

SUMMARY
Somatic mutations in genes encoding components of the RNA splicing machinery occur frequently in multiple forms of cancer. The most frequently mutated RNA splicing factors in cancer impact intronic branchsite and 3’ splice site recognition. These include mutations in the core RNA splicing factor SF3B1 as well as mutations in the U2AF1/2 heterodimeric complex which recruit the SF3b complex to the 3’ splice site. Additionally, mutations in splicing regulatory proteins SRSF2 and RBM10 are frequent in cancer and there has been recent suggestion that variant forms of small nuclear RNAs (snRNAs) may contribute to splicing dysregulation in cancer. Here we describe molecular mechanisms by which mutations in these factors alter splice site recognition and how studies of this process have yielded new insights into cancer pathogenesis and the molecular regulation of splicing. We also discuss data linking mutant RNA splicing factors to RNA metabolism beyond splicing.
eTOC blurb
Mutations in genes encoding components of the RNA splicing machinery occur frequently in multiple cancers. This review describes molecular mechanisms by which cancer-associated mutations in splicing factors alter splice site recognition and how studies of this process have yielded new insights into cancer pathogenesis and the molecular regulation of splicing.
INTRODUCTION
Widespread efforts to catalog somatic mutations in human cancer genomes initiated in the mid-2000s and provided clear evidence for widespread dysregulation in the gene expression regulatory machinery in cancer cells. One of the most surprising findings to come from this effort was the discovery of identified frequent mutations in genes encoding core as well as accessory components of the RNA splicing factor machinery in a variety of cancer types1–5. RNA splicing, the process by which non-coding segments of precursor messenger RNA (mRNA) are excised and coding transcripts are ligated to give rise to mature coding mRNA is essential in encoding protein diversity and regulating transcript stability and protein-coding potential. While the potential for mutations in genes affecting RNA splicing of the same transcript in cis is well established, the concept of mutations in trans factors regulating RNA splicing was quite new at the time of discovery of mutations in the splicing machinery.
The most commonly mutated RNA splicing factors across cancers is the core spliceosome component SF3B1 (Splicing Factor 3b Subunit 1) which is mutated in up to 80% of patients with specific clinicopathologic subtypes of myelodysplastic syndromes (MDS), 8–10% of patients with chronic lymphocytic leukemia (CLL)1–5, ~25% of patients with non-cutaneous forms of melanoma6–8, and many epithelial tumors including 1.8% of patients with breast adenocarcinomas9 (Figure 1A). Specific amino acid residues in SF3B1 within its HEAT repeat domains are recurrently affected by heterozygous mis-sense mutations and most commonly occur at the K700 residue as K700E substitutions (Figure 1A). Interestingly, some mutations in SF3B1 outside of K700 residue are statistically significantly enriched in specific cancer types, suggesting allele-specific associations of these mutations and specific cancer types. For example, SF3B1 R625 mutations occur most frequently in melanomas6,7 and prolactinomas10, while G742D mutations are most enriched in CLL1–5. While the molecular impact of mutations in SF3B1 have been heavily studied, the molecular basis for allele-specific enrichment of specific SF3B1 mutations in cancers of particular types is not well understood currently.
Figure 1. Location and frequencies of commonly mutated RNA splicing factors in cancer.
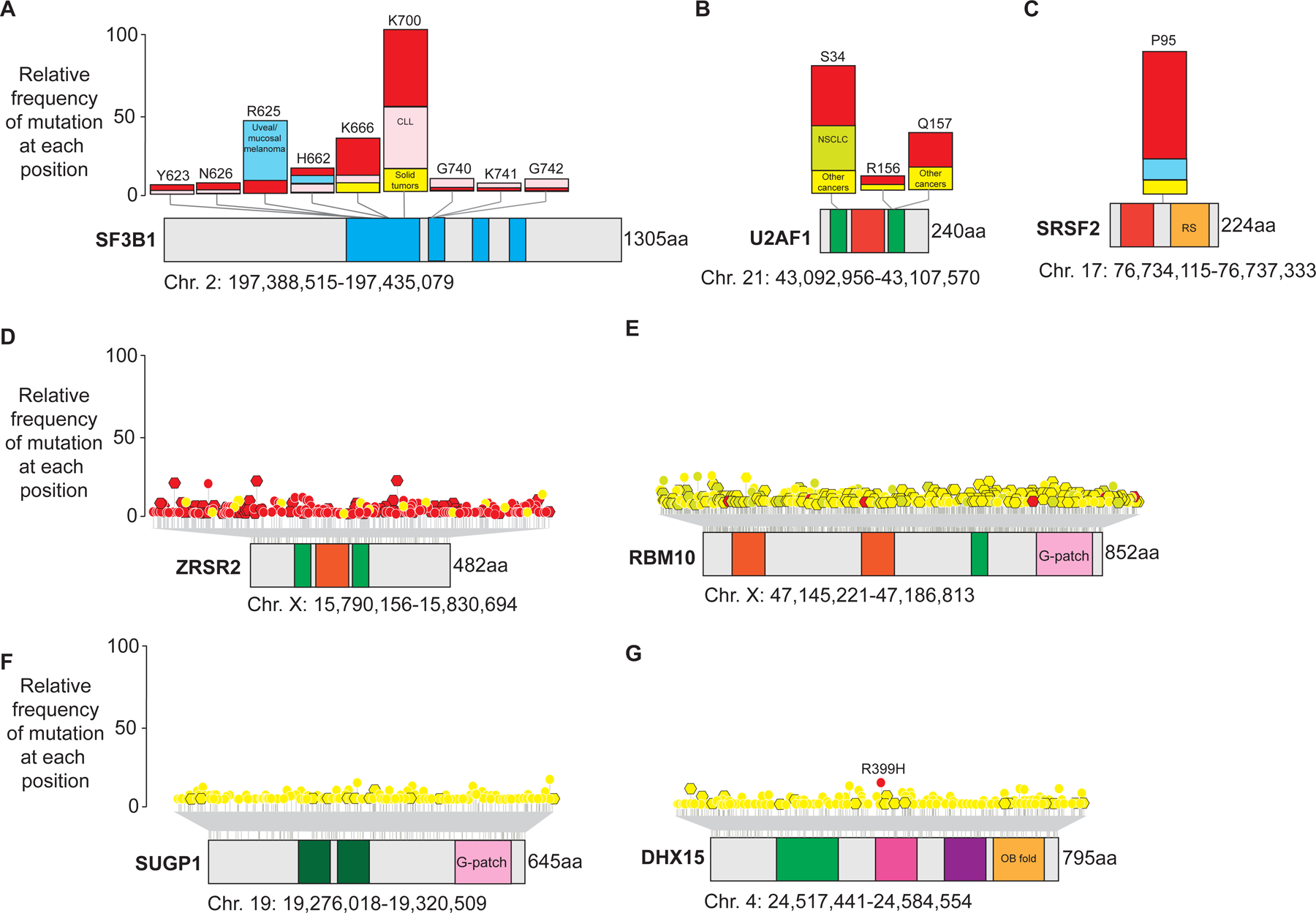
Shown are protein diagrams and annotated domains of commonly mutated RNA splicing factors across cancers. (A) SF3B1, (B) U2AF1, and (C) SRSF2 are shown in the top row and are most commonly impacted by heterozygous missense mutations at specific amino acid residues (“hotspot” mutations) as shown. The bar plots accompanying each mutant residue depict the proportion of patients with myeloid blood cancers (red), chronic lymphocytic leukemia (CLL; pink), uveal/mucosal melanoma (blue), or solid tumors (yellow) with mutations at each amino acid residue. Patients with non-small cell lung cancer center (NSCLC) are also shown. In contrast to the hotspot mutations in SF3B1, SRSF2, and U2AF1, (D) ZRSR2, (E) RBM10, and (F) SUGP1 are impacted by missense (circles with white outline) or nonsense/frameshift mutations (hexagons with black outline) throughout their open reading frames. Mutations in (G) DHX15 occur at R399 in acute myeloid leukemia but are otherwise spread throughout the coding region in other diseases. These mutations are color coded by disease as in the top row. Abbreviations: HA2: helicase associated 2; HEAT: Huntington, Elongation Factor 3, PR65/A, TOR domain; OB fold: oligonucleotide- or oligosaccharide-binding fold; RRM: RNA recognition motif; RS: Serine/Arginine rich domain; Zn: zinc finger domains.
Importantly, the earliest descriptions of SF3B1 mutations in myeloid neoplasms by Yoshida et al. noted their mutual exclusivity with heterozygous hotspot mutations in additional RNA splicing factors commonly mutated in the same diseases in U2AF1 and SRSF25 (Figure 1B–C). While the location of mutations in SF3B1 did not provide immediate clues into how these mutations may impact the function of SF3B1, mutations in U2AF1 nearly all cluster in one of its two zinc finger domains which is well-established to be essential for U2AF1 binding to the 3’ intron/exon boundary of pre-mRNA5,11,12 (Figure 1B). Mutations at the S34 residue in its first zinc finger domain account for nearly two-thirds of U2AF1 mutations and are prevalent in MDS (5–15% of patients), and AML (5–17%), and non-small cell lung adenocarcinomas (3%)5,13–15. Mutations at the Q157 residue of U2AF1 account for the majority of the remaining U2AF1 mutations and are more commonly seen in myeloid malignancies compared with other cancers14.
In addition to mutually exclusive heterozygous point mutations in SF3B1, SRSF2, and U2AF1, missense, nonsense, and frameshift mutations throughout the open reading frame of the gene encoding the RNA splicing factor ZRSR2 were also identified in the same patient cohorts with myeloid neoplasms (Figure 1D)5,16–18. Consistent with ZRSR2 mutations conferring loss-of-function, ZRSR2 is encoded on X-chromosome and there is a clear male predominance amongst ZRSR2 mutant patients. The unique genetic configuration of ZRSR2 mutations compared with other RNA splicing factors mutated in blood cancers is consistent with its particular role in splicing described below.
Additional RNA splicing factors mutated in cancer include mutations in SUGP119,20, DHX1521, and RBM1022–24 (Figure 1E–G). The impact of each of these on RNA splicing and disease pathogenesis are discussed below. Beyond mutations in the protein components of the spliceosome, a series of papers recently reported discovery of recurrent hotspot mutations in genes encoding non-protein coding RNA components of the spliceosome known as small nuclear RNAs (snRNAs) in several cancers.25–27 These have been reported to impact the U1, U2, and U11 snRNAs. For example, variants at nucleotide position three (r.3A>G) of the U1 or five (r.5A>G) in the U11 snRNA were found in approximately 50% of patients with sonic hedgehog (SHH) type medulloblastomas25. Conversely, the r.3A>C variant in the U1 snRNA mutation was present in multiple cancer subtypes, including CLL, other B cell non-Hodgkin lymphomas, hepatocellular carcinoma, and pancreatic adenocarcinoma26. More recently, c.28T>G variants within the U2 snRNA were reported in B cell malignancies, prostate, and pancreatic cancer27. Each of these snRNA variants occur at critical sites at which snRNAs interact with either pre-mRNA or other snRNAs and would therefore be expected to have clear impacts on splicing as described below.
MECHANISMS OF NORMAL RNA SPLICING
The spliceosome is a dynamic large ribonucleoprotein complex that involves the assembly of five small nuclear RNAs (snRNAs) and hundreds of proteins. Most introns are characterized by ‘GT’ dinucleotides at the 5’ splice site (5’ss) and ‘AG’ dinucleotides at the 3’ splice site (3’ss) and are spliced by the major spliceosome. Less than 0.5% of the human introns are spliced by a separate, less abundant RNA splicing machinery known as the minor spliceosome, which recognizes a conserved set of consensus sequences near the 5’ and 3’ss (Figure 2A). These minor introns contain GT-AG (75%) or AT-AC dinucleotides (remainder of introns) at their 5’−3’ ends.
Figure 2. Cis signals and trans factors required for RNA splice site recognition.

(A) Pre-mRNA contains introns and sequences embedded in the 5’ and 3’ ends of introns, the branch point site (BPS), and polypyrimidine (pY) tract are essential for the assembly of the spliceosome. The diagram shows the consensus sequence identified at these sites for the recognition and assembly of major spliceosome (left) and minor spliceosome (right), and the relative introns are called major / U2-type introns or minor / U12-type introns. U12-type introns can be further divided into GT-AG subtype and AT-AC subtype according to their splice site sequences. (B) Core components of complex E, A, and B of the major spliceosome (left), and the equivalent components of the minor spliceosome (right). The splicing factors undergoing frequent mutations in cancer mostly participate in splicing steps before the activation of the B complex. Compared to the factors involved in the early steps of the major spliceosome assembly, the regulation of minor spliceosome is less explored. (C) Categories of alternative splicing events: exon skipping, mutually exclusive exons, alternative usage of splice sites, and retained introns. The black arrows indicate the canonical splicing of the introns while the colored arrows indicate the alternative splicing events.
Each of the five snRNAs in the major spliceosome (the U1, U2, U4, U5 and U6 snRNAs) binds a set of protein factors to form a small nuclear ribonucleoprotein (snRNP) particle. These five snRNPs constitute the core components of the spliceosome, while additional auxiliary factors such Sm, Lsm, SR, and hnRNP proteins are required for splicing as well. The additional RNA splicing factors have been comprehensively reviewed recently28. In the minor spliceosome, the five snRNAs are U11, U12, U4atac, U5, and U6atac and most protein factors associated with them are shared between the major and minor spliceosome (Figure 2B). The U1 and U2 snRNAs interact with the proteins of their specific RNPs and then facilitate recognition of the 5′ss and branch site of major introns, respectively, via Watson-Crick base pairing. Similarly, the U11 snRNA associates with the 5’ss of minor introns and recruits U11 snRNP for splicing of minor introns.
In recent years, a number of studies have reported spliceosome cryo-EM structures, providing detailed insight on the precise regulation, assembly, and cycling of splicing factors across an RNA splicing reaction (reviewed in29,30). Different snRNAs and snRNPs base-pair and bind the 3’ss, 5’ss, the branchpoint sequence (BPS), and the polypyrimidine (pY) tract within the intron through several steps to conduct splicing. Spliceosome assembly starts with U1/U11 recognizing the 5’ss, SF1 binding to the BPS, and the U2 auxiliary factor (U2AF) complex binding to the pY tract and 3’ss. SF1 is then displaced by U2/U12 snRNPs. These initial steps in spliceosome assembly are crucial to determine both the inclusion/exclusion of introns and the boundaries of mRNA sequences to be spliced out of the mature RNA. The resulting distinct patterns of RNA splicing choice yield different RNA splicing event outcomes which are categorized as exon skipping, retained intron, alternative 3’ splice site, alternative 5’ splice site, and mutually exclusive exon splicing events (Figure 2C). Alternative RNA splicing in this manner allows the generation of many different mature RNA isoforms from a same pre-mRNA sequence. Additionally, RNA splicing is an important regulator of gene expression as splicing determines the usage and location of premature termination codons within pre-mRNA.
The further stepwise assembly of the spliceosome complexes (named sequentially as E, A, pre-B, B, Bact, B*, C, C* and P complex) has been reviewed extensively29,30. The two major transesterification reactions required for splicing excision of premature mRNA are catalyzed by the B* complex and the C* complex. In the first reaction, the branchpoint sequence attacks the 5’ ss, producing a lariat intermediate and the cleaved exon at the 5’ end. In the second reaction, the 5’ exon attacks the 3’ss, and then the two exons are joined while the intron spliced out as a lariat. Mutations of key RNA splicing factors, such as SF3B1 (a component of U2 snRNP) and U2AF1/2, would thereby be expected to result in a significant change in alternative splicing pattern of thousands of pre-mRNAs.
MECHANISTIC EFFECTS OF MUTATIONS IN RNA SPLICING FACTORS ON RNA SPLICING
Cancer-associated mutations in SF3B1
As part of the U2 snRNP complex, SF3B1 plays an essential role in recognizing the branchpoint sequence and spliceosomal complex A assembly during early stage of RNA splicing. It is now well established that the cancer-associated heterozygous mutations in SF3B1 within its carboxy-terminal HEAT domain (Figure 1), change branchpoint recognition to promote use of intron proximal aberrant (or cryptic) 3′ss in several hundred introns. This change in RNA splicing thereby results in aberrant expression of many genes in tandem that play known roles in tumor initiation or/and progression as well as many genes whose functional role is not well studied.
While the mechanisms by which mutant SF3B1 contributes to disease phenotypes and tumorigenesis are still being actively investigated, several cellular pathways have been suggested to play a role. For example, all recurrent cancer-associated mutations in SF3B1 result in BRD9 (bromodomain containing 9) mRNA degradation, through inclusion of a poison exon induced by aberrant branchpoint recognition (Figure 3A). This downregulation of BRD9 results in impaired formation of the non-canonical BAF chromatin remodeling complex and promotes melanomagenesis31 as well as development of MDS32. In another example, SF3B1 mutations have been repeatedly shown to reduce RNA and protein levels of the kinase MAP3K7 (also known as TAK1), due to promoting usage of a cryptic 3’ss upstream of the canonical 3’ss and resulting in non-sense mediated mRNA decay (NMD) of MAP3K7 mRNA33,34 (Figure 3B). Reduced MAP3K7 protein has a multitude of phenotypic effects depending on the cancer type. For example, in the context of pancreatic cancer, mutant SF3B1-mediated loss of MAP3K7 diminishes TGF-β-induced apoptosis and epithelial-mesenchymal transition (EMT) and increases the aggressiveness of pancreatic ductal adenocarcinoma.35 In the setting of hematopoiesis, loss of MAP3K7 or introduction of the SF3B1 mutation disrupts p38 MAPK signal transduction and impairs erythropoiesis, in part contributing to severe anemia34. A number of additional mRNAs encoding key proteins with known roles in erythropoiesis are similarly aberrantly spliced by mutant SF3B1 due to aberrant, intron proximal 3’ss usage including mRNAs encoding ABCB7, TMEM14C, and PPOX36. The downregulation of each of these proteins simultaneously is believed to contribute to the aberrant erythroid phenotype characteristic of SF3B1 mutant MDS.
Figure 3. Molecular basis for SF3B1 mutant mis-splicing.

(A) Diagram of how cancer-associated mutations promote expression of a cryptic exon containing an inframe premature termination codon (“poison exon”) in BRD9. The poison exon and its flanking regulatory sequences are derived from an intronic viral LTR (long terminal repeat). The cis elements required for mutant SF3B1 expression of the poison exon include an aberrant adenine branchpoint nucleotide, a short polypyrimidine (pY) tract, a 3’ splice site (3’ss), and four nucleotides at the very 5’ end of the cryptic exon as indicated. (B) Diagram of the recurrent SF3B1 mutant mis-splicing event in MAP3K7 resulting in use of an aberrant intron proximal 3’ss. The aberrant branchpoint adenine, short pY tract, aberrant 3’ss, and splicing enhancer required for mutant SF3B1 mis-splicing of MAP3K7 is shown. (C) One model currently proposed for the basis for SF3B1 mutant mis-splicing. Interaction of SF3B1 with the RNA helicase DHX15 via the DHX15 co-factor SUGP1 is required for normal intronic branchpoint recognition by the U2 snRNP complex. When SF3B1 is affected by cancer-associated mutations, the interaction of mutant SF3B1 with SUGP1 is reduced, resulting in usage of cryptic upstream branchpoints. Another DHX15 cofactor GPATCH8, competes with SUGP1 for DHX15 interaction and the abundance of GPATCH8 further regulates abundance of DHX15 for interaction with SUGP1.
In addition to the above mutant SF3B1 mis-spliced mRNAs involved in epigenetic regulation, signal transduction, and metabolism, Liu et al. investigated the impact of mutant SF3B1 on the post-translational regulatory networks via inactivation of a specific protein phosphatase 2A (PP2A) complex. Aberrant splicing of PPP2R5A, a regulatory B subunit of the PP2A complex, promotes both MYC activation as well as resistance to BCL2-mediated apoptosis. This occurs because the PPP2R5A-containing PP2A complex normally dephosphorylates MYC at Serine 62 and BCL2 at Serine 70. The PPP2R5A mis-splicing in SF3B1 mutant cells results in degradation of PPP2R5A transcripts and reduction in active PP2A. Loss of MYC and BCL2 phosphorylation at these sites promote MYC protein stability and anti-apoptotic effects. These changes are well known to drive B-cell transformation and connect SF3B1 mutations to the B-cell malignancy CLL. Interestingly, pharmacologic activation of PP2A using an FDA-approved PP2A activator overcomes the PPP2R5A downregulation in SF3B1 mutant cells and results in prolonged survival of Sf3b1K700E/+ mutant murine B-cell lymphomas driven by transgenic MYC expression37. Of note, the splicing alterations in MAP3K7 and PPPR25A induced by mutant SF3B1 are conserved across human and mouse cells33 (despite not being described in an initial analysis of splicing changes in Sf3b1 K700E knockin mice38). In contrast, the splicing alteration in BRD9 described above is not seen in murine cells as the poison exon and its regulatory splicing elements are derived from insertion of a viral LTR (long terminal repeat) into human and primate genomes31 (Figure 3A). Although global sequence-specific changes in RNA splicing are consistent between mouse and human cells with mutations in RNA splicing factors33,39–41, the lack of conservation in splicing of BRD9 between mouse and human SF3B1 mutant cells illustrates the challenges in the use of such models to study specific mis-spliced transcripts created by mutations in RNA splicing factors.
Interestingly, despite conferring global changes in RNA splicing and specific mis-spliced transcripts shared across different cancer types, mutations in SF3B1 can confer very distinct impacts on clinical prognosis in different cancers. For example, in the context of MDS, SF3B1 mutations are associated with favorable prognosis in MDS and the ring sideroblast subtype of MDS3 while in CLL, mutations in SF3B1 confer adverse prognosis4.
Mechanistic explanations of SF3B1 altered branchpoint selection
Unlike mutations in U2AF1 or SRSF2 which directly modify RNA recognition, hotspot mutations in SF3B1 do not appear to directly alter RNA:protein interaction. Instead, several groups have proposed that mutations in SF3B1 alter protein-protein interactions between mutant SF3B1 containing U2 snRNP complex and factors required for normal branchpoint recognition. For example, wild-type U2 snRNP interacts with DHX15, an RNA helicase essential for ATP hydrolysis, and this interaction is required for canonical branch site recognition during the initial stage of spliceosome assembly42–44. The Manley lab has recently identified that the G-patch domain protein SUGP1 facilitates interaction between DHX15 and U2 snRNP42,43.
G-patch motifs are co-factors for DEAH-box helicases as the interaction of G-patch motifs with helicases alter the conformation of helicases to promote helicase activity. G-patch domain containing proteins are increasingly appreciated as being implicated in altered RNA splicing in cancer. Zhang et al. have found that the regions of SUGP1 flanking its G-patch domain physically interact with SF3B1 at the region of SF3B1 affected by cancer-associated hotspot mutations. Interaction between SF3B1 and SUGP1 results in a conformational change in SUGP1 that exposes its G-patch domain to interact with DHX15 and creates a trimeric complex between SF3B1, SUGP1, and DHX1542. Interestingly, SUGP1 is occasionally also mutated in cancer, and some of the mutations in SUGP1 also occur at the interface of the SUGP1/SF3B1 interaction domains (Figure 1). Consistent with this, a significant proportion of aberrant RNA splicing changes seen with these particular cancer-associated mutations in SUGP1 mimic those created by mutations in SF3B1. DHX15 itself is also occasionally mutated with a specific hotspot mutation enriched in a form of acute myeloid leukemia marked by an oncogenic RUNX1-RUNX1T1 fusion (Figure 1). Ultimately, cancer-associated hotspot mutations in SF3B1, SUGP1, or DHX15, result in disruption of the SF3B1/DHX15 interaction and failure to activate DHX15 (Figure 3C). This consequently leads to aberrant use of upstream branch points and cryptic 3’ss characteristic of tumors with mutant SF3B142,43.
Intriguingly, overexpression of SUGP1 partially mitigates the splicing defects caused by mutant SF3B143. More recently, Benbarche et al. uncovered the role of another G-patch domain containing protein GPATCH8 as essential for mis-splicing mediated by SF3B1 mutations45. Through structural modeling and experimental examination, GPATCH8 was found to exert an antagonistic effect with SUGP1. Specifically, GAPTCH8 and SUGP1 compete for interaction with DHX15 (Figure 3C). Thus, depletion of GPATCH8 enhances the DHX15/SUGP1 interaction and partially corrects SF3B1 mis-splicing defects. Despite these insights, it is important to note that there is no known direct role of GPATCH8 in RNA splicing currently and the exact model of how GPATCH8 deletion is associated with correction of SF3B1 mutant mis-splicing events will require further evaluation.
It is important to note that several groups have proposed alternative hypotheses with the above model of SF3B1 mutant mis-splicing. For example, the RNA helicases-DDX42 and DDX46 have been found to compete for association with SF3B1 through their N-terminal sequences in an exclusive manner. The N-plug region of DDX42 occupies the RNA binding region of SF3B1, as indicated by cryo-EM structures46. Yang et al. thereby proposed that somatic mutations in SF3B1 abrogate its interaction with DDX42 or DDX46, resulting in pre-mature exposure of the RNA binding region to the pre-mRNA and impairing the function of U2 snRNP46. Additionally, DDX46 plays a role in proofreading branchpoint selection by anchoring onto 17S U2 snRNP through occupation of the RNA binding region within SF3B1. The helicase domains of DDX46 interact with the U2 snRNA, where the BSL (BS-interacting stem-loop) is shielded by TAT-SF1. When DDX46 unwinds the double stranded BSL, it disrupts its interaction with TAT-SF1, and facilitates the formation of the initial U2-branchsite duplex. However, mutations in SF3B1 are proposed to disrupt its interactions with DDX46, compromising branchpoint site proofreading46 (although there is no experimental evidence for this hypothesis currently). Future work identifying the structural impact of SF3B1 mutations on each of its interacting partner proteins will be critical in clarifying which of the above models best explains the stereotypical pattern of aberrant branchsite usage typical of SF3B1 mutant cancers. It is possible that mutations in SF3B1 could impair association with each of its three interacting RNA helicases (DHX15, DDX42, DDX46).
Cancer-associated mutations in U2AF1 alter 3’ss recognition
U2AF1 is a core component of the spliceosome and heterodimerizes with U2AF2 through its UHM (U2AF homology motif) domain. The U2AF1/2 heterodimer recognizes the 3’AG dinucleotide at the 3’ss within intronic pre-mRNA. U2AF2 binds the polypyrimidine tract within the intron as well as SF3B1 while U2AF1 interacts with the 3’ss boundary between the exon and intron.
As noted above, mutations in U2AF1 occur in one of its two zinc fingers (Figure 1). As is the case with mutant SF3B1, a multitude of aberrant RNA splicing events have been associated with mutant U2AF1. Recurrently described mutant U2AF1 mis-splicing events across cancers impact essential biological pathways such as DNA methylation (DNMT3B), the DNA damage response (ATR), and apoptosis (CASP8)47. Overexpression of mutant U2AF1 results in an increased number of transcripts containing unspliced intronic sequence and leads to suppressed cell growth and induced apoptosis in HeLa cells5. Additionally, U2AF1 S34F mutation induces genes expression associated with EMT and enhances tumor cell invasion in lung adenocarcinomas48. Long-read cDNA sequencing analysis of U2AF1 S34F mutations in isogenic human bronchial epithelial cells revealed downregulation of isoforms from immune-related genes, such as TNF via the NF-kB signaling pathway49.
To date, there is no single parsimonious aberrant RNA splicing event(s) associated with U2AF1 mutations to explain their occurrence in cancers. However, analyses of RNA-seq data from U2AF1 mutant cells have repeatedly revealed that U2AF1 mutations alter splicing in a sequence dependent manner based on the identities of the nucleotide flanking the AG dinucleotide at the 3’ss12 (Figure 4A). U2AF1 S34 mutants are affected by nucleotide sequence at the −3 position from the intron/exon border while Q157 mutants are affected by the nucleotide at the +1 position. Specifically, cells with U2AF1 S34F mutations tend to have exclusion of exons bearing a U at −3 of the 3’ss while exons bearing a C tend to be included. By contrast, mutations in Q157 promote inclusion of exons with a G at the +1 position while repressing usage of exons with an A at the same site12.
Figure 4. Impact of mutant splicing factors on RNA splice site recognition beyond SF3B1 mutations.

(A) Wild-type U2AF1 recognizes consensus 3’ splice site with preference sequence UAG, whereas the pathogenic mutants of U2AF1 preferentially bind alternative 3’ splice sites (3’ss) in order of abundance: CAG, GAG, AAG. (B) Wild-type SRSF2 equally engages in a stacking interaction both the second cytosine in the CCNG motif and the second guanine in the GGNG motif within exonic splicing enhancer (ESE) region. Mutation of proline 95 to histidine is thought to provide a stronger hydrogen bond with the second cytosine in the CCNG site, resulting in changes in RNA binding specificity and mis-splicing. (C) ZRSR2 primarily interacts with minor (or U12-type) introns characterized by lack of a pY tract, shorter distance from conserved sequence embedding branchpoint to 3’ss, and preferential sequences of GT, AG or AT, AC at the 5’ and 3’ splice sites, respectively. Mutations in ZRSR2 impair minor intron removal as compared to wild-type ZRSR2 counterpart. (D) Secondary structure of the mutant U1 snRNA (left) or U11snRNA (right). The red or blue circles identify the location of the hotspot mutations. The rectangles in yellow indicate the 5′ splice-site recognition site, including the third nucleotide of U1 snRNA (r.3A), the fifth nucleotide of U11 snRNA (r.5A).
Structural basis of mutant U2AF1-mediated dysregulated splicing events
Consistent with the differential nucleotide motif data above, it is now clear that mutations occurring at the two amino acid hotspots in U2AF1 lead to alteration in U2AF1’s RNA binding affinity11,15,47. Crystal structures of yeast WT U2AF1 and pathogenic mutant U2AF1 S34F and S34Y proteins elucidated the mechanism of 3’ss selection in an early splicing step. Both of U2AF1’s two zinc finger domains are required to recognize the 3’ss AG dinucleotide. While WT U2AF1 preferably interacts with −3U for efficient RNA binding and 3’ss recognition, the pathogenic mutants of U2AF1 can tolerant −3A or −3C50. An eCLIP (enhanced version of the cross-linking and immunoprecipitation) assay conducted using isogenic iPSC models WT or mutant for U2AF1 confirmed these findings within cells as U2AF1S34F RNA binding sites showed the following order of abundance: CAG, GAG, AAG and UAG. Motif enrichment analysis shows that U2AF1 WT binding sites are enriched for “UAG” whereas U2AF1S34F are enriched for “CAG.”51
In addition to impacting U2AF1/RNA interactions, mutation at U2AF1 S34 also appear to induce a conformational change in U2AF1’s binding partner U2AF2. Specifically, U2AF1 S34 mutants shift the RNA recognition motif (RRM) domain of U2AF2 from an “open” conformation to a “closed” when the U2AF heterodimer binds uridine-rich splice site52. This change in U2AF2 conformation promotes recognition of introns with weaker, uridine-poor Py tracts and may contribute to some of the pathologic splicing events seen with U2AF1 S34 mutants. Of note, rare cancer-associated U2AF2 mutations have been described and these cluster in the two RRMs of U2AF2 and modulate binding to the Py tract as well53.
Mutations in SRSF2 alter exonic splicing enhancer recognition
SRSF2 is a member of the SR family of RNA binding proteins and accessory regulators of RNA splicing. SRSF2 governs both constitutive and alternative splicing events by binding exonic splicing enhancer (ESE) sequences within pre-mRNA and physically interacting with U1 and U2 snRNPs54–56 (Figure 4B). SRSF2 hotspot mutations occur at the Proline 95 residue, which is located between the RRM and RS domains of SRSF2 (Figure 1) and alter SRSF2’s RNA binding preferences39,51,57–59. While the WT SRSF2 efficiently recognizes both C- or G-rich (GGNG or CCNG) mRNA motifs, SRSF2 mutant protein preferentially recognizes C-rich sequences (CCNG), thereby enhancing inclusion of exons with C-rich ESEs and leading to mis-splicing39,51. Structural and experimental analyses reveal WT SRSF2 equally engages the second cytosine in the CCNG motif and the second guanine in the GGNG motif using stacking interaction. However, P95H mutations in SRSF2 result in a stronger hydrogen bond formation with the second cytosine in the CCNG site compared with interactions with GGNG57,60.
Among the mis-spliced genes affected by mutant SRSF2, several contribute to myelodysplasia and hinder hematopoietic differentiation. For example, SRSF2 mutations promote an isoform of EZH2 with inclusion of a poison exon subjected to NMD, resulting in downregulation of EZH2, a histone methyltransferase and a key hematopoietic regulator39. Notably, SRSF2 mutations frequently overlap with specific additional somatic mutations, such as IDH2 mutations. Combined mutations in SRSF2 and IDH2 collectively promote leukemogenesis through collaborative effects on the epigenome and RNA splicing created by each mutation58.
Beyond RNA splicing, it was recently discovered that SRSF2 also serves as a reader of a modified cytosine in RNA (m5C). SRSF2P95 mutations impair binding to this modified cytosine and this alteration in epitranscriptomic reading may also contribute to leukemogenesis61. SRSF2 binds to leukemia-related transcripts co-localized with m5C, and such binding decreases upon knockdown of the m5C writer NSUN258.
Mutations in ZRSR2 alter minor intron recognition
ZRSR2 is an RNA splicing factor that primarily interacts with minor (or U12-type) introns. Since the pY tract is absent in minor introns, the U2AF1/2 heterodimers that interact with the pY tract and 3’ss of major introns do not associate with minor introns. Instead, U11/U12 snRNP and Urp/ZRSR2 are involved in 3’ss recognition of minor introns62.
Unlike the heterozygous hotspot mutations described in SF3B1, SRSF2 and U2AF1 described above, ZRSR2, an X-chromosome-linked gene, is affected by mutations across its protein-coding region and has a male predominance, strongly suggesting the mutations as loss-of-function (Figure 1). The impact of loss of ZRSR2 has been studied in human cell lines, a conditional knockout mouse, and patient samples and mainly results in abnormal retention of U12-type introns (Figure 4C). A number of minor-intron containing genes that require ZRSR2 for normal RNA splicing and expression are associated with clonal hematopoietic disorders. This includes the gene LTZR1, which encodes a regulator of RAS-related GTPase41,63, where retention of a minor intron results in LZTR1 downregulation and a clonal advantage to hematopoietic cells in vitro and in vivo. Interestingly, knockout of zrsr2 in zebrafish results in multiple developmental defects through downregulation of essential metabolic pathways and the aberrant retention of minor introns64, underscoring a critical role of Zrsr2 in development.
Mutations in RBM10
RBM10 is an accessory RNA splicing factor often mutated in a variety of cancers including lung, thyroid, and bladder adenocarcinomas65–71. Like ZRSR2, RBM10 is an X-linked gene affected by presumed loss-of-function mutations and appears to acts as a tumor suppressor in multiple cancer types. Additionally, RBM10 germline mutations occur in TARP syndrome, an X-linked congenital pleiotropic developmental disorder72. Loss-of-function mutations in RBM10 primarily results in the promotion of exon inclusion events. For example, RBM10 mutations are implicated in Notch signaling by promoting the inclusion of exon 9 in the Notch signaling inhibitor NUMB. This leads to Notch hyperactivation, which in turn promotes cell growth in lung cancer68.
Despite the above findings, a precise role for RBM10 in RNA splicing was not entirely clear until recently. Damianov et al. recently isolated spliceosome complexes from the chromatin compartment of live cells using a novel method73. Evaluation of U2 snRNP particles using this method revealed a striking observation that RBM10, and its closely related protein, RBM5 are bound to nearly all branch site complexes. This interaction of RBM10 with the branchsite required its zinc finger motif, a region of RBM10 commonly affected by mutations in cancer. Deletion of RBM10’s zinc finger motif disrupted U2 snRNP/branchsite interaction and resulting in expression of many alternative exons73. These data thereby identified RBM10 as a protein associated with the U2 snRNP complex and a splicing repressor. Interestingly, RBM10 also contains a G-patch motif at its C-terminus (Figure 1) and there is considerable evidence that RBM10’s G patch interacts with and activates DHX1573,74.
Variant snRNAs
As noted above, snRNAs are essential non-protein components of spliceosomes. The variants that have been described in the U1 and U11 snRNAs occur at sites that each snRNA interacts with the 5’ss and, as such, would greatly alter 5’ss usage25–27 (Figure 4D). In contrast, mutations in the 28th nucleotide of the U2 snRNA occur at the interface of the interaction between the U2 and U6 snRNAs27. The U6-U2 complex comprises the active site of the spliceosome so perturbation of this interaction could have major impacts on RNA splicing. However, it is important to note that the human genome contains numerous identical copies of the genes encoding for the U1 and U2 snRNAs and >100 pseudogenes for each gene. For example, the human reference genome has four annotated U1 snRNA genes and >100 pseudogenes. Moreover, the genomic regions flanking each gene/pseudogene are highly identical. Thus, U1 and U2 snRNAs are encoded by repetitive regions of the genome which greatly complicates identification of mutations from these loci using conventional short-read sequencing. As a result, the precise genomic coordinate of any snRNA mutation in U1 or U2 loci have not been mapped and the levels of mutant allele expression of these mutations are unclear. There is also potential evidence that U2 snRNA variants identified in tumor genomic DNA may also be present in germline27, which questions if these alterations in snRNAs may simply reflect variant sequences that are not necessarily pathologic. Future research efforts applying long-read sequencing technologies to tissues from patients thought to potentially have snRNA mutations (for example from individuals with medulloblastoma) will hopefully clarify more precise details on snRNA variants and their presence in tumor versus non-tumor tissues.
IMPACT OF SPLICING FACTOR MUTATIONS ON RNA METABOLISM BEYOND SPLICING
A number of RNA splicing factors that are commonly mutated in cancer have been proposed to play roles in RNA processing beyond strictly RNA splicing. This includes roles in the processes of NMD and R-loop formation as well as polyadenylation, mRNA translation, and stress granule formation. The mechanistic connections between mutant splicing factors and some of these processes are described below.
Role of mutant RNA splicing factors on NMD
NMD is a quality control process to degrade mRNAs with premature termination codons (reviewed recently75–77). NMD is tightly linked to RNA splicing as the initial nuclear step of NMD relies on deposition of a protein complex at the site of exon-exon junctions (the Exon Junction Complex (EJC)) created by RNA splicing. Moreover, as RNA splicing regulates the protein coding frame of an mRNA, abnormal mRNA splicing results in increased generation of non-canonical transcripts that trigger NMD. Consistent with this, Baird et al. and Cheruiyot et al. performed genome-wide CRISPR/Cas9 knock-out screens using cells expressing NMD reporters and identified U2AF1 and SF3B1 as factors promoting NMD.78,79 Similarly, it has been reported that cancer-associated point mutations in the RNA splicing factors U2AF1, SRSF2, and SF3B1 promote activation of NMD (albeit phenotypes resulting from loss of SF3B1 or U2AF1 would not be expected to be equivalent to those seen with neomorphic cancer-associated mutations in SF3B1 or U2AF1). For example, in the initial description of mutations in RNA splicing factors in myeloid blood cancers, overexpression of U2AF1S34F mutant in human cells was associated with upregulation of NMD pathway components5. Similarly, the aberrant splicing events created by the SF3B1K700E mutation in blood cancers are predicted to result in one-third to half of aberrant transcripts being NMD sensitive38,80.
Beyond activation of NMD due to increased generation of NMD substrates, at least one paper has proposed a specific role for mutant splicing factors in the enzymatic process of NMD81. As noted above, MDS-associated SRSF2 Proline 95 mutations change the RNA binding specificity of SRSF2, and thus induce hundreds of aberrant splicing events. In addition, SRSF2 mutants also enhance recruitment of several core factors of the EJC as well as several NMD factors if there is a premature termination codon upstream of the mutant SRSF2 binding site on mRNA (Figure 5A). Thus, mutations in SRSF2 result in sequence-specific changes in both RNA splicing preferences as well as NMD.
Figure 5. Connections between mutant RNA splicing factors and RNA metabolism beyond RNA splicing.

(A) Mutations in SRSF2 enhance the recruitment of the exon junction complexes (EJCs) and thus promote nonsense-mediated mRNA decay. (B) Mutations in SRSF2 and U2AF1 lead to the inhibition of RNA polymerase II (Pol II) and trigger R-loop formation. (C) Mutations in U2AF1 are associated with increased formation of stress granules in cells.
Impact of mutant RNA splicing factors on R-loops
R-loops are three-stranded structures where RNA base pairs with DNA and a single-strand of displaced DNA (recently reviewed by Petermann et al.82). While several physiologic gene regulatory roles of R-loops have been described, in the setting of cancer, R-loops are most commonly studied as a nidus for genome instability due to DNA damage in the displaced single strand of DNA. Due to the intimate role of transcription and RNA splicing, a number of RNA splicing factors have been described to protect single strand RNA from annealing to the DNA template during transcription and thereby limit R-loop formation. Interestingly, cancer-associated mutations in SF3B1, SRSF2, and U2AF1 have been reported to trigger genome-wide elevations in R-loops, replication stress, and activation of the ATR-Chk1 pathway in cells1,83–86 (Figure 5B).
Despite the connection between mutations in RNA splicing factors and increased R-loop abundance noted in several prior publications, it is important to note that the causality of R-loops in diseases where splicing factor mutations are common is not currently well understood. For example, many of the human diseases where splicing factor mutations are common are not known to exhibit genomic instability or widespread DNA damage. In contrast, it is quite possible that increased R-loop formation in splicing factor mutant cells may impact gene expression. However, knowledge of gene regulatory impact of R-loop formation in splicing factor mutant cells is currently limited. This is partly due to the limited efforts to map the genomic location of R-loops in primary human cells with mutations in the splicing machinery and reliance of studying R-loop formation in cancer cell lines with engineered mutations in the splicing machinery.
Although the molecular impact of R-loops in splicing factor mutant cancers is unclear, several studies have shown that overexpression of RNaseH1, an endonuclease that binds to R-loops in cells and cleaves the RNA in RNA:DNA hybrids87, can correct the proliferation defect of such cells in cells bearing splicing factor mutations 84,85,88. These findings suggest the possibility of targeting R-loop-related features as a strategy to eliminate cells with RNA splicing factor mutations. For example, SRSF2P95H and U2AF1S34F leukemia cells elicit an R-loop-associated poly-ADP ribose polymerase (PARP1) response and are sensitive to PARP inhibitors89. More recently, cells with SF3B1K700E mutations have also been shown to induce DNA damage via generation of genotoxic R-loops and can be targeted by PARP inhibitors86. As such, a strategy of combining PARP and ATR inhibitors has been to propose to increase DNA damage and result in selective death of RNA splicing factor-mutant leukemic cells89.
In addition to limitations in our knowledge of the functional importance of R-loops in splicing factor mutant cells, a recent study evaluating genome-wide localization of R-loops in primary human erythroid precursors from patients with or without SF3B1 mutant MDS identified a global decrease in the abundance of R loops in SF3B1 mutant cells90. Notably, each of the prior studies of R-loops in splicing factor mutant utilized R-loop detection methods and cell types distinct from one another. It is therefore unclear how results in SF3B1 mutant cells would compare to those from SRSF2 and U2AF1 mutant cells using the same cell type and R-loop detection method. Thus, it will be important for future studies of R-loops in splicing factor mutant cancers to study R-loop localization and abundance using comparable cell types, methodologies, and appropriate negative controls to address the true impact of splicing factor mutations on R-loop formation.
Stress granules
Stress granules are membraneless cell compartments containing mRNAs and RNA-binding proteins that form under stress response (reviewed recently91), the role of which in cancer has been drawing increasing attention92. Mutations in U2AF1 were recently connected to the formation of stress granules in myeloid malignancies. Biancon et al. precisely analyzed the 3’ss binding location of U2AF1 hotspot mutations by eCLIP-seq in a human leukemia cell line and identified thousands of differentially spliced events in U2AF1 S34F and Q157R mutant cells compared with wild-type93. Around 30% of mRNA targets were shared by the two mutants, and interestingly, gene ontology analysis of the shared group showed an enrichment of genes involving RNA granules. Compared to the wild-type cells, both AML cell lines and patient cells harboring the U2AF1 mutation showed increase formation of stress granules after stress induction with sodium arsenite (Figure 5C). These data thereby suggest that U2AF1 mutant AML cells may have improved cell fitness under conditions of stress. While the authors proposed that U2AF1 mutations increase the generation of transcripts and possibly proteins that are prone to stress granule formation, how exactly the alterations in RNA splicing of U2AF1 result in stress granule formation is unclear. Furthermore, it would be interesting to determine whether this phenotype of increased stress granules is seen with other splicing factor mutations.
CONCLUSIONS & UNANSWERED QUESTIONS
Studying the molecular consequences of cancer-associated mutations in RNA splicing factors on RNA splicing and gene expression has resulted in numerous discoveries with more fundamental importance in our understanding of RNA metabolism in cells. For example, studying the molecular mechanisms by which mutations in SF3B1 result in aberrant branchsite recognition has helped elucidate the importance of DHX15 in remodeling the branchsite and the requirement of a number of previously poorly studied DHX15 co-factors such as SUGP1 and GPATCH8. Similarly, studies of RBM10 have identified a specific role for this factor in U2 snRNP function which was previously unknown. Furthermore, the discovery of oncogenic neomorphic mutations in the RNA splicing machinery have spurred numerous therapeutic studies aimed at identifying synthetic lethal approaches to target cells with these mutations94,95 as well as nucleic acid based therapeutics to harness the aberrant splicing activity in these cells96. At the same time, numerous questions remain about the role of these mutations in cancer development. For example, GPATCH8 was discovered to be required for nearly one-third of the aberrant RNA splicing events mediated by mutant SF3B1. However, it is quite likely that other RNA splicing factors, possibly another of the 25 human G-patch domain containing proteins, may be required for the remainder of aberrant splicing activity of mutant SF3B1. Moreover, carrying out phenotypic screens for molecular regulators of aberrant RNA splicing activity by mutant U2AF1, SRSF2, and other splicing factors commonly mutated in cancer could be similarly enlightening.
One major question about the role of altered RNA splicing in cancer is identifying which of the many altered transcripts generated in cancer cells is important in cancer initiation and/or maintenance. There is hope that increasing use of long-read RNA-seq technology to map full-length mRNA isoforms present in cells may help more fully catalog the landscape of transcripts in cancer cells with and without mutations in splicing regulatory proteins. Similarly, application of long-read sequencing technologies may help clarify the location, expression, and somatic versus germline status of snRNAs with variant sequences in malignant and normal cells.
Finally, it is hopeful that future studies will help delineate if the impact of mutations in RNA splicing factors on cancer is attributable to their impact on RNA splicing and/or to splicing related processes such as R-loop formation, NMD, mRNA translation, and/or RNA granule formation.
ACKNOWLEDGEMENTS
O.A.-W. is supported by the Edward P. Evans Foundation, NIH/National Cancer Institute (NCI) (R01 CA251138, R01 CA242020, P50 CA254838, and R01 CA283364), and NIH/NHLBI R01 HL128239, and The Leukemia & Lymphoma Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
O.A.-W. is a founder and scientific advisor of Codify Therapeutics, holds equity in this company, and receives research funding from this company. O.A.-W. has served as a consultant for Foundation Medicine Inc., Merck, Prelude Therapeutics, Amphista Therapeutics, MagnetBio, and Janssen, and is on the Scientific Advisory Board of Envisagenics Inc., Harmonic Discovery Inc., and Pfizer Boulder; O.A.-W. has received prior research funding from H3B Biomedicine, Nurix Therapeutics, Minovia Therapeutics, and LOXO Oncology unrelated to the current manuscript. The remaining authors declare no competing interests.
REFERENCES
- 1.Cusan M, Shen H, Zhang B, Liao A, Yang L, Jin M, Fernandez M, Iyer P, Wu Y, Hart K, et al. (2023). SF3B1 mutation and ATM deletion codrive leukemogenesis via centromeric R-loop dysregulation. J Clin Invest 133. 10.1172/JCI163325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen S, Benbarche S, and Abdel-Wahab O (2021). Splicing factor mutations in hematologic malignancies. Blood 138, 599–612. 10.1182/blood.2019004260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365, 1384–1395. 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, Lawrence M, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca D, Zhang L, et al. (2011). SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. The New England journal of medicine 365, 2497–2506. 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. nature10496 [pii] 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 6.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. (2013). SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 3, 1122–1129. 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013). Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nature genetics 45, 133–135. 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. (2013). Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nature genetics 45, 933–936. 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, Liu Z, Chen S, Ki M, Erickson C, Reis-Filho JS, Durham BH, Chang Q, de Stanchina E, Sun Y, et al. (2020). Mutant SF3B1 promotes AKT and NF-kB driven mammary tumorigenesis. J Clin Invest. 10.1172/JCI138315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Xie W, Rosenblum JS, Zhou J, Guo J, Miao Y, Shen Y, Wang H, Gong L, Li M, et al. (2020). Somatic SF3B1 hotspot mutation in prolactinomas. Nat Commun 11, 2506. 10.1038/s41467-020-16052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. (2015). U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29, 909–917. 10.1038/leu.2014.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, and Bradley RK (2014). U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 10.1101/gr.181016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imielinski M, Alice HB, Peter SH, Bryan H, Trevor JP, Eran H, Jeonghee C, James S, Marzia C, Andrey S, et al. (2012). Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 150, 1107–1120. 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. (2012). Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44, 53–57. ng.1031 [pii] 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. (2014). A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9, e87361. 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. (2015). Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6, 6042. 10.1038/ncomms7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, Della Valle V, Couronné L, Scourzic L, Chesnais V, et al. (2012). Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 119, 3211–3218. 10.1182/blood-2011-12-400994. [DOI] [PubMed] [Google Scholar]
- 18.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. (2014). Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247. 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alsafadi S, Dayot S, Tarin M, Houy A, Bellanger D, Cornella M, Wassef M, Waterfall JJ, Lehnert E, Roman-Roman S, et al. (2021). Genetic alterations of SUGP1 mimic mutant-SF3B1 splice pattern in lung adenocarcinoma and other cancers. Oncogene 40, 85–96. 10.1038/s41388-020-01507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, and Rabadan R (2020). Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci U S A 117, 10305–10312. 10.1073/pnas.1922622117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, Radtke I, Chao JR, Walsh MP, Song G, et al. (2016). The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48, 1551–1556. 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. (2015). Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, 6744. 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinayanuwattikun C, Le Calvez-Kelm F, Abedi-Ardekani B, Zaridze D, Mukeria A, Voegele C, Vallee M, Purnomosari D, Forey N, Durand G, et al. (2016). Elucidating Genomic Characteristics of Lung Cancer Progression from In Situ to Invasive Adenocarcinoma. Sci Rep 6, 31628. 10.1038/srep31628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki H, Kumar SA, Shuai S, Diaz-Navarro A, Gutierrez-Fernandez A, De Antonellis P, Cavalli FMG, Juraschka K, Farooq H, Shibahara I, et al. (2019). Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574, 707–711. 10.1038/s41586-019-1650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shuai S, Suzuki H, Diaz-Navarro A, Nadeu F, Kumar SA, Gutierrez-Fernandez A, Delgado J, Pinyol M, Lopez-Otin C, Puente XS, et al. (2019). The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature. 10.1038/s41586-019-1651-z. [DOI] [PubMed] [Google Scholar]
- 27.Bousquets-Munoz P, Diaz-Navarro A, Nadeu F, Sanchez-Pitiot A, Lopez-Tamargo S, Shuai S, Balbin M, Tubio JMC, Bea S, Martin-Subero JI, et al. (2022). PanCancer analysis of somatic mutations in repetitive regions reveals recurrent mutations in snRNA U2. NPJ Genom Med 7, 19. 10.1038/s41525-022-00292-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wahl MC, and Luhrmann R (2015). SnapShot: Spliceosome Dynamics I. Cell 161, 1474–e1471. 10.1016/j.cell.2015.05.050. [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson ME, Charenton C, and Nagai K (2020). RNA Splicing by the Spliceosome. Annu Rev Biochem 89, 359–388. 10.1146/annurev-biochem-091719-064225. [DOI] [PubMed] [Google Scholar]
- 30.Wilkinson ME, Lin PC, Plaschka C, and Nagai K (2018). Cryo-EM Studies of Pre-mRNA Splicing: From Sample Preparation to Model Visualization. Annu Rev Biophys 47, 175–199. 10.1146/annurev-biophys-070317-033410. [DOI] [PubMed] [Google Scholar]
- 31.Inoue D, Chew GL, Liu B, Michel BC, Pangallo J, D’Avino AR, Hitchman T, North K, Lee SC, Bitner L, et al. (2019). Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature. 10.1038/s41586-019-1646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao M, Kondo S, Nomura M, Kato S, Nishimura K, Zang W, Zhang Y, Akashi T, Viny A, Shigehiro T, et al. (2023). BRD9 determines the cell fate of hematopoietic stem cells by regulating chromatin state. Nat Commun 14, 8372. 10.1038/s41467-023-44081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SC, North K, Kim E, Jang E, Obeng E, Lu SX, Liu B, Inoue D, Yoshimi A, Ki M, et al. (2018). Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 34, 225–241 e228. 10.1016/j.ccell.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lieu YK, Liu Z, Ali AM, Wei X, Penson A, Zhang J, An X, Rabadan R, Raza A, Manley JL, and Mukherjee S (2022). SF3B1 mutant-induced missplicing of MAP3K7 causes anemia in myelodysplastic syndromes. Proc Natl Acad Sci U S A 119. 10.1073/pnas.2111703119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simmler P, Ioannidi EI, Mengis T, Marquart KF, Asawa S, Van-Lehmann K, Kahles A, Thomas T, Schwerdel C, Aceto N, et al. (2023). Mutant SF3B1 promotes malignancy in PDAC. Elife 12. 10.7554/eLife.80683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clough CA, Pangallo J, Sarchi M, Ilagan JO, North K, Bergantinos R, Stolla MC, Naru J, Nugent P, Kim E, et al. (2022). Coordinated missplicing of TMEM14C and ABCB7 causes ring sideroblast formation in SF3B1-mutant myelodysplastic syndrome. Blood 139, 2038–2049. 10.1182/blood.2021012652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Yoshimi A, Wang J, Cho H, Chun-Wei Lee S, Ki M, Bitner L, Chu T, Shah H, Liu B, et al. (2020). Mutations in the RNA Splicing Factor SF3B1 Promote Tumorigenesis through MYC Stabilization. Cancer Discov 10, 806–821. 10.1158/2159-8290.CD-19-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. (2016). Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 30, 404–417. 10.1016/j.ccell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol J-BB, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer cell 27, 617–630. 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. (2015). Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 27, 631–643. 10.1016/j.ccell.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoue D, Polaski JT, Taylor J, Castel P, Chen S, Kobayashi S, Hogg SJ, Hayashi Y, Pineda JMB, El Marabti E, et al. (2021). Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet 53, 707–718. 10.1038/s41588-021-00828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Xie J, Huang J, Liu X, Xu R, Tholen J, Galej WP, Tong L, Manley JL, and Liu Z (2023). Characterization of the SF3B1-SUGP1 interface reveals how numerous cancer mutations cause mRNA missplicing. Genes Dev 37, 968–983. 10.1101/gad.351154.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, Raza A, Mukherjee S, and Manley JL (2019). Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol Cell 76, 82–95 e87. 10.1016/j.molcel.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Huang J, Xu K, Xing P, Huang Y, Liu Z, Tong L, and Manley JL (2022). DHX15 is involved in SUGP1-mediated RNA missplicing by mutant SF3B1 in cancer. Proc Natl Acad Sci U S A 119, e2216712119. 10.1073/pnas.2216712119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benbarche S, Pineda JMB, Galvis LB, Biswas J, Liu B, Wang E, Zhang Q, Hogg SJ, Lyttle K, Dahi A, et al. (2024). GPATCH8 modulates mutant SF3B1 mis-splicing and pathogenicity in hematologic malignancies. Mol Cell. 10.1016/j.molcel.2024.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang F, Bian T, Zhan X, Chen Z, Xing Z, Larsen NA, Zhang X, and Shi Y (2023). Mechanisms of the RNA helicases DDX42 and DDX46 in human U2 snRNP assembly. Nat Commun 14, 897. 10.1038/s41467-023-36489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ilagan J, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, and Bradley RK (2014). U2AF1 mutations alter splice site recognition in hematological malignancies. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esfahani MS, Lee LJ, Jeon YJ, Flynn RA, Stehr H, Hui AB, Ishisoko N, Kildebeck E, Newman AM, Bratman SV, et al. (2019). Functional significance of U2AF1 S34F mutations in lung adenocarcinomas. Nat Commun 10, 5712. 10.1038/s41467-019-13392-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soulette CM, Hrabeta-Robinson E, Arevalo C, Felton C, Tang AD, Marin MG, and Brooks AN (2023). Full-length transcript alterations in human bronchial epithelial cells with U2AF1 S34F mutations. Life Sci Alliance 6. 10.26508/lsa.202000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida H, Park SY, Sakashita G, Nariai Y, Kuwasako K, Muto Y, Urano T, and Obayashi E (2020). Elucidation of the aberrant 3’ splice site selection by cancer-associated mutations on the U2AF1. Nat Commun 11, 4744. 10.1038/s41467-020-18559-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wheeler EC, Vora S, Mayer D, Kotini AG, Olszewska M, Park SS, Guccione E, Teruya-Feldstein J, Silverman L, Sunahara RK, et al. (2022). Integrative RNA-omics Discovers GNAS Alternative Splicing as a Phenotypic Driver of Splicing Factor-Mutant Neoplasms. Cancer Discov 12, 836–855. 10.1158/2159-8290.Cd-21-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Warnasooriya C, Feeney CF, Laird KM, Ermolenko DN, and Kielkopf CL (2020). A splice site-sensing conformational switch in U2AF2 is modulated by U2AF1 and its recurrent myelodysplasia-associated mutation. Nucleic Acids Res 48, 5695–5709. 10.1093/nar/gkaa293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maji D, Glasser E, Henderson S, Galardi J, Pulvino MJ, Jenkins JL, and Kielkopf CL (2020). Representative cancer-associated U2AF2 mutations alter RNA interactions and splicing. J Biol Chem 295, 17148–17157. 10.1074/jbc.RA120.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graveley BR, and Maniatis T (1998). Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell 1, 765–771. [DOI] [PubMed] [Google Scholar]
- 55.Liu HX, Chew SL, Cartegni L, Zhang MQ, and Krainer AR (2000). Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol 20, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaal TD, and Maniatis T (1999). Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol 19, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, and Manley JL (2015). Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A 112, E4726–4734. 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, Lee SC, Micol JB, Zhang XJ, de Botton S, et al. (2019). Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature. 10.1038/s41586-019-1618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pangallo J, Kiladjian JJ, Cassinat B, Renneville A, Taylor J, Polaski JT, North K, Abdel-Wahab O, and Bradley RK (2020). Rare and private spliceosomal gene mutations drive partial, complete, and dual phenocopies of hotspot alterations. Blood 135, 1032–1043. 10.1182/blood.2019002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daubner GM, Clery A, Jayne S, Stevenin J, and Allain FH (2012). A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 31, 162–174. 10.1038/emboj.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma HL, Bizet M, Soares Da Costa C, Murisier F, de Bony EJ, Wang MK, Yoshimi A, Lin KT, Riching KM, Wang X, et al. (2023). SRSF2 plays an unexpected role as reader of m(5)C on mRNA, linking epitranscriptomics to cancer. Mol Cell 83, 4239–4254 e4210. 10.1016/j.molcel.2023.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen H, Zheng X, Luecke S, and Green MR (2010). The U2AF35-related protein Urp contacts the 3’ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev 24, 2389–2394. 10.1101/gad.1974810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen S, Vedula RS, Cuevas-Navarro A, Lu B, Hogg SJ, Wang E, Benbarche S, Knorr K, Kim WJ, Stanley RF, et al. (2022). Impaired Proteolysis of Noncanonical RAS Proteins Drives Clonal Hematopoietic Transformation. Cancer Discov 12, 2434–2453. 10.1158/2159-8290.CD-21-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinstein R, Bishop K, Broadbridge E, Yu K, Carrington B, Elkahloun A, Zhen T, Pei W, Burgess SM, Liu P, et al. (2022). Zrsr2 Is Essential for the Embryonic Development and Splicing of Minor Introns in RNA and Protein Processing Genes in Zebrafish. Int J Mol Sci 23. 10.3390/ijms231810668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bao Y, Zhang S, Zhang X, Pan Y, Yan Y, Wang N, Ren Y, Zuo J, Zong WX, Wang Z, and Wang Y (2023). RBM10 Loss Promotes EGFR-Driven Lung Cancer and Confers Sensitivity to Spliceosome Inhibition. Cancer Res 83, 1490–1502. 10.1158/0008-5472.CAN-22-1549. [DOI] [PubMed] [Google Scholar]
- 66.Li Z, Xue Q, Xu J, Zhang P, and Ding B (2020). The role of RBM10 mutations in the development, treatment, and prognosis of lung adenocarcinoma. Cell Cycle 19, 2918–2926. 10.1080/15384101.2020.1829801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inoue A (2021). RBM10: Structure, functions, and associated diseases. Gene 783, 145463. 10.1016/j.gene.2021.145463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bechara EG, Sebestyén E, Bernardis I, Eyras E, and Valcárcel J (2013). RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Molecular cell 52, 720–733. 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 69.Cancer Genome Atlas Research, N. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ibrahimpasic T, Xu B, Landa I, Dogan S, Middha S, Seshan V, Deraje S, Carlson DL, Migliacci J, Knauf JA, et al. (2017). Genomic Alterations in Fatal Forms of Non-Anaplastic Thyroid Cancer: Identification of MED12 and RBM10 as Novel Thyroid Cancer Genes Associated with Tumor Virulence. Clin Cancer Res 23, 5970–5980. 10.1158/1078-0432.CCR-17-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG, Cancer Genome Atlas Research, N., Buonamici S, and Yu L (2018). Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep 23, 282–296 e284. 10.1016/j.celrep.2018.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gorlin RJ, Cervenka J, Anderson RC, Sauk JJ, and Bevis WD (1970). Robin’s syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am J Dis Child 119, 176–178. [PubMed] [Google Scholar]
- 73.Damianov A, Lin CH, Huang J, Zhou L, Jami-Alahmadi Y, Peyda P, Wohlschlegel J, and Black DL (2024). The splicing regulators RBM5 and RBM10 are subunits of the U2 snRNP engaged with intron branch sites on chromatin. Mol Cell 84, 1496–1511 e1497. 10.1016/j.molcel.2024.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Luhrmann R, and Stelzl U (2012). Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 45, 567–580. 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 75.He F, and Jacobson A (2015). Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu Rev Genet 49, 339–366. 10.1146/annurev-genet-112414-054639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurosaki T, Popp MW, and Maquat LE (2019). Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol 20, 406–420. 10.1038/s41580-019-0126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lykke-Andersen S, and Jensen TH (2015). Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16, 665–677. 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- 78.Cheruiyot A, Li S, Nonavinkere Srivatsan S, Ahmed T, Chen Y, Lemacon DS, Li Y, Yang Z, Wadugu BA, Warner WA, et al. (2021). Nonsense-Mediated RNA Decay Is a Unique Vulnerability of Cancer Cells Harboring SF3B1 or U2AF1 Mutations. Cancer Res 81, 4499–4513. 10.1158/0008-5472.CAN-20-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baird TD, Cheng KC, Chen YC, Buehler E, Martin SE, Inglese J, and Hogg JR (2018). ICE1 promotes the link between splicing and nonsense-mediated mRNA decay. Elife 7. 10.7554/eLife.33178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. (2015). Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3? Splice Site Selection through Use of a Different Branch Point. Cell Rep 13, 1033–1045. 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 81.Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O, and Krainer AR (2020). Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev 34, 413–427. 10.1101/gad.332270.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petermann E, Lan L, and Zou L (2022). Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat Rev Mol Cell Biol 23, 521–540. 10.1038/s41580-022-00474-x. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, and Zou L (2017). Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol Cell 65, 832–847 e834. 10.1016/j.molcel.2017.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, Zou L, and Graubert TA (2018). Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res 78, 5363–5374. 10.1158/0008-5472.CAN-17-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. (2018). The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell 69, 412–425 e416. 10.1016/j.molcel.2017.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lappin KM, Barros EM, Jhujh SS, Irwin GW, McMillan H, Liberante FG, Latimer C, La Bonte MJ, Mills KI, Harkin DP, et al. (2022). Cancer-Associated SF3B1 Mutations Confer a BRCA-Like Cellular Phenotype and Synthetic Lethality to PARP Inhibitors. Cancer Res 82, 819–830. 10.1158/0008-5472.CAN-21-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amon JD, and Koshland D (2016). RNase H enables efficient repair of R-loop induced DNA damage. Elife 5. 10.7554/eLife.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Catto LFB, Zanelatto LC, Donaires FS, de Carvalho VS, Santana BA, Pinto AL, Fantacini D, de Souza LEB, Fonseca NP, Telho BS, et al. (2023). Telomeric repeat-containing RNA is dysregulated in acute myeloid leukemia. Blood Adv 7, 7067–7078. 10.1182/bloodadvances.2023010658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu ZS, Sinha S, Bannister M, Song A, Arriaga-Gomez E, McKeeken AJ, Bonner EA, Hanson BK, Sarchi M, Takashima K, et al. (2024). R-Loop Accumulation in Spliceosome Mutant Leukemias Confers Sensitivity to PARP1 Inhibition by Triggering Transcription-Replication Conflicts. Cancer Res 84, 577–597. 10.1158/0008-5472.CAN-23-3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rombaut D, Lefevre C, Rached T, Bondu S, Letessier A, Mangione RM, Farhat B, Lesieur-Pasquier A, Castillo-Guzman D, Boussaid I, et al. (2024). Accelerated DNA replication fork speed due to loss of R-loops in myelodysplastic syndromes with SF3B1 mutation. Nat Commun 15, 3016. 10.1038/s41467-024-46547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guzikowski AR, Chen YS, and Zid BM (2019). Stress-induced mRNP granules: Form and function of processing bodies and stress granules. Wiley Interdiscip Rev RNA 10, e1524. 10.1002/wrna.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Asadi MR, Moslehian MS, Sabaie H, Poornabi M, Ghasemi E, Hassani M, Hussen BM, Taheri M, and Rezazadeh M (2021). Stress Granules in the Anti-Cancer Medications Mechanism of Action: A Systematic Scoping Review. Front Oncol 11, 797549. 10.3389/fonc.2021.797549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Biancon G, Joshi P, Zimmer JT, Hunck T, Gao Y, Lessard MD, Courchaine E, Barentine AES, Machyna M, Botti V, et al. (2022). Precision analysis of mutant U2AF1 activity reveals deployment of stress granules in myeloid malignancies. Mol Cell 82, 1107–1122 e1107. 10.1016/j.molcel.2022.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22, 672–678. 10.1038/nm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.North K, Benbarche S, Liu B, Pangallo J, Chen S, Stahl M, Bewersdorf JP, Stanley RF, Erickson C, Cho H, et al. (2022). Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat Biotechnol 40, 1103–1113. 10.1038/s41587-022-01224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]