

Abstract
Dexmedetomidine (DEX) has been confirmed to exert neuroprotective effects in various nerve injury models by regulating ferroptosis, including spinal cord injury (SCI). Although it has been established that CDGSH iron sulfur domain 2 (CISD2) can regulate ferroptosis, whether DEX can regulate ferroptosis by CISD2 in SCI remains unclear. Lidocaine was used to induce PC12 cells and stimulate rats to establish SCI models in vitro and in vivo. MTT assays were performed to analyze cell viability. Ferroptosis was assessed by determining the levels of cellular reactive axygen species (ROS), malondialdehyde (MDA), glutathione (GSH), and Fe2+. Ferritinophagy was analyzed by LysoTracker staining, FerroOrange staining, and immunofluorescence. Western blotting was carried out to quantify the levels of several proteins. Fluorescence microscopy was also used to observe cell autophagy. The morphology of mitochondria within the tissue was observed under transmission electron microscopy (TEM). DEX treatment weakened lidocaine-induced elevation of ROS, Fe2+, and MDA and reduced GSH in PC12 cells, indicating that DEX treatment weakened lidocaine-induced ferroptosis in PC12 cells. Similarly, lidocaine promoted autophagy, Fe2+, and microtubule-associated protein 1 light chain 3 (LC3) in PC12 cells and suppressed ferritin and p62 protein levels, indicating that DEX could weaken lidocaine-induced ferritinophagy in PC12 cells. DEX treatment improved the BBB score, reduced tissue damage, increased the number of neurons, and alleviated mitochondrial damage by inhibiting ferroptosis and ferritinophagy in lidocaine-induced SCI rat models. The decreased CISD2, ferritin heavy chain 1 (FTH1), solute carrier family 7-member 11-glutathione (SLC7A11), and glutathione peroxidase 4 (GPX4) protein levels and the elevated nuclear receptor coactivator 4 (NCOA4) protein levels in rat models in the lidocaine group were weakened by DEX treatment. Moreover, CISD2 inhibition reversed the inhibitory effects of DEX treatment on lidocaine-induced ferroptosis and ferritinophagy in PC12 cells significantly. Taken together, DEX treatment could impair lidocaine-induced SCI by inhibiting ferroptosis and ferritinophagy by upregulating CISD2 in rat models.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10863-024-10034-x.
Keywords: Spinal cord injury, Dexmedetomidine, CISD2, Ferroptosis, Ferritinophagy, Lidocaine
Introduction
Spinal cord injury (SCI) has been increasing in incidence and prevalence specially in high income countries (Chay and Kirshblum 2020). SCI is usually caused by external direct or indirect factors, leading to various motor, sensory, and sphincter dysfunctions, different degrees of weakness or later spasticity, pathological reflexes, and dysautonomic manifestations according to the neurologic level of injury (Eli et al. 2021; Quadri et al. 2020). The primary injury is mainly caused by acute mechanical damage and spinal cord compression or transection caused by spinal dislocation. Secondary injuries mainly include inflammatory reactions, cell death, oxidative stress reactions, etc. (Anjum et al. 2020). The incidence rate varies from 12 to more than 65 cases per million per year (Hamid et al. 2018). It has been established that the incidence rate of SCI in China is relatively high (Chen et al. 2022). There is no effective treatment method for SCI, but it brings serious physical and psychological harm to the patient and poses a huge economic burden on society (Russo et al. 2020; Snyder et al. 2020).
The main challenge in treating SCI is to prevent the loss of remaining nerve cells to mitigate secondary injury (Cruz et al. 2015). Ferroptosis is an iron-dependent novel cell death pathway triggered by reactive oxygen species (ROS) generation and severe lipid peroxidation caused by iron overload (Tang et al. 2021). Whether ferroptosis occurs depends on the balance between iron overload-induced ROS and antioxidant systems that prevent lipid peroxidation. Once an imbalance occurs, excessive ROS production or decreased antioxidant system activity can lead to lipid peroxidation, damaging the plasma membrane and leading to ferroptosis (Wang et al. 2022a, b). Morphologically, ferroptosis mainly occurs in cells, characterized by a decrease in the size of mitochondria, an increase in the density of bilayer membranes, and a decrease or disappearance of mitochondrial cristae, while the cell membrane remains intact, the size of the nucleus is normal, and there is no concentration of chromatin (Battaglia et al. 2020; Tadokoro et al. 2020). Biochemically, the consumption of glutathione (GSH) within cells leads to a decrease in glutathione peroxidase 4 (GPX4 ) activity, which prevents lipid peroxides from being metabolized by the reduction reaction catalyzed by GPX4 and the production of hydroxyl radicals through the Fenton reaction, resulting in the production of a large amount of ROS and ultimately promoting ferroptosis (LoBianco et al. 2022; Lv et al. 2020; Yuan et al. 2021). An increasing body of evidence suggests that ferroptosis is involved in the pathological process of SCI (Ge et al. 2021; Yao et al. 2019; Li et al. 2023). Significant changes in ferroptosis markers were observed in the spinal cord tissue of SCI rats, and changes in mitochondrial characteristics of iron death were observed using transmission electron microscopy (TEM), thereby confirming the important role of ferroptosis in SCI (Ma et al. 2021). GPX4 can inhibit lipid peroxidation, and its abnormal function may lead to ferroptosis and aggravate SCI (Wei et al. 2021). It has been established that SCI can regulate ferroptosis metabolism-related proteins, decrease the activity of GPX4, induce the release of NO to induce microglia activation, and trigger neuronal ferroptosis, thus leading to motor function recovery disorders (Wei et al. 2021). The increased free iron after SCI leads to excessive ROS production, resulting in imbalanced ROS metabolism in the damaged area, conducive to oxidative stress and neuronal cell ferroptosis (Cheng 2021). These findings emphasize the need to explore the mechanisms related to ferroptosis after SCI.
An increasing body of evidence suggests that the occurrence of ferroptosis requires the involvement of autophagy mechanisms (Liu et al. 2020). Masaldan et al. showed that iron accumulation in aging cells was related to impaired ferritinophagy and inhibition of ferroptosis (Masaldan et al. 2018). Latunde Dada et al. reported that cysteine deficiency-mediated ferritin degradation releases iron through the nuclear receptor coactivator 4 (NCOA4) -mediated autophagy pathway (Latunde-Dada 2017). By inhibiting autophagy or the expression of NCOA4, ferritinophagy can be inhibited, leading to the accumulation of unstable iron and lipid ROS related to ferroptosis in cells (Santana-Codina et al. 2021). Accordingly, exploring the mechanism underlying the inhibition of ferritinophagy and subsequently inhibiting ferroptosis after SCI is of great significance.
Dexmedetomidine (DEX) is α 2 adrenergic receptor agonist widely used in clinical practice due to its biological effects such as sedation, analgesia, anti-anxiety, and sympathetic inhibition (Mahmoud and Mason 2015). DEX has been confirmed in numerous studies to exert a definite protective effect on multiple organ ischemia-reperfusion injury (Liang et al. 2019; Tao et al. 2022; Wang et al. 2022a, b). Similarly, DEX has demonstrated neuroprotective effects in various nerve injury models, including SCI. For instance, DEX was found to improve the neural function of traumatic SCI rats and reduce the apoptosis of spinal cord neurons (Liu et al. 2022a, b, c). DEX alleviated spinal cord ischemia-reperfusion injury in rabbits by reducing oxidation and cell apoptosis (Liu et al. 2022a, b, c). DEX inhibited neutrophil infiltration, microglia activation, and reactive glial proliferation to alleviate spinal cord ischemia-reperfusion injury in rats (Chen et al. 2023). Currently, the ferroptosis-associated mechanisms by which DEX improves SCI have been largely understudied, warranting further research.
Accordingly, this study evaluated whether DEX could improve ferritinophagy and ferroptosis in SCI by mediating CDGSH iron sulfur domain 2 (CISD2) expression and its related molecular mechanisms.
Materials and methods
Cell culture and treatment
PC12 cells (CRL-1721™, ATCC, Manassas, VA, USA) were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco) and 1% Penicillin-Streptomycin (10,000 U/mL) (Gibco) at 37 °C in a humidified incubator with 5% carbon dioxide. For lidocaine stimulation, PC12 cells (P9-13 passage) were treated with different concentrations of lidocaine (0, 0.25mM, 0.5mM, 1mM). For DEX treatment, PC12 cells were treated with 1mM lidocaine combined with different concentrations of DEX (0, 10 µΜ, 25µΜ, 50 µΜ).
Cell transfection
PC12 cells (1 × 105) were cultured until reaching 70–80% confluence in 12-well plates. Small interference RNA (siRNA) targeting CISD2 (si-CISD2) and its negative control (si-NC) were transfected into PC12 cells using Lipofectamine 2000 (Invitrogen).
MTT assay
Cell viability was determined by using the MTT assay kit (M1020, Solarbio, Beijing, China). PC12 cells were inoculated at a density of 1 × 104 cells per well and incubated for 12 h. Subsequently, medication was used to treat and incubate for 24 h. Ten microliters of MTT solution were added and cultured for 4 h. The formazan solution (110 µL) was added to each well to dissolve the crystals after removing the supernatant. Measurement of the absorbance values of each well at 490 nm was conducted on an enzyme-linked immunosorbent assay (ELISA).
Flow cytometry for ROS measurement in cells
A reagent kit for detecting reactive oxygen species using fluorescent probe DCFH-DA (Beyotime, China) was used in this experiment. Dilution of DCFH-DA was performed using a serum-free culture medium at a ratio of 1:1000, resulting in a final concentration of 10 µmol/L. The collected cells were suspended in diluted DCFH-DA at a cell concentration of 1 × 106/mL. To ensure sufficient contact between the probe and the cell, mixing was performed by inverting it up and down every 3–5 min. After incubation for 20 min, the cells were washed three times using a serum-free cell culture medium. The detection of fluorescence intensity was performed by flow cytometry (Biosciences, San Diego, CA, USA) using 488 nm excitation wavelength and 525 nm emission wavelength.
Detection of malondialdehyde (MDA), glutathione (GSH) and Fe2+
The levels of malondialdehyde (MDA), GSH, and ferrous iron (Fe 2+) in PC12 cells and/or spinal cord lysate were detected by the MDA assay kit (S0131, Beyotime), GSH assay kit (S0053, Beyotime) and iron assay kit (MAK025, Sigma, USA) according to relevant manufacturers’ protocols.
LysoTracker staining
The cell culture medium was removed, followed by the addition of LysoTracker Green staining solution (C1047S, Beyotime) with a final concentration of 50–75 nM and preheated at 37℃. After incubation for 30 min, the Lyso Tracker Green staining solution was removed, and a fresh cell culture medium was added. Subsequently, the fluorescence intensity of lysosomes was observed using a fluorescence microscope (TE-2000, Nikon, Tokyo, Japan).
Immunofluorescence staining
P12 cells in each group were cultured and permeated by 0.03% Triton X-100 (Beyotime). After incubation for 60 min, the cells were fixed, followed by blocking with 0.1% BSA (Beyotime). Subsequently, the primary antibodies anti-ferritin (ab75973, Abcam, Cambridge, UK) and anti-microtubule-associated protein 1 light chain 3 (LC-3) were applied to the medium. The cells were analyzed via fluorescence microscopy (Tokyo).
Western blotting
Total protein was extracted using a RIPA buffer (Solarbio) and then measured using a commercial BCA kit (Pierce, Rockford, IL, USA). Equal amounts of protein (30 µg) were then separated by 10% SDS-PAGE, transferred to polyvinylidene fluoride membranes, and blocked into 5% fat-free milk. After incubation with primary antibodies LC3I/II (1:1000; #4108, Cell Signaling Technology, Beverly, MA, USA), sequestosome 1 (p62,1:10000; ab109012, Abcam), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:1000; ab181602, Abcam), CISD2 (1:1000; #60758, Cell Signaling Technology), ferritin heavy chain 1 (FTH1,1:1000; #4393, Cell Signaling Technology), solute carrier family 7-member 11-glutathione (SLC7A11, 1:1000; ab307601, Abcam), GPX4 (1:1000; ab181602, Abcam) and nuclear receptor coactivator 4 (NCOA4) (1:1000; #66849, Cell Signaling Technology), the membranes were incubated with secondary antibody conjugated to horseradish peroxidase. After enhanced chemiluminescence (ECL) detection, protein bands were quantified using ImageJ version 1.51 software (NIH).
SCI and treatment of drugs
Healthy Sprague Dawley (SD) rats (male, adult, 230–270 g) (Experimental Animal Center of the Academy of Military Medical Sciences, Beijing, China) were selected. All rats were provided ad libitum access to food and clean water. The indoor temperature of the artificial control room was around 22 ℃, with a humidity of 50 -60% and alternating night and day cycles for 12 h. Animal experiments were conducted after one week of feeding. All experimental procedures on animals were approved by the Ethics Committee of the hospital (approval number: SYXK (Su) 2018-0003) in accordance with the guidelines provided by the Committee for Purpose of Control and Supervision of Experiments on Animals (CPCSEA). The rats were anesthetized by intraperitoneal injection of 5% phenobarbital. A longitudinal incision of about 2 cm was made above the foramen magnum of the occipital bone while in a prone position. The subcutaneous tissue and neck muscles were bluntly separated, and the atlantooccipital membrane was exposed. The atlantooccipital membrane was then penetrated using the needle tip, resulting in the clear cerebrospinal fluid gushing out and fluctuating with breathing. A sterilized, closed-end PE-10 catheter filled with physiological saline was inserted 7 cm towards the tail end, with the catheter successfully reaching the spinal canal indicated by the sudden lateral sway or hind leg twitching of the rat tail. The catheter was secured, and the incision was sutured layer by layer to the back of the lumbosacral segment of the spinal cord. On the first day after surgery, all rats were randomly divided into 3 groups according to computer sequence numbers (n = 6), namely, the sham, the 10% lidocaine, the 10% lidocaine + DEX (25 µg/kg). For the 10% lidocaine group, an intrathecal injection of 10% lidocaine was performed using a volume of 20 µL (Zhang et al. 2022; Ding et al. 2021). Positive results for the lidocaine screening test were determined if the rats experienced paralysis of both lower limbs within 30 s or exhibited avoidance reactions when both upper limbs were clamped. Rats showing no reaction in both lower limbs and the disappearance of the tail clamp reflex were selected for the experiment. Rats with quadriplegia, mobility disorders, negative lidocaine test results, or unilateral limb paralysis after surgery were excluded. For the 10% lidocaine + DEX (25 µg/kg) group, rats were intraperitoneally injected with 25 µg/kg DEX in a 0.1 mL vehicle containing 2% DMSO (Sigma) and 0.9% NaCl (Meilunbio, China). The first injection was administered two hours after the successful establishment of the model, followed by daily injections until the eight day. The sham group received an equal volume of vehicle injections. The rats were killed on the eight day after injection in vivo.
Behavioral score
On days 0, 2, 4, 6, and 8, the motor recovery of different groups of rats was evaluated using Basso, Beattie, and Bresnahan (BBB) motor scores (Basso et al. 1995) by two independent examiners blinded to the grouping. The scores ranged from 0 to 21, with 0 indicating no motor function and 21 indicating normal performance. Rats were allowed to walk freely within a 90 cm2 field for 5 min, and the movement of their hind limbs was closely observed.
Nissl staining and hematoxylin-eosin (HE) staining
The paraffin sections were dewaxed and soaked in three changes of distilled water for 5 min each. They were then stained with methyl violet staining solution for 10 min. After washing the slide, it was differentiated in Nissl (Beyotime) solution for 5 s, followed by dehydration in a gradient alcohol solution. After removal in xylene, the sections were mounted on a glass slide using neutral adhesive. The changes in the number of Nissl bodies in the anterior horn of the spinal cord were observed using an optical microscope at the same location. For HE staining, the paraffin sections were stained with hematoxylin (Solarbio) aqueous solution for 5 min. The slides were rinsed three times with distilled water, differentiated in ammonia and acidic water for 30 s, and then soaked in distilled water for 15 min. Subsequently, the sections were stained with Eosin (Solarbio) for 2 min, followed by dehydration in gradient alcohol and xylene removal. After the slides were fixed with neutral gum, the tissue cavities and cell numbers of each group were observed under a microscope.
Flow cytometry for ROS measurement in tissues
A ROS assay kit was used to determine ROS levels according to the manufacturer’s specification (Beyotime). Spinal cord samples in each group were collected and homogenized under ice-cold conditions. Each sample was incubated in the dark for 20 min in 500 µL of DCFH-DA (10 µM). After replacing the loading buffer and eliminating residual DCFH-DA, sample measurements were immediately performed using flow cytometry (Biosciences), with excitation light at 488 nm and emission light at 525 nm.
Transmission electron microscopy (TEM)
As mentioned earlier, TEM observations were conducted on the ultrastructure of spinal cord samples from different groups. The spinal cord samples from each group were incubated overnight in 2% glutaraldehyde at 4℃, followed by fixation with 1% citric acid. After immersion in uranyl acetate, they were dehydrated with gradient acetone. Subsequently, the samples were embedded in epoxy resin and cut into 70–90 nm sections, followed by re-stained with lead citrate on a copper groove grid. The ultrastructure of mitochondria was observed using TEM (Chiyoda ku, Tokyo, Japan).
Statistical analysis
Graphpad version 9.0 (Graphpad Software Inc., San Diego, CA, USA) was used for data analysis and plotting. All data are represented as mean ± standard deviation. The Shapiro-Wilk test was used to evaluate the distribution of data. If there were three or more datasets, the comparison was performed using a one-way analysis of variance (ANOVA) with Tukey’s post-test. A P-value < 0.05 was statistically significant.
Results
DEX weakened lidocaine-induced oxidative stress and ferroptosis in PC12 cells
The effect of lidocaine on PC12 cell viability was first assessed. MTT assays showed that lidocaine stimulation reduced the viability of PC12 cells in a concentration-dependent manner (Fig. 1A). Next, the effect of different doses of DEX treatment on PC12 cell viability, oxidative stress, and ferroptosis under lidocaine stimulation (1 mM) was further explored. We observed that increased DEX dose gradually mitigated the inhibitory effect of lidocaine on PC12 cell viability (Fig. 1B). We then assessed the effects of DEX (50 µM) on oxidative stress and ferroptosis in lidocaine-stimulated PC12 cells. The results exhibited that lidocaine stimulation significantly elevated ROS, MDA, and GSH levels in PC12 cells, while these increases were improved after DEX treatment (Fig. 1C-E). Consistently, ferrous iron (Fe2+) levels were upregulated by lidocaine in PC2 cells, while DEX treatment mitigated lidocaine-induced Fe2+ levels in the cells (Fig. 1F). These results indicated that DEX could mitigate lidocaine-induced oxidative stress and ferroptosis in PC12 cells.
Fig. 1.

DEX treatment impaired lidocaine-induced PC12 cell oxidative stress and ferroptosis. (A) The viability of PC12 cells stimulated with different concentrations of lidocaine was detected by MTT assays. (B) Assessment of cell viability in lidocaine-stimulated PC12 cells treated with different doses of DEX was performed by MTT assays. (C-E) The levels of ROS, MDA, and GSH in PC12 cells of different groups were assessed using ROS, MDA, and GSH assay kits. (F) The levels of Fe2+ were detected. Data are presented as the mean ± standard deviation (n = 3)
DEX treatment reduced lidocaine-induced ferritinophagy in PC12 cells
We further investigated whether DEX-mediated ferroptosis is associated with ferritinophagy in lidocaine-induced PC12 cells (degradation of ferritin in lysosomes). Lysotracker staining was performed on cells to observe changes in intracellular lysosomes. The cytoplasm exhibited an increase in red bright spots (ferroOrange). However, DEX treatment reduced the number of red bright spots, although the difference was not statistically significant (Fig. 2A and B). Moreover, lidocaine stimulation could significantly increase intracellular Fe2+ levels (ferroOrange), mitigated after DEX treatment (Fig. 2A and C). The control group without lidocaine stimulation showed high ferritin fluorescence, indicating a low ferritin degradation. Interestingly, lidocaine stimulation caused lysosome ferritin degradation, resulting in a significant decrease in fluorescence, as demonstrated by immunofluorescence assays. In addition, the levels of ferritin were partially recovered after DEX treatment (Fig. 2D-F). Furthermore, lidocaine treatment enhanced the fluorescence intensity of LC3, but it was weakened after DEX treatment (Fig. 2D-F). We also observed that lidocaine stimulation elevated the value of LC-3 II/I and reduced p62 protein levels, but these changes were reversed significantly following DEX treatment (Fig. 2G). These results revealed that DEX weakened the degradation of lysosomal ferritin and impaired ferritin phagocytosis in PC12 cells induced by lidocaine stimulation.
Fig. 2.

DEX treatment impaired lidocaine-induced ferritinophagy in PC12 cells. (A) LysoTracker and FerroOrange were shown by confocal imaging in PC12 cells in control, lidocaine, and lidocaine + DEX groups. (B) Quantification of the fluorescence intensity of FerroOrange (red) colocalized with LysoTracker (green). (C) Quantification of the FerroOrange fluorescence intensity. (D) Confocal images of ferritin (red fluorescence) and LC-3 (green fluorescence) for PC12 cells in different groups. (E) Quantification of ferritin colocalizing with autophagosomes (LC3-GFP). (F) Quantification of autophagosomes (LC3-GFP). (G) Western blotting determined the value of LC-3 II/I and p62 protein levels. Data are presented as the mean ± standard deviation (n = 3)
DEX could improve lidocaine-induced SCI by inhibiting ferroptosis in rats
To investigate the impact of DEX treatment on lidocaine-induced SCI in rats (Supplementary Fig. 1), we assessed the BBB score. We observed that the lidocaine + DEX group had a higher BBB score than the lidocaine group (Fig. 3A). After 4 weeks of SCI, we observed pathological changes, and the lidocaine + DEX group exhibited significantly lighter cavities and tissue damage than the lidocaine group (Fig. 3B). Nissl staining revealed more neurons in the spinal cord anterior horn of the lidocaine + DEX group than the lidocaine group (Fig. 3C). Given that high levels of iron can directly or indirectly promote the accumulation of ROS after SCI injury (Feng et al. 2021), we subsequently determined the ROS level in the epicenter of the injured spinal cord. Lidocaine stimulation significantly increased ROS levels, but DEX treatment weakened the increased levels of ROS (Fig. 3D). A significant increase in MDA levels and a decrease in GSH levels in the epicenter of the injured spinal cord were observed in the lidocaine group, but DEX treatment weakened both the elevated MDA and reduced GSH levels (Fig. 3E and F). Additionally, DEX significantly alleviated lidocaine-mediated elevation of Fe2+ levels in the epicenter of the injured spinal cord (Fig. 3G). These results revealed that DEX could improve lidocaine-induced SCI in rats by inhibiting ferroptosis.
Fig. 3.

DEX improved lidocaine-induced SCI by suppressing ferroptosis in rats. (A) The exercise behaviors of mice in the control, lidocaine, and lidocaine + DEX groups were evaluated using BBB scores. (B) Representative images of HE staining and the quantitation of the cavities in the epicenter of the injured spinal cord in each group. (C) Representative images of Nissl staining and the quantitation of the neurons in the spinal cord anterior horn in different groups (D) Representative images of ROS accumulation evaluated using flow cytometry and the quantitation of the percentage of ROS fluorescence. (E and F) MDA and GSH assay kits were used to measure MDA and GSH levels. (G) Assessment of Fe2+ levels was performed using the epicenter of the injured spinal cord. Data are presented as the mean ± standard deviation (n = 6)
DEX could improve lidocaine-induced SCI by suppressing ferritinophagy in rats
Scanning electron microscopy revealed that the mitochondria in the lidocaine + DEX group exhibited regular morphology and fewer vacuoles, while the mitochondria in the lidocaine group had irregular morphology, more vacuoles, and collapsed mitochondrial membranes (Fig. 4A). We found that the fluorescence intensity of ferritin (red) in the lidocaine group was weakened, while the fluorescence intensity of LC3 (green) was enhanced compared to the control group. However, after DEX treatment, these changes mediated by lidocaine were partly restored (Fig. 4B-D). A higher LC-3 II/I value and lower protein levels of p62 were observed in the lidocaine group, suppressing ferritinophagy after DEX treatment (Fig. 4E and F). Taken together, our findings suggested that DEX could improve lidocaine-induced SCI in rats by repressing ferritinophagy.
Fig. 4.

DEX improved lidocaine-induced SCI by suppressing ferritinophagy in rats. (A) Representative electron microscopy images of the epicenter of the injured spinal cord in the control, lidocaine, and lidocaine + DEX groups. (B) Fluorescence analysis showed representative images of ferritin and LC3. (C) Quantification of ferritin colocalizing with autophagosomes (LC3-GFP). (D) Quantification of autophagosomes (LC3-GFP). (E and F) Western blotting assessed the value of LC-3 II/I and the protein levels of p62. Data are presented as the mean ± standard deviation (n = 6)
Knockdown of CISD2 partly reversed the inhibitory effect of DEX on ferroptosis in lidocaine-stimulated PC12 cells
Previous studies have demonstrated low expression of CISD2 in spinal injury models (Lin et al. 2015), and CISD2 knockdown could inactivate the NRF2 pathway to induce oxidative stress, ferroptosis, and ferritinophagy (Li et al. 2022a, b, c). Besides, it has been shown that silencing of NRF2 decreased FTH1, SLC7A11, and GPX4 levels and increased NCOA4 levels, thus promoting ferritinophagy and ferroptosis (Xiu et al. 2022; Anandhan et al. 2023; Gao et al. 2019). Thus, we further explored whether DEX can improve lidocaine-induced SCI in rats by mediating CISD2, FTH1, SLC7A11, GPX4, and NCOA4 expression levels. As shown in Fig. 5A, CISD2 and FTH1, SLC7A11, and GPX4 protein levels were significantly reduced, and NCOA4 protein levels were elevated in the epicenter of the injured spinal cord in the lidocaine group, but these changes were weakened by DEX treatment. Consistently, lidocaine resulted in the downregulation of CISD2, SLC7A11, GPX4, and FTH1 in PC12 cells and the upregulation of NCOA4 in PC12 cells, but these alterations were partially suppressed following DEX treatment (Fig. 5B). Next, we explored whether DEX could mediate lidocaine-induced ferritinophagy and ferroptosis by CISD2 in PC12 cells. Importantly, the effects of DEX treatment on CISD2, SLC7A11, GPX4, FTH1, and NCOA4 in lidocaine-stimulated PC12 cells were attenuated by silencing CISD2 (Fig. 5C). DEX treatment increased PC12 cell viability under lidocaine stimulation, but this effect mediated by DEX treatment was mitigated after CISD2 knockdown (Fig. 5D).
Fig. 5.

CISD2 silencing lowered DEX-mediated inhibitory effect on ferroptosis in lidocaine-stimulated PC12 cells. (A) Detection of CISD2, FTH1, SLC7A11, GPX4, and NCOA4 protein levels in the epicenter of the injured spinal cord in the control, lidocaine, and lidocaine + DEX groups. (B-C) Detection of CISD2, FTH1, SLC7A11, GPX4, and NCOA4 protein levels in PC12 cells in control, lidocaine, lidocaine + DEX, or lidocaine + DEX + si-CISD2 groups. (D) MTT analysis of the viability of PC12 cells in different groups. (E-G) Measurement of MDA, GSH, and Fe2+ levels in PC12 cells in different groups. (H) Detection of the levels of ROS was made by flow cytometry. Data are presented as the mean ± standard deviation (n = 3)
Moreover, the inhibitory effects of DEX treatment on ROS and MDA in lidocaine-stimulated PC12 cells were reversed significantly following CISD2 silencing (Fig. 5E and H). On the contrary, the stimulatory effect of DEX treatment on GSH levels in lidocaine-stimulated PC12 cells was diminished after inhibiting CISD2 (Fig. 5F). In addition, treatment with CISD2 reversed the decrease in Fe2+ levels induced by DEX treatment in lidocaine-stimulated PC12 cells significantly (Fig. 5G). Collectively, these findings suggested that CISD2 silencing lowered the DEX-mediated inhibitory effect on ferroptosis in lidocaine-stimulated PC12 cells.
CISD2 silencing reversed the suppressive effect of DEX on ferritinophagy in lidocaine-stimulated PC12 cells
Next, we further investigated whether DEX-mediated lidocaine-induced ferritinophagy in PC12 cells through regulating CISD2 expression. DEX treatment did not affect lidocaine-induced autophagy (Lysotracker) in PC12 cells, and there was no significant difference after CISD2 knockdown (Fig. 6A and B). Furthermore, DEX treatment lowered lid ocaine-induced Fe2+ (FerroOrange) levels, but this reduction was partly alleviated after CISD2 silencing (Fig. 6A and C). Silencing CISD2 expression reversed the elevated protein levels of ferritin (red fluorescence) and the reduced protein levels of LC-3 (green fluorescence) in lidocaine-stimulated PC12 cells induced by DEX treatment significantly (Fig. 6C-G). Additionally, the reduction of LC-3 II/I and the upregulation of p62 in lidocaine-stimulated PC12 cells caused by DEX treatment was impaired after CISD2 knockdown, suppressing ferritinophagy (Fig. 6G). Overall, these results substantiated that CISD2 silencing alleviated the inhibitory effect of DEX on ferritinophagy in PC12 cells during lidocaine stimulation.
Fig. 6.

Downregulation of CISD2 lessened the suppressive effect of DEX on lidocaine-induced ferritinophagy in PC12 cells. (A) LysoTracker and FerroOrange staining of PC12 cells in control, lidocaine, lidocaine + DEX, or lidocaine + DEX + si-CISD2 groups. (B) The fluorescence intensity of FerroOrange (red) colocalized with LysoTracker (green) was quantified. (C) FerroOrange fluorescence intensity was quantified. (D) Immunofluorescence images of ferritin (red fluorescence) and LC-3 (green fluorescence) in PC12 cells of different groups. (E) Quantification of ferritin colocalizing with autophagosomes (LC3-GFP). (F) Quantification of autophagosomes (LC3-GFP). (G) Western blotting was utilized to analyze LC-3 II/I and p62 protein levels in PC12 cells from different groups. Data are presented as the mean ± standard deviation (n = 3)
Discussion
In recent years, research has revealed the neuroprotective effects of DEX, and numerous animal experiments have confirmed its significant therapeutic potential in repairing SCI (Li et al. 2022a, b, c; Zhao et al. 2019). To elucidate the neuroprotective effect and underlying mechanisms of DEX in SCI, this study employed a lidocaine-induced PC12 cell model and a lidocaine-induced rat SCI model to investigate the impact of DEX on SCI and analyze its associated molecular mechanisms. These findings provide a theoretical foundation for expanding the clinical applications of DEX and developing treatment strategies and mechanisms for SCI.
Studies have established a close relationship between ferroptosis and SCI Unlike other forms of cell death, ferroptosis is characterized by the accumulation of Fe2+ within cells, which leads to the consumption of unsaturated fatty acids in the cell membrane and subsequent lipid peroxidation-induced ROS accumulation (Li et al. 2020a, b). This mechanism involves the catalysis of highly expressed unsaturated fatty acids on the cell membrane by Fe2+ or lipoxygenase, leading to lipid peroxidation and the consumption of unsaturated fatty acids in the cell membrane (Wu et al. 2023). Additionally, it is characterized by alterations in the antioxidant system, resulting in decreased expression levels of GSH and GPX4, leading to excessive ROS accumulation and oxidative stress-induced damage to proteins, nucleic acids, and lipids, ultimately culminating in cell death (Li et al. 2022a, b, c). In this study, treatment with DEX attenuated the increase in ROS, MDA, and Fe2+ levels and decreased GSH levels induced by lidocaine in PC12 cells, suggesting that DEX may alleviate lidocaine-induced ferroptosis. Wang et al. reported that DEX regulated NRF2 or GPX4 expression levels, thereby mitigating myocardial ischemia/reperfusion-induced ferroptosis (Wang et al. 2022a, b; Yu et al. 2022). DEX also alleviated sepsis-induced myocardial iron deficiency by reducing iron concentration and enhancing GPX4 activity (Wang et al. 2020). Furthermore, DEX inhibited acyl-coA synthetase long-chain family member 4 (ACSL4) expression to counteract ferroptosis in renal ischemia-reperfusion injury (Tao et al. 2022). Studies by Liu et al. demonstrated that DEX ameliorated hemorrhagic brain injury by reducing ferroptosis-mediated damage in mice (Liu et al. 2022a, b, c). Additionally, DEX exerted inhibitory effects on ferroptosis in mice, thereby attenuating cerebral ischemia-reperfusion injury (Hu et al. 2022). These findings collectively support the suppressive effects of DEX on ferroptosis in various diseases.
Multiple studies have confirmed the involvement of autophagy in regulating ferroptosis (Zhou et al. 2020). DEX has been shown to exert neuroprotective effects by reducing cell apoptosis and autophagy (Li et al. 2020a, b; Xue et al. 2021; Zhang et al. 2021). It has been reported that DEX could decrease methotrexate-induced neurotoxicity and inflammation by improving NCOA4-mediated ferritinophagy in hippocampal HT22 cell lines (Chen et al. 2021). Herein, we observed that DEX could decrease lidocaine-mediated promoting effect on autophagy, Fe2+, and LC3 in PC12 cells and lidocaine-mediated suppressive effect on ferritin and p62 protein levels, indicating that DEX could weaken lidocaine-induced ferritinophagy in PC12 cells. Importantly, we observed that DEX treatment improved BBB scores, reduced tissue damage, increased the number of neurons, and alleviated mitochondrial damage by inhibiting ferroptosis and ferritinophagy in lidocaine-induced SCI rat models. These results demonstrate that DEX could attenuate lidocaine-induced SCI in rat models by inhibiting ferritinophagy-mediated ferroptosis.
CISD2, located on the long arm of chromosome 4, is a member of the gene family that contains the CDGSH iron-sulfur cluster-binding domain (Shen et al. 2021). Studies have revealed that CISD2 is involved in maintaining the stability of the mitochondrial outer membrane (Yeh et al. 2020; Salameh et al. 2021), and its loss can lead to breakdown and functional impairment of the mitochondrial outer membrane, resulting in autophagy and cell death (Li et al. 2021; Wang et al. 2023). Downregulation of CISD2 expression has been observed in animal models of SCI and astrocyte reactivation cell models (Kung et al. 2020a, b). Moreover, CISD2 was significantly downregulated under conditions related to central nervous system injury and diseases, such as spinal cord hemisection injury in rats (Kung et al. 2020a, b). It has been reported that silencing CISD2 could block the NRF2 pathway, inducing oxidative stress, ferroptosis, and ferritinophagy (Li et al. 2022a, b, c). Additionally, DEX has been shown to increase NRF2 expression, while NRF2 knockdown reduced FTH1 expression and increased NCOA4 expression, thereby promoting ferritinophagy-induced ferroptosis(Xiu et al. 2022; Anandhan et al. 2023). The neuroprotective effects of DEX on MTX-induced inflammation and its inhibitory effects on MTX-induced iron and ROS overproduction in HT22 cells were annulled by NCOA4 knockdown (Chen et al. 2021). DEX also enhanced SCI repair by activating the NRF2/HO-1 signaling pathway (Luo et al. 2020). Knockdown of NRF2 promoted ferritinophagy and ferroptosis by downregulating FTH1, SLC7A11, and GPX4, and upregulating NCOA4 (Xiu et al. 2022; Anandhan et al. 2023; Gao et al. 2019). Furthermore, silencing CISD2 expression accelerated ferroptosis through ferritinophagy-mediated ferritin turnover by mediating the p62-Keap1-NRF2 pathway (Li et al. 2022a, b, c). However, whether DEX can downregulate NCOA4 and upregulate FTH1, SLC7A11, and GPX4 by mediating CISD2 expression to improve ferritinophagy and ferroptosis in SCI remains unclear.
In this study, we investigated whether DEX can regulate lidocaine-induced ferroptosis and ferritinophagy by mediating the expression levels of FTH1, SLC7A11, GPX4, and NCOA4 by modulation of CISD2. Our data showed that CISD2, FTH1, SLC7A11, and GPX4 protein levels were significantly reduced, while NCOA4 protein levels were elevated in the epicenter of the injured spinal cord in the lidocaine group. However, these changes were attenuated by DEX treatment, indicating that DEX may mitigate lidocaine-induced SCI by regulating CISD2, FTH1, SLC7A11, GPX4, and NCOA4 expression in rats. Moreover, silencing CISD2 decreased FTH1, SLC7A11, and GPX4 expression and increased NCOA4 expression in PC12 cells. Importantly, CISD2 inhibition partially reversed the inhibitory effects of DEX treatment on lidocaine-induced ferroptosis and ferritinophagy in PC12 cells. These findings suggest DEX treatment could alleviate lidocaine-induced SCI by inhibiting ferroptosis and ferritinophagy by upregulating CISD2 in rat models.
Conclusion
Our findings substantiate that DEX treatment could inhibit ferritinophagy-mediated ferroptosis by upregulating FTH1, SLC7A11 and GPX4, and downregulating NCOA4 expression levels through the upregulation of CISD2, thereby alleviating lidocaine-induced SCI in rat models (Fig. 7). Although the specific lidocaine-induced SCI model used in our study is not a common cause of SCI, the discovery of its injury pathway may also appear in the most common traumatic SCI model, which provides a new research idea for further exploration of the mechanism. Moreover, findings from this study provide compelling evidence supporting the ability of DEX to alleviate SCI, suggesting that DEX may hold potential as a therapeutic strategy for the treatment of SCI.
Fig. 7.
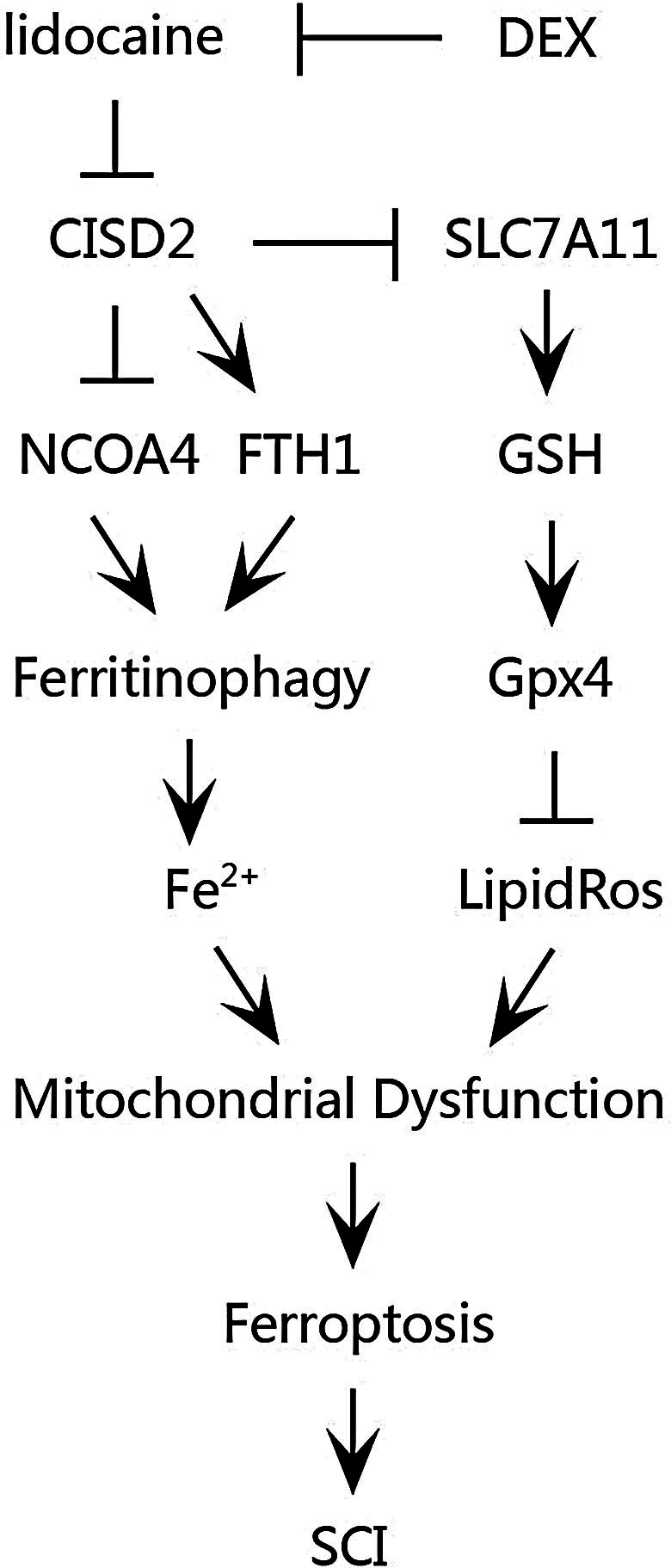
Schematic illustration of DEX in counteracting ferritinophagy-mediated ferroptosis and alleviating SCI
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This research was funded by Guangzhou Science and Technology Bureau.
Author contributions
Qiong Wang: Methodology, Investigation, Formal Analysis, Writing - Original Draft. Yonghong Tan and Yubing Guo: Resources, Data Curation, Writing - Original Draft. Xue Bai and Yingyi Xu: Visualization, Investigation. Na Zhang and Jianhua Liu: Supervision, Visualization. Xiaobao Bi and Yonghong Tan: Conceptualization, Resources, Validation, Writing - Review & Editing. All authors read and approved the final manuscript.
Funding
This work was supported by Guangzhou Municipal Science and Technology Project (NO. 202102080031).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethical approval
The animal study was reviewed and approved by the Laboratory Animal Center of Chongqing Medicine University in China (license No. SYXK (Su) 2018–0003).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yonghong Tan and Qiong Wang contributed equally to this work.
References
- Anandhan A, Dodson M, Shakya A et al (2023) NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv 9(5):eade9585. 10.1126/sciadv.ade9585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjum A, Yazid MD, Fauzi Daud M et al (2020), spinal cord Injury: pathophysiology, Multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci 21(20). 10.3390/ijms21207533 [DOI] [PMC free article] [PubMed]
- Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12(1):1–21. 10.1089/neu.1995.12.1 [DOI] [PubMed] [Google Scholar]
- Battaglia AM, Chirillo R, Aversa I et al (2020), Ferroptosis and Cancer: Mitochondria Meet the Iron Maiden Cell Death. Cells 9(6). 10.3390/cells9061505 [DOI] [PMC free article] [PubMed]
- Chay W, Kirshblum S (2020) Predicting outcomes after spinal cord Injury, physical medicine and rehabilitation clinics of North America. 31(3):331–343. 10.1016/j.pmr.2020.03.003 [DOI] [PubMed]
- Chen J, Wang J, Li C et al (2021) Dexmedetomidine reverses MTX-induced neurotoxicity and inflammation in hippocampal HT22 cell lines via NCOA4-mediated ferritinophagy. Aging 13(4):6182–6193. 10.18632/aging.202626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Qiao X, Liu W et al (2022) Epidemiology of spinal cord injury in China: a systematic review of the Chinese and English literature, spinal cord. 60(12):1050–1061. 10.1038/s41393-022-00826-6 [DOI] [PubMed]
- Chen F, Wang D, Jiang Y et al (2023) Dexmedetomidine postconditioning alleviates spinal cord ischemia-reperfusion injury in rats via inhibiting neutrophil infiltration, microglia activation, reactive gliosis and CXCL13/CXCR5 axis activation. Int J Neurosci 133(1):1–12. 10.1080/00207454.2021.1881089 [DOI] [PubMed] [Google Scholar]
- Cheng J (2021) TaoXun, ChuanhuiGuo, HailongCao, RuiGao, ShutaoSheng, Weibin %J life sciences, carnosic acid protects against ferroptosis in PC12 cells exposed to erastin through activation of Nrf2 pathway. 266(1):118905. 10.1016/j.lfs.2020.118905 [DOI] [PubMed]
- Cruz CD, Coelho A, Antunes-Lopes T et al (2015) Biomarkers of spinal cord injury and ensuing bladder dysfunction, Advanced drug delivery reviews. 82–83. 10.1016/j.addr.2014.11.007 [DOI] [PubMed]
- Ding XD, Cao YY, Li L et al (2021) Dexmedetomidine reduces the Lidocaine-Induced neurotoxicity by inhibiting Inflammasome activation and reducing pyroptosis in rats. Biol Pharm Bull 44(7):902–909. 10.1248/bpb.b20-00482 [DOI] [PubMed] [Google Scholar]
- Eli I, Lerner DP, Ghogawala Z (2021) Acute traumatic spinal cord Injury, neurologic clinics. 39(2):471–488. 10.1016/j.ncl.2021.02.004 [DOI] [PubMed]
- Feng Z, Min L, Chen H et al (2021) Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol 43. 10.1016/j.redox.2021.101984 [DOI] [PMC free article] [PubMed]
- Gao J, Sun Z, Xiao Z et al (2019) Dexmedetomidine modulates neuroinflammation and improves outcome via alpha2-adrenergic receptor signaling after rat spinal cord injury. Br J Anaesth 123(6):827–838. 10.1016/j.bja.2019.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge MH, Tian H, Mao L et al (2021) Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway, CNS neuroscience & therapeutics. 27(9):1023–1040. 10.1111/cns.13657 [DOI] [PMC free article] [PubMed]
- Hamid R, Averbeck MA, Chiang H et al (2018), Epidemiology and pathophysiology of neurogenic bladder after spinal cord injury. World J Urol 36(10):1517–1527. 10.1007/s00345-018-2301-z [DOI] [PubMed] [Google Scholar]
- Hu M, Men Y, Chen L et al (2022) Dexmedetomidine exerts its protective effect on cerebral ischemia reperfusion injury in mice by inhibiting ferroptosis. Zhong Nan Da Xue Xue Bao Yi Xue Ban 47(5):600–609. 10.11817/j.issn.1672-7347.2022.210443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung WM, Lin CC, Kuo CY et al (2020a) Wild bitter melon exerts anti-inflammatory effects by Upregulating Injury-attenuated CISD2 expression following spinal cord Injury. Behav Neurol 2020(1080521). 10.1155/2020/1080521 [DOI] [PMC free article] [PubMed]
- Kung WM, Chang CJ, Chen TY et al (2020b) Cryogen spray cooling mitigates inflammation and injury-induced CISD2 decline in rat spinal cord hemisection model. J Integr Neurosci 19(4):619–628. 10.31083/j.jin.2020.04.255 [DOI] [PubMed] [Google Scholar]
- Latunde-Dada GO (2017) Role of lipid peroxidation, iron and ferritinophagy, Biochimica et biophysica acta. Gen Subj 1861(8):1893–1900. 10.1016/j.bbagen.2017.05.019 [DOI] [PubMed] [Google Scholar]
- Li N, Wang W, Zhou H et al (2020a) Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med 160:303–318. 10.1016/j.freeradbiomed.2020.08.009 [DOI] [PubMed] [Google Scholar]
- Li H, Lu C, Yao W et al (2020b) Dexmedetomidine inhibits inflammatory response and autophagy through the circLrp1b/miR-27a-3p/Dram2 pathway in a rat model of traumatic brain injury. Aging 12(21):21687–21705. 10.18632/aging.103975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Wei S, Yang L et al (2021), CISD2 promotes resistance to Sorafenib-Induced ferroptosis by regulating Autophagy in Hepatocellular Carcinoma. Front Oncol 11:657723. 10.3389/fonc.2021.657723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu B, Ren X et al (2022a) Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. 27(1). Cellular & molecular biology letters10.1186/s11658-022-00383-z [DOI] [PMC free article] [PubMed]
- Li Y, Xu B, Ren X et al (2022b) Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell Mol Biol Lett 27(1):81. 10.1186/s11658-022-00383-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FJ, Long HZ, Zhou ZW et al (2022c) System X(c) (-)/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol 13. 10.3389/fphar.2022.910292 [DOI] [PMC free article] [PubMed]
- Li JZ, Fan BY, Sun T et al (2023) Bioinformatics analysis of ferroptosis in spinal cord injury. Neural Regeneration Res 18(3):626–633. 10.4103/1673-5374.350209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Wang Y, Liu Y (2019) Dexmedetomidine alleviates lung ischemia-reperfusion injury in rats by activating PI3K/Akt pathway, European review for medical and pharmacological sciences. 23(1):370–377. 10.26355/eurrev_201901_16785 [DOI] [PubMed]
- Lin CC, Chiang TH, Chen WJ et al (2015) CISD2 serves a novel role as a suppressor of nitric oxide signalling and curcumin increases CISD2 expression in spinal cord injuries. Injury 46(12):2341–2350. 10.1016/j.injury.2015.07.040 [DOI] [PubMed] [Google Scholar]
- Liu J, Kuang F, Kroemer G et al (2020) Autophagy-dependent ferroptosis: Machinery and Regulation. Cell Chem Biology 27(4):420–435. 10.1016/j.chembiol.2020.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YB, Liu WF, Chen WC et al (2022a) Dexmedetomidine alleviates traumatic spinal cord injury in rats via inhibiting apoptosis induced by endoplasmic reticulum stress. Neurol Res 44(3):275–284. 10.1080/01616412.2021.1979750 [DOI] [PubMed] [Google Scholar]
- Liu B, Liang Y, Huang W et al (2022b), Dexmedetomidine attenuates spinal cord ischemia-reperfusion Injury in rabbits by decreasing oxidation and apoptosis, current molecular medicine. 10.2174/1566524022666220525142954 [DOI] [PubMed]
- Liu MJ, Zhao XC, Gong HS et al (2022c) Dexmedetomidine prevents hemorrhagic brain injury by reducing damage induced by ferroptosis in mice. Neurosci Lett 788:136842. 10.1016/j.neulet.2022.136842 [DOI] [PubMed] [Google Scholar]
- LoBianco FV, Krager KJ, Johnson E et al (2022) Parthenolide induces rapid thiol oxidation that leads to ferroptosis in hepatocellular carcinoma cells. Front Toxicol 4:936149. 10.3389/ftox.2022.936149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Chen T, Kang G et al (2020) Dexmedetomidine promotes spinal cord injury repairing via activating Nrf2/HO-1 signaling pathway. J Neurosurg Sci 64(6):583–585. 10.23736/s0390-5616.19.04812-4 [DOI] [PubMed] [Google Scholar]
- Lv Q, Niu H, Yue L et al (2020) Abnormal ferroptosis in Myelodysplastic Syndrome. Front Oncol 10. 10.3389/fonc.2020.01656 [DOI] [PMC free article] [PubMed]
- Ma D, Jiang P, Jiang Y et al (2021) Effects of lipid peroxidation-mediated ferroptosis on severe Acute Pancreatitis-Induced Intestinal Barrier Injury and bacterial translocation, 2021. 6644576. 10.1155/2021/6644576 [DOI] [PMC free article] [PubMed]
- Mahmoud M, Mason KP (2015) Dexmedetomidine: review, update, and future considerations of paediatric perioperative and periprocedural applications and limitations. Br J Anaesth 115(2):171–182. 10.1093/bja/aev226 [DOI] [PubMed] [Google Scholar]
- Masaldan S, Clatworthy SAS, Gamell C et al (2018) Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis, Redox biology. 14:100–115. 10.1016/j.redox.2017.08.015 [DOI] [PMC free article] [PubMed]
- Quadri SA, Farooqui M, Ikram A et al (2020) Recent update on basic mechanisms of spinal cord injury. Neurosurg Rev 43(2):425–441. 10.1007/s10143-018-1008-3 [DOI] [PubMed] [Google Scholar]
- Russo GS, Mangan JJ, Galetta MS et al (2020) Update on Spinal Cord Injury Management, clinical spine surgery. 33(7):258–264. 10.1097/bsd.0000000000000956 [DOI] [PubMed]
- Salameh M, Riquier S, Guittet O et al (2021) New insights of the NEET protein CISD2 reveals distinct features compared to its close mitochondrial homolog mitoNEET. Biomedicines 9(4). 10.3390/biomedicines9040384 [DOI] [PMC free article] [PubMed]
- Santana-Codina N, Gikandi A, Mancias JD (2021) The role of NCOA4-Mediated ferritinophagy in Ferroptosis, advances in experimental medicine and biology. 1301:41–57. 10.1007/978-3-030-62026-4_4 [DOI] [PubMed]
- Shen ZQ, Huang YL, Teng YC et al (2021) CISD2 maintains cellular homeostasis, Biochimica et biophysica acta. Mol cell Res 1868(4):118954. 10.1016/j.bbamcr.2021.118954 [DOI] [PubMed] [Google Scholar]
- Snyder R, Verla T, Ropper AE (2020) Practical application of recent advances in Diagnostic, Prognostic, and therapeutic modalities for spinal cord Injury, World neurosurgery. 136:330–336. 10.1016/j.wneu.2020.01.011 [DOI] [PubMed]
- Tadokoro T, Ikeda M, Ide T et al (2020) Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity, JCI insight. 5(9). 10.1172/jci.insight.132747 [DOI] [PMC free article] [PubMed]
- Tang D, Chen X, Kang R et al (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31(2):107–125. 10.1038/s41422-020-00441-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao WH, Shan XS, Zhang JX et al (2022), Dexmedetomidine attenuates ferroptosis-mediated renal Ischemia/Reperfusion Injury and inflammation by inhibiting ACSL4 via α2-AR. Front Pharmacol 13:782466. 10.3389/fphar.2022.782466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yuan W, Hu A et al (2020) Dexmedetomidine alleviated sepsis–induced myocardial ferroptosis and septic heart injury. Mol Med Rep 22(1):175–184. 10.3892/mmr.2020.11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang M, Liu Y et al (2022a) Integrated regulation of stress responses, autophagy and survival by altered intracellular iron stores. Redox Biol 55. 10.1016/j.redox.2022.102407 [DOI] [PMC free article] [PubMed]
- Wang Z, Yao M, Jiang L et al (2022b) Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 154:113572. 10.1016/j.biopha.2022.113572 [DOI] [PubMed]
- Wang J, Hu J, Wang M et al (2023), CISD2 promotes proliferation of Colorectal Cancer cells by inhibiting autophagy in a Wnt/β-Catenin-signaling-dependent pathway, biochemical genetics. 61(2):615–627. 10.1007/s10528-022-10267-8 [DOI] [PubMed]
- Wei N, Lu T, Yang L et al (2021) Lipoxin A4 protects primary spinal cord neurons from Erastin๊nduced ferroptosis by activating the Akt/Nrf2/HOsignaling pathway. 11(8):2118–2126. 10.1002/2211-5463.13203 [DOI] [PMC free article] [PubMed]
- Wu ZF, Liu XY, Deng NH et al (2023), Outlook of ferroptosis-targeted lipid peroxidation in Cardiovascular Disease, current medicinal chemistry. 30(31):3550–3561. 10.2174/0929867330666221111162905 [DOI] [PubMed]
- Xiu Z, Zhu Y, Han J et al (2022), Caryophyllene Oxide induces Ferritinophagy by regulating the NCOA4/FTH1/LC3 pathway in Hepatocellular Carcinoma. Front Pharmacol 13:930958. 10.3389/fphar.2022.930958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Wu Z, Xu Y et al (2021) Dexmedetomidine post-conditioning ameliorates long-term neurological outcomes after neonatal hypoxic ischemia: the role of autophagy, life sciences. 270:118980. 10.1016/j.lfs.2020.118980 [DOI] [PubMed]
- Yao X, Zhang Y, Hao J et al (2019) Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis, neural regeneration research. 14(3):532–541. 10.4103/1673-5374.245480 [DOI] [PMC free article] [PubMed]
- Yeh CH, Chou YJ, Kao CH et al (2020), Mitochondria and Calcium Homeostasis: Cisd2 as a big player in Cardiac Ageing. Int J Mol Sci 21(23). 10.3390/ijms21239238 [DOI] [PMC free article] [PubMed]
- Yu P, Zhang J, Ding Y et al (2022) Dexmedetomidine post-conditioning alleviates myocardial ischemia-reperfusion injury in rats by ferroptosis inhibition via SLC7A11/GPX4 axis activation, human cell. 35(3):836–848. 10.1007/s13577-022-00682-9 [DOI] [PubMed]
- Yuan Y, Zhai Y, Chen J et al (2021), Kaempferol ameliorates oxygen-glucose Deprivation/Reoxygenation-Induced neuronal ferroptosis by activating Nrf2/SLC7A11/GPX4 Axis, Biomolecules. 11(7). 10.3390/biom11070923 [DOI] [PMC free article] [PubMed]
- Zhang L, Xiao F, Zhang J et al (2021), Dexmedetomidine Mitigated NLRP3-Mediated Neuroinflammation via the ubiquitin-autophagy pathway to improve Perioperative Neurocognitive Disorder in mice. Front Pharmacol 12:646265. 10.3389/fphar.2021.646265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jian Y, Wu L et al (2022) Analysis of the long non-coding RNA and mRNA expression profiles associated with lidocaine-induced neurotoxicity in the spinal cord of a rat model. Neurotoxicology 90:88–101. 10.1016/j.neuro.2022.03.002 [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhai M, Yang X et al (2019) Dexmedetomidine attenuates neuronal injury after spinal cord ischaemia-reperfusion injury by targeting the CNPY2-endoplasmic reticulum stress signalling. J Cell Mol Med 23(12):8173–8183. 10.1111/jcmm.14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Liu J, Kang R et al (2020) Ferroptosis is a type of autophagy-dependent cell death. Sem Cancer Biol 66:89–100. 10.1016/j.semcancer.2019.03.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.