

Abstract
During lytic infection, herpes simplex virus type 1 (HSV-1) represses host transcription, recruits RNA polymerase II (RNAP II) to viral replication compartments, and alters the phosphorylation state of the RNAP II large subunit. Host transcription repression and RNAP II modifications require expression of viral immediate-early (IE) genes. Efficient modification of the RNAP II large subunit to the intermediately phosphorylated (IIi) form requires expression of ICP22 and the UL13 kinase. We have further investigated the mechanisms by which HSV-1 effects global changes in RNAP II transcription by analyzing the RNAP II holoenzyme. We find that the RNAP II general transcription factors (GTFs) remain abundant after infection and are recruited into viral replication compartments, suggesting that they continue to be involved in viral gene transcription. However, virus infection modifies the composition of the RNAP II holoenzyme, in particular triggering the loss of the essential GTF, TFIIE. Loss of TFIIE from the RNAP II holoenzyme requires viral IE gene expression, and viral IE proteins may be redundant in mediating this effect. Although viral IE proteins do not associate with the RNAP II holoenzyme, they interact with RNAP II in complexes of lower molecular mass. As the RNAP II holoenzyme containing TFIIE is necessary for activated transcription initiation and RNAP II large subunit phosphorylation in uninfected cells, virus-induced modifications to the holoenzyme may affect both of these processes, leading to aberrant phosphorylation of the RNAP II large subunit and repression of host gene transcription.
Herpes simplex virus type 1 (HSV-1) is a 152-kb double-stranded DNA virus, the genome of which is transcribed and replicated within the host cell's nucleus (reviewed in reference 54). During lytic infection, the HSV-1 genes are transcribed by the host's RNA polymerase II (RNAP II) transcription machinery (20, 71). The HSV-1 genes are expressed in a regulated cascade and are classified into three groups based on their order of expression: immediate-early (IE), delayed-early (DE), and late (L). The five IE genes (encoding ICP4, ICP0, ICP27, ICP22, and ICP47) are expressed immediately after infection, and all IE gene products except ICP47 are regulatory proteins involved in controlling expression of the DE and L genes. Their synthesis reaches a peak between 2 and 4 h postinfection, but IE proteins persist throughout infection.
Infection with HSV-1 results in dramatic alterations to host gene transcription. Within 6 h postinfection, RNAP II transcription of many, if not most, cellular genes is repressed to less than 40% of uninfected levels (36, 61, 64, 66), and transcription levels decline for at least another 6 h. At the same time, RNAP II transcription of HSV-1 genes is induced to high levels (20, 64). We have shown that repression of host gene transcription does not require DE or L gene transcription or viral DNA replication. Also, virion components do not trigger host transcription repression in the absence of viral IE gene expression (64). The IE proteins may be redundant in their effects on host gene transcription, as transcription is repressed after infection with viruses bearing null mutations in individual IE genes (64).
The preferential transcription of viral DE and L genes over host cell genes cannot be explained by sequence differences between host and viral gene promoters (reviewed in reference 61). Each of the viral gene promoters displays features of typical RNAP II promoters. IE gene promoters contain TATA boxes, start sites, and TAATGARAT elements that bind cellular complexes containing Oct-1. The virion transactivator VP16, in association with Oct-1 and HCF, binds to these elements and stimulates transcription of each IE gene (31, 41). The viral DE gene promoters are simple RNAP II promoters, containing TATA boxes, start sites, and promoter-proximal cis-acting sequences that bind basal cellular transcription factors such as Sp1. L gene promoters are even simpler than those of DE genes, comprising only TATA boxes and sequences surrounding transcription start sites. Neither DE nor L gene promoters contain specific binding sites for viral regulatory proteins that are necessary for DE or L gene transcription during infection (60). The sequence independence of host and viral gene transcription regulation is demonstrated by studies of cellular genes expressed within the viral genome. For example, the β-globin gene, with 1.2 kb of upstream promoter DNA, is expressed as a viral DE gene after infection of MEL cells with the recombinant β-globin-HSV; however, the endogenous cellular β-globin gene is transcriptionally repressed (59, 61). These and similar data have led to the hypothesis that the global shift in RNAP II promoter recognition that occurs after HSV-1 infection may be due to sequence-independent mechanisms, such as recruitment of transcription proteins into viral compartments, differences between host and viral chromatin, and modification of the host's RNAP II transcription machinery (61).
We have reported previously that RNAP II is recruited into viral replication compartments following HSV-1 infection (51). However, considering that repression of host transcription occurs after infection with ICP4 mutant viruses, and that replication compartments do not form during these infections, relocation of RNAP II to viral replication compartments is unlikely to be a requirement for host transcription repression (64). Nonetheless, RNAP II recruitment to viral replication compartments may be a requirement for efficient activation of viral DE and L gene transcription.
We have also reported that HSV-1 infection alters the phosphorylation state of the RNAP II large subunit (51). RNAP II consists of at least 10 subunits, the largest of which contains sites of catalysis and of DNA and nascent RNA binding and the carboxy-terminal domain (CTD). The CTD is a highly conserved structure of 52 repeats of the sequence YSPTSPS and is essential for cell viability (reviewed in reference 10). Due to cyclic phosphorylation of serine, threonine, and tyrosine residues on the CTD, RNAP II is present in two forms in vivo—RNAP IIO (hyperphosphorylated) and RNAP IIA (hypophosphorylated). RNAP IIA is recruited to preinitiation complexes, and RNAP IIO is the form of the enzyme involved in transcription elongation. The transition from IIA to IIO occurs during transcription initiation and is due to the activities of CTD kinases including cyclin-dependent kinase 7 (cdk7, a subunit of TFIIH) and cdk9 (a subunit of P-TEFb) (9, 10, 35, 69). The CTD has several functions in vivo, including mediating responses to transcription activators and stimulating transcription elongation. The CTD may also play a role in pre-mRNA processing by recruiting mRNA processing factors to nascent transcripts (26).
We have reported that within 5 h postinfection, RNAP IIO, the hyperphosphorylated form of RNAP II, is replaced by an intermediately phosphorylated, transcriptionally active form, RNAP III. The viral IE protein ICP22 and the viral kinase UL13 are required for efficient production of RNAP III, but the hyperphosphorylated RNAP IIO form is lost after infection with both wild-type and individual IE null mutant viruses (33, 50, 51). Given the importance of the CTD and its phosphorylation cycle to transcription regulation, we have hypothesized that HSV-1-induced alterations to CTD phosphorylation may contribute to the shift in transcription from host to viral genes.
In order to initiate transcription, RNAP II requires a number of accessory factors—the general transcription factors (GTFs). These include TFIID, TFIIB, TFIIF, TFIIH, and TFIIE. It is now thought that most or all of the GTFs participate in stable, preassembled RNAP II complexes (the ∼2-MDa holoenzymes) that are thought to be the physiologically relevant forms of RNAP II that enter most (but not all) preinitiation complexes in vivo (7, 22, 24, 38, 67). Genetic studies in yeast show that artificial recruitment of a holoenzyme subunit to promoter DNA is sufficient to activate transcription and remodel chromatin (17, 19). Although the composition of the RNAP II holoenzyme varies somewhat depending on the method of isolation, holoenzymes from human cells contain at least RNAP II, TFIIE, TFIIF, and TFIIH, and some contain TFIIB and TFIID. Besides the GTFs, RNAP II holoenzymes contain a number of factors that mediate transcription activation, modify chromatin structure, and interact with mRNA processing factors (reviewed in reference 24).
In this study, we examine virus-induced modifications to the RNAP II holoenzyme. We find that HSV-1 infection leads to depletion of TFIIE from the RNAP II holoenzyme and a reduction in holoenzyme activity in vitro. TFIIE depletion requires IE gene expression but occurs in the absence of individual IE proteins, suggesting that two or more IE proteins may be redundant in their effects on RNAP II holoenzyme composition. Since TFIIE is an essential holoenzyme GTF and stimulates RNAP II large subunit phosphorylation mediated by TFIIH, its absence from the holoenzyme may contribute to aberrant phosphorylation of the RNAP II large subunit and altered transcription patterns during HSV-1 infection.
MATERIALS AND METHODS
Cells, viruses, and infections.
HeLa S3 cells (American Type Culture Collection, Manassas, Va.) were used for infections. Cells were propagated in Dulbecco modified Eagle medium containing 10% fetal bovine serum. HeLa S3 cells were grown either as monolayers or as suspension cells in spinner flasks.
Infections were performed at a multiplicity of infection (MOI) of 10 PFU per cell. Infections of monolayer cells were performed as described elsewhere (50). Suspension cells were infected by addition of virus at 10 PFU/cell and incubated for 12 h.
The following HSV-1 strains were used: the wild-type strain F22, the ICP4 mutant strain d120 (13), the ICP0 mutant strain n212 (5), the ICP27 mutant strain d27-1 (49), and the ICP22 mutant d22-lacZ (33). The F22 virus contains a FLAG epitope-tagged ICP22 gene and is a derivative of the wild-type virus KOS1.1. The 12-codon FLAG epitope was inserted between codons 6 and 7 of the ICP22 gene in KOS1.1. F22 is phenotypically wild type by viral transcription and growth in ICP22 permissive and restrictive cell lines. Details of the construction and characterization of F22 will be described elsewhere (C. A. Spencer and S. A. Rice, unpublished data). Growth and titering of virus strains have been described previously (33, 50).
UV-inactivated virus stock was prepared as described previously (51) from a stock of the wild-type HSV-1 strain KOS1.1. The titer of UV-inactivated virus was 4 to 5 orders of magnitude reduced from that of the parent stock. Infections with UV-inactivated virus were carried out using the stock's preirradiation titer to obtain an MOI of 10.
Western blotting and immunofluorescence.
Preparation of whole-cell extracts and immunoblotting were performed as described previously (51). Blots were probed with the following primary antibodies: the anti-RNAP II large subunit antibodies ARNA-3 (29) (Research Diagnostics) at a 1:2,000 dilution and 8WG16 (68) at a 1:7,500 dilution, the anti-p56E antibody TFIIE-α C-17 (Santa Cruz) at a 1:1,000 dilution, an anti-TATA-binding protein (anti-TBP) antibody (Promega) at a 1:1,000 dilution, the anti-MO15 antibody MO1.1 (NeoMarkers) at a 1:400 dilution, the anti-RAP74 antibody 7B3 (Austral Biologicals) at a 1:300 dilution, the anti-MLH antibody hMLH1 C-20 (Santa Cruz) at a 1:100 dilution, the anti-FLAG antibody M2 (Eastman Kodak) at 1:1,000, the anti-ICP4 antibody H1101 at 1:2,000, the anti-ICP27 antibody H1119 at 1:8,000, and the anti-ICP0 antibody H1112 at 1:2,500 (Goodwin Institute). Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse (or anti-rabbit) immunoglobulin G (IgG) (Jackson ImmunoResearch). Blots were visualized using the ECL Western blotting detection system (Amersham).
Immunostaining was performed as described previously (65). Primary antibodies were the anti-ICP4 antibody H1101 conjugated to either Texas Red or Oregon Green fluorochrome (Molecular Probes) and used at a 1:200 dilution, 8WG16 at 1:200, anti-TBP at 1:200, TFIIE-α C-17 at 1:50, and anti-p62H (Upstate Biotechnology) at 1:100. Secondary antibodies were goat anti-mouse Alexa 488 at a 1:250 dilution, goat anti-rabbit Alexa 488 at a 1:250 dilution (Molecular Probes), or lissamine rhodamine-conjugated goat anti-mouse IgG at a 1:200 dilution (Jackson ImmunoResearch). DNA was visualized by staining cells with 4′,6′-diamidino-2-phenylindole (DAPI) at 5 μg/ml. Coverslips were mounted in glycerol, and cells were visualized with a Zeiss LSM510 confocal microscope using sequential laser scans for each fluorochrome. Images for Fig. 8 were converted to tagged-image format files (TIFFs) and were assembled and labeled using Adobe Photoshop software. Control stains included secondary antibodies only, ICP4 stains only, and primary or secondary stains only in the absence of ICP4 staining. Bleedthrough staining was undetectable using confocal microscopy and sequential laser scans.
FIG. 8.

Intracellular distribution of RNAP II and GTFs in HSV-1-infected cells. HeLa S3 cells were infected with wild-type virus for 6 h, fixed, and stained with anti-ICP4 antibody conjugated to either Texas Red or Oregon Green in order to visualize viral replication compartments. Cells were costained with DAPI to visualize DNA and with antibodies recognizing the RNAP II large subunit, TBP, p56E, or p62H (a subunit of TFIIH) and secondary antibodies conjugated to either rhodamine (TBP, red stain) or Alexa 488 (green stain). Staining patterns were visualized by confocal microscopy.
Nuclear extract preparation.
Nuclear extracts were prepared from 109 uninfected or infected HeLa S3 cells by following the methods of Shapiro et al. (58). HeLa S3 cells were grown to a density of approximately 106/ml in spinner flasks and were infected with wild-type or mutant HSV-1 strains at an MOI of 10. At 12 h postinfection, cells were harvested and nuclei were isolated as described previously (58). Nuclear extracts were dialyzed into nuclear dialysis buffer (20 mM HEPES [pH 7.9], 100 mM KCl, 0.2 mM EDTA, 0.2 mM EGTA, 2 mM dithiothreitol [DTT], and 20% [vol/vol] glycerol) and typically contained protein concentrations between 8 and 15 mg per ml. Extracts were frozen in liquid nitrogen until use.
Sepharose CL-2B gel filtration chromatography.
Approximately 2 mg of nuclear extracts was fractionated on 25-ml Sepharose CL-2B columns as described previously (45). The CL-2B columns were equilibrated and run with CL-2B buffer (20 mM HEPES [pH 7.9], 100 mM KCl, 20% glycerol) at 4°C. Fractions of 500 μl were collected and precipitated with acetone. Precipitated proteins were resuspended in sodium dodecyl sulfate (SDS) loading buffer and analyzed by immunoblotting. Bands on Western blots were quantitated by scanning autoradiographs with an LKB Ultroscan XL Enhanced Laser Densitometer. Densitometer readings were normalized against identical standards (20 μg of nuclear extract) on each gel. Normalized readings were plotted using DeltaGraph 4.0 software, and plots were assembled and labeled using Quark Xpress software (Fig. 3, 5, and 9).
FIG. 3.

Elution profiles of RNAP II and GTFs from Sepharose CL-2B gel filtration chromatography. Nuclear extracts from uninfected cells or cells infected with HSV-1 were chromatographed and analyzed by immunoblotting, as described in the legend to Fig. 2. The intensity of each band was quantitated by densitometry. Densitometer readings were normalized against identical standards on each gel (20 μg of infected or uninfected nuclear extract) and expressed as relative densitometry units. Relative densitometry units were plotted against fraction number. Positions of 2-MDa and 670-kDa molecular standards are indicated. Horizontal lines below graphs indicate fractions at ∼2 MDa and ∼670 kDa that were pooled for GST-TFIIS affinity chromatography and in vitro transcriptions (Fig. 4, 6, 7, and 10).
FIG. 5.
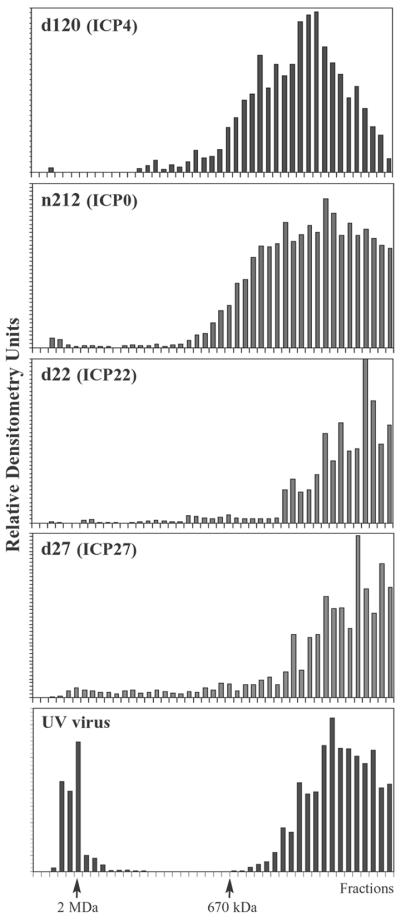
Elution profiles of TFIIE (p56E subunit) from Sepharose CL-2B gel filtration chromatography. Nuclear extracts were prepared from cells infected with viruses bearing null mutations in one of the IE genes ICP4, ICP0, ICP22, and ICP27, as well as with UV-inactivated virus, as indicated. Nuclear extracts were fractionated on Sepharose CL-2B columns, and fractions were analyzed by immunoblotting, as described for Fig. 2. Relative densitometry units were determined and plotted as described for Fig. 3. Positions of 2-MDa and 670-kDa molecular standards are indicated.
FIG. 9.

Elution profiles of viral IE proteins from Sepharose CL-2B gel filtration chromatography. Nuclear extracts prepared from cells infected with wild-type HSV-1 were chromatographed on Sepharose CL-2B columns, and fractions were analyzed by immunoblotting. Blots were probed with antibodies that recognize ICP4, ICP0, ICP22, and ICP27 as indicated. Relative densitometry units were determined and plotted as described for Fig. 3. Positions of 2-MDa and 670-kDa molecular standards are indicated.
Sepharose CL-2B columns were calibrated with Blue Dextran 2000 (2 MDa; Pharmacia) and thyroglobulin (670 kDa; Bio-Rad). To control for protein binding to RNA and DNA, an infected and an uninfected nuclear extract were incubated at 4°C for 20 min in 20 μg of ethidium bromide/ml, 100 μg of RNase A/ml, and 500 U of DNase I/ml prior to CL-2B chromatography.
Affinity purification of the RNAP II holoenzyme.
TFIIS affinity chromatography was carried out as described by Pan et al. (45). Affinity columns were prepared by binding glutathione S-transferase (GST) or GST-TFIIS fusion protein to glutathione-Sepharose 4B beads as described by the manufacturer (Amersham Pharmacia). Affinity matrices of 50 μl were dispensed into 250-μl columns (Promega) and washed with 20 bed volumes of ACB (10 mM HEPES [pH 7.9], 50 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT, 10% glycerol). Columns were blocked with ACB plus 0.1% bovine serum albumin (BSA). Affinity matrices were incubated with either pooled 2-MDa fractions (fractions surrounding the 2-MDa peak) or pooled 670-kDa fractions (fractions at and below 670 kDa) from Sepharose CL-2B chromatography (fractions indicated in Fig. 2 and 3). The flowthrough was reapplied once. Columns were washed with 20 bed volumes of ACB. Bound proteins were then eluted successively with 4 bed volumes of ACB plus 0.3 M NaCl and ACB plus 0.5 M NaCl. Proteins in the eluates were concentrated by acetone precipitation and analyzed by immunoblotting.
FIG. 2.

Gel filtration chromatography of RNAP II and GTFs. Nuclear extracts from uninfected cells (A) or cells infected with wild-type HSV-1 (B) were chromatographed on Sepharose CL-2B columns. Fractions were concentrated and analyzed by immunoblotting. Blots were probed with antibodies that recognize the RNAP II large subunit, the p56E subunit of TFIIE, the TBP subunit of TFIID, and the cdk7 subunit of TFIIH. Blots were also probed with an antibody that recognizes the DNA repair protein MLH1. Lane 1 contains 20 μg of uninfected (A) or infected (B) nuclear extract. Fractions 21 to 31 surrounded ∼2 MDa; fractions 42 to 52 were at and below ∼670 kDa. Ten fractions between approximately 1 MDa and 670 kDa are not shown in this figure. Fractions 24 through 28 (2 MDa Pool) were pooled for GST-TFIIS affinity chromatography and in vitro transcription assays (Fig. 4, 6, and 10A), as were fractions 43 through 52 (670 kDa Pool) (Fig. 7 and 10B).
In vitro transcriptions.
The DNA template for in vitro transcriptions consisted of the 2.4-kb SmaI fragment of adenovirus (Ad), containing the Ad major late promoter (MLP) (14), cloned into vector pSG5 (Stratagene) to create plasmid pSG5Ad. pSG5Ad was digested with BamHI, 540 bp downstream of the Ad MLP transcription start site. Reaction mixtures contained either crude nuclear extracts (46 μg) or concentrates from the ∼2-MDa or ∼670-kDa peaks of Sepharose CL-2B columns. Fractions surrounding the 2-MDa or 670-kDa peaks (as indicated in Fig. 2 and 3) were pooled and concentrated 50- or 10-fold in UltraFree-0.5 concentrators (Millipore Corp.). Ten microliters of concentrate was used in each reaction. An aliquot of each nuclear extract and concentrate was analyzed by immunoblotting, to estimate the relative quantities of RNAP II in each reaction. Transcription reactions were carried out as follows.
DNA template (600 ng) was preincubated at 4°C for 15 min in a solution containing nuclear extract (or column concentrate), 10 mM HEPES (pH 7.6), 50 mM KCl, 8% glycerol, 0.04 mM EDTA, and 0.1 mM DTT. After preincubation, magnesium chloride was added to 8 mM, plus ATP, GTP, and CTP to 0.4 mM, and 10 μCi of 5′-[α-32P]UTP. Reactions were terminated after 45 min at 30°C by addition of 300 mM Tris HCl (pH 7.4)–300 mM sodium acetate–0.5% SDS–2 mM EDTA. Reaction products were treated with 50 μg of proteinase K for 20 min at 42°C, then phenol extracted and precipitated. RNA was separated on 5% acrylamide-urea gels, and radioactivity was visualized by autoradiography. Signals were quantitated by phosphorimaging (Fujix Bas100 analyzer).
Artwork for autoradiographs.
Autoradiographs (Fig. 1, 2, 4, 6, 7, and 10) were scanned on a Hewlett-Packard desk scanner, and scans were cropped and saved as TIFFs using Adobe Photoshop software. Images were assembled and labeled using Quark Xpress software.
FIG. 1.

Abundance of RNAP II and GTFs in mock-infected and HSV-1-infected cells. Whole-cell lysates were prepared from mock-infected cells or cells infected with wild-type virus for the times indicated. Samples representing equal numbers of cells were analyzed by immunoblotting, and blots were probed with antibodies that recognize subunits of the factors indicated. The RNAP II large subunit shows three phosphorylation variants: IIo, IIi, and IIa.
FIG. 4.

Affinity chromatography of pooled ∼2-MDa fractions from gel filtration chromatography. Nuclear extracts from uninfected or infected cells were fractionated by Sepharose CL-2B gel filtration chromatography. Fractions surrounding the 2-MDa peak were pooled (fractions indicated in Fig. 2 and 3) and chromatographed on GST and GST-TFIIS columns. Bound proteins were eluted sequentially with buffers containing 0.3 and 0.5 M NaCl. Eluates were concentrated and analyzed by immunoblotting with antibodies that recognize the large subunit of RNAP II, the p56E subunit of TFIIE, the TBP subunit of TFIID, and the cdk7 subunit of TFIIH. Each blot contained a lane of uninfected (A) or infected (B) nuclear extract (20 μg each) to indicate positions of GTFs, as well as a lane containing one-quarter of the flowthrough (Fl-Thru) and a lane containing one-half of the wash.
FIG. 6.

In vitro transcription activities of nuclear extracts (NE), RNAP II holoenzymes (Holo), and pooled low-molecular-mass fractions (Low). In vitro transcription reactions were carried out using a linear DNA template containing the Ad MLP. Reactions were performed with nuclear extracts (46 μg) or 10 μl of concentrated fractions from gel filtration chromatography, adjusted to contain similar quantities of RNAP II. Transcription was template dependent and sensitive to 2 μg of α-amanitin/ml, indicating transcription by RNAP II. Arrow indicates runoff transcripts originating at the Ad MLP. Lanes 1 and 3, nuclear extracts prepared from infected and uninfected cells. Lanes 2 and 4, concentrated fractions from the ∼2-MDa peak of Sepharose CL-2B gel filtration chromatography (fractions indicated in Fig. 2 and 3). Transcription signals were at background levels in lane 4. Lanes 5 and 6, crude uninfected nuclear extracts and pooled concentrates from the ∼670-kDa peak of Sepharose CL-2B chromatography (fractions shown in Fig. 2 and 3). Lanes 7 and 8, crude infected nuclear extracts and pooled concentrates from the ∼670-kDa peak of Sepharose CL-2B chromatography (fractions indicated in Fig. 2 and 3).
FIG. 7.

TFIIS affinity chromatography of pooled low-molecular-mass fractions from gel filtration chromatography. Nuclear extracts prepared from uninfected or infected cells were fractionated by Sepharose CL-2B gel filtration chromatography, as described for Fig. 2. Fractions at and below ∼670 kDa (indicated in Fig. 2 and 3) were pooled and chromatographed on GST and GST-TFIIS columns. Bound proteins were eluted sequentially with buffers containing 0.3 and 0.5 M NaCl. Eluates were concentrated and analyzed by immunoblotting with antibodies that recognize the large subunit of RNAP II, the p56E subunit of TFIIE, the TBP subunit of TFIID, and the cdk7 subunit of TFIIH. Each blot contained a lane of uninfected (A) or infected (B) nuclear extract (20 μg each) to indicate positions of GTFs, as well as a lane containing one-quarter of the flowthrough (Fl-Thru) and a lane containing one-half of the wash.
FIG. 10.

TFIIS affinity chromatography showing viral IE proteins in low-molecular-mass fractions containing RNAPII. Nuclear extracts, prepared from cells infected with wild-type HSV-1, were fractionated by Sepharose CL-2B gel filtration chromatography, as described for Fig. 2. (A) Fractions surrounding the 2-MDa peak (indicated in Fig. 2, 3, and 9) were pooled and subjected to GST and GST-TFIIS affinity chromatography. Bound proteins were eluted sequentially with buffers containing 0.3 and 0.5 M NaCl. Eluates were concentrated and analyzed by immunoblotting with antibodies that recognize the IE proteins ICP4, ICP0, ICP22, and ICP27. Each blot contained a lane of nuclear extract (20 μg) to indicate the position of each IE protein. The Fl-Thru lane contained one-quarter of the flowthrough, and the wash lane contained one-half of the wash. (B) Fractions at and below ∼670 kDa (indicated in Fig. 2, 3, and 9) were pooled and subjected to GST and GST-TFIIS affinity chromatography. Bound proteins were eluted and analyzed as described for panel A. The black dot indicates an ICP22 band above the ICP27 band, visible from a previous probing of the Western blot.
RESULTS
HSV-1 infection alters the composition of the RNAP II holoenzyme.
We have previously reported that HSV-1 infection brings about rapid modifications in RNAP II large subunit phosphorylation and repression of host transcription (51, 64). Since transcription initiation and large subunit phosphorylation are carried out by the RNAP II holoenzyme in uninfected cells, we reasoned that these changes may reflect virus-induced modifications to the composition of the RNAP II holoenzyme.
To assess the abundance of RNAP II GTFs during infection, we prepared whole-cell lysates from mock-infected cells or cells infected with the wild-type virus KOS1.1 and analyzed their GTF contents by immunoblotting. None of the GTFs examined displayed significant alterations in abundance or migration on SDS-polyacrylamide gel electrophoresis (PAGE) gels, up to 9 h postinfection (Fig. 1). We also examined the abundance of p62H (a subunit of TFIIH), cdk8, cdk9, and cyclin T1 after infection and found no significant changes (data not shown). As seen previously, the RNAP II large subunit was aberrantly phosphorylated after infection, leading to the loss of the hyperphosphorylated IIo form and the appearance of the intermediately migrating IIi form (Fig. 1). Treatment of lysates with calf intestinal phosphatase collapsed hyperphosphorylated forms of the RNAP II large subunit to the hypophosphorylated (IIa) form and eliminated the apparent abundance differences, which are due to the polydispersed migration of phosphorylated forms (reference 51; also data not shown).
We next determined the GTF composition of RNAP II holoenzymes from uninfected cells. In order to analyze RNAP II holoenzymes, we used a combination of Sepharose CL-2B gel filtration chromatography and TFIIS affinity chromatography. These methods have been used by others to purify and characterize RNAP II holoenzymes from mammalian cells (37, 45, 72). We first prepared nuclear extracts from uninfected HeLa cells and then separated proteins using Sepharose CL-2B gel filtration chromatography. Column fractions were concentrated and analyzed by immunoblotting, using antibodies that recognize the large subunit of RNAP II, the p56 subunit of TFIIE, the TBP subunit of TFIID, and the cdk7 subunit of TFIIH. Gel filtration chromatography showed that RNAP II, TFIID (TBP), TFIIE (p56E), and TFIIH (cdk7) from uninfected nuclear extracts eluted in fractions surrounding 2 MDa (Fig. 2A). These factors also eluted in fractions below 670 kDa, similar to patterns seen previously (43, 45). Sepharose CL-2B efficiently separates proteins from approximately 1 to 10 MDa; proteins below approximately 1 MDa elute together over a broad peak. Hence, our molecular mass estimates for macromolecules and complexes below ∼1 MDa are based on the elution peak of the 670-kDa thyroglobulin marker. Treatment of uninfected nuclear extracts with ethidium bromide, DNase I, and RNase A prior to CL-2B chromatography did not alter the elution profiles of RNAP II and the GTFs (data not shown), indicating that their presence in high-molecular-mass fractions was not due to interactions with nucleic acids in the nuclear extracts. In addition, the DNA repair protein MLH1 did not elute in the 2-MDa peak (Fig. 2A), demonstrating that the presence of RNAP II GTFs in high-molecular-mass complexes is not a general feature of proteins that interact with DNA.
In order to assess the relative quantities of GTFs in the 2-MDa peak from uninfected nuclear extracts, we scanned Western blots by densitometry, normalized bands to identical standards (nuclear extracts) on each gel, and plotted these as relative densitometry units (Fig. 3, top panels). The relative densitometry units in eight fractions surrounding the 2-MDa peak were added and expressed as a percentage of total densitometry units over the entire chromatography run (Table 1). Approximately 14% of RNAP II, 15% of TFIIE (p56), and 29% of TFIIH (cdk7) were present in the ∼2-MDa fractions. In contrast, approximately 73% of total TFIID (TBP) was present in these fractions. Since TBP participates in a number of high-molecular-mass complexes, including RNAP I and RNAP III holoenzymes (7, 56, 70), the presence of TBP in the ∼2-MDa fractions likely represents a mixture of TBP-containing holoenzyme complexes of all three nuclear RNA polymerases.
TABLE 1.
Quantitative comparison of GTF levels in the 2-MDa peak from Sepharose CL-2B chromatography
| GTF | % of total GTF present in ∼2-MDa fractions from:
|
Infected/uninfected GTF ratio (%) | |
|---|---|---|---|
| Uninfected nuclear extracts | Infected nuclear extracts | ||
| RNAP II | 14 | 14 | 100 |
| TBP (TFIID) | 73 | 32 | 43 |
| cdk7 (TFIIH) | 29 | 19 | 66 |
| p56E (TFIIE) | 15 | 0.6 | 4 |
To determine whether GTF components of the ∼2-MDa peak from uninfected extracts copurify with RNAP II, we pooled fractions surrounding the 2-MDa peak from CL-2B columns (fractions indicated in Fig. 2 and 3) and subjected these to GST-TFIIS affinity chromatography. The RNAP II transcription elongation factor TFIIS is used as an immobilized ligand for RNAP II holoenzyme affinity purification (45). TFIIS binds directly to RNAP II, although it may also interact with other members of the RNAP II holoenzyme during affinity chromatography (1, 45, 63). Holoenzymes purified by GST-TFIIS chromatography are ∼2 MDa in mass and contain RNAP II and all GTFs including TFIID. Pan et al. (45) have shown that RNAP II holoenzymes elute in the 0.3 M elution. Fractions surrounding the 2-MDa peak were pooled and passed over GST-TFIIS affinity columns. The columns were washed, and bound proteins were eluted successively with buffers containing 0.3 and 0.5 M NaCl. Eluates were concentrated and analyzed by immunoblotting. GST-TFIIS chromatography of pooled 2-MDa fractions showed that TFIIE (p56E), TFIID (TBP), and TFIIH (cdk7) coeluted with RNAP II in holoenzymes from uninfected cells (Fig. 4A). Holoenzyme subunits did not elute from control GST-Sepharose columns.
In summary, RNAP II holoenzymes from uninfected cells contain RNAP II, TFIIE, TFIIH, and TFIID, as seen previously (43–45, 72).
We next wanted to analyze the GTF contents of RNAP II holoenzymes isolated from cells infected with HSV-1. We infected HeLa cells with wild-type HSV-1, prepared nuclear extracts, and fractionated the extracts using Sepharose CL-2B gel filtration chromatography. Fractions from CL-2B columns were concentrated and analyzed by immunoblotting, using the antibodies that recognize the RNAP II large subunit, p56E, TBP, and cdk7. A typical elution profile is shown in Fig. 2B. Although RNAP II, TBP, and cdk7 continued to elute in the ∼2-MDa fractions after infection, p56E appeared to be depleted in these high-molecular-mass fractions from HSV-1-infected nuclear extracts.
In order to quantitate the relative abundance of each of these factors in the ∼2-MDa fractions, we scanned Western blots by densitometry, normalized band intensities to identical standards on each gel, and plotted values as relative densitometry units (Fig. 3, bottom panels). The relative densitometry units in eight fractions surrounding the 2-MDa peak were added and expressed as a percentage of total relative densitometry units over the entire chromatography run (Table 1). As seen in Fig. 3, the Sepharose CL-2B elution profile of RNAP II from infected extracts was similar to that from uninfected extracts. Approximately 14% of the total RNAP II eluted in the 2-MDa peak in both infected and uninfected nuclear extracts (Table 1). The CL-2B elution profile of TFIIH (cdk7) from infected extracts was similar to that for uninfected extracts (Fig. 2B and Fig. 3, lower panels) but showed a shift towards lower-molecular-mass fractions. Approximately 19% of total cdk7 from infected extracts eluted in the 2-MDa range, compared to approximately 29% from uninfected extracts. A more pronounced change was apparent for TFIID (TBP). After infection, approximately 32% of the total TBP eluted in the ∼2-MDa fractions, compared to 73% eluting in these fractions prior to infection. As noted above, TBP participates in a number of high-molecular-mass complexes including RNAP I, II, and III holoenzymes. Depletion of TBP in high-molecular-mass fractions after HSV-1 infection may indicate disruption of any of these complexes. The greatest change was apparent for TFIIE (p56E). Although p56E was abundant in the 2-MDa peak from uninfected nuclear extracts (15% of total p56E), p56E eluting in the 2-MDa peak was reduced to <1% of total p56E in infected nuclear extracts (Fig. 2B and 3; Table 1).
In order to determine whether GTF components of the ∼2-MDa fractions from HSV-1-infected extracts copurified with RNAP, we pooled fractions surrounding the ∼2-MDa peak from CL-2B columns (pooled fractions indicated in Fig. 2 and 3) and performed GST-TFIIS affinity chromatography. GST-TFIIS chromatography showed that TFIID (TBP) and TFIIH (cdk7) copurified with RNAP II in holoenzymes from infected cells (Fig. 4B). TFIIE (p56E), although present at low levels in the pooled fractions, did not elute in either the 0.3 or the 0.5 M salt elution from GST-TFIIS columns. RNAP II and GTF subunits did not elute from control GST-Sepharose columns. In addition, the GST-TFIIS affinity elution patterns of RNAP II holoenzymes were altered after infection, with a portion of complexes eluting in the 0.5 M fraction. In summary, RNAP II holoenzymes from HSV-1-infected cells contain RNAP II, TFIIH, and TFIID but are depleted of the GTF TFIIE.
Loss of TFIIE from the RNAP II holoenzyme requires expression of viral IE genes.
The observation that TFIIE is depleted in the postinfection RNAP II holoenzyme was surprising, since TFIIE is a consistent component of mammalian RNAP II holoenzymes and is an essential GTF, required for TFIIH-mediated CTD phosphorylation and transcription activation. This raised the possibility that loss of TFIIE from the holoenzyme may contribute to some of the alterations in RNAP II and transcription patterns that occur after HSV-1 infection, including aberrant phosphorylation of the RNAP II CTD and selective repression of host gene transcription. We previously showed that CTD modification and host transcription repression require expression of viral IE proteins and that these proteins may be redundant in their capacity to modify RNAP II and host transcription. In order to determine whether loss of TFIIE from the holoenzyme has similar requirements for IE gene expression, we first analyzed the TFIIE content of RNAP II holoenzymes isolated from cells infected with the ICP4-null mutant virus d120 (13). Because ICP4 is required for transcription of DE and L genes and hence for viral DNA replication, infections with ICP4-null viruses are arrested at the IE stage (20). We prepared nuclear extracts from cells infected with the d120 virus, fractionated the extracts by Sepharose CL-2B gel filtration chromatography, and analyzed the fractions by immunoblotting (Fig. 5). The levels of p56E were low in the 2-MDa peak, similar to patterns seen in nuclear extracts prepared from cells infected with wild-type HSV-1 (Fig. 3). These data indicate that expression of ICP4 is not required for loss of TFIIE from the 2-MDa RNAP II holoenzyme. In addition, DE and L gene expression, as well as viral DNA replication, are not required for this effect.
These results suggested that loss of TFIIE from the RNAP II holoenzyme may require expression of other viral IE proteins and/or the presence of virion components. In order to test the requirements for other IE gene products, we analyzed TFIIE elution patterns in extracts prepared from cells infected with the following null mutants: n212, which bears a nonsense mutation in the ICP0 gene (5); d22-lacZ, which bears a deletion of the ICP22 gene (33); and d27-1, which bears a deletion of the ICP27 gene (49). We infected cells with each of these mutant viruses, prepared nuclear extracts, fractionated extracts by Sepharose CL-2B gel filtration chromatography, and analyzed fractions by immunoblotting (Fig. 5). The elution profiles of TFIIE were similar in extracts prepared from cells infected with each of the mutant viruses in that all showed low levels of TFIIE in fractions surrounding 2 MDa.
To assess the relative quantities of TFIIE in the ∼2-MDa fractions from cells infected with each of the viral mutants, we added the relative densitometry units in eight fractions surrounding the 2-MDa peak and expressed these as a percentage of total densitometry units over the entire chromatography run. This analysis showed that the TFIIE content in the ∼2-MDa peak was 0.2% for d120-infected cells, 1.0% for n212 infected cells, 0.6% for d22-lacZ-infected cells, and 2.0% for d27-1-infected cells. These values are similar to those seen in wild-type virus-infected cells (0.6%) and are below those seen in uninfected cells (15%).
We next examined the contribution of virion components to depletion of TFIIE from the RNAP II holoenzyme fractions by analyzing TFIIE elution profiles in cells infected with UV-inactivated virus. UV-inactivated virus was prepared as described previously (51) and showed a 4- to 5-log reduction in virus titer. UV-inactivated HSV-1 is used to distinguish the relative contributions of virion proteins and viral gene expression. Treatment of HSV-1 with 254-nm UV light cross-links viral DNA and prevents viral gene transcription but does not damage virion proteins (18, 25, 51). We infected cells with UV-inactivated virus, prepared nuclear extracts, fractionated the extracts by Sepharose CL-2B gel filtration chromatography, and analyzed the fractions by immunoblotting (Fig. 5). UV-inactivated virus was unable to deplete TFIIE from the ∼2-MDa fractions. Approximately 19% of total TFIIE was present in the ∼2-MDa peak, compared with results for uninfected cells (15%) and cells infected with wild-type virus (0.6%) (Table 1). These data suggest that virion components are not sufficient to cause loss of TFIIE from the RNAP II holoenzyme. They do not eliminate the possibility, though, that virion components contribute to TFIIE depletion, in conjunction with IE gene expression.
In summary, HSV-1 infection results in loss of TFIIE from the RNAP II holoenzyme, and this loss requires viral IE gene expression but not viral DE or L gene expression or viral DNA replication.
RNAP II holoenzymes from infected cells are transcriptionally inactive.
One possible consequence of the loss of TFIIE from the RNAP II holoenzyme is that RNAP II holoenzymes may be inactive after HSV-1 infection. As a first step in analyzing the transcriptional consequences of RNAP II holoenzyme modification, we performed in vitro transcription assays on nuclear extracts and RNAP II holoenzymes from infected and uninfected cells. We prepared nuclear extracts from infected or uninfected cells as described in Materials and Methods. Nuclear extracts were fractionated by Sepharose CL-2B gel filtration chromatography, and fractions surrounding the 2-MDa peak (shown in Fig. 2 and 3) were pooled and concentrated 50-fold. Crude nuclear extracts or holoenzyme concentrates containing comparable amounts of RNAP II (as assessed by immunoblotting) were used for in vitro transcription assays, along with a linear DNA template bearing the Ad MLP 540 bp upstream of the linear DNA end. Radioactive RNAs were analyzed by acrylamide-urea gel electrophoresis and autoradiography (Fig. 6). Infected nuclear extracts were consistently less active than uninfected nuclear extracts during in vitro transcription assays (Fig. 6, lanes 1 and 3). Phosphorimaging revealed that infected nuclear extracts yielded 10 to 40% less promoter-specific transcription than uninfected nuclear extracts (depending on the extract). We do not know the basis for this lower activity. DNA templates were not detectably degraded during the transcription assays with either infected or uninfected nuclear extracts (data not shown). Interestingly, nuclear extracts prepared from cells infected with the ICP4 mutant virus, d120, were as transcriptionally active as uninfected nuclear extracts (M. L. Long and C. A. Spencer, unpublished data), suggesting that one or more DE or L gene products may inhibit RNAP II transcription in vitro. However, extracts prepared from cells infected with the d120 virus also contain high levels of the viral IE proteins ICP0, ICP22, and ICP27; therefore, their high transcriptional activity may be due to the presence of these proteins.
RNAP II holoenzymes from uninfected cells supported in vitro transcription (Fig. 6, lane 2), albeit at lower levels than the parent crude nuclear extract (∼30% of nuclear extract activity). Although we do not know the reason for this lower activity, it may be due to a loss of activity during the 2 to 3 h required to concentrate the pooled column fractions. We were unable to detect transcriptional activity above background levels in RNAP II holoenzyme concentrates prepared from infected nuclear extracts (Fig. 6, lane 4), even though the amounts of RNAP II were comparable in the infected and uninfected holoenzyme concentrates. Studies are in progress to determine the biochemical basis for low transcriptional activities in infected nuclear extracts and RNAP II holoenzymes, and whether loss of TFIIE from the RNAP II holoenzyme contributes to this loss of activity.
Low-molecular-mass RNAP II-containing fractions are transcriptionally active and exhibit RNAP II-GTF interactions.
Our in vitro transcription assays suggested that RNAP II holoenzymes may be inactive after HSV-1 infection. If holoenzymes are also inactive in vivo after HSV-1 infection, it is possible that free RNAP II and GTFs, or low-molecular-mass complexes containing these factors, may be the transcriptionally relevant species after infection with HSV-1. To test the transcription activity of low-molecular-mass material, we performed in vitro transcription assays on concentrated fractions from Sepharose CL-2B columns. Nuclear extracts were prepared from uninfected or wild-type virus-infected cells, extracts were fractionated on Sepharose CL-2B columns, fractions at and below ∼670 kDa were pooled (as indicated in Fig. 2 and 3), and pooled fractions were concentrated 10-fold. Crude nuclear extracts or ∼670-kDa concentrates containing comparable amounts of RNAP II (as assayed by immunoblotting) were used in in vitro transcription assays, as described above for RNAP II holoenzymes. The transcription activities of ∼670-kDa concentrates from both infected and uninfected cells were greater (∼3- to 4-fold) than those of the nuclear extracts from which they were derived (Fig. 6, lanes 5 to 8). In addition, the ∼670-kDa concentrates from infected nuclear extracts directed synthesis of multiple bands, suggesting inaccurate transcription initiation and/or premature transcription termination. These data suggest that RNAP II and GTFs in infected nuclear extracts are transcriptionally active; however, this activity may be masked or repressed within the context of a crude nuclear extract. The biochemical basis for these different activities in crude versus fractionated material will be addressed in future studies.
As low-molecular-mass material derived from nuclear extracts supports high levels of transcription activity, we wanted to determine whether the RNAP II and GTFs in these fractions exist as free factors or whether they interact in low-molecular-mass complexes. In order to examine the interactions of GTFs with RNAP II in low-molecular-mass fractions, we pooled fractions from CL-2B gel filtration chromatography (fractions indicated in Fig. 2 and 3), representing material from ∼670 kDa and lower peaks, and subjected these to GST-TFIIS affinity chromatography. GST-TFIIS affinity chromatography of pooled low-molecular-mass fractions from uninfected extracts showed that TFIID (TBP), TFIIH (cdk7), and TFIIE (p56E), coeluted with RNAP II (Fig. 7A). Similarly, TFIID, TFIIH, and TFIIE coeluted in GST-TFIIS affinity chromatography of pooled low-molecular-mass fractions from extracts prepared from cells infected with HSV-1 (Fig. 7B). None of these factors eluted from control GST-Sepharose columns (Fig. 7, lower panels). In summary, although TFIIE is absent from RNAP II holoenzymes after HSV-1 infection, it is capable of interacting with RNAP II in complexes of lower molecular mass. As these complexes are in the pooled fractions of ∼670 kDa, it is likely that RNAP II interacts with the GTFs in a number of different complexes. This is consistent with observations that free RNAP II is capable of binding individual GTFs and GTF subunits in vitro (4, 32).
If TFIIE continues to be involved in transcription of viral genes after HSV-1 infection, despite its absence from the RNAP II holoenzyme, it may be visibly recruited to viral replication compartments. We previously reported that RNAP II is recruited to viral replication compartments after HSV-1 infection and that this recruitment coincides with repression of host gene transcription and induction of viral gene transcription, at approximately 4 to 6 h postinfection (51, 64). To examine the intracellular localization of GTFs after infection, we infected HeLa cells for 6 h, fixed them, and immunostained them with antibodies recognizing RNAP II and GTFs. Cells were also stained with an antibody against the viral IE protein ICP4 to visualize viral replication compartments (Fig. 8). Infected cells displayed typical intranuclear viral replication compartments, which are sites of viral DNA synthesis and transcription (11, 12, 47, 51, 65). Immunostains showed that the GTFs TFIID (TBP), TFIIH (p62H), and TFIIE (p56E) were present in viral replication compartments, along with RNAP II. In summary, these data show that TFIIE and other GTFs interact with RNAP II in complexes of low molecular mass, both before and after infection with HSV-1. In addition, TFIIE localizes to viral replication compartments along with RNAP II and other GTFs, consistent with its potential role in transcription of viral genes.
Viral IE proteins ICP4, ICP0, and ICP27 interact with RNAP II in low-molecular-mass complexes.
Our data suggest that RNAP II holoenzymes may not be involved in viral or host gene transcription after infection; however, free RNAP II and GTFs (or various low-molecular-mass complexes containing RNAP II and GTF subunits) may be the transcriptionally relevant forms. One way in which RNAP II and GTFs could be assembled on viral DNA in the absence of the RNAP II holoenzyme is for viral regulatory proteins to interact with RNAP II and GTFs and to recruit these individual components (or small complexes) to viral promoters. One candidate for such a regulatory protein is ICP4, which is able to interact with TFIID and TFIIB in vitro and to stimulate the assembly of transcription preinitiation complexes (6, 23, 62).
In order to detect the association of viral IE proteins with RNAP II and GTFs, we first analyzed the IE protein content in fractions from CL-2B gel filtration chromatography. Nuclear extracts from cells infected with wild-type HSV-1 were fractionated on CL-2B columns, and fractions were analyzed by immunoblotting. Western blots were scanned by densitometry, values were normalized to identical standards on each gel, and values were plotted as relative densitometry units. Each of the viral IE proteins was present in the ∼2-MDa fractions from CL-2B chromatography (Fig. 9). The CL-2B elution profiles of the IE proteins were not affected by prior treatment of nuclear extracts with ethidium bromide, DNase I, and RNase A, indicating that their presence in high-molecular-mass fractions was not due to interactions with nucleic acids in the nuclear extracts (data not shown). The densitometry units in fractions surrounding the 2-MDa peak were expressed as a percentage of the total densitometry units over the entire run. These calculations indicated that approximately 2% of total ICP4, 1% of total ICP0, 64% of total ICP22, and 40% of total ICP27 eluted in high-molecular-mass fractions from CL-2B columns.
To determine whether the IE proteins in high-molecular-mass fractions interact with RNAP II holoenzyme complexes, we pooled fractions surrounding the 2-MDa peak from CL-2B columns (indicated in Fig. 9) and subjected these to GST-TFIIS affinity chromatography. None of the IE proteins eluted in the 0.3 or 0.5 M elutions or from GST columns (Fig. 10A). These data indicate that viral IE proteins, although present in high-molecular-mass complexes, do not interact with the RNAP II holoenzyme after HSV-1 infection.
Although IE proteins appeared not to interact with RNAP II holoenzyme complexes, we wanted to examine their interactions with RNAP II in complexes of lower molecular mass. To do this, we pooled fractions of ∼670 kDa and lower from CL-2B columns (fractions indicated in Fig. 9) and subjected these to GST-TFIIS affinity chromatography. Small but detectable amounts of ICP4, ICP27, and ICP0 coeluted with RNAP II from pooled low-molecular-mass fractions but not from GST columns (Fig. 10B). ICP22 was not detectable in GST-TFIIS eluates. In addition, ICP22 consistently displayed a mobility shift and altered immunostaining after TFIIS or GST chromatography, perhaps due to phosphatase activity during the chromatography run. Since RNAP II and IE proteins interact in fractions of approximately 670 kDa, it is likely that these associations represent a mixture of individual IE proteins with free RNAP II or RNAP II–GTF complexes of low molecular mass. These data do not distinguish between direct and indirect interactions of RNAP II with ICP4, ICP0, or ICP27. It is possible that IE proteins may interact directly with TFIIS; however, no IE proteins were detected in TFIIS affinity chromatography of high-molecular-mass fractions which also contained viral IE proteins (Fig. 10A). Further biochemical analysis will be required to precisely define the composition of each of the RNAP II-containing low-molecular-mass complexes and to determine whether the associations of GTF subunits or viral IE proteins with free RNAP II occur in vivo.
DISCUSSION
In this study, we show that HSV-1 infection modifies the composition of the RNAP II holoenzyme. A number of biochemical methods have been used to isolate RNAP II holoenzymes from mammalian cells. Although the composition of these large multisubunit complexes varies depending on the method of isolation, they contain at least RNAP II and the GTFs TFIIH, TFIIF, and TFIIE (8, 34, 39, 40, 43, 45, 46, 55). TFIIB and TFIID are also components of RNAP II holoenzymes isolated using one- or two-step purification procedures; hence, their association with RNAP II complexes is thought to be less stable than that of other GTFs. RNAP II holoenzymes, unlike free RNAP II and GTFs, are responsive to transcription activators and are thought to be responsible for most transcription initiation in yeast and mammalian cells (2, 17, 30, 43, 67).
Using a two-step purification procedure (gel filtration chromatography and TFIIS affinity purification), we show that TFIIE (p56E), TFIID (TBP), and TFIIH (cdk7) copurify with RNAP II in high-molecular-mass holoenzyme complexes prior to infection. TBP and cdk7 also copurify with RNAP II holoenzyme complexes after infection; however, p56E levels are low in the ∼2-MDa peak from CL-2B columns and undetectable in eluates from TFIIS chromatography.
The observation that the interaction of the RNAP II holoenzyme with TFIIE is disrupted after infection is intriguing, given the biochemical functions ascribed to TFIIE (21, 42, 53, 57). TFIIE consists of a 180-kDa heterotetramer of 56- and 34-kDa subunits and is essential for most RNAP II transcription in vivo and in vitro. TFIIE interacts physically with TFIIH and stimulates the CTD kinase and ATPase activities of TFIIH. Interestingly, certain mutant recombinant TFIIEs, when added to in vitro kinase assays containing RNAP II, TFIIH, and other purified GTFs, lead to partial CTD phosphorylation, yielding forms that resemble the HSV-1 modified form, RNAP III (42). These mutant TFIIE proteins are also defective for RNAP II transcription in vitro. In addition to stimulating CTD phosphorylation, TFIIE plays roles in open complex formation and promoter clearance (15, 27). These roles may involve the ability of TFIIE to stimulate TFIIH helicase activity, as well as binding to single-stranded and double-stranded DNA immediately upstream of the transcription start site (52). The requirement for TFIIE can be circumvented by premelting the promoter DNA (27, 28).
It is conceivable that disruption of TFIIE from the postinfection holoenzyme could contribute to the changes in CTD phosphorylation and RNAP II transcription that occur after infection with HSV-1. In the absence of TFIIE, the CTD kinase activity of holoenzyme-associated TFIIH may be impaired, leading to aberrant phosphorylation of RNAP II. Aberrant phosphorylation of RNAP II could in turn contribute to repression of host gene transcription by interfering with the ability of RNAP II transcription initiation complexes to make the transition to productive elongation (10, 15, 28, 57). It is possible that viral proteins such as ICP22 and the UL13 kinase may partially compensate for TFIIE's stimulatory function in CTD phosphorylation, thereby facilitating viral gene transcription. In addition, disruption of TFIIE from the holoenzyme could interfere with the ability of RNAP II transcription initiation complexes to open DNA during transcription elongation on cellular genes, thereby contributing to host transcription repression after infection. Specific structural features of the viral genome or the presence of viral regulatory proteins could compensate for an RNAP II holoenzyme that is impaired in promoter opening. Finally, loss of TFIIE from the RNAP II holoenzyme could lead to defects in host RNA processing, since CTD hyperphosphorylation is required for interactions of the CTD with mRNA processing factors (3).
Another possibility is that the RNAP II holoenzyme may not be involved in HSV-1 gene transcription after infection. In this scenario, free RNAP II and GTFs, or small complexes containing these factors, may be the transcriptionally relevant forms of these factors on viral DNA templates in vivo. This hypothesis would be consistent with the known role of the RNAP II holoenzyme as the form of the enzyme responsible for activator-responsive transcription initiation. Since viral DE and L gene promoters contain no known class-specific enhancer elements that are responsible for transcription activation by IE proteins (61), it is possible that DE and L gene transcription represents a kind of basal transcription, resembling in vitro transcription on linear or supercoiled templates. In the absence of a requirement for activator-driven transcription of viral genes, free RNAP II and GTFs (including TFIIE) may be recruited to these basal viral promoters in a stepwise manner, perhaps assisted by their individual interactions with viral regulatory proteins. In this scenario, the postinfection RNAP II holoenzyme would be incompetent for transcription initiation on cellular genes due to its lack of TFIIE and its inability to hyperphosphorylate the RNAP II CTD. However, viral genes would circumvent the requirement for the RNAP II holoenzyme by recruiting free RNAP II and GTFs to promoters that do not utilize classical transcription activators and are maintained in a nucleosome-free conformation. This hypothesis is consistent with our immunofluorescence staining data showing that RNAP II and GTFs including TFIIE appear to concentrate within replication compartments. Data from our laboratory (C. A. Spencer, unpublished data) and from others (12, 48, 65) show that ICP4 and ICP27 also localize to viral replication compartments, consistent with their possible roles as recruiters of RNAP II and GTFs to viral DNA. ICP0 also localizes to viral replication compartments, although this association is less defined than that of ICP4 and ICP27 (16).
Our data show that loss of TFIIE from the RNAP II holoenzyme requires viral IE gene expression and that two or more viral IE proteins may be redundant in bringing about this modification. We previously showed that repression of host transcription and loss of the hyperphosphorylated form of the RNAP II large subunit also require viral IE gene expression and that viral IE genes may be redundant in mediating these changes. Hence, loss of TFIIE from the RNAP II holoenzyme correlates with loss of RNAP IIO and repression of host transcription, suggesting a functional link between these virus-induced events.
In this study, we report that the RNAP II holoenzyme is depleted of TFIIE and is transcriptionally inactive after HSV-1 infection. In addition, free RNAP II or RNAP II in low-molecular-mass complexes may interact with the viral IE proteins ICP4, ICP0, and ICP27. It will be of interest to determine the biochemical basis for the transcriptional-activity differences between infected and uninfected RNAP II holoenzymes, as well as their intrinsic CTD kinase activities. In addition, analysis of RNAP II complexes in uninfected cells expressing one or more IE genes may provide clues as to the mechanisms by which HSV-1 infection modifies the RNAP II holoenzyme and redirects the host's transcription machinery from host to viral genes.
ACKNOWLEDGMENTS
We thank Steve Rice for virus strains d27-1 and d22-lacZ and for valuable advice and comments on the manuscript. We also thank Neal DeLuca for the d120 virus, Priscilla Schaffer for the n212 virus, Jack Greenblatt for the GST and GST-TFIIS constructs, and Xuejun Sun for advice on confocal microscopy.
This work was supported by grants from the Canadian Institutes of Health Research and the Alberta Cancer Board. C.A.S. is a Senior Scholar of the Alberta Heritage Foundation for Medical Research.
REFERENCES
- 1.Agarwal K, Baek K H, Jeon C J, Miyamoto K, Ueno A, Yoon H S. Stimulation of transcript elongation requires both the zinc finger and RNA polymerase II binding domains of human TFIIS. Biochemistry. 1991;30:7842–7851. doi: 10.1021/bi00245a026. [DOI] [PubMed] [Google Scholar]
- 2.Barberis A, Pearlberg J, Simkovich N, Farrell S, Reinagel P, Bamdad C, Sigal G, Ptashne M. Contact with a component of the polymerase II holoenzyme suffices for gene activation. Cell. 1995;81:359–368. doi: 10.1016/0092-8674(95)90389-5. [DOI] [PubMed] [Google Scholar]
- 3.Bentley D. Coupling RNA polymerase II transcription with pre-mRNA processing. Curr Opin Cell Biol. 1999;11:347–351. doi: 10.1016/S0955-0674(99)80048-9. [DOI] [PubMed] [Google Scholar]
- 4.Bushnell D A, Bamdad C, Kornberg R D. A minimal set of RNA polymerase II transcription protein interactions. J Biol Chem. 1996;271:20170–20174. doi: 10.1074/jbc.271.33.20170. [DOI] [PubMed] [Google Scholar]
- 5.Cai W, Astor T L, Liptak L M, Cho C, Coen D M, Schaffer P A. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol. 1993;67:7501–7512. doi: 10.1128/jvi.67.12.7501-7512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrozza M J, DeLuca N A. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol Cell Biol. 1996;16:3085–3093. doi: 10.1128/mcb.16.6.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang M, Jaehning J A. A multiplicity of mediators: alternative forms of transcription complexes communicate with transcriptional regulators. Nucleic Acids Res. 1997;25:4861–4865. doi: 10.1093/nar/25.24.4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho H, Maldonado E, Reinberg D. Affinity purification of a human RNA polymerase II complex using monoclonal antibodies against transcription factor IIF. J Biol Chem. 1997;272:11495–11502. doi: 10.1074/jbc.272.17.11495. [DOI] [PubMed] [Google Scholar]
- 9.Conaway J W, Conaway R C. Transcription elongation and human disease. Annu Rev Biochem. 1999;68:301–319. doi: 10.1146/annurev.biochem.68.1.301. [DOI] [PubMed] [Google Scholar]
- 10.Dahmus M E. Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J Biol Chem. 1996;271:19009–19012. doi: 10.1074/jbc.271.32.19009. [DOI] [PubMed] [Google Scholar]
- 11.de Bruyn Kops A, Knipe D M. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell. 1988;55:857–868. doi: 10.1016/0092-8674(88)90141-9. [DOI] [PubMed] [Google Scholar]
- 12.de Bruyn Kops A, Uprichard S L, Chen M, Knipe D M. Comparison of the intranuclear distributions of herpes simplex virus proteins involved in various viral functions. Virology. 1998;252:162–178. doi: 10.1006/viro.1998.9450. [DOI] [PubMed] [Google Scholar]
- 13.DeLuca N A, McCarthy A M, Schaffer P A. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol. 1985;56:558–570. doi: 10.1128/jvi.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dignam J D, Lebovitz R M, Roeder R G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dvir A, Conaway R C, Conaway J W. A role for TFIIH in controlling the activity of early RNA polymerase II elongation complexes. Proc Natl Acad Sci USA. 1997;94:9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett R D. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays. 2000;22:761–770. doi: 10.1002/1521-1878(200008)22:8<761::AID-BIES10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 17.Farrell S, Simkovich N, Wu Y, Barberis A, Ptashne M. Gene activation by recruitment of the RNA polymerase II holoenzyme. Genes Dev. 1996;10:2359–2367. doi: 10.1101/gad.10.18.2359. [DOI] [PubMed] [Google Scholar]
- 18.Fenwick M L, Owen S A. On the control of immediate early (α) mRNA survival in cells infected with herpes simplex virus. J Gen Virol. 1988;69:2869–2877. doi: 10.1099/0022-1317-69-11-2869. [DOI] [PubMed] [Google Scholar]
- 19.Gaudreau L, Schmid A, Blaschke D, Ptashne M, Horz W. RNA polymerase II holoenzyme recruitment is sufficient to remodel chromatin at the yeast PHO5 promoter. Cell. 1997;89:55–62. doi: 10.1016/s0092-8674(00)80182-8. [DOI] [PubMed] [Google Scholar]
- 20.Godowski P J, Knipe D M. Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc Natl Acad Sci USA. 1986;83:256–260. doi: 10.1073/pnas.83.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrich J A, Tjian R. Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell. 1994;77:145–156. doi: 10.1016/0092-8674(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 22.Greenblatt J. RNA polymerase II holoenzyme and transcriptional regulation. Curr Opin Cell Biol. 1997;9:310–319. doi: 10.1016/s0955-0674(97)80002-6. [DOI] [PubMed] [Google Scholar]
- 23.Grondin B, DeLuca N. Herpes simplex virus type 1 ICP4 promotes transcription preinitiation complex formation by enhancing the binding of TFIID to DNA. J Virol. 2000;74:11504–11510. doi: 10.1128/jvi.74.24.11504-11510.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hampsey M, Reinberg D. RNA polymerase II as a control panel for multiple coactivator complexes. Curr Opin Genet Dev. 1999;9:132–139. doi: 10.1016/S0959-437X(99)80020-3. [DOI] [PubMed] [Google Scholar]
- 25.Hill T M, Sinden R R, Sadler J R. Herpes simplex virus types 1 and 2 induce shutoff of host protein synthesis by different mechanisms in Friend erythroleukemia cells. J Virol. 1983;45:241–250. doi: 10.1128/jvi.45.1.241-250.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirose Y, Manley J L. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000;14:1415–1429. [PubMed] [Google Scholar]
- 27.Holstege F C, Tantin D, Carey M P, van der Vliet C, Timmers H T. The requirement for the basal transcription factor IIE is determined by the helical stability of promoter DNA. EMBO J. 1995;14:810–819. doi: 10.1002/j.1460-2075.1995.tb07059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holstege F C, van der Vliet P C, Timmers H T. Opening of an RNA polymerase II promoter occurs in two distinct steps and requires the basal transcription factors IIE and IIH. EMBO J. 1996;15:1666–1677. [PMC free article] [PubMed] [Google Scholar]
- 29.Kramer A, Haars R, Kabisch R, Will H, Bautz F A, Bautz E K. Monoclonal antibody directed against RNA polymerase II of Drosophila melanogaster. Mol Gen Genet. 1980;180:193–199. doi: 10.1007/BF00267369. [DOI] [PubMed] [Google Scholar]
- 30.Kuchin S, Treich I, Carlson M. A regulatory shortcut between the Snf1 protein kinase and RNA polymerase II holoenzyme. Proc Natl Acad Sci USA. 2000;97:7916–7920. doi: 10.1073/pnas.140109897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaBoissiere S, O'Hare P. Analysis of HCF, the cellular cofactor of VP16, in herpes simplex virus-infected cells. J Virol. 2000;74:99–109. doi: 10.1128/jvi.74.1.99-109.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leuther K K, Bushnell D A, Kornberg R D. Two-dimensional crystallography of TFIIB- and IIE-RNA polymerase II complexes: implications for start site selection and initiation complex formation. Cell. 1996;85:773–779. doi: 10.1016/s0092-8674(00)81242-8. [DOI] [PubMed] [Google Scholar]
- 33.Long M C, Leong V, Schaffer P A, Spencer C A, Rice S A. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J Virol. 1999;73:5593–5604. doi: 10.1128/jvi.73.7.5593-5604.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maldonado E, Shiekhattar R, Sheldon M, Cho H, Drapkin R, Rickert P, Lees E, Anderson C W, Linn S, Reinberg D. A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Nature. 1996;381:86–89. doi: 10.1038/381086a0. [DOI] [PubMed] [Google Scholar]
- 35.Marshall N F, Peng J, Xie Z, Price D H. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 36.Mayman B A, Nishioka Y. Differential stability of host mRNAs in Friend erythroleukemia cells infected with herpes simplex virus type 1. J Virol. 1985;53:1–6. doi: 10.1128/jvi.53.1.1-6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson S D, Wickens M, Bentley D L. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- 38.McNeil J B, Agah H, Bentley D. Activated transcription independent of the RNA polymerase II holoenzyme in budding yeast. Genes Dev. 1998;12:2510–2521. doi: 10.1101/gad.12.16.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakajima T, Uchida C, Anderson S F, Lee C G, Hurwitz J, Parvin J D, Montminy M. RNA helicase A mediates association of CBP with RNA polymerase II. Cell. 1997;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 40.Neish A S, Anderson S F, Schlegel B P, Wei W, Parvin J D. Factors associated with the mammalian RNA polymerase II holoenzyme. Nucleic Acids Res. 1998;26:847–853. doi: 10.1093/nar/26.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Hare P. The virion transactivator of herpes simplex virus. Semin Virol. 1993;4:145–155. [Google Scholar]
- 42.Okamoto T, Yamamoto S, Watanabe Y, Ohta T, Hanaoka F, Roeder R G, Ohkuma Y. Analysis of the role of TFIIE in transcriptional regulation through structure-function studies of the TFIIE β subunit. J Biol Chem. 1998;273:19866–19876. doi: 10.1074/jbc.273.31.19866. [DOI] [PubMed] [Google Scholar]
- 43.Ossipow V, Fonjallaz P, Schibler U. An RNA polymerase II complex containing all essential initiation factors binds to the activation domain of PAR leucine zipper transcription factor thyroid embryonic factor. Mol Cell Biol. 1999;19:1242–1250. doi: 10.1128/mcb.19.2.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ossipow V, Tassan J P, Nigg E A, Schibler U. A mammalian RNA polymerase II holoenzyme containing all components required for promoter-specific transcription initiation. Cell. 1995;83:137–146. doi: 10.1016/0092-8674(95)90242-2. [DOI] [PubMed] [Google Scholar]
- 45.Pan G, Aso T, Greenblatt J. Interaction of elongation factors TFIIS and elongin A with a human RNA polymerase II holoenzyme capable of promoter-specific initiation and responsive to transcriptional activators. J Biol Chem. 1997;272:24563–24571. doi: 10.1074/jbc.272.39.24563. [DOI] [PubMed] [Google Scholar]
- 46.Parvin J D, Young R A. Regulatory targets in the RNA polymerase II holoenzyme. Curr Opin Genet Dev. 1998;8:565–570. doi: 10.1016/s0959-437x(98)80012-9. [DOI] [PubMed] [Google Scholar]
- 47.Phelan A, Dunlop J, Patel A H, Stow N D, Clements J B. Nuclear sites of herpes simplex virus type 1 DNA replication and transcription colocalize at early times postinfection and are largely distinct from RNA processing factors. J Virol. 1997;71:1124–1132. doi: 10.1128/jvi.71.2.1124-1132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Randall R E, Dinwoodie N. Intranuclear localization of herpes simplex virus immediate-early and delayed-early proteins: evidence that ICP4 is associated with progeny virus DNA. J Gen Virol. 1986;67:2163–2177. doi: 10.1099/0022-1317-67-10-2163. [DOI] [PubMed] [Google Scholar]
- 49.Rice S A, Knipe D M. Genetic evidence for two distinct transactivation functions of the herpes simplex virus α protein ICP27. J Virol. 1990;64:1704–1715. doi: 10.1128/jvi.64.4.1704-1715.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rice S A, Long M C, Lam V, Schaffer P A, Spencer C A. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J Virol. 1995;69:5550–5559. doi: 10.1128/jvi.69.9.5550-5559.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rice S A, Long M C, Lam V, Spencer C A. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J Virol. 1994;68:988–1001. doi: 10.1128/jvi.68.2.988-1001.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robert F, Forget D, Li J, Greenblatt J, Coulombe B. Localization of subunits of transcription factors IIE and IIF immediately upstream of the transcriptional initiation site of the adenovirus major late promoter. J Biol Chem. 1996;271:8517–8520. doi: 10.1074/jbc.271.15.8517. [DOI] [PubMed] [Google Scholar]
- 53.Roeder R G. Nuclear RNA polymerases: role of general initiation factors and cofactors in eukaryotic transcription. Methods Enzymol. 1996;273:165–171. doi: 10.1016/s0076-6879(96)73016-1. [DOI] [PubMed] [Google Scholar]
- 54.Roizman B, Sears A E. Herpes simplex viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 2231–2295. [Google Scholar]
- 55.Scully R, Anderson S F, Chao D M, Wei W, Ye L, Young R A, Livingston D M, Parvin J D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci USA. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seither P, Iben S, Grummt I. Mammalian RNA polymerase I exists as a holoenzyme with associated basal transcription factors. J Mol Biol. 1998;275:43–53. doi: 10.1006/jmbi.1997.1434. [DOI] [PubMed] [Google Scholar]
- 57.Serizawa H, Conaway J W, Conaway R C. An oligomeric form of the large subunit of transcription factor (TF) IIE activates phosphorylation of the RNA polymerase II carboxyl-terminal domain by TFIIH. J Biol Chem. 1994;269:20750–20756. [PubMed] [Google Scholar]
- 58.Shapiro D J, Sharp P A, Wahli W W, Keller M J. A high-efficiency HeLa cell nuclear transcription extract. DNA. 1988;7:47–55. doi: 10.1089/dna.1988.7.47. [DOI] [PubMed] [Google Scholar]
- 59.Smibert C A, Smiley J R. Differential regulation of endogenous and transduced β-globin genes during infection of erythroid cells with a herpes simplex virus type 1 recombinant. J Virol. 1990;64:3882–3894. doi: 10.1128/jvi.64.8.3882-3894.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smiley J R, Johnson D C, Pizer L I, Everett R D. The ICP4 binding sites in the herpes simplex virus type 1 glycoprotein D (gD) promoter are not essential for efficient gD transcription during virus infection. J Virol. 1992;66:623–631. doi: 10.1128/jvi.66.2.623-631.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smiley J R, Smibert C, Everett R D. Regulation of cellular genes by HSV products. In: Wagner E, editor. Herpesvirus transcription and its regulation. Boca Raton, Fla: CRC Press, Inc.; 1991. pp. 151–179. [Google Scholar]
- 62.Smith C A, Bates P, Rivera-Gonzalez R, Gu B, DeLuca N A. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J Virol. 1993;67:4676–4687. doi: 10.1128/jvi.67.8.4676-4687.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sopta M, Carthew R W, Greenblatt J. Isolation of three proteins that bind to mammalian RNA polymerase II. J Biol Chem. 1985;260:10353–10360. [PubMed] [Google Scholar]
- 64.Spencer C A, Dahmus M E, Rice S A. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J Virol. 1997;71:2031–2040. doi: 10.1128/jvi.71.3.2031-2040.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spencer C A, Kruhlak M J, Jenkins H L, Sun X, Bazett-Jones D P. Mitotic transcription repression in vivo in the absence of nucleosomal chromatin condensation. J Cell Biol. 2000;150:13–26. doi: 10.1083/jcb.150.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stenberg R M, Pizer L I. Herpes simplex virus-induced changes in cellular and adenovirus RNA metabolism in an adenovirus type 5-transformed human cell line. J Virol. 1982;42:474–487. doi: 10.1128/jvi.42.2.474-487.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson C M, Young R A. General requirement for RNA polymerase II holoenzymes in vivo. Proc Natl Acad Sci USA. 1995;92:4587–4590. doi: 10.1073/pnas.92.10.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson N E, Aronson D B, Burgess R R. Purification of eukaryotic RNA polymerase II by immunoaffinity chromatography. Elution of active enzyme with protein stabilizing agents from a polyol-responsive monoclonal antibody. J Biol Chem. 1990;265:7069–7077. [PubMed] [Google Scholar]
- 69.Wada T, Orphanides G, Hasegawa J, Kim D K, Shima D, Yamaguchi Y, Fukuda A, Hisatake K, Oh S, Reinberg D, Handa H. FACT relieves DSIF/NELF-mediated inhibition of transcriptional elongation and reveals functional differences between P-TEFb and TFIIH. Mol Cell. 2000;5:1067–1072. doi: 10.1016/s1097-2765(00)80272-5. [DOI] [PubMed] [Google Scholar]
- 70.Wang Z, Luo T, Roeder R G. Identification of an autonomously initiating RNA polymerase III holoenzyme containing a novel factor that is selectively inactivated during protein synthesis inhibition. Genes Dev. 1997;11:2371–2382. doi: 10.1101/gad.11.18.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weinheimer S P, McKnight S L. Transcriptional and post-transcriptional controls establish the cascade of herpes simplex virus protein synthesis. J Mol Biol. 1987;195:819–833. doi: 10.1016/0022-2836(87)90487-6. [DOI] [PubMed] [Google Scholar]
- 72.Yankulov K, Todorov I, Romanowski P, Licatalosi D, Cilli K, McCracken S, Laskey R, Bentley D L. MCM proteins are associated with RNA polymerase II holoenzyme. Mol Cell Biol. 1999;19:6154–6163. doi: 10.1128/mcb.19.9.6154. [DOI] [PMC free article] [PubMed] [Google Scholar]