

Abstract
Background and Objective
Orodispersible tablets (ODT) rapidly dissolve in the oral cavity and can improve patient’s convenience. This pharmacokinetic study assessed the bioequivalence of a novel 20 mg ODT formulation of bilastine compared with bilastine 20 mg tablets in healthy volunteers under fasting conditions.
Methods
A phase I, single-center, open-label, two-period, two-sequence crossover randomized clinical trial was conducted. The study comprised two periods, in which participants were administered a single oral dose of bilastine 20 mg in the form of ODT as the test product, or conventional tablets as the reference product, and a washout of 7 days between each period. Blood samples were collected for up to 72 h. Bioequivalence was established if the 90% confidence intervals of the Cmax and AUC0–t were within the acceptance range (80−125%). Safety was evaluated at the follow-up visit (days 4−7 after the second dose) and throughout the study.
Results
A total of 42 healthy volunteers were randomized, and 41 completed the study. Pharmacokinetic parameters were comparable for both formulations after a single dose of 20 mg. Bilastine ODT and conventional tablets were bioequivalent as the 90% confidence intervals of the test over reference ratios were within the predefined range (80−125%). Both formulations were well tolerated and showed a similar safety profile.
Conclusions
Bilastine ODT was bioequivalent to the reference treatment formulated as conventional tablets when administered as a single oral dose of 20 mg under fasting conditions. Both formulations showed a similar tolerability and safety profile, with no serious adverse events or significant analytical alterations reported.
Trial Registration: 2019-004071-39.
Date of authorization: 10 December 2019.
Key Points
| The pharmacokinetic analysis showed that orodispersible tablets of bilastine at a dose of 20 mg had similar exposure in the body as the traditional tablet form. |
| Both the orodispersible tablet and conventional tablet forms of bilastine were well tolerated by participants, with no significant safety concerns identified during the study. |
Introduction
Bilastine is a non-sedating, second-generation, long-acting histamine H1 receptor inverse agonist [1] with a higher affinity for H1 receptors than cetirizine and fexofenadine and no affinity for other receptors [2, 3].
Clinical studies demonstrated the efficacy and safety of bilastine for the treatment of allergic rhino-conjunctivitis and urticaria. Bilastine was significantly superior to placebo and showed comparable efficacy to cetirizine [4], desloratadine [5], and fexofenadine [6] in relieving allergic rhinitis symptoms. In chronic idiopathic urticaria, bilastine and levocetirizine were equally efficacious and superior to placebo in reducing pruritus and wheals [7]. Bilastine also led to a significant reduction of histamine-induced wheal and flare responses [8, 9], with greater inhibition compared with desloratadine and rupatadine in healthy subjects [9]. In terms of safety, bilastine was well tolerated and showed a similar safety profile to placebo in several clinical studies [4, 5, 7, 10]. Importantly, bilastine is a non-sedating antihistamine according to the positron emission tomography criteria [11]. This second-generation antihistamine exhibits nearly no penetration through the blood-brain barrier, being considered a non-brain penetrating antihistamine, and does not cause somnolence [12], driving [13], psychomotor [14] or flying-related performance impairment [15], changes in heart-rate corrected QT interval (QTc) on electrocardiogram (ECG) at therapeutic and supratherapeutic doses [16], and does not require dose adjustments in patients with renal impairment [17].
On the basis of the available clinical evidence, bilastine has received approval in Europe for the symptomatic treatment of allergic rhino-conjunctivitis and urticaria in adults and children aged 6 years and older. The approved dosage for adults and adolescents (12 years of age and older) is 20 mg once daily, while for children aged 6–11 years with a body weight of at least 20 kg, the dosage is 10 mg or 4 mL once daily [18, 19].
The pharmacokinetic profile of bilastine has been described in Caucasian and Japanese adult subjects. The absorption of bilastine is rapid and proportional to dose, with a time to reach maximum plasma concentration of around 1.3 h and a mean elimination half-life of 14.5 h in healthy volunteers [7, 20]. The oral bioavailability of bilastine is 60.67% [20], and is reduced by grapefruit juice or foods [21]. A pharmacokinetic analysis showed that bilastine follows a two-compartmental model with first-order absorption and elimination [22]. Bilastine undergoes minimal metabolic transformation and is primarily eliminated unchanged from the body, approximately 67% in feces and 33% in urine. The pharmacokinetic profile of bilastine is not modified by age or sex [23], and its bioavailability is increased when co-administered with P-glycoprotein inhibitors and decreased with organic anion-transporting polypeptide inhibitors [21].
Bilastine is commercially available in tablets, orodispersible tablets (ODT), and oral solution formulations. ODT formulations are widely used in drug therapy since they offer advantages over conventional formulations, such as improved adherence, ease of administration, and convenience. These advantages are particularly beneficial for specific patient subsets, such as children, the elderly, or individuals with dysphagia. This study aimed to assess the pharmacokinetic profile and bioequivalence of bilastine formulated as ODT or conventional tablets after a single oral dose of 20 mg in healthy volunteers.
Methods
Participants
Eligible participants were healthy volunteers aged between 18 and 55 years; with no clinically significant abnormalities in medical history, physical examination, laboratory tests, vital signs or electrocardiogram (ECG); free from organic or psychic conditions that, according to the investigator, could interfere with the follow-up and/or study outcomes; with a body mass index between 18.5 and 29 kg/m2 and who gave their consent to participate. Main exclusion criteria were: treatment with prescribed pharmacological therapy within the previous 4 weeks or any type of medication within the 48 h before receiving the study treatment; history of sensitivity to any drug; pregnancy or breastfeeding; history of gastrointestinal tract surgery (except appendectomy), which might affect the absorption process; participation in another clinical study within the previous 3 months; use of drugs that could induce or inhibit hepatic microsomal enzymes and/or P-glycoproteins; and consumption of citrus, berries, or any fruit juice within the 7 days before receiving the study treatment and throughout the study or frequent excessive consumption of food or beverages containing xanthine.
Participants were required to refrain from intense physical activity and from consuming alcoholic or stimulant beverages, poppy seeds, grapefruits, citrus juices, or citrus fruits within 7 days before the start of each admission day until the last blood extraction. They were also instructed not to take concomitant medications, except for symptomatic treatment with drugs with no known interaction with bilastine or hormonal contraceptive drugs, and to avoid smoking at any time during the study.
The reference product was bilastine 20 mg conventional tablets (Bilaxten®, manufacturer: FAES FARMA, S.A, batch number: 3586, expiry date: January 2024) with the following chemical structure: 2-[4-(2-(4-(1-(2-ethoxyethyl)-1H-benzimidazole-2-yl) piperidine-1-yl) ethyl) phenyl]-2-methyl propanoic acid. The test product was bilastine 20 mg ODT (Bilaxten®, manufacturer: FAES FARMA, S.A, batch number: F02919, expiry date: October 2020), with the same chemical structure. The formulation, manufacturing, packaging, and labeling of the investigational products followed the principles of Good Manufacturing Practices.
Before administering bilastine 20 mg ODT (test), participants were instructed to wet their mouth by swallowing 20 mL of water. They were subsequently directed to place the tablet on their tongue and facilitate its complete dissolution within the oral cavity. For the administration of bilastine 20 mg conventional tablets (reference), participants were instructed to swallow the tablet with 150 mL of water. Immediately after administering both formulations, the investigator conducted a mouth inspection to ensure the correct administration of study treatments in both periods.
Study Design
This was a phase I, single-center, open-label, single-dose, two-period, two-sequence crossover randomized clinical trial to assess the bioequivalence of bilastine 20 mg ODT and bilastine 20 mg conventional tablets under fasting conditions. Since at the time the study was designed and conducted (December 2019–January 2020), there was no ODT formulation of bilastine 20 mg approved for use as a reference product, bilastine 20 mg tablets was used as a reference product. The study was conducted at the Hospital Universitario de La Princesa (Madrid, Spain) after receiving approval from the Ethics Committee of the hospital and the Spanish Agency of Medicines and Medical Devices (AEMPS). The study adhered to the ethical principles of the Declaration of Helsinki and followed the criteria for the investigation of bioequivalence set by the European Medicines Agency (EMA) [24, 25]. All participants provided written informed consent.
The study comprised a screening visit (days − 20 to − 1), during which participants provided informed consent, inclusion and exclusion criteria and clinical history were reviewed, and physical examination, vital signs, 12-lead ECG, blood and urine analysis, ethanol breath test (if suspected), and pregnancy test for women were performed. After the screening visit, two study periods (days 0–4 and days 7–11), with a washout period of 7 days between each period, and a safety follow-up visit, were conducted (Fig. 1). On admission days (days 0 and 7), eligibility criteria were assessed again, and a pregnancy test, abuse drug test, and ethanol breath test (if suspected) were performed. On days 1 and 8, participants were administered a single oral dose of bilastine 20 mg as ODT or conventional tablets and blood samples for pharmacokinetic analyses were collected. Treatment sequences were allocated by randomization following a 1:1 ratio and a block size of six. The analysts responsible for determining bilastine concentrations were blinded to treatment allocation. Blood sampling and evaluation of adverse events (AEs) were also performed 24, 48, and 72 h after drug administration (days 2, 3, and 4 in period 1, and days 9, 10, and 11 in period 2). At the safety follow-up visit (between 4 and 7 days after the second dose administration), participants underwent a complete physical examination and blood and urine samples were collected for safety analyses. AEs were also registered at the safety follow-up visit. Additionally, the investigator inquired the participants about the occurrence of AEs throughout the study.
Fig. 1.

Study design. ODT orodispersible tablets, R reference, T treatment
Blood samples were collected at room temperature in 4 mL Sodium Heparin Vacutainer® tubes by direct venipuncture or from an indwelling cannula. Sampling time points were as follows: 0 (before dosing), 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 7, 12, 24, 48, and 72 h.
Volunteers were admitted to hospital within 10 h before to 12 h after dosing in each period and they returned to the Clinical Trial Units after 24, 48, and 72 h of treatment administration. Treatment administration was performed after an overnight fast of at least 10 h. Volunteers were not allowed to eat for at least 5 h or to drink water for 3 h after treatment administration.
Efficacy Assessment
The primary objective of the study was to assess the bioequivalence of bilastine 20 mg ODT (test) and bilastine 20 mg conventional tablets (reference) by comparing the relative bioavailability of both formulations administered in a single oral dose and under fasting conditions. To this end, the primary endpoint of the study was the relative bioavailability of the test and reference formulations.
The pharmacokinetic profile of both formulations was compared by calculating the following variables from plasma concentrations of bilastine: the area under the plasma concentration–time curve from dosing to the last observation (AUC0–t, primary variable), the maximum plasma concentration observed (Cmax, primary variable), the time to reach Cmax (tmax), the AUC extrapolated to infinity (AUC0–∞), the percentage of the extrapolated area under the plasma concentration–time curve (AUCext), the plasma concentration half-life (t1/2), the apparent total plasma clearance (Cl/F), the apparent volume of distribution (Vz/F), and the mean residence time (MRT).
The AUC0–t was determined by the linear trapezoidal rule. The AUC0–∞ was calculated as AUC0–t + Ct/Ke, where Ct is the last quantifiable plasma concentration and Ke is the elimination rate constant. The t1/2 was determined as 0.693/Ke. Vz/F was calculated as Cl/k, where Cl is the apparent total plasma clearance calculated as dose/AUC, adjusted for bioavailability. The mean MRT for each group (bilastine 20 mg ODT and tablets) was calculated by first determining the MRT for each individual subject using the formula MRT = area under the first moment curve (AUMC)/AUC, and then mean MRT across all subjects in each group.
Safety Assessment
The secondary objective of the study was to evaluate the safety and tolerability of both formulations. Safety was monitored at the safety follow-up visit (between 4 and 7 days after the second dose administration). Moreover, the investigator interviewed participants concerning the incidence of AEs throughout the duration of the study. To this end, the incidence of AEs, and findings from physical examinations, ECGs, vital signs, and laboratory tests, were recorded. AEs were characterized by type, onset, duration, intensity, evolution, and causality with study treatments. The AEs recorded during the study were classified by system organ class (SOC) and preferred term (PT) according to the Medical Dictionary for Regulatory Activities (MedDRA) (version 22.1). All abnormal clinical findings were followed up until they returned to normal or a rational explanation was found.
Bioanalytical Method
Blood samples were centrifuged at 1900g and 4 °C for 10 min, and 0.7 mL of the supernatant was transferred to a 1.5 mL tube. The samples were immediately frozen at − 20 °C until shipment. Bilastine was extracted using protein precipitation with a 50/50 mixture of methanol and acetonitrile. Bilastine-d6 was used as the internal standard. The concentration of bilastine in plasma was determined using reversed-phase ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC-MS/MS) at a central laboratory. The analysis was performed using a Sciex API 5000 mass spectrometer equipped with a Turbo Ion Spray. The assay was validated over a range of 0.20–400.80 ng/mL, with a lower limit of quantification in plasma of 0.2 ng/mL.
Statistical Analysis
Pharmacokinetic and statistical analyses were performed using the statistical package WinNonlin Professional Edition (Certara, Sheffield, UK), version 8.1. Statistical significance was set at a p value ≤ 0.05. The pharmacokinetic analysis was based on data from volunteers who completed the two study periods, whereas the safety analysis included data from volunteers who received at least one dose of bilastine and with at least one visit after the administration.
Pharmacokinetic parameters were analyzed using the analysis of variance (ANOVA) model. The model was adjusted for sequence, subject (sequence), period, and treatment. The 90% confidence intervals of the log-transformed ratios (test over reference) for the pharmacokinetic parameters were obtained from the adjusted model. To test for bioequivalence, the Food and Drug Administration (FDA) and EMA criteria were followed, which establishes that the standard 90% confidence intervals for AUC0–t and Cmax with log transformation should be within the acceptance interval of 80−125% [24, 26]. Intrasubject variability of AUC and Cmax between test and reference, expressed as percent coefficient of variation (CV), was approximated by the intrasubject standard deviation (square root of the error mean square) of ln (AUC) and ln (Cmax) [24].
The sample size was calculated to reject a difference between the test and reference formulations of 20%, with an 80% power and an alpha error of 0.05, and considering the bioequivalence acceptance limits defined by the EMA and FDA [24, 26]. Previous pharmacokinetic studies showed a bilastine CV of 28.17−35.48% for AUC and of 28.23−40.14% for Cmax [8, 17, 27]. Additionally, the Sponsor conducted an open-label, crossover, randomized, controlled, single-dose, three-period clinical trial (BODT-0219-BE) to evaluate the bioequivalence of bilastine 20 mg ODT (test product), with or without water, versus bilastine 20 mg conventional tablets (reference product), in healthy fasting volunteers between June and July of 2019. In that study, the intersubject CV for AUC ranged between 14.0 and 21.0%, and between 25.2 and 33.4% for Cmax. Considering an intrasubject variability of around 30%, an expected ratio between both formulations of 0.95−1.10, and assuming 5−10% losses, a minimum number of 42 subjects was required [28].
Baseline characteristics were described as mean, standard deviation (SD), median, and extremes (min, max) for the numerical variables, and as the number of patients and percentages for the categorical variables. AEs were summarized descriptively.
Results
Patient Population
From December 2019 to January 2020, 42 healthy volunteers signed the informed consent and were randomized (21 to each treatment sequence). Of them, 41 subjects completed the two periods of the study, and one subject was excluded from pharmacokinetic and bioequivalence analyses due to the intake of prohibited concomitant treatment. For this volunteer, individual pharmacokinetic parameters were considered. The safety analysis included data from 42 patients (Fig. 2). Other protocol deviations were minor and did not interfere with pharmacokinetic or safety assessments. These deviations were associated with scheduled blood sampling, for which the pharmacokinetic analysis was based on actual sampling times. A total of ten (10) subjects received concomitant pharmacological treatment during the study, but they were not excluded from the analysis since none of the treatments were prohibited concomitant medications and had no interactions with bilastine.
Fig. 2.

Flow chart showing subject distribution. PK pharmacokinetic, R reference, T test
The overall cohort comprised 16 (38.1%) men and 26 (61.9%) women who received at least one dose of bilastine. Mean age was 30.6 years (Table 1). Baseline data, and findings of medical history and physical examinations, were compatible with a healthy status.
Table 1.
Baseline characteristics of study participants
| Parameter | N = 42 |
|---|---|
| Age (years), mean ± SD | 30.6 ± 9.1 |
| Median (min, max) | 29.5 (18.0, 53.0) |
| Sex, no. (%) | |
| Women | 26 (61.9%) |
| Men | 16 (38.1%) |
| BMI (kg/m2), mean ±SD | 23.9 ± 2.8 |
| Median (min, max) | 24.5 (19.5, 28.4) |
Data are expressed as mean ± SD, median and range, or as number and percentage.
BMI body mass index
Pharmacokinetic Evaluation
Bilastine pre-dose concentration was below the lower limit of quantification in all the subjects at the two periods. Figure 3 shows serum concentration–time profiles for up to 72 h after oral administration of bilastine 20 mg using linear and semi-logarithmic scales. The concentration–time profiles were almost identical for bilastine ODT and conventional tablets.
Fig. 3.
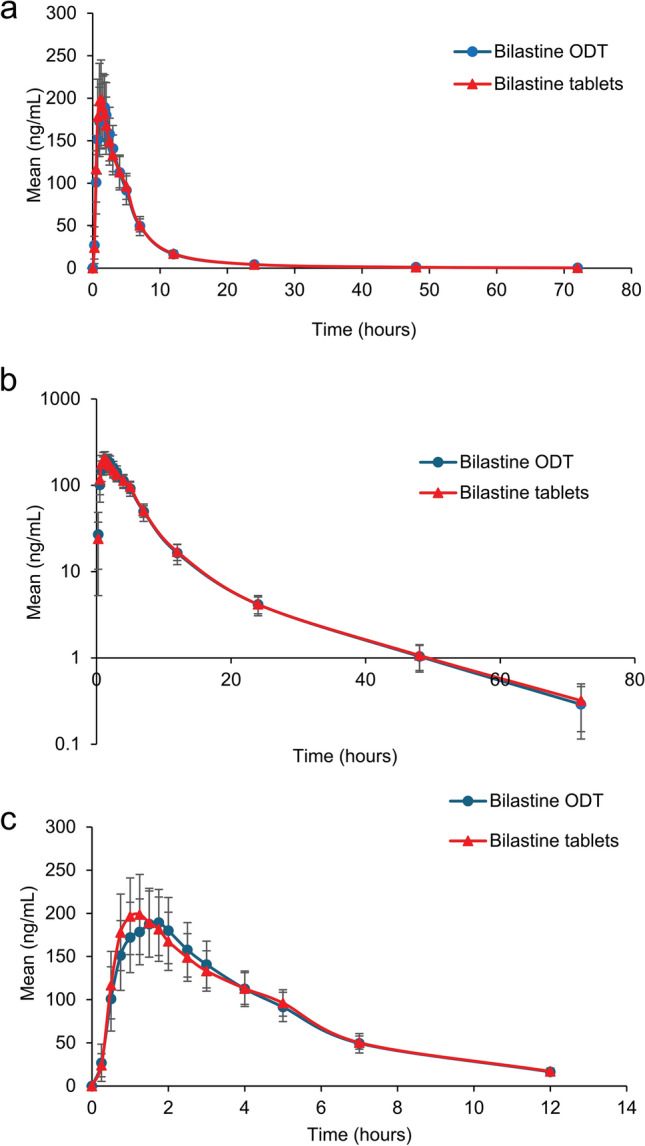
Concentration–time profiles for bilastine orodispersible tablets and bilastine conventional tablets; the graph shows mean concentration of bilastine depicted on linear (a) and semi-logarithmic (b) scales, and (c) over 12 h
The pharmacokinetic parameters obtained for bilastine 20 mg as ODT and conventional tablets are presented in Table 2. No sequence or period effect was observed for AUC0–t and Cmax. The percentage of the extrapolated portion of the AUC was 0.94% (0.83% for ODT and 1.04% for conventional tablets). Mean Cmax in the test and reference treatments were 227.55 ng/mL and 229.54 ng/mL, respectively, at a median of 1 h (range 0.50−2.50 for ODT and 0.50−5.00 for conventional tablets) after drug administration. The terminal half-life was around 11.43 h (range 2.69−33.61 h) for both formulations (11.28 h for ODT and 11.57 h for conventional tablets) (Table 2).
Table 2.
Pharmacokinetic parameters after oral administration of bilastine orodispersible tablets or conventional tablets
| Parameter | Bilastine ODT | Bilastine tablets |
|---|---|---|
| Primary endpoints | ||
| AUC0-t (h × ng/mL) | 1163.22 ± 369.47 | 1181.64 ± 362.96 |
| Cmax (ng/mL) | 227.55 ± 81.70 | 229.54 ± 90.57 |
| Secondary endpoints | ||
| AUC0–∞ (h × ng/mL) | 1172.79 ± 371.48 | 1192.62 ± 362.20 |
| AUCext (%) | 0.83 ± 0.66 | 1.04 ± 1.45 |
| t1/2 (h) | 11.28 ± 4.97 | 11.57 ± 6.25 |
| MRT (h) | 7.50± 1.95 | 7.71 ± 2.9 |
| Cl/F (mL/h) | 18808.32 ± 5987.27 | 18278.78 ± 5497.78 |
| Vz/F (mL) | 303490.82 ± 159414.52 | 312742.58 ± 272359.72 |
| tmax (h) | 1.18 ± 0.54 | 1.24 ± 0.97 |
| Median (range) | 1.00 (0.50−2.50) | 1.00 (0.50−5.0) |
Data are expressed as mean ± SD unless otherwise specified (N = 41)
AUC area under the plasma concentration–time curve from dosing to the last observation (0−t) or to infinite (0−∞), AUCext percentage of the extrapolated area under the plasma concentration-time curve, Cl/F apparent total plasma clearance, Cmax maximum plasma concentration, MRT mean residence time, ODT orodispersible tablet, tmax time to reach maximum plasma concentration, t1/2 plasma concentration half-life, Vz/F apparent volume of distribution
Pharmacokinetic parameters were comparable for both formulations, with differences between ODT and conventional tablets of − 1.56% for AUC0–t and − 0.87% for Cmax. Intersubject variability for ODT and conventional tablets was 35.91% and 39.46% for Cmax and 31.76% and 30.72% for AUC0–t, respectively. The calculated intrasubject CV between test and reference was 20.64% for AUC0–t and 38.24% for Cmax.
Bioequivalence analyses are summarized in Table 3. Bioequivalence between both formulations was demonstrated for Cmax and AUC0–t because the 90% confidence intervals for the corresponding mean ratios (test over reference) was within the 80−125% range predefined for bioequivalence.
Table 3.
Bioequivalence analysis
| Parameter | Ratio (90% CI) Test/reference |
CV | Statistical power ANOVA |
|---|---|---|---|
| Primary endpoints | |||
| Ln (AUC0–t) | 97.85 (90.69−105.58) | 20.64% | 0.998 |
| Ln (Cmax) | 101.15 (88.15−116.05) | 38.24% | 0.849 |
| Secondary endpoints | |||
| Ln (AUC0–∞) | 97.64 (90.66−105.16) | ||
| Ln (t1/2) | 100.25 (90.27−111.33) | ||
| Ln (tmax) | 101.14 (83.2−122.94) | ||
Data are expressed as the 90% confidence intervals of the log-transformed ratios (test over reference) for the pharmacokinetic parameters (N = 41).
AUC area under the plasma concentration–time curve from dosing to the last observation (0−t) or to infinite (0−∞), CI confidence interval, Cmax maximum plasma concentration, CV coefficient of variation, tmax time to reach maximum plasma concentration, t1/2 plasma concentration half-life
Safety Evaluation
A total of 31 AEs were reported during the study, of which 28 were treatment-emergent AEs (TEAEs). Two AEs in the reference group (thirst and cold sore lip) and two (headache and nausea) in the test group were considered possibly related to either treatment. All AEs resolved by the end of the trial, and no serious AEs (SAEs) were reported (Table 4). No clinically significant alterations were detected in physical exploration, vital signs, or ECG readings throughout the study. Likewise, there were no major changes in laboratory parameters compared with baseline.
Table 4.
Safety profile over the study period
| Parameter | Bilastine ODT | Bilastine tablets |
|---|---|---|
| TEAEs | 14 | 14 |
| Subjects with at least one TEAE | 13 (31.0%) | 10 (24.4%) |
| Subjects with at least one drug-related TEAE | 2 (4.8%) | 2 (4.9%) |
| Relationship | ||
| Related | 2 (14.3%) | 2 (14.3%) |
| Not related | 12 (85.7%) | 12 (85.7%) |
| Severity | ||
| Mild | 10 (71.4%) | 10 (71.4%) |
| Moderate | 4 (28.6%) | 3 (21.4%) |
| Severe | 0 | 1 (7.1%) |
| System organ class | ||
| Gastrointestinal disorders | 4 | 2 |
| General disorders and administration site conditions | 0 | 2 |
| Infections and infestations | 4 | 3 |
| Musculoskeletal and connective tissue disorders | 2 | 3 |
| Nervous system disorders | 2 | 2 |
| Reproductive system and breast disorders | 0 | 1 |
| Respiratory, thoracic and mediastinal disorders | 2 | 0 |
| Vascular disorders | 0 | 1 |
| SAEs | 0 | 0 |
Data are expressed as n (%) of patients with AEs unless otherwise specified (N = 42)
AE adverse event, ODT orodispersible tablets, TEAE treatment-emergent adverse event, SAE serious adverse event
Discussion
This bioequivalence and safety study was conducted to compare the pharmacokinetic profile of bilastine formulated either as 20 mg ODT or as conventional 20 mg tablets in healthy volunteers after a single oral dose. Pharmacokinetic and safety results demonstrated that both formulations of bilastine were bioequivalent and showed a comparable safety profile.
Bilastine is an approved second-generation non-sedating histamine H1 receptor inverse agonist that received marketing authorization in 2010 [1]. It is an efficacious and safe treatment option for allergic rhino-conjunctivitis and urticaria without sedative or cardiotoxic effects [11, 16]. Despite second-generation antihistamines being the first-line treatment option in allergic rhino-conjunctivitis and urticaria, adherence to treatment remains an important factor in the management of these patients [29]. ODT have emerged as an alternative to conventional formulations, offering advantages such as easy administration and improved patient convenience. These benefits are particularly relevant for certain patient subsets such as children, the elderly, or those with swallowing disorders. Additionally, ODT can be taken without water, making them convenient in situations where water may not be readily available. However, no study has evaluated whether the pharmacokinetic behavior of ODT and conventional tablets of bilastine 20 mg is similar in adults.
To this end, a randomized, two-period, two-sequence crossover bioequivalence clinical trial was conducted following the design recommended by the EMA [24]. While a three-period study is generally recommended when the ODT test product is an extension of another oral formulation, the EMA guideline allows assuming bioequivalence of ODT taken with water if bioequivalence between ODT taken without water and the reference formulation with water is demonstrated in a two-period study.
The study population comprised 42 healthy volunteers (26 women and 16 men), of whom 41 received a single oral dose of bilastine 20 mg in each treatment period after 7 days of washout between each administration. The selected dosage of bilastine corresponds to the therapeutic dose for adults on the basis of its safety profile and kinetic linearity [20]. Considering that the half-life of bilastine is around 14 h, sampling was extended to 72 h, and a washout period of 7 days between each administration was deemed sufficient to ensure that bilastine concentration was undetectable at the beginning of the second period. The number of blood samples collected and the sampling schedule were calculated on the basis of the pharmacokinetic characteristics of oral bilastine [20] and the regulatory guidelines on bioavailability and bioequivalence [24, 26].
Pharmacokinetic parameters derived from the adjusted model were comparable for both formulations of bilastine and similar to those previously reported for bilastine tablets [8]. Remarkably, bilastine 20 mg ODT administered without water were bioequivalent to the reference treatment formulated as conventional tablets of 20 mg administered with water, since the 90% confidence intervals for the corresponding mean ratios (test over reference) were within the acceptance range for bioequivalence. These results align with those observed before, in which different pediatric formulations of bilastine 10 mg were proven bioequivalent [30]. Similarly, bioequivalence was demonstrated between levocetirizine 5 mg formulated as immediate-release tablets and as ODT [31]. The maximum plasma concentration in both bilastine formulations (around 1.2 h after dosing) is considered fast and consistent with that previously reported in the studies conducted by Togawa [8], Lasseter [17], and Sádaba [20], with values ranging from 1.1 to 2.25 h post-dose. The shorter time to reach maximum plasma concentration for the ODT tablet suggests that the drug is more rapidly absorbed compared with the conventional tablet. A short time to reach maximum plasma concentration is particularly beneficial if the drug is intended to provide a rapid symptom relief (such as for allergic rhinitis symptoms). A narrower median time to reach maximum plasma concentration indicates less variability in the time needed for the drug to reach its maximum plasma concentration among different subjects. This lower variability suggests more consistent absorption, which may improve the predictability of therapeutic response and more homogeneous outcomes.
Both formulations were safe and well tolerated in healthy volunteers at the dose and formulations administered, as no clinically significant analytical alterations were found. Previous studies have consistently shown that bilastine, administered at the therapeutic dose of 20 mg in adults, is safe and well tolerated [4–7]. Furthermore, at this dosage, bilastine does not cause sedation or impair driving [13], psychomotor [14], or flying-related performance. It also exhibits no cardiotoxicity [16] and does not accumulate in individuals with renal impairment [17]. Similarly, oral pediatric formulations of bilastine exhibit a similar tolerability and safety profile [30].
Taken together, these results demonstrate the similar pharmacokinetic behavior of bilastine ODT and conventional tablets in adults. It is important to highlight that the ODT formulation, which does not require water intake, can be very useful in older patients (some elderly individuals may have difficulty swallowing pills, and ODT provide a convenient alternative), individuals with swallowing difficulties (patients with swallowing problems, dysphagia, or those experiencing discomfort while swallowing may find it easier to take tablets that dissolve in the mouth), and pediatric patients (young children often struggle with swallowing pills and may prefer or find it easier to take tablets that dissolve in the mouth). Therefore, the bioequivalence of the two formulations supports the use of bilastine ODT as a convenient alternative, especially for patient populations where ease of administration is important.
Conclusions
Bilastine ODT administered without water was proven to be bioequivalent to the reference treatment formulated as conventional tablets when administered as a single oral dose of 20 mg under fasting conditions. Bioequivalence was established since the 90% confidence intervals for the mean ratios of Cmax and AUC0–t were within the acceptance range for bioequivalence (80−125%). The time to reach the maximum plasma concentration of bilastine was similar for ODT and conventional tablets and consistent with previously published data. Both formulations showed a comparable safety and tolerability profile, with no SAEs or significant analytic alterations attributable to any of the formulations studied.
Acknowledgements
The sponsor (FAES FARMA) was involved in the study design and collection, analysis and interpretation of data as well as data checking of information provided in the manuscript. Ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors.
Declarations
Conflict of interest
D. Ochoa, M Román, S. Martin Vilchez, and S. Luquero-Bueno have been a consultant or investigator in clinical trials sponsored by the following pharmaceutical companies: Abbott, Alter, Aptatargets, Chemo, FAES, Farmalíder, Ferrer, Galenicum, GlaxoSmithKline, Gilead, Italfarmaco, Janssen-Cilag, Kern, Normon, Novartis, Servier, Teva, and Zambon. C. Sánchez, I. Gilaberte, and P. Arranz are full-time employees of FAES FARMA.
Medical writing assistance
Carla Granados from Trialance SCCL provided medical writing assistance supported by FAES FARMA according to Good Publication Practice guidelines.
Funding
This work was funded by FAES FARMA (Leioa, Spain)
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Ethics approval
The study was conducted at Hospital Universitario de La Princesa (Madrid, Spain) after receiving approval from the Ethics Committee of the hospital and the Spanish Agency of Medicines and Medical Devices (AEMPS), and was conducted in accordance with the ethical principles based on the Declaration of Helsinki of 1964 and its later amendments.
Consent to participate
All participants provided written informed consent.
Consent for publication
No identifying data of study subjects were used. All subjects were informed via the informed consent form about the potential publication of study results, ensuring that their identities were not disclosed.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Manuel Román, Dolores Ochoa, Samuel Martin, and Sergio Luquero. The first draft of the manuscript was written by Carlos Sánchez and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
References
- 1.Mizuguchi H, Wakugawa T, Sadakata H, Kamimura S, Takemoto M, Nakagawa T, et al. Elucidation of inverse agonist activity of bilastine. Pharmaceutics. 2020;12:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corcóstegui R, Labeaga L, Innerárity A, Berisa A, Orjales A. Preclinical pharmacology of bilastine, a new selective histamine H1 receptor antagonist: receptor selectivity and in vitro antihistaminic activity. Drugs R D. 2005;6:371–84. [DOI] [PubMed] [Google Scholar]
- 3.Corcóstegui R, Labeaga L, Innerárity A, Berisa A, Orjales A. In vivo pharmacological characterisation of bilastine, a potent and selective histamine H1 receptor antagonist. Drugs R D. 2006;7:219–31. [DOI] [PubMed] [Google Scholar]
- 4.Kuna P, Bachert C, Nowacki Z, Van Cauwenberge P, Agache I, Fouquert L, et al. Efficacy and safety of bilastine 20 mg compared with cetirizine 10 mg and placebo for the symptomatic treatment of seasonal allergic rhinitis: a randomized, double-blind, parallel-group study. Clin Exp Allergy. 2009;39:1338–47. [DOI] [PubMed] [Google Scholar]
- 5.Bachert C, Kuna P, Sanquer F, Ivan P, Dimitrov V, Gorina MM, et al. Comparison of the efficacy and safety of bilastine 20 mg vs desloratadine 5 mg in seasonal allergic rhinitis patients. Allergy. 2009;64:158–65. [DOI] [PubMed] [Google Scholar]
- 6.Horak F, Zieglmayer P, Zieglmayer R, Lemell P. The effects of bilastine compared with cetirizine, fexofenadine, and placebo on allergen-induced nasal and ocular symptoms in patients exposed to aeroallergen in the Vienna Challenge Chamber. Inflamm Res. 2010;59:391–8. [DOI] [PubMed] [Google Scholar]
- 7.Zuberbier T, Oanta A, Bogacka E, Medina I, Wesel F, Uhl P, et al. Comparison of the efficacy and safety of bilastine 20 mg vs levocetirizine 5 mg for the treatment of chronic idiopathic urticaria: a multi-centre, double-blind, randomized, placebo-controlled study. Allergy. 2010;65:516–28. [DOI] [PubMed] [Google Scholar]
- 8.Togawa M, Yamaya H, Rodríguez M, Nagashima H. Pharmacokinetics, pharmacodynamics and population pharmacokinetic/pharmacodynamic modelling of bilastine, a second-generation antihistamine, in healthy Japanese subjects. Clin Drug Investig. 2016;36:1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonijoan R, Coimbra J, García-Gea C, Puntes M, Gich I, Campo C, et al. Comparative efficacy of bilastine, desloratadine and rupatadine in the suppression of wheal and flare response induced by intradermal histamine in healthy volunteers. Curr Med Res Opin. 2017;33:129–36. [DOI] [PubMed] [Google Scholar]
- 10.Church MK. Safety and efficacy of bilastine: a new H(1)-antihistamine for the treatment of allergic rhinoconjunctivitis and urticaria. Expert Opin Drug Saf. 2011;10:779–93. [DOI] [PubMed] [Google Scholar]
- 11.Farré M, Pérez-Mañá C, Papaseit E, Menoyo E, Pérez M, Martin S, et al. Bilastine vs. hydroxyzine: occupation of brain histamine H1-receptors evaluated by positron emission tomography in healthy volunteers. Br J Clin Pharmacol. 2014;78:970–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawauchi H, Yanai K, Wang DY, Itahashi K, Okubo K. Antihistamines for allergic rhinitis treatment from the viewpoint of nonsedative properties. Int J Mol Sci. 2019;20:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conen S, Theunissen EL, Van Oers ACM, Valiente R, Ramaekers JG. Acute and subchronic effects of bilastine (20 and 40 mg) and hydroxyzine (50 mg) on actual driving performance in healthy volunteers. J Psychopharmacol. 2011;25:1517–23. [DOI] [PubMed] [Google Scholar]
- 14.García-Gea C, Martínez-Colomer J, Antonijoan RM, Valiente R, Barbanoj MJ. Comparison of peripheral and central effects of single and repeated oral dose administrations of bilastine, a new H1 antihistamine: a dose-range study in healthy volunteers with hydroxyzine and placebo as control treatments. J Clin Psychopharmacol. 2008;28:675–85. [DOI] [PubMed] [Google Scholar]
- 15.Valk PJL, Simons R, Jetten AM, Valiente R, Labeaga L. Cognitive performance effects of bilastine 20 mg during 6 hours at 8000 ft cabin altitude. Aerosp Med Hum Perform. 2016;87:622–7. [DOI] [PubMed] [Google Scholar]
- 16.Tyl B, Kabbaj M, Azzam S, Sologuren A, Valiente R, Reinbolt E, et al. Lack of significant effect of bilastine administered at therapeutic and supratherapeutic doses and concomitantly with ketoconazole on ventricular repolarization: results of a thorough QT Study (TQTS) with QT-concentration analysis. J Clin Pharmacol. 2012;52:893–903. [DOI] [PubMed] [Google Scholar]
- 17.Lasseter KC, Sologuren A, La Noce A, Dilzer SC. Evaluation of the single-dose pharmacokinetics of bilastine in subjects with various degrees of renal insufficiency. Clin Drug Investig. 2013;33:665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medicines & Healthcare products Regulatory Agency (MHRA). Summary of product characteristics Ilaxten (bilastine). https://www.medicines.org.uk/emc/product/4551/smpc#gref.
- 19.Spanish Agency for Medicines and Medical Devices. Ministry of Health and Consumer Affairs. Bilaxten® (bilastina). Summaries of product characteristics. https://cima.aemps.es/cima/dochtml/p/73027/Prospecto_73027.html.
- 20.Sádaba B, Gómez-Guiu A, Azanza JR, Ortega I, Valiente R. Oral availability of bilastine. Clin Drug Investig. 2013;33:375–81. [DOI] [PubMed] [Google Scholar]
- 21.Crean C, Valiente R, Sologuren A. Effect of grapefruit juice on the pharmacokinetics of bilastine [abstract]. J Clin Pharmacol. 2007;47:1198. [Google Scholar]
- 22.Jauregizar N, de la Fuente L, Lucero ML, Sologuren A, Leal N, Rodríguez M. Pharmacokinetic-pharmacodynamic modelling of the antihistaminic (H1) effect of bilastine. Clin Pharmacokinet. 2009;48:543–54. [DOI] [PubMed] [Google Scholar]
- 23.Roupe K, Sologuren A, Crean C, et al. Effect of age and gender on the pharmacokinetics and pharmacodynamics of bilastine. J Clin Pharmacol. 2007;47(9):1198. [Google Scholar]
- 24.Investigation of bioequivalence—scientific guideline | European Medicines Agency. https://www.ema.europa.eu/en/investigation-bioequivalence-scientific-guideline. [DOI] [PubMed]
- 25.Committee for medicinal products for human use (CHMP). Questions & Answers: positions on specific questions addressed to the pharmacokinetics working party EMA/618604/2008 Rev. 10, 07 October 2014. 2014;44.
- 26.FDA Guidance for Industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations—ECA Academy. https://www.gmp-compliance.org/guidelines/gmp-guideline/fda-guidance-for-industry-bioavailability-and-bioequivalence-studies-for-orally-administered-drug-products-general-consideration.
- 27.Sadaba B, Ramon J, Gomez-Guiu A, Rodil R. Therapeutics and clinical risk management critical appraisal of bilastine for the treatment of allergic rhinoconjunctivitis and urticaria. Ther Clin Risk Manag. 2013:9–197. [DOI] [PMC free article] [PubMed]
- 28.Hauschke D, Steinijans VW, Diletti E, Burke M. Sample size determination for bioequivalence assessment using a multiplicative model. J Pharmacokinet Biopharm. 1992;20:557–61. [DOI] [PubMed] [Google Scholar]
- 29.Simon FER, Simons KJ. H1 Antihistamines: current status and future directions. World Allergy Organ J. 2008;1:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sádaba B, Azanza JR, García-Bea A, Labeaga L, Campo C, Valiente R. Bioequivalence evaluation of three pediatric oral formulations of bilastine in healthy subjects: results from a randomized, open label, crossover study. Eur J Drug Metab Pharmacokinet. 2020;45:265–72. [DOI] [PubMed] [Google Scholar]
- 31.Ino H, Shiramoto M, Eto T, Haranaka M, Irie S, Terao T, et al. Levocetirizine oral disintegrating tablet: a randomized open-label crossover bioequivalence study in healthy Japanese volunteers. Clin Pharmacol Drug Dev. 2020;9:805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.