

Abstract
Aging is a common risk factor in neurodegenerative disorders. Investigating neuronal aging in an isogenic background stands to facilitate analysis of the interplay between neuronal aging and neurodegeneration. Here we perform direct neuronal reprogramming of longitudinally collected human fibroblasts to reveal genetic pathways altered at different ages. Comparative transcriptome analysis of longitudinally aged striatal medium spiny neurons (MSNs) in Huntington’s disease identified pathways involving RCAN1, a negative regulator of calcineurin. Notably, RCAN1 protein increased with age in reprogrammed MSNs as well as in human postmortem striatum and RCAN1 knockdown rescued patient-derived MSNs of Huntington’s disease from degeneration. RCAN1 knockdown enhanced chromatin accessibility of genes involved in longevity and autophagy, mediated through enhanced calcineurin activity, leading to TFEB’s nuclear localization by dephosphorylation. Furthermore, G2–115, an analog of glibenclamide with autophagy-enhancing activities, reduced the RCAN1–calcineurin interaction, phenocopying the effect of RCAN1 knockdown. Our results demonstrate that targeting RCAN1 genetically or pharmacologically can increase neuronal resilience in Huntington’s disease.
Aging is a major risk factor in most forms of neurodegenerative diseases and age-related changes affect many cellular processes leading to disease pathology1–5. While longitudinal studies in human individuals have been performed to assess the risk of aging in late-onset disorders, it is unfeasible to model this aging process with longitudinally collected human neurons. Therefore, there is a need to establish a human neuron platform that allows for studies of aging effects in an isogenic background. For generating aged human neurons, direct fate conversion of adult fibroblasts to neurons has been shown to propagate chronological age-related characteristics such as epigenetic cellular age stored in starting fibroblasts, thereby generating neurons that mimic the epigenetic age of fibroblast donors6. For producing disease-relevant neuronal subtypes, ectopic expression of neurogenic microRNAs, miR-9/9* and miR-124 (miR-9/9*−124), in human fibroblasts induces a chromatin reconfiguration landscape upon which subtype-defining transcription factors (TFs) guide the conversion to specific types of neurons7,8. As such, striatal MSNs directly reprogrammed from fibroblasts of patients with Huntington’s disease (HD) (HD-MSNs) recapitulate hallmarks of adult-onset HD pathologies including mutant HTT (mHTT) aggregation and neurodegeneration9–11. Thus, directly reprogrammed human MSNs serve as a patient-derived neuron model that captures age-dependent adult-onset degenerative pathology of HD6,9–11.
Directly reprogrammed MSNs retain age-associated epigenetic signatures stored in starting fibroblasts6. In this study, we utilized MSNs directly reprogrammed from isogenic, longitudinally collected fibroblasts to identify age-associated transcriptome changes in MSNs. We then applied these findings in HD by testing whether the age-associated genes can be perturbed to protect HD-MSNs from neurodegeneration. Through comparative transcriptome analyses between reprogrammed MSNs from longitudinal young and old ages, we reveal age-associated increase in RCAN1 protein both in reprogrammed MSNs and in postmortem human striatum. RCAN1 is an inhibitory interactor of calcineurin (CaN)12,13, a calcium- and calmodulin-dependent serine/threonine phosphatase, which in turn regulates phosphorylation and nuclear localization of target TFs14–16. In human brains, RCAN1 is widely expressed in various cell types within the nervous system but most highly expressed in neurons of older adults17–19. Whether RCAN1 would directly contribute to the age-dependent onset of neurodegeneration remains to be carefully dissected in a human neuron model of neurodegeneration. Interestingly, RCAN1 resides in chromosome 21 where the increased gene dosage in trisomy 21 Down syndrome could be linked to increased susceptibility to Alzheimer’s disease20–24.
We show that dampening the age-associated increase in RCAN1 protects HD patient-derived MSNs from neurodegeneration. This protective effect of RCAN1 knockdown (KD) results from the enhanced CaN activity, leading to dephosphorylation and nuclear localization of TFEB, an autophagy regulator25–27, and increased accessibility of chromatin regions that harbor TFEB binding sites. Moreover, we reveal that a G2 compound analog28 is a small molecule that can phenocopy the protective effect of RCAN1 KD by reducing the RCAN1–CaN interaction and, in turn, phosphorylation of TFEB. Collectively, our study highlights RCAN1 as an effective genetic or pharmacological target that can confer neuronal resilience against the age-associated neurodegeneration of HD.
Results
Transcriptome analysis of longitudinally aged-MSNs
We investigated age-related differences in reprogrammed MSNs from longitudinally collected male fibroblasts from three independent healthy individuals and carried out comparative transcriptome analysis between the age groups (Fig. 1a). We designate fibroblasts initially collected during middle age as ‘young’ and samples subsequently collected approximately 20 years later from three independent individuals as ‘old’ groups (Coriell Institute for Medical Research, National Institute on Aging (NIA) Aging Cell Culture Repository): AG10049 (48 years), AG16030 (68 years); AG10047 (53 years), AG14048 (71 years); and AG04456 (49 years), AG14251 (68 years) (Supplementary Table 1). Consistent with the previous results6,7,9,29,30, longitudinally derived young- and old-MSNs were electrically active, displaying inward and outward currents with action potentials observed in more than 80% of the reprogrammed cells (Extended Data Fig. 1a–c). We also validated the MSN identity of reprogrammed neurons from all groups by assessing the expression of an MSN marker (DARPP-32) (Fig. 1b,c and Extended Data Fig. 1d). RNA sequencing (RNA-seq) analysis in both longitudinally collected fibroblasts and corresponding reprogrammed MSNs revealed differentially expressed genes (DEGs) between young and old samples (false discovery rate (FDR) < 0.05, | fold-change (FC) | ≥ 1.5) (Extended Data Fig. 1e,f). DEGs up- or downregulated in both old-fibroblasts and old-MSNs were commonly enriched in age-associated pathways such as ECM–receptor interaction, protein digestion, cell adhesion molecules and focal adhesion (Fig. 1d). Interestingly, downregulated DEGs manifested in old-MSNs over young-MSNs (FDR < 0.05, | FC | ≥ 1.5) were uniquely enriched in the calcium signaling pathway, yet not detected in fibroblasts, suggesting MSN-specific alteration in gene expressions (Fig. 1e and Extended Data Fig. 1g). Finally, ingenuity pathway analysis revealed upstream effectors of DEGs in old-fibroblasts or old-MSNs (FDR < 0.05, | FC | ≥ 1.5) (Extended Data Fig. 2a,b). Among these, RCAN1 was identified as an MSN-specific upstream effector of the calcium signaling pathway as well as inhibitor of CaN (tacrolimus and cyclosporin A (CsA)) (Fig. 1f), suggesting that MSNs reprogrammed from older individuals behave as if CaN function has been compromised.
Fig. 1 |. Identification of RCAN1 as an age-associated factor in reprogrammed MSNs from longitudinally collected fibroblasts.
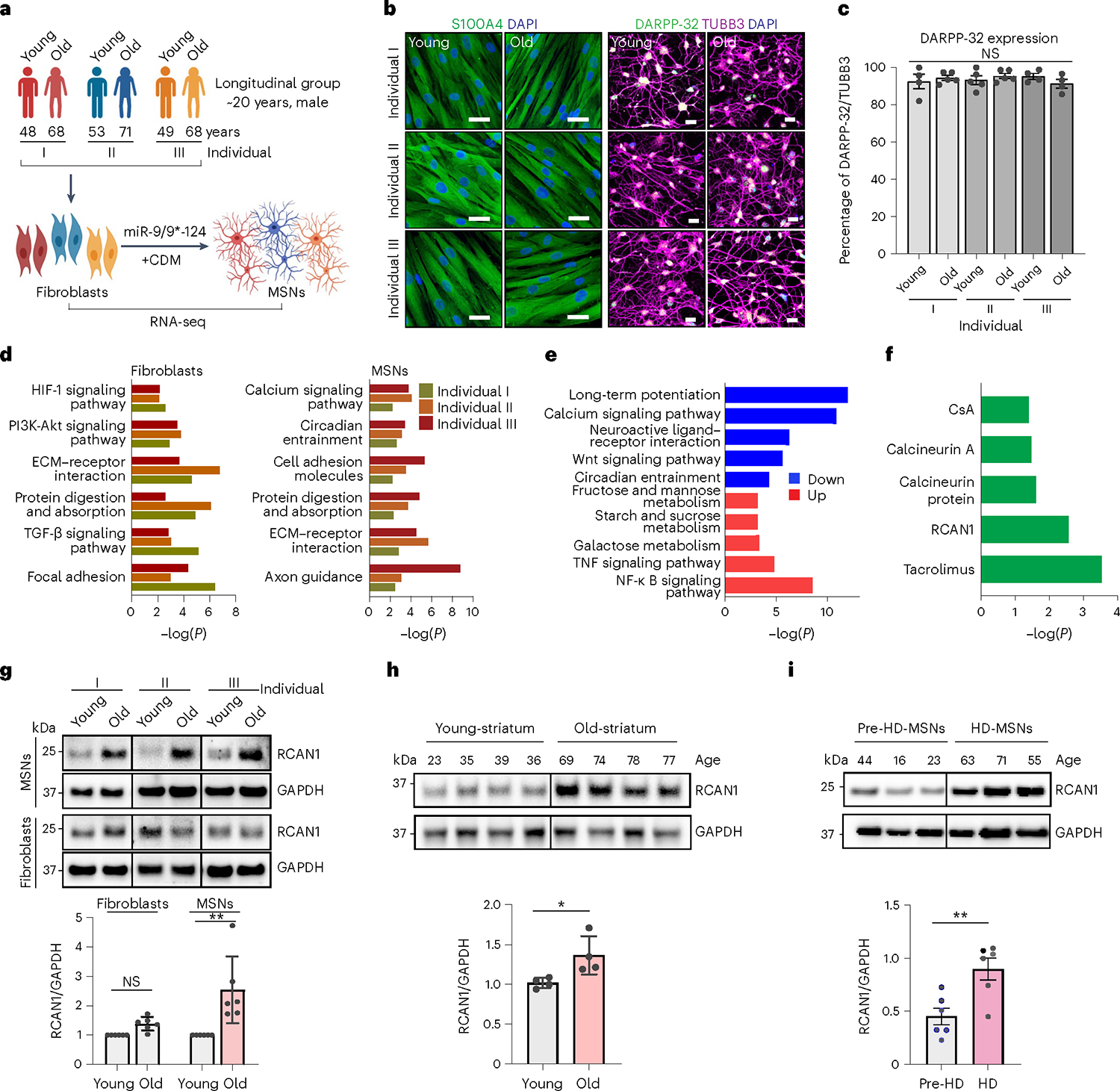
a, Experimental scheme of RNA-seq in fibroblasts and reprogrammed MSNs (young and old) from three independent longitudinal groups from individuals I, II and III. MSNs were reprogrammed by overexpressing miR-9/9* and miR-124 (miR-9/9*−124) as well as MSN-defining TFs CTIP2, DLX1, DLX2 and MYT1L (CDM). b, Representative images of fibroblasts marked by S100A4 (left) and corresponding reprogrammed MSNs marked by DARPP-32 used for RNA-seq. c, Quantification of DARPP-32-positive cells from reprogrammed MSNs from all samples (individual I: young, n = 4; old, n = 5; individual II: young, n = 5; old, n = 5; individual III: young, n = 4; old, n = 4). An average of 300 cells were counted from four or more randomly chosen fields. Scale bars, 20 μm. d, Gene Ontology (GO) enrichment analysis of all DEGs in two replicates of old-fibroblasts (left) and old-MSNs (right) from three independent individuals (FDR < 0.05, | FC | ≥ 1.5). e, GO enrichment analysis of up-/downregulated genes commonly manifested in old-MSNs compared with young-MSNs (FDR < 0.05, | FC | ≥ 1.5). f, Upstream regulator analysis of up-/downregulated genes in old-MSNs in e. g, Representative immunoblotting (top) and quantification (bottom) of RCAN1 in longitudinal MSNs and fibroblasts (young and old) from three independent individuals (n = 6, **P = 0.0077). The quantification is normalized to values from young samples per line. h, Representative immunoblotting (top) and quantification (bottom) of RCAN1 expression in eight human striatum samples from age 23, 35, 39 and 36 (young) and age 69, 74, 78 and 77 (old) individuals (n = 8, *P = 0.0489). i, Representative immunoblotting (top) and quantification (bottom) of RCAN1 expression in three reprogrammed MSNs from presymptomatic patient-derived fibroblasts (44-, 16- and 23-year-old pre-HD-MSN: Pre-HD.42, Pre-HD.45, Pre-HD.40/50) and three reprogrammed MSNs from symptomatic patient-derived fibroblasts (63-, 71- and 55-year-old HD-MSN: HD.47, HD.40, HD.45) (n = 6, **P = 0.0063). Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test (c), two-tailed paired t-test (g) and two-tailed unpaired t-test (h,i); **P < 0.01, *P < 0.05, NS, not significant, and mean ± s.e.m. (c,g,h,i). The sample size (n) corresponds to the number of biologically independent samples (c,g,h,i). DAPI, 4’,6-diamidino-2-phenylindole.
Age-associated upregulation of RCAN1 in MSNs
To further investigate whether RCAN1 expression may be related to aging, we assessed the RCAN1 expression between longitudinal male fibroblasts and corresponding reprogrammed MSNs. As assessed by immunoblots, RCAN1 protein was expressed at a higher level in old-MSNs compared with the younger samples, whereas this difference was not detected between young- and old-fibroblasts (Fig. 1g). This MSN-enriched upregulation of RCAN1 with aging was consistent in reprogrammed MSNs from other multiple individuals (~30-year difference in age, two males and one female for each young/old individual) (Extended Data Fig. 2c) and human striatum (~40-year age difference, eight female striatum samples) (Fig. 1h) between young and old samples at the protein level, but not in transcripts (Extended Data Fig. 2d). Moreover, RCAN1 expression was significantly increased in HD-MSNs from older symptomatic patients compared with HD-MSNs from younger presymptomatic patients (pre-HD-MSNs) (~35-year age difference, one male and two females for each HD-MSN sample) (Fig. 1i), suggesting the global age-associated upregulation of RCAN1 in MSNs. Altogether, our results indicate that RCAN1 is an age-associated factor whose protein expression undergoes upregulation in aged-MSNs.
To compare RCAN1 turnover between old-MSNs and young-MSNs, we compared RCAN1’s half-life between young- and old-MSNs in the presence of cycloheximide, an inhibitor of protein synthesis. Notably, RCAN1 in young-MSNs displayed a faster half-life than in old-MSNs from three independent individuals, suggesting slower RCAN1 protein turnover in old-MSNs (Extended Data Fig. 2e). Moreover, treatment with the proteasome inhibitor (MG132) or lysosome inhibitor (CQ) significantly increased the RCAN half-life in young-MSNs but not in old-MSNs. These results suggest the presence of proteasome- or lysosome-dependent regulation of RCAN1 protein stability in young-MSNs, which may be ineffective in old-MSNs. Previous studies suggest that HDAC3 can increase the protein stability of RCAN1 through posttranslational modifications31,32. Interestingly, we observed that there was an age-associated increase of HDAC3 in old-MSNs compared with young-MSNs (Extended Data Fig. 2f), suggesting a potential involvement of HDAC3 in age-associated increase in RCAN1 stability.
Next, we asked whether the effect of aging on the RCAN1 level would be consistent in the context of HD. Comparing RCAN1 expression in reprogrammed neurons between age-matched healthy control old-MSNs and HD-MSNs, we found, surprisingly, that HD-MSNs exhibited even higher levels of RCAN1 than control old-MSNs (Extended Data Fig. 2g). These results suggest that RCAN1 is an age-associated factor whose expression increases with aging in MSNs, and its upregulation becomes even more pronounced in HD patient-derived MSNs. We then tested whether it would be feasible to reduce the RCAN1 expression to a level similar to young-MSNs by short hairpin RNA (shRNA), given that RCAN1 still plays important roles in neurons such as in synaptic plasticity33. We observed that through RCAN1 shRNA, the age-associated upregulation of RCAN1 in old-MSNs could be lowered to a level comparable to young-MSNs (Extended Data Fig. 2h).
Validation of RCAN1 as a disease modifier gene in HD
Genome-wide association studies have identified Genetic Modifiers of HD (GeM-HD) comprising polymorphic gene variants associated with accelerated or delayed onset of HD34. These genes were discovered as modifiers that can affect the age of symptomatic onset. Interestingly, RCAN1 is also included in the list of candidate HD-modifier genes. Thus, as a parallel investigation, we knocked down 246 candidate genes to identify genes whose reduction-of-function would protect HD-MSNs from degeneration, thereby identifying genes that contribute to HD-MSN degeneration (Extended Data Fig. 3a and Supplementary Table 2). Reprogrammed HD-MSNs (Extended Data Fig. 3b) were cultured in 96-well plates to be assayed for neuronal death using Sytox-Green as previously described (approximately 3,000 cells counted per well) (Extended Data Fig. 3c)9,10. For this assay, we first used HD-MSNs from the GM04194 line (CAG repeat size 46; HD.46), which showed a twofold increase in cell death at around 50% compared with control MSNs from the healthy individual (GM02171) (Extended Data Fig. 3d). We then added lentivirus carrying gene-specific shRNAs to HD-MSNs and the average cell death level for each gene KD was compared with the average level with scrambled control shRNA (shCtrl) (Extended Data Fig. 3e). Interestingly, in this unbiased testing, we also identified RCAN1 whose KD led to the most significant reduction in neuronal death compared with other identified genes RTCA and UBE2D4 (pink zone, plus or minus 10% of healthy control level). This protective effect was further validated in HD-MSNs from other independent patients with HD (Extended Data Fig. 3f). We also tested whether KD of the identified genes would lower mHTT aggregation and found that among the genes tested, only RCAN1 KD significantly decreased the amount of mHTT inclusion bodies (Extended Data Fig. 3g).
To confirm the specificity of shRCAN1 for neuronal survival, we prolonged RCAN1 expression by overexpressing RCAN1 complementary DNA in HD-MSNs in the presence of shRCAN1 (Extended Data Fig. 4a). Continuous RCAN1 expression reversed the neuroprotective effect of shRCAN1 in HD-MSNs from multiple patients (Fig. 2a). Furthermore, since caspase activation signals have been detected in brains of patients with HD35–41, we also assessed caspase 3/7 activation and annexin V signal (an apoptotic marker via its ability to bind to phosphatidylserine on the extracellular surface) in HD-MSNs as previously described10. RCAN1 KD significantly reduced caspase activation and annexin V signals while the rescuing effect of shRCAN1 was abolished by RCAN1 cDNA (Fig. 2b,c). Moreover, the clearance of HTT aggregation following RCAN1 KD was also reversed by overexpressing RCAN1 in the presence of shRCAN1 (Fig. 2d). Therefore, our results overall indicate that RCAN1 is an age-associated disease modifier whose KD leads to clearance of mHTT aggregation and neuroprotection of HD-MSNs.
Fig. 2 |. RCAN1 KD protects HD-MSNs from degeneration and induces chromatin accessibility changes.
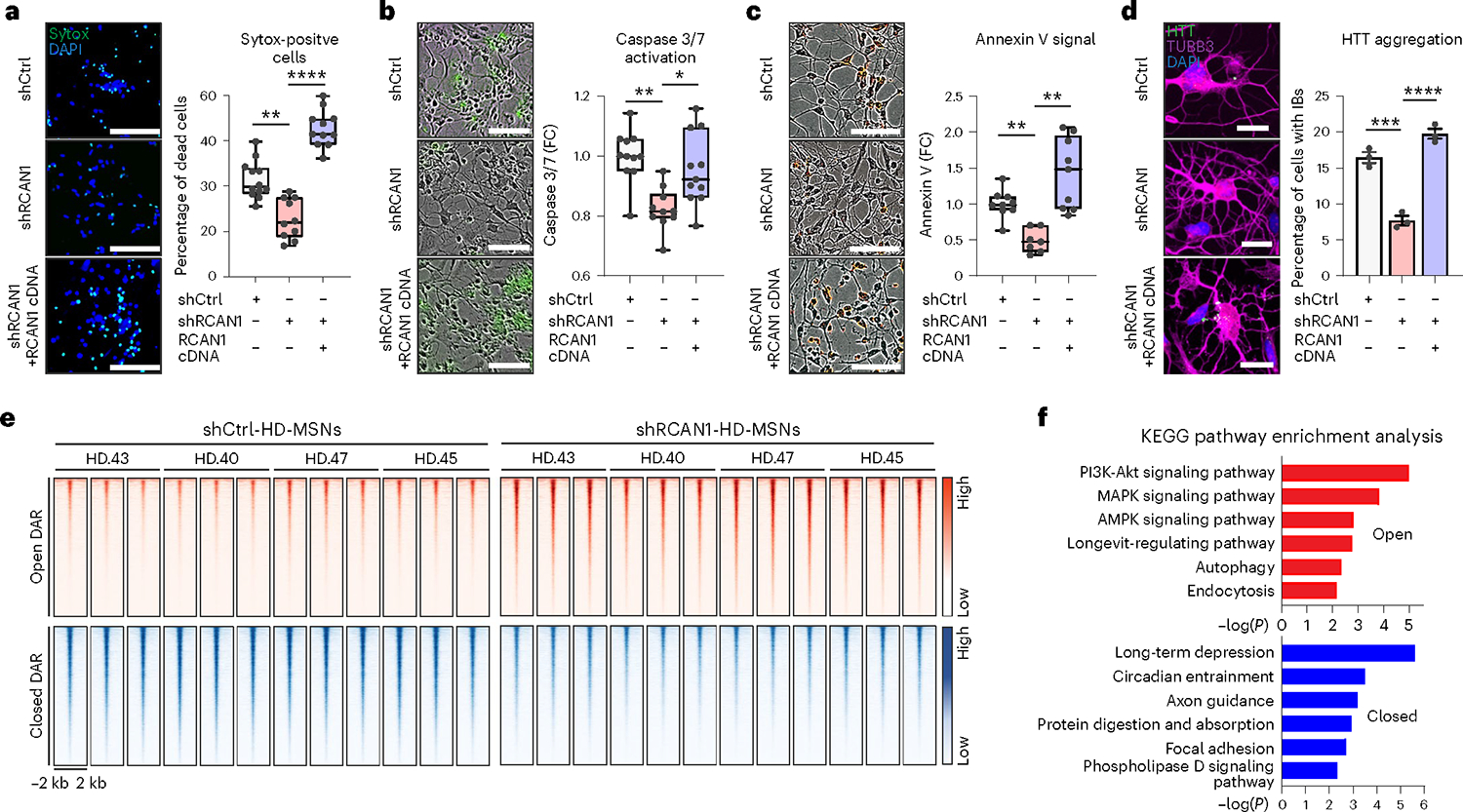
a–c, Representative images (left) and quantification (right) of Sytox-positive cells (n = 12, 10, 11; **P = 0.0021, ****P < 0.0001) (a), caspase 3/7 activation (green; n = 11, 10, 11; **P = 0.0023, *P = 0.0258) (b) and annexin V signal (red; n = 9, n = 7, n = 9; shCtrl versus shRCAN1 **P = 0.0012, shCtrl versus shRCAN1&RCAN1 cDNA **P = 0.0092) (c) in three independent HD-MSNs (HD.40, HD.43, HD.47) transduced with shCtrl, shRCAN1 or RCAN1 cDNA. Scale bars, 100 μm (a–c). d, Representative images (left) and quantification (right) of cells with HTT inclusion bodies (IBs) in three independent HD-MSNs (HD.40, HD.43, HD.47; n = 3) transduced with shCtrl, shRCAN1 or RCAN1. Cells were immunostained with anti-HTT and TUBB3 antibodies. An average of 120 cells each were counted from three or more randomly chosen fields. Scale bars, 20 μm. Mean ± s.e.m. ***P = 0.0003, ****P < 0.0001. e,f, Analysis of ATAC-seq in four independent HD-MSNs (HD.43, HD.40, HD.47, HD.45) transduced with shCtrl or shRCAN1. e, Heatmaps of signal intensity in chromatin peaks (FDR < 0.05, | FC | ≥ 1.5) of open and closed DARs in shRCAN1-HD-MSNs compared with shCtrl-HD-MSNs. f, KEGG pathway enrichment analysis of genes associated with open (top) and closed (bottom) DARs in shRCAN1-HD-MSNs. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test (a–d). In box-and-whisker plots, the center line denotes the median value, while the box contains the 25th to 75th percentiles of the dataset. The whiskers mark minimal value to maximal value (a–c). ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 (a–d). The sample size (n) corresponds to the number of biological replicates (a–d).
RCAN1 KD induces chromatin accessibility changes
RCAN1 primarily functions to inhibit its interacting partner, CaN, calcium- and calmodulin-dependent protein serine/threonine phosphatase, which in turn regulates phosphorylation of target TFs14–16. Due to this potential link between RCAN1 KD and changes in TF activities, we investigated whether RCAN1 KD would lead to changes in chromatin accessibilities in HD-MSNs by performing the assay for transposase-accessible chromatin-sequencing (Omni-ATAC-seq)42 analyses comparatively between shCtrl- and shRCAN1-expressing HD-MSNs from multiple HD samples (Fig. 2e) (ND30013 (HD.43), ND33947 (HD.40), GM04198 (HD.47), GM04230 (HD.45); three biological replicates per independent line) (Supplementary Table 1). Of the total number of 102,747 peaks detected across samples, we identified 15,767 differentially accessible regions (DARs) (FDR < 0.05, | FC | ≥ 1.5) between shCtrl- and shRCAN1-HD-MSNs (15.345% of the total peaks). Of the total DARs, 6,050 DARs corresponded to chromatin regions that became more accessible (open) and 9,717 to those less accessible (closed) in shRCAN1-HD-MSNs compared with shCtrl-HD-MSNs. Focusing on DARs ±2 kilobases (kb) around the transcription start site, we identified 505 genes with increased and 1,173 genes with decreased ATAC-seq signals with shRCAN1 compared with shCtrl (FDR < 0.05, | FC | ≥ 1.5) (Fig. 2e).
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis revealed that genes associated with DARs opened by shRCAN1 in HD-MSNs were enriched with longevity-regulating pathway, PI3K-AKT/MAPK/AMPK signaling pathway, autophagy and endocytosis, suggesting that RCAN1 KD led to chromatin changes proximal to genes associated with aging in HD-MSNs (Fig. 2f, top). Interestingly, the role of autophagy in clearing mHTT aggregates and neuroprotection was previously shown by the discovery of miR-29b-3p-STAT3 axis, Beclin1 and autophagy-related FYVE protein (ALFY), which modifies the amount of mHTT aggregation10,43,44. Genes associated with closed DARs in shRCAN1-HD-MSNs were, however, enriched in other pathways, including long-term depression, circadian entrainment, axon guidance, protein digestion and absorption, focal adhesion and phospholipase D signaling pathway (Fig. 2f, bottom). Altogether, these results demonstrate that the protective effect of RCNA1 KD is accompanied by increased chromatin accessibility to genes involved in longevity and autophagy.
CaN inhibition reverses RCAN1 KD-mediated neuroprotection
Next, we asked whether RCAN1 KD-induced changes in chromatin would occur through CaN. Because RCAN1 normally inhibits CaN, we first tested whether inhibiting CaN simultaneously while knocking down RCAN1 would revert HD-MSNs to degeneration. RCAN1 KD decreased caspase 3/7 activation and annexin V signals in HD-MSNs, whereas inhibiting CaN using shRNA specific for calcineurin A (shCaN) or CsA, a well-established inhibitor of CaN45, abolished the neuroprotective effect of RCAN1 KD (Fig. 3a). Additionally, shRCAN1 failed to reduce HTT inclusion bodies when RCAN1 KD-HD-MSNs were treated with shCaN or CsA (Fig. 3b). These results demonstrate that the protective effect of RCAN1 KD occurs through CaN activity in HD-MSNs.
Fig. 3 |. RCAN1 KD- and CaN KD-induced chromatin changes.
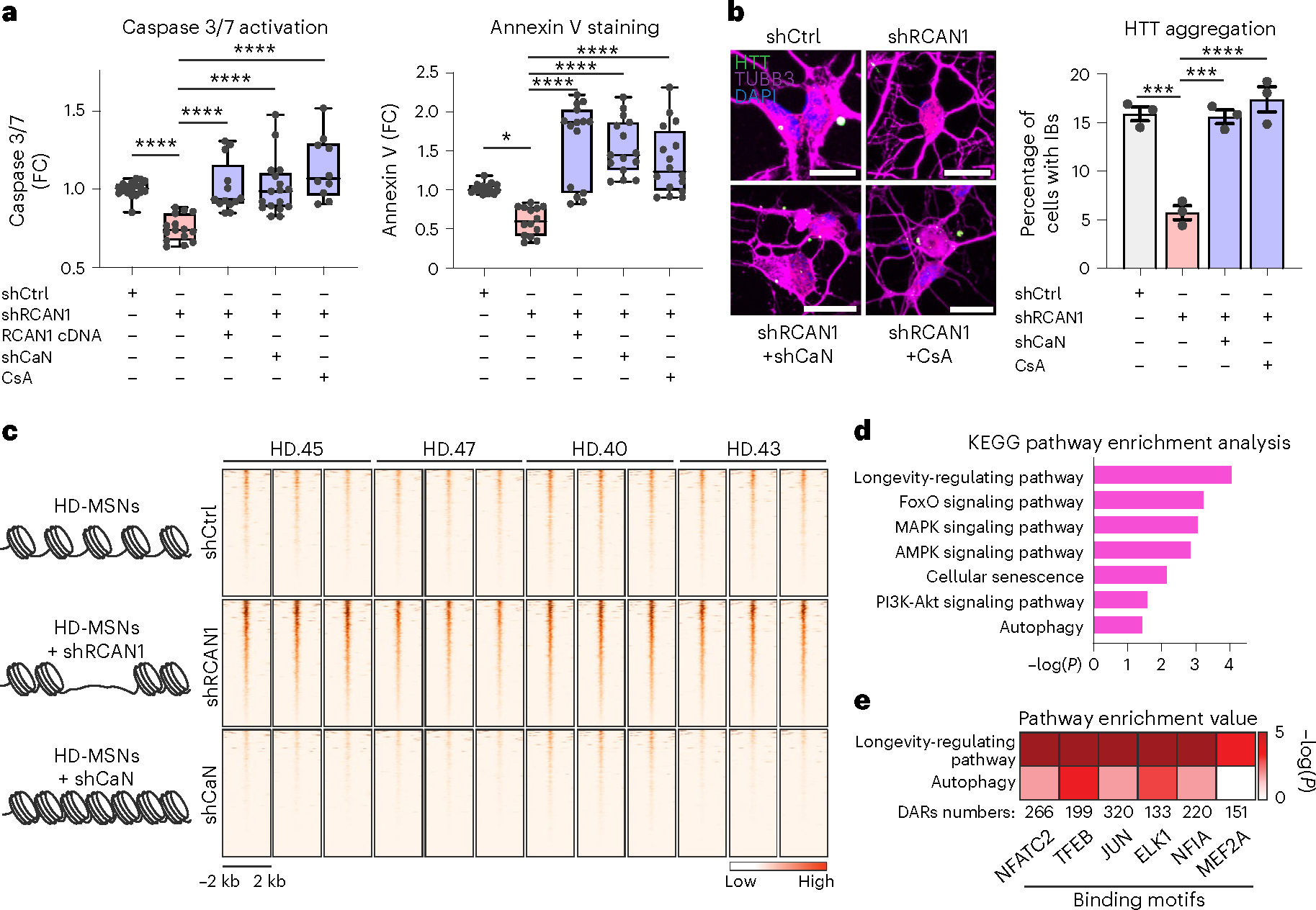
a, Quantification of caspase 3/7 activation (left, n = 18, 15, 13, 17, 10) and annexin V signal (right, n = 18, 14, 15, 15, 16) in four independent HD-MSNs (HD43, HD40, HD47, HD45) transduced with shCtrl, shRCAN1, RCAN1 or shCaN. Cells were also treated with 10 μM CsA, a CaN inhibitor. In box-and-whisker plots, the center line denotes the median value, while the box contains the 25th to 75th percentiles of the dataset. The whiskers mark minimal value to maximal value. *P = 0.0175, ****P < 0.0001. b, Representative images (left) and quantification (right) of cells with HTT IBs in three independent HD-MSNs (HD.43, HD.40, HD.47; n = 3) transduced with shCtrl, shRCAN1 or shCaN. Cells were treated with 10 μM CsA. Cells were immunostained with anti-HTT and TUBB3 antibodies. An average of 117 cells each were counted from three or more randomly chosen fields. Scale bars, 20 μm. Mean ± s.e.m. shCtrl versus shRCAN1 ***P = 0.0002; shRCAN1 versus shRCAN1+shCaN ***P = 0.0003, ****P < 0.0001. c–e, Analysis of ATAC-seq from four independent HD-MSNs (HD43, HD40, HD47, HD45; three replicates each) transduced with shCtrl (control), shRCAN1 (rescuing) or shCaN (detrimental). c, Heatmaps of signal intensity in overlapping chromatin peaks of open DAR (FDR < 0.05, FC ≥ 1.5) in shRCAN1-HD-MSNs and closed DAR (FDR < 0.05, FC ≤ −1.5) in shCaN-HD-MSNs compared with shCtrl-HD-MSNs. d,e, KEGG pathway enrichment analysis (d) and pathway enrichment analysis (e) of genes associated with open DARs in shRCAN1-HD-MSNs and closed DARs in shCaN-HD-MSNs in c. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test in a and b; ****P < 0.0001, ***P < 0.001, *P < 0.05 (a,b). The sample size (n) corresponds to the number of biologically independent samples (a,b).
We then leveraged the finding that CaN KD reverses the neuroprotection by RCAN1 KD to infer DARs opened by RCAN1 KD that are instead closed in response to CaN KD. We performed a comparative ATAC-seq analysis between shRCAN1-HD-MSNs (rescuing condition) and shCaN-HD-MSNs (nonrescuing condition) where DARs in opposite directions were defined by comparisons with shCtrl-HD-MSNs (Fig. 3c and Extended Data Fig. 4b,c). In total, 467 ATAC-seq peaks that opened with RCAN1 KD compared with shCtrl were overlapped with chromatin regions that instead closed with shCaN compared with shCtrl (FDR < 0.05, | FC | ≥ 1.5) (Fig. 3c). These overlapping DARs ±2 kb around the transcription start site identified 286 genes whose pathway analysis identified terms associated with longevity-regulating pathway, FoxO signaling, cellular senescence and autophagy (Fig. 3d). Therefore, these results demonstrate that RCAN1 KD-induced neuroprotection through CaN is accompanied by chromatin changes proximal to genes involved in aging and autophagy.
RCAN1 KD opens chromatin regions with TFEB binding sites
CaN, a Ser/Thr phosphatase, has been shown to partner with various TFs (NFATC2, TFEB, JUN, ELK1, NF1A and MEF2A)46–51 to regulate their phosphorylation and activities14–16. We searched sequence motifs using the JASPAR TF database52 within DARs corresponding to regions that became more accessible with shRCAN1 (FDR < 0.05, FC ≥ 1.5) and closed with shCaN (FDR < 0.05, FC ≤ −1.5) and found that binding sites for TFs including NFATC2, TFEB, JUN, ELK1, NF1A and MEF2A enriched in the opposite DARs (Fig. 3e). We analyzed the pathway enrichment value (−log(P)) from KEGG pathway enrichment analysis for associated genes and the DAR number. Among the TF sites within the identified DARs, TFEB binding site was most significantly enriched with genes associated with both longevity and autophagy (Fig. 3e), suggesting TFEB as a critical TF associated with the protective role of RCAN1 KD in HD-MSNs.
RCAN1 KD promotes TFEB nuclear localization
We further confirmed the TFEB binding site enrichment by separately extracting the DARs containing TFEB binding sites corresponding to regions that became more accessible in shRCAN1-HD-MSNs (FDR < 0.05, FC ≥ 1.5) and closed in shCaN-HD-MSNs (FDR < 0.05, FC ≤ −1.5) compared with shCtrl-HD-MSNs (Fig. 4a). Genes proximal to these oppositely accessible DARs containing TFEB binding sites were enriched with longevity and autophagy pathways (Fig. 4b, top). These genes include RB1CC1, an autophagy inducer53,54, and MAPK1, whose function has been shown to decline with brain aging55,56 (Fig. 4b, bottom).
Fig. 4 |. Enhancing TFEB function by RCAN1 KD via its nuclear localization.
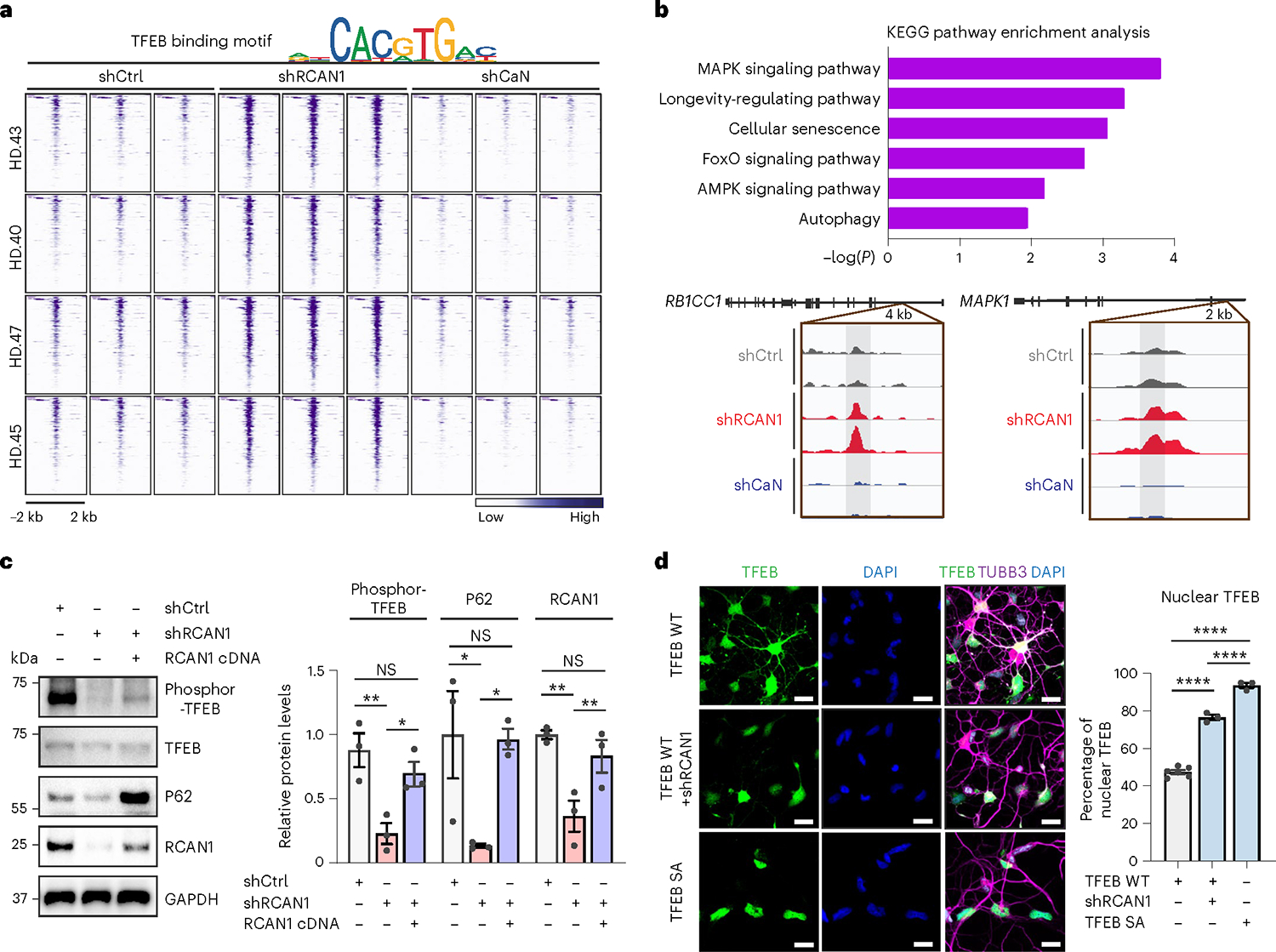
a, Heatmap representation of open DARs with shRCAN1 (rescuing) and closed DARs with shCaN (detrimental) harboring TFEB binding motifs, compared with shCtrl. Motif analysis from ATAC-seq was from four independent HD-MSNs (HD.43, HD.40, HD.47, HD.45; three biologically independent samples each) (FDR < 0.05, FC ≥ 1.5). Top legend depicts representative motifs for TFEB binding sites. b, KEGG pathway enrichment analysis (top) of TFEB binding motif-containing genes associated with DARs in a. Integrative Genomics Viewer snapshots (bottom) showing peaks enriched in shRCAN1-HD-MSNs (red) and reduced in shCaN-HD-MSNs (blue) within RB1CC1 and MAPK1 in comparison with shCtrl (gray). c, Representative immunoblotting (left) and quantification (right) of the expression of phosphor-TFEB (Ser142) from three independent HD-MSNs (HD.43, HD.40, HD.47; n = 3) transduced with shCtrl, shRCAN1 or RCAN1. Phosphor-TFEB (Ser142): **P = 0.0095, *P = 0.0495; P62: shCtrl versus shRCAN1 *P = 0.0497, shRCAN1 versus shRCAN1&RCAN1 cDNA *P = 0.0496; RCAN1: shCtrl versus shRCAN1 **P = 0.0023, shRCAN1 versus shRCAN1&RCAN1 cDNA **P = 0.0034. d, Representative image (left) and quantification (right) of nuclear TFEB from three independent HD-MSNs (HD.43, HD.40, HD.47) transduced with TFEB WT, shRCAN1 or TFEB phosphor-mutant (S142/211A (SA)). Cells were immunostained with anti-TFEB and TUBB3 antibodies. An average of 130 cells each were counted from three or more randomly chosen fields (n = 6, 3, 3). Scale bars, 20 μm. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test in c and d; ****P < 0.0001, **P < 0.01, *P < 0.05 and mean ± s.e.m. (c,d). The sample size (n) corresponds to the number of biologically independent samples (c,d).
TFEB is known as a regulator of lysosomal biogenesis and autophagy25–27, which may also regulate longevity57–62. Phosphorylation keeps TFEB localization in the cytoplasm, whereas its dephosphorylation allows TFEB shuttling into the nucleus26,63,64. Since RCAN1 inhibits CaN function and CaN has been shown to dephosphorylate TFEB46, we tested whether RCAN1 KD would lead to TFEB dephosphorylation and nuclear localization in HD-MSNs. RCAN1 KD reduced the level of phosphorylated TFEB, which was reversed by overexpressing RCAN1 as assessed by immunoblots in HD-MSNs from multiple patients with HD (Fig. 4c). Also, while expressing exogenous TFEB led to the localization of TFEB in both cytoplasm and nucleus, RCAN1 KD significantly increased nuclear localization of TFEB, which was mimicked by phosphor-mutant (S142/211A) TFEB containing mutations at the serine residues S142 and S211 (CaN’s dephosphorylation sites in TFEB) to alanine26,46,65 (Fig. 4d and Extended Data Fig. 5a). Our results thus indicate that RCAN1 KD enhances TFEB activity by promoting its nuclear localization. To further investigate the link between RCAN1 and CaN in terms of TFEB localization, we checked whether RCAN1 would regulate TFEB localization through CaN. We found that the increased nuclear localization of TFEB (wild-type (WT) cDNA) by RCAN1 KD was reversed by the treatment with CsA, a CaN inhibitor (Extended Data Fig. 5b), indicating that CaN function is necessary for the effect of RCAN1 KD on the nuclear localization of TFEB.
RCAN1 KD promotes neuronal survival through TFEB
Given the link between RCAN1 and TFEB activity, we asked whether RCAN1 KD would enhance autophagy function in HD-MSNs. We performed CYTO-ID assay (a fluorescence-based live-cell assay for accumulated autophagic vacuoles10), immunoblotting assay for p62/SQSTM1 expression (a marker widely used to monitor autophagic activity due to its binding to LC3 and degradation by autophagy66) and autophagic flux assay with tandem monomeric mCherry-GFP-tagged LC3 (previously shown to distinguish prefusion autophagic compartments from mature acidic autolysosomes based on the differential pH sensitivity of GFP versus mCherry67,68). RCAN1 KD increased CYTO-ID signal compared with shCtrl in HD-MSNs from multiple patients with HD, which was reversed by overexpressing RCAN1 (Fig. 5a). The level of p62/SQSTM1 protein was also significantly decreased by RCAN1 KD (Fig. 5b). Moreover, RCAN1 KD increased the average number of both prefusion autophagosomes (mCherry-positive:GFP-positive) and postfusion autolysosomes (mCherry-positive:GFP-negative) per cell, which was reversed by overexpressing RCAN1 (Fig. 5c).
Fig. 5 |. RCAN1 KD promotes neuronal resilience through enhancing TFEB nuclear localization.
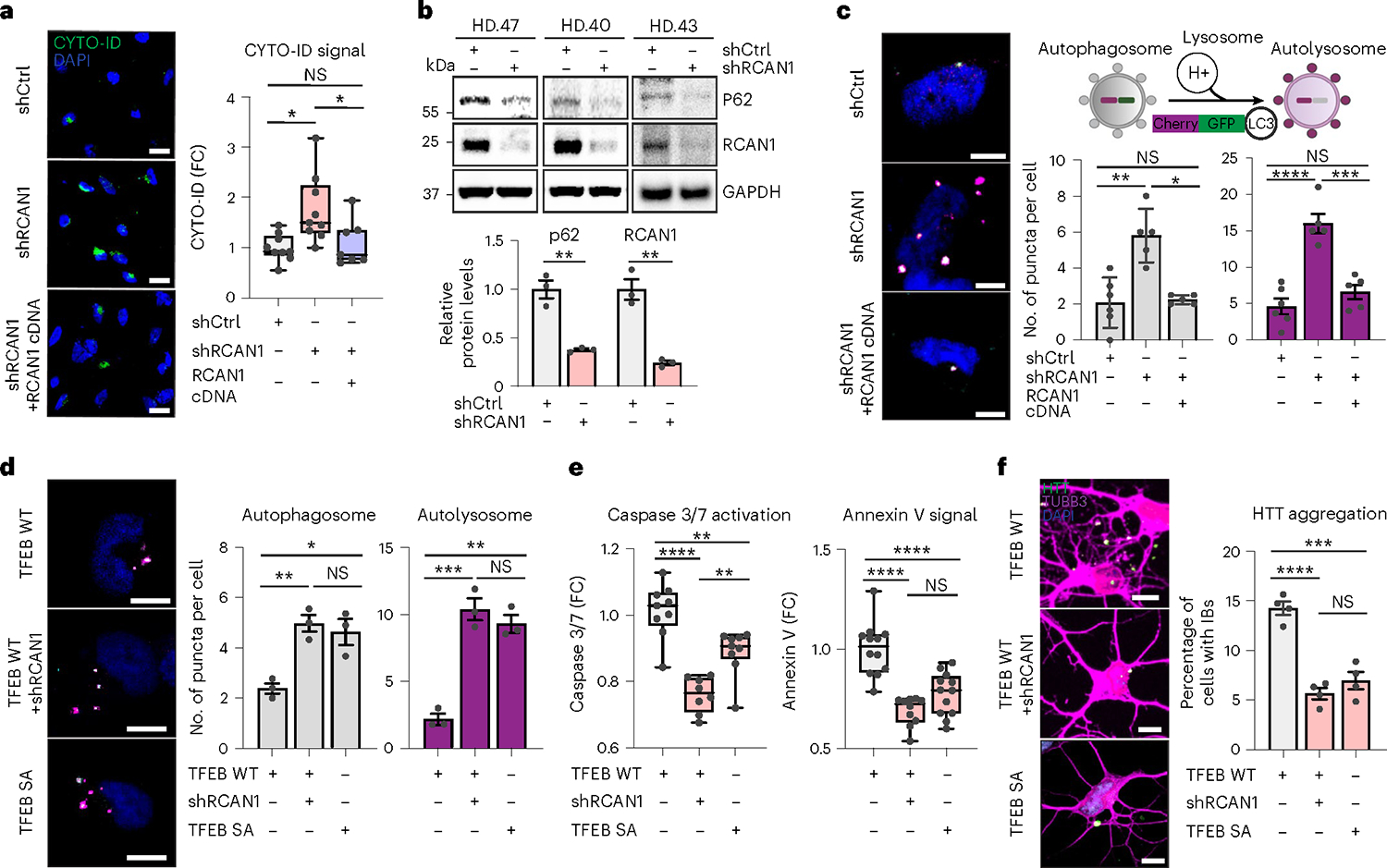
a, Representative images (left) and quantification (right) of CYTO-ID-positive cells from three independent HD-MSNs (HD.43, HD.40, HD.47) transduced with shCtrl, shRCAN1 or RCAN1 (n = 9, 9, 7). shCtrl versus shRCAN1 *P = 0.0103, shRCAN1 versus shRCAN1&RCAN1 cDNA *P = 0.0406. Scale bars, 20 μm. b, Immunoblotting (top) and quantification (bottom) of the expression of p62 (**P = 0.0027) and RCAN1 (**P = 0.0021) from three independent HD-MSNs (HD.43, HD.40, HD.47) transduced with shCtrl or shRCAN1 (n = 3). c, Autophagic flux measurements using tandem monomeric mCherry-GFP-LC3 (right top). Representative image (left) and quantification (right bottom) of autophagosome and autolysosome from cells having reporter signal puncta from three independent HD-MSNs (HD.43, HD.40, HD.47) transduced with shCtrl, shRCAN1 or RCAN1 (n = 6, 5, 5). **P = 0.0012, *P = 0.0132, ***P = 0.0002, ****P < 0.0001. d, Representative image (left) and quantification (right) of autophagosome and autolysosome from cells having puncta from three independent HD-MSNs (HD.43, HD.40, HD.47) transduced with TFEB WT, shRCAN1 or TFEB Phosphor-mutant (SA) (n = 3). Autophagosome *P = 0.0106, **P = 0.0060; autolysosome **P = 0.0014, ***P = 0.0006. e, Quantification of caspase 3/7 activation (left, n = 9, 8, 9) and annexin V signal (right, n = 12, 10, 12) from three independent HD-MSNs (HD.40, HD.43, HD.47) transduced with TFEB WT, shRCAN1 or TFEB SA. Caspase 3/7 activation: TFEB WT versus TFEB SA **P = 0.0028; TFEB WT + shRCAN1 versus TFEB SA **P = 0.0026, TFEB WT versus TFEB WT + shRCAN1 ****P < 0.0001. f, Representative images (left) and quantification (right) of HTT IBs from four independent HD-MSNs (HD.47, HD.40, HD.43, HD.45) transduced with TFEB WT, shRCAN1 or TFEB SA. Cells were immunostained with anti-HTT and TUBB3 antibodies. An average of 120 cells each were counted from three or more randomly chosen fields (n = 4). ***P = 0.0001, ****P < 0.0001. Scale bars, 10 μm (c,d,f). Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test (a,c,d,e,f) and two-tailed unpaired t-test (b); ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, NS, not significant (a–f) and mean ± s.e.m. (b,c,d,f). In box-and-whisker plots, the center line denotes the median value, while the box contains the 25th to 75th percentiles of the dataset. The whiskers mark minimal value to maximal value (a,e). The sample size (n) corresponds to the number of biologically independent samples (a–f).
We then assessed whether RCAN1 KD would decrease HD-associated phenotype through TFEB. When HD-MSNs expressed TFEB cDNA, RCAN1 KD mimicked the effect of TFEB phosphor-mutant (TFEB S142/211A), which increased the average number of both prefusion autophagosomes (mCherry-positive:GFP-positive) and postfusion autolysosomes (mCherry-positive:GFP-negative) per cell (Fig. 5d). Similarly, phospho-mutant of TFEB decreased caspase 3/7 activation, annexin V signal and the formation of mHTT inclusion bodies, which was replicated by RCAN1 KD in the presence of WT TFEB (Fig. 5e,f). Also, the reduction of neuronal death caused by knocking down RCAN1 was reversed when we knocked down TFEB (Extended Data Fig. 5c). Altogether, these results indicate that RCAN1 KD promotes HD-MSN resilience against degeneration largely through enhancing TFEB activity.
Neuroprotection by G2 analog via reducing RCAN1–CaN binding
We then wondered whether the genetic effect of RCAN1 KD on TFEB dephosphorylation for neuroprotection could be replicated by small molecules. We first tested various small molecules known to increase autophagy including G2–115, metformin, carbamazepine and rapamycin28,69,70. Among the compounds tested, we found that G2–115 significantly reduced the phosphorylation of TFEB compared with other autophagy inducers (Fig. 6a). This effect was consistent in multiple HD-MSN lines in which G2–115 decreased the phosphorylation of endogenous TFEB (Fig. 6b). G2 was the original analog of glibenclamide identified as sufficient for the autophagic-enhancing activity that promoted degradation of misfolded α1-antitrypsin Z variant in mammalian cell models of α1-antitrypsin deficiency28,69. Importantly, G2 analog G2–115 was recently shown to reduce HD-MSN death and mHTT inclusion bodies in HD-MSNs10. Due to changes in TFEB phosphorylation, we tested whether G2–115 would affect the interaction between RCAN1 and CaN. Strikingly, G2–115 reduced the binding of RCAN1 to CaN in a dose-dependent manner when Flag-tagged RCAN1 was pulled down, and then CaN was probed with various concentrations of G2–115 (Fig. 6c). When endogenous CaN was pulled down in fibroblasts or HD-MSNs in the presence of lysosome inhibitor chloroquine (to keep the consistent level of RCAN1), G2–115 interrupted RCAN1–CaN interaction (Fig. 6d and Extended Data Fig. 6a). This effect was specific to G2–115 as other autophagy inducers did not affect RCAN1–CaN interaction (Extended Data Fig. 6b). We also performed the NanoBit binding assay in HEK293 cells transfected with the interaction domains of RCAN1 (amino acids 89–197) and CaN (amino acids 1–391)16 fused with luciferase subunits that can generate luminescent signals when they interact. This assay confirmed the specificity of G2–115 in reducing RCAN1–CaN binding among other autophagy inducers and was repeated at various concentrations of G2–115 (Fig. 6e and Extended Data Fig. 6c). Importantly, we tested whether G2–115 would affect the TFEB localization as its dephosphorylation would allow TFEB shuttling into the nucleus26,63,64. G2–115 significantly increased the nuclear localization of endogenous TFEB as determined in HD-MSNs from multiple patients (Fig. 6f and Extended Data Fig. 6e). Altogether, our results indicate that G2–115 enhances TFEB functions by specifically reducing the interaction between RCAN1 and CaN.
Fig. 6 |. G2–115 promotes TFEB function by reducing RCAN1–CaN interaction and promoting TFEB nuclear localization.
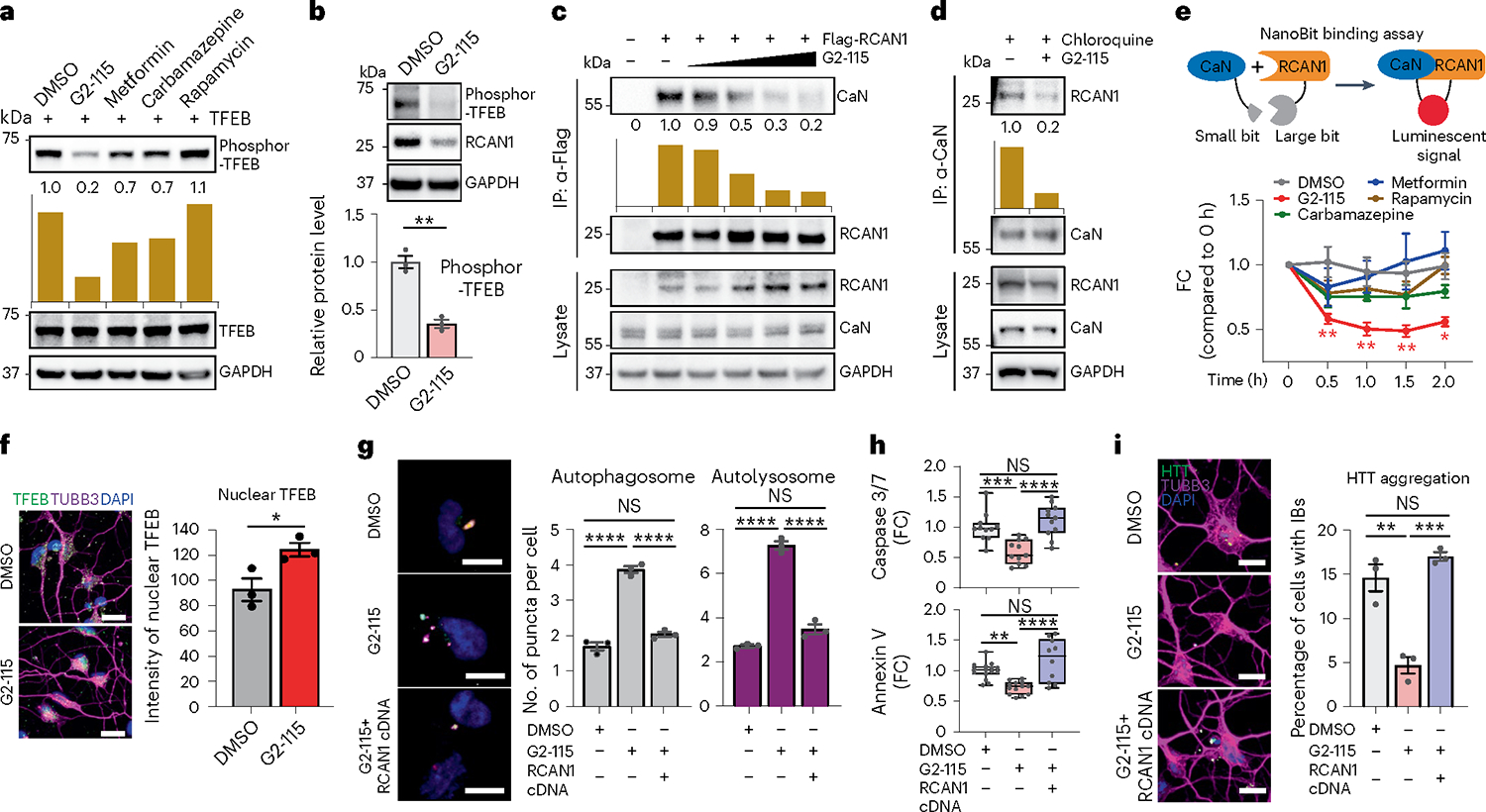
a,b, Immunoblotting of fibroblasts treated with DMSO, G2–115 (0.5 μM), metformin (8 mM), carbamazepine (100 μM) and rapamycin (500 nM) (a) and HD-MSNs (HD.47, HD.40, HD.45) treated with DMSO and G2–115 (0.5 μM) (n = 3, **P = 0.0013) (b) with anti-phosphor-TFEB (Ser142). c, Immunoprecipitation of Flag-RCAN1-transfected HEK293 cells with anti-Flag followed by CaN immunoblotting after G2–115 (0.25; 0.5; 2.5; 5 μM) treatment. d, Immunoprecipitation of chloroquine (lysosome inhibitor)-treated fibroblasts with anti-CaN followed by RCAN1 immunoblotting after DMSO, G2–115 (0.5 μM) and chloroquine (60 μM) treatment. e, Experimental scheme of NanoBit binding assay (top). Binding assay of HEK293 cells transfected with RCAN1-large-Bit and CaN-small-Bit (bottom) after G2–115 (2.0 μM), metformin (8 mM), carbamazepine (100 μM) and rapamycin (500 nM) treatment (n = 3 independent experiments). DMSO versus G2–115: 0.5 h **P = 0.0041; 1.0 h **P = 0.0037; 1.5 h **P = 0.0016; 2.0 h *P = 0.0111. f, Representative image (left) and quantification (right) of nuclear TFEB in HD-MSNs (HD.43, HD.45, HD.40) treated with DMSO and G2–115 (0.5 μM) (n = 3, *P = 0.0380). Scale bars, 20 μm. g, Representative images (left) of control and RCAN1-transduced HD-MSNs expressing the tandem monomeric mCherry-GFP-LC3 reporter. Quantification (right) of autophagosomes and autolysosomes from HD-MSNs with puncta (HD.40, HD.47, HD.43; n = 3; ****P < 0.0001; more than 50 cells per line) treated with DMSO and G2–115 (0.5 μM). Scale bars, 20 μm. h, Quantification of caspase 3/7 activation (top) from HD-MSNs (HD.40, HD.47, HD.45; n = 11) and annexin V signal (bottom) from HD-MSNs (HD.43, HD.47, HD.45; n = 15, 13, 10) treated with DMSO and G2–115 (0.5 μM) after control and RCAN1 transduction. In box-and-whisker plots, the center line denotes the median value, while the box contains the 25th to 75th percentiles of the dataset. The whiskers mark minimal value to maximal value. ****P < 0.0001, ***P = 0.0009, **P = 0.0028. i, Representative image (left) and quantification (right) of HTT IBs in control and RCAN1-transduced HD-MSNs (HD.43, HD.40, HD.47) treated with DMSO and G2–115 (0.5 μM) (n = 3; **P = 0.0015, ***P = 0.0005) followed by HTT and TUBB3 staining. An average of 300 cells were counted from three or more randomly chosen fields. Scale bars, 10 μm. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test (g,h,i) and two-tailed unpaired t-test (b,e,f); ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, NS, not significant, and mean ± s.e.m. (b,e,f,g,i). Sample size (n): number of biologically independent samples (b,f,g,h,i). IP, immunoprecipitation.
To further test the rescuing effect of G2–115 potentially through reducing RCAN1 function, we first measured the autophagy activity by autophagic flux assay in multiple independent HD-MSNs. G2–115 increased the average number of both prefusion autophagosomes (mCherry-positive and GFP-positive) and postfusion autolysosomes (mCherry-positive and GFP-negative) per cell, which was reversed by overexpressing RCAN1 (Fig. 6g). G2–115 also increased CYTO-ID signal in HD-MSNs, which was reversed by overexpressing RCAN1 (Extended Data Fig. 6d), suggesting that the effect of G2–115 occurs through reducing RCAN1 activity in HD-MSNs. Additionally, neuronal cell death and the formation of HTT inclusion body were decreased by G2–115 and this effect was reversed by RCAN1 overexpression (Fig. 6h,i). Collectively, our results indicate that RCAN1–CaN is a chemically modifiable target that G2–115 can act on to increase neuronal resilience of HD-MSNs to neurodegeneration.
Discussion
In the current study, we performed a comparative transcriptome analysis in longitudinally collected fibroblasts and corresponding reprogrammed MSNs derived from healthy individuals. MicroRNA-based direct fate conversion of adult fibroblasts to neurons has been shown to retain chronological age signature stored in starting fibroblasts6, and hallmarks of adult-onset HD pathologies are also captured in directly reprogrammed striatal MSNs from HD patient-derived fibroblasts9–11. Therefore, directly reprogrammed human MSNs from longitudinally collected fibroblasts are the established human neuron platform that allows for studies of aging in human neurons in an isogenic background.
This analysis identified RCAN1 as an age-associated regulator whose expression was upregulated in aged-MSNs and human striatum. Mechanistically, we provide evidence that reducing RCAN1 function protects HD-MSNs by opening chromatin regions proximal to genes involved in longevity-regulating pathway. Knocking down RCAN1 relieves its repression on its interactor CaN, which in turn dephosphorylates and promotes nuclear localization of TFEB and increased accessibility of chromatin regions that harbor TFEB binding sites. Moreover, we reveal the mechanism of the G2 analog that can mimic the protective effect of RCAN1 KD by reducing the RCAN1–CaN interaction and phosphorylation of TFEB. Through this mode, the G2 analog can help clearance of mHTT inclusion bodies and the survival of HD-MSNs. Therefore, RCAN1 functions as an age-associated modifier that promotes neurodegeneration, and genetic or pharmacological intervention on RCAN1 activity can be potentially harnessed to promote neuronal resilience against the age-associated onset of neurodegeneration in HD (Extended Data Fig. 6f).
Remarkably, reduced RCAN1 expression enhances chromatin accessibility of genes involved in longevity and autophagy to promote neuronal resilience against neurodegeneration in HD-MSNs. Interestingly, the increase in RCAN1 expression in the striatum of older adults compared with young individuals was captured in reprogrammed MSNs from longitudinally collected fibroblasts. The increase in RCAN1 gene dosage is also implicated in Down syndrome as a gene in chromosome 21 trisomy, which increases the susceptibility to Alzheimer’s disease20–24. Future studies should be directed to further investigate the changes in RCAN1 expression in different ages of HD patient brains, or whether the changes in RCAN expression may be related to other neurodegenerative disorders.
Based on previous reports, the role of CaN in neurodegeneration seems multifaceted and not entirely straightforward. While there is evidence that CaN inhibition can provide neuroprotective effects in some contexts, such as the overactivation of CaN (and its downstream effector, NFAT) associated with calcium dysregulation and Aβ exposure71, interestingly, there is also evidence suggesting that prolonged inhibition could be detrimental to neurons. For instance, patients who receive CsA as an immunosuppressant experience clinical symptoms associated with neurotoxicity72. Moreover, studies have shown that genetic KD or chemical inhibition of CaN was shown to exacerbate Aβ42-induced neurotoxicity in a Drosophila model of Alzheimer’s disease73. The precise consequences may depend on factors such as the specific neuronal subtype, the disease context and the degree or duration of inhibition. Pertinent to the topic of the current study, where we model neuronal aging by taking a snapshot of different human lifespan stages through neuronal reprogramming and perturbing the age-associated pathway in a patient-based neuron model of HD, our results are consistent in that knocking down CaN or chemically inhibiting CaN increased neurodegeneration. In this scenario, our study offers a slightly different perspective: dampening the age-related increase in RCAN1 would balance the CaN function and promote neuronal survival. Therefore, the detrimental effect of CaN KD could be specific to aged human neurons. Another factor could be the dependence on the neuronal subtype specificity. We observed in our previous study9 that when HD fibroblasts were reprogrammed into the cortical lineage, the neurodegeneration phenotype was not as pronounced as in HD-MSNs. Therefore, future studies should be directed to investigate whether a similar increase in RCAN1 would be observed in cortical cells and whether knocking down CaN would induce degeneration in HD cortical neurons.
Glibenclamide, a sulfonylurea drug, has been used broadly in clinics as an oral hypoglycemic agent. A glibenclamide analog, G2, promoted autophagic degradation of misfolded α1-antitrypsin Z variant in mammalian cell models of α1-antitrypsin deficiency disorder28,69. We found a unique feature of G2–115 as an autophagy inducer that promotes TFEB activity by reducing the RCAN1–CaN interaction to promote clearance of HTT inclusion bodies and neuronal survival. However, it remains unclear how G2–115 mechanistically interferes with RCAN1–CaN interaction. Previous studies provided the structural information for RCAN1–CaN binding that delineates the structural basis of RCAN1 and CaN interaction16. RCAN1 inhibits the activity of CaN directly by binding and blocking both substrate-binding sites and the active site of CaN, in which RCAN1–CaN binding is disrupted when SPPASPP and TxxP motifs in the N-terminal domain of RCAN1 are mutated16. Our study was not designed to address whether G2–115 reduces the RCAN1–CaN interaction by interfering with RCAN1 stability through a secondary pathway or by binding directly to the interaction site in the N-terminal domain of RCAN1. Further investigations into the specific mechanism of how G2–115 reduces RCAN1–CaN interaction may provide new insights into how small compounds can be used to increase the resilience against neurodegeneration in HD. Additionally, screening additional small molecules that directly interfere with the interaction between RCAN1 and CaN, and replicating the neuroprotective effect of RCAN1 inhibition, may offer a new therapeutic target that may alleviate neurodegeneration in HD.
Methods
Plasmids, shRNAs and cell lines
Research described in this study complies with all relevant ethical regulations and was approved by the Washington University School of Medicine. The construction of all plasmids used for MSN reprogramming in this study has been previously described6,9–11,29,43,74–77, and they are publicly available at Addgene as pTight-9–124-BclxL (no. 60857), rtTA-N144 (no. 66810), pmCTIP2-N106 (no. 66808), phDLX1-N174 (no. 60859), phDLX2-N174 (no. 60860) and phMYT1L-N174 (no. 66809). For the overexpression of RCAN1, the RCAN1 genomic sequence was cloned and ligated into the pcDNA, pcDNA-Flag-HA and N174-lentiviral vector. For the overexpression of TFEB WT and S142/211A, pcDNA3.1-TFEB-WT-MYC (no. 99955) was obtained from Addgene, mutagenized and ligated into the N174-lentiviral vector. Lentiviral shCtrl (SHC002), human RCAN1 shRNAs (TRCN0000256296) and human PPPC3A (CaN) shRNA (TRCN0000342619) were obtained from Sigma. To visualize free autophagosomes and autolysosomes, FUW mCherry-GFP-LC3 (no. 110060) was obtained from Addgene. Adult dermal fibroblasts from longitudinal healthy individuals (Coriell NINDS and NIGMS Repositories: AG10049, AG16030, AG10047, AG14048, AG04456, AG14251), healthy controls (Coriell NINDS and NIGMS Repositories: GM03440, GM00495, AG11732, AG10047, AG12956, GM02187, GM08379, GM02171), symptomatic HD patients (Coriell NINDS and NIGMS Repositories: ND33947, GM04230, ND30013, GM04198, GM04194, GM02147) and presymptomatic HD patients (Coriell NINDS and NIGMS Repositories: GM04717, GM04861, GM04857, GM04855, GM04829) were acquired from the Coriell Institute for Medical Research.
Human brain tissue
Our research complies with all relevant ethical regulations and was approved by the institutional review board at the Washington University School of Medicine. For the analysis of human striatum samples, we obtained formalin-fixed paraffin-embedded (FFPE) striatal sections from cognitively normal individuals aged 23, 35, 36 and 39 years (young group) and aged 69, 74, 77 and 78 years (old group) with written, informed consent from the Tissue Procurement Core of Washington University School of Medicine. These samples were deidentified and exempt from human subjects as verified within our institution. FFPE striatal sections were scraped off unstained slides and proteins were extracted using the Qproteome FFPE Tissue Kit (Qiagen, no. 37623), following the manufacturer’s recommended protocol.
Antibodies
Primary antibodies used for immunostaining and immunoblot included rabbit anti-MAP2 (CST, no. 4542, 1:200), rabbit anti-DARPP-32 (19A3) (CST, no. 2306, 1:200), rabbit anti-GABA (Sigma-Aldrich, A2052, 1:500), mouse anti-NCAM1 (ERIC) (Santa Cruz, sc-106, 1:100), rabbit anti-NEUN (Millipore, ABN78, 1:1,000), mouse anti-ACTL6B (Antibodies Incorporated, 75–311, 1:100), mouse anti-tubulin β III (Covance, MMS-435P, 1:1,000), rabbit anti-tubulin β III (Covance, PRB-435P-100, 1:5,000), rabbit anti-RCAN1/DSCR1 (Sigma-Aldrich, D6694, 1:1,000), rabbit anti-pan-Calcineurin A (Cell Signaling, 2614, 1:1,000), rabbit anti-TFEB (Cell Signaling, 4240, 1:1,000), rabbit anti-phosphor-TFEB (Ser142) (Millipore, ABE1971, 1:1,000), rabbit anti-phosphor-TFEB (Ser211) (Cell Signaling, 37681, 1:1,000), rabbit anti-p62/SQSTM1 (Abcam, ab109012, 1:1,000), mouse anti-Flag (Sigma-Aldrich, F1804, 1:5,000), mouse anti-HDAC3 (Santa Cruz, sc-376957, 1:200) and rabbit anti-GAPDH (Santa Cruz, sc-32233, 1:5,000) antibodies. The secondary antibodies included goat anti-rabbit IgG (H + L) secondary antibody, HRP (Invitrogen, 65–6120, 1:5,000); goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody, HRP (Invitrogen, A16078, 1:5,000); Alexa Fluor 594 goat anti-chicken IgG (H + L) (Invitrogen, A32759, 1:1,000); Alexa Fluor 488 goat anti-mouse IgG (H + L) (Invitrogen, A11029, 1:1,000); Alexa Fluor 488 goat anti-rabbit IgG (H + L) (Invitrogen, A11034, 1:1,000); Alexa Fluor 594 goat anti-rabbit IgG (H + L) (Invitrogen, A11037, 1:1,000); and Alexa Fluor 594 goat anti-mouse IgG (H + L) (Invitrogen, A11032, 1:1,000).
Primary cell culture
Adult human fibroblasts were cultured in fibroblast medium composed of DMEM (high glucose and no glutamine) supplemented with 15% FBS (Gibco), 0.01% β-mercaptoethanol, 1% nonessential amino acids, 1% sodium pyruvate, 1% GlutaMAX, 1% 1 M HEPES buffer solution and 1% penicillin/streptomycin solution (all from Invitrogen). Cells were only maintained for up to 15 passages.
Lentiviral preparation
Lentiviral production was carried out separately for each plasmid, but they were transduced together as a single cocktail as previously described10,74. Briefly, the supernatant was collected 72 h after transfection of Lenti-X 293T Cell Line (Clonetech) with each plasmid, in addition to psPAX2 and pMD2.G, using polyethyleneimine (Polyscience). Collected lentiviruses were filtered through 0.45-μm PES membranes, mixed for a single cocktail and incubated with the Lenti-X concentrator overnight to concentrate the viruses tenfold. Concentrated lentiviruses are resuspended in 1/10 of the original volume with 1 × PBS after spinning down at 1,500g for 45 min at 4 °C. In centrifuge tubes, we added 7 ml of 20% sucrose cushion solution (20% sucrose, 100 mM NaCl, 20 mM HEPES (pH 7.4), 1 mM EDTA in distilled water) and then overlayed the resuspended lentiviruses on the sucrose solution. After centrifugation at 70,000g for 2 h at 4 °C, viral pellets were resuspended in 10% sucrose solution (10% sucrose, 25 mM HEPES (pH 7.3) in DPBS) and stored at −80 °C. Typical titers of lentivirus ranged from 1 × 107 to 2.5 × 108 infection-forming units per milliliter.
MSN reprogramming
Direct neuronal reprogramming of human fibroblasts to MSNs was performed as previously described6,9–11,29,43,74–77. Briefly, human fibroblasts were seeded onto Costar six-well cell culture vessels (Corning) at a density of 300,000 cells per well. The following day, each plate was transduced with the lentiviral cocktail of pTight-9/9*−124-BclxL, rtTA, CTIP2, DLX1, DLX2 and MYT1L in the presence of polybrene (8 μg ml−1, Sigma-Aldrich). Then, 4 ml of the lentiviral cocktail/fibroblast medium was added to each well and spinfected at 1,000g for 30 min at 37 °C using a swinging bucket rotor. Next, 1 d after transduction (PID 1), cells were washed with PBS and added to fresh fibroblast medium (2 ml per well) supplemented with doxycycline (Dox, 1 μg ml−1) (Sigma-Aldrich). After 2 d (PID 3), the medium was changed to fresh fibroblast medium supplemented with Dox and puromycin (3 μg ml−1) (Life Technologies). After 2 d (PID 5), cells were replated onto poly-ornithine/laminin/fibronectin-coated glass coverslips previously treated with nitric acid and added to fibroblast medium supplemented with Dox (1 μg ml−1). The following day (PID 6), medium was changed to Neurobasal-A Medium (Gibco, catalog no. 10888022) containing B-27 Plus Supplement (Gibco, catalog no. A3582801) and GlutaMAX Supplement (Gibco, catalog no. 35050061) and supplemented with Dox (1 μg ml−1), valproic acid (1 mM), dibutyryl cAMP (200 μM), BDNF (10 ng ml−1), NT-3 (10 ng ml−1), retinoic acid (1 μM), RevitaCell Supplement (RVC, 1×), ascorbic acid (200 μM) and antibiotics (puromycin, 3 μg ml−1; blasticidin, 3 μg ml−1; geneticin, 300 μg ml−1). Dox was replenished every 2 d and half-volume medium changes were performed every 4 d. At PID 14, medium was switched to Brainphys (Stemcell, catalog no. 05793) containing NeuroCult SM1 neuronal supplement and N2 Supplement-A and supplemented with Dox (1 μg ml−1), valproic acid (1 mM), dibutyryl cAMP (200 μM), BDNF (10 ng ml−1), NT-3 (10 ng ml−1), retinoic acid (1 μM), RVC (1×), ascorbic acid (200 μM) and puromycin (3 μg ml−1). The addition of blasticidin and geneticin was halted after PID 10 and puromycin was continuously added until PID 30. The addition of RVC and ascorbic acid was also terminated after PID 21.
Whole-cell recording
Reprogrammed MSNs were transduced with pSynapsin-RFP and cocultured with human astrocytes. Whole-cell patch-clamp recordings were performed at 7–8 weeks after the initiation of neuronal reprogramming. Recordings were carried out in the extracellular solution (140 nM NaCl, 3 nM KCl, 10 nM glucose, 10 nM HEPES, 1.5 nM CaCl2 and 1 nM MgCl2; pH 7.3; 295 ~ 310 mOsM). Electrode pipettes were pulled from borosilicate glass (World Precision Instruments), and typically the resistance ranged between 5 and 8 MΩ filled with intracellular solution (125 nM K-Gluconate, 4 nM NaCl, 2 nM MgCl2, 1 nM EGTA, 10 nM HEPES, 4 nM Na2-ATP, 0.4 nM Na3-GTP, 5 nM Creatine phosphate; pH 7.5; 290–300 mOsM). Data were acquired using pCLAMP 10 software with a multiClamp 700B amplifier and Digidata 1550 digitizer (Molecular Devices). Liquid junction potential was calculated to be 15.2 mV and corrected in calculating resting membrane potential. In voltage-clamp mode, membrane potentials were typically kept at −70 mV, and the inward sodium currents (INa) and the outward potassium currents (IK) were recorded with voltage steps ranging from −20 mV to +80 mV. In current-clamp mode, action potentials were elicited by the injection of step currents that modulated membrane potential by holding at −70~80 mV, with step up from +10 mV to +100 mV.
Reduction-of-function testing of GeM-HD modifiers
To streamline the modifier gene identification, we adapted 96-well culture plates to be assayed for neuronal death using Sytox-Green as a cell death marker as previously described9 (approximately 3,000 cells counted in each well). For this assay, we used HD-MSNs from the GM04194 line (CAG repeat size 46; HD.46), which showed the twofold increase in cell death at around 50% compared with control MSNs from the healthy individual (GM02171). We carried out individual KD of 246 genes selected among 308 suggested modifier genes based on their expression level in the HD-MSN transcriptome9 (Supplementary Table 2). We performed reduction-of-function testing by adding lentivirus carrying gene-specific shRNAs to HD-MSNs at PID 21, a time-point when miRNA-induced cells start acquiring the neuronal identity8, to avoid interference with early stages of neuronal reprogramming. Cells were then cultured to reprogramming day 35 (PID 35), a timepoint when HD-MSNs start undergoing spontaneous neuronal death compared with control MSNs9, and the average cell death level for each gene KD was compared with the average level detected with shCtrl.
Sytox assay in live cells
First, 0.1 μM Sytox gene nucleic acid stain and 1 μl ml−1 Hoechst 33342 solution were added into the cell medium. Samples were incubated for at least 15 min at 37 °C before imaging. Images were taken using a Leica DMI 4000B inverted microscope with Leica Application Suite (LAS) Advanced Fluorescence.
Apoptosis assay in live cells
Cells were treated with 1 × Essen Bioscience IncuCyte Caspase-3/7 Green Reagent (final concentration 5 μM) and 1 × Essen Bioscience IncuCyte Annexin V Green or Red Reagent at PID 22 or 26. Image scheduling, collection and analysis were conducted with the IncuCyte S3 Live Cell Analysis System and IncuCyte S3 v.2017A software. Treated plates were imaged every 2 h for 7 d. At each time-point, over two images were taken per well in brightfield, FITC and TRITC channels. Images were analyzed for the number of green or red objects per well. For the apoptotic index, the number of green or red objects (that is, fluorescence cells) divided by phase area (μm2) per well was quantified by the IncuCyte S3 Live Cell Analysis System.
Immunostaining analysis
Cells were fixed with 4% paraformaldehyde for 20 min at room temperature and then permeabilized with PBS containing 0.2% Triton X-100 for 10 min at room temperature. Cells were then blocked with blocking buffer (5% BSA and 1% goat serum in PBS) for 1 h at room temperature. Primary antibodies were incubated in blocking buffer at 4 °C overnight. Cells were washed with PBS for 5 min three times and then incubated with secondary antibodies in blocking buffer for 1 h at room temperature. Cells were washed with PBS two times and incubated with DAPI for 10 min. Images were captured using a Leica SP5X white-light laser confocal system with LAS Advanced Fluorescence 2.7.3.9723.
Immunoblot analysis
Cells were lysed with RIPA buffer containing 1 × protease inhibitor/1 × phosphatase inhibitor or SDS buffer (2% SDS, 10% glycerol, 12.5 mM EDTA, 50 mM Tris-HCL pH 6.8). The concentrations of whole-cell lysates were measured using the Pierce BCA protein assay kit. Equal amounts of whole-cell lysates were resolved by SDS–PAGE and transferred to a nitrocellulose membrane (GE Healthcare Life Sciences, no. 10600006) using a transfer apparatus according to the manufacturer’s protocols (Bio-Rad). After incubation with blocking buffer (5% BSA, 0.1% Tween-20 in TBS (TBS-T)) for 1 h, the membrane was incubated with primary antibodies at 4 °C overnight. After washing with TBS-T three times for 5 min, the membrane was then incubated with a horseradish peroxidase-conjugated secondary antibody for 30 min at room temperature. The membrane was washed with TBS-T three times for 10 min and developed with the ECL system (Thermo Scientific, no. 34580) according to the manufacturer’s protocols. The quantification of immunoblot images from young and old groups was performed using ImageJ 1.52a. Unprocessed original images of immunoblots are shown in the Source data.
RNA preparation and quantitative PCR with reverse transcription
Total RNA was extracted using the RNeasy Micro Kit (Qiagen) and reverse transcription was performed using the SuperScript IV First Strand Synthesis System for RT–PCR (Invitrogen) according to the manufacturer’s protocol. Quantitative PCR was performed using SYBR Green PCR master mix (Applied Biosystems) and StepOnePlus Real-Time PCR system (Applied Biosystems, 4376600), according to the manufacturer’s protocol, against target genes. Quantitative PCR analysis was done with the following primers: RCAN1: 5′-TGGAGCTTCATTGACTGCGA-3′ and 5′-CTCAAATTTGGCCCGGCAC-3′; PPPC3A: 5′-GCGCATCTTATGAAGGAGGGA-3′ and 5′-TGACTGGCGCATCAATATCCA-3′; and GAPDH: 5’-ATGTTCGTCATGGGTGTGAA-3′ and 5′-TGTGGTCATGAGTCCTTCCA-3′.
Omni-ATAC-seq preparation
Omni-ATAC was performed as outlined by Corces et al.42. Briefly, each sample was treated with DNase for 30 min before collection. Approximately 50,000 cells were collected for library preparation. The transposition reaction was completed with Nextera Tn5 Transposase (Illumina Tagment DNA Enzyme and Buffer Kit, Illumina) for 30 min at 37 °C, and library fragments were amplified under optimal amplification conditions. Final libraries were purified by the DNA Clean & Concentrator 5 Kit (Zymo). Libraries were sequenced on an Illumina NovaSeq S4 XP (Genome Technology Access Center at Washington University in St. Louis).
ATAC-seq analysis
For ATAC-seq analysis in directly reprogrammed neurons, the raw data containing FASTQ files were uploaded to Partek Flow Software (Partek). ATAC-seq reads were aligned to the hg38 human genome assembly using BWA-0.7.17, and uniquely mapped reads were used for downstream analysis. Differential peaks were identified using Partek’s Gene Specific Analysis (10.0.20.1231) algorithm with a cut-off of FC ≥ 1.5 and FDR < 0.05 and were regarded as peaks gained or lost. Gained peaks in shCtrl-HD-MSNs were combined and defined as open (more accessible) chromatin regions. Conversely, all reduced peaks in shCtrl-HD-MSNs were defined as closed chromatin regions.
mCherry-GFP-LC3 quantification
FUW mCherry-GFP-LC3 was a gift from Anne Brunet (Addgene plasmid no. 110060; http://n2t.net/addgene:110060; RRID: Addgene_110060). The concentrated lentivirus of mCherry-GFP-LC3 was added to reprogrammed MSNs at PID 20. For imaging of cells expressing mCherry-GFP-LC3, cells were washed once with PBS, fixed and stained by anti-TUBB3 antibody at PID 26, after validation of the expression of GFP and mCherry by microscopy. Images were captured using a Leica SP5X white-light laser confocal system with LAS Advanced Fluorescence 2.7.3.9723. Pseudocolors have been adopted for the accessibility of colorblind readers.
Protein binding assay
The NanoBiT Protein:Protein Interaction System (Promega, no. N2014) was used for the binding assay of RCAN1–CaN interaction according to the manufacturer’s protocol. HEK293 cells plated in a 96-well plate were transfected with 25 ng (per well) of pBiT1.1-N-RCAN1 (89–197) and pBiT2.1-C-CaN (1–391) using polyethyleneimine (Polysciences, 24765) with Opti-MEM (Life Technologies, 31985). At 48 h after transfection, 25 μl of Nano-Glo Live Cell Assay reagent was added to each well after autophagy inducers were treated. After the initial measurement, luminescence values were measured every 30 min using the Synergy H1 Hybrid plate reader (BioTek).
Pathway enrichment analysis
The molecular pathways associated with DEGs identified in the transcriptome datasets and ATAC-seq datasets were analyzed using KEGG by Partek pathway analytical methods. The computational platform of the Partek pathway algorithm was used to identify the significant pathways for the DEGs according to the KEGG database. The pathways with P < 0.05 were selected as statistically significant pathways.
Statistics and reproducibility
Data are expressed as mean ± s.e.m. from at least three independent experiments unless otherwise indicated. Statistical comparisons were performed by a paired/unpaired t-test with a two-tailed distribution or one-way analysis of variance (ANOVA) with Tukey’s post hoc test using GraphPad Prism v.9.1. Statistical significance was set at P < 0.05, with the following standard abbreviations used to reference P values: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Detailed statistical information for each experiment is provided in the corresponding figure legend. For all quantified data, multiple cell cultures were counted from at least three biological replicates from multiple independent patient samples, as indicated in Supplementary Table 1. Studies were performed in a blinded manner and automated whenever possible with the aid of cell counting software tools and confirmation from multiple investigators with cell score quantification results as needed. For scoring phenotypes that required cell visualizations, at least three laboratory members counted coded cell samples in a blinded manner. For most of the cell death assays, we used the automated fluorescence measurement in the IncuCyte S3 Live Cell Analysis System with standardized procedures and used the data points generated by the analysis software. Data distribution was assumed to be normal, but this was not formally tested. Data in graphs are expressed as mean values and error bars represent s.e.m. unless noted otherwise. The center line in box-and-whisker plots denotes the median value, while the box contains the 25th to 75th percentiles of the dataset. The whiskers mark minimal to maximal values. No data were excluded from the analyses. Data collection was not randomized but was always done in parallel with controls and quantified in a blinded manner when necessary. Allocation of primary patient cells acquired from Coriell Biorepository into the ‘HD group’ was done randomly. Samples of the control group (healthy controls) were age-matched and sex-matched with HD samples and were acquired and available through Coriell Biorepository. Samples in the ‘pre-HD group’ were collected 13–17 years before the reported age of HD symptoms, ranging from 13 to 44 years of age, and sex-matched with HD samples from symptomatic patient samples. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to or larger than those reported in previous publications9,10. All data quantified for cellular phenotypes were from 3 to 4 independent healthy controls or patients with HD or presymptomatic HD individuals. For transcriptome analyses, fibroblasts and reprogrammed MSNs from longitudinally collected fibroblasts from three independent cognitively normal, healthy individuals were used. These fibroblasts were initially collected during middle age as ‘young’, and samples subsequently collected approximately 20 years later from three independent individuals were denoted as ‘old’ groups (Coriell NINDS and NIGMS Repositories: AG10049 (48 years) and AG16030 (68 years); AG10047 (53 years) and AG14048 (71 years); AG04456 (49 years) and AG14251 (68 years); Supplementary Table 1). All fibroblasts and reprogrammed MSN samples were used with at least two biological replicates per individual. For ATAC-seq analyses, shCtrl-, shRCAN1- and shCaN-expressing HD-MSNs from four independent patients with postsymptomatic HD were used with three biological replicates per individual (ND30013 (HD.43), ND33947 (HD.40), GM04198 (HD.47), GM04230 (HD.45); Supplementary Table 1). No sample size calculation was done, as the sample sizes used in the study were adequate to yield consistent differential gene expression and chromatin accessibilities with significant adjusted P values (<0.05).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Gene expression profiling in longitudinally collected fibroblasts and corresponding reprogrammed MSNs.
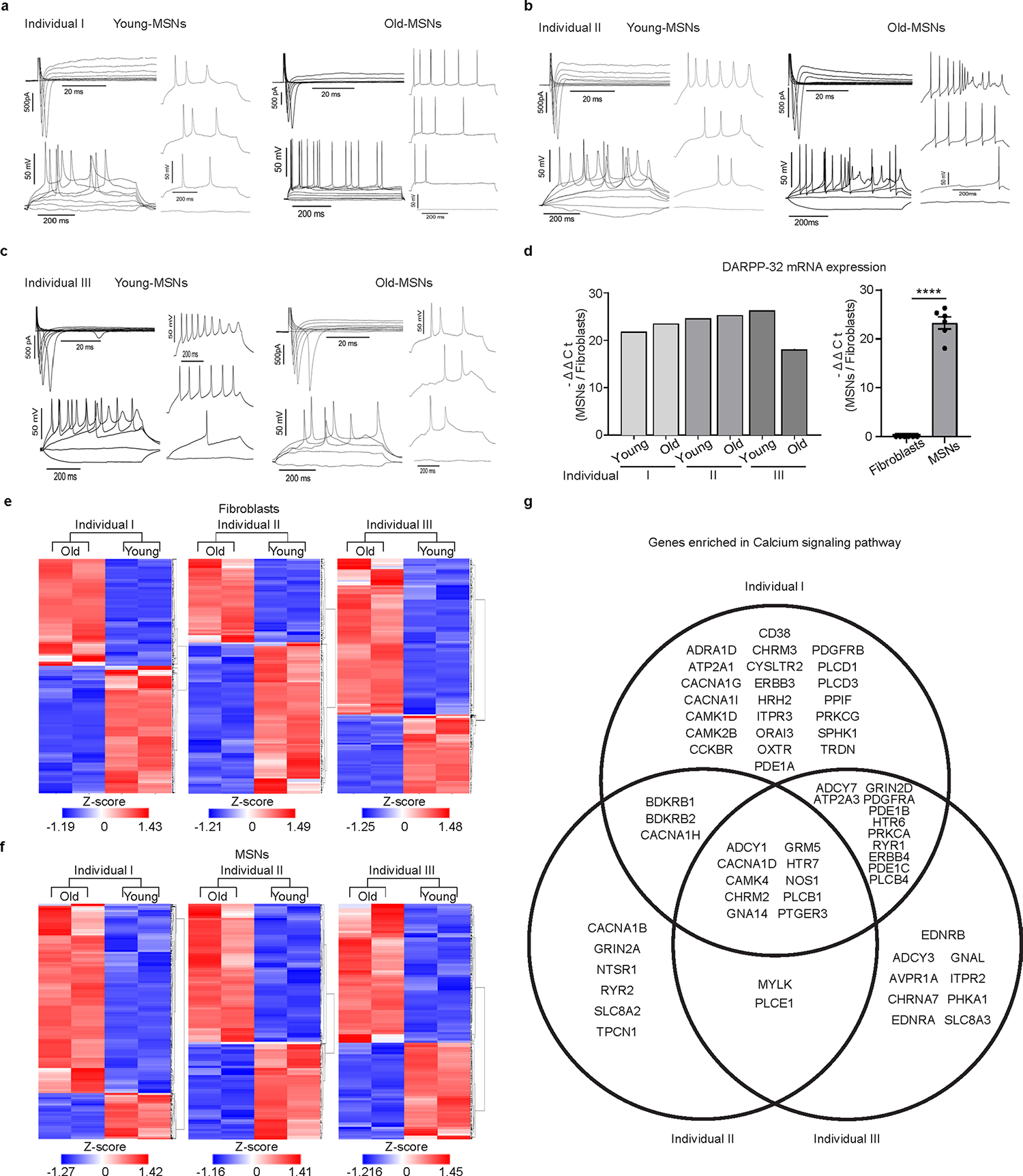
a-c, Whole-cell recording from reprogrammed MSNs from three independent longitudinal groups (individual I (a); II (b); III (c)) co-cultured with human astrocytes showing the inward/outward currents and multiple action potentials (APs). d, RT-qPCR analysis of DARPP-32 expression in longitudinally aged MSNs (n = 6, ****p < 0.0001, The sample size (n) corresponds to the number of biologically independent samples). Statistical significance was determined using two-tailed unpaired t-test and mean±s.e.m. e and f, Heatmap of Differentially Expressed Genes (DEGs) in fibroblasts (e) and MSNs (f) (FDR < 0.05, | FC | ≥ 1.5). g, Venn diagram of the genes enriched in calcium signaling pathway from old HD-MSNs.
Extended Data Fig. 2 |. Age-associated RCAN1 in longitudinally aged MSN.
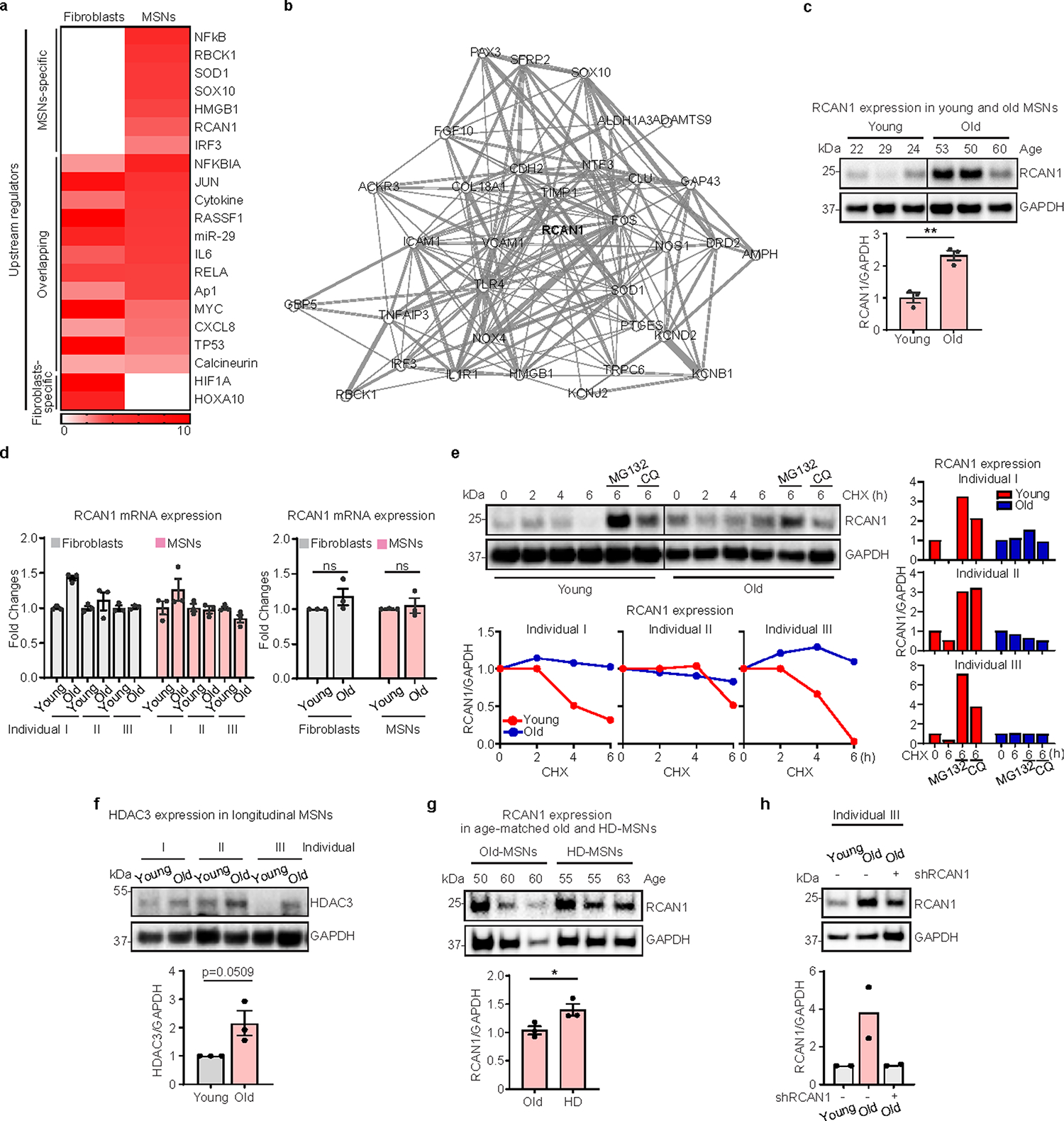
a, Upstream regulator analysis of up- or down-regulated genes in old fibroblasts and MSNs. b, Gene network of upstream regulators and DEGs. c, Representative immunoblotting (top) and quantification (bottom) of RCAN1 expression in six MSNs from 22, 29, 24 (young) and 53, 50, 60 (old) years old-individuals (young n = 3, old n = 3, **p = 0.0038). d, Quantification of RCAN1 mRNA from six independent fibroblasts and MSNs from three longitudinal individuals (I, II, and III) (Fibroblasts n = 12, MSNs n = 12). e, Representative immunoblotting (top, left) and quantification of relative RCAN1 expression normalized to GAPDH (bottom. left) in Young / Old-MSNs from three longitudinal individuals treated with cyclohexamide (CHX). Comparison of RCAN1 expression in CHX-treated MSNs (Young and Old) from three longitudinal individuals in the presence of DMSO, MG132 or, CQ (right) (n = 6). f. Representative immunoblotting (top) and quantification (bottom) of RCAN1 expression in age-matched control-Old-MSNs and HD-MSNs (n = 6). g. Representative Immunoblotting (top) and quantification (bottom) of HDAC3 expression in MSNs from three longitudinal individuals (n = 6, *p = 0.0467). h. Representative immunoblotting (top) and quantification (bottom) of RCAN1 expression in individual III’s MSNs (n = 2). Statistical significance was determined using two-tailed unpaired t-test (c,f,g) and one-way ANOVA with Tukey’s post hoc test (d,h). *p < 0.05, **p < 0.01, ns, not significant, and mean±s.e.m (c,d,f,g). The sample size (n) corresponds to the number of biologically independent samples (c-h).
Extended Data Fig. 3 |. Identification of modifier genes whose reduction protects HD-MSNs from degeneration.
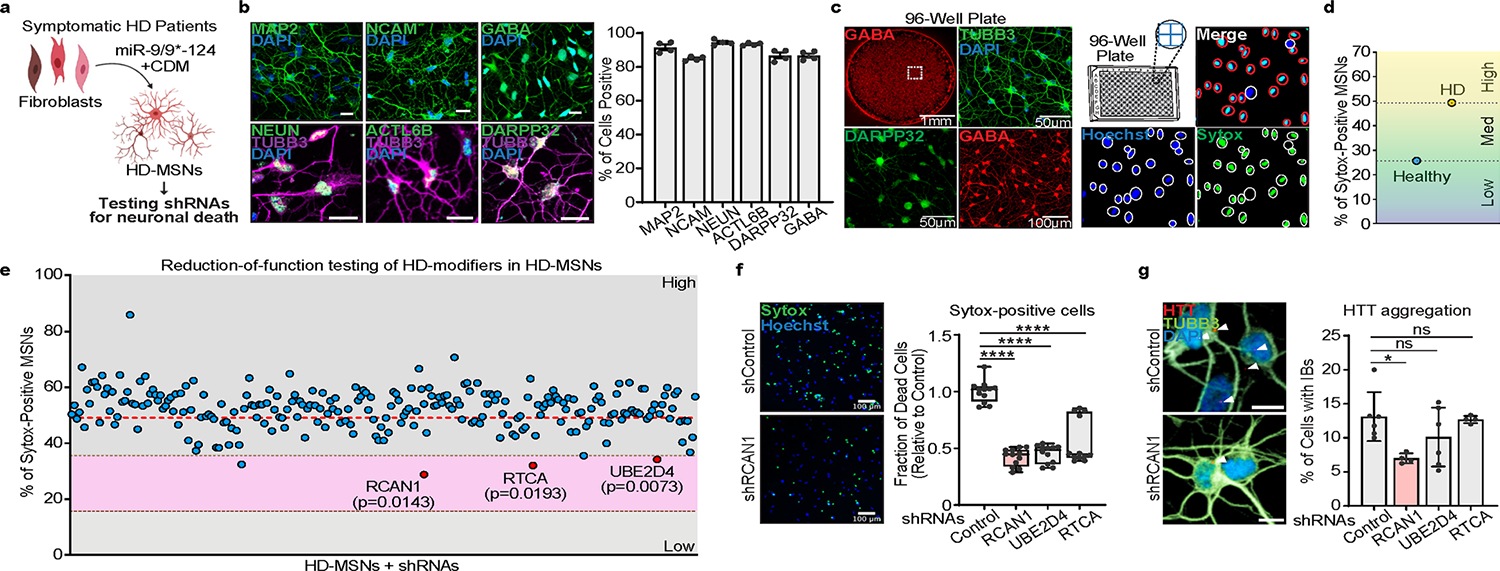
a, Experimental scheme of genetic modifiers testing in HD-MSNs. b, Representative images (left) and quantification (right) of MAP2-, NCAM-, NEUN-, ACTL6B-, DARPP-32-, and GABA-positive cells from four independent HD-MSNs (HD.43, HD.40, HD.47, HD.45). An average of 300 cells per each were counted from three or more randomly chosen fields (n = 4). Scale bars represent 20 μm. c, High-content imaging of Sytox green dye accumulation in HD-MSNs (HD.46) in a 96-well format. Representative images of HD-MSNs in each well of a 96-well plate, immunostained with anti-GABA, TUBB3, and DARPP-32 antibodies (left). Example pictures for high content image analysis to measure cell death levels (right): Hoechst for whole cell population and Sytox-green for dead cells. d, Quantification of Sytox-positive cells from HD-MSNs (HD.46) and healthy control (Ctrl.17) at post-induction day 35 (n = 2). e, Quantification of Sytox-positive cells in HD-MSNs (HD.46) transduced with shRNAs of modifier genes. The genes whose reduction significantly lowered cell death levels were marked (red) within the pink area (± 10 % of cell death level from healthy control) compared to control shRNA. Statistical significance was determined using unpaired t-test and mean±s.e.m (n = 2, RCAN1: p = 0.0143); RTCA: p = 0.0198); UBE2D4: p = 0.0073). f, Representative image (left) and quantification (right) of Sytox-positive cells from three independent HD-MSNs (HD.46, HD.44, HD.43) transduced with shRNAs of each gene (n = 12). Scale bars represent 100 μm. Box-and-whiskers plot: The center line denotes the median value while the box contains the 25th to 75th percentiles of dataset. The whiskers mark minimal value to maximal value. ****p < 0.0001. g, Representative image (left) and quantification (right) of cells with HTT inclusion bodies (IBs) in HD-MSNs (HD.40) transduced with shRNAs of each gene. Cells were immunostained with anti-HTT and TUBB3 antibodies. An average of 100 cells per each were counted from four to six randomly chosen fields (n = 6, 4, 6, 4). Scale bars represent 10μm. Statistical significance was determined using unpaired t-test (e) and one-way ANOVA with Tukey’s post-hoc test (f,g); ****p < 0.0001, *p < 0.05, ns, not significant, and mean±s.e.m. The sample size (n) corresponds to the number of biologically independent samples (b,d,e,f,g).
Extended Data Fig. 4 |. Validation of reprogrammed neurons of rescuing or non-rescuing condition for ATAC-sequencing.
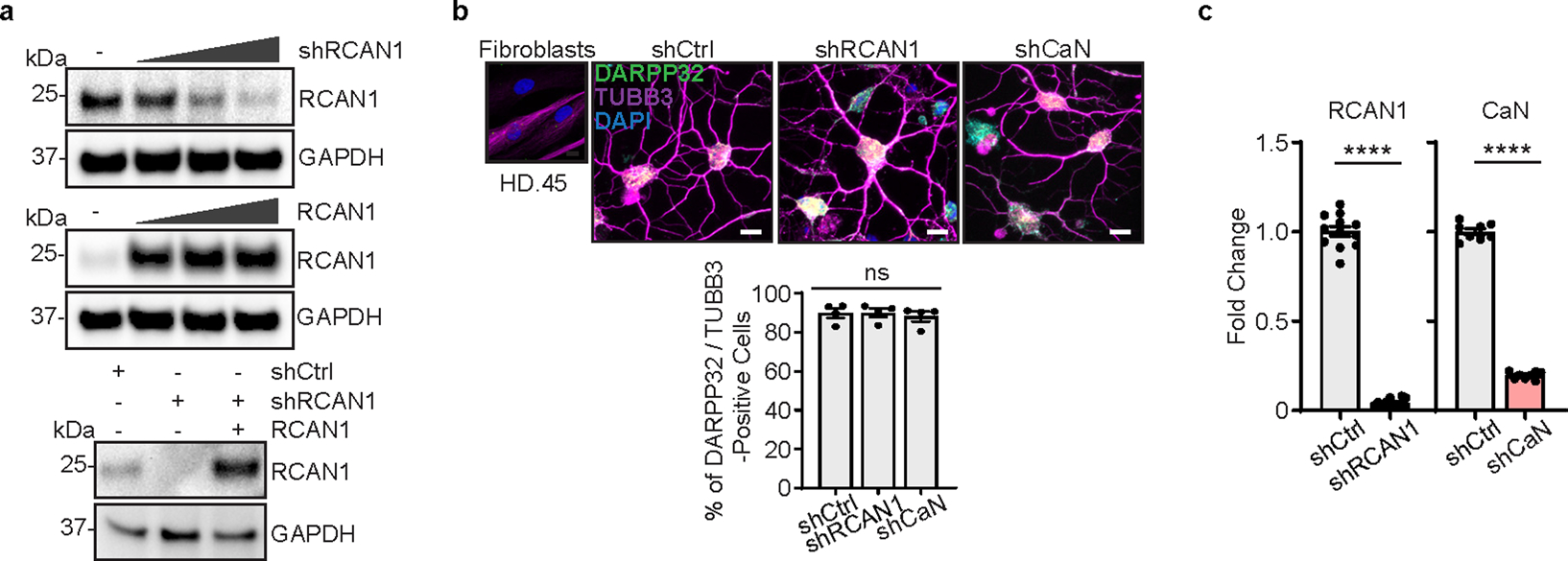
a, RCAN1 expression in fibroblasts transduced with shRCAN1 (top) or RCAN1 (middle) in a dose-dependent manner. RCAN1 expression in HD-MSNs (HD.43) transduced with shCtrl, shRCAN1, or RCAN1 (bottom). b, Representative image (top) and quantification (bottom) of DARPP-32-positive cells from four independent HD-MSNs transduced with shCtrl, shRCAN1, or shCaN (HD.43, HD.40, HD.47, HD.45). Cells were immunostained with anti-DARPP-32 and TUBB3 antibodies. An average of 183 cells of each were counted from three or more randomly chosen fields (n = 4). Scale bars represent 10 μM. c, RT-qPCR analysis of the expression of RCAN1 and CaN in (b) (n = 12, 12, 8, 8). Statistical significance was determined using one-way ANOVA with Tukey’s post-hoc test (b) and two-tailed unpaired t-test (c); ****p < 0.0001, ns, not significant, and mean±s.e.m (b,c). The sample size (n) corresponds to the number of biologically independent samples (b,c).
Extended Data Fig. 5 |. RCAN1 promotes nuclear localization of TFEB for HD survival.
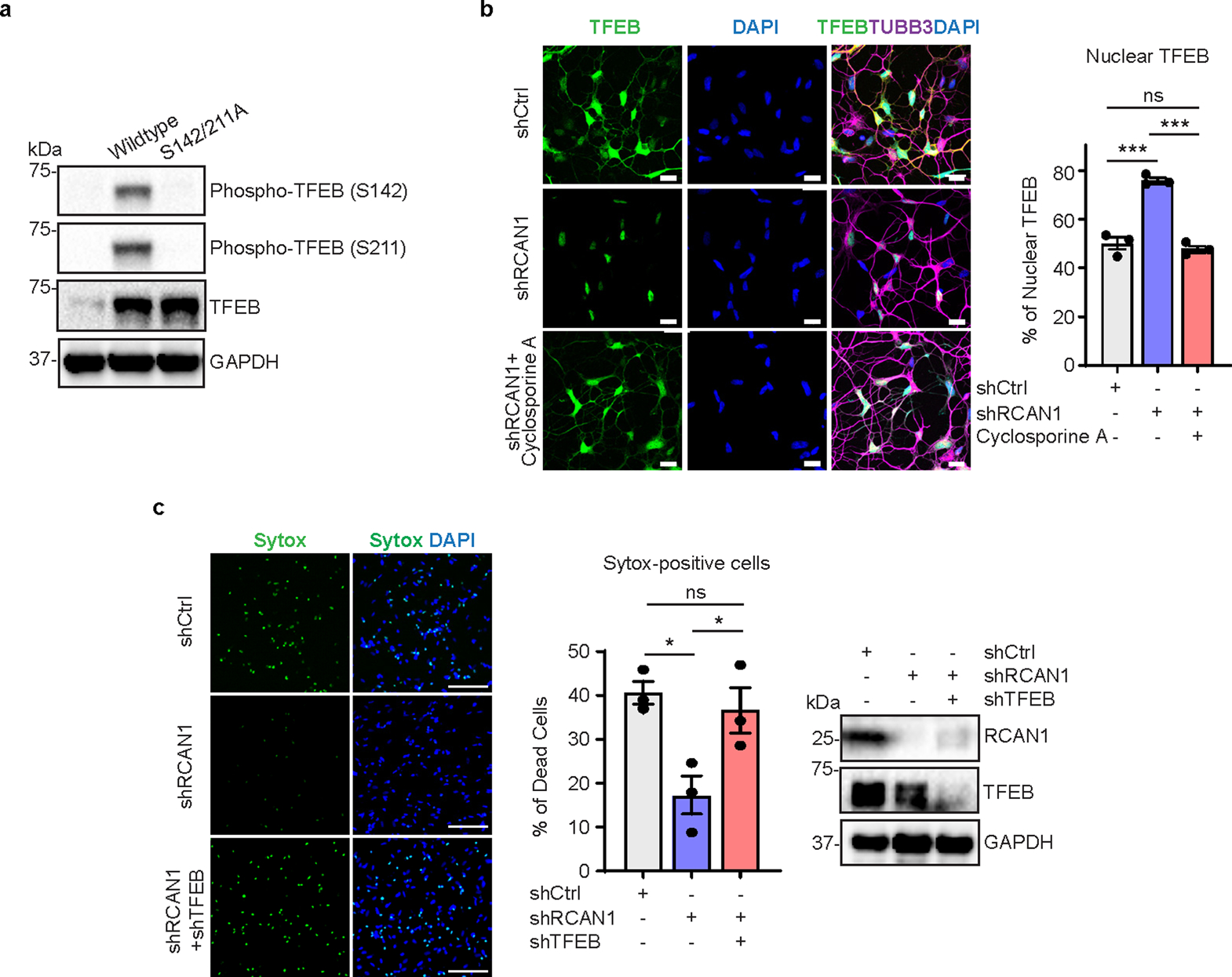
a, Expression of phosphor-TFEB in fibroblasts transduced with Control, TFEB wildtype, or phosphor-mutant (S142/211 A). b. Representative image (left) and quantification of nuclear TFEB from three independent HD-MSNs (HD.45, HD.45b, HD.47) transduced with shCtrl or shRCAN1. Cells were treated with DMSO or Cyclosporin A (CaN inhibitor) (n = 3). shCtrl versus shRCAN1 ***p = 0.0002, shRCAN1 versus shRCAN1+Cyclosporin A ***p = 0.0001. c, Representative image (left) and quantification of Sytox-positive cells (middle) from three independent HD-MSNs (HD.45, HD.45b, HD.47) transduced with shCtrl, shRCAN1, or shTFEB. Expression of RCAN1 and TFEB in HD-MSNs transduced with shCtrl, shRCAN1, or shTFEB (right) (n = 3). Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test (b,c); ***p < 0.001, *p < 0.05, ns, not significant, and mean±s.e.m (b,c). The sample size (n) corresponds to the number of biologically independent samples (b,c).
Extended Data Fig. 6 |. Neuroprotective role of G2–115 through reducing RCAN1-CaN interaction.
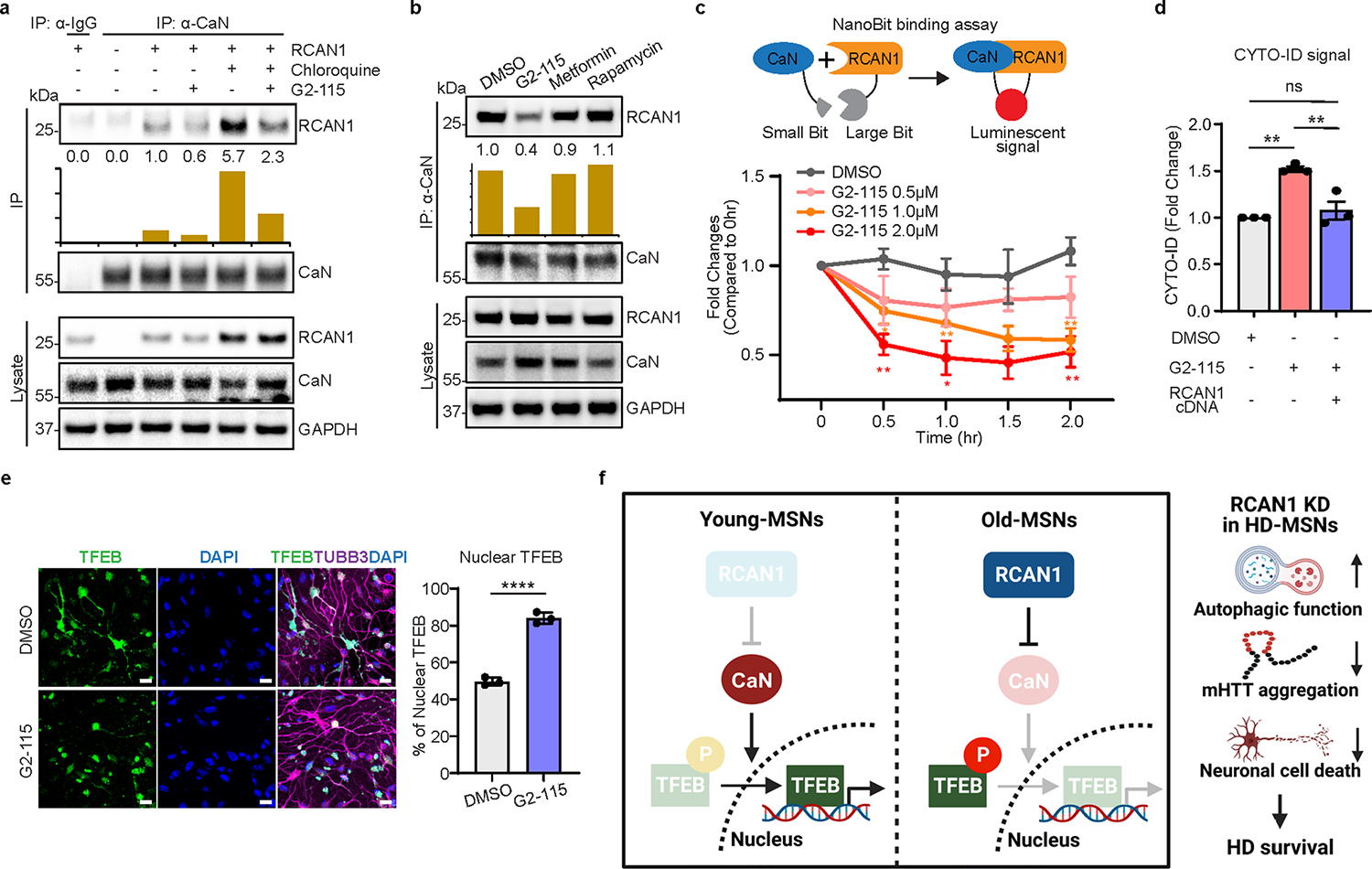
a, Immunoprecipitation analysis of RCAN1-transduced fibroblasts with anti-CaN antibody followed by immunoblotting with anti-RCAN1 antibody. Cells are treated with 0.5 μM of G2–115 and 60 μM of chloroquine (lysosome inhibitor). b, Immunoprecipitation analysis of RCAN1-transduced fibroblasts with anti-CaN followed by immunoblotting with anti-RCAN1 antibody. Cells were treated with DMSO or 0.5 μM of G2–115, 8 mM of metformin, and 500 nM of rapamycin. c, Experimental scheme of NanoBit binding assay (top). Binding assay of HEK293 cells transfected with RCAN1 fused to large Bit and CaN fused to small Bit. Cells were treated with 0.5, 1.0, 1.5, and 2.0 μM of G2–115 in a dose-dependent manner (bottom). (n = 3, The sample size (n) corresponds to the number of independent experiments). DMSO versus G2–115 1.0 μM 0.5 hr *p = 0.0379, 1.0 hr **p = 0.0091, 2.0 hr **p = 0.0082, DMSO versus G2–115 2.0 μM 0.5 hr **p = 0.0044, 1.0 hr *p = 0.0230, 2.0 hr **p = 0.0084. d, Quantification of CYTO-ID-positive cells from three independent HD-MSNs (HD.45, HD.45b, HD.47) treated with DMSO or G2–115. Cells were transduced with RCAN1 (n = 3). DMSO versus G2–115 **p = 0.0025, G2–115 versus G2–115 + RCAN1 cDNA **p = 0.0060. e, Representative image (left) and quantification (right) of nuclear TFEB from three-independent HD-MSNs (HD.45, HD.45b, HD.47) treated with DMSO or G2–115. Cells were immunostained with anti-TFEB and TUBB3 antibodies. An average of 107 cells per each were counted from three or more randomly chosen fields (n = 3, ****p < 0.0001). Scale bars represent 20 μm. f, Graphical work model to illustrate the function of RCAN1-CaN-TFEB cascade in Young/Old-MSNs (left) and the neuroprotective role of RCAN1 KD for HD survival. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test in (d) and two-tailed unpaired t-test (c,e); ****p < 0.0001, **p < 0.01, *p < 0.05, ns, not significant, and mean±s.e.m (c-e). The sample size (n) corresponds to the number of biologically independent samples (d,e).
Supplementary Material
Acknowledgements
We thank the Genome Technology Access Center at Washington University for deep-sequencing experiments. An image of the experimental scheme was created with BioRender.com. This study was supported by a Hereditary Disease Foundation (HDF) Grant, the Cure Alzheimer’s Fund (CAF), the CHDI Foundation Research Agreement, National Institute on Aging (NIA) Grant no. RF1AG056296, NIA grant no. R01AG078964, National Institute of Neurological Disorders and Stroke (NINDS) Grant no. R01NS107488, the Farrell Foundation Fund and the Mallinckrodt Scholar Award (A.S.Y.).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s43587-023-00538-3.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43587-023-00538-3.
Peer review information Nature Aging thanks Scott Zeitlin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
RNA-seq data and ATAC-seq data presented in the current study will be available through the Gene Expression Omnibus (GEO) at NCBI with accession IDs GSE241430 and GSE210996. Raw data that support all findings of the study are available as Source data and supplementary tables provided with this paper. All other data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Wyss-Coray T Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rose MR Adaptation, aging, and genomic information. Aging (Albany NY) 1, 444–450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Machiela E & Southwell AL Biological aging and the cellular pathogenesis of Huntington’s disease. J. Huntingtons Dis. 9, 115–128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niccoli T & Partridge L Ageing as a risk factor for disease. Curr. Biol. 22, R741–752 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Hou Y et al. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Huh CJ et al. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 10.7554/eLife.18648 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abernathy DG et al. MicroRNAs induce a permissive chromatin environment that enables neuronal subtype-specific reprogramming of adult human fibroblasts. Cell Stem Cell 21, 332–348.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cates K et al. Deconstructing stepwise fate conversion of human fibroblasts to neurons by microRNAs. Cell Stem Cell 28, 127–140. e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Victor MB et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 21, 341–352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh YM et al. Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy. Nat. Neurosci. 10.1038/s41593-022-01185-4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh YM, Lee SW & Yoo AS Modeling Huntington disease through microRNA-mediated neural reprogramming identifies age-associated autophagy dysfunction driving the onset of neurodegeneration. Autophagy 10.1080/15548627.2023.2175572 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klee CB, Crouch TH & Krinks MH Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc. Natl Acad. Sci. USA 76, 6270–6273 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rusnak F & Mertz P Calcineurin: form and function. Physiol. Rev. 80, 1483–1521 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Hoeffer CA et al. The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J. Neurosci. 27, 13161–13172 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan PG, Chen L, Nardone J & Rao A Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Li Y et al. The structure of the RCAN1:CN complex explains the inhibition of and substrate recruitment by calcineurin. Sci. Adv. 10.1126/sciadv.aba3681 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell AN et al. Brain expression of the calcineurin inhibitor RCAN1 (Adapt78). Arch. Biochem. Biophys. 467, 185–192 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Porta S, Marti E, de la Luna S & Arbones ML Differential expression of members of the RCAN family of calcineurin regulators suggests selective functions for these proteins in the brain. Eur. J. Neurosci. 26, 1213–1226 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Cook CN, Hejna MJ, Magnuson DJ & Lee JM Expression of calcipressin1, an inhibitor of the phosphatase calcineurin, is altered with aging and Alzheimer’s disease. J. Alzheimers Dis. 8, 63–73 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Fuentes JJ et al. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 9, 1681–1690 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Fuentes JJ et al. A new human gene from the Down syndrome critical region encodes a proline-rich protein highly expressed in fetal brain and heart. Hum. Mol. Genet. 4, 1935–1944 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Rothermel B et al. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J. Biol. Chem. 275, 8719–8725 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Ermak G, Morgan TE & Davies KJ Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer’s disease. J. Biol. Chem. 276, 38787–38794 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Harris CD, Ermak G & Davies KJ RCAN1–1L is overexpressed in neurons of Alzheimer’s disease patients. FEBS J. 274, 1715–1724 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Sardiello M et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Settembre C et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Settembre C et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15, 647–658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded α1-antitrypsin Z. PLoS ONE 14, e0209748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Victor MB et al. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 84, 311–323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoo AS et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han KA et al. Histone deacetylase 3 promotes RCAN1 stability and nuclear translocation. PLoS ONE 9, e105416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noh EH et al. Covalent NEDD8 conjugation increases RCAN1 protein stability and potentiates its inhibitory action on calcineurin. PLoS ONE 7, e48315 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudilot A, Trillaud-Doppia E & Boehm J RCAN1 regulates bidirectional synaptic plasticity. Curr. Biol. 30, 1167–1176 e1162 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Genetic Modifiers of Huntington’s Disease, C. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hickey MA & Chesselet MF Apoptosis in Huntington’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 255–265 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Khan S et al. Implication of caspase-3 as a common therapeutic target for multineurodegenerative disorders and its inhibition using nonpeptidyl natural compounds. BioMed Res. Int. 2015, 379817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ona VO et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature 399, 263–267 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Portera-Cailliau C, Hedreen JC, Price DL & Koliatsos VE Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J. Neurosci. 15, 3775–3787 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soles-Tarres I et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) protects striatal cells and improves motor function in Huntington’s disease models: role of PAC1 receptor. Front. Pharmacol. 12, 797541 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganz J et al. A novel specific PERK activator reduces toxicity and extends survival in Huntington’s disease models. Sci. Rep. 10, 6875 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leitman J et al. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS ONE 9, e90803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox LM et al. Huntington’s disease pathogenesis is modified in vivo by Alfy/Wdfy3 and selective macroautophagy. Neuron 105, 813–821.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashkenazi A et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azzi JR, Sayegh MH & Mallat SG Calcineurin inhibitors: 40 years later, can’t live without. J. Immunol. 191, 5785–5791 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Medina DL et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brun M, Glubrecht DD, Baksh S & Godbout R Calcineurin regulates nuclear factor I dephosphorylation and activity in malignant glioma cell lines. J. Biol. Chem. 288, 24104–24115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jain J et al. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature 365, 352–355 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Shalizi A et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311, 1012–1017 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Li H, Rao A & Hogan PG Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 21, 91–103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang CC et al. Calcineurin-mediated dephosphorylation of c-Jun Ser-243 is required for c-Jun protein stability and cell transformation. Oncogene 27, 2422–2429 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Fornes O et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li L et al. RB1CC1-enhanced autophagy facilitates PSCs activation and pancreatic fibrogenesis in chronic pancreatitis. Cell Death Dis. 9, 952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao J et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 11, 939–953 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhen X, Uryu K, Cai G, Johnson GP & Friedman E Age-associated impairment in brain MAPK signal pathways and the effect of caloric restriction in Fischer 344 rats. J. Gerontol. A Biol. Sci. Med. Sci. 54, B539–548 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Liu Y et al. Age-related decline in mitogen-activated protein kinase activity in epidermal growth factor-stimulated rat hepatocytes. J. Biol. Chem. 271, 3604–3607 (1996). [PubMed] [Google Scholar]
- 57.Pyo JO et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4, 2300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Melendez A et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Jia K & Levine B Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3, 597–599 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Lapierre LR et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 4, 2267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen M et al. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4, e24 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakamura S et al. Mondo complexes regulate TFEB via TOR inhibition to promote longevity in response to gonadal signals. Nat. Commun. 7, 10944 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Settembre C et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roczniak-Ferguson A et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5, ra42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martina JA, Chen Y, Gucek M & Puertollano R MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoshii SR & Mizushima N Monitoring and measuring autophagy. Int. J. Mol. Sci. 10.3390/ijms18091865 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leeman DS et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pankiv S et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Hidvegi T et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science 329, 229–232 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Thellung S, Corsaro A, Nizzari M, Barbieri F & Florio T Autophagy activator drugs: a new opportunity in neuroprotection from misfolded protein toxicity. Int. J. Mol. Sci. 10.3390/ijms20040901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hudry E et al. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 3176–3192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bechstein WO Neurotoxicity of calcineurin inhibitors: impact and clinical management. Transpl. Int. 13, 313–326 (2000). [DOI] [PubMed] [Google Scholar]
- 73.Lee S et al. The calcineurin inhibitor Sarah (Nebula) exacerbates Aβ42 phenotypes in a Drosophila model of Alzheimer’s disease. Dis. Model. Mech. 9, 295–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Richner M, Victor MB, Liu Y, Abernathy D & Yoo AS MicroRNA-based conversion of human fibroblasts into striatal medium spiny neurons. Nat. Protoc. 10, 1543–1555 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Lu YL & Yoo AS Mechanistic insights into microRNA-induced neuronal reprogramming of human adult fibroblasts. Front. Neurosci. 12, 522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCoy MJ et al. LONGO: an R package for interactive gene length dependent analysis for neuronal identity. Bioinformatics 34, i422–i428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Church VA et al. Generation of human neurons by microRNA-mediated direct conversion of dermal fibroblasts. Methods Mol. Biol. 2239, 77–100 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data and ATAC-seq data presented in the current study will be available through the Gene Expression Omnibus (GEO) at NCBI with accession IDs GSE241430 and GSE210996. Raw data that support all findings of the study are available as Source data and supplementary tables provided with this paper. All other data that support the findings of this study are available from the corresponding author upon reasonable request.