

Abstract
The growing advances in spatial transcriptomics (ST) stand as the new frontier bringing unprecedented influences in the realm of translational oncology. This has triggered systemic experimental design, analytical scope, and depth alongside with thorough bioinformatics approaches being constantly developed in the last few years. However, harnessing the power of spatial biology and streamlining an array of ST tools to achieve designated research goals are fundamental and require real‐world experiences. We present a systemic review by updating the technical scope of ST across different principal basis in a timeline manner hinting on the generally adopted ST techniques used within the community. We also review the current progress of bioinformatic tools and propose in a pipelined workflow with a toolbox available for ST data exploration. With particular interests in tumor microenvironment where ST is being broadly utilized, we summarize the up‐to‐date progress made via ST‐based technologies by narrating studies categorized into either mechanistic elucidation or biomarker profiling (translational oncology) across multiple cancer types and their ways of deploying the research through ST. This updated review offers as a guidance with forward‐looking viewpoints endorsed by many high‐resolution ST tools being utilized to disentangle biological questions that may lead to clinical significance in the future.
Keywords: biomarker profiling, mechanism elucidation, oncology, single‐cell resolution, spatial transcriptomics, tumor microenvironment
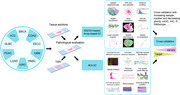
Proposed biomarker development pipeline using spatial transcriptome. Schematic illustration of biomarker profiling workflow based on ST. Cancer types are listed on the left for demonstrative purposes (see abbreviations). Sampling strategies are divided into full tissue sections and tissue microarrays (TMA) depending on ST technologies as exploratory tools (ISS/ISH‐ST, array‐based ST, and ROI‐ST). Bioinformatic explorations (listed in boxes) are grouped accordingly to different ST technologies being applied. Downstream validation is for demonstration but involves increasing number of samples and decreasing number of targets. Abbreviations: BRCA, breast carcinoma; COAD, colon adenocarcinoma; ESSC, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal cancer; LUAD, lung adenocarcinoma; PDAC, pancreatic ductal carcinoma; DLBC, lymphoid diffuse large B‐cell lymphoma; HCC, hepatocellular carcinoma.
1. INTRODUCTION
Influenced by the wide‐spread application of single‐cell transcriptomics (mainly via single‐cell RNA sequence, scRNA‐seq), understanding the cellular dynamics within the complex tissue microenvironment is not limited to decipher each cell's identity via its “omics” profiles but goes further to put them under a histological context. In various research settings—including organ development, embryonic morphogenesis, cognitive science (such as brain theory and neuroscience), and pathologies (including infection‐related pathogenesis, neurodegenerative diseases, and oncogenesis)—the three‐dimensional (3D) interplay of cells dictates biological functions.
In tissues, multiple cellular niches play significant roles such as intercellular modulation via direct contact or short‐ranged paracrine that reprograms biological processes or chemical/molecular dispersion under organ‐specific conditions in a patterned manner. As a result, the spatially compiled multiparameter data with high resolution become useful resources to open up a new field in many biology settings and same as the case in tumor biology. Since the application of ST grew exponentially in the past 3 years and true single‐cell ST methods were gradually becoming available, to update their progress, adaptation of bioinformatics tools in real practice and application frontiers in tumor biology, we provide this systemic review.
Here, we propose the rationale of applying spatial transcriptomics (ST), the building blocks of spatial biology for hypothesis‐free discovery and summarize the historical development path of various ST technologies on their relevant ground basis. With particular interests in tissue biology including cancer, we review the major bioinformatics tools being adopted in the past few years and present an analytical schema compatible with different ST approaches. We also highlight major ST works contributing toward tumor biology and translational oncology wherein spatial single‐cell resolved ST is specifically emphasized. These altogether shed light on future direction and application of ST on a multilayered perspective.
2. THE APPLICATION RATIONALE OF ST IN TUMOR BIOLOGY
Tissues, composed of millions of cells and other cellular functional units, are indispensable resources to gain biological insights under various pathophysiological settings. In tumor biology, pathological evaluation offers as a gold‐standard approach throughout decades and therefore becomes the “central dogma” for clinical decision making. 1 In‐depth characterization of tissues essentially by compiling multilayers of molecular information now goes much beyond conventional genetics/epigenetics, transcriptomics, proteomics, and metabolomics treating tissues as a whole but rather can be conducted in a decomposed manner at subhistological or individual cell dimensions. Through this magnifying lens, multiomics profiling can be revisited and potentially redefine our comprehension toward disease conditions making novel breakthroughs in translational oncology. Prior to this, scRNA‐seq together with other single‐cell omics (including multiplex single‐cell proteomics and single‐cell analysis of accessible chromatin) has already pioneered in this field but rapid development and maturation of ST further enables in situ profiling of dynamic changes at the transcriptome‐wide scale highlighting the prerequisite of adding locational information in exploring histological specimens. 2 In the following section, we give a holistic overview of various ST technologies categorized by their base principles and technical strength with a particular emphasis on those that are already available in general research communities. The original technical papers and review summaries are referable in related publications and will not be elaborated here. 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 (Figure 1)
FIGURE 1.
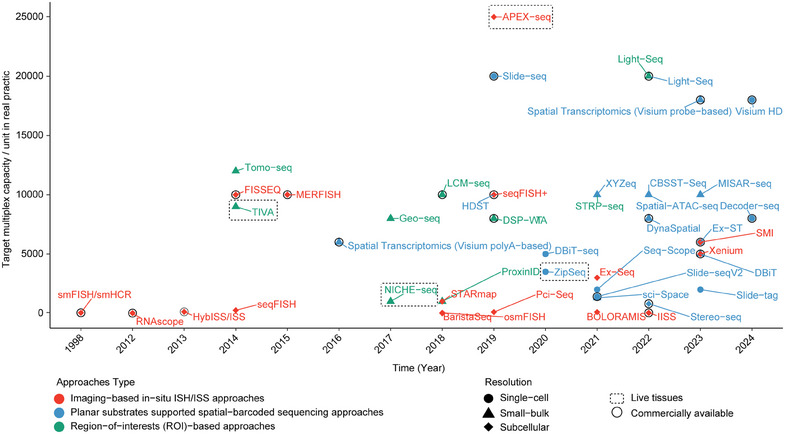
The development roadmap of spatial transcriptomics (ST). Individual ST technologies are presented in a timeline graph between 1998 and 2024 (current). Colors annotate different categories based on their analytical principles including imaging‐based in situ ISH/ISS (red) and planar substrates supported spatial‐barcoded sequencing (blue). From application perspective, other regions‐of‐interest (ROI) based technologies (light‐guided and image‐free) are grouped in green color. Spatial resolutions are presented in different shapes (triangle: small bulk, circle: single‐cell, diamond: subcellular). Black circles indicate technologies that are commercially available. Dashed rectangles indicate application scenario (live tissues). Horizontal axis shows the multiplexity of technologies. Note: data summarized may not include all relevant technologies and target plexity is a rough estimate based on references.
3. THE ONCOMING ERA OF TECHNICAL AND ANALYTICAL DEVELOPMENT IN ST
3.1. Upstream technology development and current progress
The development paths of various ST can generally be traced back according to their detection principles, which either reply upon conventional next‐generation sequencing (NGS), or in situ hybridization (ISH)/in situ sequencing (ISS), the latter of which share similar principle in signal readouts but differ in their intermediate procedures. From the application aspect, since a subset of technologies requires laser‐captured microdissection (LCM) such as geographical position sequencing (Geo‐seq) and ProximID 10 , 11 or photocleavable linkers to extract part of tissue materials/information to generate NGS‐based transcriptomic profiles (transcriptome in vivo analysis: TIVA, NICHE‐seq, Light‐seq, and GeoMx digital spatial profiler whole transcriptome analysis: DSP‐WTA) 12 , 13 , 14 , 15 or rather employs image‐free spatial information reconstruction approaches (tomo‐seq and STRP‐seq) that are often used under specific settings, we group these technologies in a category termed region‐of‐interests (ROI)‐based spatial approach. 6 , 16 , 17
Though a common feature of those technologies is their limited resolution at a subhistological level that often requires at least a few hundred of cells for downstream profiling, due to their high‐plex potential capable of covering the entire transcriptome, some (such as commercialized GeoMx DSP, nanoString, WA) have already been widely adopted and will be discussed in the below section.
3.1.1. A history of imaging‐based ST technologies
Historically, imaging‐based ST has long track histories and can date back when single RNA molecules could be detected and visualized using conventional fluorescent in situ hybridization (FISH) or single‐molecule FISH (smFISH) at single‐cell resolution and was more recently developed by Nilsson's laboratory using padlock probe‐mediated transcript hybridization followed by rolling‐circle amplification (RCA) for signal amplification and visualization. 18 , 19 , 20 This was subsequently combined with cyclic fluorescence and imaging, a process called in situ serial decoding to allow multiplex target detection. 20 To facilitate unbiased RNA characterization, Church's laboratory applied di‐nucleotide‐specific fluorescent oligonucleotides to generate a codebook that directly ligates with adapter primers targeting the paired anchoring sequences predesigned within the RCA product complex (ISS‐based). 21 , 22 This technique, referred to as fluorescence in situ sequencing (FISSEQ), was initially designed to detect RNA molecules in an un‐targeted way and later, this RCA process was more frequently used as an efficient strategy for in situ signal amplification with or without introducing target‐specific padlock oligonucleotide structures to detect localized RNA transcripts in a targeted manner. Owing to the superior signal‐to‐noise ratio, the RCA‐mediated in situ amplification technologies have vastly revolutionized in the past decade with similar methods being developed such as HybISS/ISS, STARmap, BaristaSeq, Ex‐Seq, and BOLORAMIS with multiplexity ranging from 30 to over 10,000. 21 , 23 , 24 , 25 , 26 Recently, the technical advancement also made the commercial form available, which is now being used to address biological questions under various settings including cancer (Xenium in situ; 10X Genomics, CA). 27 , 28 , 29
An alternatively imaging approach to increase detection sensitivity was also introduced following the invention of a novel RNA ISH method: RNA Scope that utilizes paired target RNA‐specific oligonucleotide probes (18‐base each to form a double Z structure) as the docking point to allow overhanging preamplifer oligonucleotides to bind and the free‐floating overhangs contain multiple fluorescence‐oligo hybridization sites allowing signal amplification for visualization. 30 Similarly, to resolve high‐plex gene profiling, cyclic hybridization is also necessary for producing fluorescence‐based sequential codes. Using this concept, higher plexity can be reached with different analytical scopes and those are exemplified by low target throughput (less than 100) techniques such as smHCR, osmFISH, median throughput methods (100–1,000) such as sequential FISH (seqFISH), and high‐throughput techniques (over 10,000) such as seqFISH+ (an upgrade version of seqFISH) and multiplexed error‐robust FISH (MERFISH). 6 , 8 , 31 , 32 , 33 , 34 , 35 Theoretically, by applying multiple target‐specific fluorescence‐conjugated DNA probes in each hybridization round, signals can be successfully detected and quantitatively resolved using specific decoding strategies. In brief, the latest high‐throughput versions such as seqFISH+ and MERFISH use target‐specific saddle probe sets as landing points to capture fluorescence‐labeled readout probes generating exquisitely designed coding schemas via binary 69‐bit harming distance 4 or combinatory pseudocolor readout sequences. 8 , 34 , 36 With super‐resolved imaging systems, tens of thousands of transcripts can be successfully discerned and quantified and these prototypes are now invented commercially as MERSCOPE (Vizgen, MA) and seqFISH (Spatial Genomics, CA), respectively. Other successful counterparts with various detection capacities such as spatial molecular imager (SMI CosMX, nanoString, WA), Molecular Cartography (Resolve Biosciences, GmbH), Rebus Esper (Rebus Biosystems, CA), and SEERNA ISS (Dynamic Biosystems, Suzhou) also started to appear at the application frontier. 2 , 37 , 38 , 39 A very important application advantage shared across these imaging‐based ST platforms is their capability typically reaching at single cell or even subcellular resolution with analytes (tissues or cells) being unaffected, an approach amenable for parallel pathology examination and downstream multiplex staining.
3.1.2. Spatial barcode‐based ST technologies
Due to the broader application of NGS, recently, spatial barcoding‐based technologies also open a new avenue in the spatial biology field and in particular this was predominantly boosted by the development and application of ST developed by Lundeberg's laboratory, a technology later transformed into its commercial version as Visium ST (10X Genomics). 40 Essentially, the earlier versions of ST and many other successors label planarly indexed x–y units or use single‐cell label indentation (spot arrays or microbeads, DNA nanoballs, microfluidic chambers, or other microscale molecules) with predefined oligo sequences per x–y/analytical unit to create spatial oligo barcodes allowing transcript profiles to project into designated space. 3 , 41 , 42 , 43 , 44 , 45 , 46 , 47 In fact, many of these STs employ oligo (dT) primers aligned with unique molecular identifier together with spatial barcode oligos and use the classic polyA capturing method for gene identification and quantification at per spatial coordinate. Since these ST techniques require polyA capture, fresh frozen (FF) samples are primarily used. More recently, to extend the analytical scope and sample compatibility, probe‐based STs were developed such as the upgraded versions of Visium ST including Visium V2 and Visium HD (10X Genomics). Apart from their downstream workflow being nearly identical, the major technical difference lies in its upstream design of genes‐specific DNA oligos that hybridize to complementary RNA in situ and flanking ployA sequence for probe capture followed by library construction and sequencing. Having such an advantage, designing transcriptome‐wide probe sets are becoming feasible.
For ST, despite the analytical resolution spanning from 0.5 to 100 µm per spatial unit, the major challenge remains as to leverage the spatial resolution and transcripts being detected. The major advantage of those technologies is that they do not require extra instrumentation and can be standardized in a typical bench‐side workflow. Therefore, many have been commercialized and wide‐spread across research areas such as neuroscience, development biology and disease pathogenesis including cancer. 48 , 49 , 50 , 51 Some technologies translating into commercial products include Visium/Visium HD based on ST and HDST (10X Genomics), 40 Curio Seeker based on Slide‐seq (Curio Bioscience, CA), 42 , 52 STOmics based on Stereo‐seq (BGI, Shenzhen), 47 DBiT‐seq based on DBiT (AtlasXomics, CT), 46 Dynaspatial based on Decoder‐seq (Dynamic Biosystems), 53 and BMKMANU S1000 (BMKgene, Qingdao). 54 Technology‐wise, since most of these technologies are based on direct sequencing of reverse‐transcribed (RT) oligo products, they are compatible for exploratory studies in an unbiased manner and as for resolution, a bunch of them have already reached near single‐cell or subcellular level with multiplexity roughly from few hundred to over 10,000. 2 , 6 , 41 , 55 More recently, head‐to‐head comparison was conducted aligning 11 sequencing‐based ST methods to assess their transcript capture efficiency, feature gene detection sensitivity and molecular diffusion. 56 Within those, considering the resolution as the major deterministic factor, Stereo‐seq, Slide‐seq V2, Visium V2, and DynaSpatial outperformed others with regard to their capturing efficiency. 56
3.1.3. ROI‐based ST technologies
ROI‐based ST often requires NGS sequencing as data output and was primarily introduced when specific tissue regions could be collected using LCM in low plex manner (few targets detected via polymerase chain reaction PCR). 57 Comprehensive coverage of transcriptome was later achieved and some were compatible with single‐cell resolution such as LCM‐seq, Geo‐seq, and ProximID. 10 , 11 , 58 Instead of sectioning tissues, the alternative approach to detect transcripts with in predefined regions was to employ light‐assisted methods to select ROI. Such include NICHE‐seq that uses photoactivatable fluorescent proteins to allow visual inspection and selection of tissue regions 13 and others such as Light‐seq, which utilizes light‐directed spatial barcode indentation through photo‐crosslinking at target tissue regions being analyzed via ex situ NGS. 15 Recently, digital spatial profiling (DSP) was developed and broadly propagated within the community and this technique employs photocleavable linkers and micrometer‐sized digital mirror device for ROI selection coupled with downstream NGS profiling. Due to its probe‐based detection theory via ISH, it is widely adaptable in formalin fixed paraffin‐embedded (FFPE) samples and more advantageously compatible with RNA–protein coprofiling. 14 Though limited by their spatial resolution typically ranging from 10 to hundreds of cells to start with, these STs are mainly pathologically informed, an important preanalytical factor to be considered.
3.2. Application consideration using different ST approaches
Overall, these ROI‐based ST together with aforementioned ISS/ISH and spatial‐barcoded ST have been gradually reshaping our understanding toward tissue molecular biology. In real practice, as for spatial‐barcoded ST, since they are kit‐based and often do not rely on heavy instrumentation, a few of them have been rapidly propagated and used across research fields (Figure 1). Also noted is that although ST technologies developed through different routes are versatile, imaging‐based ST having the longest track‐history, remains as a generally adopted ST approach and even in the high‐plex ST application field, some are already proven to be robust and becoming technically feasible (Figure 1). Another key advantage of image ST is within its wide compatibility for clinically archived FFPE samples, an analytical hinderance for FF samples. Nowadays, even some of spatial barcoded ST are emerging to be FFPE‐compatible (Visium probe‐based and Visium HD) 59 image ST is still the mainstay for FFPE‐based application. However, recent commercialization of spatial‐barcoded ST (probe‐based) may accelerate in short time. Of note, despite their spatial resolution, most probe‐based ST (mainly include image‐based and some ROI‐based ST) are suited for human and mouse exclusively, a species‐limiting factor to be taken into account, especially the ISS/ISH‐based methods that often employ probe‐based detection. For these single‐cell or even subcellularly resolved ST, comparative analysis was already conducted. These include head‐to‐head comparison between Xenium in situ, Merscope and SMI on FFPE samples and additional Molecular Cartography and HiPlex RNA Scope on a set of FF samples. 60 , 61 Despite data being preliminary, considering detection sensitivity (readout detectability), false discovery dates (noise control) and cell type imputation being the key analytical parameters, Merscope and Xenium in situ under many tested scenarios may have more potentials for broader application and have been gradually manifested by others. 62
Since median gene detection capability at per spatial unit is a major concern in ST, this has been the battlefield for many benchmarking studies. In real‐world practice, one technical challenge is to retain gene features to be detected when increasing analytical resolution (from few hundred micrometers down to 0.5–10 µm in size). Based on the whole transcriptome analysis covering over 18,000 genes, median gene features per spatially indexed unit can typically vary from 8,000 to 10,000 genes per analytical units for ROI‐based ST (in case of DSP with ROI diameters of 50–500 µm), 1000–3000 genes for spatial‐barcoded ST (in case of Visium polyA‐based with spot size of 55 µm) and 10 to a few hundred for spatial‐barcoded ST (in case of Stereo‐seq and Visium HD with 0.5–2 µm per bin) in resolution. Technically, a word of warning is that at near single‐cell resolution, challenges still remain as to characterize over 1000 genes per analytical unit, a consensus quality control (QC) measure widely adopted in scRNA‐seq nowadays. To fill these gaps, a body of computational tools have been developed and will be discussed in below sections.
4. HARNESSING THE EVOLVING BIOINFORMATIC TOOLBOX GROWN OUT FROM ST
4.1. A brief walkthrough of analysis in ST
The multifaceted technical progress in ST makes bioinformatic tools demanding and is being actively developed. Generally, for ST technologies that usually incorporate thousands of indexed spatial units (primarily led spatial‐barcoded ST such as Visium polyA and Stereo‐seq), a typical spatial data analysis procedure may involve raw data processing/QC, sample integration and normalization across genes/samples, dimension reduction/clustering, and cell annotation. On this basis, exploratory work can be carried out including spatial variable gene (SVG) identification, data deconvolution/mapping, gene expression imputation, cell–cell/gene–gene interaction, dynamic analysis (trajectory/RNA velocity), and other complex spatial analysis such as highly ordered cellular neighborhoods (CN), spatial context profiling, or spatial copy number inferring (applicable mainly on ST with single‐cell resolution). 2 , 4 , 63 , 64 , 65 , 66 , 67 , 68 Since the data format derived from ROI‐based ST contain sufficient reads per gene in individual ROIs that mimics large‐scale expression data, they apply another route following conventional high‐throughput analysis used in RNA‐seq incorporating methods such as dimension reduction/clustering, variable gene identification, and pathway enrichment (usually based on Limma or DEseq2, GO and KEGG), unbiased expression module identification (GSEA/GSVA, weighted correlation network analysis WGCNA or other machine learning approaches). Another commonly used approach in ROI‐based ST is spatial deconvolution and many methods have been used such as conventional ssGSEA developed for bulk RNA‐seq analysis.
4.2. The toolbox and application of bioinformatics in ST
Regardless of diversified data formats, gene detection efficiencies and spatial resolution derived from various ST platforms, in particular those from discrete ROI‐based ST technologies, the preprocessing for image registration and alignment, data binning needed for microarray‐ST as well as the cell segmentation used in ISH/ISS‐based ST, the mainstay of downstream analysis still shares a few in common and thus is summarized in a comprehensive but nonexhaustive list presented in Figure 2. The detailed methods and underlying algorithm principles can be referred elsewhere without detailed discussion in this section. 64 , 65 , 69
FIGURE 2.
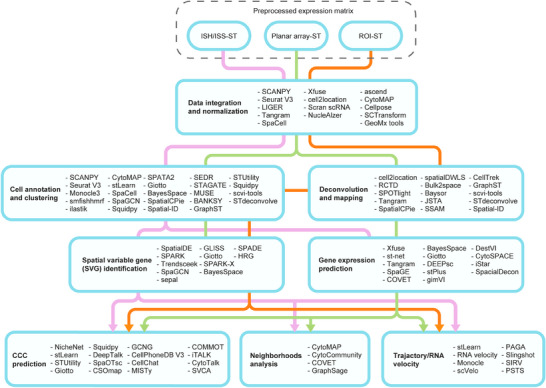
The spatial transcriptome data analysis toolbox. Graphical demonstration of typical ST data analysis pipeline based on three mainstream technical or strategical grounds (ISH/ISS‐ST, planar array‐ST, and ROI‐ST). Workflows start with preprocessed expression matrix and three different colored lines (light blue, pale green and orange) depict data analysis normally involved for three ST outputs respectively. The workflow directions are based on stepwise analysis approaches and each box represents a domain of spatial profiling methodology. Notes: The analysis toolbox is summarized based on literature searches and may not include all available methods. ISH, in‐site hybridization; ISS, in situ sequencing; ROI, region‐of‐interest.
Basically, microarray‐ST and ISH/ISS differ slightly from common scRNA‐seq data in that they contain extra spatial indices for individual data points generated across tissue‐covered regions being analyzed. Therefore, by taking advantages from computational methods developed for scRNA‐seq data analysis, many existing methods can be intuitively transferred and implemented such as Seurat V3 (integrated toolbox), SCTransform, Scran, and harmony for data integration/normalization 70 , 71 , 72 , 73 , 74 ; Squidpy (Scanpy), monocle 3, and scvi‐tools for clustering and annotation 75 , 76 ; Trendsceek and HRG for SVG identification 77 , 78 ; CellPhoneDB and CellChat for cellular crosstalk profiling 79 , 80 ; and Slingshot, scVelo, and RNA velocity for transcriptional dynamics measures and cell state tracking (trajectory/RNA velocity). 81 , 82 , 83 Simultaneously, inspired by efflux of available datasets within the community, by incorporating ST and histological information, researchers adopted various machine learning, topology‐based approaches and deep learning algorithms to continuously develop tools for ST analysis. 63 , 64 , 65 Analysis frameworks including stLearn, Squidpy, Giotto, SPATA2, Tangram, STUtility, CytoMAP, Spacemake and others suitable for multitasking in ST data analysis. 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 Besides, many other bioinformatic tools are also established for fit‐for‐purpose analysis tools including LIGER, BayesSpace, SpatialDE, RCTD, cell2location, spatialDWLS, SpaGE, gimVI, SpaOTsc, SIRV, and many more summarized in Figure 2. 92 , 93 , 94 , 95 , 96 , 97 , 98 , 99 , 100
4.3. Comparison of computational tools used across ST
Given the robust performance of data integration, normalization, and cluster identification using these ST‐adaptable tools, 101 , 102 many have been focusing on data enhancement, spatial clustering, spatial resolution enhancement, and cell type annotation/deconvolution as ample amount of ST data are realistically not down to single‐cell resolution (such like 55 µm in diameter in case of Visium polyA). A recent work systemically summarized 13 computational methods (conventional nonspatial methods: Louvain, Leiden and spatially designed: spaGCN, BayesSpace, stLearn, and many others) used for clustering of ST data. 103 On leveraging their performance on clustering accuracy, spatial continuity, marker gene detection, scalability and robustness, the major determinant relies significantly on the spatial resolution of technologies per se. Taking clustering accuracy as the key parameter, at 55 µm (Visium), GraphST, SCAN‐IT, and BASS stand as the preferred methods; however, at single‐cell resolution (MERFISH), the best performing methods switch to CCST, SpaceFlow, and SCAN‐IT. 103 Other side‐by‐side comparisons for spatial clustering have concluded that Seurat‐LVM, SpaGCN, and Seurat‐LV had overll the most accurate performance. 102 Previously as the most extensive used STs were Visium polyA (spatial‐barcoded AT) and DSP (ROI‐based ST) both of which have limited spatial resolution for data interpretability to define cell types, many methods have been developed accordingly. Systematic comparisons were also conducted between methods giving the conclusion that some scRNA‐seq reference‐based methods (cell2location, CARD, RCTD, Tangram, and EnDecon) may outperform others. 101 , 104 Other approaches including also exist such like STdeconvolve, SMART and CARDfree that are scRNA‐seq reference‐free. 105 , 106 , 107 With increasing resolution, including those with cellular or subcellular resolution (most ISH/ISS‐based), methods employing label transfer from scRNA‐seq, such as Spatial‐ID and JSTA are being applied. 108 , 109 , 110 In addition, for imaging‐based ST reaching over single‐cell resolution, known genes are often predesigned into analytical panels and thus cell types are directly definable based on this. 27 , 59 , 111 In real practice, it is also worth noting that for spatial‐barcoded ST with near single‐cell resolution, typical analytical points often require data binning to incorporate enough reads for profiling. However, the major challenge lies in that cells are irregular shaped disallowing precise transcripts allocation into designated cells. Under such scenario, spatial deconvolution remains as a standard tool for cell type inference but herein the concept is to take the cell type with the highest fraction for the cell bin being analyzed. Finding localized gene expression pattern (SVG) is another typical work implemented in spatial analysis wherein many have been developed and testified including regression‐based SpatialDE, Trendscreek, and SPARK‐X, multiple machine learning‐based spaGCN, sepal, GLISS, and many others. 63 , 77 , 95 , 112 , 113 , 114 , 115 A recent benchmarking work was carried out evaluating eight SVG identification methods (SpatialDE, SPARK‐X, Giotto, MERINGUE, and others). Under multiparameterized comparison, they found acceptable proportion of genes were detected across SVG methods but with relatively low overlapping, however relative coexpression of those SVG across methods are generally matched. 116 Under adjusted false discovery rate, SpatialDE, nnSVG, and MERINGUE are the top performers though certain issues still exist such as reproducibility (SpatialDE) and SVD prediction accuracy that exists almost across all methods except SPARK‐X and SOMDE. 116 To boost the number of genes characterized via ST, another popular direction in ST method development is gene inference and by model construction using scRNA‐seq and high‐plex ST data, methods such as Tangram, spaGE, gimVI, iSpatial, and iStar are proven methodologies for this type of analysis. 89 , 97 , 101 , 117 , 118 A recent work characterized 12 leading methods for gene expression imputation with ST datasets covering Visium polyA, Stereo‐seq, and Slide‐seq. 119 Overall, methods incorporating spatial information generally have higher prediction accuracy and the newly developed graph neural network‐based method Impeller outpaced other competitors for gene imputation regardless of the ST platforms being used. 119 Another added advantage of ST is to identify true interactors (cell–cell/gene–gene) under predefined spatial setting, a major advantage to apply in spatial biology since it gives histologically visible information that is completely lost in scRNA‐seq data. Those particularly involve tissue‐informed characterization of the signaling crosstalk (such as ligand‐receptor pairs) within direct or short‐ranged physical distance under cellular contexts. In theory, spatial cellular cross‐talk can be treated as interacting cell clusters often delineated via pathological annotation or within molecular‐informed cellular niches. Therefore, once a spatial count matrix is derived from particular histological regions (a subhistological cell clusters such as tumor epithelium‐enriched regions, tumor–immune interfaces, or tertiary lymphoid structures [TLS]), many existing approaches can be deployed. These mainly include conventional methods such as popular cell surface protein permutation‐based CellPhone DB and CellChat with proven performance in some benchmark studies. 79 , 80 , 111 , 120 intracellular gene–gene interaction‐based methods: NicheNet and CytoTalk 121 , 122 and more recently developed COMMOT using collective optimal transport. 123 Besides, cell–cell communication analysis can also be conducted using probabilistic‐based and machine learning‐based tools: SVCA, GCNG, and MISTy. 124 , 125 , 126 Similarly, most of spatial cellular fate dynamic tracking (trajectory‐based analysis) also employs well‐utilized scRNA‐seq tools such as stLearn, RNA velocity, scVelo, Monocle, Slingshot, PAGA, 81 , 82 , 83 , 86 , 127 many of which are extensively cross‐compared 128 and meanwhile others (SIRV) integrating ST with single‐cell also emerged. 100 A note to take in is that for existing targeted spatial single‐cell technologies, trajectory analysis is often employed to explore specific cell or cell network alteration across various status (such as cross‐group comparison between normal and disease settings or drug responder versus nonresponder). Given the ST‐defined individual cells in local space (similar to those achieved via spatial phenotyping techniques such as high‐plex immunohistochemistry IHC and Imaging mass cytometry IMC), high‐ordered cellular architectures such as CNs, colocalization patterns, and local enrichment profiles can also be achieved. 66 , 129 Though systematically sparse and relying on individual computational efforts, some methods have been developed such as CytoMAP toolbox and recently developed CytoCommunity and GraphSage. 130 , 131 Moreover, we already see those applications being applied under a few research settings. 111 Last, from our extensive experience, it is equally import to state that for most of ROI‐based ST such as widely used GeoMx DSP (pipelined in GeoMx tools), 84 , 132 though many analytical approaches can be potentially adopted such as ROI‐level clustering, SVGs, cellular decomposition, and trajectory analysis, 133 , 134 , 135 the data exploration is rather context‐dependent and normally follows methods being used in bulk RNA‐seq analysis and explained in the above section. However, the in‐depth data covered by entire transcriptomics in ROI‐based ST such as DSP facilitate biological exploration using sophisticated tools such as WGCNA that relies on network topology to untangle gene regulatory modules associated phenotypes. 136 , 137
4.4. A conclusive remark toward bioinformatics tools in ST
From a user's perspective, our survey of spatial bioinformatics is yet not explicit and depending on the ST technologies or various computational tools, user experience‐based data benchmarking will still be in demand considering data formats as input in conjunction with thorough understanding of pathology and explainable biological phenomenon under investigation. We summarized the latest update of those bioinformatic tools that are available for open publics and their application in representative publications in Table 1. Though currently there are no generalizable standards as to what methods to be applied under a given condition, certain computational methods may emerge as the mainstream along with cumulating ST data publicly available. Powered by the ever‐increasing resolution down to single‐cell or even subcellular level, spatial deconvolution may be surpassed and under these resolutions, higher molecular capture efficiency may allow thousands of genes to be detected and quantified within a particular cell. Upon such, benchmarking on current and future computational methods may be reinstigated redefining our analytical paradigm for ST data.
TABLE 1.
Representative bioinformatic tools frequently used for spatial transcriptomics.
| Analysis | Method | Year developed | Main ST application | Application examples in ST |
|---|---|---|---|---|
| Data integration and normalization | Squidpy (Scanpy) | 2022 | Spatial‐barcoded ST, ISS/ISH‐ST | 138 |
| Seurat V3 | 2019 |
Spatial‐barcoded ST, ISS/ISH‐ST |
71 | |
| LIGER | 2023 | Spatial‐barcoded ST | 139 | |
| Tangram | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST | 89 | |
| cell2location | 2022 | Spatial‐barcoded ST | 93 | |
| SpaCell | 2020 | Spatial‐barcoded ST | 140 | |
| Xfuse | 2021 | Spatial‐barcoded ST | 141 | |
| stLearn | 2020 | Spatial‐barcoded ST | 142 | |
| Ascend | ||||
| SCTransform | 2019 | Spatial‐barcoded ST, ISS/ISH‐ST | 143 | |
| Harmony | 2019 | Spatial‐barcoded ST, | 73 | |
| GeoMx tools | ROI‐ST | |||
| Cell annotation and clustering | spaGCN | 2020 | Spatial‐barcoded ST, ISS/ISH‐ST | 114 |
| BayesSpace | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST | 94 | |
| stLearn | 2020 | Spatial‐barcoded ST, ISS/ISH‐ST | 142 | |
| BANKSY | 2022 | Spatial‐barcoded ST, ISS/ISH‐ST | 144 | |
| BASS | 2022 | Spatial‐barcoded ST, ISS/ISH‐ST | 145 | |
| SCAN‐IT | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST | 146 | |
| STAGATE | 2022 | Spatial‐barcoded ST, ISS/ISH‐ST | 147 | |
| GraphST | 2023 | Spatial‐barcoded ST, ISS/ISH‐ST | 148 | |
| SEDR | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST | 149 | |
| Deconvolution and mapping | cell2location | 2022 |
Spatial‐barcoded ST, ROI‐ST |
93 |
| RCTD | 2021 |
Spatial‐barcoded ST, ROI‐ST |
96 | |
| SPOTlight | 2021 |
Spatial‐barcoded ST, ROI‐ST |
150 | |
| Tangram | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST, ROI‐ST | 89 | |
| SpatialDWLS | 2021 |
Spatial‐barcoded ST, ROI‐ST |
92 | |
| STdevonvolve | 2022 |
Spatial‐barcoded ST, ROI‐ST |
106 | |
| CellTrek | 2022 |
Spatial‐barcoded ST, ROI‐ST |
151 | |
| CARD | 2022 |
Spatial‐barcoded ST, ROI‐ST |
105 | |
| JSTA | 2021 | ISS/ISH‐ST | 110 | |
| Bulk2space | 2022 |
Spatial‐barcoded ST, ISS/ISH‐ST |
152 | |
| Spatial variable gene (SVG)) identification | SpatialDE | 2018 | Spatial‐barcoded ST, ISS/ISH‐ST | 95 |
| SPARK | 2020 | Spatial‐barcoded ST, ISS/ISH‐ST | 112 | |
| Trendsceek | 2018 | Spatial‐barcoded ST, ISS/ISH‐ST | 77 | |
| spaGCN | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST | 114 | |
| sepal | 2021 | Spatial‐barcoded ST | 113 | |
| GLISS | 2020 | Spatial‐barcoded ST | 153 | |
| STAGATE | 2022 | Spatial‐barcoded ST, ISS/ISH‐ST | 147 | |
| Spatial gene expression prediction | gimVI | 2019 | ISS/ISH‐ST | 154 |
| iStar | 2024 | 118 | ||
| Tangram | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST | 89 | |
| SpaGE | 2020 | ISS/ISH‐ST | 97 | |
| COVET | 2024 | ISS/ISH‐ST | 155 | |
| Cell–cell communication | CellPhoneDB v3 | 2020 | Spatial‐barcoded ST, ISS/ISH‐ST, ROI‐ST | 79 |
| COMMOT | 2023 | Spatial‐barcoded ST, ISS/ISH‐ST | 123 | |
| spaOTsc | 2020 | Spatial‐barcoded ST, ISS/ISH‐ST | 99 | |
| MISTy | 2022 | Spatial‐barcoded ST | 126 | |
| GCNG | 2020 | ISS/ISH‐ST | 125 | |
| stLearn | 2020 | Spatial‐barcoded ST | 142 | |
| DeepTalk | 2024 | Spatial‐barcoded ST, ISS/ISH‐ST | 156 | |
| CellChat | 2021 | Spatial‐barcoded ST, ISS/ISH‐ST, ROI‐ST | 80 | |
| SpaTalk | 2022 | Spatial‐barcoded ST | 157 | |
| Trajectory and RNA velocity | stLearn | 2020 | Spatial‐barcoded ST | 142 |
| Monocle | 2017 | Spatial‐barcoded ST | 158 | |
| PSTS | 2023 | Spatial‐barcoded ST | 142 | |
| SIRV | 2021 | ISS/ISH‐ST | 159 | |
| PAGA | 2019 |
Spatial‐barcoded ST, ISS/ISH‐ST |
127 | |
| scVelo | 2020 |
Spatial‐barcoded ST, ISS/ISH‐ST |
81 | |
| Neighborhoods analysis | CytoMAP | 2020 | Histo‐cytometry | 85 |
| CytoCommunity | 2024 | ISS/ISH‐ST | 130 | |
| COVET | 2024 | ISS/ISH‐ST | 155 |
5. ST‐DRIVEN RESEARCH IN TRANSLATIONAL ONCOLOGY
5.1. A foresight on ST in cancer research
The way to study tissue oncogenesis has already tweaked the research paradigm, in which much is influenced by ST. This is simply because tumors on the whole reside in a changing ecosystem often referred as tumor microenvironment (TME), a complex milieu that opens to questions such as inter/intratumor heterogeneity (ITH), spatial cellular context‐dependent mechanisms and predictive, prognostic, or therapeutic biomarker identification based on high‐plex spatial molecular information. Some have already been deciphered using dissociation‐based techniques (single‐cell multiomics) bearing potentials to translate into clinics, 160 but many more are yet to be unveiled. We foresee promises being accelerated by integrating high‐plex spatial omics with conventionally pathological techniques and thereby overview the current progress in this field. Our discussion is much toward the available technologies utilized throughout research and commercial institutions and summarized under such basis (Figure 3). Those mainly include but not restricted to Visium‐ST, Stereo‐seq, GeoMx DSP, and ISH/ISS‐based techniques (MERSCOPE, Xenium in situ, and SMI) and largely a surge is witnessed in the past few years. 49 , 161 , 162 Another point to add is that we purposely focus on research adopting ST as the major exploratory tool throughout their studies, highlighting the added benefit from this technical point of view. Since low‐resolution ST are already widely used, we presented oncology‐related application using ST with near/true single‐cell resolution and summarized in Table 2.
FIGURE 3.
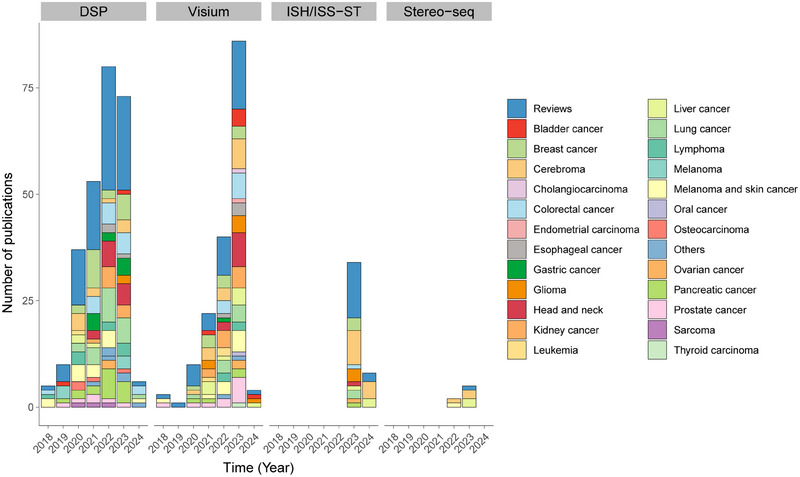
Overview of growth pattern of spatial transcriptome application in cancer research. Data are summarized in stacked bar charts and collected between 2018 and 2024 (current) with mainstream platforms commercially available (DSP, Visium, MERSCOPE, CosMx, Xenium, Stereo‐seq). Major cancer types are color‐annotated and reviews are separately categorized. ISS/ISH‐based ST (MERSCOPE, CosMx, and Xenium) are compiled together in one group and others are listed separately. Notes: Analysis is based on manual searches and may not include all related publications. Development of bioinformatic tools and abstracts are purposely excluded.
TABLE 2.
Application of near single‐cell spatial transcriptomics in oncology.
| ST technology | Cancer type | Year | Multiplexity | References |
|---|---|---|---|---|
| MERFISH | BC | 2023 | 500 | 163 |
| Stereo‐seq | CRC | 2024 | Transcriptome‐wide | 164 |
| SMI, Xenium MERFISH | OV | 2023 |
SMI: 960 Xenium: 280 MERFISH:140 |
165 |
| MERFISH | HCC | 2023 | 50 | 166 |
| MERFISH | LC | 2024 | 479 | 167 |
| MERFISH | GBM | 2021 | 135 | 168 |
|
Stereo‐seq SMI |
HCC | 2024 |
Transcriptome‐wide SMI: 960 |
169 |
| Stereo‐seq | LC | 2023 | Transcriptome‐wide | 170 |
| Stereo‐seq | CRC | 2023 | Transcriptome‐wide | 171 |
| Stereo‐seq | HCC/ICC | 2023 | Transcriptome‐wide | 172 |
| Stereo‐seq | CSCC | 2022 | Transcriptome‐wide | 173 |
|
Stereo‐seq Molecular Cartography |
Melanoma | 2022 |
Stereo‐seq: Transcriptome‐wide Molecular Cartography: |
174 |
| Stereo‐seq | CRC | 2023 | Transcriptome‐wide | 175 |
| SMI | OV | 2024 | 960 | 176 |
|
SMI Xenium |
HGG | 2023 |
SMI: 960 Xenium: 339 |
177 |
| SMI | HNC | 2023 |
SMI: 960 |
178 |
| Xenium | GBM | 2024 | 358 | 179 |
| Xenium | LC | 2023 | 302 | 29 |
| Xenium | BC | 2024 | 280 | 111 |
|
Xenium Visium HD |
CRC | 2024 |
422 Transcriptome‐wide |
59 |
| Xenium | BC | 2023 | 280 | 180 |
| Xenium | GBM | 2023 | 298 | 181 |
| Slide‐seq V2 | NBM | 2024 | Transcriptome‐wide | 182 |
| Slide‐seq V2 | PC | 2023 | Transcriptome‐wide | 183 |
Notes: References only incorporate real experiments using ST technologies excluding bioinformatics tool development, ST technology validation, reviews, or publications using public ST data.
Abbreviation: BC, breast cancer; CRC, colorectal cancer; OV, ovarian cancer; HCC, hepatocellular carcinoma; GBM, glioblastoma; LC, lung cancer; ICC, intrahepatic cholangiocarcinoma; CSCC, cervical squamous cell carcinoma; HGG, high grade glioma; HNC, head and neck cancer; NBM, neuroblastoma; PC, prostate cancer.
5.1.1. Mechanistic elucidation in heterogenous cancer tissues
One favorable advantage of ST is its compatibility for both archived FFPE and FF tissues and in many circumstances only small handful of tissue is required as analytical inputs granting flexible fit‐for‐purpose experimental designs. In breast cancer, an active application field of ST, Andersson et al. 184 used ST to find infiltration niche of CXCL10+ M2‐like macrophages with IFIT1+ T cells with HER2‐positive breast tumors yielding tertiary lymphoid‐like structures as a universal indicator across tissue types. Using ST combined with single‐cell T cell receptor‐seq, Mao et al. 185 elucidated spatial‐driven M1/M2 characteristics associated with breast tumor that orchestrate with cancer‐associated fibroblasts (CAFs) to form immunosuppressive microenvironment. The spatially informed breast cancer transcriptome obtained via ST also reveals an imbalance in oxygen distribution within claudin‐low tumor, with hypoxia in the tumor center and normoxia in the periphery. 186 Another advantage of ST is to investigate pathogen‐host responses where regions of infection can be precisely spotted. 187 Galeano Nino et al. 188 combined multiple spatial techniques with scRNA‐seq to map the spatial, cellular, and molecular interactions of host cells within the TME, revealing the presence and dynamics of intratumoral microbial communities and their potential impact on tumor heterogeneity. Similar works were also carried out using ROI‐ST (GeoMx‐DSP) to uncover bacteria burdens associated with lung cancer epithelium wherein Wnt/β‐catenin, HIF1A and VEGFA‐related signaling were mechanistically identified. 189 In immunotherapy‐favored cancers, using ROI‐ST, a study involving 152 non‐small cell lung cancer (NSCLC) patients revealed that the spatially enriched 163+ tumor‐associated macrophages (TAM) in the TME are associated with immunotherapy resistance, driven by the upregulation of CD27, ITGAM, and CCL5 expression within the tumor cavity. 190 Using joint scRNA‐seq and ST to delineate oncogenic transcriptional programs, Zhu et al. 191 found that UBE2C+ cancer cells during the lung adenocarcinoma (LUAD) invasion, as a progression hallmark companied by multiple localized immune cell alteration and activated TGF‐β signaling. By dissecting TME into tumor and immune‐enriched areas, Zhang et al. 133 established a ST landscape of NSCLC with brain metastasis, wherein reduced antigen presentation, B/T cell function and reprogrammed neutrophils and M2‐like macrophages, immature microglia, and reactive astrocytes were observed. Recently, Wang et al. 192 elucidated spatial transcriptome scale molecular features during LUAD tumor progression and the establishment on holistic tissue architecture captured major events in hypoxia‐induced macrophages and other molecular characteristics in certain subtypes. In melanoma, comprehensive spatial exploration established mechanistic hallmarks within tumor precursor regions and tumor‐stomal boundary that involve gradient cytokines to stimulate immune cell recruitment. 193 In tumors bearing less mutational burdens with complex TME with immune checkpoint inhibitors (ICIs) often being ineffective, Liu et al. 194 combined ST with scRNA‐seq and multiplex immunofluorescence staining, revealing the existence of a tumor–immune barrier structure in hepatocellular carcinoma (HCC): a spatial niche composed of SPP1+ macrophages and CAFs near the tumor border. Mechanistic deciphering of this spatial niche revealed that a hypoxic microenvironment promotes SPP1+ macrophages as an advert regulator for effective check point blockade. 194 More recently, Sun et al. 195 established a comprehensive HCC primary and metastatic landscape wherein they used ROI‐ST to analyze Wnt‐mutation (Wnt‐mut) associated spatial transcriptome and found upregulated exhausted T cells associated with mutant phenotypes and wide‐type Wnt (Wnt‐wt) had enriched iCAF population. The same group also applied subcellular resolved ST (Stereo‐seq) to identify key regulatory events at the tumor–immune interface that involve trafficking of amloid A1 and A2 and activation of tumor‐stemming JAK–STAT3 via CXCL6. 172 Zooming into the tumor and invasive margins, they found increasing patterns of angiogenetic signal, extracellular matrix remodeling and TP53 activity in Wnt‐wt tumors. 195 In pancreatic ductal adenocarcinoma (PDAC), single‐cell transcriptomics combined with ROI‐ST covering the whole transcriptome delineated neoadjuvant chemotherapy and radiotherapy (RT)‐associated spatiotemporal dynamics of treatment‐refractory patients, a mechanism mediated by three multicellular communities, that are each reprogrammed. 196 Another study incorporated ST to investigate treatment‐associated spatiotemporal changes in different therapeutic arms under neoadjuvant settings. 197 They discovered tumor‐intrinsic and transitional cellular programs mediated via TIGHT+ exhausted and regulatory T cells and NECTIN‐regulatory in tumor‐proximal niches highlighting TIGHT–NECTIN axis as potential targets. 197 A recent study incorporated multiparameterized spatial single‐cell proteomics and ROI‐ST to elucidate mechanistic linkage in PDAC patients stratified by homologues recombinant deficiency (HRD). 198 They discovered tumor‐infiltrating macrophages that are elevated in response to oncogenic transformation and HRD status highlighting a potential therapeutic target CD52 in treating PDAC patients in combination with PARP inhibitors (PARPi). 198 In gastrointestinal cancer, using ROI‐ST on FFPE samples, an early work demonstrated the utility of the technology to profile biomarkers in anti‐PD‐1 antibody driven TME alteration in colorectal cancer (CRC). 199 Qi et al. 200 used scRNA‐seq and ST to identify and orthogonally validate a tumor‐associated FAP1+ fibroblasts and SPP1+ macrophages in CRC, a mechanism involving chemerin, TGF‐β and IL‐1 to form immune‐resistant cellular niches with impaired T cell functioning. In gastric cancer (GC), Kumar et al. 201 used ROI‐ST to cross‐validate INHBA+ CAF and tumor‐expressing KLF2 in the diffused subtype complimenting their finding from scRNA‐seq. In gynecological cancer, Yeh et al. 202 recently applied ISH‐ST to uncover spatial alterations in high‐grade serous ovarian cancer (HGSC), wherein distinct infiltrated T/nature killer cellular states were spotted in subset of treatment sensitive tumor epithelium collaborating with transcriptional change in PTPN1 and ACTR8. In another application scenario, Liu et al. 203 tracked spatiotemporal dynamics of esophageal squamous cell carcinoma from precancerous lesions to low‐grade/high‐grade intraepithelial neoplasia using ROI‐ST on transcriptome‐wide scale capturing major events at tumor sites mediated through inversely correlated TAGLN2 and CRNN as progressive hallmarks. In urological cancers, a recent work incorporating ST and scRNA‐seq identified PDGFRα+/ITGA11+ fibroblasts that mediate lymphovascular invasion and lymphatic metastasis via ITGA11–SELE ligand‐receptor crosstalk in early stage bladder cancer. 204 In brain cancers, Vo et al. 205 used Sonic hedgehog medulloblastoma derived organoid (SHH‐DPOX) to resolve cellular heterogeneity within the TME in response to Palbociclib treatment (a CDK4/6 inhibitor). Using Visium ST they spotted species lineage associated coexpression at the immune‐infiltration regions and Palbociclib‐treated models induced regression of clonogenicity of MB tumor together with activated neurodifferentiation in the tumor center but not at the tumor–immune boundary. 205 More importantly, in the drug‐treated group, a spatially niche was also identified coordinated expression of astrocytes and tumor‐associated microglia together with tumor‐infiltrating macrophages (TMA) suggesting a functional interplay between them. 205 Likewise, many other works used ROI‐ST to target specific regions within different cancer tissues (TLSs or perirenal fat enriched regions) to elucidate transcriptional mechanisms linking with disease prognosis. 134 , 206 Many more can be referred in recent reviews. 4 , 161 , 207
Toward this end, however it is still well‐worth to mention that with ST technologies being constantly developed and improved, the real‐world application scenarios are much beyond early prototyping studies where model systems were often used for proof‐of‐concept studies and from extensive experience including ours. On top of this, due to the heavy investment upfront to generate large scale data across patient cohorts and intensive computational efforts for data integration (especially single‐cell spatial data that easily generate tens of millions of cells), more systemic works will still be needed. This is particularly indispensable when researching under such heterogenous tumor molecular mechanistic contexts. Those include some preliminary works being carried out by us and others using cutting‐edge technologies such as Xenium in situ, CosMx, and MERSCOPE as in their commercial forms and with propagate in short time. 27 , 28 , 166 , 169
5.1.2. Empowering biomarker profiling using advanced ST in translational oncology
Of more clinical relevance, under many experimental contexts, the ultimate goal following mechanistic elucidation biomarker discovery that enables ultimate clinical implementation. This is significantly inspired by evolving tumor biology and the rapid progress in cancer drug development and their associated resistance mechanisms not only through tumor‐centric targeting approaches (modulation of cancer cell plasticity) but also versatile strategies involving microenvironment modulation. 208 , 209 Nowadays, the widespread application of machine learning/deep‐learning assisted by digital pathology in fusion with ST has emerged as a novel tool toward this direction. The multidimensional layer of omics embedded in situ brings unforeseen analytical potentials to catalyze novel biomarker translation. Leveraging ST profiling across whole‐slide histopathological images, deep learning algorithms can further develop meticulously trained models capable of capturing tumor biomarkers of clinical significance. 210 , 211 As said before, at this stage, most of ST‐based technologies are yet expensive to scale up in sample number to allow systemic profiling, but some especially those supporting FFPE or tissue microarray (TMA) application are in play since for those, clinical metadata are often at hand.
On this ground, we and others have already started to attempt in this field. TLSs, an indicator within the TME often associates with favored prognosis but in‐depth studies are lacking. Gan et al. 212 applied ST to unveil TLS associated expression signatures in combined hepatocellular–cholangiocarcinoma (cHCC–CCA) patients and generated a scoring system considering spatial distribution of TLSs where intratumor TLSs (iTLS) stands as a predictive indicator for prognosis. Under another setting, using ROI‐ST, Kiuru et al. 213 profiled melanoma TME and identified S100A8 expressed on keratinocytes as an early oncogenic biomarker and validated the finding in larger cohorts. Another systemic work using Visium ST profiled the heterogenous stroma TME in a set of HGSC. 214 Their findings, arising from the complex TME identified a subset of CAF at the tumor‐stroma interface with significant intercellular crosstalk of APOE–LPR5 as a predictive biomarker for short‐term survival. 214 Similarly, a recent work focusing on cHCC–CCA also employed Visium ST to discover TLS‐associated gene expression pattern that predict disease prognosis. 212 Based on the ST‐derived data, they developed a TLS score by leveraging the contribution of either intratumor or extratumor regions (iTLS and eTLS) wherein the iTLS had better prognostic values that were not observed in eTLS counting, again stressing the needs of taking spatial parameters for prediction model generation. 212 Monkman et al. 215 applied ROI‐ST to discover T cells and macrophage‐dominated immune traffics that potentially relate to ICI responses in NSCLC. They built spatially chartered proteogenomic biomarker panels using sPLS‐DA model to discriminate ICI response and overall survival. 215 These phenomena are also reflected on some preliminary spatial biomarker profiling works being carried out in ICI‐related studies in other thoracic and GCs. 216 , 217 Notably, works from above studies acquired extensive resources from archived FFPE samples in which many were conducted in a TMA format suggesting a trend in practice of those ST technologies. Likewise, in our hands, using ROI‐ST, Guo et al. 136 explored 45 mismatch repair‐deficient endometrial cancer (MMRd‐EC) finding a 14‐gene biomarker signature associated with tumor cells that defines three EC subtypes with varying CD8 T cells infiltration status. In the more deadly lung cancer subtype (small cell lung cancer, SCLC), ST was applied to define TME‐based molecular subtypes. In a retrospective study, Yang et al. 218 used ROI‐ST to discover two immune microenvironment‐defined molecular subtypes (ID: immune‐deficient and IE: immune‐enriched) in primary SCLC (TMA cohort of 29 patients) using transcriptome‐wide spatial profiling and found the clinical utility of this TME‐based subtyping in predicting patient survival outcomes and response to immunotherapy. Using the same ST technique, they used multiple ROI selection strategy to evaluate ITH of 25 SCLC patients using DEPTH algorism and identified three transcriptional subtypes featured by distinct molecular mechanisms. 219 More importantly, this classification correlated with CD8 T cell infiltration status and by grouping patients based on spatially defined ITH scores, they found high‐plex (HC) and low complex (LC) defined patients that differentiate between clinical outcomes. 219
Interestingly, we witnessed a gradual growing tendency of spatial multiomics being incorporated into clinical trial‐based studies undertaken by research communities and ourselves (data not to disclose), though most are retrospectively designed. For the sake of functional interpretation, these are mainly informed via spatial proteomics tools. 131 , 220 However, exploratory works in novel drug development (netrin‐1), 160 , 221 investigator initialed trials, as well as real‐world studies incorporating ST are gradually emerging and some studies have already become low‐hanging fruit. The forward‐looking assumption is made since the true spatial single‐cell ST performed using clinically archived samples is tangible. This may be gradually incorporated into careful designed clinical project pipelines wherein 10−100 proteins can be characterized under single‐cell spatial resolution, but beyond that, have extended analytical scope since the transcriptional state of over 10,000 genes at spatial single‐cell resolution is now in reach. The preliminary works on breast cancer have already shown promises as the growing prosperity of neoadjuvant therapies in breast cancer allows trial designs and biopsy sampling longitudinally. For example, in a phase I/II single‐arm study assessing pembrolizumab + sequential RT under neoadjuvant settings in triple‐negative breast cancer (TNBC), using multiplex IHC (mIHC), researchers examined 40 proteins to define cellular lineages within the TME and mapped all high‐ordered cellular niches (defined as districts) in space finding key regulatory mechanisms predisposed or induced in response to therapies. 131 In a multicentric randomized study (NeoTRIP clinical trial, NCT02620280), researchers explored the effect of atezolizumab + chemotherapy versus chemotherapy alone in early TNBC using a longitudinal approach. 220 They identified major events triggered via TCF1+ CD8 T cells and MHCII+ cancer cells as drug sensitive predictors and other immune‐dictated coregulatory multicellular modules involving B cells and granzyme B+ cytotoxic T cells underscoring their values in personalized clinical decision making. 220 These recent works, although being exploratory, may be a weathercock in the coming future under clinical settings. Nevertheless, it is equally important to stress that, the success path for cancer biomarker identification is wrapped in a coordinated delivery package querying into omics‐based profiling from upstream and corroborating in independent cohorts and relevant functional assays downstream.
5.2. Streamlining translational oncology via ST from bench to clinics
The plethora of new ST constantly drives the growth of translational oncology gradually redefining our understanding into this field. However, we still see the progress made in current era as being a puberty that is yet to flourish in the future. Similar to the development curve witnessed in dissociation‐based single‐cell omics whereby application is moving into clinical settings evidenced by some novel‐conceptualized clinical trials. For example, EXALT‐1/2, which are hematological malignancies‐oriented interventional study, incorporated scRNA‐seq and AI‐driven drug screening to guide clinical decision‐making. 160 , 222 A single‐arm proof‐of‐concept phase II trial combining tumor‐targeting B‐RAF inhibitor and MEK1/2 inhibitor with PD‐1 inhibitor (PDR001) also launched in metastatic CRC utilizing scRNA‐seq to analyze pretreatment and on‐treatment biopsy samples. 160 These and a few more prospective studies point at a promising potential of integrating novel omics into clinical settings. One key advantage of spatial multiomics is their tight coherence with standard pathology, a major benefit for data exploration and interpretation perspective under controlled settings. This is somewhat impractical using conventional dissociation based single‐cell techniques since most are relying upon fresh tissues, which are difficult to manipulate. More importantly, since spatial organization of cells are more important to decipher biological questions, the spatially coordinated cells bear eminent potentials for translational medicine, similar to what was observed in ICI modulation wherein spatial PD‐L1 distribution and abundance are key determinants for therapeutic responses in multiple cancers. 223 Other successful biomarkers such as “immunoscore” derived from quantitative measurement of tumor‐infiltrated CD3+/CD8+ T cells has already placed into the application frontline in clinics in CRC and potentially many others. 224 In addition, the development of novel checkpoints or drug combinations require investigation of TME in a depth of field to underpin deterministic biological mechanisms. 225 These are already evidenced at phenotypical level. Such as oncolytic virus (OV) being tested in clinics. Linking OV with immune‐activation in recurrent glioblastoma has been observed with novel CAN‐3110, an oncolytic herpes virus (oHSV). 226 Other supporting evidence also revealed altered neutrophil‐to‐lymphocyte ratio during OV (H101 an oncolytic adenovirus) pretreatment as good prognostic indicator in advanced refractory HCC. 227 Moreover, the recent success of antibody–drug conjugates (ADC) in many late‐stage solid tumors have also ushered the needs of patient stratification biomarkers 228 A few good examples include the success application of T‐DXd (a HER2–ADC) that is being approved for metastatic HER2+ breast cancer and sacituzumab govitecan (SG), a trophoblast cell‐surface antigen 2 (TROP2)‐targeting ADC. 228 Interestingly, ample amount of clinical evidence suggest that the bystander effect induced by those ADC often reprograms TME and such include T‐DXd being effective in HER2‐low patients and SG nondiscriminatively targets tumors with low‐TROP2 expression. 228 Since many of these ADCs are being explored with combinatorial regimes with other drugs such as Atezolizumab (anti‐PD‐L1 antibody) in metastatic BC (mBC, NCT04740918) and late‐stage/locally advanced urothelial cancer (la/mUC) with combination of enfortumab vedotin (a Nectin‐4 ADC) with pembrolizumab (anti‐PD‐1 antibody), both of which demonstrated promising clinical benefits, adding high‐plex spatial data would be beneficial to address TME‐related mechanisms. 229 This would ultimately deliver extra clinical benefit via balancing immunogenicity by removing undesirable adverse effects while inflaming or reverting immune excluded tumor stands as the main battlefield in oncoimmunology. 230 Beyond those facts, the multiparametrized data obtained with spatial dimension certainly have added values for patient stratification and may direct novel biomarker identification in a single or multimarker collaborated manner. 231
The developed ST techniques in current era are already reaching into the single‐cell level with ultrahigh‐plexity conducted in many prototype studies allowing true single‐cell spatial phenotype identification with simultaneously thousands of transcriptomic information aligned. While determining pivotal cell subtypes such as tumor‐reactive proimmune cells, rare malignant cell clones with pluripotency and plasticity and other key regulatory cells within the TME is becoming possible, careful design of experiments or orthogonal validation across ST platforms for fit‐for‐purpose analysis lay ahead of the biological questions. Herein, we propose a systemic workflow based on our user's experience to research communities (Figure 4). Practically, a balance between the cost efficiency and layers of data dimension (spatial resolution and target numbers) to be acquired need to be compromised. It may not be a huge burden to query into mechanism‐related issues since not many samples are required, but more for translational purposes. In addition, considering the timeline for research output, possibility of sample procurement, preexperimental QC measures, using clinical archived samples may be a wider future direction and many ST are now compatible with this type of samples. Therefore, a schema can start with a rather small sample set within which each assigned phenotypic group contains 3−6 samples for single‐cell ST technologies (planar array‐ST and ISS/ISH‐based) and these samples sometimes can be pooled into one glass‐slide to save costs or conducted using TMA in some circumstances. This pan‐tissue exploratory method allows limit‐free analysis for cell–cell association, biomarker‐specific cell type identification and orchestrated cell community identification based on manually defined cell types within contexts. Selected candidates can then be cross‐compared against RNAscope, mIHC, cyclic IF, and IMC for high‐plex profiling and resultant conclusions can be extrapolated and cross‐validated using clinically benchmarking technologies such as IHC and FISH. Meanwhile, deeper mechanistic analysis can be done via ROI‐ST targeting cells at specific ROI. The alternative strategy to deploy involves thorough evaluation of on‐study samples by experienced pathologists and for biomarker profiling, patients in each assigned metadata group often start with at least 10 samples and are made into TMA formats. Under such a background, using ROI‐ST, particular ROIs can be analyzed across patients within a well‐controlled environment under pathological assistance (tumor‐centric, tumor–immune interface or a particular cell type with TME). This often supports transcriptome‐wide discovery and parallel mechanistic elucidation and one can use well‐defined deconvolution methods to deduce major cell type alteration in abundance. Orthogonal validation using RNAscope, mIHC, cyclic IF, and IMC will subsequently be required in follow‐up analysis and cross‐validation using IHC or FISH on expanded cohorts can follow. However, in both circumstances, full pathological engagement is paramount throughout a study to lead to successful interpretation.
FIGURE 4.
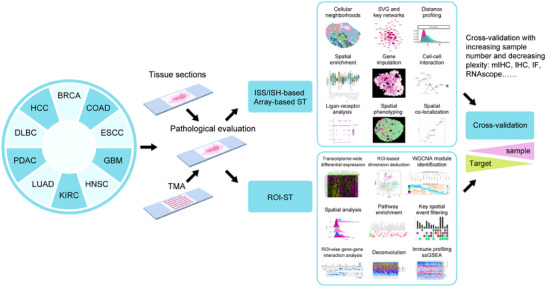
Proposed biomarker development pipeline using spatial transcriptome. Schematic illustration of biomarker profiling workflow based on ST. Cancer types are listed on the left for demonstrative purposes (see abbreviations). Sampling strategies are divided into full tissue sections and tissue microarrays (TMA) depending on ST technologies as exploratory tools (ISS/ISH‐ST, array‐based ST, and ROI‐ST). bioinformatic explorations (listed in boxes) are grouped accordingly to different ST technologies being applied. Downstream validation is for demonstration but involves increasing number of samples and decreasing number of targets. BRCA, breast carcinoma; COAD, colon adenocarcinoma; ESSC, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal cancer; LUAD, lung adenocarcinoma; PDAC, pancreatic ductal carcinoma; DLBC, lymphoid diffuse large B‐cell lymphoma; HCC, hepatocellular carcinoma.
6. CONCLUSIVE REMARKS AND FUTURE DIRECTION
Finally, our conclusive remarks are drawn in view of the current progress made on technical development as well as translational oncology being conducted. We anticipate a further improvement of existing technologies with joint multidimensional omics in real practice and these include high‐plex spatial proteogenomics, epigenetics assayed via transposase‐accessible chromatin, spatial T/B cell receptors (Spatial V(D)J recombination), and metabolomics obtained at bona‐fide single‐cell or even subcellular resolution. 232 Alongside with these layers of spatial information, reconstruction of tissue architecture in three‐dimensional using ST is also underway. 233 Moreover, the growing needs of researching into intratumoral microbiota open a new avenue to explore since many microorganisms are found in solid tumors that play either cancer‐initiating or inhibitory roles. 234 However, their exact mechanisms cannot be elucidated without application of ST and tracking the colocalization pattern of diversely distributed microbes within host tissues holds promises to address those questions. Some of those technologies are being developed such as spatial‐host‐microbiome sequencing (SHM‐seq) or many other ISS‐based methods. 235 , 236 More importantly, alongside with broader affordability and data accessibility, benchmarking on technologies or bioinformatics are always needed as references for preexperimental consideration. Aggregating the multilayered spatial omics can ultimately become the analytical keystone undoubtfully in cancer research, a gap to be filled just around the corner.
AUTHOR CONTRIBUTIONS
Conceptualization, data collection, and writing original draft: Nan Wang. Data analysis, data collection, and writing original draft: Weifeng Hong. Presentation, data collection and writing original draft: Yixing Wu. Guidance and review—editing: Zhe‐Sheng Chen. Supervision and review: Minghua Bai. Conceptualization, review—editing and supervision: Weixin Wang. Supervision, review—editing and fundings: Ji Zhu. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
N. W. and W. X. W. are full‐time employees of Cosmos Wisdom Biotech Co. Ltd. The commercial participant offers spatial multiomics as standard services. Author Zhe‐Sheng Chen is an Editorial board member of Medcomm. Author Zhe‐Sheng Chen was not involved in the journal's review of or decisions related to this manuscript. All authors declare no competing interests.
ETHICS STATEMENT
There is no ethical approval related to this review and no human and animal data are included.
ACKNOWLEDGMENTS
We thank the funding bodes and R&D team at Cosmos Wisdom Biotech Co. Ltd for technical support. We also thank Miss Jiajia Ying for graphical editing. This study is supported by Key Research Development Program of Zhejiang (2022C03015) and National Health Commission Research Foundation (WKJ‐ZJ‐2305).
Wang N, Hong W, Wu Y, et al. Next‐generation spatial transcriptomics: unleashing the power to gear up translational oncology. MedComm. 2024;5:e765. 10.1002/mco2.765
Contributor Information
Minghua Bai, Email: baimh2004@126.com.
Weixin Wang, Email: weixin.wang@cwmda.com.
Ji Zhu, Email: zhuji@zjcc.org.cn.
DATA AVAILABILITY STATEMENT
All data collections including all technical aspects and research references are manually curated, presented in relevant figures and available upon request.
REFERENCES
- 1. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18(6):359‐376. [DOI] [PubMed] [Google Scholar]
- 2. Bressan D, Battistoni G, Hannon GJ. The dawn of spatial omics. Science. 2023;381(6657):eabq4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robles‐Remacho A, Sanchez‐Martin RM, Diaz‐Mochon JJ. Spatial transcriptomics: emerging technologies in tissue gene expression profiling. Anal Chem. 2023;95(42):15450‐15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elhanani O, Ben‐Uri R, Keren L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell. 2023;41(3):404‐420. [DOI] [PubMed] [Google Scholar]
- 5. Yue L, Liu F, Hu J, et al. A guidebook of spatial transcriptomic technologies, data resources and analysis approaches. Comput Struct Biotechnol J. 2023;21:940‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asp M, Bergenstrahle J, Lundeberg J. Spatially resolved transcriptomes‐next generation tools for tissue exploration. Bioessays. 2020;42(10):e1900221. [DOI] [PubMed] [Google Scholar]
- 7. Wang N, Li X, Wang R, Ding Z. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnol J. 2021;16(9):e2100041. [DOI] [PubMed] [Google Scholar]
- 8. Park HE, Jo SH, Lee RH, et al. Spatial transcriptomics: technical aspects of recent developments and their applications in neuroscience and cancer research. Adv Sci (Weinh). 2023;10(16):e2206939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou R, Yang G, Zhang Y, Wang Y. Spatial transcriptomics in development and disease. Mol Biomed. 2023;4(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J, Suo S, Tam PP, Han JJ, Peng G, Jing N. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo‐seq. Nat Protoc. 2017;12(3):566‐580. [DOI] [PubMed] [Google Scholar]
- 11. Boisset JC, Vivie J, Grun D, Muraro MJ, Lyubimova A, van Oudenaarden A. Mapping the physical network of cellular interactions. Nat Methods. 2018;15(7):547‐553. [DOI] [PubMed] [Google Scholar]
- 12. Lovatt D, Ruble BK, Lee J, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11(2):190‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Medaglia C, Giladi A, Stoler‐Barak L, et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA‐seq. Science. 2017;358(6370):1622‐1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merritt CR, Ong GT, Church SE, et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol. 2020;38(5):586‐599. [DOI] [PubMed] [Google Scholar]
- 15. Kishi JY, Liu N, West ER, et al. Light‐Seq: light‐directed in situ barcoding of biomolecules in fixed cells and tissues for spatially indexed sequencing. Nat Methods. 2022;19(11):1393‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Junker JP, Noel ES, Guryev V, et al. Genome‐wide RNA Tomography in the zebrafish embryo. Cell. 2014;159(3):662‐675. [DOI] [PubMed] [Google Scholar]
- 17. Schede HH, Schneider CG, Stergiadou J, et al. Spatial tissue profiling by imaging‐free molecular tomography. Nat Biotechnol. 2021;39(8):968‐977. [DOI] [PubMed] [Google Scholar]
- 18. Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of single RNA transcripts in situ. Science. 1998;280(5363):585‐590. [DOI] [PubMed] [Google Scholar]
- 19. Larsson C, Grundberg I, Soderberg O, Nilsson M. In situ detection and genotyping of individual mRNA molecules. Nat Methods. 2010;7(5):395‐397. [DOI] [PubMed] [Google Scholar]
- 20. Ke R, Mignardi M, Pacureanu A, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10(9):857‐860. [DOI] [PubMed] [Google Scholar]
- 21. Lee JH, Daugharthy ER, Scheiman J, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc. 2015;10(3):442‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee JH, Daugharthy ER, Scheiman J, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343(6177):1360‐1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang X, Allen WE, Wright MA, et al. Three‐dimensional intact‐tissue sequencing of single‐cell transcriptional states. Science. 2018;361(6400):eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X, Sun YC, Church GM, Lee JH, Zador AM. Efficient in situ barcode sequencing using padlock probe‐based BaristaSeq. Nucleic Acids Res. 2018;46(4):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alon S, Goodwin DR, Sinha A, et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science. 2021;371(6528):eaax2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gyllborg D, Langseth CM, Qian X, et al. Hybridization‐based in situ sequencing (HybISS) for spatially resolved transcriptomics in human and mouse brain tissue. Nucleic Acids Res. 2020;48(19):e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Janesick A, Shelansky R, Gottscho AD, et al. High resolution mapping of the tumor microenvironment using integrated single‐cell, spatial and in situ analysis. Nat Commun. 2023;14(1):8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seo ES, Lee B, Hwang I, Kim J‐Y, Park K, Park W‐Y. Decoding spatial organization maps and context‐specific landscapes of breast cancer and its microenvironment via high‐resolution spatial transcriptomic analysis. bioRxiv. 2023:2023.2010.2025.563904. [Google Scholar]
- 29. Haga Y, Sakamoto Y, Kajiya K, et al. Whole‐genome sequencing reveals the molecular implications of the stepwise progression of lung adenocarcinoma. Nat Commun. 2023;14(1):8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang F, Flanagan J, Su N, et al. RNAscope: a novel in situ RNA analysis platform for formalin‐fixed, paraffin‐embedded tissues. J Mol Diagn. 2012;14(1):22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shah S, Lubeck E, Schwarzkopf M, et al. Single‐molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development. 2016;143(15):2862‐2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Codeluppi S, Borm LE, Zeisel A, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods. 2018;15(11):932‐935. [DOI] [PubMed] [Google Scholar]
- 33. Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single‐cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11(4):360‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eng CL, Lawson M, Zhu Q, et al. Transcriptome‐scale super‐resolved imaging in tissues by RNA seqFISH. Nature. 2019;568(7751):235‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348(6233):aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia C, Fan J, Emanuel G, Hao J, Zhuang X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle‐dependent gene expression. Proc Natl Acad Sci USA. 2019;116(39):19490‐19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chiba K, Lorbeer FK, Shain AH, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two‐step mechanism. Science. 2017;357(6358):1416‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He S, Bhatt R, Brown C, et al. High‐plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol. 2022;40(12):1794‐1806. [DOI] [PubMed] [Google Scholar]
- 39. Groiss S, Pabst D, Faber C, et al. Highly resolved spatial transcriptomics for detection of rare events in cells. bioRxiv. 2021:2021.2010.2011.463936. [Google Scholar]
- 40. Stahl PL, Salmen F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78‐82. [DOI] [PubMed] [Google Scholar]
- 41. Cheng M, Jiang Y, Xu J, et al. Spatially resolved transcriptomics: a comprehensive review of their technological advances, applications, and challenges. J Genet Genomics. 2023;50(9):625‐640. [DOI] [PubMed] [Google Scholar]
- 42. Rodriques SG, Stickels RR, Goeva A, et al. Slide‐seq: A scalable technology for measuring genome‐wide expression at high spatial resolution. Science. 2019;363(6434):1463‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vickovic S, Eraslan G, Salmen F, et al. High‐definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16(10):987‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srivatsan SR, Regier MC, Barkan E, et al. Embryo‐scale, single‐cell spatial transcriptomics. Science. 2021;373(6550):111‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cho CS, Xi J, Si Y, et al. Microscopic examination of spatial transcriptome using Seq‐Scope. Cell. 2021;184(13):3559‐3572 e3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y, Yang M, Deng Y, et al. High‐spatial‐resolution multi‐omics sequencing via deterministic barcoding in tissue. Cell. 2020;183(6):1665‐1681 e1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen A, Liao S, Cheng M, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball‐patterned arrays. Cell. 2022;185(10):1777‐1792 e1721. [DOI] [PubMed] [Google Scholar]
- 48. Duan H, Cheng T, Cheng H. Spatially resolved transcriptomics: advances and applications. Blood Sci. 2023;5(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moses L, Pachter L. Museum of spatial transcriptomics. Nat Methods. 2022;19(5):534‐546. [DOI] [PubMed] [Google Scholar]
- 50. Seferbekova Z, Lomakin A, Yates LR, Gerstung M. Spatial biology of cancer evolution. Nat Rev Genet. 2023;24(5):295‐313. [DOI] [PubMed] [Google Scholar]
- 51. Wu Y, Cheng Y, Wang X, Fan J, Gao Q. Spatial omics: Navigating to the golden era of cancer research. Clin Transl Med. 2022;12(1):e696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stickels RR, Murray E, Kumar P, et al. Highly sensitive spatial transcriptomics at near‐cellular resolution with Slide‐seqV2. Nat Biotechnol. 2021;39(3):313‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cao J, Zheng Z, Sun D, et al. Decoder‐seq enhances mRNA capture efficiency in spatial RNA sequencing. Nat Biotechnol. 2024. [DOI] [PubMed] [Google Scholar]
- 54. Song X, Guo P, Xia K, et al. Spatial transcriptomics reveals light‐induced chlorenchyma cells involved in promoting shoot regeneration in tomato callus. Proc Natl Acad Sci USA. 2023;120(38):e2310163120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liao J, Lu X, Shao X, Zhu L, Fan X. Uncovering an organ's molecular architecture at single‐cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 2021;39(1):43‐58. [DOI] [PubMed] [Google Scholar]
- 56. You Y, Fu Y, Li L, et al. Systematic comparison of sequencing‐based spatial transcriptomic methods. Nat Methods. 2024;21(9):1743‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emmert‐Buck MR, Bonner RF, Smith PD, et al. Laser capture microdissection. Science. 1996;274(5289):998‐1001. [DOI] [PubMed] [Google Scholar]
- 58. Zhou Q, Xu X, Li M, et al. Laser capture microdissection transcriptome (LCM RNA‐seq) reveals BcDFR is a key gene in anthocyanin synthesis of non‐heading Chinese cabbage. BMC Genomics. 2024;25(1):425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oliveira MF, Romero JP, Chung M, et al. Characterization of immune cell populations in the tumor microenvironment of colorectal cancer using high definition spatial profiling. bioRxiv. 2024;2024.2006.2004.597233. [Google Scholar]
- 60. Rademacher A, Huseynov A, Bortolomeazzi M, et al. Comparison of spatial transcriptomics technologies using tumor cryosections. bioRxiv. 2024;2024.2004.2003.586404. [Google Scholar]
- 61. Wang H, Huang R, Nelson J, et al. Systematic benchmarking of imaging spatial transcriptomics platforms in FFPE tissues. bioRxiv. 2023:2023.2012.2007.570603. [Google Scholar]
- 62. Hartman A, Satija R. Comparative analysis of multiplexed in situ gene expression profiling technologies. bioRxiv. 2024. [Google Scholar]
- 63. Zeng Z, Li Y, Li Y, Luo Y. Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 2022;23(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hu J, Schroeder A, Coleman K, Chen C, Auerbach BJ, Li M. Statistical and machine learning methods for spatially resolved transcriptomics with histology. Comput Struct Biotechnol J. 2021;19:3829‐3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fang S, Chen B, Zhang Y, et al. Computational approaches and challenges in spatial transcriptomics. Genomics Proteomics Bioinformatics. 2023;21(1):24‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schurch CM, Bhate SS, Barlow GL, et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell. 2020;182(5):1341‐1359 e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Elyanow R, Zeira R, Land M, Raphael BJ. STARCH: copy number and clone inference from spatial transcriptomics data. Phys Biol. 2021;18(3):035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bhate SS, Barlow GL, Schurch CM, Nolan GP. Tissue schematics map the specialization of immune tissue motifs and their appropriation by tumors. Cell Syst. 2022;13(2):109‐130 e106. [DOI] [PubMed] [Google Scholar]
- 69. Palla G, Fischer DS, Regev A, Theis FJ. Spatial components of molecular tissue biology. Nat Biotechnol. 2022;40(3):308‐318. [DOI] [PubMed] [Google Scholar]
- 70. Hafemeister C, Satija R. Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single‐cell data. Cell. 2019;177(7):1888‐1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lun AT, Bach K, Marioni JC. Pooling across cells to normalize single‐cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single‐cell data with harmony. Nat Methods. 2019;16(12):1289‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol. 2015;33(5):495‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wolf FA, Angerer P, Theis FJ. SCANPY: large‐scale single‐cell gene expression data analysis. Genome Biol. 2018;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gayoso A, Lopez R, Xing G, et al. A Python library for probabilistic analysis of single‐cell omics data. Nat Biotechnol. 2022;40(2):163‐166. [DOI] [PubMed] [Google Scholar]
- 77. Edsgard D, Johnsson P, Sandberg R. Identification of spatial expression trends in single‐cell gene expression data. Nat Methods. 2018;15(5):339‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wu Y, Hu Q, Wang S, et al. Highly Regional Genes: graph‐based gene selection for single‐cell RNA‐seq data. J Genet Genomics. 2022;49(9):891‐899. [DOI] [PubMed] [Google Scholar]
- 79. Efremova M, Vento‐Tormo M, Teichmann SA, Vento‐Tormo R. CellPhoneDB: inferring cell‐cell communication from combined expression of multi‐subunit ligand‐receptor complexes. Nat Protoc. 2020;15(4):1484‐1506. [DOI] [PubMed] [Google Scholar]
- 80. Jin S, Guerrero‐Juarez CF, Zhang L, et al. Inference and analysis of cell‐cell communication using CellChat. Nat Commun. 2021;12(1):1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38(12):1408‐1414. [DOI] [PubMed] [Google Scholar]
- 82. Street K, Risso D, Fletcher RB, et al. Slingshot: cell lineage and pseudotime inference for single‐cell transcriptomics. BMC Genomics. 2018;19(1):477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. La Manno G, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature. 2018;560(7719):494‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kleino I, Frolovaite P, Suomi T, Elo LL. Computational solutions for spatial transcriptomics. Comput Struct Biotechnol J. 2022;20:4870‐4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stoltzfus CR, Filipek J, Gern BH, et al. CytoMAP: a spatial analysis toolbox reveals features of myeloid cell organization in lymphoid tissues. Cell Rep. 2020;31(3):107523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pham D, Tan X, Xu J, et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell‐cell interactions and spatial trajectories within undissociated tissues. bioRxiv. 2020;2020.2005.2031.125658. [Google Scholar]
- 87. Dries R, Zhu Q, Dong R, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021;22(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kueckelhaus J, Jv Ehr, Ravi VM, et al. Inferring spatially transient gene expression pattern from spatial transcriptomic studies. bioRxiv. 2020;2020.2010.2020.346544. [Google Scholar]
- 89. Biancalani T, Scalia G, Buffoni L, et al. Deep learning and alignment of spatially resolved single‐cell transcriptomes with Tangram. Nat Methods. 2021;18(11):1352‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bergenstrahle J, Larsson L, Lundeberg J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics. 2020;21(1):482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sztanka‐Toth TR, Jens M, Karaiskos N, Rajewsky N. Spacemake: processing and analysis of large‐scale spatial transcriptomics data. Gigascience. 2022;11:giac064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dong R, Yuan GC. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biol. 2021;22(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kleshchevnikov V, Shmatko A, Dann E, et al. Cell2location maps fine‐grained cell types in spatial transcriptomics. Nat Biotechnol. 2022;40(5):661‐671. [DOI] [PubMed] [Google Scholar]
- 94. Zhao E, Stone MR, Ren X, et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol. 2021;39(11):1375‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Svensson V, Teichmann SA, Stegle O. SpatialDE: identification of spatially variable genes. Nat Methods. 2018;15(5):343‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cable DM, Murray E, Zou LS, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2022;40(4):517‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Abdelaal T, Mourragui S, Mahfouz A, Reinders MJT. SpaGE: spatial gene enhancement using scRNA‐seq. Nucleic Acids Res. 2020;48(18):e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liu J, Gao C, Sodicoff J, Kozareva V, Macosko EZ, Welch JD. Jointly defining cell types from multiple single‐cell datasets using LIGER. Nat Protoc. 2020;15(11):3632‐3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cang Z, Nie Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat Commun. 2020;11(1):2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Abdelaal T, Lelieveldt BPF, Reinders MJT, Mahfouz A. SIRV: Spatial inference of RNA velocity at the single‐cell resolution. bioRxiv. 2021;2021.2007.2026.453774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Li B, Zhang W, Guo C, et al. Benchmarking spatial and single‐cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat Methods. 2022;19(6):662‐670. [DOI] [PubMed] [Google Scholar]
- 102. Cheng A, Hu G, Li WV. Benchmarking cell‐type clustering methods for spatially resolved transcriptomics data. Brief Bioinform. 2023;24(1):bbac475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yuan Z, Zhao F, Lin S, et al. Benchmarking spatial clustering methods with spatially resolved transcriptomics data. Nat Methods. 2024;21(4):712‐722. [DOI] [PubMed] [Google Scholar]
- 104. Yan L, Sun X. Benchmarking and integration of methods for deconvoluting spatial transcriptomic data. Bioinformatics. 2023;39(1):btac805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ma Y, Zhou X. Spatially informed cell‐type deconvolution for spatial transcriptomics. Nat Biotechnol. 2022;40(9):1349‐1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Miller BF, Huang F, Atta L, Sahoo A, Fan J. Reference‐free cell type deconvolution of multi‐cellular pixel‐resolution spatially resolved transcriptomics data. Nat Commun. 2022;13(1):2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang C, Sin D, Ng R. SMART: reference‐free deconvolution for spatial transcriptomics using marker‐gene‐assisted topic models. bioRxiv. 2023;2023.2006.2020.545793. [Google Scholar]
- 108. Shen R, Liu L, Wu Z, et al. Spatial‐ID: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding. Nat Commun. 2022;13(1):7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Garrido‐Trigo A, Corraliza AM, Veny M, et al. Macrophage and neutrophil heterogeneity at single‐cell spatial resolution in human inflammatory bowel disease. Nat Commun. 2023;14(1):4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Littman R, Hemminger Z, Foreman R, et al. Joint cell segmentation and cell type annotation for spatial transcriptomics. Mol Syst Biol. 2021;17(6):e10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang N, Song Y, Hong W, et al. Spatial single‐cell transcriptomic analysis in breast cancer reveals potential biomarkers for PD‐1 blockade therapy. In: Research Square, 2024.
- 112. Sun S, Zhu J, Zhou X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat Methods. 2020;17(2):193‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Andersson A, Lundeberg J. sepal: identifying transcript profiles with spatial patterns by diffusion‐based modeling. Bioinformatics. 2021;37(17):2644‐2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hu J, Li X, Coleman K, et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods. 2021;18(11):1342‐1351. [DOI] [PubMed] [Google Scholar]
- 115. Li K, Yan C, Li C, et al. Computational elucidation of spatial gene expression variation from spatially resolved transcriptomics data. Mol Ther Nucleic Acids. 2022;27:404‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chen C, Kim HJ, Yang P. Evaluating spatially variable gene detection methods for spatial transcriptomics data. Genome Biol. 2024;25(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lopez R, Nazaret A, Langevin M, et al. A joint model of unpaired data from scRNA‐seq and spatial transcriptomics for imputing missing gene expression measurements. ArXiv. 2019;abs/1905.02269. [Google Scholar]
- 118. Zhang D, Schroeder A, Yan H, et al. Inferring super‐resolution tissue architecture by integrating spatial transcriptomics with histology. Nat Biotechnol. 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Duan Z, Riffle D, Li R, Liu J, Min MR, Zhang J. Impeller: a path‐based heterogeneous graph learning method for spatial transcriptomic data imputation. Bioinformatics. 2024;40(6):btae339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dimitrov D, Turei D, Garrido‐Rodriguez M, et al. Comparison of methods and resources for cell‐cell communication inference from single‐cell RNA‐Seq data. Nat Commun. 2022;13(1):3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 2020;17(2):159‐162. [DOI] [PubMed] [Google Scholar]
- 122. Hu Y, Peng T, Gao L, Tan K. CytoTalk: De novo construction of signal transduction networks using single‐cell transcriptomic data. Sci Adv. 2021;7(16):eabf1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cang Z, Zhao Y, Almet AA, et al. Screening cell‐cell communication in spatial transcriptomics via collective optimal transport. Nat Methods. 2023;20(2):218‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Arnol D, Schapiro D, Bodenmiller B, Saez‐Rodriguez J, Stegle O. Modeling cell‐cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep. 2019;29(1):202‐211 e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yuan Y, Bar‐Joseph Z. GCNG: graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 2020;21(1):300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tanevski J, Flores ROR, Gabor A, Schapiro D, Saez‐Rodriguez J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol. 2022;23(1):97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wolf FA, Hamey FK, Plass M, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single‐cell trajectory inference methods. Nat Biotechnol. 2019;37(5):547‐554. [DOI] [PubMed] [Google Scholar]
- 129. Ruf B, Bruhns M, Babaei S, et al. Tumor‐associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell. 2023;186(17):3686‐3705 e3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hu Y, Rong J, Xu Y, et al. Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes. Nat Methods. 2024;21(2):267‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shiao SL, Gouin KH 3rd, Ing N, et al. Single‐cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell. 2024;42(1):70‐84 e78. [DOI] [PubMed] [Google Scholar]
- 132. Wang N, Li X, Ding Z. High‐plex spatial profiling of RNA and protein using digital spatial profiler. Methods Mol Biol. 2023;2660:69‐83. [DOI] [PubMed] [Google Scholar]
- 133. Zhang Q, Abdo R, Iosef C, et al. The spatial transcriptomic landscape of non‐small cell lung cancer brain metastasis. Nat Commun. 2022;13(1):5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ling Y, Zhong J, Weng Z, et al. The prognostic value and molecular properties of tertiary lymphoid structures in oesophageal squamous cell carcinoma. Clin Transl Med. 2022;12(10):e1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Tripodo C, Bertolazzi G, Cancila V, Morello G, Iannitto E. Pseudotemporal ordering of spatial lymphoid tissue microenvironment profiles trails Unclassified DLBCL at the periphery of the follicle. Front Immunol. 2023;14:1207959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Guo J, Tang B, Fu J, et al. High‐plex spatial transcriptomic profiling reveals distinct immune components and the HLA class I/DNMT3A/CD8 modulatory axis in mismatch repair‐deficient endometrial cancer. Cell Oncol (Dordr). 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Palla G, Spitzer H, Klein M, et al. Squidpy: a scalable framework for spatial omics analysis. Nat Methods. 2022;19(2):171‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Vahid MR, Brown EL, Steen CB, et al. High‐resolution alignment of single‐cell and spatial transcriptomes with CytoSPACE. Nat Biotechnol. 2023;41(11):1543‐1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tan X, Su A, Tran M, Nguyen Q. SpaCell: integrating tissue morphology and spatial gene expression to predict disease cells. Bioinformatics. 2020;36(7):2293‐2294. [DOI] [PubMed] [Google Scholar]
- 141. Bergenstrahle L, He B, Bergenstrahle J, et al. Super‐resolved spatial transcriptomics by deep data fusion. Nat Biotechnol. 2022;40(4):476‐479. [DOI] [PubMed] [Google Scholar]
- 142. Pham D, Tan X, Balderson B, et al. Robust mapping of spatiotemporal trajectories and cell‐cell interactions in healthy and diseased tissues. Nat Commun. 2023;14(1):7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Atta L, Clifton K, Anant M, Aihara G, Fan J. Gene count normalization in single‐cell imaging‐based spatially resolved transcriptomics. Genome Biol. 2024;25(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Singhal V, Chou N, Lee J, et al. BANKSY unifies cell typing and tissue domain segmentation for scalable spatial omics data analysis. Nat Genet. 2024;56(3):431‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Li Z, Zhou X. BASS: multi‐scale and multi‐sample analysis enables accurate cell type clustering and spatial domain detection in spatial transcriptomic studies. Genome Biol. 2022;23(1):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Cang Z, Ning X, Nie A, Xu M, Zhang J. SCAN‐IT: Domain segmentation of spatial transcriptomics images by graph neural network. BMVC : proceedings of the British Machine Vision Conference British Machine Vision Conference . 2021;32:406. [PMC free article] [PubMed] [Google Scholar]
- 147. Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto‐encoder. Nat Commun. 2022;13(1):1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Long Y, Ang KS, Li M, et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat Commun. 2023;14(1):1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Fu H, Xu H, Chong K, et al. Unsupervised spatially embedded deep representation of spatial transcriptomics. bioRxiv. 2021;2021.2006.2015.448542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Elosua‐Bayes M, Nieto P, Mereu E, Gut I, Heyn H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single‐cell transcriptomes. Nucleic Acids Res. 2021;49(9):e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wei R, He S, Bai S, et al. Spatial charting of single‐cell transcriptomes in tissues. Nat Biotechnol. 2022;40(8):1190‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Liao J, Qian J, Fang Y, et al. De novo analysis of bulk RNA‐seq data at spatially resolved single‐cell resolution. Nat Commun. 2022;13(1):6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhu J, Sabatti C. Integrative spatial single‐cell analysis with graph‐based feature learning. bioRxiv. 2020;2020.2008.2012.248971. [Google Scholar]
- 154. Lopez R, Nazaret A, Langevin M, et al. A joint model of unpaired data from scRNA‐seq and spatial transcriptomics for imputing missing gene expression measurements. bioRxiv. 2019;abs/1905.02269. [Google Scholar]
- 155. Haviv D, Remsik J, Gatie M, et al. The covariance environment defines cellular niches for spatial inference. Nat Biotechnol. 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yang W, Wang P, Xu S, et al. Deciphering cell‐cell communication at single‐cell resolution for spatial transcriptomics with subgraph‐based graph attention network. Nat Commun. 2024;15(1):7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Shao X, Li C, Yang H, et al. Knowledge‐graph‐based cell‐cell communication inference for spatially resolved transcriptomic data with SpaTalk. Nat Commun. 2022;13(1):4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Qiu X, Mao Q, Tang Y, et al. Reversed graph embedding resolves complex single‐cell trajectories. Nat Methods. 2017;14(10):979‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Abdelaal T, Lelieveldt BPF, Reinders MJT, Mahfouz A. SIRV: Spatial inference of RNA velocity at the single‐cell resolution. bioRxiv. 2021:2021.2007.2026.453774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sheridan C. Can single‐cell biology realize the promise of precision medicine? Nat Biotechnol. 2024;42(2):159‐162. [DOI] [PubMed] [Google Scholar]
- 161. Wang Q, Zhi Y, Zi M, et al. Spatially resolved transcriptomics technology facilitates cancer research. Adv Sci (Weinh). 2023;10(30):e2302558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Coulson A. How is spatial transcriptomics influencing cancer research and diagnostics? Biotechniques. 2022;73(5):215‐217. [DOI] [PubMed] [Google Scholar]
- 163. Erfanian N, Nasseri S, Miraki Feriz A, Safarpour H, Namaei MH. Characterization of Wnt signaling pathway under treatment of Lactobacillus acidophilus postbiotic in colorectal cancer using an integrated in silico and in vitro analysis. Sci Rep. 2023;13(1):22988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ferri‐Borgogno S, Burks JK, Seeley EH, et al. Molecular, metabolic, and subcellular mapping of the tumor immune microenvironment via 3D targeted and non‐targeted multiplex multi‐omics analyses. Cancers (Basel). 2024;16(5):846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yeh CY, Aguirre K, Laveroni O, et al. Mapping ovarian cancer spatial organization uncovers immune evasion drivers at the genetic, cellular, and tissue level. bioRxiv. 2023;2023.2010.2016.562592. [Google Scholar]
- 166. Magen A, Hamon P, Fiaschi N, et al. Intratumoral dendritic cell‐CD4(+) T helper cell niches enable CD8(+) T cell differentiation following PD‐1 blockade in hepatocellular carcinoma. Nat Med. 2023;29(6):1389‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Chen JH, Nieman LT, Spurrell M, et al. Human lung cancer harbors spatially organized stem‐immunity hubs associated with response to immunotherapy. Nat Immunol. 2024;25(4):644‐658. [DOI] [PubMed] [Google Scholar]
- 168. Hara T, Chanoch‐Myers R, Mathewson ND, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal‐like states in glioblastoma. Cancer Cell. 2021;39(6):779‐792 e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Li Z, Pai R, Gupta S, et al. Presence of onco‐fetal neighborhoods in hepatocellular carcinoma is associated with relapse and response to immunotherapy. Nat Cancer. 2024;5(1):167‐186. [DOI] [PubMed] [Google Scholar]
- 170. Chen C, Guo Q, Liu Y, et al. Single‐cell and spatial transcriptomics reveal POSTN(+) cancer‐associated fibroblasts correlated with immune suppression and tumour progression in non‐small cell lung cancer. Clin Transl Med. 2023;13(12):e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Qin P, Chen H, Wang Y, et al. Cancer‐associated fibroblasts undergoing neoadjuvant chemotherapy suppress rectal cancer revealed by single‐cell and spatial transcriptomics. Cell Rep Med. 2023;4(10):101231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wu L, Yan J, Bai Y, et al. An invasive zone in human liver cancer identified by Stereo‐seq promotes hepatocyte‐tumor cell crosstalk, local immunosuppression and tumor progression. Cell Res. 2023;33(8):585‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Ou Z, Lin S, Qiu J, et al. Single‐Nucleus RNA Sequencing and Spatial Transcriptomics Reveal the Immunological Microenvironment of Cervical Squamous Cell Carcinoma. Adv Sci (Weinh). 2022;9(29):e2203040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Karras P, Bordeu I, Pozniak J, et al. A cellular hierarchy in melanoma uncouples growth and metastasis. Nature. 2022;610(7930):190‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Zhang R, Feng Y, Ma W, et al. Spatial transcriptome unveils a discontinuous inflammatory pattern in proficient mismatch repair colorectal adenocarcinoma. Fundam Res. 2023;3(4):640‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Denisenko E, de Kock L, Tan A, et al. Spatial transcriptomics reveals discrete tumour microenvironments and autocrine loops within ovarian cancer subclones. Nat Commun. 2024;15(1):2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Moffet JJD, Fatunla OE, Freytag L, et al. Spatial architecture of high‐grade glioma reveals tumor heterogeneity within distinct domains. Neurooncol Adv. 2023;5(1):vdad142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Derry JMJ, Burns C, Frazier JP, et al. Trackable intratumor microdosing and spatial profiling provide early insights into activity of investigational agents in the intact tumor microenvironment. Clin Cancer Res. 2023;29(18):3813‐3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Duchatel RJ, Jackson ER, Parackal SG, et al. PI3K/mTOR is a therapeutically targetable genetic dependency in diffuse intrinsic pontine glioma. J Clin Invest. 2024;134(6):e170329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Seo ES, Lee B, Hwang I, Kim J‐Y, Park K, Park W‐Y. Decoding spatial organization maps and context‐specific landscapes of breast cancer and its microenvironment via high‐resolution spatial transcriptomic analysis. bioRxiv. 2023;2023.2010.2025.563904. [Google Scholar]
- 181. Mangoli A, Wu S, Liu HQ, et al. Ataxia‐telangiectasia mutated (Atm) disruption sensitizes spatially‐directed H3.3K27M/TP53 diffuse midline gliomas to radiation therapy. bioRxiv. 2023;2023.2010.2018.562892. [Google Scholar]
- 182. Patel AG, Ashenberg O, Collins NB, et al. A spatial cell atlas of neuroblastoma reveals developmental, epigenetic and spatial axis of tumor heterogeneity. bioRxiv. 2024;2024.2001.2007.574538. [Google Scholar]
- 183. Hirz T, Mei S, Sarkar H, et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single‐cell and spatial transcriptomic analyses. Nat Commun. 2023;14(1):663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Andersson A, Larsson L, Stenbeck L, et al. Spatial deconvolution of HER2‐positive breast cancer delineates tumor‐associated cell type interactions. Nat Commun. 2021;12(1):6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Mao X, Zhou D, Lin K, et al. Single‐cell and spatial transcriptome analyses revealed cell heterogeneity and immune environment alternations in metastatic axillary lymph nodes in breast cancer. Cancer Immunol Immunother. 2023;72(3):679‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sun H, Li Y, Zhang Y, et al. The relevance between hypoxia‐dependent spatial transcriptomics and the prognosis and efficacy of immunotherapy in claudin‐low breast cancer. Front Immunol. 2022;13:1042835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Nejman D, Livyatan I, Fuks G, et al. The human tumor microbiome is composed of tumor type‐specific intracellular bacteria. Science. 2020;368(6494):973‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Galeano Nino JL, Wu H, LaCourse KD, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. 2022;611(7937):810‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Wong‐Rolle A, Dong Q, Zhu Y, et al. Spatial meta‐transcriptomics reveal associations of intratumor bacteria burden with lung cancer cells showing a distinct oncogenic signature. J Immunother Cancer. 2022;10(7):e004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Larroquette M, Guegan JP, Besse B, et al. Spatial transcriptomics of macrophage infiltration in non‐small cell lung cancer reveals determinants of sensitivity and resistance to anti‐PD1/PD‐L1 antibodies. J Immunother Cancer. 2022;10(5):e003890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Zhu J, Fan Y, Xiong Y, et al. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single‐cell RNA sequencing and spatial transcriptomics. Exp Mol Med. 2022;54(11):2060‐2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Wang Y, Liu B, Min Q, et al. Spatial transcriptomics delineates molecular features and cellular plasticity in lung adenocarcinoma progression. Cell Discov. 2023;9(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Nirmal AJ, Maliga Z, Vallius T, et al. The spatial landscape of progression and immunoediting in primary melanoma at single‐cell resolution. Cancer Discov. 2022;12(6):1518‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Liu Y, Xun Z, Ma K, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78(4):770‐782. [DOI] [PubMed] [Google Scholar]
- 195. Sun Y, Wu P, Zhang Z, et al. Integrated multi‐omics profiling to dissect the spatiotemporal evolution of metastatic hepatocellular carcinoma. Cancer Cell. 2024;42(1):135‐156 e117. [DOI] [PubMed] [Google Scholar]
- 196. Hwang WL, Jagadeesh KA, Guo JA, et al. Single‐nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet. 2022;54(8):1178‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Cui Zhou D, Jayasinghe RG, Chen S, et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo‐resistant pancreatic cancer. Nat Genet. 2022;54(9):1390‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Hong WF, Zhang F, Wang N, et al. Dynamic immunoediting by macrophages in homologous recombination deficiency‐stratified pancreatic ductal adenocarcinoma. Drug Resist Updat. 2024;76:101115. [DOI] [PubMed] [Google Scholar]
- 199. Wang N, Wang R, Li X, et al. Tumor microenvironment profiles reveal distinct therapy‐oriented proteogenomic characteristics in colorectal cancer. Front Bioeng Biotechnol. 2021;9:757378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Qi J, Sun H, Zhang Y, et al. Single‐cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun. 2022;13(1):1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Kumar V, Ramnarayanan K, Sundar R, et al. Single‐cell atlas of lineage states, tumor microenvironment, and subtype‐specific expression programs in gastric cancer. Cancer Discov. 2022;12(3):670‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Yeh CY, Aguirre K, Laveroni O, et al. Mapping ovarian cancer spatial organization uncovers immune evasion drivers at the genetic, cellular, and tissue level. bioRxiv. 2023;2023.2010.2016.562592. [Google Scholar]
- 203. Liu X, Zhao S, Wang K, et al. Spatial transcriptomics analysis of esophageal squamous precancerous lesions and their progression to esophageal cancer. Nat Commun. 2023;14(1):4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Zheng H, An M, Luo Y, et al. PDGFRalpha(+)ITGA11(+) fibroblasts foster early‐stage cancer lymphovascular invasion and lymphatic metastasis via ITGA11‐SELE interplay. Cancer Cell. 2024;42(4):682‐700 e612. [DOI] [PubMed] [Google Scholar]
- 205. Vo T, Balderson B, Jones K, et al. Spatial transcriptomic analysis of Sonic hedgehog medulloblastoma identifies that the loss of heterogeneity and promotion of differentiation underlies the response to CDK4/6 inhibition. Genome Med. 2023;15(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Ning K, Li Z, Liu H, et al. Perirenal fat thickness significantly associated with prognosis of metastatic renal cell cancer patients receiving anti‐VEGF therapy. Nutrients. 2022;14(16)3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Xie F, Xi N, Han Z, et al. Progress in research on tumor microenvironment‐based spatial omics technologies. Oncol Res. 2023;31(6):877‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Scott EC, Baines AC, Gong Y, et al. Trends in the approval of cancer therapies by the FDA in the twenty‐first century. Nat Rev Drug Discov. 2023;22(8):625‐640. [DOI] [PubMed] [Google Scholar]
- 209. Niu X, Liu W, Zhang Y, et al. Cancer plasticity in therapy resistance: mechanisms and novel strategies. Drug Resist Updat. 2024;76:101114. [DOI] [PubMed] [Google Scholar]
- 210. Zhang L, Chen D, Song D, et al. Clinical and translational values of spatial transcriptomics. Signal Transduct Target Ther. 2022;7(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Wang Y, Mashock M, Tong Z, et al. Changing technologies of RNA sequencing and their applications in clinical oncology. Front Oncol. 2020;10:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Gan X, Dong W, You W, et al. Spatial multimodal analysis revealed tertiary lymphoid structures as a risk stratification indicator in combined hepatocellular‐cholangiocarcinoma. Cancer Lett. 2024;581:216513. [DOI] [PubMed] [Google Scholar]
- 213. Kiuru M, Kriner MA, Wong S, et al. High‐plex spatial RNA profiling reveals cell type‒specific biomarker expression during melanoma development. J Invest Dermatol. 2022;142(5):1401‐1412 e1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ferri‐Borgogno S, Zhu Y, Sheng J, et al. Spatial transcriptomics depict ligand‐receptor cross‐talk heterogeneity at the tumor‐stroma interface in long‐term ovarian cancer survivors. Cancer Res. 2023;83(9):1503‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Monkman J, Kim H, Mayer A, et al. Multi‐omic and spatial dissection of immunotherapy response groups in non‐small cell lung cancer. Immunology. 2023;169(4):487‐502. [DOI] [PubMed] [Google Scholar]
- 216. Park S, Hong C, Cheong J‐H, et al. Abstract 2262: spatial architecture and cellular interactions of tumor immune microenvironment to discover biomarkers and predict immune checkpoint inhibitor response in gastric cancer. Cancer Res. 2023;83(7_Supplement):2262‐2262.37145144 [Google Scholar]
- 217. Moutafi M, Martinez‐Morilla S, Garcia‐Milian R, et al. Abstract 2027: Spatial omics and multiplexed imaging to discover new biomarkers of response or resistance to immune checkpoint inhibitors (ICI) in advanced non‐small cell lung cancer (NSCLC). Cancer Res. 2022;82(12_Supplement):2027‐2027. [Google Scholar]
- 218. Yang L, Zhang Z, Dong J, et al. Multi‐dimensional characterization of immunological profiles in small cell lung cancer uncovers clinically relevant immune subtypes with distinct prognoses and therapeutic vulnerabilities. Pharmacol Res. 2023;194:106844. [DOI] [PubMed] [Google Scholar]
- 219. Zhang Z, Sun X, Liu Y, et al. Spatial transcriptome‐wide profiling of small cell lung cancer reveals intra‐tumoral molecular and subtype heterogeneity. Adv Sci (Weinh). 2024:e2402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Wang XQ, Danenberg E, Huang CS, et al. Spatial predictors of immunotherapy response in triple‐negative breast cancer. Nature. 2023;621(7980):868‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Cassier P, Eberst L, Garin G, et al. 439O ‐ A first in human, phase I trial of NP137, a first‐in‐class antibody targeting netrin‐1, in patients with advanced refractory solid tumors. Ann Oncol. 2019;30:v159. [Google Scholar]
- 222. Kornauth C, Pemovska T, Vladimer GI, et al. Functional precision medicine provides clinical benefit in advanced aggressive hematologic cancers and identifies exceptional responders. Cancer Discov. 2022;12(2):372‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Doroshow DB, Bhalla S, Beasley MB, et al. PD‐L1 as a biomarker of response to immune‐checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345‐362. [DOI] [PubMed] [Google Scholar]
- 224. Bruni D, Angell HK, Galon J. The immune contexture and immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20(11):662‐680. [DOI] [PubMed] [Google Scholar]
- 225. Murciano‐Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment‐targeting combinations. Cell Res. 2020;30(6):507‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Ling AL, Solomon IH, Landivar AM, et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature. 2023;623(7985):157‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Yi L, Ning Z, Xu L, et al. The combination treatment of oncolytic adenovirus H101 with nivolumab for refractory advanced hepatocellular carcinoma: an open‐label, single‐arm, pilot study. ESMO Open. 2024;9(2):102239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Jiang M, Li Q, Xu B. Spotlight on ideal target antigens and resistance in antibody‐drug conjugates: strategies for competitive advancement. Drug Resist Updat. 2024;75:101086. [DOI] [PubMed] [Google Scholar]
- 229. Powles T, Valderrama BP, Gupta S, et al. Enfortumab vedotin and pembrolizumab in untreated advanced urothelial cancer. N Engl J Med. 2024;390(10):875‐888. [DOI] [PubMed] [Google Scholar]
- 230. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175(2):313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Lu S, Stein JE, Rimm DL, et al. Comparison of biomarker modalities for predicting response to PD‐1/PD‐L1 checkpoint blockade: a systematic review and meta‐analysis. JAMA Oncol. 2019;5(8):1195‐1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Engblom C, Thrane K, Lin Q, et al. Spatial transcriptomics of B cell and T cell receptors reveals lymphocyte clonal dynamics. Science. 2023;382(6675):eadf8486. [DOI] [PubMed] [Google Scholar]
- 233. Schott M, Leon‐Perinan D, Splendiani E, et al. Open‐ST: High‐resolution spatial transcriptomics in 3D. Cell. 2024;187(15):3953‐3972 e3926. [DOI] [PubMed] [Google Scholar]
- 234. Yang L, Li A, Wang Y, Zhang Y. Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy. Signal Transduct Target Ther. 2023;8(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Lotstedt B, Strazar M, Xavier R, Regev A, Vickovic S. Spatial host‐microbiome sequencing reveals niches in the mouse gut. Nat Biotechnol. 2023;42(9):1394‐1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Magoulopoulou A, Qian X, Pediatama Setiabudiawan T, et al. Spatial resolution of mycobacterium tuberculosis bacteria and their surrounding immune environments based on selected key transcripts in mouse lungs. Front Immunol. 2022;13:876321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data collections including all technical aspects and research references are manually curated, presented in relevant figures and available upon request.