

Summary
Protein glycation is a universal, non-enzymatic modification that occurs when a sugar covalently attaches to a primary amine. These spontaneous modifications may have deleterious or regulatory effects on protein function, and their removal is mediated by the conserved metabolic kinase Fructosamine-3-kinase (FN3K). Despite its crucial role in protein repair, we currently have a poor understanding of how FN3K engages or phosphorylates its substrates. By integrating structural biology and biochemistry, we elucidated the catalytic mechanism for FN3K-mediated protein deglycation. Our work identifies key amino acids required for binding and phosphorylating glycated substrates and reveals the molecular basis of an evolutionarily conserved protein repair pathway. Additional structural-functional studies revealed unique structural features of human FN3K as well as differences in the dimerization behavior and regulation of FN3K family members. Our findings improve our understanding of the structure of FN3K and its catalytic mechanism, which opens new avenues for therapeutically targeting FN3K.
Keywords: Kinase, Deglycation, Protein-repair
Graphical Abstract

eTOC Blurb
Lokhandwala et al. report the crystal structure of human FN3K, a kinase enzyme involved in protein deglycation and repair. Structural and functional analysis identified FN3K residues that influence substrate specificity and provided insight into the process of FN3K-mediated deglycation. These findings advance our understanding of early glycation repair mechanisms.
Introduction
In living organisms, proteins are constantly exposed to reactive cellular metabolites that can alter their structure and function.1–3 Reducing sugars such as glucose can spontaneously react with free amine groups on proteins to form an early glycation product known as a Schiff base. 4–6 Over time, early glycation products can undergo Amadori rearrangements to form more stable fructosamine or amadori products. The formation of initial glycation products such as Schiff’s base and ketosamines are reversible. However, Schiff base and fructosamine products can participate in various irreversible reactions, eventually forming a heterogeneous group of modified protein species known as advanced glycation end products (AGEs) (Figure S1A).7,8 In the past few decades, it has become well-accepted that this chemical process is linked to aging, diabetes, neurodegenerative disorders, osteoarthritis, cancer, and atherosclerosis.9–12 To counter the effects of glycation, organisms have evolved deglycation mechanisms, including a highly conserved protein repair enzyme that reverses protein glycation.13–15 In contrast to AGEs, which are thought to be irreversible, early glycation events can be repaired by Fructosamine-3-kinase (FN3K).
FN3K homologs are present in all taxa suggesting that deglycation is an ancient protein repair mechanism. Simple eukaryotes and prokaryotes contain a single copy of the FN3K gene, whereas complex eukaryotes, including mammals, encode two copies, FN3K and FN3K-related protein (FN3K-RP).16 FN3K and FN3K-RP are unique “hybrid” kinase/deglycase enzymes that mediate deglycation by phosphorylating the third carbon of the sugar moiety.13,17 Due to the proximity of the sugar’s keto group and the phosphate, this phosphoester linkage is labile and readily decomposes through β-elimination to regenerate an unmodified amino acid, inorganic phosphate, and 3-deoxyglucosone (3DG) (Figures 1A and S1B).14,18,19 Human FN3K-related protein (HsFN3KRP) shares 65% sequence identity with human FN3K (HsFN3K) but can only phosphorylate ribulosamines whereas HsFN3K can phosphorylate both ribulosamines and fructosamines.15, 16,20 The conservation of FN3K and FN3KRP underscores their important biological role in regulating glycation events; however, we currently have a poor mechanistic understanding of how FN3K engages and phosphorylates its substrates at a molecular level.
Figure 1: Crystal structure of HsFN3K.
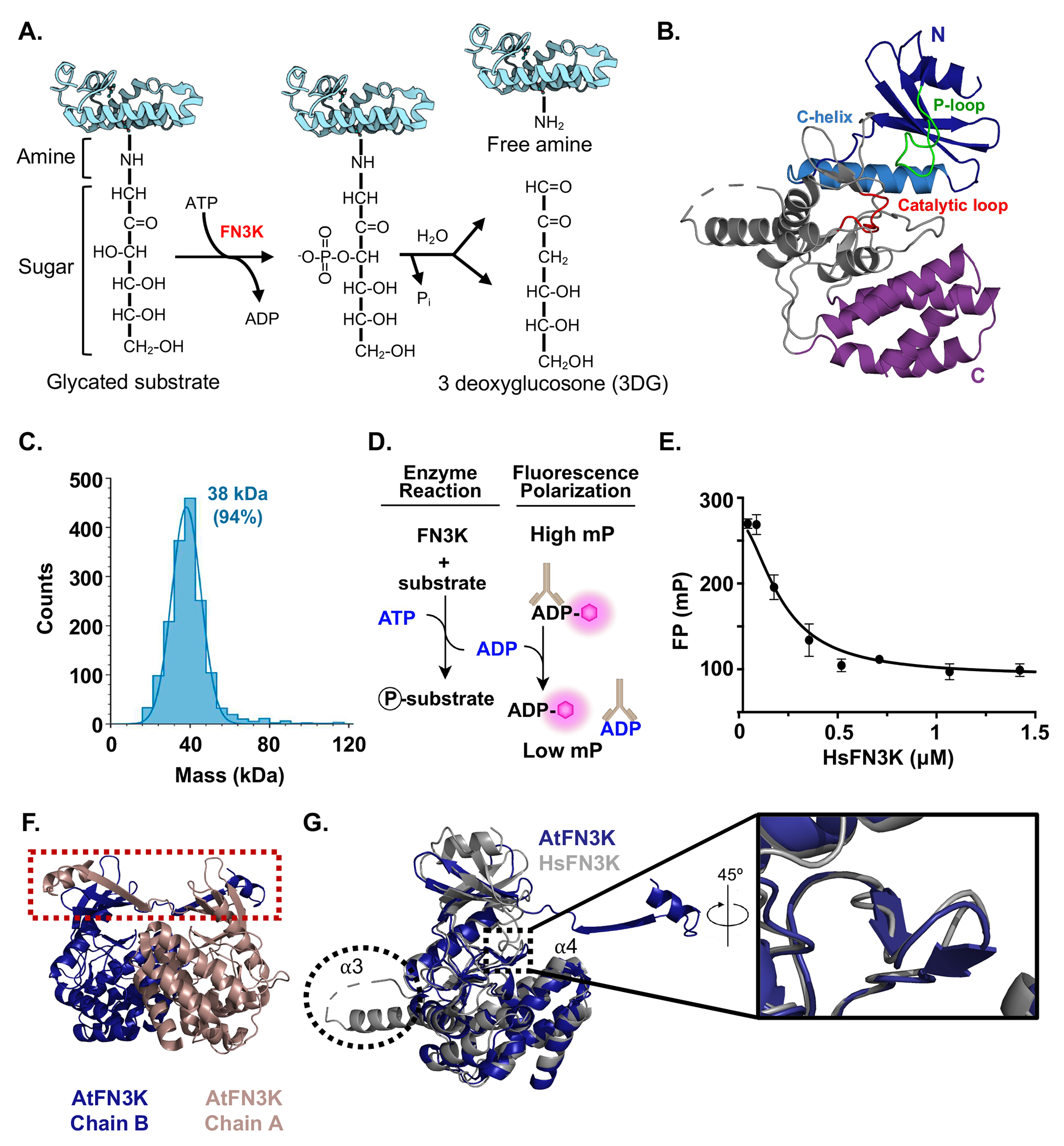
A) A schematic of FN3K deglycation reaction. B) Ribbon diagram of HsFN3K. The N-terminal lobe is shown in blue; the C-helix in marine blue, and the P-loop in green. The C-terminal lobe is divided into two regions, the catalytic core shown in grey, which contains the catalytic loop shown in red, and the helical sub-domain shown in purple. C) Mass photometry of HsFN3K under reducing conditions. Calculated MW is 38 kDa for 94% of the sample. D) Schematic of the fluorescence polarization assay used to measure kinase activity. E) Kinase activity of HsFN3K. HsFN3K was titrated in the indicated concentration range and ADP production was detected by fluorescence polarization. Each data point represents means of triplicates; error bars indicate standard deviation. F) A ribbon diagram of the AtFN3K dimer (PDB:6OID). The β-strand exchange is highlighted in the red box. G) Alignment of HsFN3K and AtFN3K. The extended α3 helix in HsFN3K is circled and the AtFN3K β-hairpin is highlighted in the inset. See also Figures S1 and S2.
From a clinical perspective, dysregulation of HsFN3K and HsFN3K-RP is implicated in human diseases associated with elevated levels of glycation.21–23 Early studies showed that Fn3k-deficient mice exhibit elevated glycation levels, yet these mice displayed no other adverse phenotypes.24 This is in contrast to other protein repair enzymes25; however, it is unclear if HsFN3K-RP serves a redundant, compensatory role in Fn3k-deficient mice. In humans, polymorphisms in FN3K are associated with variations in HbA1c levels and the onset of type 2 diabetes mellitus (T2DM).23 Recently, there has been a growing interest in targeting FN3K for age-related macular degeneration (AMD) and cancer therapy.26–29 For the former, enzymatic treatment with FN3K reduced AGE-related retinal autofluorescence and could serve as a potential treatment option against AMD. For the latter, it has been shown that the transcriptional activity of nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of oxidative stress, depends on deglycation by HsFN3K. Depletion of HsFN3K reduced tumor growth in NRF2-driven lung and liver tumors,27 suggesting that HsFN3K inhibition holds therapeutic potential for treating NRF2-driven tumors. These studies highlight the importance of regulating HsFN3K activity to maintain cellular homeostasis. It is worth noting that in the process of recovering lysine from its glycated derivative, HsFN3K produces 3-deoxyglucosone (3DG), a glycating agent and generator of oxidative stress.30–32 Therefore, inhibiting FN3K activity may result in the accumulation of glycated proteins, while activating FN3K might result in the accumulation of 3DG. This highlights the paradoxical role of FN3K in protecting cells against glycation and presents a potential double-edged sword when developing FN3K therapeutics. To realize the full therapeutic potential of FN3K, we need to develop a comprehensive understanding of FN3K enzymatic activity and cellular regulation.
To gain deeper insights into FN3K-mediated deglycation, we sought to molecularly define how FN3K binds and phosphorylates its substrates and carries out its catalytic function. FN3K belongs to a large superfamily of protein kinase-like (PKL) enzymes that include eukaryotic protein kinases (ePKs), small molecule kinases, and atypical kinases.33,34 FN3K is more similar to small molecule kinases such as aminoglycoside kinases (APH) than ePKs.33,34 While numerous studies have mechanistically interrogated APH activity, there are key functional differences between APH and FN3K enzymes that limit our ability to translate the findings from APH studies to FN3K. To address this limitation, we determined the crystal structure of HsFN3K to 2.9 Å. By combining the structural insight gained from the HsFN3K structure with functional binding and catalytic studies, we have determined unique structural features conserved in vertebrate FN3Ks. Through molecular docking, we identified key catalytic residues and elucidated the mechanism of action for FN3K deglycation. Our improved understanding of the structure of FN3K and its catalytic mechanism opens new avenues for designing small molecule inhibitors to treat AGE-related disorders and NRF2-driven tumors.
Results
Crystal structure of human FN3K.
Here we reveal the crystal structure of human FN3K (HsFN3K) to 2.9Å resolution (Figures 1B, and Table S1). FN3K has been previously reported to form an inactive dimer under non-reducing conditions, therefore, we conducted all experiments under strong reducing conditions.35 HsFN3K purified as a monomer (Figure 1C) and retained catalytic activity in the presence of a chemical substrate, 1-Deoxy-1-morpholino-fructose (DMF) (Figures 1D, and 1E). Our initial attempts to determine the structure of HsFN3K were unsuccessful as has been previously reported.35 To overcome this, we reductively methylated HsFN3K, which allowed HsFN3K to form needle-like crystals and did not impair its catalytic activity (Figure S2).
HsFN3K crystallized with one molecule in the asymmetric unit. No stable dimer is observed in the crystal structure, which is in line with the fact that monomeric HsFN3K was used for crystallization screening. The crystal structure revealed a two-domain architecture consisting of an N-terminal (1-127) and a C-terminal (163-309) lobe connected by a long linker (128-162) (Figure 1B). The overall HsFN3K organization is a canonical protein-kinase-like fold (PKL). The N-terminal domain comprises residues critical for ATP binding, whereas the C-terminal domain comprises residues required for catalysis (Figure 1B). The P-loop depicted in green plays a critical role in coordinating ATP. In our apo structure, the absence of the nucleotide caused this loop to close over the ATP binding pocket. Due to the high mobility of this region, the electron density is weak, which is consistent with other kinase structures in the absence of the nucleotide.36,37
Though the overall HsFN3K organization is a canonical PKL fold, it displays notable sequence divergence from the closely related small molecule kinases aminoglycoside kinase/phosphotransferase (APH) ~23% identity, choline kinases ~22% identity, and Arabidopsis thaliana FN3K (AtFN3K) ~38% identity). A detailed comparison of the HsFN3K and AtFN3K structures revealed several differences (Figures 1F, and G). First, AtFN3K crystallized as an inactive, strand-exchange dimer, whereas HsFN3K crystallized as a monomer (Figure 1F). Second, there is a small β-hairpin present in AtFN3K (N132-Q141) that is not found in HsFN3K (Figure 1G). Though these residues have not been reported to play a role in catalysis, they do point toward the substrate binding cavity. Third, HsFN3K exhibits an extension from residues 116-138 that is absent from plant and bacterial FN3Ks (Figures 1G and S3). Based on the sequence alignment of FN3Ks from various taxa, this extension is found across vertebrate FN3Ks (Figures 2A and S4). The HsFN3K crystal structure revealed that the first 11 amino acids of this extension contribute to an extended α3 helix and that the remaining residues contribute to a large loop connecting α3 and α4 helix. Eight residues in this loop are not modeled due to weak electron density. The biological purpose of the extended loop, if any exists, remains unclear.38 Altogether, these findings indicate that HsFN3K possesses unique structural features that could be related to its specific regulation.
Figure 2. Loop insertion in HsFN3K is dispensable for catalytic activity.

A) Multiple sequence alignment of FN3K orthologs. B) HsFN3K deletion and domain swap constructs. C) The top panel highlights the distinct structural features of HsFN3K from AtFN3K. The 23 amino acid extension (116-138) is shown in green. The HsFN3K loop region (147-156) that corresponds to a β-hairpin (132-140) in AtFN3K (blue) is shown in orange. The lower panel depicts the swap constructs made by replacing the HsFN3K loop region (orange and gray) with the corresponding AtFN3K region (blue). D) Kinase activity of MBP-HsFN3K WT, Δ116-138, and SWAP1. Each data point represents means of triplicates; error bars indicate standard deviation. E) Reaction velocity as a function of DMF concentration for MBP-HsFN3K WT and Δ116-138. Error bars represent the standard error of the mean of at least two independent experiments. F) Kinetic parameters of the Michaelis-Menten fit as determined in (E). See also Figures S3–S5.
Loop insertion in HsFN3K is dispensable for catalytic activity.
To investigate the role of unique structural features of HsFN3K on its catalytic activity, we generated four deletion and domain swap mutants (Figures 2A–B and S5A). HsFN3K Δ116-138 and Δ147-156 correspond to deleting the 23 amino acid extension and the 9 amino acids equivalent to a β-hairpin in AtFN3K, respectively. FN3K SWAP1 replaced the loop segment (147-156) of HsFN3K with the corresponding β-hairpin of AtFN3K (132-141). In case neighboring residues in AtFN3K support the β-hairpin formation, we generated FN3K SWAP2, which replaced the entire loop segment (139-162) with the corresponding residues in AtFN3K (123-146) (Figure 2C). All four constructs readily expressed in bacteria as MBP fusion constructs, however, MBP-HsFN3K Δ147-156 and SWAP2 formed soluble aggregates that eluted in the void volume by size-exclusion chromatography (SD200 10/300, void volume corresponds to ≥ ~1,600 kDa) (Figure S5B). In contrast, MBP-HsFN3K Δ116-138 and SWAP1 expressed and purified similar to wildtype MBP-HsFN3K allowing us to assess their catalytic activity. MBP-HsFN3K Δ116-138 showed a 3.3-fold decrease in activity compared to the wildtype, and MBP-HsFN3K SWAP1 completely abolished catalytic activity (Figures 2D–2F). Together our data establish that the loop insertion (116-138) is dispensable for catalytic activity, whereas the HsFN3K loop spanning residues 147-156 has an essential role for both catalytic activity and protein stability.
Docking model of the HsFN3K-substrate complex reveals residues for deglycation.
To further identify molecular features of HsFN3K that govern catalysis, we computationally docked ATP, Mg ion, and DMF into the putative substrate binding pocket of HsFN3K (Figure 3A). Since molecular docking generates a list of energetically favorable docking poses, the top 10 poses were further evaluated based on the following criteria: (1) The hydroxyl group on C3 of DMF should be within the coordinating distance of either D217 or D234 or both, as these are presumed to play a role in catalysis based on sequence alignment, (2) the hydroxyl group on C3 of DMF should be located near the ATP binding pocket in order to attack the gamma phosphate and (3) The C=O moiety at C2 of DMF should orient toward the solvent in order to conjugate with a lysine residue of an FN3K substrate. Based on our docking model, DMF was oriented in a cleft between the HsFN3K catalytic core and the C-terminal helical sub-domain. Our docking model showed MgATP bound to HsFN3K in a highly conserved pocket similar to ePK and small molecule kinases.38,39 The docking model illustrated that the conserved F39 residue stabilizes the adenine ring of ATP through π-stacking interactions. The conserved E55 residue forms a salt bridge with K41 and polarizes it for electrostatic interaction with the phosphate groups of ATP similar to other kinases (Figures 3B and 3D).40 The C3 hydroxyl group of DMF points directly toward the gamma phosphate of ATP and is in a ready position for in-line attack on the phosphorus atom. Both C3 and C4 hydroxyl groups of DMF are within the hydrogen bonding distance of the gamma phosphate, suggesting a direct interaction between DMF and ATP (Figure S6A). HsFN3K residues D234 and N222 are highly conserved and correspond to residues important for Mg2+ ion coordination in other kinase family members.40,41 In our docking model, D234 and N222 coordinate the Mg2+ ion. In addition, D234 forms an ionic interaction with K41 and polarizes it for interaction with ATP. As D234 is 3.9 Å away from the C3 hydroxyl group of DMF in the docking model, it is close enough to serve as the catalytic base. However, given the role it plays in coordinating MgATP in the active site, it is unlikely to act as the base. On the other hand, the D217 side chain is situated 2.7 Å from the C3 hydroxyl group of DMF (Figure S6A). D217 belongs to the well-conserved Brenner motif and most likely functions as a catalytic base (Figures 3C–D).39,42–44.
Figure 3. Identification of residues critical for enzyme activity and ligand binding.

A) Docking model of the HsFN3K-ATP-DMF complex with ATP and DMF. B) Detailed view of the nucleotide-binding pocket of HsFN3K with docked ATP and conserved residues involved in nucleotide binding shown as sticks. C) A detailed view of the catalysis pocket of HsFN3K. D) Logo plots for FN3K amino acids involved in nucleotide and substrate binding. The sequence numbering is according to HsFN3K. The amino acids starred at the bottom were mutated to alanine and their kinase activity was determined. E) Kinase activity of WT, F39A, E55A, D217A, D234A, N222A and MBP K41A. Each data point represents means of triplicates; error bars indicate standard deviation. See also Figure S6.
To experimentally validate our docking model, residues predicted to coordinate MgATP and DMF binding were mutated to alanine and assessed for catalytic activity. E55A and F39A mutants retained activity similar to wildtype HsFN3K establishing that these residues are dispensable for catalysis (Figures 3E, S6B and S6C). HsFN3K mutants D234A, N222A, and K41A completely abolished activity, which supports their role in MgATP binding (Figure 3E). Of note, the kinase assay was performed with the MBP-tagged HsFN3K K41A as cleavage of the MBP-tag prevented us from purifying this mutant. D217A reduced enzyme activity 58-fold compared to wildtype HsFN3K, but it did not completely abolish activity (Figure S6C). Given that this assay measures the conversion of ATP to ADP, it is possible that the D217A mutation retains a low level of ATP hydrolysis. To address this, we performed direct substrate phosphorylation assays. HsFN3K WT was able to phosphorylate glycated lysozyme; however, no substrate phosphorylation was observed for D217A (Figure S6D). Thus, we cannot rule out the possibility that D217A retains a low level of ATP hydrolysis. Together, these results establish the critical role of D234, N222, K41, and D217 in FN3K-mediated deglycation.
Binding and kinase assays suggest that both D217 and D234 are important for catalysis.
Based on our model, we predicted that D234 is involved in MgATP and substrate binding whereas D217 acts as the catalytic base for HsFN3K. To directly assess ATP and substrate binding, we performed a differential scanning fluorimetry (DSF) assay to measure ATP and DMF-mediated thermal stabilization of HsFN3K. HsFN3K has a melting temperature (Tm) of 56.9 ± 0.7°C (Figure 4A). The addition of ATP increased the Tm by ~6°C (63.2 ± 0.5°C), indicating that ATP binding stabilizes the HsFN3K structure. The addition of DMF alone did not alter the Tm of HsFN3K, but the addition of both ATP and DMF resulted in a ~2°C (65.2 ± 0.6°C) shift compared to HsFN3K in the presence of ATP alone (Figure 4A). Given that ATP was required for substrate binding, we also assessed whether an ATP mimetic would perturb substrate recognition. Similar to ATP, adenylyl-imidodiphosphate (AMP-PNP) readily bound to HsFN3K, however, the presence of this ATP mimetic prevented substrate engagement (Figure S7). Together, these data establish that binding to both ATP and substrate stabilizes HsFN3K, ATP binding is a prerequisite for substrate binding, and the active site chemistry is tightly regulated as subtle changes in ATP binding perturbed substrate binding. These experimental observations are also corroborated by our docking model showing direct engagement of C3 and C4 hydroxyl groups of DMF with ATP gamma phosphate (Figure S6A). In the absence of ATP, these direct hydrogen bonding interactions are severed, and thus, DMF binding is abolished.
Figure 4. HsFN3K D217 and D234 are important for catalysis.

DSF thermal unfolding curves of HsFN3K A) WT, B) D217A, and C) D234A in the absence (black) or presence of ATP (blue), DMF (teal), or both ATP and DMF (red). MST binding curves for HsFN3K D) WT E) D217A, and F) D234A interacting with ATP (blue), DMF (teal), ATP and DMF (red). Each data point represents means of triplicates; error bars indicate standard deviation. See also Figures S7–S9.
To identify whether D217 or D234 contributed to ATP and/or substrate binding, we performed the DSF assays with alanine substitution mutants. D217A retained the ability to bind ATP but lost binding to DMF (Figure 4B). In contrast, the D234A mutant displayed no observable Tm shift in the presence of ATP or DMF individually or together (Figure 4C). To quantitatively assess HsFN3K binding to ATP and DMF, we fluorescently labeled HsFN3K, D217A, and D234A and performed microscale thermophoresis (MST). HsFN3K bound to ATP with a binding affinity (KD) of 3.9 ± 3.4 μM (Figure 4D). HsFN3K bound to DMF with a binding affinity (KD) of 3.7 ± 0.7 μM in the presence of ATP but did not bind DMF in the absence of ATP, which is in agreement with our DSF binding results (Figure 4D). D217A bound ATP similar to wildtype HsFN3K with a binding affinity (KD) of 3.5 ± 2.3 μM but lost the ability to bind DMF (Figure 4E). D234A impaired binding to both ATP and DMF (Figure 4F). As we have established that ATP binding to FN3K is a prerequisite for substrate binding, we posit that the D234A mutation abolishes substrate binding due to its impaired MgATP binding. To further probe the role of D217 and D234 in enzyme activity, these residues were mutated to glutamate (E), glutamine (Q), or asparagine (N). The glutamate mutant conserves the charge but not the position, the asparagine mutant is isosteric but without appreciable charge, and the glutamine mutant has a similar charge to asparagine and positioning similar to glutamate. All D234 mutants lacked any detectable activity, underscoring the critical nature of this residue in catalysis. The D217E mutation was the only mutation to retain kinase activity, whereas D217Q was catalytically dead and D217N was misfolded (Figure S8). These data establish that D217 coordinates substrate binding and positions the sugar in the active site for catalysis. Further, the flexible motion of the D217 side chain and proximity to the C3 hydroxyl group suggest that D217 serves as the catalytic base. In this case, D234 would be primarily responsible for binding MgATP and DMF (Figure S9).
The substrate binding pocket is highly conserved across the FN3K family.
Conservation analysis of HsFN3K showed that the substrate binding pocket is highly conserved among the FN3K family of kinases (Figures 5A, and S10). To identify key residues involved in substrate recognition, we focused our search on highly conserved amino acids in close proximity to DMF based on our structural analysis of the HsFN3K-DMF complex. We initially focused on aromatic amino acids as they are often preferred in the carbohydrate-binding sites of proteins.45 Tryptophan, Tyrosine, and Histidine residues are frequently found at carbohydrate binding sites in the order of Tryptophan>Tyrosine>Histidine.45 FN3K contains a highly conserved Trp219 (W219) residue on the catalytic loop between β5 and β6. Our docking model revealed that the C3 hydroxyl group of DMF is poised to form a hydrogen bond (2.9 Å) with the W219 side chain. The C5 hydroxyl group of DMF is 3.8 Å from H291 while the C6 hydroxyl group is 3.2 Å away from H288, indicating they could interact with H288 and H291 side chains. In addition, the nitrogen atom on the morpholine ring of DMF is within hydrogen bond distance (2.9Å) with H288, while the morpholine ring itself is snugly sandwiched in a hydrophobic pocket with its oxygen atom positioned right at the protein-solvent interface (Figure 5A). To investigate the role of these residues in catalysis and substrate recognition, HsFN3K W219, H288, and H291 were mutated to alanine and assessed for catalytic activity and substrate binding. The W219A mutant showed a complete loss of enzyme activity, indicating it has an essential role in DMF binding as supported by our docking model. In contrast, H288A and H219A retained some catalytic activity, but these mutants were 13-fold and 11-fold less active than the wildtype HsFN3K, respectively (Figures 5B–5D). The Km for DMF for both H288A and H291A mutants increased by 3-fold and 3.5-fold, respectively, indicating their role in substrate binding (Figure 5C and 5D). While all three HsFN3K mutants show impaired catalytic activity, they retained the ability to bind ATP as determined by DSF (Figures 5E–5G). In agreement with our kinase assay, W219A did not bind DMF (Figures 5E, and S11), whereas H288A and H291A bound DMF resulting in a modest Tm shift of 0.6°C and 3.6°C, respectively (Figures 5F, 5G, and Table. S3). Collectively, our results determine that W219 is required for substrate binding and H288 and H291 contribute to substrate binding. Furthermore, the highly conserved nature of these amino acids suggests that they are key substrate-binding residues in other FN3K orthologs.
Figure 5. The substrate binding region is highly conserved in the FN3K family.

A) Conservation analysis of HsFN3K amino acid residues. The zoom panel displays the HsFN3K substrate binding with predicted substrate binding residues shown as sticks. B) Kinase activity of WT, W219A, H288A, and H291A Each data point represents means of triplicates; error bars indicate standard deviation. C) Reaction velocity as a function of DMF concentration for WT, H288A, and H291A was measured using ADP-Glo assay. The inset shows the H288A and H291A data again for better visualization. Error bars represent the standard error of the mean of at least two independent experiments. D) Kinetic parameters of the Michaelis-Menten fit as determined in (C). DSF thermal unfolding curves of HsFN3K E) W219A, F) H288A, G) H291A in the absence (black) or presence of ATP (blue), DMF (teal), or both ATP and DMF (red). Each data point represents means of triplicates; error bars indicate standard deviation. See also Figures S10 and S11.
HsFN3K has less propensity to dimerize than AtFN3K.
Previous reports have shown that AtFN3K predominantly forms a dimer in solution, especially under non-reducing conditions.35 To explore the propensity of HsFN3K to form a dimer in solution, we purified HsFN3K under non-reducing conditions. Size-exclusion chromatography revealed that HsFN3K elutes as a mixture of monomers and dimers in the absence of reducing reagents, with the monomeric species remaining the predominant population (Figure 6A). Mass measurements of the HsFN3K dimer species by liquid chromatography-mass spectrometry (LC-MS) confirmed the molecular weight to be dimeric (70.34 kDa). Moreover, the addition of a reducing reagent to the HsFN3K dimer species disrupted dimerization and resulted in a predominantly monomeric population by LC-MS (Figures S12A–D). These data establish that HsFN3K exists in a monomeric state under reducing conditions and that the dimerization is facilitated by disulfide bond formation. This is distinct from AtFN3K, which can dimerize when all reactive cysteine residues are mutated to alanine.35
Figure 6. HsFN3K has less propensity to dimerize than AtFN3K.

A) Size-exclusion chromatography profile of HsFN3K purified under non-reducing conditions (black) and reducing conditions (grey). The inset shows the SDS-PAGE of dimer and monomer fractions in the presence or absence of DTT. B) The Kcat values were determined from the ADP-Glo kinase assay of the HsFN3K monomer and dimer fractions. Error bars indicate standard error of the mean of at least two independent experiments. C) The top panel shows the surface representation of the AtFN3K dimer, and the bottom panel shows the surface representation of the AtFN3Kmut dimer. Residues involved in the dimer interface were identified with Protein Interfaces, Surfaces, and Assemblies (PISA) and highlighted in orange and green, respectively. D) Conservation analysis of AtFN3K dimer interface. The AtFN3K dimer is shown as a surface representation and the dimer interface residues are colored based on their conservation across different FN3K orthologs. See also Figures S10, S12, and S13.
To further test the sensitivity of HsFN3K kinase activity to redox conditions, we evaluated the catalytic activity of the HsFN3K monomer and dimer purified under reducing (2 mM DTT) or non-reducing conditions, respectively. We found that the HsFN3K monomer (Kcat = 1.18 ± 0.031 min−1) was 2.4-fold more active than the HsFN3K dimer (Kcat = 0.5 ± 0.012 min−1) (Figures 6B, and S12E–F). This is in contrast to AtFN3K, which displays a 40-fold increased catalytic activity in the presence of reducing reagents.35 Together, these biochemical studies highlight key differences in dimerization behavior between HsFN3K and AtFN3K.
To investigate the molecular features governing dimer formation, we analyzed contacts within the ATFN3K dimer interface,47 which revealed that AtFN3K dimerization is supported by two interchain disulfide bonds and 36 hydrogen bonds. As AtFN3K has been reported to dimerize in the absence of disulfide bonds, we posit that these hydrogen bonds are the primary determinants of dimerization. We also modeled HsFN3K residues onto the AtFN3K dimer interface (AtFN3Kmut) to compare the dimer-forming contacts between the HsFN3K and AtFN3K proteins. We found that the modeled AtFN3Kmut dimer has 9 fewer hydrogen bonds and 1 less disulfide bond than the AtFN3K dimer (Figure S13). The spatial arrangement of residues at the dimer interface (Figure 6C), shifted from clustered (orange) to dispersed (green) due to the loss of hydrogen bonds. This change in spatial arrangement, coupled with the overall loss of hydrogen bonds, likely explains the preference for homodimerization in AtFN3K.46 These findings also coincide with our functional studies indicating that HsFN3K has a lower propensity for dimerization than AtFN3K.
Finally, we performed a conservation analysis of the AtFN3K dimer interface to investigate whether dimer-forming residues are present in other FN3K orthologs. Of the 22 residues that mediate homodimerization of AtFN3K, six are conserved across the FN3K family of kinases. Three of the conserved residues are on the P-loop (G31, I33, and N34). The dimer interface residues at the β-strand exchange region are not well-conserved except for residues A36 and D42 (Figure 6D). Most of the AtFN3K contacts are found in this region, which could explain why AtFN3K can dimerize in the absence of disulfide bond formation, but HsFN3K cannot. Further, C236 residue which is involved in one of the disulfide linkages at the AtFN3K dimer interface is conserved in plants but absent in other species which further supports AtFN3K’s higher redox sensitivity than HsFN3K (Figure S10C). These findings provide valuable insights into the dimerization behavior and regulation in HsFN3K and AtFN3K enzymes, highlighting the functional differences between these orthologs and suggesting that the redox sensitivity and the tendency to form a dimer will vary across the larger FN3K family of kinases.
Discussion
Here, we present the crystal structure of human FN3K (2.9Å), an enzyme that mediates protein repair through deglycation. The structure of HsFN3K allowed us to visualize a mammalian fructosamine kinase in a monomeric conformation. Our structure-function studies enabled us to assess the ability of human FN3K to mediate deglycation and ultimately determine the mechanism of action for FN3K. Structural analysis of HsFN3K identified both similarities and key differences between the molecular assemblies of HsFN3K and AtFN3K. We observed that both FN3Ks adopt the same overall protein fold and that the structural differences, particularly those of functional relevance, were located within loop regions, especially the loop region where HsFN3K exhibited a loss of secondary structure residues (147-156) corresponds to a small β-hairpin in AtFN3K. We also determined that the 23-amino acid extension in HsFN3K, which is conserved in mammalian FN3Ks but not in AtFN3K, exhibited minimal effect on FN3K enzymatic activity. Because this region does not play a significant role in catalysis, it remains unclear if this extension confers novel function(s) for FN3Ks. As previous studies have shown loop regions can impart diverse functions and substrate promiscuity to enzymes,47,48 we cannot rule out a functional role for this extension in a cellular context, despite it being dispensable for catalysis. Additional studies, especially emerging research to better understand the physiological substrates of FN3K, will be critical in understanding the functional importance, if any, of this region for FN3K-mediated protein repair. Based on the high degree of structural similarity between HsFN3K and AtFN3K, and on the observation that FN3K family members have an overlapping substrate preference for ribulosamines, we anticipate that the FN3K family members share a common protein fold. This finding will facilitate efficient homology modeling of additional FN3Ks to better predict how mutations contribute to the overlapping, but distinct substrate preference between FN3K and FN3K-RPs.
To date, there is no structure of an FN3K enzyme bound to its substrate. Nearly everything we know about the mechanism of action for FN3K is derived from studying its substrates and reaction products. Despite numerous efforts, our attempts to crystallize HsFN3K in the presence of ATP and DMF were unsuccessful. Through our biochemical studies, we demonstrated that ATP is a prerequisite for FN3K to bind substrates. Thus, our structural and functional studies reveal that FN3K follows a sequential bi-bi mechanism wherein the enzyme first binds ATP followed by the fructosamine substrate. Further, DSF studies exhibited a shift in temperature upon the formation of a ternary complex between FN3K, ATP, and DMF. We also discovered that ATP and substrate coordination are tightly regulated as the presence of an ATP analog, AMP-PNP, impairs substrate binding (Figure S7). These findings provide a potential explanation as to why the HsFN3K-substrate complex has remained refractory to structural efforts.
In the absence of a HsFN3K-substrate complex structure, computational modeling of the HsFN3K-ATP-DMF complex allowed us to visualize this interaction and identify key residues relating to the mechanism of phosphoryl transfer. Previous kinase studies have shown that the aspartate within the well-conserved Brenner motif often serves as the catalytic base.39,42–44 Based on this, HsFN3K D217 would serve as the putative base for catalysis. However, our docking model identified another highly conserved aspartate, D234, which is also positioned to potentially serve as a proton shuttle. This in conjunction with our functional studies, allowed us to propose a model for FN3K-mediated deglycation where D217 coordinates the substrate and D234 coordinates MgATP in the active site. Given the common role of the Brenner motif aspartate and the role of D234 in binding MgATP, we posit that D217 will serve as the catalytic base to deprotonate the hydroxyl group of the third carbon of DMF. After deprotonation, the oxygen group on the DMF acts as a nucleophile attacking the γ-phosphate of ATP (Figure S9). Phosphorylation of sugar destabilizes its linkage to the amine, and it decomposes to generate inorganic phosphate and 3DG. Alternatively, FN3K could use a dissociative phosphoryl transfer mechanism similar to aminoglycoside antibiotic kinase APH-(3′)-IIIa. In this case, D217 would act as a proton trap to orient the substrate hydroxyl group for attack on the γ phosphate of ATP, and the phosphate transfer would occur before substrate deprotonation.49 In turn, D234 would support catalysis by aiding in the formation of a metaphosphate transition state intermediate for phosphoryl transfer to occur before substrate deprotonation. While our data does not establish if FN3K utilizes an associative or dissociative phosphoryl transfer mechanism, the fact that ATP is required for substrate binding and the presence of AMP-PNP disrupts substrate binding suggests that the transition state chemistry is tightly regulated for FN3K.
Finally, early studies demonstrated that HsFN3K and HsFN3K-RP are constitutively expressed in cells and their transcriptional levels are insensitive to several diabetes-associated stimuli.50 This raised the question as to how HsFN3K activity is regulated, if at all. It was recently reported that dimerization may regulate the activity of FN3K, particularly through a redox-sensing mechanism. Structural and functional studies with AtFN3K identified reactive cystines that confer redox sensitivity by modulating both dimer formation and catalytic activity.35 The P-loop redox-active cysteine is present in both AtFN3K and HsFN3K, and Shrestha et al. showed that HsFN3K can form higher-order oligomers when cells are exposed to a thiol-oxidizing agent.35 While we too found that HsFN3K can dimerize under oxidizing conditions, HsFN3K remains predominantly monomeric in solution. Consistent with this finding, our biochemical studies established that the dimer species of HsFN3K exhibited a modest decrease in enzyme activity compared to the monomeric species. This is in stark contrast to AtFN3K, which shows a 40-fold change in activity in the presence of a reducing agent. We further assessed the conservation of dimer-forming amino acids across the FN3K family and observed that many of the AtFN3K dimer contacts are weakly conserved, suggesting that dimerization may not be a universal regulatory mechanism for FN3K family members. AtFN3K localizes to the chloroplast and HsFN3K has been reported to localize to the mitochondria and found in red blood cells and serum.13,51 It is possible that FN3Ks from different cellular compartments evolved distinct regulatory mechanisms. This may also explain the different glycation patterns observed in plant and human proteomes.52–56 Thus, the propensity of FN3Ks to dimerize and undergo redox regulation of their catalytic activity will vary from species to species. The functional consequences of this and whether this regulation is cellular compartment-specific will require additional studies.
In conclusion, we have structurally and functionally characterized HsFN3K, a conserved protein repair enzyme, to reveal key amino acid residues required for the HsFN3K-substrate binding and catalysis. This work advances our molecular understanding of FN3K-mediated deglycation and reveals insight into the differential regulation of FN3K activity in FN3K orthologs. Collectively, these studies will provide new opportunities for structure-based drug design to therapeutically target HsFN3K in diseases associated with elevated glycation
STAR★Methods
Resource availability
Lead contact
Requests of information and requests for reagents should be directed and will be fulfilled by the lead contact, Jennifer M. Binning (Jennifer.binning@moffitt.org)
Materials availability
All unique reagents generated in this study will be made available by the lead contact.
Data and code availability
The HsFN3K crystal structure has been deposited in the Protein Data Bank under the accession number: 8UE1 and is publicly available as of the date of publication. The accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| DH5a Competent Cells (E. coli) | NEB | C2987H |
| BL21(DE3) Competent Cells (E.coli) | NEB | C2527 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Luria Broth | RPI | L24400-5000.0 |
| IPTG | Chem Impex | 00194-100g |
| Ampicillin | Fisher Scientific | BP1760-25 |
| Carbenicillin | Gold Biotechnology | 50-153-2751 |
| Chloramphenicol | Fisher Scientific | AC227921000 |
| L-(+)-Arabinose | Sigma Aldrich | A3256-100G |
| Chaperone Plasmid Set | Takara Biotech | 3340 |
| Sodium Chloride | RPI | S23025-3000.0 |
| Tris Base Ultra-Pure | RPI | T60040-5000.0 |
| HEPES | RPI | H75030-1000.0 |
| Imidazole | Fisher Scientific | AAA102210E |
| DTT | RPI | D11000-50.0 |
| TCEP | Thermo Scientific | PI20491 |
| NP-40 | Sigma Aldrich | I3021-100ML |
| Fluorescein-5-maleimide | Fisher Scientific | 50-187-4590 |
| DMAB | Acros Organics | AC177310250 |
| Paraformadehyde | Thermo Scientific | 28906 |
| Magnesium Chloride | RPI | M24000-500.0 |
| Adenosine-5-triphosphate disodium salt | Chem Impex | 00015-25G |
| Adenosine-5-diphosphate disodium salt | RPI | A11220-10.0 |
| Adenylyl-imidodiphosphate salt | Sigma-Aldrich | SIAL-10102547001 |
| Polyethylene-glycol-3350 | Hampton Research | HR2-527 |
| 4-15% Mini Protean Stain free protein gels | Bio-Rad | 4568086 |
| Coomassie Brilliant Blue- R250 | Thermo Scientific | 20278 |
| 4X SDS-Sample buffer | Fisher Scientific | BP-110R |
| Glycine | RPI | G36050-5000.0 |
| Acetic acid | RPI | 50-197-7981 |
| SDS | Fisher Scientific | BP166-500 |
| SYPRO Orange | Thermo Scientific | S6650 |
| Pro-Q Diamond Phosphoprotein Gel Stain | Invitrogen | P33301 |
| Lysozyme | Hampton Research | HR7-110 |
| D-Ribose | Fischer Scientific | AAA1789409 |
| Critical Commercial Assays | ||
| Transcreener-ADP2 Fp assay | Sigma-Aldrich | 3010-1K |
| ADP-Glo Assay | Promega | V6930 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| QIAquick PCR & Gel Cleanup Kit | Qiagen | 28506 |
| Deposited Data | ||
| HsFN3K Crystal structure | This paper | PDB: 8UE1 |
| Recombinant DNA | ||
| Plasmid pMBP-His6TEV | This paper | N/A |
| pMBP-His6TEV-FN3K | This paper | N/A |
| pMBP-His6TEV-FN3K Δ116-138 | This paper | N/A |
| pMBP-His6TEV-FN3K Δ147-156 | This paper | N/A |
| pMBP-His6TEV- FN3K SWAP1 | This paper | N/A |
| pMBP-His6TEV- FN3K SWAP2 | This paper | N/A |
| pMBP-His6TEV- FN3K F39A | This paper | N/A |
| pMBP-His6TEV- FN3K K41A | This paper | N/A |
| pMBP-His6TEV- FN3K E55A | This paper | N/A |
| pMBP-His6TEV- FN3K D217A | This paper | N/A |
| pMBP-His6TEV- FN3K D217E | This paper | N/A |
| pMBP-His6TEV- FN3K D217Q | This paper | N/A |
| pMBP-His6TEV- FN3K D217N | This paper | N/A |
| pMBP-His6TEV- FN3K W219A | This paper | N/A |
| pMBP-His6TEV- FN3K N222A | This paper | N/A |
| pMBP-His6TEV- FN3K D234A | This paper | N/A |
| pMBP-His6TEV- FN3K D234E | This paper | N/A |
| pMBP-His6TEV- FN3K D234Q | This paper | N/A |
| pMBP-His6TEV- FN3K D234N | This paper | N/A |
| pMBP-His6TEV- FN3K H288A | This paper | N/A |
| pMBP-His6TEV- FN3K H291A | This paper | N/A |
| Software and Algorithms | ||
| Discover MP 2.4.2 | Refeyn | https://www.refeyn.com/software-release-updates |
| Pymol v2.0.7 | 57 | http://www.pymol.org/ |
| Phyre2 | 58 | http://www.sbg.bio.ic.ac.uk/~phyre2/ |
| PHENIX | 59 | https://www.phenix-online.org/ |
| Coot | 60 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHASER | 61 | http://www.ccp4.ac.uk/html/phaser.html |
| WebLogo 3 | 62 | https://weblogo.berkeley.edu/logo.cgi |
| HKL-3000 | 63 | http://www.hkl-xray.com/ |
| Prism | Prism | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Mass Photometry | Refeyn | N/A |
| EnVision 2102 Multilabel microplate reader | Perkin Elmer | 2102-0010 |
| BioTek Synergy microplate reader | Agilent Technologies | 11-120-533 |
| StepOne Real Time PCR System | Applied Biosystems | 4376600 |
| AKTA Pure Chromatography system | Cytiva Life sciences | 29018224 |
| Monolith NT.115- Microscale thermophoresis | Nanotemper | G008 |
| 384-well Black round bottom microplate | Corning | 4514 |
| 384-well White round bottom microplate | Corning | 4512 |
| 96-well plates | ThermoFisher Scientific | 4346907 |
| HisTrap FF, His-tagged protein purification column | Cytiva Life sciences | 17-5255-01 |
| HiTrap Q HP anion exchange column | Cytiva Life sciences | 17115401 |
| HiTrap S HP cation exchange column | Cytiva Life sciences | 17115101 |
| Superdex™ 200 Increase 10/300 | Cytiva Life sciences | 28-9909-44 |
Experimental model and study participant details
For protein production, Escherichia coli (E.coli) strain BL21(DE3) (C2527 NEB) was used.
For cloning purposes, Escherichia coli (E.coli) strain DH5α (C2987H NEB) was used
Method details
Plasmids:
HsFN3K was codon-optimized for E. coli expression (Integrated DNA Technologies). All constructs were generated utilizing standard PCR and restriction-based cloning methods. HsFN3K was cloned into a modified pET15b vector containing an N-terminal maltose-binding protein and His-tag. The pGro7 plasmid, encoding GroEL/GroES chaperones, was obtained from Takara Biotech (Cat# 3340).
Protein expression and purification of human FN3K:
HsFN3K was co-expressed with GroEL/ES in E. coli BL21(DE3) cells (C2527 NEB). The cells were grown in Luria-Bertani (LB) media at 37°C to an optical density of 0.6–0.8 and induced with 0.5 mM IPTG and 0.5 mg/mL of Arabinose for 16 hours at 18 °C. Cells were harvested at 5000 × g, lysed, and clarified at 40,000 × g in lysis buffer containing 20 mM Tris pH 7.5, 500 mM NaCl, 20 mM Imidazole, 5 mM BME. All HsFN3K constructs were subjected to Ni-NTA affinity purification and eluted with a linear gradient of imidazole (20 mM - 500 mM imidazole in 50 mM Tris pH 8.0, 25 mM NaCl, 2mM DTT). Subsequently, HsFN3K was purified by anion-exchange chromatography using a Q HP column (Cytiva, 17115401). The protein was eluted from the ion-exchange column with a linear gradient of NaCl (25 mM - 1000 mM NaCl in 50 mM Tris pH 8.0, 2 mM DTT. The fusion tag was removed with recombinant tobacco etch virus (rTEV) protease. Size exclusion chromatography (SD200) was used as the final purification step, and HsFN3K was stored in 50 mM HEPES pH 7.0, 250 mM NaCl, and 2 mM DTT or 2 mM TCEP for protein used in crystallization trials. HsFN3K was concentrated using an Amicon centrifugal filter (Millipore, UFC901024). A final concentration of 7 mg/ml was determined by measuring absorbance at 280nm and HsFN3K was stored at −80°C for all subsequent experiments.
Protein crystallography and data collection:
HsFN3K was subjected to reductive methylation as previously described.57 Briefly, 1 mg/ml FN3K was treated with 20 μl of freshly prepared 1 M dimethylamine–borane complex (Acros Organics) and 40 μl 1 M formaldehyde per milliliter of protein solution. The reaction was incubated at 4°C for 2 hrs and then this process was repeated a second time. Finally, 10 μl of 1 M dimethylamine–borane complex was added to the reaction and incubated at 4°C overnight. The reaction was quenched by size exclusion chromatography in buffer containing 20 mM Tris (pH 7.5) and 100 mM NaCl. HsFN3K was concentrated to 7 mg/mL and flash-frozen for subsequent crystallization trials.
Initial crystallization hits for HsFN3K were identified using a commercial crystallization screen Qiagen (JCSG Core I-IV) and in-house optimized native crystals were grown at 20 °C using the hanging-drop vapor diffusion method. 7 mg/ml HsFN3K was diluted 1:1 ratio with a reservoir solution containing 100 mM HEPES pH 7.5, 300 mM NaCl, and 20% PEG-3350. Crystals were cryoprotected in a reservoir solution containing 20% ethylene glycol and vitrified in liquid nitrogen. Diffraction data was collected at the SER-CAT 22-ID beamline at the Argonne National Laboratory on an Eiger 16M detector at 100K. The data was collected at an X-ray energy of 12680.0 eV and wavelength of 0.9792 Å. One hundred and eighty frames were recorded at a crystal-to-detector distance of 350 mm using an oscillation range of 1°. All diffraction data were indexed, integrated, scaled, and merged with the XDS package (Version January 31, 2020, BUILT=20200131).58 or HKL3000 (v721.3) and CCP4 (V7.0.058).59,60 The structure was solved using molecular replacement with Phaser-MR implemented in the Phenix suite of programs.61,62 The HsFN3K sequence was threaded onto the AtFN3K structure (6OID) using the Protein Homology/analogy Recognition Engine (Phyre2).49, and the resulting HsFN3K model was used as the search model for molecular replacement. The initial solution was refined using Phenix and manual model building was performed using Coot.52,53 Final validation was performed using MOLPROBITY server.63
Structural analysis:
All structural Figures were prepared using PyMOL (Schrödinger; version 2.5.4).64 Buried surface area and interface residues for AtFN3K and AtFN3Kmut were determined using the PISA web server.65 Conservation analysis was done using the ConSurf server.66–69
Ligand docking:
The HsFN3K structure was solved in the absence of ATP and substrate. To computationally open the ATP-binding loop of HsFN3K and dock in MgATP, Alphafold3 was used to model in MgATP into HsFN3K active site.70 We subsequently used the model of HsFN3K with bound MgATP to computationally dock DMF into the active site of HsFN3K using Autodock suite version 4.2.6 via the Molecular Graphics Laboratory Tools 1.5.7 (MGLTools) graphical user interface.71–73 Lamarckian Genetic Algorithm (LGA) docking parameters were employed and the rest of the parameters were applied as default values.
Sequence alignment:
All FN3K and FN3K homolog sequences were obtained from the NCBI database https://www.ncbi.nlm.nih.gov/). Sequences were aligned using Clustal Omega.74,75Logo plots were generated using WebLogo 3.76
Mass Photometry:
Purified HsFN3K was diluted in a buffer containing 50 mM HEPES pH 7.0, 250 mM NaCl, and 1 mM TCEP to a final concentration of 10 nM for Mass photometry (Refeyn TwoMP) analysis. Microscope coverslips were used in sample preparation by washing three times with Milli-Q water followed by two isopropanol washes. The coverslip was then dried using an air can. After which 2 μl of 10 nM protein was applied, and the coverslip was loaded onto the instrument. A movie of 60 seconds was recorded. Data was analyzed using DiscoverMP (Refeyn TwoMP).
Intact protein LC-MS and data analysis:
HsFN3K was expressed and purified as mentioned above in the absence of reducing agents. For intact LC-MS analysis, 25 pmol of protein was injected and measured using ultra high-performance liquid chromatography high resolution mass spectrometry (UHPLC-HRMS) on a Vanquish UHPLC interfaced with an electrospray Q Exactive Focus mass spectrometer (ThermoFisher Scientific, San Jose, CA) using Full scan MS. Chromatographic separation was performed using an Agilent Bio-HPLC PLRP-S revere phase HPLC column1000 Å, 5 μm x 50 mm x 1.0 mm, Santa Clara, CA). Mobile phase A was water with 0.2% (v/v) formic acid and mobile phase B was acetonitrile with 0.2% (v/v) formic acid. A post-column valve diverted the effluent from the column away from the mass spectrometer for the first 2 min. The flow rate was set to 0.400 mL/min and the column temperature was set to 70 °C. The autosampler was cooled to 5 °C and a 5 μL volume of sample was injected. Full scan MS was performed in positive ion mode scanning ions from 700 to 3000 m/z. The following MS parameters were used: resolution 17,500; AGC target 3E6 and maximum injection time 200 ms. Xcalibur Quan Browser (version 4.2.47 ThermoFisher Scientific, San Jose, CA) was used to extract the intact protein and the summed scans from the total ion chromatograms (TIC). The summed scans were deconvoluted to determine the intact protein molecular weight using BioPharm Finder (ThermoFisher Scientific, San Jose, CA).
Transcreener fluorescence polarization assay:
HsFN3K kinase activity was measured using the Transcreener ADP Fluorescent polarization assay (BellBrook Labs). The kinase reaction was carried out in 10 μL containing 200 μM 1-Deoxy-1-morpholino-fructose (DMF), 100 μM ATP, 2 mM MgCl2, and 0-5 μM of HsFN3K at room temperature. The reaction was quenched after 30 minutes by adding 10 μl of ADP detection mixture containing 4nM of ADP Alexa Fluor 633 tracer, 241 μg/ml of ADP2 antibody, 10 mM EDTA, and 0.01% Brij-35. The final mixture was incubated for 1 hour, and fluorescent polarization measurements were made using a Perkin Elmer plate reader using Cy5 excitation/emission filters (λex. = 630/30 nm; λem. = 680/30 nm), Cy5 FP epi-mirror and Cy5 emission dichroic. This assay was performed in triplicates and curves were fitted using 4-parameter logistic fit and the EC50 values were determined using GraphPad Prism.
ADP-Glo assay to measure kinetics:
The classical kinetic parameters of HsFN3K and mutants were measured using the ADP-Glo assay as previously described.77 The kinase reactions were carried out in 5 μL containing 0.5 μM of HsFN3K enzyme, 200 μM ATP, 10 mM MgCl2, and 0-100 μM of DMF at room temperature. For determining Km values for ATP, the kinase reaction was carried out in 5 μL containing 0.5 μM of HsFN3K, 1 mM DMF, 10 mM MgCl2, and 0-200 μM of ATP at room temperature. Time points were quenched by adding 5 μL of the ADP-glo reagent, and the reactions were incubated for 40 minutes at room temperature. This was followed by adding 10 μL of ADP detection reagent to measure the ADP produced. The reactions were incubated for another 40 minutes before reading luminescence on a BioTek Synergy microplate reader. The luminescence was correlated to the amount of ADP formed by generating ATP to ADP conversion curves following the manufacturer’s instructions. The amount of ADP formed was plotted versus time to obtain the slope of the linear portion representing initial velocity (v0). To obtain Km and Vmax the initial velocity was plotted as a function of DMF/ATP concentration in GraphPad Prism and data was fitted using Michaeli-Menten’s fit. Errors on v0 are shown as the standard error of the mean from individual fits used to obtain v0 The kinetic experiments for D217A and D217E were performed at 7 μM of the enzyme.
Differential scanning fluorimetry assays:
For Tm measurements, 20 μL reaction mixtures contained 5 μM HsFN3K and 5x SYPRO dye in 50 mM HEPES pH 7.0, 250 mM NaCl, 1mM TCEP, and 2 mM MgCl2. To measure changes to Tm upon the binding of nucleotide and substrate, ATP, DMF, or ATP+DMF were added to the FN3K mixture at a final concentration of 250 μM. The fluorescent measurements were made using a StepOnePlus Real-Time PCR machine (Applied Biosystems). The mixture was heated from 20°C to 95°C at a rate of 1°C per minute. Melting curves were generated from the first derivative of the fluorescent readings. DSF scans of all samples were performed in triplicates and the data were plotted using GraphPad Prism.
Isothermal titration calorimetry (ITC):
Isothermal titration calorimetry (ITC) experiments were performed on a MicroCal iTC200 instrument (Malvern Panalytical). FN3K and ATP were buffer exchanged into 50 mM HEPES pH 7.0, 150 mM NaCl, 3 mM MgCl2, and 1 mM TCEP. The syringe contained 500 μM of ATP and the cell contained 50 μM FN3K. ITC titrations were carried out at 25 °C using an initial 0.4 μL injection and 20 subsequent injections of 2 μL at 150-second intervals. The experiment was performed in duplicate and the resulting ITC data were processed and fit to a one-site binding model to determine n (number of binding sites) and KD (dissociation constant) using ORIGIN 7.0 software.
Microscale thermophoresis (MST):
Purified lysine-cysteine-lysine (KCK) tagged HsFN3K was labeled with fluorescein-5-maleimide dye. 10 nM labeled FN3K was titrated with increasing ligand concentration (15 nM to 500 μM for ATP and 7.6 nM to 250 μM for DMF). The experiments were performed in triplicate in 20 mM HEPES pH 7.0, 150 mM NaCl, 4 mM MgCl2, and 1 mM DTT buffer supplemented with 0.02% NP-40. KD measurements with DMF were performed in buffer containing 2 mM ATP. The MST data were acquired on a Monolith NT.115 Pico instrument (NanoTemper Technologies) at 25°C using standard capillaries. The instrument was set to medium MST power and 20% LED power. The experiment was performed in triplicates and the data was analyzed using the NTAnalysis software (NanoTemper Technologies).
Substrate Phosphorylation:
HsFN3K-mediated in vitro phosphorylation was carried out on glycated lysozyme. Glycated lysozyme was prepared as previously described.78 Briefly, crystallization grade lysozyme was incubated with ribose or glucose at a 1:1 molar ratio for 3 or 28 days, respectively, at 50°C. The phosphorylation assays were performed for 1 hour at 25°C in a 10 μl final volume containing 10 or 20 μM of FN3K, 2mg/mL of glycated lysozyme, and 1x kinase buffer (5 mM Hepes pH 7, 5 mM MgCl2, 100 μM ATP). The reaction was quenched with Laemelli sample loading dye and analyzed by SDS-PAGE. The gel was fixed with 50% methanol and 10% acetic acid solution and then stained with Pro-Q Diamond Phosphoproteinstain. Destaining was performed using 20% acetonitrile in 50 mM sodium acetate, pH 4 according to the manufacturer’s instruction. The gel was imaged using the BioRad ChemiDoc Pro-Q channel (ex.602nm). Finally, the gels were then stained with Coomassie Blue to confirm loading.
Quantification and statistical analysis:
Data acquired for the kinase and DSF assay experiments were analyzed by the software GraphPad Prism 10. Data acquired for MST assays were analyzed using the NTAnalysis software (NanoTemper Technologies). Data acquired for ITC was analyzed by ORIGIN 7.0 software. All error bars represent standard deviation (SD) or standard error of the mean (SEM). All experimental data are representative of at least two independent experiments.
Supplementary Material
Highlights.
Crystal structure of HsFN3K, an enzyme that repairs fructosamines.
The HsFN3K loop (116-138) extension is dispensable for kinase activity.
HsFN3K is primarily monomeric and has a lower redox sensitivity than plant FN3Ks.
Structural and biochemical studies reveal residues critical for deglycation.
Acknowledgments
We thank members of the Binning and Luca lab for their suggestions and technical assistance. We also thank Austin Graves and Sara Wilson (Refeyn) for their assistance with mass photometry experiments. We also acknowledge the staff at Southeast Regional Collaborative Access Team (SER-CAT) for help with X-ray data collection. The authors also acknowledge the Chemical Biology Core and Lancia Darville and John Koomen in the Proteomics and Metabolomics Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, which is supported in part by a National Cancer Institute (NCI) support grant (P30-CA076292). The icons for the graphical abstract and Figure S1A were created using Biorender.com. This study was supported by the U.S. National Institutes of Health grant R35 GM143004 to J.M.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
References
- 1.Chondrogianni N, Petropoulos I, Grimm S, Georgila K, Catalgol B, Friguet B, Grune T, and Gonos ES (2014). Protein damage, repair and proteolysis. Mol Aspects Med 35, 1–71. 10.1016/j.mam.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Martínez A, Portero-Otin M, Pamplona R, and Ferrer I (2010). Protein Targets of Oxidative Damage in Human Neurodegenerative Diseases with Abnormal Protein Aggregates. Brain Pathology 20, 281–297. 10.1111/j.1750-3639.2009.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies MJ (2016). Protein oxidation and peroxidation. Biochemical Journal 473, 805–825. 10.1042/BJ20151227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunn HF, and Higgins PJ (1981). Reaction of Monosaccharides with Proteins: Possible Evolutionary Significance. Science (1979) 213, 222–224. 10.1126/science.12192669. [DOI] [PubMed] [Google Scholar]
- 5.Maksimovic I, Maksimovic I, Zheng Q, Trujillo MN, Galligan JJ, David Y, David Y, David Y, and David Y (2020). An Azidoribose Probe to Track Ketoamine Adducts in Histone Ribose Glycation. J Am Chem Soc 142, 9999–10007. 10.1021/jacs.0c01325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vistoli G, De Maddis D, Cipak A, Zarkovic N, Carini M, and Aldini G (2013). Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radio Res 47, 3–27. 10.3109/10715762.2013.815348. [DOI] [PubMed] [Google Scholar]
- 7.Singh R, Barden A, Mori T, and Beilin L (2001). Advanced glycation end-products: a review. Diabetologia 44, 129–146. 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 8.Huebschmann AG, Regensteiner JG, Vlassara H, and Reusch JEB (2006). Diabetes and Advanced Glycoxidation End Products. Diabetes Care 29, 1420–1432. 10.2337/dc05-2096. [DOI] [PubMed] [Google Scholar]
- 9.Kim C-S, Park S, and Kim J (2017). The role of glycation in the pathogenesis of aging and its prevention through herbal products and physical exercise. J Exerc Nutrition Biochem 21, 55–61. 10.20463/jenb.2017.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan S. Du, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, et al. (1995). Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nat Med 1, 693–699. 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 11.Pun PBL, and Murphy MP (2012). Pathological Significance of Mitochondrial Glycation. Int J Cell Biol 2012, 1–13. 10.1155/2012/843505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner DP (2017). The Role of Advanced Glycation End-Products in Cancer Disparity. In, pp. 1–22. 10.1016/bs.acr.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delpierre G, Rider MH, Collard F, Stroobant V, Vanstapel F, Santos H, and Van Schaftingen E (2000). Identification, cloning, and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes 49, 1627–1634. 10.2337/diabetes.49.10.1627. [DOI] [PubMed] [Google Scholar]
- 14.Szwergold BS, Howell S, and Beisswenger PJ (2001). Human Fructosamine-3-Kinase. Diabetes 50, 2139–2147. 10.2337/diabetes.50.9.2139. [DOI] [PubMed] [Google Scholar]
- 15.Fortipied J, Gemayel R, Stroobant V, and van Schaftingen E (2005). Plant ribulosamine/erythrulosamine 3-kinase, a putative protein-repair enzyme. Biochemical Journal 388, 795–802. 10.1042/BJ20041976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delplanque J, Delpierre G, Opperdoes FR, and Van Schaftingen E (2004). Tissue distribution and evolution of fructosamine 3-kinase and fructosamine 3-kinase-related protein. Journal of Biological Chemistry 279, 46606–46613. 10.1074/jbc.M407678200. [DOI] [PubMed] [Google Scholar]
- 17.Delpierre G, Collard F, Fortipied J, and Van Schaftingen E (2002). Fructosamine 3-kinase is involved in an intracellular deglycation pathway in human erythrocytes. Biochemical Journal 365, 801–808. 10.1042/bj20020325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collard F, Wiame E, Bergans N, Fortpied J, Vertommen D, Vanstapel F, Delpierre G, and Van Schaftingen E (2004). Fructosamine 3-kinase-related protein and deglycation in human erythrocytes. Biochemical Journal 382, 137–143. 10.1042/BJ20040307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delpierre G, Vanstapel F, Stroobant V, and Van Schaftingen E (2000). Conversion of a synthetic fructosamine into its 3-phospho derivative in human erythrocytes. [PMC free article] [PubMed] [Google Scholar]
- 20.Collard F, Delpierre G, Stroobant V, Matthijs G, and Van Schaftingen E (2003). A Mammalian Protein Homologous to Fructosamine-3-Kinase Is a Ketosamine-3-Kinase Acting on Psicosamines and Ribulosamines but not on Fructosamines. Diabetes 52, 2888–2895. 10.2337/diabetes.52.12.2888. [DOI] [PubMed] [Google Scholar]
- 21.Alderawi A, Caramori G, Baker EH, Hitchings AW, Rahman I, Rossios C, Adcock I, Cassolari P, Papi A, Ortega VE, et al. (2020). FN3K expression in COPD: A potential comorbidity factor for cardiovascular disease. BMJ Open Respir Res 7. 10.1136/bmjresp-2020-000714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown TR, Su B, Brown KA, Schwartz MA, Tobia AM, and Kappler F (2003). Modulation of in vivo 3-deoxyglucosone levels. Biochem Soc Trans 31, 1433–1437. 10.1042/bst0311433. [DOI] [PubMed] [Google Scholar]
- 23.Mohás M, Kisfali P, Baricza E, Mérei Á, Maász A, Cseh J, Mikolás E, Szijártó IA, Melegh B , and Wittmann I (2009). A Polymorphism within the Fructosamine-3-kinase Gene is Associated with HbA 1c Levels and the Onset of Type 2 Diabetes Mellitus. Experimental and Clinical Endocrinology & Diabetes 118, 209–212. 10.1055/s-0029-1238319. [DOI] [PubMed] [Google Scholar]
- 24.Veiga-Da-Cunha M, Jacquemin P, Delpierre G, Godfraind C, Théate I, Vertommen D, Clotman F, Lemaigre F, Devuyst O, and Van Schaftingen E (2006). Increased protein glycation in fructosamine 3-kinase-deficient mice. Biochemical Journal 399, 257–264. 10.1042/BJ20060684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim E, Lowenson JD, MacLaren DC, Clarke S, and Young SG (1997). Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proceedings of the National Academy of Sciences 94, 6132–6137. 10.1073/pnas.94.12.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beeraka NM, Zhang J, Mandal S, Vikram PR,H, Liu J, B M N, Zhao D, Vishwanath P, B M G, and Fan R (2023). Screening fructosamine-3-kinase (FN3K) inhibitors, a deglycating enzyme of oncogenic Nrf2: Human FN3K homology modelling, docking and molecular dynamics simulations. PLoS One 18, e0283705. 10.1371/journal.pone.0283705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanghvi VR, Leibold J, Mina M, Mohan P, Berishaj M, Li Z, Miele MM, Lailler N, Zhao C , de Stanchina E, et al. (2019). The Oncogenic Action of NRF2 Depends on De-glycation by Fructosamine-3-Kinase. Cell 178, 807–819.e21. 10.1016/j.cell.2019.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanghvi VR, Mohan P, Singh K, Cao L, Berishaj M, Wolfe AL, Schatz JH, Lailler N, de Stanchina E, Viale A, et al. (2021). Nrf2 activation confers resistance to eif4a inhibitors in cancer therapy. Cancers (Basel) 13, 1–13. 10.3390/cancers13040639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Bruyne S, Van den Broecke C, Vrielinck H, Khelifi S, De Wever O, Bracke K, Huizing M, Boston N, Himpe J, Speeckaert M, et al. (2020). Fructosamine-3-Kinase as a Potential Treatment Option for Age-Related Macular Degeneration. J Clin Med 9, 2869. 10.3390/jcm9092869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niwa T, and Tsukudhi S (2001). 3-Deoxyglucosone and AGEs in uremic complications: Inactivation of glutathione peroxidase by 3-deoxyglucosone. Kidney Int 59, S37–S41. 10.1046/j.1523-1755.2001.59780037.x. [DOI] [PubMed] [Google Scholar]
- 31.Niwa T, Takeda N, Yoshizumi H, Tatematsu A, Ohara M, Tomiyama S, and Niimura K (1993). Presence of 3-Deoxyglucosone, a Potent Protein Crosslinking Intermediate of Maillard Reaction, in Diabetic Serum. Biochem Biophys Res Commun 196, 837–843. 10.1006/BBRC.1993.2325. [DOI] [PubMed] [Google Scholar]
- 32.Kusunoki H, Miyata S, Ohara T, Liu B-F, Uriuhara A, Kojima H, Suzuki K, Miyazaki H, Yamashita Y, Inaba K, et al. (2003). Relation Between Serum 3-Deoxyglucosone and Development of Diabetic Microangiopathy. Diabetes Care 26, 1889–1894. 10.2337/diacare.26.6.1889. [DOI] [PubMed] [Google Scholar]
- 33.Kannan N, Taylor SS, Zhai Y, Venter JC, and Manning G (2007). Structural and functional diversity of the microbial kinome. PLoS Biol 5, 17. 10.1371/journal.pbio. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oruganty K, Talevich EE, Neuwald AF, and Kannan N (2016). Identification and classification of small molecule kinases: Insights into substrate recognition and specificity Genome evolution and evolutionary systems biology. BMC Evol Biol 16, 1–14. 10.1186/s12862-015-0576-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shrestha S, Katiyar S, Sanz-Rodriguez CE, Kemppinen NR, Kim HW, Kadirvelraj R, Panagos C, Keyhaninejad N, Colonna M, Chopra P, et al. (2020). A redox-active switch in fructosamine-3-kinases expands the regulatory repertoire of the protein kinase superfamily. Sci Signal 13. 10.1126/scisignal.aax6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burk DL, Hon WC, Leung AKW, and Berghuis AM (2001). Structural analyses of nucleotide binding to an aminoglycoside phosphotransferase. Biochemistry 40, 8756–8764. 10.1021/bi010504p. [DOI] [PubMed] [Google Scholar]
- 37.Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C, Kronkaitis A, Li J, White A, Mische S, et al. (2005). Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. Journal of Biological Chemistry 280, 6130–6137. 10.1074/jbc.M409123200. [DOI] [PubMed] [Google Scholar]
- 38.Knighton DR, Zheng J, Ten Eyck LF, Ashford VA, Xuong N-H, Taylor SS, and Sowadski JM (1991). Crystal Structure of the Catalytic Subunit of Cyclic Adenosine Monophosphate-Dependent Protein Kinase. Science (1979) 253, 407–414. 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 39.Johnson LN, Noble MEM, and Owen DJ (1996). Active and Inactive Protein Kinases: Structural Basis for Regulation. Cell 85, 149–158. 10.1016/S0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 40.Meharena HS, Fan X, Ahuja LG, Keshwani MM, McClendon CL, Chen AM, Adams JA, and Taylor SS (2016). Decoding the Interactions Regulating the Active State Mechanics of Eukaryotic Protein Kinases. PLoS Biol 14. 10.1371/journal.pbio.2000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehr DD, Thompson PR, and Wright GD (2001). Molecular mechanism of aminoglycoside antibiotic kinase APH(3’)-IIIa: Roles of conserved active site residues. Journal of Biological Chemistry 276, 23929–23936. 10.1074/jbc.M100540200. [DOI] [PubMed] [Google Scholar]
- 42.Brenner S. (1987). Phosphotransferase sequence homology. Nature. [DOI] [PubMed] [Google Scholar]
- 43.Kannan N, and Neuwald AF (2005). Did Protein Kinase Regulatory Mechanisms Evolve Through Elaboration of a Simple Structural Component? J Mol Biol 351, 956–972. 10.1016/JJMB.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 44.Krupa A, Preethi G, and Srinivasan N (2004). Structural Modes of Stabilization of Permissive Phosphorylation Sites in Protein Kinases: Distinct Strategies in Ser/Thr and Tyr Kinases. J Mol Biol 339, 1025–1039. 10.1016/JJMB.2004.04.043. [DOI] [PubMed] [Google Scholar]
- 45.Hudson KL, Bartlett GJ, Diehl RC, Agirre J, Gallagher T, Kiessling LL, and Woolfson DN (2015). Carbohydrate-Aromatic Interactions in Proteins. J Am Chem Soc 137, 15152–15160. 10.1021/jacs.5b08424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brinda KV, Kannan N, and Vishveshwara S (2002). Analysis of homodimeric protein interfaces by graph-spectral methods. Protein Engineering, Design and Selection 15, 265–277. 10.1093/protein/15.4.265. [DOI] [PubMed] [Google Scholar]
- 47.Tokuriki N, and Tawfik DS (2009). Protein Dynamism and Evolvability. Science (1979) 324, 203–207. 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 48.Nestl BM, and Hauer B (2014). Engineering of Flexible Loops in Enzymes. ACS Catal 4, 3201–3211. 10.1021/cs500325p. [DOI] [Google Scholar]
- 49.Boehr DD, Thompson PR, and Wright GD (2001). Molecular Mechanism of Aminoglycoside Antibiotic Kinase APH(3’)-IIIa. Journal of Biological Chemistry 276, 23929–23936. 10.1074/jbc.M100540200. [DOI] [PubMed] [Google Scholar]
- 50.Conner JR, Beisswenger PJ, and Szwergold BS (2004). The expression of the genes for fructosamine-3-kinase and fructosamine-3-kinase-related protein appears to be constitutive and unaffected by environmental signals. Biochem Biophys Res Commun 323, 932–936. 10.1016/J.BBRC.2004.08.181. [DOI] [PubMed] [Google Scholar]
- 51.Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science (1979) 356. 10.1126/science.aa!3321. [DOI] [PubMed] [Google Scholar]
- 52.Bilova T, Lukasheva E, Brauch D, Greifenhagen U, Paudel G, Tarakhovskaya E, Frolova N, Mittasch J, Balcke GU, Tissier A, et al. (2016). A snapshot of the plant glycated proteome structural, functional, and mechanistic aspects. Journal of Biological Chemistry 291, 7621–7636. 10.1074/jbc.M115.678581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q, Monroe ME, Schepmoes AA, Clauss TRW, Gritsenko MA, Meng D, Petyuk VA, Smith RD, and Metz TO (2011). Comprehensive identification of glycated peptides and their glycation motifs in plasma and erythrocytes of control and diabetic subjects. J Proteome Res 10, 3076–3088. 10.1021/pr200040j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bechtold U, Rabbani N, Mullineaux PM, and Thornalley PJ (2009). Quantitative measurement of specific biomarkers for protein oxidation, nitration and glycation in Arabidopsis leaves. Plant Journal 59, 661–671. 10.1111/j.1365-313X.2009.03898.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Q, Petyuk VA, Schepmoes AA, Orton DJ, Monroe ME, Yang F, Smith RD, and Metz TO (2008). Analysis of non-enzymatically glycated peptides: Neutral-loss-triggered MS3 versus multi-stage activation tandem mass spectrometry. Rapid Communications in Mass Spectrometry 22, 3027–3034. 10.1002/rcm.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shumilina J, Kusnetsova A, Tsarev A, Janse van Rensburg HC, Medvedev S, Demidchik V, Van den Ende W, and Frolov A (2019). Glycation of plant proteins: Regulatory roles and interplay with sugar signalling? Int J Mol Sci 20. 10.3390/ijms20092366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rayment I. (1997). [12] Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol 276, 171–179. 10.1016/S0076-6879(97)76058-0. [DOI] [PubMed] [Google Scholar]
- 58.Kabsch W. (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: The integration of data reduction and structure solution - From diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 60.Otwinowski Z, and Minor W (1997). [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 61.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan S, Chan HCS, Filipek S, and Vogel H (2016). PyMOL and Inkscape Bridge the Data and the Data Visualization. Structure 24, 2041–2042. 10.1016/j.str.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 65.Krissinel E, and Henrick K (2007). Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol 372, 774–797. 10.1016/JJMB.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 66.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–W350. 10.1093/NAR/GKW408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ashkenazy H, Erez E, Martz E, Pupko T, and Ben-Tal N (2010). ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38. 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, and Ben-Tal N (2005). ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33. 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–W350. 10.1093/NAR/GKW408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, and Olson AJ (2010). Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov 5, 597–607. 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Forli S, and Olson AJ (2012). A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J Med Chem 55, 623–638. 10.1021/jm2005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, and Lopez R (2010). A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38. 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: A sequence logo generator. Genome Res 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, Scherer CA, Scholl C, Fröhling S, Carr SA, Stern AM, et al. (2012). STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proceedings of the National Academy of Sciences 109, 2860–2865. 10.1073/pnas.1120589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yeboah FK, Alli I, Yaylayan VA, Yasuo K, Chowdhury SF, and Purisima EO (2004). Effect of Limited Solid-State Glycation on the Conformation of Lysozyme by ESI-MSMS Peptide Mapping and Molecular Modeling. Bioconjug Chem 15, 27–34. 10.1021/bc034083v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The HsFN3K crystal structure has been deposited in the Protein Data Bank under the accession number: 8UE1 and is publicly available as of the date of publication. The accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| DH5a Competent Cells (E. coli) | NEB | C2987H |
| BL21(DE3) Competent Cells (E.coli) | NEB | C2527 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Luria Broth | RPI | L24400-5000.0 |
| IPTG | Chem Impex | 00194-100g |
| Ampicillin | Fisher Scientific | BP1760-25 |
| Carbenicillin | Gold Biotechnology | 50-153-2751 |
| Chloramphenicol | Fisher Scientific | AC227921000 |
| L-(+)-Arabinose | Sigma Aldrich | A3256-100G |
| Chaperone Plasmid Set | Takara Biotech | 3340 |
| Sodium Chloride | RPI | S23025-3000.0 |
| Tris Base Ultra-Pure | RPI | T60040-5000.0 |
| HEPES | RPI | H75030-1000.0 |
| Imidazole | Fisher Scientific | AAA102210E |
| DTT | RPI | D11000-50.0 |
| TCEP | Thermo Scientific | PI20491 |
| NP-40 | Sigma Aldrich | I3021-100ML |
| Fluorescein-5-maleimide | Fisher Scientific | 50-187-4590 |
| DMAB | Acros Organics | AC177310250 |
| Paraformadehyde | Thermo Scientific | 28906 |
| Magnesium Chloride | RPI | M24000-500.0 |
| Adenosine-5-triphosphate disodium salt | Chem Impex | 00015-25G |
| Adenosine-5-diphosphate disodium salt | RPI | A11220-10.0 |
| Adenylyl-imidodiphosphate salt | Sigma-Aldrich | SIAL-10102547001 |
| Polyethylene-glycol-3350 | Hampton Research | HR2-527 |
| 4-15% Mini Protean Stain free protein gels | Bio-Rad | 4568086 |
| Coomassie Brilliant Blue- R250 | Thermo Scientific | 20278 |
| 4X SDS-Sample buffer | Fisher Scientific | BP-110R |
| Glycine | RPI | G36050-5000.0 |
| Acetic acid | RPI | 50-197-7981 |
| SDS | Fisher Scientific | BP166-500 |
| SYPRO Orange | Thermo Scientific | S6650 |
| Pro-Q Diamond Phosphoprotein Gel Stain | Invitrogen | P33301 |
| Lysozyme | Hampton Research | HR7-110 |
| D-Ribose | Fischer Scientific | AAA1789409 |
| Critical Commercial Assays | ||
| Transcreener-ADP2 Fp assay | Sigma-Aldrich | 3010-1K |
| ADP-Glo Assay | Promega | V6930 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| QIAquick PCR & Gel Cleanup Kit | Qiagen | 28506 |
| Deposited Data | ||
| HsFN3K Crystal structure | This paper | PDB: 8UE1 |
| Recombinant DNA | ||
| Plasmid pMBP-His6TEV | This paper | N/A |
| pMBP-His6TEV-FN3K | This paper | N/A |
| pMBP-His6TEV-FN3K Δ116-138 | This paper | N/A |
| pMBP-His6TEV-FN3K Δ147-156 | This paper | N/A |
| pMBP-His6TEV- FN3K SWAP1 | This paper | N/A |
| pMBP-His6TEV- FN3K SWAP2 | This paper | N/A |
| pMBP-His6TEV- FN3K F39A | This paper | N/A |
| pMBP-His6TEV- FN3K K41A | This paper | N/A |
| pMBP-His6TEV- FN3K E55A | This paper | N/A |
| pMBP-His6TEV- FN3K D217A | This paper | N/A |
| pMBP-His6TEV- FN3K D217E | This paper | N/A |
| pMBP-His6TEV- FN3K D217Q | This paper | N/A |
| pMBP-His6TEV- FN3K D217N | This paper | N/A |
| pMBP-His6TEV- FN3K W219A | This paper | N/A |
| pMBP-His6TEV- FN3K N222A | This paper | N/A |
| pMBP-His6TEV- FN3K D234A | This paper | N/A |
| pMBP-His6TEV- FN3K D234E | This paper | N/A |
| pMBP-His6TEV- FN3K D234Q | This paper | N/A |
| pMBP-His6TEV- FN3K D234N | This paper | N/A |
| pMBP-His6TEV- FN3K H288A | This paper | N/A |
| pMBP-His6TEV- FN3K H291A | This paper | N/A |
| Software and Algorithms | ||
| Discover MP 2.4.2 | Refeyn | https://www.refeyn.com/software-release-updates |
| Pymol v2.0.7 | 57 | http://www.pymol.org/ |
| Phyre2 | 58 | http://www.sbg.bio.ic.ac.uk/~phyre2/ |
| PHENIX | 59 | https://www.phenix-online.org/ |
| Coot | 60 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHASER | 61 | http://www.ccp4.ac.uk/html/phaser.html |
| WebLogo 3 | 62 | https://weblogo.berkeley.edu/logo.cgi |
| HKL-3000 | 63 | http://www.hkl-xray.com/ |
| Prism | Prism | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Mass Photometry | Refeyn | N/A |
| EnVision 2102 Multilabel microplate reader | Perkin Elmer | 2102-0010 |
| BioTek Synergy microplate reader | Agilent Technologies | 11-120-533 |
| StepOne Real Time PCR System | Applied Biosystems | 4376600 |
| AKTA Pure Chromatography system | Cytiva Life sciences | 29018224 |
| Monolith NT.115- Microscale thermophoresis | Nanotemper | G008 |
| 384-well Black round bottom microplate | Corning | 4514 |
| 384-well White round bottom microplate | Corning | 4512 |
| 96-well plates | ThermoFisher Scientific | 4346907 |
| HisTrap FF, His-tagged protein purification column | Cytiva Life sciences | 17-5255-01 |
| HiTrap Q HP anion exchange column | Cytiva Life sciences | 17115401 |
| HiTrap S HP cation exchange column | Cytiva Life sciences | 17115101 |
| Superdex™ 200 Increase 10/300 | Cytiva Life sciences | 28-9909-44 |