

Abstract
Hsp70 are ubiquitous, versatile molecular chaperones that cyclically interact with substrate protein(s). The initial step requires synergistic interaction of a substrate and a J-domain protein (JDP) cochaperone, via its J-domain, with Hsp70 to stimulate hydrolysis of its bound ATP. This hydrolysis drives conformational changes in Hsp70 that stabilize substrate binding. However, because of the transient nature of substrate and JDP interactions, this key step is not well understood. Here we leverage a well characterized Hsp70 system specialized for iron-sulfur cluster biogenesis, which like many systems, has a JDP that binds substrate on its own. Utilizing an ATPase-deficient Hsp70 variant, we isolated a Hsp70-JDP-substrate tripartite complex. Complex formation and stability depended on residues previously identified as essential for bipartite interactions: JDP-substrate, Hsp70-substrate and J-domain-Hsp70. Computational docking based on the established J-domain-Hsp70(ATP) interaction placed the substrate close to its predicted position in the peptide-binding cleft, with the JDP having the same architecture as when in a bipartite complex with substrate. Together, our results indicate that the structurally rigid JDP-substrate complex recruits Hsp70(ATP) via precise positioning of J-domain and substrate at their respective interaction sites - resulting in functionally high affinity (i.e., avidity). The exceptionally high avidity observed for this specialized system may be unusual because of the rigid architecture of its JDP and the additional JDP-Hsp70 interaction site uncovered in this study. However, functionally important avidity driven by JDP-substrate interactions is likely sufficient to explain synergistic ATPase stimulation and efficient substrate trapping in many Hsp70 systems.
Keywords: molecular chaperones, Hsp70 ATPase cycle, substrate delivery, Ssq1, Hsc20
Introduction
Via cyclic interaction with substrate proteins, Hsp70 molecular chaperones play key roles in general protein homeostasis, as well as many critical cellular pathways. Regardless of the particular substrate or cellular pathway, the substrate-Hsp70 interaction is driven by the same conformational changes of Hsp70 - induced by ATP hydrolysis, reversed by nucleotide exchange [1–4]. In the ATP bound conformation Hsp70’s peptide binding cleft, located in the β subdomain of the substrate binding domain (SBDβ), is open – thus available for rapid binding of substrate, but also allowing its rapid release. ATP hydrolysis leads to trapping of the substrate – covering of the cleft by the “lid” (i.e., the SBDα subdomain), minimizing escape. In this ADP-conformation only the linker connects the nucleotide binding domain (NBD) and SBD, while in the ATP conformation, the linker and SBDβ dock onto the NBD, as can SBDα, as well.
A key to this efficient nonequilibrium binding of substrates by Hsp70 is the timing of ATP hydrolysis such that the transiently interacting substrate is trapped in the cleft [5]. However, though the presence of substrate in the cleft does result in an increase in ATPase activity, it is very modest. Efficient substrate trapping requires a J-domain protein (JDP) cochaperone [6, 7]. Together J-domains and substrates, synergistically stimulate Hsp70’s ATPase activity [8–11]. J-domains interact at the NBD-linker-SBDβ interface of Hsp70(ATP) [12]. The invariant, His, Pro, Asp (HPD) motif critical for ATPase stimulation is in a loop following helix II, whose residues are also critical for Hsp70 binding. As many JDPs bind proteins that are also Hsp70 substrates, this step of the Hsp70 substrate interaction cycle is often referred to as substrate delivery [3]. However, experimental information about this step is scant [11, 13–17], primarily because of the transient nature of the interactions – both between the J-domain and Hsp70(ATP), and between substrate and Hsp70(ATP). To investigate this step in the cycle we have taken advantage of a specialized JDP/Hsp70 system in which both the JDP and the Hsp70 bind a single substrate [18, 19]. We reasoned that if delivery by JDPs to Hsp70 is a mechanistically important feature of JDP/Hsp70 systems a specialized one would be most likely to have evolved such characteristics, as it lacks constraints conferred by having multiple substrates.
The JDP/Hsp70 system we chose for analysis is in mitochondria of Saccharomyces cerevisiae, functioning in the biogenesis of iron-sulfur clusters (Fe-S), prosthetic groups critical for activity of many proteins [20]. The substrate of the JDP Hsc20 and the specialized Hsp70 Ssq1 is Isu1, a small (15 kDa), single domain scaffold protein on which clusters are synthesized, and then - upon interaction with Hsc20/Ssq1 - transferred onto recipient proteins [18]. Hsc20 is a simple, 21 kDa JDP. The 11 kDa C-terminal Isu1 binding domain (CTD) is juxtaposed to the J-domain forming a rigid L-shaped structure [21–23]. The Hsc20-Isu1 binding interface involves evolutionary conserved surface-exposed hydrophobic and charged residues [23]. While Hsc20 functions as a specialized JDP in Fe-S biogenesis in bacteria and mitochondria, Ssq1 emerged as a Fe-S specialized Hsp70 in the S. cerevisiae lineage [24]. It is the result of a duplication of the multifunctional mitochondrial (mt)Hsp70 present in all mitochondria [25]. Ssq1 maintains structural and functional features of its ancestor, but evolved unusually high specificity toward its substrate Isu1 and JDP partner Hsc20 [24, 26, 27]. Ssq1 recognizes a single site on Isu1 that is distinct from the site of Hsc20 binding – LPPVK pentapeptide on a surface exposed loop [28].
Here, building on our understanding of the bipartite interactions amongst JDP Hsc20, Hsp70 Ssq1 and substrate Isu1 [23, 29–32], we report the isolation and analysis of a tripartite Ssq1-Hsc20-Isu1 complex. Our results are consistent with precise positioning of the native substrate Isu1 via the rigid JDP - driving avidity based simultaneous J-domain and substrate interaction with Hsp70 for efficient substrate trapping.
Results
Tripartite Ssq1-Hsc20-Isu1 complex is dependent on established bipartite interaction sites
To test whether a tripartite Ssq1-Hsc20-Isu1 complex could be isolated we took advantage of well chracterized Hsp70 mutational variants, in which an evolutionary conserved Thr residue involved in ATP hydrolysis is replaced by Ala – such variants bind ATP but their ATPase is strongly reduced [33]. We carried out pulldown assays using Ssq1T239A (henceforth called Ssq1*), which, as expected, is defective in ATP hydrolysis (Figure S1). Isu1 tagged at the C-terminus with glutathione-S-transferase (Isu1GST), allowing it to be pulled down by glutathione resin, was used (Figure 1(A)) [34]. As expected, little Ssq1* was pulled down when added to Isu1GST alone. However, when Hsc20 was included, Ssq1*, as well as Hsc20, were pulled down (Figures 1(B) and S2(A)). When wild-type Ssq1 rather than Ssq1* was used, it was pulled down, but Hsc20 was not - consistent with the ability of catalytic amounts of Hsc20 to drive stable binding of Isu1 by Ssq1 after ATP hydrolysis (Figure S2(C)) [25].
Figure 1. Reconstitution of tripartite Ssq1*-Hsc20-Isu1 complex.
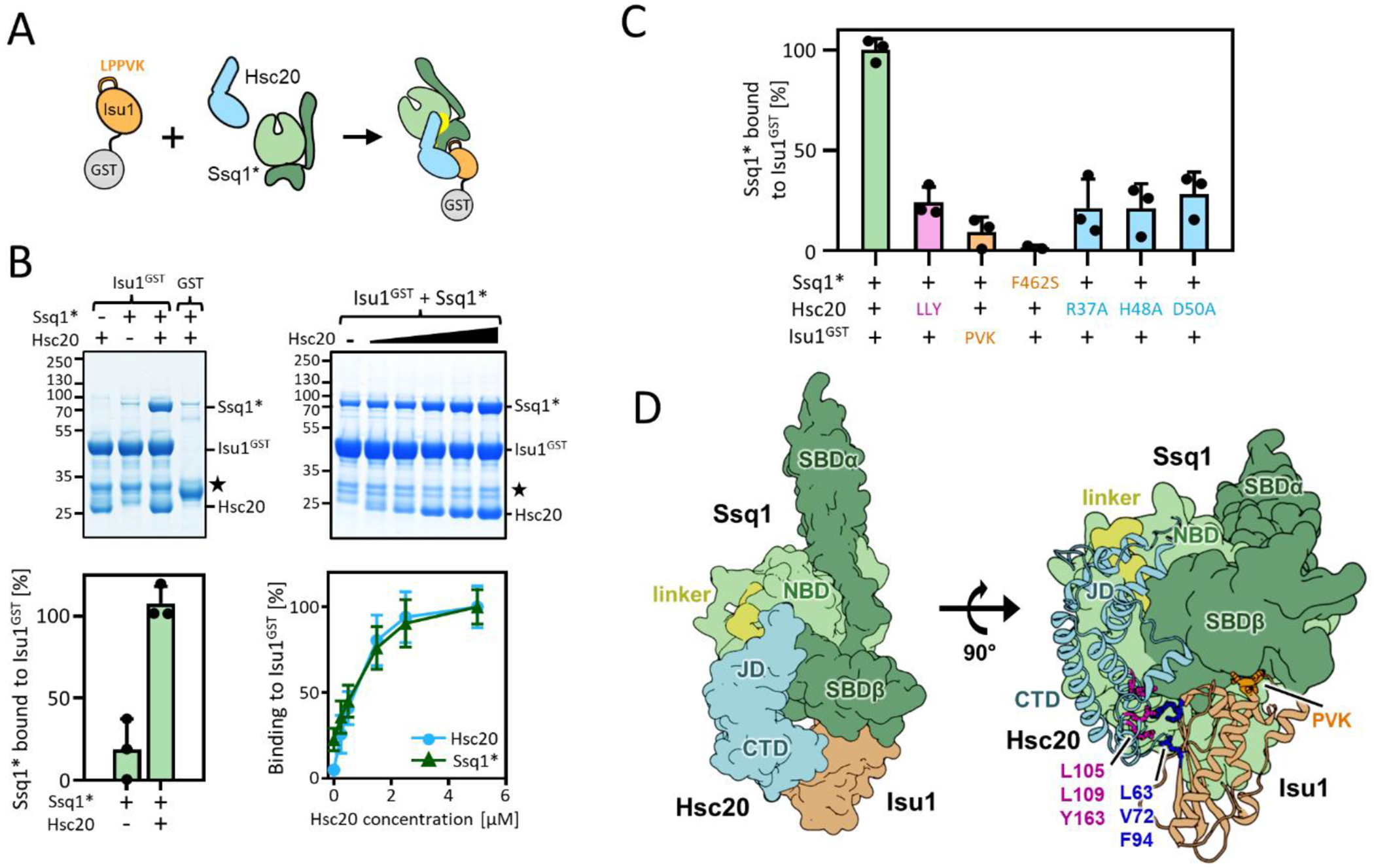
(A-C) To assess tripartite complex formation pulldown assays were performed. Isu1GST (2.5 μM) was incubated in the presence of Hsc20 and/or Ssq1* (2.5 μM and 5 μM, respectively unless otherwise indicated). Glutathione resin was added to pull-down GST and associated proteins, which were then separated by SDS-PAGE and stained with Coomassie blue (entire gels and controls in indicated Supplemental Figures). Molecular weight markers in kDa on left. ★; Isu1GST degradation product. Amounts of pulled down Ssq1 were quantified by densitometry and corrected for background binding to GST alone for three independent experiments. Error bars represent SD. Maximum binding set at 100%. (A) Schematic of basic pulldown assay. (B) Dependence of Ssq1* pulldown on presence of Hsc20. Representative gel at top, quantitation at bottom (for details see Supplmental Materials and Methods). (left) in presence and absence of Hsc20. (right) with increasing concentrations of Hsc20 (0.0, 0.25, 0.5, 1.5, 2.5, 5.0 μM). Entire gels and controls, Figure S2. (C) Disruption of individual protein-protein interactions affects formation of tripartite complex. +, WT protein; see Table 1 for key to residues of indicated substitutions and Figures S3 and S4 for gels. (D) Computational model of the tripartite Ssq1(ATP)-Hsc20-Isu1 complex, generated by docking model of Isu1 to the previously published [32] model of Hsc20-Ssq1 complex and refining the obtained dominant structural state by all-atom MD simulations (8.6 μs). (left) Surface representation shows overall architecture of the complex: Ssq1 in the ATP bound conformation with SBDβ and SBDα (dark green) docked to NBD (light green) and interdomain linker (yellow) placed inside the NBD. Hsc20 (cyan) interacts with Ssq1 via J-domain (JD) at the NBD/SBDβ, linker interface and with Isu1 via C-terminal domain (CTD). (right) cartoon representation of Hsc20 and Isu1 in the tripartite complex show Hsc20-Isu1 and Ssq1-Isu1 interfaces; L63, V72 and F94 of Isu1 in contact with L105, L109 and Y163 of Hsc20 and LPPVK (PVK) of Isu1 in proximity to the substrate binding cleft of SBDβ. (Hsc20-Ssq1 interface, Figure S5(A).
To preliminarily assess the dependence of Ssq1* association with Isu1GST on Hsc20 concentration, the amount of Hsc20 was titrated, while holding Ssq1* and Isu1GST concentrations fixed. Proportionally increasing amounts of Ssq1* (as well as Hsc20) were pulled down, until saturation was reached when Hsc20 reached the concentration of Ssq1 in the assay (Figures 1(B) and S2(B)), consistent with a stable tripartite Ssq1*-Hsc20-Isu1GST complex. To ensure that complex formation does not depend of the presence of the GST tag on Isu1 we carried out pull-down experiments using Hsc20 or Ssq1* tagged by GST at the C- and N-termini, respectively. In both cases the untagged components were pulled down as efficiently as when Isu1 was tagged (Figure S3).
We then asked if amino acid substitutions known to disrupt individual protein-protein interactions (Table 1) affect the efficiency of Ssq1* pulldown by Isu1GST. When Hsc20 was replaced by a variant having substitutions of residues at the Hsc20-Isu1 binding site (Hsc20LLY) [23], the amount of Ssq1*pulled down was reduced by ~70% (Figures 1(C), and S4(A)). To test the effect of disruption of the interaction of Isu1 in the peptide binding cleft of Ssq1*, two variants were used - Isu1GST having a triple alanine substitution in the LPPVK Ssq1* binding site (Isu1GST-PVK) and Ssq1* having the key phenylalanine in the substrate binding cleft changed to serine (Ssq1*-F462S) (Figures 1(C) and S4(B) and (C)) [29, 35]. In both cases Hsc20 was pulled down efficiently, but less than 10% of Ssq1* was recovered with Isu1GST. Importance of the J-domain-Ssq1 interaction was assessed using alanine-substitutions in Hsc20’s J-domain, two in the HPD (H48A and D50A) and one in Helix II (R37A) (Figures 1(C) and S5) [31, 32]. As expected, Hsc20 was pulled down by Isu1GST, but Ssq1* was reduced by ~70 % in each case, indicating the importance of the J-domain-Ssq1 interaction. Taken together, the results support the idea that formation of a tripartite Ssq1*-Hsc20-Isu1 complex requires multivalent interactions between its constituents, as disruption of any of the established interactions interfere with its formation.
Table 1.
Protein variants used that have disturbed interaction between tripartite complex subunits
| Protein | Variant | Location | Affected interaction |
|---|---|---|---|
| Ssq1 | F462S | substrate binding cleft | Isu1 (Ref. 34) |
|
| |||
| Hsc20 | R37A | J-domain helix II | Ssq1 (Ref. 32) |
| H48A | J-domain HPD | Ssq1 (Ref. 31) | |
| D50A | J-domain HPD | Ssq1 (Ref. 31) | |
| LLY (L105A, L109A, Y163A) | CTD | Isu1 (Ref. 23) | |
| K132A | CTD | Ssq1 (this manuscript) | |
| K172A | CTD | Ssq1 (this manuscript) | |
| KK (K132A, K172A) | CTD | Ssq1 (this manuscript) | |
|
| |||
| Isu1 | PVK (P135A, V136A, K137A) | LPPVK (Ssq1 substrate) | Ssq1 (Ref. 29) |
The dependence on Hsc20’s known bivalent interaction sites for pulldown of Ssq1* by Isu1GST, motivated us to generate a structural model of the tripartite complex - using molecular docking and all-atom molecular dynamics (MD) simulations. Isu1 was docked to the previously characterized Hsc20-Ssq1(ATP) structural model in which the J-domain positions at the conserved NBD-linker-SBDβ interface of Ssq1 [32]. In the obtained dominant structural state - accounting for 54% of the structures, with no other states accounted for more than 8% - the J-domain binding interface was the same as in the initial Hsc20-Ssq1(ATP) complex (Figures 1(D) and S6(A)) [32]. Hsc20 also maintained its original, rigid structure [23]. Isu1 positioned having two contact points: (1) the known Hsc20-Isu1 binding interface; (2) the LPPVK of Isu1 located in proximity to the SBDβ substrate binding cleft of Ssq1 Figures 1(D) and S6(B) [23, 29, 30]. Furthermore, the spatial arrangement of Hsc20 and Isu1 in the tripartite complex closely resembled that in the modeled bipartite complex (Figure S6(C)). Overall, the modeling suggests that upon formation of the tripartite complex none of the components undergo major conformational changes.
Hydrogen-deuterium exchange analysis of tripartite complex
To obtain the tripartite complex in quantities suitable for biophysical analysis we co-expressed the three components in Escherichia coli. The major peaks obtained from size exclusion chromatography contained Ssq1, Hsc20, Isu1 (peak A) and Hsc20, Isu1 (peak B) (Figures 2 and S7). Native mass spectrometry (MS) revealed a molecular weights of 107.6 kDa for the tripartite complex and 35.8 kDa for the Hsc20-Isu1 complex consistent with a 1:1:1 and 1:1 stoichiometries.
Figure 2. Isolation of the tripartite Ssq1*-Hsc20-Isu1 and bipartite Hsc20-Isu1 complexes from E. coli co-expressing their constituents.
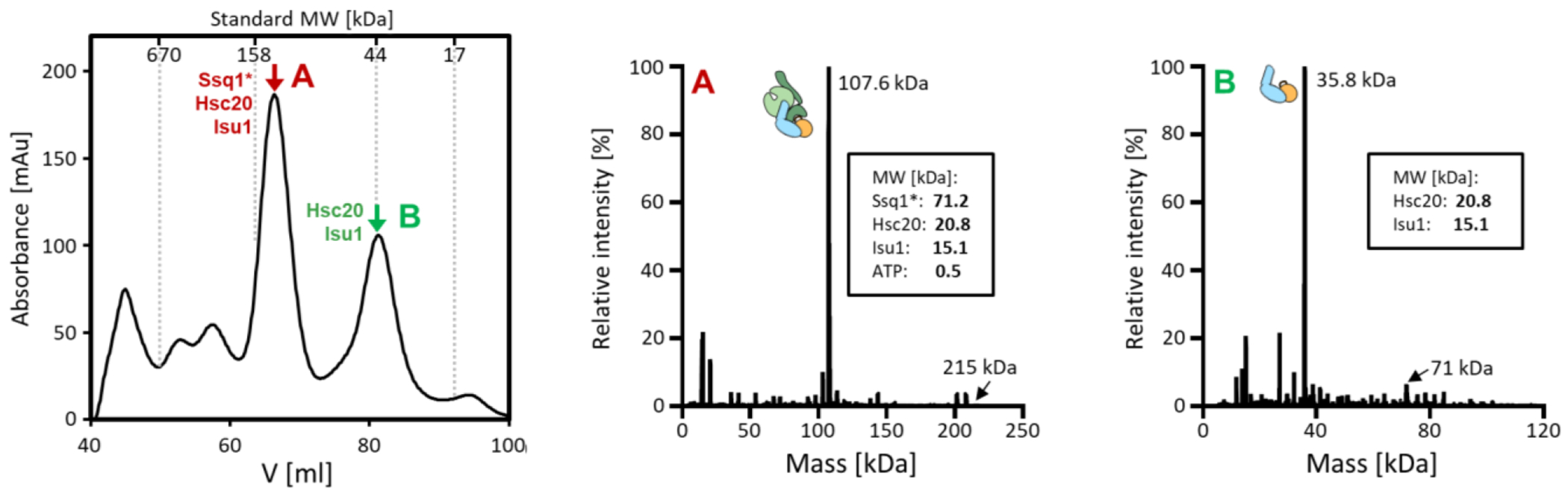
(left) Size exclusion chromatography of Ni-NTA purified complexes; peak A, tripartite and peak B, bipartite. For identification of proteins in peak A and B see Figure S7.
(middle, right) Deconvoluted native mass spectrometry (MS) spectra of the tripartite (middle) and bipartite (right) complexes- predicted molecular weights are indicated. For the original native MS spectra see FigureS7.
To experimentally assess dynamic features of Ssq1*, alone and in the tripartite complex, we used hydrogen-deuterium exchange (HDX) in conjunction with MS [36, 37], monitoring the exchange of main chain amide protons over time upon addition of deuterium (Figure 3(A)). Expected differences emerged from a global comparison of the 30 s exchange time point (Figures 3(B), (C) and S8(A)); for other time points see Figures S8(B) and S9(A) and (B). Compared to Ssq1* alone, deuterium uptake for most of the NBD, SBDβ and interdomain linker of Ssq1 in the tripartite complex was slower - indicating conformational stabilization, as would be expected with binding of Hsc20’s J-domain at the NBD-linker-SBDβ interface and LPPVK of Isu1 at the binding cleft of SBDβ (Figures 3(B–D) and S8–S9). A different picture emerged from analysis of SBDα. First, the rates of exchange for its N- and C- terminal α-helices were the same for the tripartite complex and Ssq1* alone. Second, the exchange rate for the N-terminal helix was very rapid, consistent with this portion of the lid being highly dynamic, regardless of whether in the complex or not.
Figure 3. HDX-MS analysis of Ssq1 alone and in the tripartite Ssq1*-Hsc20-Isu1 complex.
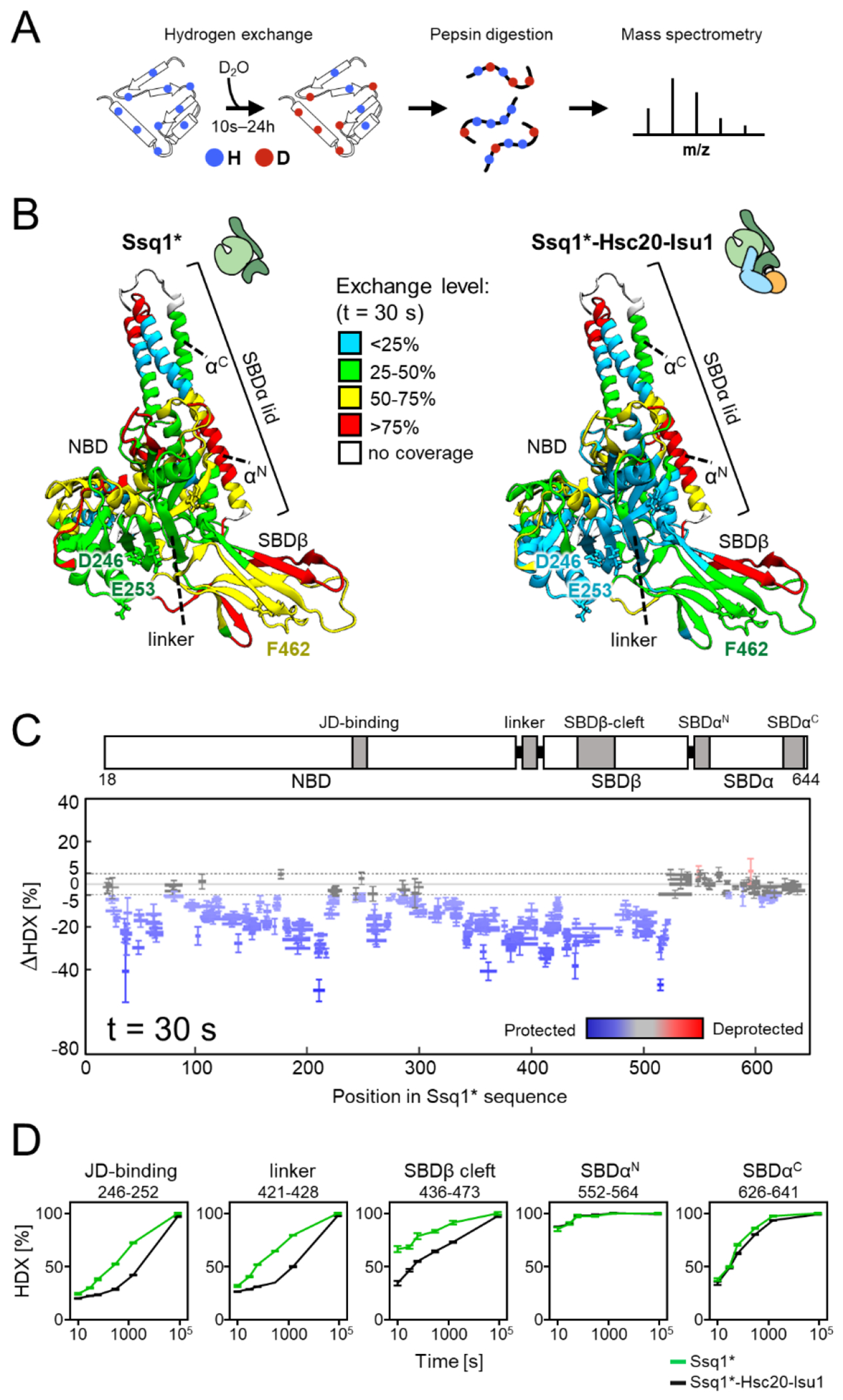
(A) Scheme of HDX-MS experiment. Proteins, alone or in complexes, were incubated in D2O buffer allowing deuterium incorporation into the polypeptide backbone. After quenching exchange at a number of points in time and subsequent proteolysis, peptides were subjected to MS analysis to determine the increase in mass resulting from deuterium uptake.
(B-C) Deuterium uptake of Ssq1* alone and in the tripartite Ssq1*-Hsc20-Isu1 complex. Data shown are for 30 s exchange time point. (B) Relative fractional deuterium uptake mapped on the structural model of Ssq1*(ATP). Nucleotide binding domain (NBD), substrate binding subdomains (SBDβ, having substrate binding cleft; SBDα “lid”), interdomain linker (linker); N-terminal (αN) and C-terminal (αC) lid helices. D246, E253 – residues of the NBD interacting with Hsc20; F462 residue of the substrate binding cleft of SBDβ interacting with LPPVK of Isu1. (C) Difference in deuterium uptake between Ssq1* alone and in the tripartite complex. Horizontal lines indicate peptides observed; differences in deuterium uptake colored – blue (retarded), red (accelerated). Error bars represent uncertainty in difference in deuterium uptake for a given peptide. (D) Kinetics of relative deuterium uptake into selected peptides of Ssq1* alone (green), tripartite complex (black).
We also carried out HDX on Hsc20 and Isu1 alone, in complex with each other and in the tripartite complex. The exchange rates for Hsc20 alone (Figures S10 and S11) were consistent with its rigid structure. The junction region between the two domains displayed the slowest rates, which were retarded even further upon interaction with Isu1 and Ssq1*. As expected, rates of exchange for CTD and J-domain segments involved in interaction with Isu1 and Ssq1*, respectively, were also slower in the tripartite complex (Figure S11). In striking contrast to Ssq1* and Hsc20, very fast exchange rates were observed for Isu1 (Figure S12), consistent with its conformational flexibility reported previously in NMR experiments [38, 39]. Although this feature curtailed detailed analysis of HDX patterns, slower exchange rates were observed for Isu1 when in complex with Hsc20 and in the tripartite complex, consistent with structural stabilization.
Kinetics of tripartite complex formation
Equilibrium pull-down and HDX-MS experiments allow monitoring interaction of individual proteins and their conformational dynamics, but do not provide information about the kinetics of complex formation. Because a biologically meaningful tripartite complex must form rapidly, we analyzed the complex formation using biolayer interferometry (BLI), which allows time resolved measurements. Isu1GST was immobilized on anti-GST antibody-coated BLI sensors, which were then introduced into solutions containing ATP and one or more test proteins, that is the association phase (Figure 4(A)). When Hsc20 or Ssq1* was added individually the results were consistent with the pulldown experiments: Hsc20 was rapidly recruited to the sensor; no binding signal was observed for Ssq1* alone. In addition, consistent with efficient formation of a tripartite complex, the binding signal of Ssq1* and Hsc20 together was higher than Hsc20 alone, with the magnitude of the difference being proportional to the amount of Hsc20 added (Figure S13). This signal increased rapidly, reaching 50% of maximum in less than 9 seconds. When the sensor was moved to a solution containing no protein (i.e., the dissociation phase), the signal decreased.
Figure 4. Kinetics of the tripartite complex formation.
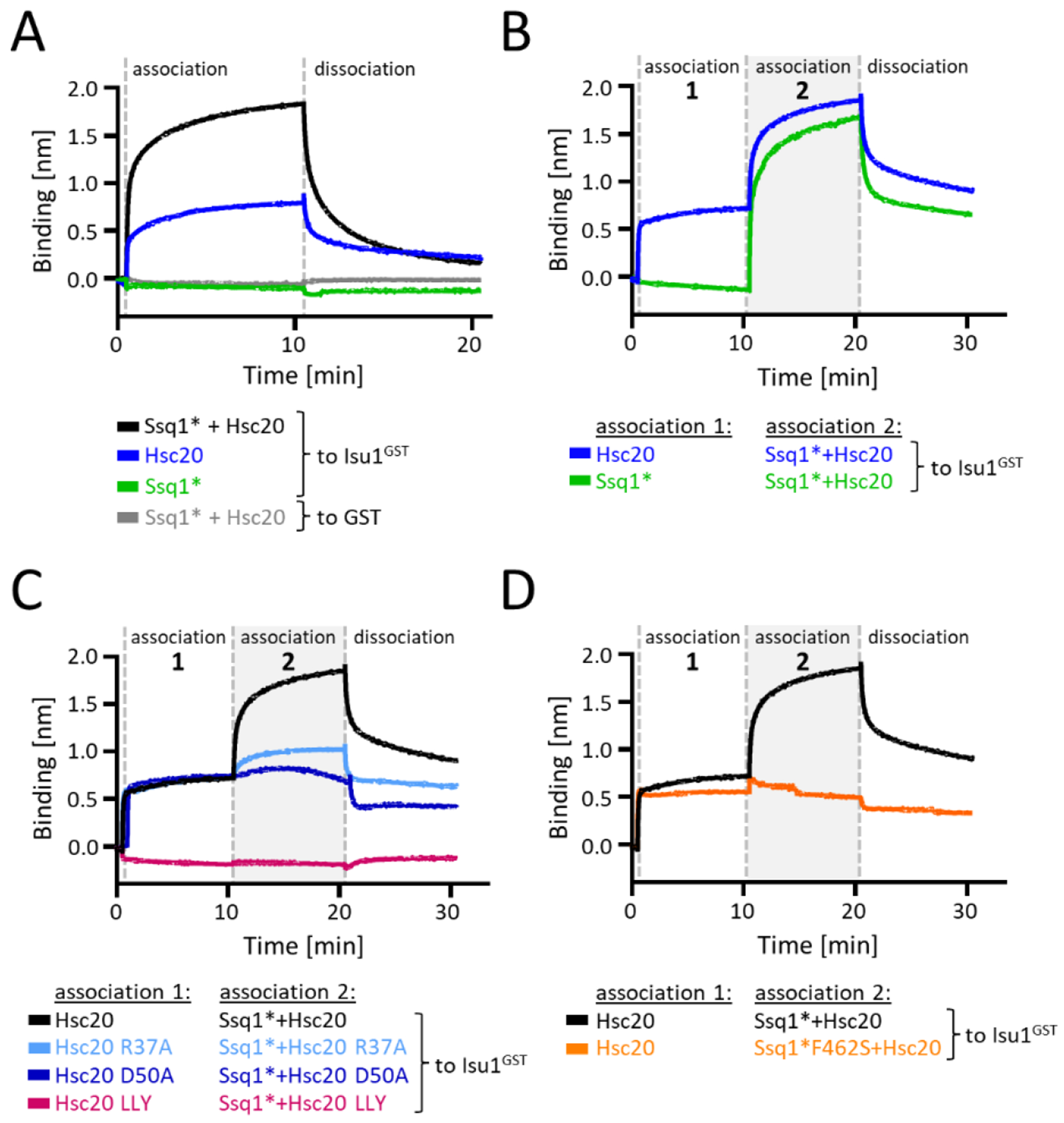
(A-D) BLI analysis of Isu1GST interaction with Ssq1* and Hsc20, and their variants. BLI sensors were loaded with Isu1GST or GST (background control). Association: loaded sensors were inserted into solutions containing Ssq1* and/or Hsc20. If two step was performed (B-D), 10 min later sensors were moved to a solution containing indicated combination of Ssq1* and/or Hsc20 for an additional 10 min. Dissociation phase was initiated by placing sensors into solutions without proteins. (A) One step association analysis of Ssq1* and Ssq1*/Hsc20 using Isu1GST and, as a control, GST. (B) Two step analysis of Isu1GST interaction with Ssq1* and Hsc20, assessing effect of order of addition of Hsc20 and Ssq1*. (C) Two step analysis of interaction of Isu1GST with Ssq1* and indicated variants defective in Ssq1* interaction: Hsc20 (R37A or D50A); Isu1LLY (LLY). (D) Two step analysis of Isu1GST interaction with Hsc20 and Ssq1* variant (F426S) defective in interaction with Isu1.
On closer inspection, two phases of binding were apparent– a rapid initial phase followed by slower binding. We therefore divided association into two steps, first one component, then both together to ensure binding equilibrium of the first is maintained (Figure 4(B)). When Isu1GST loaded sensor was placed in Hsc20 solution (first step) rapid binding was observed, followed by slower binding when the sensor was moved to a mixture that contained Ssq1* as well (second step). We interpret this result as indicating rapid formation of the Hsc20-Isu1 complex (first step), followed by slower binding of Ssq1* to the Hsc20-Isu1 complex (second step). When, the order of addition was reversed, no binding of Ssq1* was observed in the first step, as expected, while efficient formation of the tripartite complex was observed in the second step when Hsc20 was also present.
Next, we tested how disruption of bipartite protein-protein interactions affect the kinetics of tripartite complex formation (Figure 4(C) and (D)). Consistent with our pulldown results: (1) no signal above background was observed for an Hsc20 variant defective in Isu1 interaction; (2) no signal above that observed for Hsc20 alone for variants defective in J-domain-Ssq1 or Ssq1-Isu1 interaction. We conclude that the tripartite complex forms rapidly, but disruption of any of the bipartite interactions between its constituents severely impedes its formation.
An additional interaction site between Hsc20 and Ssq1 stabilizes the tripartite complex
The surprising stability of the tripartite complex led us to reinspect the structural model. Positively charged K132 and K172 of Hsc20’s CTD and negatively charged D364 of Ssq1’s NBD (Figures 5(A) and S14) stood out as potentially forming an additional interaction site. A peptide encompassing D364 had retarded deuterium uptake in the tripartite complex compared to Ssq1* alone (Figure 5(B)) supporting this idea. Therefore, we carried out pulldown and BLI experiments. When Hsc20 K132A and K172A variants were tested for tripartite complex formation the amount of Ssq1* pulled down with Isu1GST was reduced by ~40% and ~60%, respectively (Figures 5(C) and S15). When the double variant was used, the amount of pulled down Ssq1* was reduced to the background level. In the BLI assay the signal for each single variant was substantially less than for wild-type (WT) Hsc20. When the double Hsc20 variant was used, no signal was detected above that observed when no Hsc20 was present (Figure 5(D)) . We conclude that the Hsc20 CTD/Ssq1 NBD interaction plays a role in formation of the Ssq1*-Hsc20-Isu1 tripartite complex and may in part explain its unusual stability.
Figure 5. Novel interaction between CTD of Hsc20 and NBD of Ssq1 is required for the tripartite complex formation.
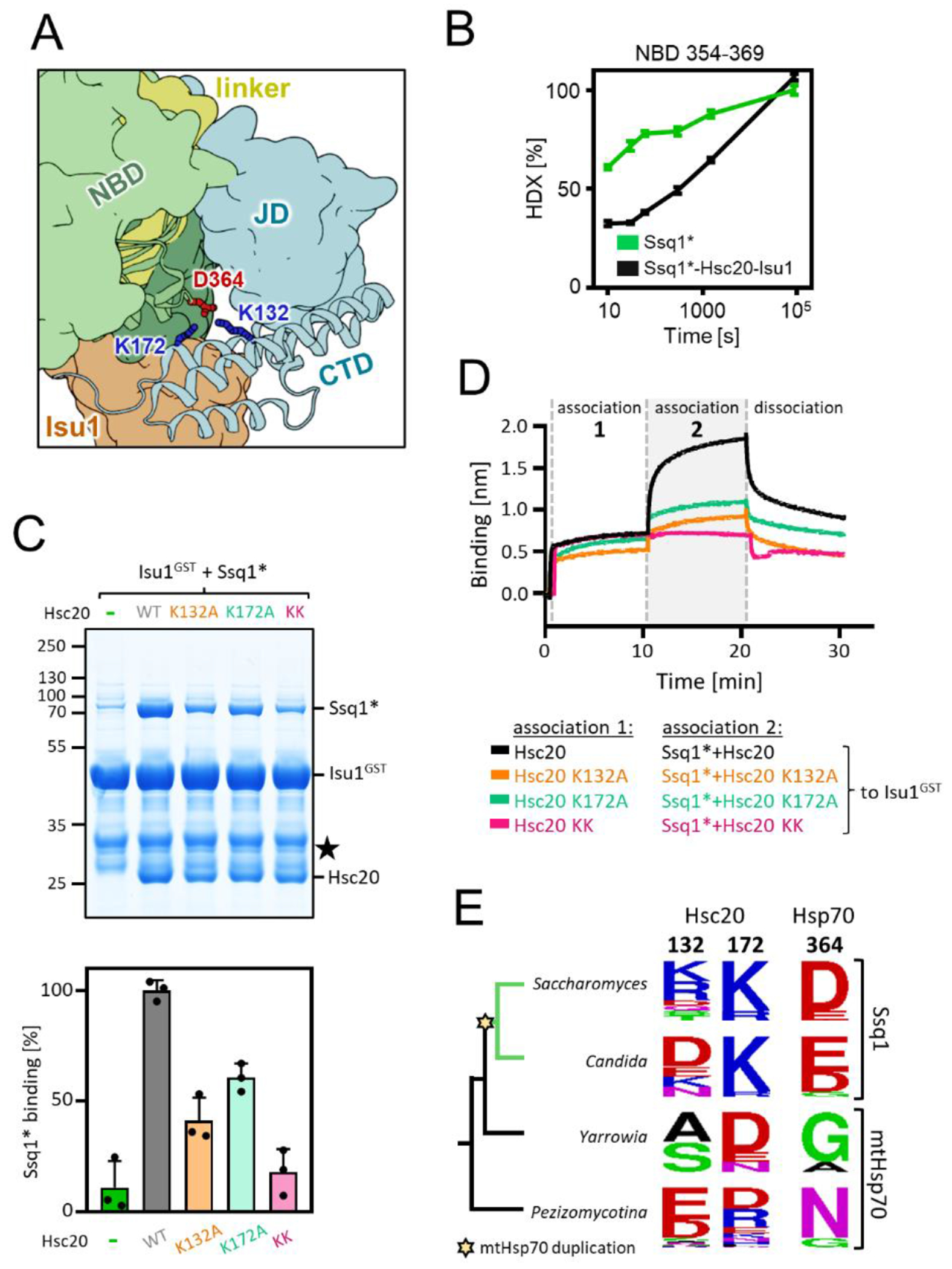
(A) Fragment of the structural model of the tripartite complex showing interaction between K132 and K172 of Hsc20’s CTD and D364 of Ssq1’s NBD- the novel interaction site.
(B) Kinetics of relative deuterium uptake into the D364-containing Ssq1* peptide (354-369). Ssq1* alone (green); tripartite complex (black). (C) (top) Isu1GST (2.5 μM) was incubated with Ssq1* (5 μM) and Hsc20 WT or K132A, K172A or double K132A/K172A (KK) variants (2.5 μM). Glutathione resin was added to pull-down GST and associated proteins, which were then separated by SDS-PAGE and stained with Coomassie blue; entire gel, loading controls and quantification in Figure S12. Molecular weight markers in kDa on left ★ - marks the Isu1GST degradation product. (bottom) Amounts of Ssq1* pulled down were quantified by densitometry and corrected for background binding to GST alone; Ssq1* levels for Isu1GST interacting with Hsc20 and Ssq1* were set at 100%. Error bars represent SD. (D) BLI analysis of Isu1GST interaction with Ssq1* and Hsc20 variants defective in the novel Hsc20-Ssq1 interaction. Association-1, BLI sensors loaded with Isu1GST were placed in solution containing Hsc20 WT or variants K132A, K172A or double K132A/K172A (KK) (1 μM); association-2, the sensors were placed in solution containing Ssq1* (1 μM) and Hsc20 WT or variant (1 μM). Dissociation, the sensors were placed in solution without proteins.
(E) Sequence conservation of Hsc20 and Hsp70 positions involved in the novel Hsc20-Ssq1 interaction in S. cerevisiae across fungi post-duplication species (green branches) in which Hsc20 functions with Ssq1 and non-duplication species (black branches) in which Hsc20 functions with mtHsp70. Star indicates emergence of Ssq1 as result of mtHsp70 gene duplication.
To gain insight into the origin and evolution of the additional interaction site we first analyzed Ssq1, mtHsp70, and Hsc20 sequences from a wide variety of eukaryotes. Finding that positions homologous to D364 of Ssq1 and K132/K172 of Hsc20 are not universally conserved (Figure. S16(A)), we focused on fungi closely related to S. cerevisiae, including both species in which Hsc20 functions with Ssq1 (i.e., post-duplication) and those in which Hsc20 functions with mtHsp70 (i.e., non-duplication) (Figure 5(E)). In non-duplication species positions 364 of Ssq1 and 132/172 of Hsc20 are not conserved. However, in post-duplication species Ssq1s have mostly acidic amino acids (D, E) at position 364 and Hsc20s have mostly basic amino acids (K, R) at position 172. This pattern of conservation suggests that a new electrostatic interaction between an acidic residue at position 364 of Ssq1 and a basic residue at Hsc20 position 172 evolved in the last common ancestor of post-duplication species. Indeed, maximum likelihood phylogeny predicted, with 90% probability, that the common ancestor of Ssq1 had D364 while its Hsc20 partner had K172 (Figure S16(B) and (C)). The conservation pattern of Hsc20 position 132 in post-duplication species is more complex. It is occupied by basic amino acids in most species belonging to the Saccharomyces clade (Figure 5(E)), but is variable in the sister Candida clade. This pattern suggests that the strength of this new electrostatic interaction varies among Hsc20/Ssq1 systems - strong in S. cerevisiae and other species in which positions 172 and 132 are occupied by basic amino acids, but weaker in others.
Discussion
Here we report the biochemical isolation of a tripartite complex consisting of Hsp70 Ssq1, JDP Hsc20 and their substrate protein Isu1. Formation of this 1:1:1 complex requires not only the sites necessary for the subsequent hydrolysis of bound ATP that leads to substrate “trapping” (J-domain-Hsp70 and substrate-Hsp70), but also JDP-substrate interaction sites. This dependence underscores the importance of avidity in driving the Hsp70-substrate interaction cycle by promoting simultaneous interaction of J-domain and substrate. Though the specialized system analyzed here evolved to be highly specialized, allowing isolation of the complex, it likely represents an initial step in the substrate binding cycle key to JDP/Hsp70 systems more generally.
While the dramatic effects on complex formation/stability of substitutions at the binding sites make the importance of these interactions evident, the conformation of Ssq1* in the complex is less straightforward. As expected, the results of HDX point to the docking of the NBD and SBDβ, a requirement for J-domain interaction [32, 40]. The positioning of the SBDα lid is less clear, not only for the Ssq1*(ATP)-Hsc20-Isu1 complex discussed here, but for ATP-bound Hsp70s more generally. The relatively high rate of exchange revealed in the HDX experiments and the similarity of HDX data for Ssq1* in isolation and in the complex, suggests that the SBDα lid is undocked and dynamic in both. Thus, we conclude that the lid does not play a critical role in stabilization of the tripartite complex via interaction with Isu1. Although lid docking has been well established for some ATP-bound Hsp70s [4], undocked lids have been reported for others [41–43].
Our results are informative as to how synergy of ATPase stimulation by J-domain and substrate is accomplished in this specialized system [25, 44], as well as the role of avidity in forming this remarkably stable complex. The precision of the architecture positioning the J-domain and the substrate’s Hsp70 binding site is striking. Hsc20 is a rigid molecule forming a structurally rigid complex with Isu1 [23]. When the J-domain of Hsc20 interacts with Ssq1*(ATP) via long distance electrostatic interactions [32], the bound Isu1 is positioned such that the LPPVK is in very close proximity to the substrate binding cleft of Ssq1*(ATP). This positioning results in extremely high local concentration, enabling avidity, that is functionally high affinity. The “fourth Interaction”, that between the CTD of Hsc20 and the NBD of Ssq1 illustrates the evolvability of JDP/Hsp70 systems. We think that this additional interaction, though weak, increases avidity and allows Hsc20/Ssq1 system to function at the very low concentrations present in mitochondria. While mtHsp70 is one of the most abundant mitochondrial protein, Ssq1 functions at an approximately 1000-fold lower concentration [45]. On the other hand, our evolutionary analysis shows that this interaction is restricted to S. cerevisiae and closely related species, suggesting that other Hsp20/Ssq1 and Hsc20/mtHsp70 systems function without this additional interaction. It should also be kept in mind that additional interaction(s) between JDP and its Hsp70 partner may not only increase avidity but further regulate the timing of hydrolysis [46–48].
It is easy to envision how precise positioning of the Hsc20-Isu1-Ssq1 system promotes avidity driven substrate trapping, via synergistic stimulation of ATP hydrolysis. Other systems likely achieve sufficient avidity by increasing local concentration of substrate and J-domains using less precise positioning [48–52]. JDP rigidity, though advantageous in the case of a single substrate, would likely constrain adaptability to function with many substrates. Having a more flexible, yet restricted distance between J-domain and JDP bound substrate sufficiently increases local concentrations. In the case of JDPs that do not bind substrate on their own, avidity could be achieved with help of additional “adapter” protein(s) or additional domains of the JDP itself that position substrate or JDP (or both) in close proximity to the Hsp70 partner. Such mechanisms likely operate for JDP/Hsp70 systems involved in protein import into the mitochondrial matrix and at the tunnel exit of the ribosome [53].
Materials and Methods
Protein Purification
Mutants of SSQ1 were constructed by site-directed PCR mutagenesis. Recombinant mature Ssq1 (residues 19-657) WT, Ssq1T239A (Ssq1*) and Ssq1*-F462S with a polyhistidine (His) tag at the C-terminus were purified as described in [54]. Recombinant mature Hsc20, also termed Jac1 (residues 10-184) WT and alanine substitution variants (Table 1) with the C-terminal His tag were purified as described in Supplementary Methods. Recombinant mature Isu1 (residues 36-165) with the C-terminal His tag and Isu1GST with C-terminal GST tag were purified as described in [26] and [23], respectively.
GST Pulldown Assay
Pull-down experiments were performed as described in [34]. In short, in a 150 µL reaction, 2.5 μM Isu1GST or purified GST (control for background binding) was incubated with Ssq1* and/or Hsc20 (typically 5 μM and 2.5 μM, respectively) in buffer A (40 mM HEPES–KOH, pH 7.5, 5% (v/v) glycerol, 100 mM KCl, 1 mM dithiothreitol, and 10 mM MgCl2) for 15 min at 25°C to allow complex formation. An aliquot of the reaction (7.5 μL) was taken as a loading control “input 5%”. 40 μl of glutathione-immobilized agarose beads (pre-equilibrated with 0.1% (w/v) bovine serum albumin, 0.1% (v/v) Triton X-100 and 10% (v/v) glycerol in buffer A) were added to each reaction and incubated at 4°C for 1 h with rotation. The beads were washed one time with 500 µL of buffer A and then three times with 200 µL of buffer A. Proteins bound to the beads were incubated with 20 µL of two-fold concentrated Laemmli sample buffer (125 mM Tris– HCl, pH 6.8, 5% (w/v) sodium lauryl sulfate (SDS), 10% (v/v) 2-mercaptoethanol, 20% (v/v) glycerol) for 10 min at 100°C and aliquots were loaded on SDS-PAGE (Bolt™ 12%, Bis-Tris, 1.0 mm, Mini Protein Gels, Invitrogen) and visualized by Coomassie blue staining.
Structural model of the tripartite Ssq1(ATP)-Hsc20-Isu1 complex
All simulations were performed using GROMACS 2021.3 [55], if not stated otherwise. The CHARMM36 force field [56] was used for proteins, ions and ATP-Mg, and the TIP3P model was used for water. In each of the simulation boxes, the numbers of Na+ and Cl- ions were adjusted to 0.15 M. If not stated otherwise, temperature was kept at 298.15 K with the v-rescale algorithm [57] using a coupling constant of 0.1 ps. Pressure was kept at 1 bar using the Parrinello-Rahman algorithm [58] with a coupling time of 5 ps. Periodic boundary conditions were applied and the Particle Mesh Ewald summation [59] was used to calculate long-range electrostatic interactions, with a cut-off radius of 1 nm and a Fourier grid spacing of 0.12 nm. Van der Waals interactions were calculated with Lennard-Jones potential with a cut-off radius of 1 nm. All bonds involving hydrogen were constrained using the LINCS algorithm. Leap-frog Verlet algorithm was used to integrate equations of motion with a time step of 2 fs.
A structural model of the tripartite complex was obtained by docking the homology model of Isu1 to the structural model of the Ssq1(ATP)-Hsc20 complex using ClusPro [60]: a) without restraints on protein–protein interactions; b) with attraction restraint on Isu1 LPPVK - Ssq1 F462 interaction; c) with attraction restraint on Isu1 L63,V72, F94 - Hsc20 L105, L109, Y163 interaction; d) with both restraints simultaneously. From the 382 structures obtained from docking we selected 72 that had Isu1 located near Hsc20 CTD. Redundancy among these 72 models was removed using gmx cluster tool from GROMACS package, with single-linkage clustering method and 0.5 nm root mean square deviation (RMSD) cutoff on Isu1 position, resulting in 11 distinct models of the Hsc20-Ssq1(ATP)-Isu1 complex. Next, each of these models was placed in a 16 x 16 x 16 nm dodecahedron box, solvated with ~90000 water molecules and energy-minimized. The systems were then simulated in equilibrium MD for at least 500 ns - total simulation time was 8.6 μs. Trajectories were superimposed on Cα atoms of Ssq1 NBD domain, and clustered based on backbone of Hsc20 and Isu1 using gmx cluster with Jarvis-Patrick method and 0.3 nm RMSD cutoff.
Purification of tripartite Ssq1*-Hsc20-Isu1 complex from Escherichia coli co-expressing its components
E. coli Rosetta 2 strain with plasmid pLysSRARE2 (chloramphenicol resistance) was transformed with plasmid pRSF HSC20-ISU1 (kanamycin resistance) harboring Hsc20 sequence without tag and Isu1 sequence tagged with six histidine codons at C-terminus and plasmid pETDuet1 SSQ1*-HEP1 (ampicillin resistance) harboring Ssq1 sequence tagged at C-terminus with six histidine codons and no tagged Hep1 sequence. Hep1 stabilizes Ssq1 when expressed in bacterial cells [54, 61], but does not form stable complex with Ssq1 and is not present in the tripartite complex preparation. The strain was grown in LB medium (with 50 μg/mL ampicillin, 25 μg/mL kanamycin and 10 μg/mL chloramphenicol) at 37°C. Expression was induced by addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at A600 = 0.45. After 3 hours at 30°C, cells were harvested and lysed in a French Press in buffer B (25 mM HEPES-KOH pH 7.5, 150 mM KCl, 1 mM PMSF, 10% (v/v) glycerol 2 mM magnesium acetate and 30 mM imidazole pH 7.5). After clarifying spin, the supernatant was loaded on a Novagen His-Bind Resin column (2.5 mL) equilibrated in buffer B. The column was washed with buffer B (50 mL) and subsequently with buffer B with the addition of 10 mM MgCl2 and 1 mM ATP (25 mL). Proteins were eluted with 30 mM - 500 mM imidazole gradient in buffer B (20 column volumes). Fractions containing Ssq1*, Hsc20 and Isu1 and fractions containing Hsc20 and Isu1 were concentrated using Amicon® Ultra – centrifugal filters with 100 kDa and 30 MWCO, respectively. Concentrated tripartite complex was incubated at 4°C with 1 mM dithiothreitol for 10 min before addition of 2 mM ATP and 10 mM MgCl2 and further 20 min incubation. Then the complex was subjected to size exclusion chromatography in buffer C (25 mM HEPES-KOH pH 8.0, 100 mM KCl, 5% (v/v) glycerol, 1 mM DTT and 10 mM MgCl2) using HiLoad 16/600 Superdex 200 prep grade column. Fractions containing all three proteins were concentrated using Amicon® Ultra – 4 centrifugal filter with 100 kDa MWCO, and stored at −70°C.
Hydrogen/deuterium exchange mass spectrometry (HDX-MS)
Protein samples (5 µL) were diluted 10-fold in D2O buffer D (25 mM HEPES-KOD, 100 mM KCl, 5% (v/v) glycerol, 10 mM MgCl2, 1 mM DTT, pD= 8.0) and incubated for 10 s, 1 min, 5 min, 25 min, 120 min, and additionally 30 s for samples containing Ssq1*, at room temperature. The exchange was quenched by transferring 50 µL of reaction mixture to ice-cold eppendorf tube containing 10 ul of quench buffer (2 M glycine, 4 M guanidium hydrochloride, 1 M Tris(2-carboxyethyl)phosphine (TCEP), pD=2.4). Samples were then vortexed and flash frozen in liquid nitrogen and kept frozen at −80 °C until MS data acquisition. Reactions were performed in quadruplicates (in triplicates for reactions containing Ssq1*). Control samples with maximal deuteration level were incubated in D2O buffer for 24 h and quenched as described, while minimal deuteration controls were prepared by adding the protein to the quenched reaction conditions. Deuteration levels were not corrected for back exchange.
Samples were injected into a nanoACQUITY UPLC system (Waters) and digested online using immobilized pepsin resin column (Poroszyme) with 0.07% (v/v) formic acid in water as mobile phase (200 µL/min flow rate). Peptides were loaded onto the 2.1 mm × 5 mm C18 trapping column (ACQUITY BEH C18 VanGuard precolumn, 1.7 µm resin, Waters) and eluted onto a reversed phase C18 column (Acquity UPLC BEH, 2.1 × 50 mm, 1.7 µm resin, Waters) using a 7–35% gradient of acetonitrile in 0.1% (v/v) formic acid at 90 µL/min flow rate. Temperature of all fluidics, columns, and valves, except the pepsin digestion column kept at 13°C, was maintained at 0.5°C using HDX Manager (Waters). Outlet of the C18 column was directly coupled to the ion source of SYNAPT G2-HDMS mass spectrometer (Waters) operating in IMS mode. Leucine-enkephalin (Sigma) was used for carrying out lock mass and activation. Mass spectra were acquired in IMS mode over the m/z range of 50–2000. The spectrometer parameters were set as described in [62].
Peptides were identified using ProteinLynx Global Server software (Waters) using a randomized database, based on a peptide list obtained using non-deuterated proteins, processed as described above and measured in MSE mode. The peptide list was filtered by minimum intensity (3000) and minimum product per amino acid (0.3) and analyzed using DynamX 3.0 (Waters). All MS spectra were manually inspected. Percentage of peptide deuteration was calculated using HaDeX tool [63], using minimal and maximal deuteration controls described above as 0% and 100%, respectively. Difference in number of exchanging protons was calculated using HaDeX. Exchange levels and differential deuterium exchange of peptides exhibiting exchange kinetics representative for the protein region (Supplementary Excel files) were then mapped on Ssq1, Hsc20 and Isu1 structural models using VMD [64].
Bio Layer Interferometry (BLI)
BLI measurements were performed in buffer E (40 mM HEPES–KOH, pH 7.5, 5% (v/v) glycerol, 100 mM KCl, 1 mM dithiothreitol, 10 mM MgCl2, 0.05% (v/v) Triton X-100) using a single channel BLItz instrument (Pall ForteBio) operating at room temperature. Sensograms were recorded as a function of time. After the initial equilibration step (30 s) Isu1GST or GST (10 μM) was immobilized on anti-GST biosensors in the presence of bovine serum albumin at 0.5 mg/ml (10 min). The sensors were then washed with buffer C (10 min). Following a baseline step (30 s), the sensors were immersed into solutions containing analytes to measure association, followed by immersion into protein-free solutions to record dissociation (10 min). To assess the background binding, sensors with immobilized GST were used.
Supplementary Material
Acknowledgements
This work was supported by National Science Center, Poland (OPUS 21 2021/41/B/NZ1/00449) (to J.M.) and the work of M.J., I.G., A.G-R., B.T., R.D. by Foundation for Polish Science, Poland (TEAM POIR.04.04.00-00-4114) (to J.M.) and by the National Institutes of Health (R35 GM127009) (to E.A.C.)
This research was supported by PLGrid infrastructure.
Abbreviations:
- JDP
J-domain protein
- SBD
substrate binding domain of Hsp70
- NBD
nucleotide binding domain of Hsp70
- CTD
C-terminal domain of Hsc20
- Ssq1*
Ssq1(T239A) variant defective in ATPase activit
- GST
glutathione-S-transferase
- MD
molecular dynamics simulations
- HDX
hydrogen-deuterium exchange
- MS
mass spectrometry
- BLI
bio-layer interferometry
- RMSD
root-mean-square deviation
- LB
Luria Broth media
Footnotes
Credit authorship contribution statement
Marcin Jelen: Investigation. Igor Grochowina: Investigation, Software, Formal analysis, Data Curation, Visualization. Aneta Grabinska-Rogala: Investigation. Szymon J. Ciesielski Investigation, Methodology. Katarzyna Dabrowska: Investigation, Formal analysis. Bartlomiej Tomiczek: Formal analysis, Software. Lukasz Nierzwicki: Formal analysis, Software. Wojciech Delewski: Investigation. Brenda Schilke: Investigation, Methodology. Jacek Czub: Supervision, Formal analysis. Michal Dadlez: Supervision, Formal analysis. Rafal Dutkiewicz: Supervision, Investigation, Methodology, Writing- Original Draft. Elizabeth A. Craig: Conceptualization, Supervision, Writing – Review & Editing, Funding acquisition. Jaroslaw Marszalek: Conceptualization, Supervision, Writing – Original Draft, Writing – Reviewing & Editing, Funding acquisition.
DECLARATION OF COMPETING INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Rosenzweig R, Nillegoda NB, Mayer MP & Bukau B (2019) The Hsp70 chaperone network. Nat Rev Mol Cell Biol 20, 665–80. [DOI] [PubMed] [Google Scholar]
- 2.Mayer MP & Gierasch LM (2019) Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J Biol Chem 294, 2085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clerico EM, Meng W, Pozhidaeva A, Bhasne K, Petridis C & Gierasch LM (2019) Hsp70 molecular chaperones: multifunctional allosteric holding and unfolding machines. Biochem J 476, 1653–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Liang C & Zhou L (2020) Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Sci 29, 378–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barducci A & De Los Rios P (2015) Non-equilibrium conformational dynamics in the function of molecular chaperones. Curr Opin Struct Biol 30, 161–9. [DOI] [PubMed] [Google Scholar]
- 6.Zhang R, Malinverni D, Cyr DM, Rios PL & Nillegoda NB (2023) J-domain protein chaperone circuits in proteostasis and disease. Trends Cell Biol 33, 30–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craig EA & Marszalek J (2017) How Do J-Proteins Get Hsp70 to Do So Many Different Things? Trends Biochem Sci 42, 355–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, et al. (1999) Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci USA 96, 5452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horne BE, Li TF, Genevaux P, Georgopoulos C & Landry SJ (2010) The Hsp40 J-domain Stimulates Hsp70 When Tethered by the Client to the ATPase Domain. J Biol Chem 285, 21679–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wittung-Stafshede P, Guidry J, Horne BE & Landry SJ (2003) The J-domain of Hsp40 couples ATP hydrolysis to substrate capture in Hsp70. Biochemistry 42, 4937–44. [DOI] [PubMed] [Google Scholar]
- 11.Han W & Christen P (2004) cis-Effect of DnaJ on DnaK in ternary complexes with chimeric DnaK/DnaJ-binding peptides. Febs Lett 563, 146–50. [DOI] [PubMed] [Google Scholar]
- 12.Mayer MP (2021) The Hsp70-Chaperone Machines in Bacteria. Front Mol Biosci 8, 694012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misselwitz B, Staeck O & Rapoport TA (1998) J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol Cell 2, 593–603. [DOI] [PubMed] [Google Scholar]
- 14.Mayer MP, Laufen T, Paal K, McCarty JS & Bukau B (1999) Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J Mol Biol 289, 1131–44. [DOI] [PubMed] [Google Scholar]
- 15.Rüdiger S, Schneider-Mergener J & Bukau B (2001) Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J 20, 1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han W & Christen P (2003) Mechanism of the targeting action of DnaJ in the DnaK molecular chaperone system. J Biol Chem 278, 19038–43. [DOI] [PubMed] [Google Scholar]
- 17.Acebron SP, Fernandez-Saiz V, Taneva SG, Moro F & Muga A (2008) DnaJ recruits DnaK to protein aggregates. J Biol Chem 283, 1381–90. [DOI] [PubMed] [Google Scholar]
- 18.Marszalek J & Craig EA (2022) Interaction of client-the scaffold on which FeS clusters are build-with J-domain protein Hsc20 and its evolving Hsp70 partners. Front Mol Biosci 9, 1034453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dutkiewicz R & Nowak M (2018) Molecular chaperones involved in mitochondrial iron-sulfur protein biogenesis. J Biol Inorg Chem 23, 569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lill R & Freibert SA (2020) Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu Rev Biochem 89, 471–99. [DOI] [PubMed] [Google Scholar]
- 21.Cupp-Vickery JR & Vickery LE (2000) Crystal structure of Hsc20, a J-type Co-chaperone from Escherichia coli. J Mol Biol 304, 835–45. [DOI] [PubMed] [Google Scholar]
- 22.Bitto E, Bingman CA, Bittova L, Kondrashov DA, Bannen RM, Fox BG, et al. (2008) Structure of human J-type co-chaperone HscB reveals a tetracysteine metal-binding domain. J Biol Chem 283, 30184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciesielski SJ, Schilke BA, Osipiuk J, Bigelow L, Mulligan R, Majewska J, et al. (2012) Interaction of J-Protein Co-Chaperone Jac1 with Fe-S Scaffold Isu Is Indispensable In Vivo and Conserved in Evolution. J Mol Biol 417, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schilke B, Williams B, Knieszner H, Pukszta S, D’Silva P, Craig EA, et al. (2006) Evolution of mitochondrial chaperones utilized in Fe-S cluster biogenesis. Curr Biol 16, 1660–5. [DOI] [PubMed] [Google Scholar]
- 25.Kleczewska M, Grabinska A, Jelen M, Stolarska M, Schilke B, Marszalek J, et al. (2020) Biochemical Convergence of Mitochondrial Hsp70 System Specialized in Iron-Sulfur Cluster Biogenesis. Int J Mol Sci 21, 3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutkiewicz R, Schilke B, Knieszner H, Walter W, Craig EA & Marszalek J (2003) Ssq1, a mitochondrial Hsp70 involved in iron-sulfur (Fe/S) center biogenesis. Similarities to and differences from its bacterial counterpart. J Biol Chem 278, 29719–27. [DOI] [PubMed] [Google Scholar]
- 27.Pukszta S, Schilke B, Dutkiewicz R, Kominek J, Moczulska K, Stepien B, et al. (2010) Co-evolution-driven switch of J-protein specificity towards an Hsp70 partner. EMBO Rep 11, 360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vickery LE & Cupp-Vickery JR (2007) Molecular chaperones HscA/Ssq1 and HscB/Jac1 and their roles in iron-sulfur protein maturation. Crit Rev Biochem Mol Biol 42, 95–111. [DOI] [PubMed] [Google Scholar]
- 29.Dutkiewicz R, Schilke B, Cheng S, Knieszner H, Craig EA & Marszalek J (2004) Sequence-specific interaction between mitochondrial Fe-S scaffold protein Isu and Hsp70 Ssq1 is essential for their in vivo function. J Biol Chem 279, 29167–74. [DOI] [PubMed] [Google Scholar]
- 30.Majewska J, Ciesielski SJ, Schilke B, Kominek J, Blenska A, Delewski W, et al. (2013) Binding of the Chaperone Jac1 Protein and Cysteine Desulfurase Nfs1 to the Iron-Sulfur Cluster Scaffold Isu Protein Is Mutually Exclusive. J Biol Chem 288, 29134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delewski W, Paterkiewicz B, Manicki M, Schilke B, Tomiczek B, Ciesielski SJ, et al. (2016) Iron-Sulfur Cluster Biogenesis Chaperones: Evidence for Emergence of Mutational Robustness of a Highly Specific Protein-Protein Interaction. Mol Biol Evol 33, 643–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomiczek B, Delewski W, Nierzwicki L, Stolarska M, Grochowina I, Schilke B, et al. (2020) Two-step mechanism of J-domain action in driving Hsp70 function. Plos Comput Biol 16, e1007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barthel TK, Zhang J & Walker GC (2001) ATPase-Defective Derivatives of Escherichia coli DnaK That Behave Differently with Respect to ATP-Induced Conformational Change and Peptide Release. J Bacteriol 183, 5482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutkiewicz R, Nowak M, Craig EA & Marszalek J (2017) Fe-S Cluster Hsp70 Chaperones: The ATPase Cycle and Protein Interactions. Methods Enzymol 595, 161–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knieszner H, Schilke B, Dutkiewicz R, D’Silva P, Cheng S, Ohlson M, et al. (2005) Compensation for a defective interaction of the hsp70 ssq1 with the mitochondrial Fe-S cluster scaffold isu. J Biol Chem 280, 28966–72. [DOI] [PubMed] [Google Scholar]
- 36.Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, et al. (2019) Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Methods 16, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engen JR & Wales TE (2015) Analytical Aspects of Hydrogen Exchange Mass Spectrometry. Annu Rev Anal Chem (Palo Alto Calif) 8, 127–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramelot TA, Cort JR, Goldsmith-Fischman S, Kornhaber GJ, Xiao R, Shastry R, et al. (2004) Solution NMR structure of the iron-sulfur cluster assembly protein U (IscU) with zinc bound at the active site. J Mol Biol 344, 567–83. [DOI] [PubMed] [Google Scholar]
- 39.Kim JH, Tonelli M & Markley JL (2012) Disordered form of the scaffold protein IscU is the substrate for iron-sulfur cluster assembly on cysteine desulfurase. Proc Natl Acad Sci USA 109, 454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kityk R, Kopp J & Mayer MP (2018) Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol Cell 69, 227–37 e4. [DOI] [PubMed] [Google Scholar]
- 41.Zhuravleva A & Gierasch LM (2015) Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc Natl Acad Sci USA 112, E2865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng W, Clerico EM, McArthur N & Gierasch LM (2018) Allosteric landscapes of eukaryotic cytoplasmic Hsp70s are shaped by evolutionary tuning of key interfaces. Proc Natl Acad Sci USA 115, 11970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voithenberg L. V. v., Barth A, Trauschke V, Demarco B, Tyagi S, Koehler C, et al. (2021) Comparative analysis of the coordinated motion of Hsp70s from different organelles observed by single-molecule three-color FRET. Proc Nat Acad Sci USA 118, e2025578118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silberg JJ, Tapley TL, Hoff KG & Vickery LE (2004) Regulation of the HscA ATPase reaction cycle by the co-chaperone HscB and the iron-sulfur cluster assembly protein IscU. J Biol Chem 279, 53924–31. [DOI] [PubMed] [Google Scholar]
- 45.Voisine C, Schilke B, Ohlson M, Beinert H, Marszalek J & Craig EA (2000) Role of the mitochondrial Hsp70s, Ssc1 and Ssq1, in the maturation of Yfh1. Mol Cell Biol 20, 3677–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu HY, Ziegelhoffer T, Osipiuk J, Ciesielski SJ, Baranowski M, Zhou M, et al. (2015) Roles of intramolecular and intermolecular interactions in functional regulation of the Hsp70 J-protein co-chaperone Sis1. J Mol Biol 427, 1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faust O, Abayev-Avraham M, Wentink AS, Maurer M, Nillegoda NB, London N, et al. (2020) HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature 587, 489–94. [DOI] [PubMed] [Google Scholar]
- 48.Wentink AS, Nillegoda NB, Feufel J, Ubartaite G, Schneider CP, De Los Rios P, et al. (2020) Molecular dissection of amyloid disaggregation by human HSP70. Nature 587, 483–8. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez F, Arsene-Ploetze F, Rist W, Rudiger S, Schneider-Mergener J, Mayer MP, et al. (2008) Molecular basis for regulation of the heat shock transcription factor sigma32 by the DnaK and DnaJ chaperones. Mol Cell 32, 347–58. [DOI] [PubMed] [Google Scholar]
- 50.Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, et al. (2015) Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 524, 247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sousa R, Liao HS, Cuellar J, Jin S, Valpuesta JM, Jin AJ, et al. (2016) Clathrin-coat disassembly illuminates the mechanisms of Hsp70 force generation. Nat Struct Mol Biol 23, 821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amin-Wetzel N, Saunders RA, Kamphuis MJ, Rato C, Preissler S, Harding HP, et al. (2017) A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 171, 1625–37 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Craig EA (2018) Hsp70 at the membrane: driving protein translocation. BMC Biol 16, 11 - [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manicki M, Majewska J, Ciesielski S, Schilke B, Blenska A, Kominek J, et al. (2014) Overlapping binding sites of the frataxin homologue assembly factor and the heat shock protein 70 transfer factor on the Isu iron-sulfur cluster scaffold protein. J Biol Chem 289, 30268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. [Google Scholar]
- 56.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, et al. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bussi G, Donadio D & Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- 58.Parrinello M & Rahman A (1981) Polymorphic Transitions in Single-Crystals - a New Molecular-Dynamics Method. J Appl Physics 52, 7182–90. [Google Scholar]
- 59.Darden T, York D & Pedersen L (1993) Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J Chem Physics 98, 10089–92. [Google Scholar]
- 60.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. (2017) The ClusPro web server for protein-protein docking. Nat Protoc 12, 255–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goswami AV, Samaddar M, Sinha D, Purushotham J & D’Silva P (2012) Enhanced J-protein interaction and compromised protein stability of mtHsp70 variants lead to mitochondrial dysfunction in Parkinson’s disease. Hum Mol Genet 21, 3317–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Das A, Thapa P, Santiago U, Shanmugam N, Banasiak K, Dabrowska K, et al. (2022) A heterotypic assembly mechanism regulates CHIP E3 ligase activity. EMBO J 41, e109566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puchala W, Burdukiewicz M, Kistowski M, Dabrowska KA, Badaczewska-Dawid AE, Cysewski D, et al. (2020) HaDeX: an R package and web-server for analysis of data from hydrogen-deuterium exchange mass spectrometry experiments. Bioinformatics 36, 4516–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humphrey W, Dalke A & Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14, 33–8, 27-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.