Netter H J, Chassot S, Chang S F, Cova L, Will H. Sequence heterogeneity of heron hepatitis B virus genomes determined by full-length DNA amplification and direct sequencing reveals novel and unique features. J Gen Virol. 1997;78:1707–1718. doi: 10.1099/0022-1317-78-7-1707. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto H, Imai M, Kametani M, Nakamura T, Mayumi M. Genomic heterogeneity of hepatitis B virus in a 54-year-old woman who contracted the infection through materno-fetal transmission. Jpn J Exp Med. 1987;57:231–236. [PubMed] [Google Scholar]
- 23.Pathak V K, Temin H M. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc Natl Acad Sci USA. 1990;87:6019–6023. doi: 10.1073/pnas.87.16.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pugh J C, Yaginuma K, Koike K, Summers J. Duck hepatitis B virus (DHBV) particles produced by transient expression of DHBV DNA in a human hepatoma cell line are infectious in vitro. J Virol. 1988;62:3513–3516. doi: 10.1128/jvi.62.9.3513-3516.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pult I, Chouard T, Wieland S, Klemenz R, Yaniv M, Blum H E. A hepatitis B virus mutant with a new hepatocyte nuclear factor 1 binding site emerging in transplant-transmitted fulminant hepatitis B. Hepatology. 1997;25:1507–1515. doi: 10.1002/hep.510250633. [DOI] [PubMed] [Google Scholar]
- 26.Summers J, Mason W S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 27.Summers J, Smith P M, Huang M, Yu M. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J Virol. 1991;65:1310–1317. doi: 10.1128/jvi.65.3.1310-1317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Triyatni M, Ey P, Tran T, Le Mire M, Qiao M, Burrell C, Jilbert A. Sequence comparison of an Australian duck hepatitis B virus strain with other avian hepadnaviruses. J Gen Virol. 2001;82:373–378. doi: 10.1099/0022-1317-82-2-373. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y Y, Summers J. Low dynamic state of viral competition in a chronic avian hepadnavirus infection. J Virol. 2000;74:5257–5265. doi: 10.1128/jvi.74.11.5257-5265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]