

Abstract
Immunoglobulin G4-related disease (IgG4-RD) is a multiorgan, fibro-inflammatory condition that presents with painless organ swelling, lymphoplasmacytic infiltration, and obliterative phlebitis, often showing a favorable response to corticosteroid therapy. The most affected organs include the pancreas, kidneys, retroperitoneum, lacrimal glands, and salivary glands. Diagnosis relies on serological, imaging, and histopathological findings, with glucocorticoids as the primary treatment. Despite its reversible nature and good prognosis in many cases, long-term complications such as organ dysfunction or malignancy can still occur. International collaborative efforts have enhanced the understanding, diagnosis, and management of IgG4-RD, emphasizing the importance of comprehensive diagnostic criteria and appropriate therapeutic strategies. Herein, we present an interesting case of a geriatric male who was referred to our clinic because of concern for pancreatic cancer. We diagnosed the patient with autoimmune pancreatitis, a manifestation of IgG4-RD. The patient experienced a dramatic response to steroid therapy and is currently on maintenance therapy.
Keywords: immunoglobulin G4-related disease, IgG4-RD, IgG4, autoimmune pancreatitis type 1, Küttner’s tumor, chronic sclerosing sialadenitis
Introduction
Immunoglobulin G4-related disease (IgG4-RD) or hyper-IgG4 disease is a systemic fibro-inflammatory disorder characterized by elevated serum IgG4 levels, abundant IgG4-bearing plasma cell infiltration, and fibrosis in affected organs.1 -3 IgG4-RD can affect any organ, but the most involved organs are the pancreas, lacrimal glands, salivary glands, kidneys, and retroperitoneum. 3 IgG4-RD can form tumefactive lesions in various organs, mimicking malignancy, and thus predisposing patients to significant perioperative morbidity and mortality. 4 Herein, we report a unique case of Küttner’s tumor and autoimmune pancreatitis (AIP) type 1 as metachronous manifestations of IgG4-RD. The patient responded well to steroid therapy, sparing him of unnecessary invasive procedures and possibly surgeries. The patient is currently maintained on low-dose prednisone and mycophenolate to prevent relapse and organ dysfunction. Understanding the multifaceted nature of IgG4-RD is crucial for accurate diagnosis and management of this complex systemic disease.
Case Summary
A 66-year-old male with a medical history of non–insulin-dependent diabetes mellitus and gastroesophageal reflux disease was referred to our interventional gastroenterology (GI) clinic due to concern for pancreatic cancer. His surgical history was notable for a left neck mass resection 2 years prior to presentation. The histopathology revealed chronic sclerosing sialadenitis of the submandibular gland (Küttner’s tumor). Immunostaining showed increased IgG4-positive plasma cells (>100 PHF) and an IgG4/IgG ratio (100%). In situ hybridization of kappa and lambda light chains revealed the presence of polyclonal plasma cells.
The patient was initially seen in the general GI clinic for evaluation of epigastric pain and iron-deficiency anemia (IDA). Esophagogastroduodenoscopy (EGD) showed diffuse, mildly erythematous mucosa in the stomach without bleeding. The histopathology revealed Helicobacter pylori (H pylori) gastritis and moderate reactive gastropathy. There was no evidence of metaplasia or dysplasia, and the patient was successfully treated for the H pylori gastritis. Owing to persistent abdominal pain, computed tomography (CT) of the abdomen and pelvis was performed, which showed mildly enlarged retroperitoneal and upper abdominal lymph nodes and dilatation of the main pancreatic duct within the distal pancreatic body and tail. Follow-up abdominal magnetic resonance imaging (MRI) without contrast showed similar findings, along with pancreatic atrophy. External blood work results were significant for hemoglobin of 11.8 g/dL, hemoglobin A1c of 8.0%, elevated erythrocyte sedimentation rate (ESR), 118 mm/h, and elevated IgG4 serum level, 1520 mg/dL (Table 1). Tumor markers, including carcinoembryonic antigen (CEA), carbohydrate antigen 19-9 (CA 19-9), and alpha-fetoprotein (AFP) were unremarkable.
Table 1.
External Blood Test Results Compared With Normal Ranges.
| Laboratory test | Result | Reference range |
|---|---|---|
| Hemoglobin | 11.8 | 11.1-15.9 g/dL |
| Hematocrit | 36.5 | 41.0-53.0% |
| White blood cell count | 7.1 × 103/mm3 | 3.4-10.8 ×103/mm3 |
| Sodium | 142 | 134-144 mEq/L |
| Potassium | 4.1 | 3.5-5.2 mEq/L |
| Blood glucose | 218 | 70-100 mg/dL |
| Creatinine | 1.2 | 0.60-1.30 mg/dL |
| Blood urea nitrogen | 22 | 7-23 mg/dL |
| Total protein | 8.4 | 6.0-8.5 g/dL |
| Albumin | 3.9 | 3.9-4.9 g/dL |
| Hemoglobin A1c | 8.0 | 4.8-5.6% |
| Thyroid-stimulating hormone | 1.590 | 0.450-4.50 uIU/mL |
| Erythrocyte sedimentation rate | 118 | 0-30 mm/h |
| Carcinoembryonic antigen | 2.0 | 0.0-4.7 ng/mL |
| Carbohydrate antigen 19-9 | 11 | 0-35 unit/mL |
| Alpha-fetoprotein | <1.8 | 0.0-8.4 g/mL |
| Immunoglobulin G subclass 4 | 1520 | 2-96 mg/dL |
On examination in the office, the patient appeared comfortable with normal vital signs. His abdomen was soft, obese, and mildly tender on deep palpation. He had a well-healed left-sided neck scar, where the Küttner’s tumor was removed. The remainder of the physical examination was normal. Owing to a high index of suspicion for AIP, we started the patient on prednisone 30 mg daily, and the serum IgG4 level decreased from 1520 to 201 mg/dL over 2 months (Table 2). Steroids were tapered over the same duration. A repeat CT scan of the abdomen, with and without contrast, showed no focal pancreatic mass, abnormal enhancement, or peripancreatic collection (Figure 1). The pancreatic duct measured 3.4 mm at the pancreatic head.
Table 2.
IgG4 Serum Level Trend Over 2 Months of Steroids Therapy.
| Duration of steroids therapy (months) | IgG4 serum level (mg/dL) |
|---|---|
| 0 | 1520 |
| 1 | 625 |
| 2 | 201 |
Figure 1.
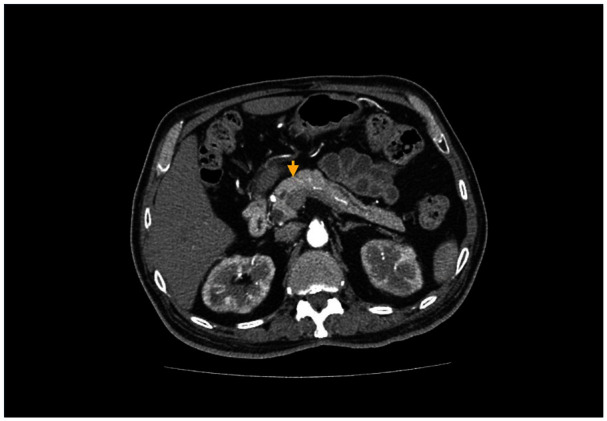
CT scan of the abdomen, with and without contrast, demonstrated a normal-appearing pancreas without masses, abnormal enhancement, or ductal dilatation.
The patient was referred to the rheumatology clinic for the co-management of autoimmune pancreatitis. The antinuclear antibody (ANA) test and 11 other rheumatological markers, complement levels, testosterone level, uric acid, lipase level, and hepatitis panel were all unremarkable. Given the dramatic response to steroid therapy (Table 2), further diagnostic studies, including endoscopic retrograde cholangiopancreatography (ERCP) and endoscopic ultrasound (EUS), were deferred. The patient is currently maintained on prednisone 5 mg daily and mycophenolate mofetil 250 mg twice daily and remains asymptomatic.
Discussion
First reported in the pancreas in 2001, IgG4-RD is an immune-mediated syndrome that can affect multiple organs simultaneously or at different times.2 -6 The pathological hallmark of this disease is dense lymphoplasmacytic infiltration, IgG4-positive plasma cells, and storiform fibrosis.2,7 Owing to its varied clinical manifestations and unclear etiopathogenesis, the exact prevalence of IgG4-RD remains unknown. The pathogenesis of IgG4-RD involves immune system dysregulation, particularly the activation of Th2 cells and regulatory T cells, which promote B-cell class switching to IgG4 production.2,5,8 This immune response triggers fibrosis through cytokines like interleukin (IL)-4, IL-13, and transforming growth factor beta (TGF-β), causing organ-specific inflammation and tissue damage. 2 Although genetic susceptibility and environmental factors may contribute, the exact pathogenic mechanisms of IgG4-RD remain incompletely understood, with possible autoimmune components and molecular mimicry playing roles in disease development.5,8
The AIP is a novel entity that was first proposed by Yoshida et al in 1995 and later refined by Hamano et al after studying a cohort of Japanese patients with sclerosing pancreatitis.5,8 -11 These patients were found to have elevated serum IgG4 levels, which correlated with disease activity.2,3,9 The AIP type 1 can present in isolation, or with simultaneous or metachronous extra-pancreatic lesions.12,13 In this case report, we describe an interesting case of AIP type 1 and chronic sclerosing sialadenitis as metachronous manifestations of IgG4-RD. Understanding the multifaceted nature of IgG4-RD is crucial for accurate diagnosis and management of this complex systemic disease.
The IgG4-RD diagnosis relies on a combination of clinical, serological, imaging, and histopathological findings, with glucocorticoids being the primary treatment for remission induction.2,6 In 2011, the all Japan IgG4-RD working group met to formulate a comprehensive diagnostic criteria for IgG4-RD. 13 In the same year, an international symposium was convened in Boston, United States, to craft guidelines for the histopathological diagnosis of IgG4-RD. 4 These hallmark events paved the way for the recognition of IgG4-RD as a distinct clinical entity worldwide. The original Japanese diagnostic criteria for IgG4-RD include (1) diffuse or focal swelling in single or multiple organs; (2) serum IgG4 levels >135 mg/dL; and (3) biopsy showing lymphoplasmacytic infiltrate and fibrosis, ratio of IgG4/IgG positive cells >40%, and >10 IgG4-positive plasma cells/high power field. 2 The 2020 revised comprehensive diagnostic (RCD) criteria include organ-specific criteria to enhance the sensitivity and specificity of the original 2011 criteria. 13
Küttner’s tumor and type 1 AIP are both recognized manifestations of IgG4-RD and share similarities in histopathological features. Küttner’s tumor presents with firm swelling of the submandibular glands, characterized by severe fibrosis, atrophic acinus, and elevated IgG4-positive plasmacytes. 14 AIP, on the contrary, is typified by lymphoplasmacytic infiltration, storiform fibrosis, and elevated IgG4-positive plasma cells in the pancreas, often challenging to differentiate from pancreatic neoplasms. Sun et al 12 described a unique case of a 53-year-old male who presented with a 4-year history of abdominal distension and diarrhea. The patient was found to have a pancreatic body mass and a left neck mass on imaging, raising concern for pancreatic cancer with metastasis to the neck. Both masses were resected, histopathology revealed AIP type 1 with Küttner’s tumor, and the patient responded well to steroids.
The signs and symptoms of IgG4-RD depend on the affected organs. The AIP may present as painless obstructive jaundice, abdominal pain, or weight loss. 11 Long-term complications include the formation of pancreatic stones and exocrine and endocrine insufficiency, whereas progression to pancreatic cancer is less common. 5 Although our patient’s presentation was highly suspicious for IgG4-RD, additional tests were performed to rule out infections, immune-mediated disorders, and tumors. Serum IgG4 levels have traditionally been considered a key diagnostic marker for IgG4-RD despite being normal in over 50% of cases.5,11
Imaging studies play a crucial role in the preoperative diagnosis of various manifestations of IgG4-RD. Abdominal CT and MRI can help visualize the pancreatic gland, with characteristic findings such as diffuse or focal enlargement of the pancreas, irregular narrowing of the pancreatic duct, a capsule-like rim, and homogeneous delayed enhancement, suggestive of AIP.5,11 The ERCP may show narrowing of the main pancreatic duct, extending over more than one third of the length of the pancreas. This characteristic feature, along with the presence of side branches emerging from the stricture area and the absence of upstream dilatation, helps distinguish AIP from pancreatic cancer. For detecting focal salivary masses in Küttner’s tumor, ultrasonography has a sensitivity and accuracy of nearly 100%.5,11,15
The EGD with EUS or pancreatoscopy facilitates direct visualization of the pancreatic duct and allows for targeted biopsy, which is essential for definitive diagnosis. Sometimes, significant thickening of the walls of the extrahepatic bile duct or gallbladder can be observed during EUS. 8 Histopathological examination of biopsy specimens reveals the characteristic features of IgG4-RD, such as dense lymphoplasmacytic infiltrates, acinar atrophy, and obliterative phlebitis, with an increased IgG4-to-IgG positive plasma cells ratio of more than 40%.16,17 The Mayo Clinic HISORt criteria for diagnosing AIP include histology with specific inflammatory patterns as mentioned above, imaging showing pancreatic abnormalities, elevated serum IgG4 levels, involvement of other organs like biliary strictures or salivary gland involvement, and a positive response to steroid therapy with improvement of symptoms. 18 Given our patient’s dramatic response to steroids, biopsy was deferred, and as a result, the patient only met 2/3 of the Mayo Clinic HISORt criteria.
Systemic steroid therapy, specifically prednisone, has been used as a mainstay treatment for IgG4-RD due to its prompt response. The treatment protocol in the United States calls for a 4-week course of 40 to 80 mg a day initial high dose, and gradually tapering until discontinuation. 2 Japanese treatment guidelines, however, recommend a 2- to 4-week course of prednisone with 0.6 mg/kg body weight, tapering 5 mg every 2 weeks, and maintenance at 2.5 to 5 mg for up to 3 years for the treatment of AIP. 2 In patients with AIP, corticosteroids are notably indicated for patients with pancreatic involvement, such as bile duct stenosis causing obstructive jaundice, as well as abdominal and back pain. In addition, maintenance therapy in patients with AIP has shown improvement in exocrine and endocrine function, but long-term investigation is still needed. 8 Recent studies have shown comparable efficacy with rituximab in cases where steroids are contraindicated or not desired due to side-effect profile. Rituximab is used as a single agent to induce remission by initiating complement-mediated B-cell lysis. This second-line immunosuppressive agent may also be used in patients with recurrent or refractory disease. 16 With that said, the use of this drug for IgG4 diseases is still understudied; therefore, the decision to treat with rituximab must be made on an individual case-to-case basis. 2
Endoscopic biliary stenting is often performed in patients with obstructive jaundice due to bile duct stenosis in the setting of AIP. Stenosis of the pancreatic duct by periductal fibrosis, referred to as an “icicle sign,” is also commonly observed in AIP. 3 With that said, stents are usually placed in the proximal main pancreatic duct and the distal common bile duct. It is important to insert a biliary stent and perform a biopsy with ERCP because it is difficult to differentiate AIP from malignancy in pancreas or bile duct and to reduce empirical use of steroid treatment. 11 After diagnosis of AIP in cases of biliary stent insertion, early stent removal with ERCP should be considered to mitigate complications like migration or stent dysfunction. 11 Overall, IgG4-RD has a good prognosis when steroids are initiated promptly and patients monitored over time for any potential complications. However, cessation of steroid therapy may lead to symptom recurrence.
Conclusion
The IgG4-RD presents with specific histopathological features, including lymphoplasmacytic infiltration, storiform fibrosis, and obliterative phlebitis, and often shows a favorable response to corticosteroid therapy. Given its tendency to form mass-like lesions in the affected organs, this condition can predispose patients to significant perioperative morbidity and mortality. We describe a unique case of a geriatric patient who was referred to our clinic due to concern for pancreatic cancer. The patient was diagnosed with AIP, which was a manifestation of IgG4-RD, and responded well to steroid therapy.
Footnotes
Author Contributions: L.B. conceptualized the idea of this case report and crafted the outline. R.B., H.F., A.S., K.V., and G.M. assisted in literature review, collection of pertinent patient information, and writing of the manuscript. K.A. and Y.C. revised the manuscript and proofread the final version. All authors reviewed the final version of this manuscript and approved it for publication.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval: Our institution does not require IRB approval/waiver for case reports.
Informed Consent: Verbal informed consent was obtained from the patient for his anonymized information to be published in this article.
Data Availability Statement: Further inquiries can be directed to the corresponding author.
ORCID iD: Lefika Bathobakae  https://orcid.org/0000-0002-2772-6085
https://orcid.org/0000-0002-2772-6085
References
- 1. Harne PS, Soni U, Albustamy A, et al. Liposarcoma masquerading as immunoglobulin G4–related disease. ACG Case Reports J. 2024;11(1):e01249. doi: 10.14309/crj.0000000000001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beyer G, Schwaiger T, Lerch MM, et al. IgG4-related disease: a new kid on the block or an old acquaintance? United Eur Gastroenterol J. 2014;2(3):165-172. doi: 10.1177/2050640614532457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang N, Zhu P, Xiang Y, et al. IgG4-related autoimmune pancreatitis and sclerosing cholangitis: a case report and literature review. Med (United States). 2024;103(17):E37922. doi: 10.1097/MD.0000000000037922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deshpande V, Zen Y, Chan JKC. et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181-1192. doi: 10.1038/modpathol.2012.72 [DOI] [PubMed] [Google Scholar]
- 5. Uchida K, Okazaki K. Current status of type 1 (IgG4-related) autoimmune pancreatitis. J Gastroenterol. 2022;57(10):695-708. doi: 10.1007/s00535-022-01891-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wallace ZS, Naden RP, Chari S, et al. The 2019 American College of Rheumatology/European league against rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020;72(1):7-19. doi: 10.1002/art.41120 [DOI] [PubMed] [Google Scholar]
- 7. Marcus KS, Hoffman HT, Rajan Kd A. Not all Küttner tumors are IgG4-related disease (IgG4-RD). Head Neck Pathol. 2021;15(4):1322-1327. doi: 10.1007/s12105-020-01268-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamisawa T, Okamoto A. Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol. 2006;41(7):613-625. doi: 10.1007/s00535-006-1862-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamano I, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. New Engl J Med. 2001;344(10):732-738. Accessed September 23, 2024. www.nejm.org [DOI] [PubMed] [Google Scholar]
- 10. Fan RY, Sheng JQ. Immunoglobulin G4-related autoimmune pancreatitis and sialadenitis: a case report. World J Gastroenterol. 2015;21(31):9448-9452. doi: 10.3748/wjg.v21.i31.9448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim SH, Lee YC, Chon HK. Challenges for clinicians treating autoimmune pancreatitis: current perspectives. World J Clin Cases. 2023;11(1):30-46. doi: 10.12998/wjcc.v11.i1.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun L, Zhou Q, Brigstock DR, et al. Focal autoimmune pancreatitis and chronic sclerosing sialadenitis mimicking pancreatic cancer and neck metastasis. World J Gastroenterol. 2014;20(46):17674-17679. doi: 10.3748/wjg.v20.i46.17674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Umehara H, Okazaki K, Kawa S, et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021;31(3):529-533. doi: 10.1080/14397595.2020.1859710 [DOI] [PubMed] [Google Scholar]
- 14. Baba D, Sotome K, Maeda I, et al. A case report of Kuttner tumor mimicking a malignant tumor, leading to overtreatment. Clin Case Reports. 2021;9(5):e04120. doi: 10.1002/ccr3.4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamiński B, Błochowiak K. Mikulicz’s disease and Küttner’s tumor as manifestations of IgG4-related diseases: a review of the literature. Reumatologia. 2020;58(4):243-250. doi: 10.5114/REUM.2020.98437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Putra J, Ornstein DL. Küttner tumor: IgG4-related disease of the submandibular gland. Head Neck Pathol. 2016;10(4):530-532. doi: 10.1007/s12105-016-0729-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Godbehere J, Scotta GB, Tahir F, et al. Küttner tumor of the parotid gland—a diagnostic rarity. Ear, Nose Throat J. 2021;100(3):NP166-NP168. doi: 10.1177/0145561319868450 [DOI] [PubMed] [Google Scholar]
- 18. Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352-358. doi: 10.1097/MPA.0b013e3182142fd2 [DOI] [PubMed] [Google Scholar]