

Abstract
Mu opioid receptors (MORs) are expressed in the dorsal striatum, a brain region that mediates goal-directed (via the dorsomedial striatum) and habitual (via the dorsolateral striatum, DLS) behaviours. Our previous work indicates that glutamate transmission is depressed when MORs are activated in the dorsal striatum, inducing MOR-mediated long-term synaptic depression (MOR-LTD) or short-term depression (MOR-STD), depending on the input. In the DLS, MOR-LTD is produced by MORs on anterior insular cortex (AIC) inputs and MOR-STD occurs at thalamic inputs, suggesting input-specific MOR plasticity mechanisms. Here, we evaluated the mechanisms of induction of MOR-LTD and MOR-STD in the DLS using pharmacology and optogenetics combined with patch-clamp electrophysiology. We found that cAMP/PKA signalling and protein synthesis are necessary for MOR-LTD expression, similar to previous studies of cannabinoid-mediated LTD in DLS. MOR-STD does not utilize these same mechanisms. We also demonstrated that cannabinoid-LTD occurs at AIC inputs to DLS. However, while cannabinoid-LTD requires mTOR signalling in DLS, MOR-LTD does not. We characterized the role of presynaptic HCN1 channels in MOR-LTD induction as HCN1 channels expressed in AIC are necessary for MOR-LTD expression in the DLS. These results suggest a mechanism in which MOR activation requires HCN1 to induce MOR-LTD, suggesting a new target for pharmacological modulation of synaptic plasticity, providing new opportunities to develop novel drugs to treat alcohol and opioid use disorders.
Keywords: cannabinoids, HCN1 channel, insular cortex, long-term depression, mu opioid receptor, opioid, striatum, synaptic plasticity
Graphical Abstract
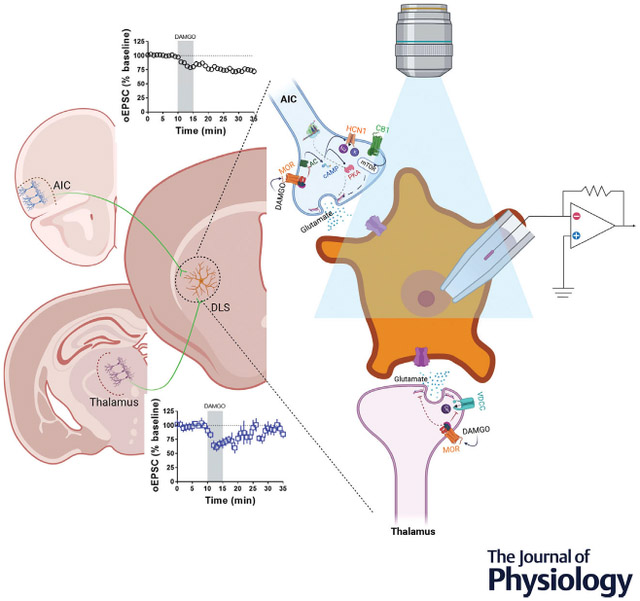
We tested the hypothesis that mu opioid receptor (MOR) plasticity utilizes different mechanisms at distinct glutamatergic synapses in the dorsolateral striatum (DLS). Glutamatergic synapses from the anterior insular cortex (AIC) expressed MOR-mediated long-term depression (MOR-LTD), and synapses from the thalamus expressed MOR-mediated short-term depression (MOR-STD). Using brain slice patch-clamp electrophysiology combined with optogenetics and pharmacology, we found that MOR from AIC inputs required a mechanism to induce LTD different from MOR from thalamic inputs. Interestingly, MOR-LTD shares similar mechanisms with cannabinoid-LTD (CB-LTD). However, MOR-LTD does not require mTOR signalling, while CB-LTD does. We characterized the role of presynaptic HCN1 channels in MOR-LTD induction as HCN1 channels expressed in AIC are necessary for MOR-LTD expression in the DLS. These results suggest a mechanism in which MOR activation needs HCN1 to induce MOR-LTD, suggesting a new target for pharmacological modulation of synaptic plasticity, providing new opportunities to develop novel drugs to treat alcohol and opioid use disorders.
Introduction
The opioid system is expressed throughout the brain (Le Merrer et al., 2009) and promotes synaptic plasticity in many brain regions (Bao et al., 2007; Dacher & Nugent, 2011a, 2011b; Drake et al., 2007; Iremonger & Bains, 2009), including the dorsal striatum (DS) (Atwood, Kupferschmidt et al., 2014; Hawes et al., 2017; Lovinger, 2010; Munoz et al., 2018; Muñoz et al., 2020). The DS is the primary input nucleus of the basal ganglia and can be subdivided into two structures: the dorsomedial striatum (DMS), which controls goal-directed learning, and the dorsolateral striatum (DLS), which regulates habit formation (Burton et al., 2015; Corbit & Janak, 2016; Hilário & Costa, 2008; Lovinger, 2010; O’Tousa & Grahame, 2014). Drug and alcohol use shift from being outcome-driven to becoming more stimulus-driven and habitual in nature during addiction development (Corbit & Janak, 2016; Everitt & Robbins, 2016; Hopf & Lesscher, 2014; Shen et al., 2018). This transition from flexible goal-directed drug use to more habitual use is paralleled by a transition in the neural activity from the DMS to the DLS (Corbit & Janak, 2016; Renteria et al., 2020; Shen et al., 2018), thereby shifting the balance of action control from the DMS to the DLS. Previous reports have demonstrated the role of glutamatergic synaptic plasticity in behaviour, showing that the disruption of glutamatergic long-term synaptic depression (LTD) at orbitofrontal cortex-DMS synapses prevents habit learning (Gremel et al., 2016), and the disruption of glutamatergic LTD in the DLS promotes habit learning (DePoy et al., 2013; Nazzaro et al., 2012). Therefore, changes in the expression of excitatory synaptic plasticity affect specific DS-related behaviours.
Our previous work established that mu opioid receptor (MOR) activation induces long-term synaptic depression (MOR-LTD) or short-term depression (MOR-STD) of glutamate release in both the DLS and DMS (Atwood, Kupferschmidt et al., 2014; Munoz et al., 2018; Muñoz et al., 2020). We demonstrated that MOR-mediated synaptic inhibition is expressed at multiple DS synapses (cortical, thalamic, amygdala and cholinergic), but only a subset of corticostriatal synapses expressed MOR-LTD (Atwood, Kupferschmidt et al., 2014) that was disrupted by prior in vivo ethanol exposure (Munoz et al., 2018; Muñoz et al., 2020). As with alcohol, we also found that mice that were exposed to the opioid oxycodone in vivo had ablated corticostriatal MOR-LTD when measured in brain slices (Atwood, Kupferschmidt et al., 2014; Munoz et al., 2018; Muñoz et al., 2020). MOR plasticity at other non-corticostriatal DS synapses was unaffected by alcohol exposure (Munoz et al., 2018; Muñoz et al., 2020). Of particular interest, in DLS, alcohol and opioid-sensitive MOR-LTD of glutamatergic input is completely restricted to anterior insular cortex (AIC) inputs (Atwood, Kupferschmidt et al., 2014; Munoz et al., 2018). Altogether, these data led us to hypothesize that MOR plasticity utilizes different mechanisms at distinct glutamatergic synapses in DS. As we previously demonstrated that MOR-LTD and cannabinoid-LTD (CB-LTD) are mutually occlusive in DLS (Atwood, Kupferschmidt et al., 2014), we predicted that these two forms of LTD would utilize similar mechanisms. cAMP/PKA signalling mediates CB-LTD in the nucleus accumbens, amygdala, cerebellum and hippocampus (Atwood, Lovinger et al., 2014). In addition, local protein translation in the presynaptic region is enhanced by the activation of the CB1 receptor, which is important for the induction of inhibitory LTD in the hippocampus (Younts et al., 2016) and likely also in the DS (Yin et al., 2006). CB-LTD at GABAergic synapses in the hippocampus is also mediated by the mammalian target of rampamycin (mTOR) signalling (Younts et al., 2016). Therefore, we sought to test whether MOR-LTD in DLS, and specifically at AIC-DLS synapses, is mediated by cAMP/PKA, protein translation and mTOR signalling. As cAMP also regulates hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, these channels can pre-synaptically regulate glutamate release, and the type 1 HCN (HCN1) channel may be localized to corticostriatal neurons, we also explored a potential role for HCN1 in mediating MOR-LTD in DLS (He et al., 2014; Huang et al., 2017; Moosmang et al., 1999; Postea & Biel, 2011).
In the current study, we probed glutamatergic synapses in the DLS using ex vivo mouse brain slice electro-physiology. We used broad electrical stimulation in C57BL/6J mice to test all glutamatergic inputs. We used an optogenetics approach to probe specific glutamate synapses in DLS, using Emx1-Ai32 mice to test all cortical inputs, VGluT2-Ai32 mice to test all thalamic inputs, and C57BL/6J mice stereotaxically injected with an AAV-ChR2 vector into the AIC to specifically probe AIC-DLS synapses. In combination with these tools, we pharmacologically and genetically probed the mechanism involved in the expression of MOR-LTD in the DLS. We demonstrate that cAMP/PKA signalling and protein synthesis are necessary for MOR-LTD expression, but not for MOR-STD. We describe a novel role of presynaptic AIC HCN1 channels in the induction of MOR-LTD in DLS. We also report that mTOR signalling is required for CB-LTD of glutamate transmission in the DLS, but this is not required for MOR-LTD, indicating a divergence in the mechanisms of these two similar forms of synaptic plasticity.
Methods
Ethical approval
Animal care and experimental protocols for this study were approved by the Institutional Animal Care and Use Committee (IACUC; approval reference 19107) at the Indiana University School of Medicine and all guidelines for ethical protocols and care of experimental animals established by the NIH (National Institutes of Health, Maryland, USA) were followed.
Methods details
All experiments were performed similarly to our previous dorsal striatal electrophysiological studies with some experiment-specific modifications (Atwood, Kupferschmidt et al., 2014; Fritz et al., 2018; Fritz et al., 2019; Munoz et al., 2018). These methods are described in brief below.
Animals and materials
Male C57BL/6J mice were obtained from the Jackson Laboratory (JAX #000664, Bar Harbor, Maine, USA). Emx1Cre-Ai32, VGluT2Cre-Ai32 and HCN1-flox mice were bred and genotyped in-house (original stock strains: Ai32: JAX #01 2569; Emx1Cre: JAX #0 05628; VGluT2Cre: JAX #01 6963; HCN1f/f: JAX #02 8299). All mutant mice used in these studies were backcrossed to C57BL/6J mice for a minimum of seven generations. The mice used in these studies were between postnatal day (PND) 60 and 100 at the time of experimentation (with the exception of the HCN1-flox AAV-cre-injected mice ~PND 98–154). Animals were group-housed in a standard 12 h light/dark cycle (lights on at 0800) at 50% humidity. Food and water were available ad libitum.
Reagents
We used the MOR agonist [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin (DAMGO; H-2535, Bachem), GABAA receptor antagonist picrotoxin (PTX, P1675, Sigma-Aldrich), PKI (6221, Tocris), KT5720 (1288, Tocris), Forskolin (F3917, Sigma-Aldrich), Cyclo-heximide (0970, Tocris), ZD7288 (1000, Tocris), WIN55,212-2 (1038, Tocris) and Torin 2 (4248, Tocris). Other reagents used for making solutions were purchased from Sigma-Aldrich or Fisher Scientific.
Brain slice preparation
Mice were killed via decapitation under deep isoflurane anaesthesia, and the brain was quickly excised and placed in an ice-cold cutting solution containing (in mM): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 10 glucose saturated with a mixture of 95% O2 and 5% CO2, and sliced to a thickness of 280 μm on a vibratome (Leica VT1200S, Germany). Slices were transferred to an artificial cerebrospinal fluid (aCSF) solution containing (in mM): 124 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 10 glucose, 2 CaCl2 (310–320 mOsm) saturated with 95% O2/5% CO2 at 30°C for 1 h before being moved to room temperature. When ready for recording, slices were transferred to a recording chamber continuously perfused with aCSF solution saturated with 95% O2/5% CO2.
Electrophysiology recordings
Whole-cell recordings of excitatory postsynaptic currents (EPSCs) in medium spiny neurons (MSNs) were carried out at 29–32°C and aCSF was continuously perfused at a rate of 1–2 ml/min. Recordings were performed in the voltage clamp configuration using a Multiclamp 700B amplifier and a Digidata 1550B (Molecular Devices, San Jose, CA). Slices were visualized on an Olympus BX51WI microscope (Olympus Corporation of America). MSNs were identified by their size, membrane resistance and capacitance. Picrotoxin (50 μM) was added to the aCSF for recordings to isolate excitatory transmission. Patch pipettes were prepared from filament-containing borosilicate micropipettes (World Precision Instruments) using a P-1000 micropipette puller (Sutter Instruments, Novato, CA), having a 2.0–3.5 MΩ resistance. The internal solution contained (in mM): 120 CsMeSO3, 5 NaCl, 10 TEA-Cl, 10 HEPES, 5 lidocaine bromide, 1.1 EGTA, 0.3 Na-GTP and 4 Mg-ATP (pH 7.2 and 290–310 mOsm). MSNs were voltage clamped at −60 mV for the duration of the recordings. For electrically evoked recordings, a twisted tungsten bipolar stimulating electrode (PlasticsONE, Roanoke, VA) was placed at the border of the white matter of the external capsule. Electrically evoked excitatory postsynaptic currents (eEPSCs) were generated by a DS3 Isolated Current Stimulator (Digitimer, Ft. Lauderdale, FL) every 20 s and the stimulus intensity was adjusted to produce stable eEPSCs of 200–600 pA in amplitude prior to the initiation of experimental recording. To induce CB-LTD, high-frequency stimulation (HFS; four pulses of 100 Hz, 10 s inter-pulse interval) coupled with depolarization (0 mV) was delivered after 10 min of baseline recording (Atwood, Kupferschmidt et al., 2014; Munoz et al., 2018). To induce hippocampal long-term potentiation (LTP), a concentric bipolar-stimulating tungsten electrode (Micro-Probes for Life Science, MD) was placed into the stratum radiatum in area CA1 ~500 μm from the recording site. The stimulus intensity for these experiments ranged from 200 to 1800 pA due to differences in recording conditions. The internal solution contained (in mM): 120 K-gluconate, 10 NaCl, 5 MgCl2, 2 phosphocreatine, 25 HEPES, 0.2 EGTA, 0.3 Na-GTP and 5 Mg-ATP (pH 7.35 and 290–300 mOsm) and the CA1 pyramidal neurons were held at −90 mV. After the baseline was established, LTP was induced by switching the mode to current-clamp and delivering HFS (four pulses of 100 Hz, 1 min inter-pulse interval) as previously reported (Duffy & Nguyen, 2003). The spontaneous EPSC (sEPSC) recordings were made for 2 min and the analysis of frequency (Hz), decay constant (ms), rise constant (ms) and amplitude (pA) were measured to determine the effects of the blockade of striatal HCNs and the deletion of HCN1 in AIC. The decay constant of sEPSCs was fitted as single exponential and both the rise and decay phases were fitted between 10% and 90% of the maximal amplitude. Data were acquired using Clampex 10.3 (Molecular Devices, San Jose, CA). Series resistance was monitored and only cells with a stable series resistance (less than 25 MΩ and that did not change more than 15% during recording) were included for data analysis. Recordings were made 2–7 h after killing.
Viral injections
C57BL/6J mice were anaesthetized with isoflurane and stereotaxically injected with the adeno-associated viral (AAV) vector, AAV9.hSyn.ChR2(H134R)-eYFP (Addgene #26973) to drive ChR2 expression in AIC neurons. Bilateral injections were made into AIC: A/P: +2.4, M/L: ±2.3, D/V: −2.25 (50 nl/injection, 12.5 nl/min infusion rate).
To produce AIC projection neuron HCN1 knockout mice, HCN1-flox mice were anaesthetized with isoflurane and stereotaxically injected with AAV9.hSyn.HI.eGFP.Cre.WPRE.SV40 (Addgene #105540) or AAV9.hSyn.eGFP.WPRE.bGH as control (Addgene #105539). Bilateral injections were made into AIC at coordinates: A/P: +2.4, M/L: ±2.3, D/V: −2.25 (75 nl/injection, 12.5 nl/min infusion rate). HCN1-flox mice were allowed to recover for at least 4 weeks to allow for adequate ablation of HCN1 channel expression before the brain slices were made for electro-physiological recordings. Prior to recording, brain slices were imaged via an Olympus MVX10 microscope (Olympus Corporation of America) to verify GFP or GFP-tagged cre-recombinase expression in injected HCN1-flox mice.
Optogenetic recordings
AAV-ChR2 injection in C57BL/6J mice was performed to target ChR2 expression to inputs from AIC to DLS. Optically evoked EPSCs (oEPSCs) in MSNs from Emx1-Ai32, VGluT2-Ai32 and C57BL/6J mice injected with AAV-ChR2 were produced in brain slices using 470 nm blue light (5 ms exposure time) delivered via field illumination through the microscope objective. Light intensity was adjusted to produce stable oEPSCs of 200–600 pA amplitude prior to experimental recording. oEPSCs were evoked every 30 s. AIC-DLS CB-LTD was produced using optical high-frequency stimulation (oHFS) coupled with depolarization (four blue light pulses of 50 Hz, 10 s inter-pulse interval). Prior to recording, brain slices were imaged via an Olympus MVX10 microscope (Olympus Corporation of America) to verify YFP-tagged ChR2, GFP-tagged cre-recombinase, or GFP expression in injected C57BL/6J mice. Injection site locations were determined from matching images to the Reference Allen Mouse Brain Atlas (Figs 2 and 12). Animals that did not have viral expression in the AIC were excluded from the study.
Figure 2. Locations of AIC injections for DLS electrophysiology.
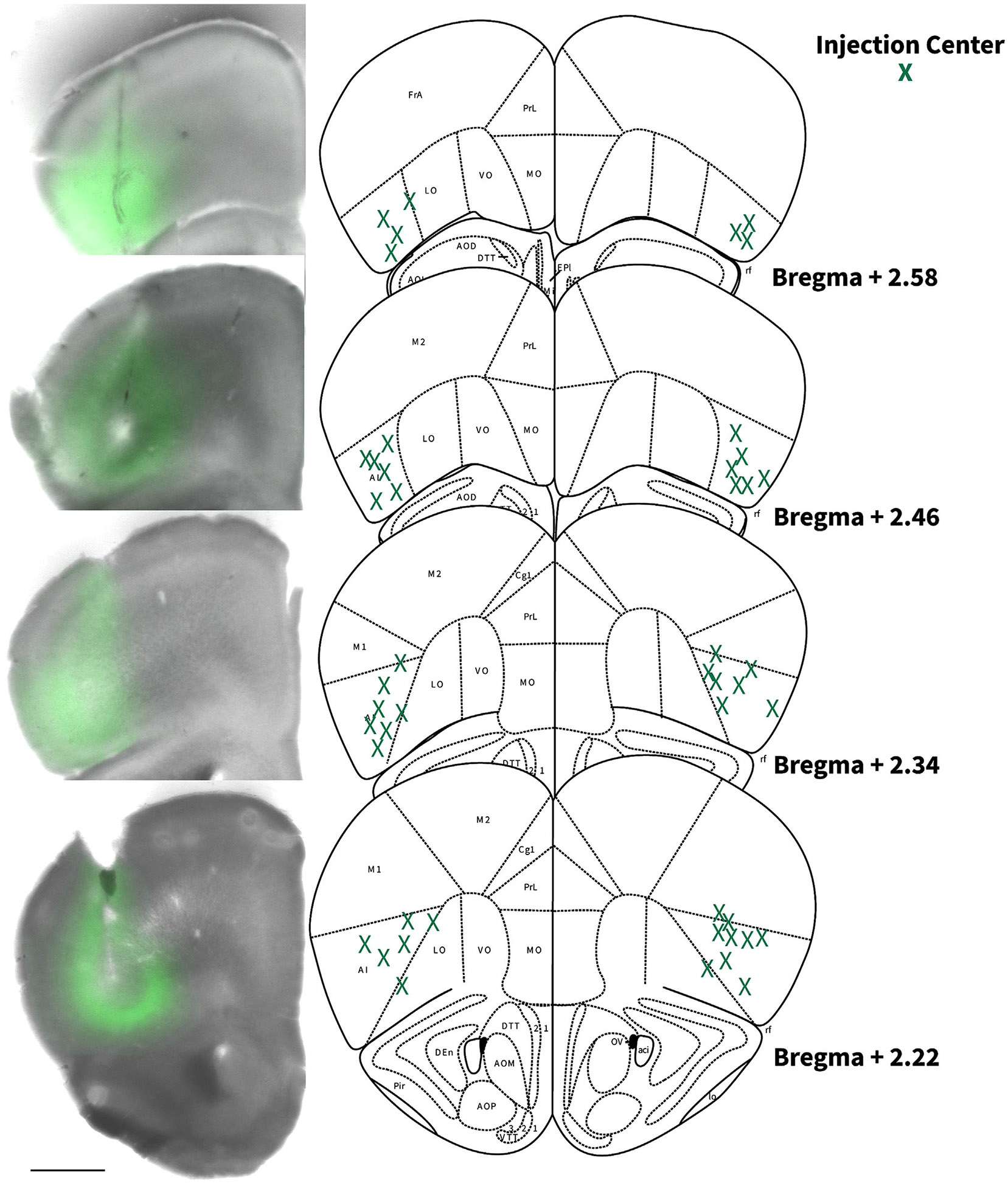
Coronal brain hemi slice images and schematic figures modified from the Allen Mouse Brain Atlas showing bilateral injection centres of AAV-ChR2-EYFP in the AIC of C57BL/6J mice verified by histology.
Figure 12. Locations of AIC injections for DLS electrophysiology.
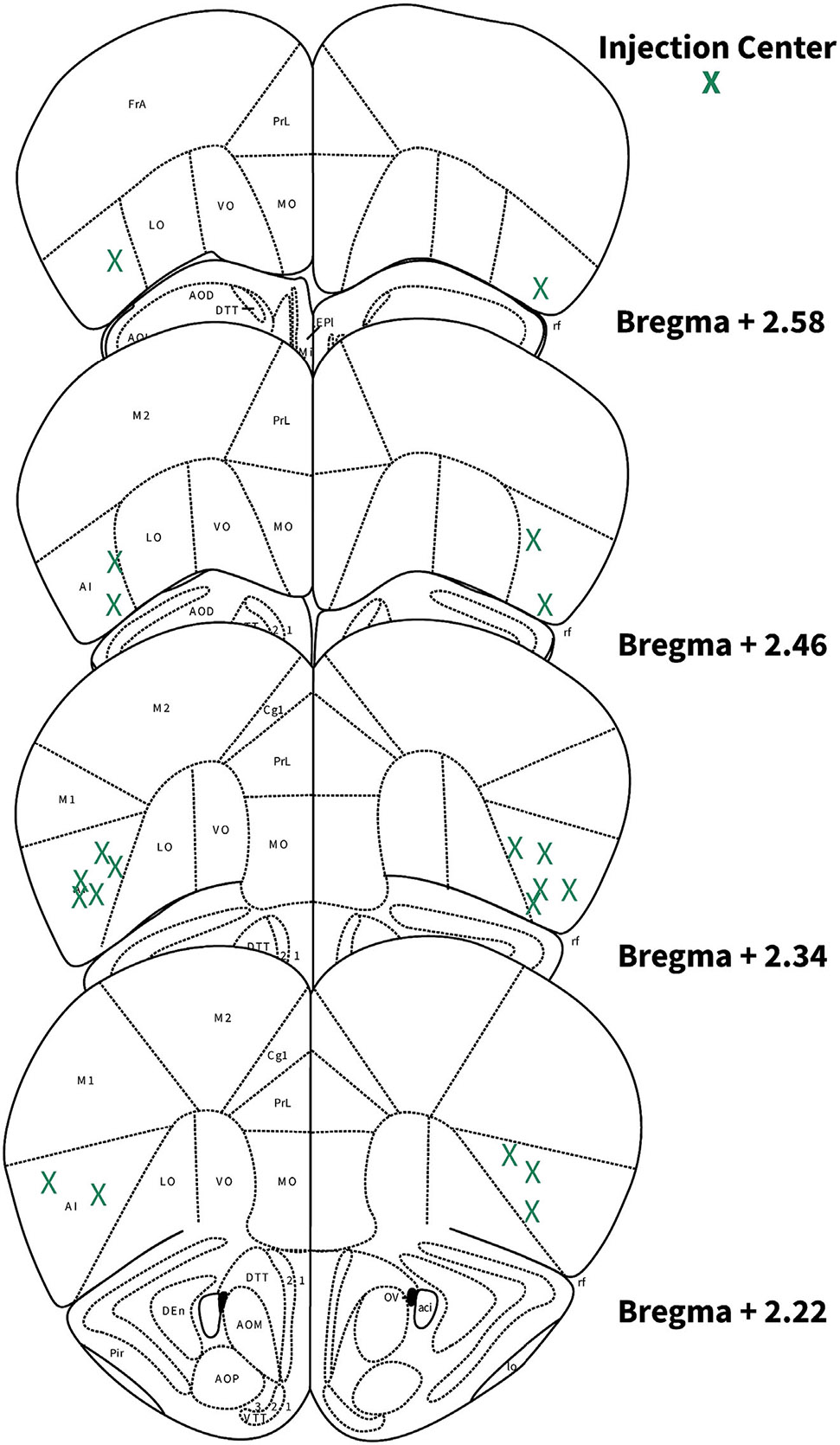
Schematic images of coronal brain slices modified from the Allen Mouse Brain Atlas showing the bilateral injection centres of AAV-cre and EGFP in the AIC of HCN1-flox mice as verified by histology.
Quantitative polymerase chain reaction
Anterior insular cortex tissues were taken from HCN1-flox/AAV-cre (+) and HCN1-flox/AAV-GFP (−) mice. RNA was isolated from brain tissue using the RNeasy Plus Universal Mini Kit (Qiagen #73404) according to the manufacturer’s protocol. Total RNA (50 ng/μl) was converted to complementary DNA (cDNA) using the High Capacity cDNA Reverse Transcription kit (Catalogue number: 468814, Applied Biosystems Inc. (ABI), Foster City, CA) and amplified using a CFX Connect Real-time PCR detection System (Bio-Rad Laboratories). The TaqMan probe used in the current study was Hcn1 (PrimePCR Assay: Hcn1.Mmu, Unique Assay ID:qMmuCED0045731, Catalogue #10025636, ThermoFisher). Quantitative PCR was performed using SsoAdvanced Universal Probes Supermix (Catalogue #: 172–5280, Bio-Rad Laboratories). The relative amount of each transcript was determined via normalization across all samples to the endogenous control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Catalogue: Mm.PT.39a.1, PrimeTime Std qPCR Assay, IDT, IA, USA) to account for variability in the initial concentration and quality of the total RNA and in the conversion efficiency of the reverse transcription reaction as recommended by ABI. In addition, before initiation of the analysis, cDNA was diluted 1:10 and amplified using the respective TaqMan probes to ensure that the amount of cDNA used was in the linear range. RNA samples from each individual animal were run in duplicate.
To quantify the relative expression levels of the different genes for each mouse genotype, we calculated the difference (ΔCt) between the cycle threshold of Hcn1 and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). From these data, the ΔΔCt ([ΔCt(Cre (+))–ΔCt(GFP (−))]) was computed and converted to a relative quantitative value using the formula 2−ΔΔCt. The results were tabulated as means ± SD and compared between genotypes using unpaired Student’s t tests.
Quantification and statistical analysis
Sample size.
The target number of samples in each group for electrophysiological experiments was determined from our previously published studies (Atwood, Kupferschmidt et al., 2014; Munoz et al., 2018; Muñoz et al., 2020).
Replication.
Sample sizes indicated in figures for electro-physiological experiments represent biological replicates. Due to the inherent biological variability between neurons recorded (biochemical makeup and innervation patterns), neurons were considered the unit of biological replication. One neuron was recorded per brain slice and all experiments involved recordings from at least three mice.
Data analyses.
eEPSCs and oEPSCs were analysed using Clampfit 10.1 (Molecular Devices, San Jose, CA) and sEPSCs with MiniAnalysis 6.0 (Synaptosoft Inc). Data are presented as means ± SD in the main text and as means ± SEM (time courses) or as box plots representing medians and interquartile ranges in the figures. The analyses of normally distributed data were performed using two-tailed unpaired or two-tailed paired Student’s t tests following an F test to confirm similar variances. Non-normally distributed data were analysed using two-tailed Welch’s t test for unpaired data (Fig. 7D). One-way ANOVA following Dunnett’s multiple comparisons test was performed to compare controls from treatments in Figs 4G, N, U and 9M. Repeated measures one-way ANOVA following Tukey’s multiple comparisons test was performed for Fig. 9C, F, I and L. Data that were analysed using this test are indicated in the figure legends. Statistical analyses were performed with Prism 9 (GraphPad, La Jolla, CA). The level of significance was set at P < 0.05 for all analyses. Representative traces are the average baseline EPSC (1–10 min), average of the acute DAMGO response (12–20 min) and average post-treatment EPSC of the final 10 min of recording. Exclusion of individual data points was determined using a ROUT outlier calculator (Q = 1%) included in the Prism 9 software package.
Figure 7. Protein translation is required for MOR-mediated LTD expression.
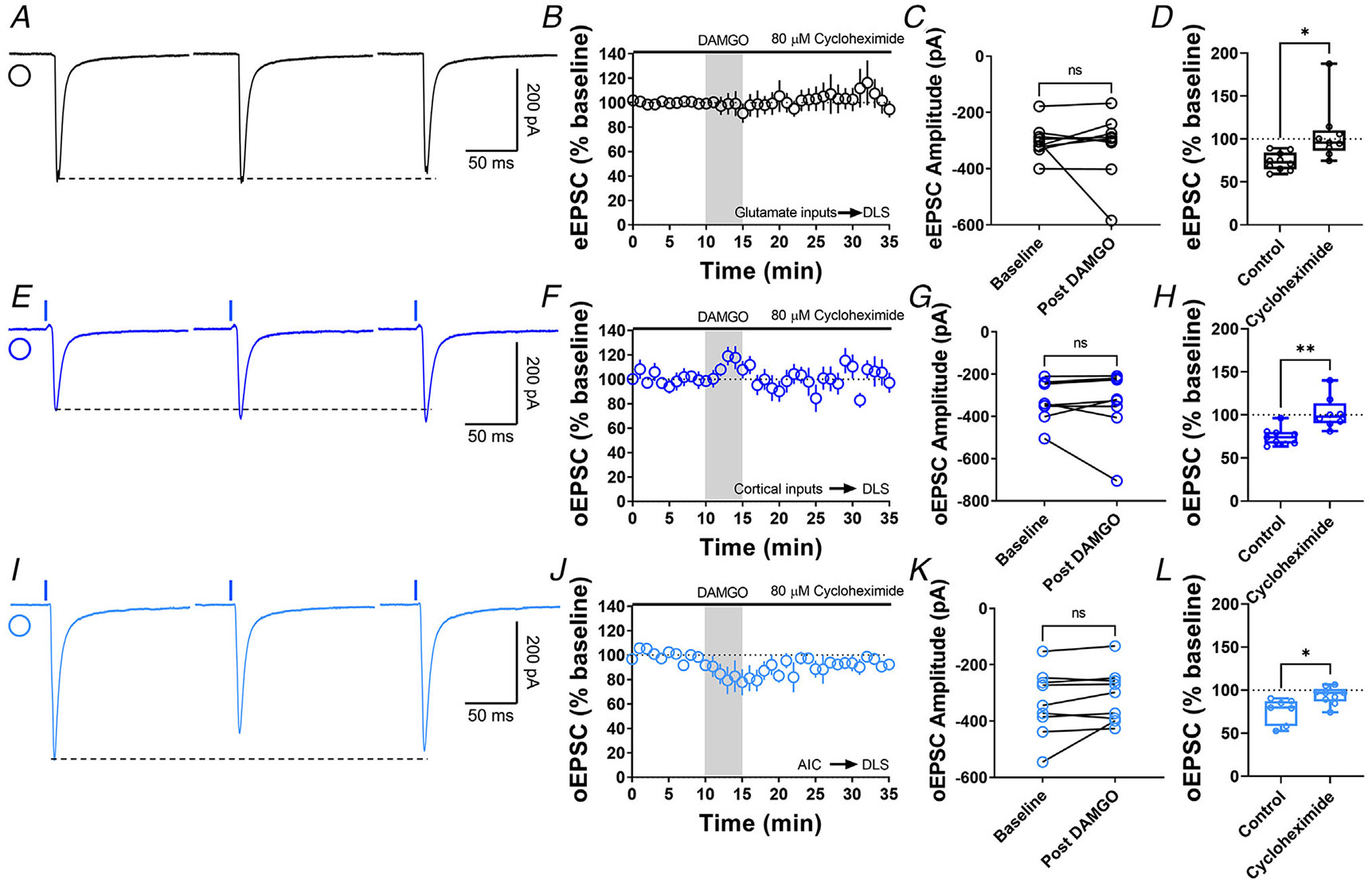
A, representative eEPSC traces showing the effects of 80 μM cycloheximide before, during and after DAMGO (0.3 μM, 5 min) application. B–D, protein translation inhibition disrupted glutamatergic MOR-LTD (105 ± 11%, unpaired Welch’s t test, P = 0.0228, t10 = 2.72), showing no changes in eEPSC amplitude after DAMGO bath application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.651, t8 = 0.471, n = 9 neurons from four mice). E, representative oEPSC traces showing the effects of 80 μM cycloheximide before, during and after DAMGO (0.3 μM, 5 min) application in brain slices from Emx1-Ai32 mice. F–H, protein translation inhibition blocked corticostriatal MOR-LTD (102 ± 7%, unpaired t test, P = 0.00163, t15 = 3.83) with no effects in oEPSC amplitude after DAMGO bath application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.633, t7 = 0.499, n = 8 neurons from three mice). I, representative AIC-DLS oEPSC traces showing the effects of 80 μM cycloheximide before, during and after DAMGO (0.3 μM, 5 min) application. J–L, protein translation is needed to induce MOR-LTD (94 ± 3%, unpaired t test, P = 0.0129, t14 = 2.85) from AIC inputs. DAMGO did not change in oEPSC amplitude (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.163, t8 = 1.54, n = 9 neurons from three mice). Time course data represent means ± SEM. Box plots show average of the final 10 min of recording and represent median and interquartile ranges. ns = not significant, *P < 0.05, **P < 0.01.
Figure 4. Inhibition of presynaptic PKA blocks MOR-mediated LTD.
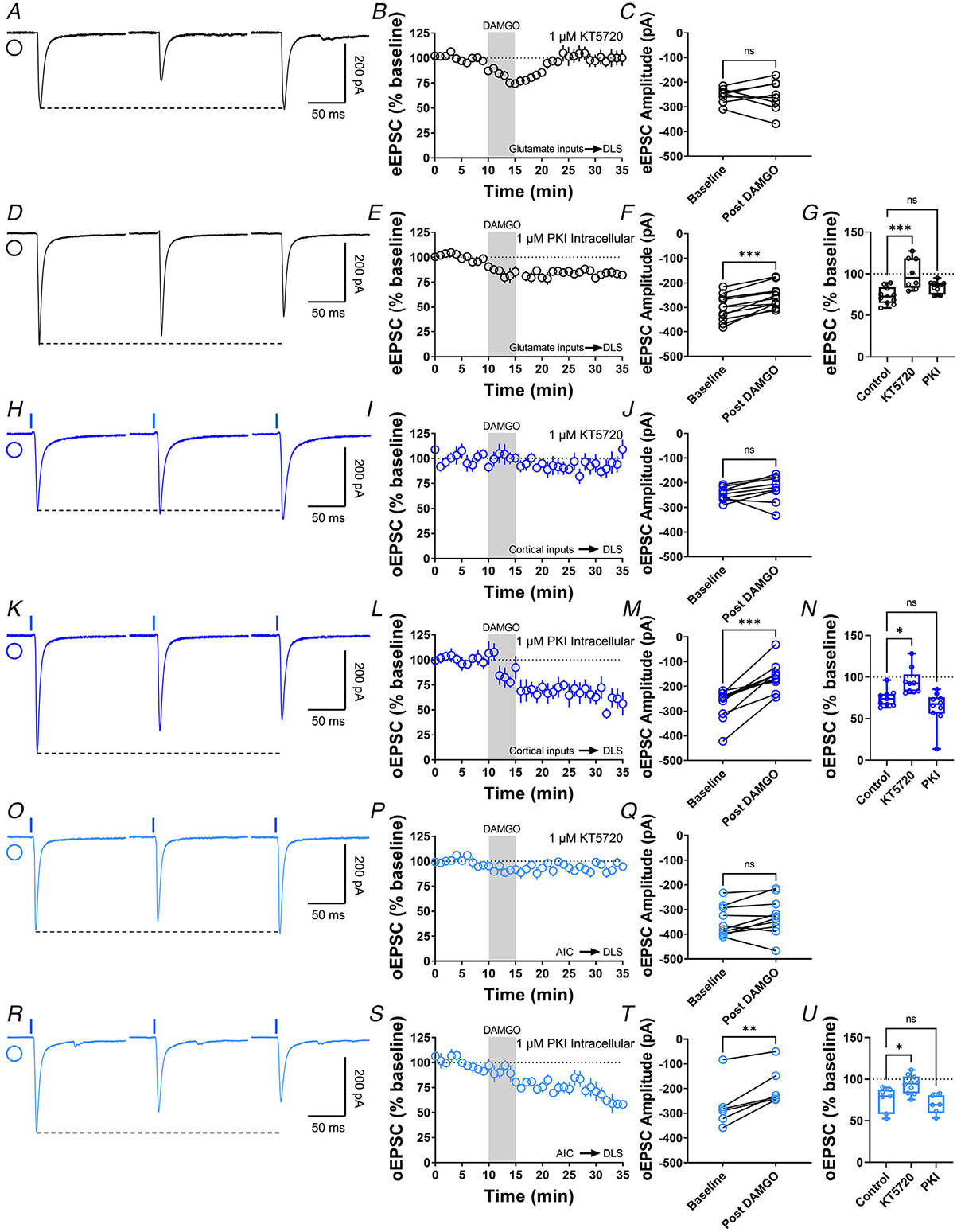
A, representative eEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the preincubation of PKA-selective inhibitor, KT5720 (1 μM, ≥1 hr). B–C, the preincubation of KT5720 blocked glutamatergic MOR-LTD, the eEPSC amplitudes did not change after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.865, t7 = 0.176, n = 8 neurons from three mice). D, representative eEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the intracellular dialysis of PKI (1 μM, ≥30 min). E–F, the inhibition of postsynaptic PKA did not alter MOR-LTD. eEPSC amplitudes were reduced after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.000120, t10 = 6.07, n = 11 neurons from five mice). G, presynaptic PKA inhibition disrupted MOR-LTD (KT5720: 101 ± 7%, P = 0.000193 vs. PKI: 84 ± 2%, P = 0.121; F(2,26) = 10.65, one-way ANOVA Dunnet’s multiple comparison test). H, representative oEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the preincubation of PKA-selective inhibitor, KT5720 (1 μM, ≥1 h). I–J, PKA inhibition blocked corticostriatal MOR-LTD, with no changes in oEPSC amplitudes after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.183, t8 = 1.46, n = 9 neurons from four mice). K, representative oEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the intracellular dialysis of PKI (1 μM, ≥30 min). L–M, the inhibition of postsynaptic PKA did not alter MOR-LTD. oEPSC amplitudes were reduced after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.000118, t9 = 6.45, n = 10 neurons from four mice). N, presynaptic PKA inhibition disrupted cortical MOR-LTD (KT5720: 94 ± 5%, P = 0.0305 vs. PKI: 64 ± 6%, P = 0.235; F(2,25) = 8.805, One-way ANOVA Dunnet’s multiple comparison test). O, representative AIC-DLS oEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the preincubation of PKA-selective inhibitor, KT5720 (1 μM, ≥1 h). P–Q, KT5720 blocked AIC-expressed MOR-LTD, with no changes in oEPSC amplitudes after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.141, t9 = 1.613, n = 10 neurons from three mice). R, representative AIC-DLS oEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the intracellular dialysis of PKI (1 μM, ≥30 min). S–T, postsynaptic PKA inhibition did not alter specific AIC MOR-mediated LTD. oEPSC amplitudes were reduced after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.00488, t5 = 4.8, n = 6 neurons from three mice). U, presynaptic PKA inhibition disrupted AIC MOR-LTD (KT5720: 94 ± 4%, P = 0.0171 vs. PKI: 69 ± 5%, P = 0.518; F(2,20) = 8.411, one-way ANOVA Dunnet’s multiple comparison test). Time course data represent means ± SEM. Box plots show average of the final 10 min of recording and represent median and interquartile ranges. ns = not significant, **P<0.01, ***P<0.001.
Figure 1. MOR activation induces glutamatergic LTD in the DLS.
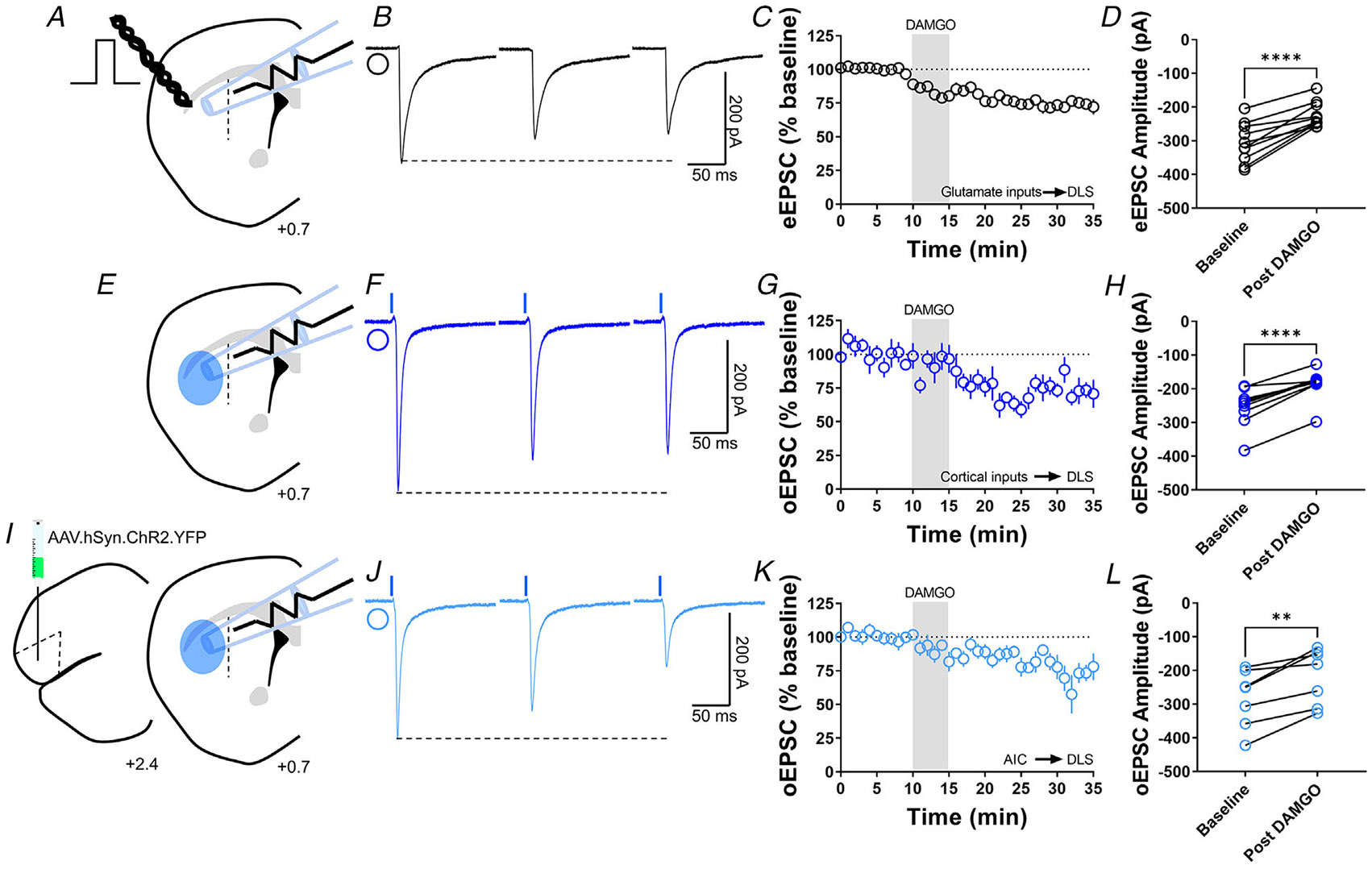
A, schematic representation of coronal brain slice showing the recording of EPSCs evoked by focal electric stimulation in the DLS of C57BL/6J mice. B, representative electrically evoked EPSC traces before, during and after DAMGO (0.3 μM, 5 min) application. C, the activation of MOR by DAMGO induced glutamatergic LTD in DLS MSNs of C57BL/6J mice (final 10 min of recording average: 74 ± 3%). D, eEPSC amplitudes in MSNs within DLS were significantly reduced after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P < 0.0001, t9 = 6.688, n = 10 neurons from five mice). E, schematic representation of coronal brain slice showing the recording of EPSCs evoked by focal optical stimulation (470 nm blue light for 5 ms exposure) in the DLS of Emx1-Ai32 mice. F, representative optically evoked EPSC traces before, during and after DAMGO (0.3 μM, 5 min) application. G, the activation of MOR by DAMGO induced corticostriatal LTD in DLS MSNs of Emx1-Ai32 (final 10 min of recording average: 75 ± 3%). H, oEPSC amplitudes were significantly reduced after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P < 0.0001, t8 = 7.73, n = 9 neurons from four mice). I, schematic figure of the injection paradigm showing an AAV vector encoding for ChR2 (AAV.hSyn.ChR2.YFP) in AIC in C57BL/6J mice, this AAV was injected 2 weeks prior to recordings. Also, the next schematic representation of coronal brain slice shows the recording of oEPSCs (470 nm blue light for 5 ms exposure) in the DLS. J, representative AIC-DLS oEPSC traces before, during and after DAMGO (0.3 μM, 5 min) application. K, DAMGO induced AIC-DLS LTD (final 10 min of recording average: 76 ± 5%). L, DAMGO application significantly reduced oEPSC amplitudes (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.00403, t6 = 4.5, n = 7 neurons from four mice). Data represent means ± SEM. **P < 0.01, ****P < 0.0001.
Figure 9. MOR-STD is not affected by PKA inhibition, cAMP activation or protein translation inhibition.
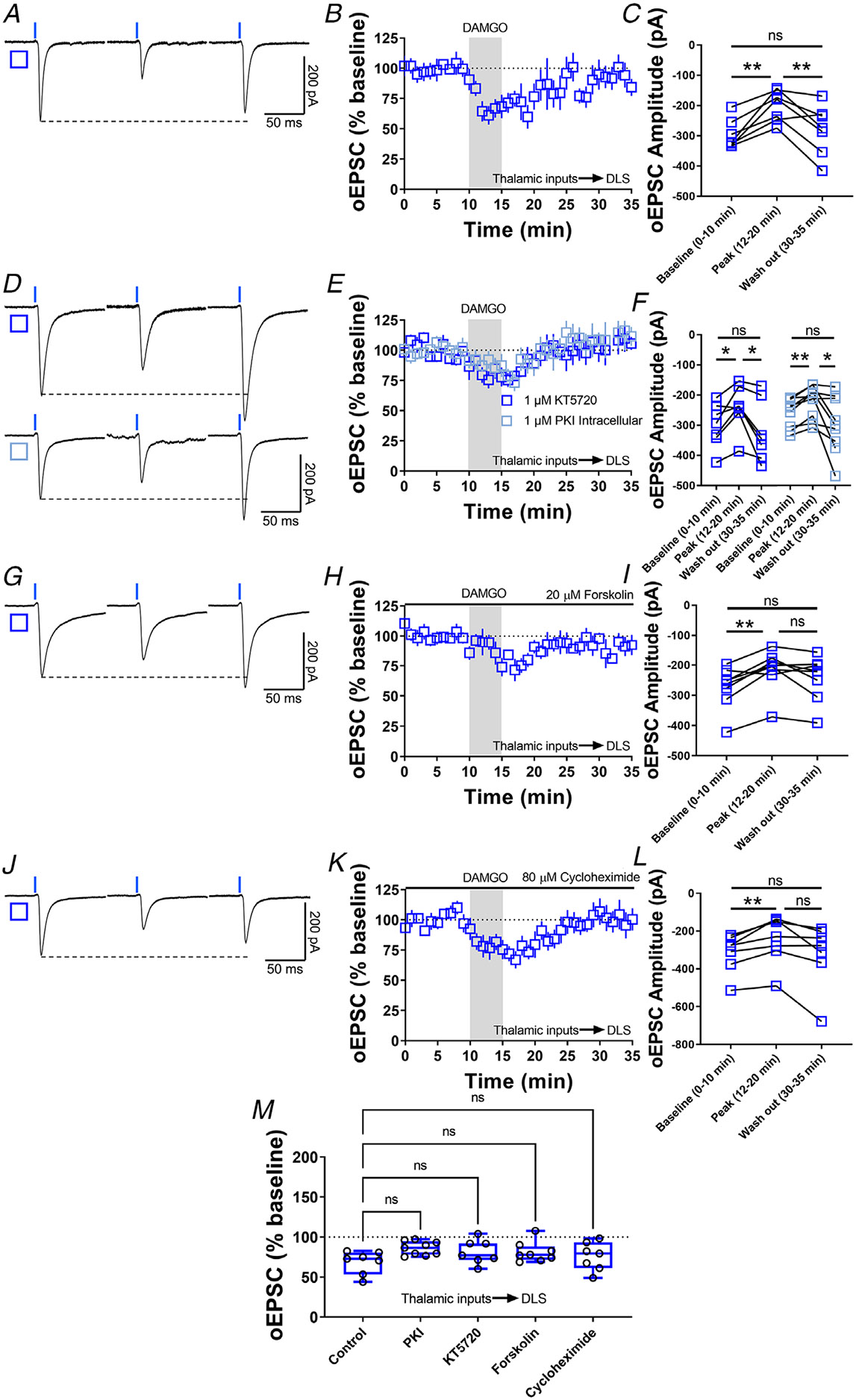
A, representative optically evoked EPSC traces before, during and after DAMGO (0.3 μM, 5 min) application in brain slices from VGluT2-Ai32 mice. B–C, thalamocortical inputs produced MOR-STD in DLS from VGluT2-Ai32 mice (10 min baseline vs. peak 12–20 min of recording P = 0.00275; 12–20 min Peak vs. 30–35 min Wash out of recording, P = 0.0889, repeated measure one-way ANOVA Tukey’s test, n = 7 neurons from three mice). D, representative oEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application after the preincubation of PKA-selective inhibitor KT5720 (1 μM, ≥1 h, blue open square) and after the intracellular dialysis of PKI (1 μM, ≥30 min, cyan open square). E–F, the inhibition of pre (1 μM KT5720) and postsynaptic PKA (1 μM PKI) did not affected MOR-STD (KT5720: 10 min baseline vs. peak 12–20 min of recording, P = 0.0353; 12–20 min Peak vs. 30–35 min Wash out of recording, P = 0.0353, repeated measure one-way ANOVA Tukey’s test, n = 7 neurons from three mice) (PKI: 10 min baseline vs. peak 12–20 min of recording, P = 0.00218; 12–20 min Peak vs. 30–35 min Wash out of recording, P = 0.0429, repeated measure one-way ANOVA Tukey’s test, n = 9 neurons from three mice). G, representative oEPSC traces showing the effects of 20 μM forskolin before, during and after DAMGO (0.3 μM, 5 min) application in brain slices from VGluT2-Ai32 mice. H–I, MOR-STD was unaffected after the activation of AC by 20 μM forskolin (10 min baseline vs. peak 12–20 min of recording, P = 0.00260; 12–20 min Peak vs. 30–35 min Wash out of recording, P = 0.168, repeated measure one-way ANOVA Tukey’s test, n = 8 neurons from four mice). J, representative oEPSC traces showing the effects of 80 μM cycloheximide before, during and after DAMGO (0.3 μM, 5 min) application in brain slices from VGluT2-Ai32 mice. K–L, protein translation is not necessary for the expression of MOR-STD (10 min baseline vs. peak 12–20 min of recording, P = 0.00973; 12–20 min Peak vs. 30–35 min Wash out of recording, P = 0.0822, repeated measure one-way ANOVA Tukey’s test, n = 7 neurons from three mice). M, the box plot illustrates the average peak current (12–20 min) and summarize the findings on MOR-STD mechanism in the DLS (one-way ANOVA Dunnett’s multiple comparisons test). Time course data represent means ± SEM. Box plots represent median and interquartile ranges and show average peak responses (12–20 min). ns = not significant, *P < 0.05, **P < 0.01.
Results
MOR activation express glutamatergic LTD in the DLS
We previously showed that the activation of MOR in DLS produces LTD of eEPSCs as well as oEPSCs (Munoz et al., 2018). Using three approaches, we confirmed that 5 min application of the MOR agonist DAMGO (0.3 μM) induces MOR-LTD. We found that DAMGO produced a persistent decrease in eEPSC amplitude in DLS MSNs (74 ± 10%; Fig. 1A-D) in brain slices from C57Bl/6J mice. To generally probe corticostriatal synapses, we used 470 nm light delivered through the microscope objective to evoke oEPSCs in brain slices from Emx1-Ai32 mice (Fig. 1E), where the activation of MOR again induced MOR-LTD (75 ± 10%; Fig. 1F-H). Finally, to specifically stimulate the release of glutamate from AIC inputs to the DLS, we infused an AAV vector expressing ChR2 in the AIC (Figs 1, 2), as we previously did (Munoz et al., 2018). As shown previously, activation of MORs produced glutamatergic LTD of AIC-DLS transmission (76 ± 15%; Fig. 1J-L). These data replicate our previous work that MORs mediated LTD of AIC-DLS synapses. We previously showed that stimulation-induced CB-LTD and MOR-LTD were mutually occlusive in the DLS (Atwood, Kupferschmidt et al., 2014). In order to determine whether we could infer potential MOR-LTD mechanisms from what is known about CB-LTD, we investigated whether CB-LTD also occurred at AIC-DLS synapses. We used the same AAV approach described above (Fig. 3A) and tested whether we could produce stimulation-induced LTD (that is likely mediated by endocannabinoid signalling) and also tested responses to the CB1 receptor agonist WIN55,212-2 (1 μM) to chemically induce CB-LTD. We found that stimulation-induced LTD (65 ± 7%; Fig. 3B, C, D and F) and chemically induced CB-LTD (68 ± 7%; Fig. 3B, C, E and F) both occur at AIC-DLS synapses. Next, we explored the signalling cascades that mediate MOR plasticity in the DLS using what is known about CB-LTD as a guide for our experiments.
Figure 3. Cannabinoid-LTD occurs at AIC-DLS synapses.
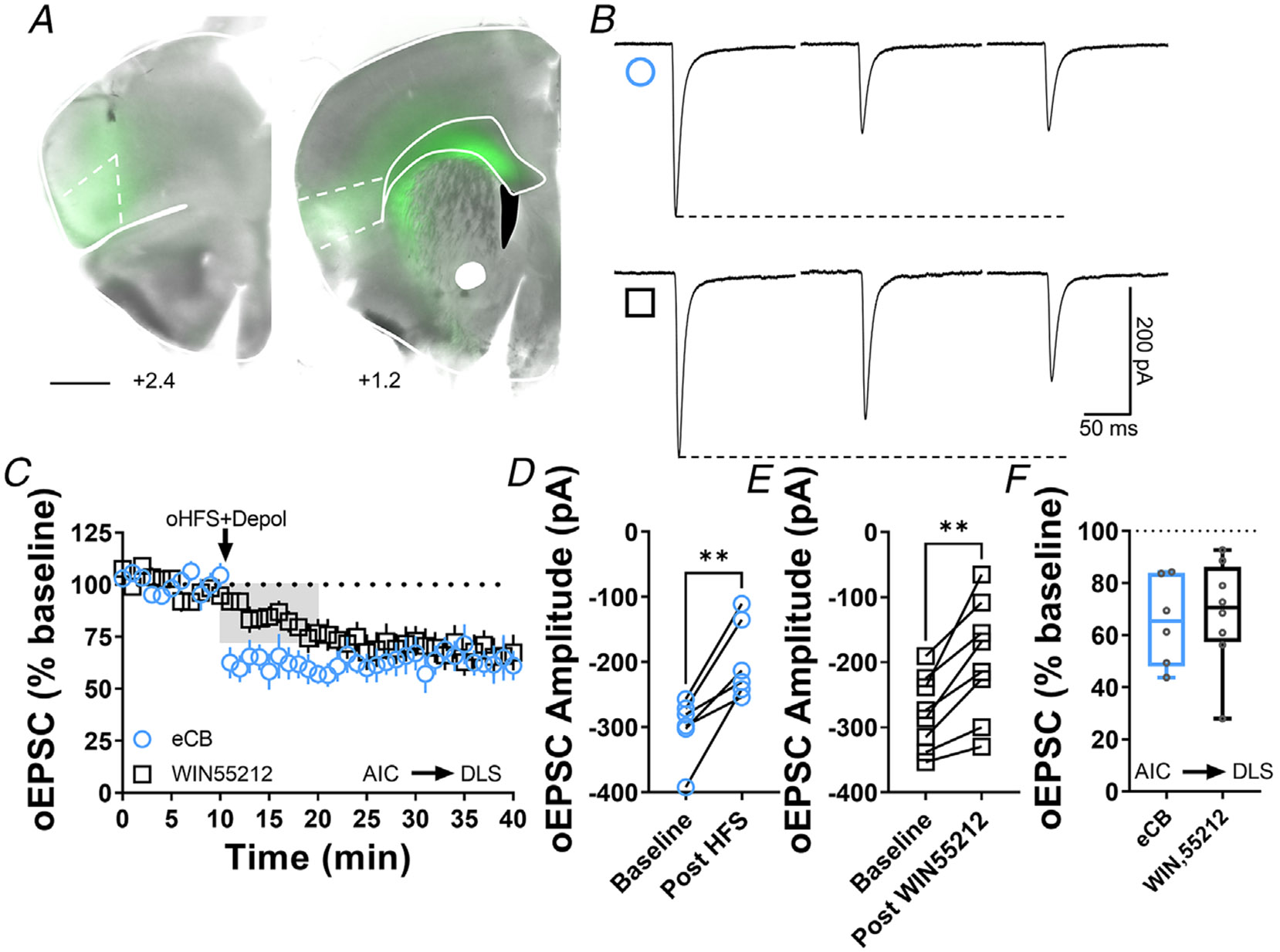
A, coronal brain slice showing the AAV-ChR2 infection of AIC and dorsal striatal terminal expression (bar scale = 1000 μm). B, representative oEPSC traces showing the effects of pairing optical high-frequency stimulation with postsynaptic depolarization (oHFS+Depol, cyan open circle) to physiologically induce LTD, presumably mediated by endocannabinoids and the application of the CB1 agonist, WIN55,212-2 (1 μM, 10 min, black open square) to chemically induce CB1-LTD in brain slices of AAV-ChR2 C57Bl/6J mice. C, both oHFS and WIN55,212-2 induced AIC-DLS LTD. Data represent means ± SEM. D, oHFS significantly reduced oEPSC amplitudes (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.00350, t5 = 5.2, n = 6 neurons from four mice). E, WIN55,212-2 significantly reduced oEPSC amplitudes (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.00179, t7 = 4.9, n = 8 neurons from four mice). F, the box plot shows the final 10 min of recording average of stimulation-induced LTD (65 ± 7%) and CB1-LTD (68 ± 7%). **P < 0.01.
Inhibition of presynaptic PKA blocks MOR-mediated LTD
The activation of opioid receptors produces an inhibition of adenylyl cyclase (AC) mediated by Gαi, decreasing cAMP levels, which leads to an attenuation of cAMP/protein kinase A (PKA) signalling (Atwood, Lovinger et al., 2014; Reeves et al., 2022). However, little is known about PKA’s role in MOR-mediated LTD. Using KT5720 (1 μM), a specific and cell-permeable inhibitor of PKA, which brain slices were incubated in for 1 h prior to recordings, we demonstrated that the inhibition of PKA blocks the maintenance of MOR-LTD of eEPSCs in the DLS (101 ± 19%; Fig. 4A-C and G). Cortico-striatal MOR-LTD was also blocked by PKA inhibition (94 ± 16%; Fig. 4H-J and N), as was MOR-LTD at AIC-DLS synapses (94 ± 11%; Fig. 4O-Q and U). We found that these effects were due to inhibiting presynaptic PKA activity as at least 30 min treatment with the membrane-impermeable PKA inhibitor PKI (1 μM), intracellularly infused through the recording micropipette into the postsynaptic MSN, did not affect MOR-LTD in the DLS (eEPSCs: 84 ± 7%; Fig. 4D-G) (corticostriatal oEPSCs: 64 ± 20%; Fig. 4K-N) (AIC-DLS oEPSCs: 69 ± 11%; Fig. 4R-U). Since this was a negative effect, we needed to test whether PKI was able to induce any effect. Therefore, we evaluated PKI in a brain region where the inhibition of postsynaptic PKA disrupts synaptic plasticity (Duffy & Nguyen, 2003). We found that intracellular PKI application blocked the expression of LTP in hippocampal CA1 pyramidal neurons (Control: 205 ± 49% vs. PKI: 88 ± 9%, Fig. 5). As we expected, our data demonstrate the role of presynaptic PKA in MOR-LTD.
Figure 5. Postsynaptic PKA is required for CA1 hippocampal LTP.
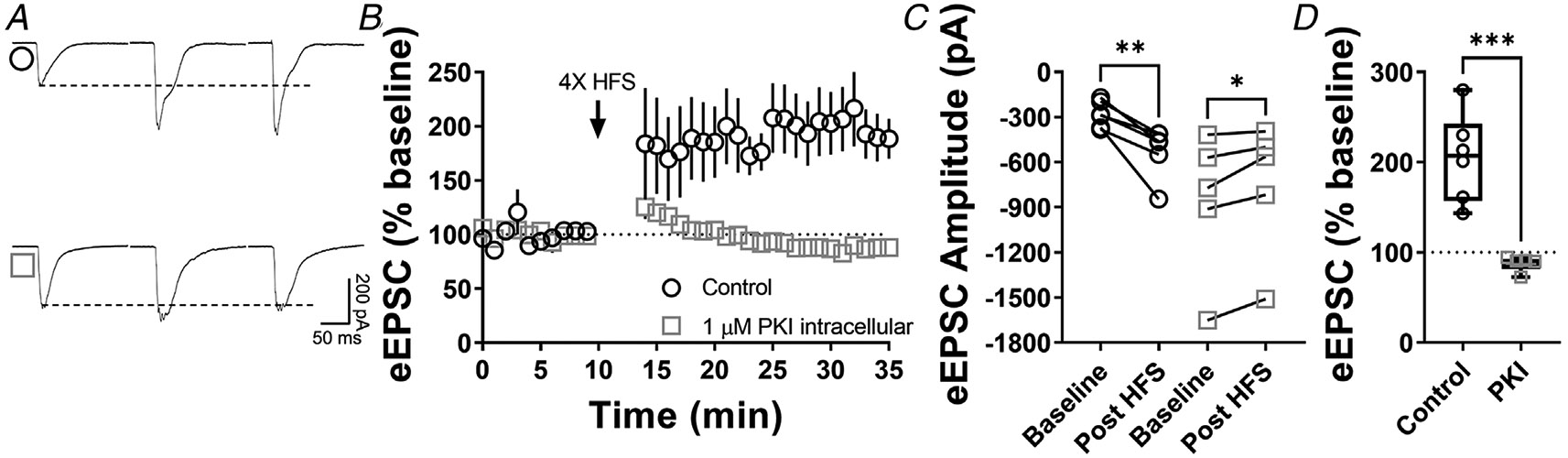
A, representative eEPSC traces before and after high-frequency stimulation (HFS) (four pulses of 100 Hz, 1 min inter-pulse interval), showing the effects of PKI (1 μM) intracellular diffusion into CA1 pyramidal neurons within hippocampal brain slices of C57BL/6J mice. B–C, postsynaptic PKA signalling is necessary to the induction of LTP in CA1 (0–10 min baseline vs. final 10 min of recording; Control: paired t test, P = 0.00587, t5 = 4.6, n = 6 neurons from four mice; PKI: paired t test, P = 0.0282, t4 = 3.36, n = 5 neurons from four mice). D, box plot shows the average disruption of hippocampal LTP by inhibiting postsynaptic PKA (Control: 205 ± 49 % vs. PKI: 88 ± 9%, P = 0.000551, t9 = 5.22, unpaired t test). Time course data represent means ± SEM. Box plots represent median and interquartile ranges. *P < 0.05, **P < 0.01, ***P < 0.001.
The activation of adenylyl cyclase disrupts MOR-mediated LTD
Demonstrating a role for PKA signalling in MOR-LTD implicates AC signalling as mediating MOR-LTD. To more specifically test this, we persistently activated AC using 20 μM forskolin throughout recording. In the presence of forskolin, DAMGO produced a transient increase of the EPSC amplitudes (Fig. 6A, B, E, F, I and J), followed by a blockade MOR-LTD at all excitatory synapses (105 ± 14%; Fig. 6A-D) and at corticostriatal (105 ± 6%; Fig. 6E-H) and AIC inputs (98 ± 14%; Fig. 6I-L). Altogether, these findings indicate that cAMP/PKA signalling pathway is involved in the induction and maintenance of MOR-LTD in the DLS.
Figure 6. The activation of adenylyl cyclase disrupts MOR-mediated LTD.
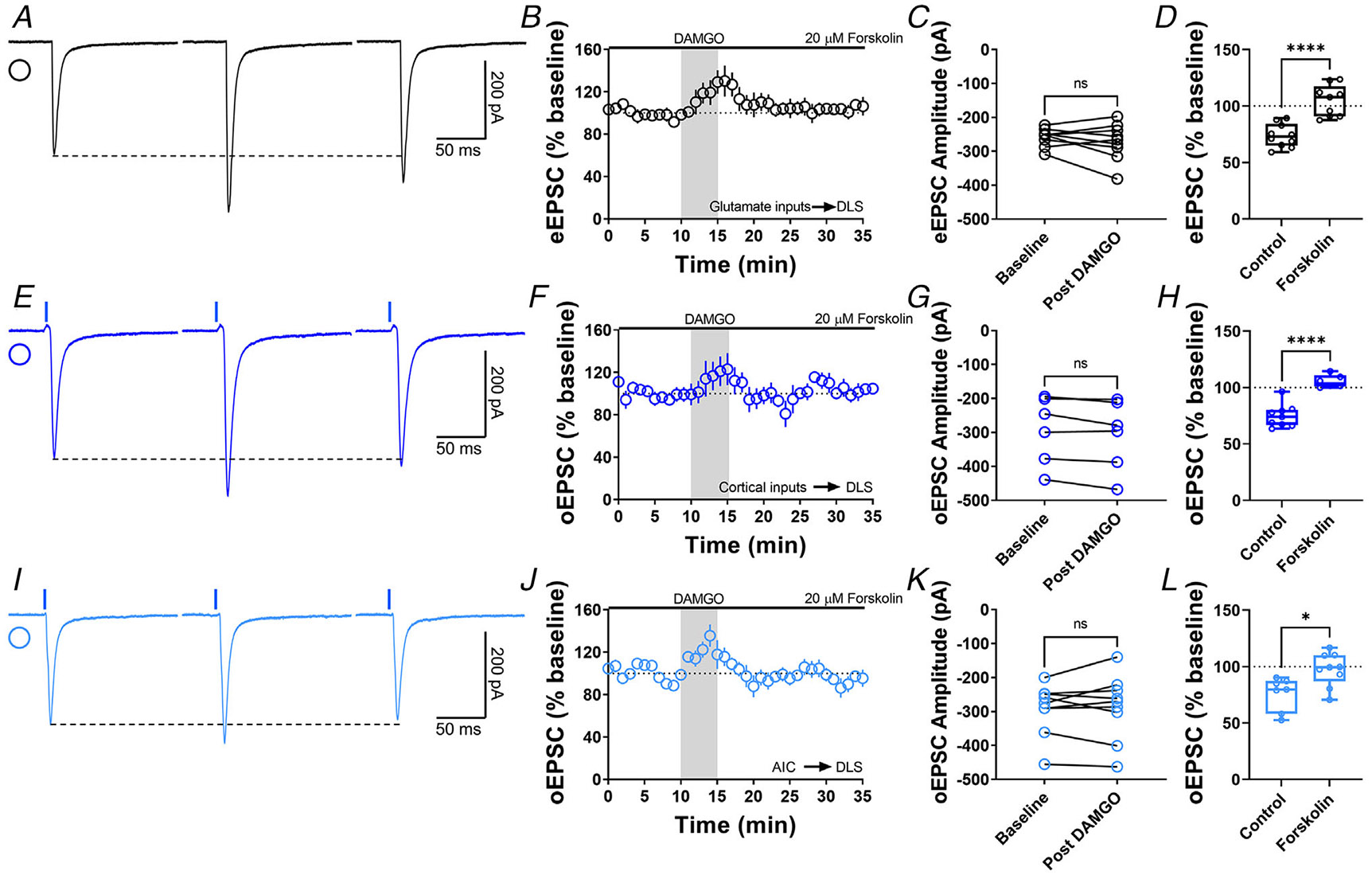
A, representative eEPSC traces showing the effects of 20 μM forskolin before, during and after DAMGO (0.3 μM, 5 min) application. B–D, AC activation disrupted glutamatergic MOR-LTD (105 ± 5%, unpaired t test, P < 0.0001, t17 = 5.54), showing no changes in eEPSC amplitude after DAMGO bath application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.324, t8 = 1.05, n = 9 neurons from four mice). E, representative oEPSC traces showing the effects of 20 μM forskolin before, during and after DAMGO (0.3 μM, 5 min) application in brain slices from Emx1-Ai32 mice. F–H, AC activation disrupted corticostriatal MOR-LTD (105 ± 2%, unpaired t test, P < 0.0001, t13 = 6.65), with no effects in oEPSC amplitude after DAMGO bath application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.0576, t5 = 2.455, n = 6 neurons from four mice). I, representative AIC-DLS oEPSC traces showing the effects of 20 μM forskolin before, during and after DAMGO (0.3 μM, 5 min) application. J–L, AC activation blocked specifically AIC MOR-LTD (98 ± 5%, unpaired t test, P = 0.0116, t14 = 2.9), without changes in oEPSC amplitude after DAMGO bath application (0–10 min base-line vs. final 10 min of recording; paired t test, P = 0.702, t8 = 0.396, n = 9 neurons from three mice). Time course data represent means ± SEM. Box plots show average of the final 10 min of recording and represent median and interquartile ranges. ns = not significant, *P < 0.05, ****P < 0.0001.
Protein translation is required to produce MOR-mediated LTD
Next, we tested the role of protein translation in MOR-LTD. Using bath application of 80 μM cyclo-heximide (selective inhibitor of protein synthesis) during the entire recording, we found that the inhibition of protein translation blocked MOR-LTD generally (105 ± 33%; Fig. 7A-D) and specifically at cortical (102 ± 18%; Fig. 7E-H) and AIC inputs (94 ± 10%; Fig. 7I-L).
It had been reported that protein translation-mediated CB-LTD involves mTOR signalling in the hippocampus (Younts et al., 2016). To test whether mTOR is also required for CB-LTD in the DLS, we used bath application of 100 nm Torin 2 (a selective mTOR inhibitor). We demonstrated that the inhibition of mTOR signalling disrupted CB-LTD in the DLS (Control: 76 ± 8% vs. Torin 2: 95 ± 5%; Fig. 8A-D). However, MOR-LTD does not require mTOR signalling in the DLS (79 ± 7%; Fig. 8E-H). These data suggest that protein translation is necessary for MOR-mediated LTD, but mTOR signalling is not involved and indicate that CB- and MOR-LTD, while qualitatively similar, do not utilize identical signalling mechanisms.
Figure 8. mTOR pathway is involved in cannabinoid-LTD but not in MOR-LTD in the DLS.
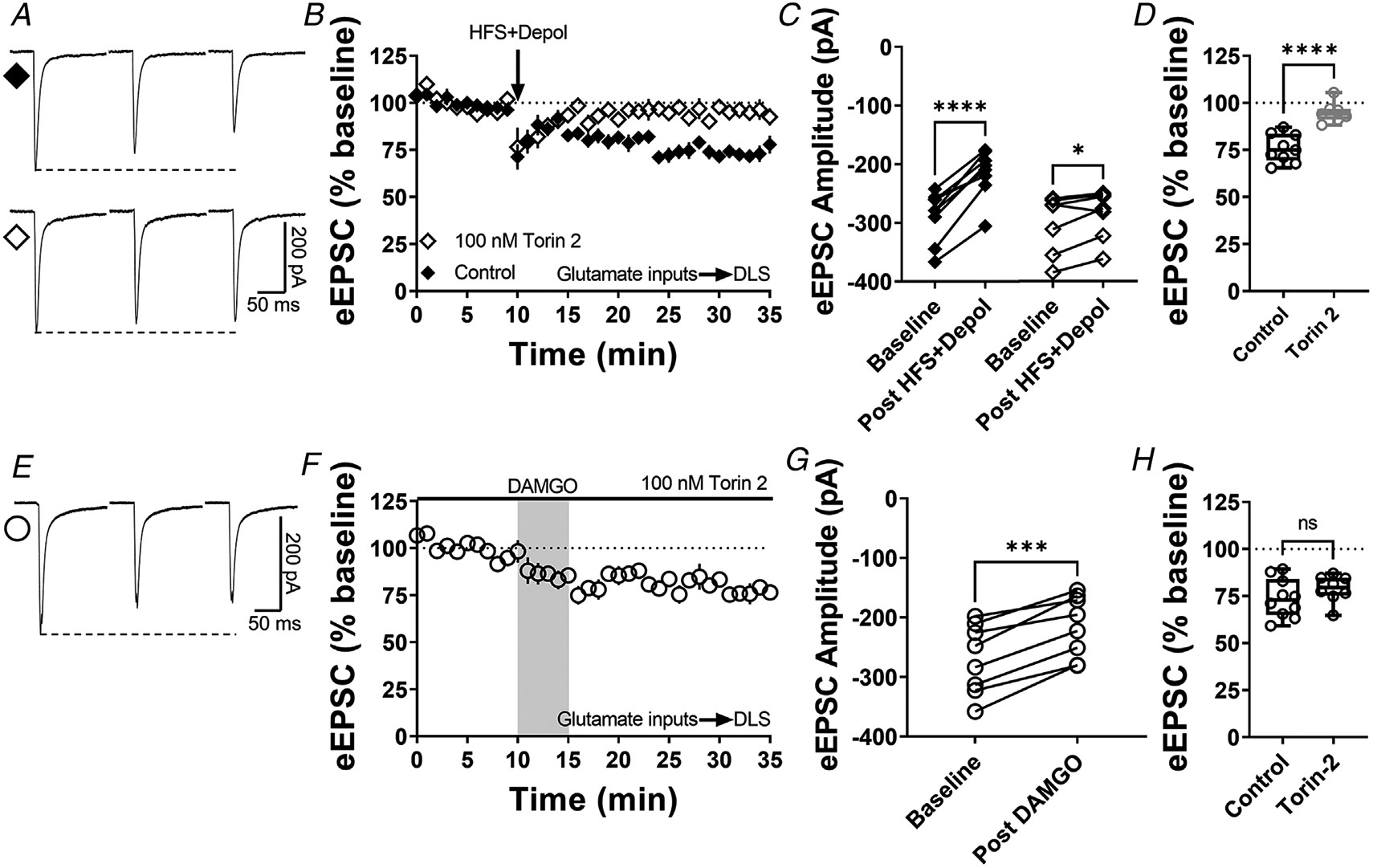
A, representative eEPSC traces before and after high-frequency stimulation (HFS) coupled with depolarization (four pulses of 100 Hz, 10 s inter-pulse interval), showing the effects of torin 2 (100 nM) bath application in brain slices of C57BL/6J mice. B–C, mTOR inhibition blocked this stimulation-induced cannabinoid-LTD in DLS (0–10 min baseline vs. final 10 min of recording; Control: paired t test, P < 0.0001, t8 = 8.6, n = 9 neurons from four mice; torin 2: paired t test, P = 0.0396, t6 = 2.62, n = 7 neurons from three mice). D, torin 2 disrupted stimulation-induced LTD in the DLS (control: 76 ± 3% vs. torin 2: 95 ± 2, unpaired t test, P < 0.0001, t14 = 5.64). E, representative eEPSC traces before and after DAMGO bath application (0.3 μM, 5 min), showing the effects of torin 2 (100 nM) bath application in brain slices of C57BL/J. F–G, mTOR inhibition did not affect the expression of MOR-LTD in DLS (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.000140, t7 = 7.48, n = 8 neurons from three mice). H, torin 2 did not block MOR-LTD in the DLS (control: 74 ± 3% vs. torin 2: 79 ± 3, unpaired t test, P = 0.270, t16 = 1.19). Time course data represent means ± SEM. Box plots show average of the final 10 min of recording and represent median and interquartile ranges. ns = not significant, *P < 0.05, ***P < 0.001, ****P < 0.0001.
MOR-STD from thalamocortical inputs is not affected by PKA inhibition, cAMP activation or protein translation inhibition
We also confirmed that thalamostriatal inputs expressed MOR-STD in the DLS using VGluT2-Ai32 transgenic mice (12–20 min: 68 ± 5%; 30–35 min: 93 ± 7%; Fig. 9A-B and M) as we previously showed (Atwood, Kupferschmidt et al., 2014; Munoz et al., 2018). These data replicate our previous work that MORs mediated STD of thalamostriatal synapses in the DLS. Unlike corticostriatal inputs, these thalamostriatal inputs were not affected by broad inhibition of PKA (12–20 min: 81 ± 15%; 30–35 min: 108 ± 26%). Inhibition of post-synaptic PKA had no effect on MOR-STD (12–20 min: 86 ± 9%; 30–35 min: 115 ± 31%; Fig. 9D-F and M), just as it had no effect on MOR-LTD. MOR-mediated STD at thalamic synapses was also not affected by the application of forskolin (12–20 min: 81 ± 13%; 30–35 min: 89 ± 15%; Fig. 9G-I and M) or cycloheximide treatment (12–20 min: 76 ± 18%; 30–35 min: 101 ± 23%; Fig. 9J-M). Altogether, these data indicate that MOR-STD at thalamic inputs to DLS does not utilize the same mechanisms that MOR-LTD does.
Presynaptic HCN1 channels are indispensable for MOR-mediated LTD
Given that we identified cAMP signalling as important for MOR-LTD expression (Fig. 6), we also explored the possibility of HCN1 channels as key factors for MOR-LTD. To do so, we applied the general HCN blocker ZD7288 (25 μM) to brain slices for at least 30 min before DAMGO application and during the entire recording. HCN inhibition blocked the expression of MOR-LTD (104 ± 17%; Fig. 10A-D), suggesting a novel role for HCN channels in DLS glutamatergic synaptic plasticity. Next, we characterized the effects of HCN channels in glutamatergic DLS synaptic transmission by bath-applying 25 μM ZD7288 for at least 15 min before recording sEPSCs. We found that the inhibition of HCN channels caused an enhancement in the frequency and a decrease in the amplitude of sEPSCs (Fig. 10E-G), without affecting rise and decay times (Fig. 10H and I). These data suggest that HCN channels in the DLS both modulate synaptic transmission and mediate MOR-LTD.
Figure 10. Inhibition of striatal HCN channels disrupts MOR-mediated LTD.
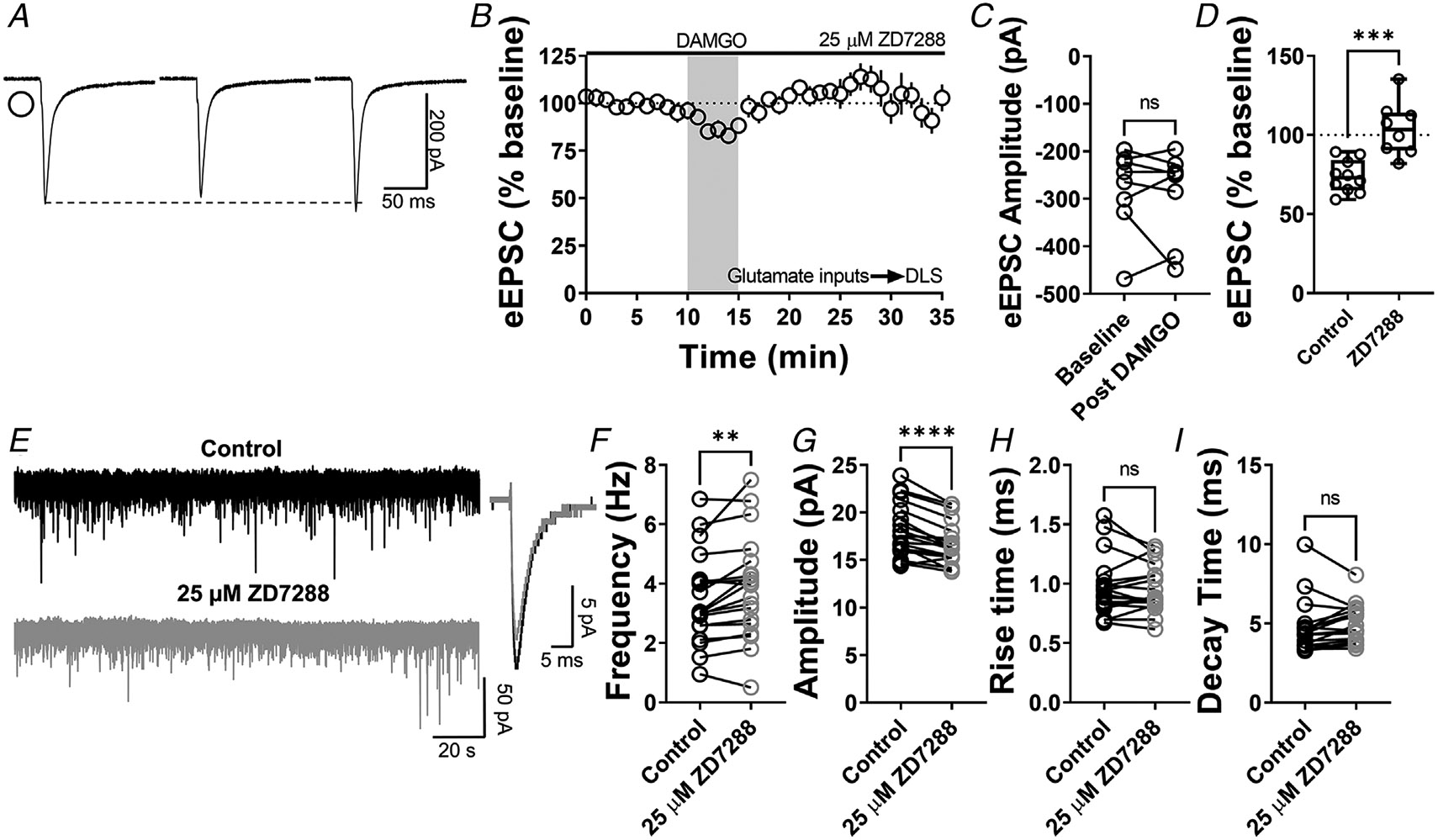
A, representative eEPSC traces showing the effects of 25 μM ZD7288 before, during and after DAMGO (0.3 μM, 5 min) application. B–D, HCN channel inhibition disrupted glutamatergic MOR-LTD (104 ± 6%, unpaired t test, P = 0.000498, t16 = 4.67) in the DLS, showing no changes in eEPSC amplitude after DAMGO bath application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.635, t7 = 0.496, n = 8 neurons from four mice). E, representative synaptic current traces from C57BL/6J mice showing the spontaneous excitatory postsynaptic currents (sEPSC) in absence (black trace) or presence of 25 μM ZD7288 (grey trace, 15 min) application. F–I, HCN channel inhibition increased frequency (paired t test, P = 0.00600, t18 = 3.113, n = 19 neurons from three mice) and decreased amplitude (paired t test, P < 0.0001, t18 = 5.88, n = 19 neurons from three mice), without changes in rise (paired t test, P = 0.608, t18 = 0.522, n = 19 neurons from three mice) and decay times (paired t test, P = 0.205, t18 = 1.314, n = 19 neurons from three mice) of the sEPSC events in DLS. Time course data represent means ± SEM. Box plots show average of the final 10 min of recording and represent median and interquartile ranges. ns = not significant, **P < 0.001, ***P < 0.001, ****P < 0.0001.
To further characterize this new role of HCN channels and specifically establish that presynaptic HCN1 channels are involved in the mechanism of expression of MOR-LTD, we used conditional HCN1 knockout mice (HCN1-flox) injected with AAV-cre vector into AIC to reduce the expression of the HCN1 channel specifically in the AIC, and used AAV-GFP vector-infused mice as controls (Figs 11A, B and 12). We used qPCR to confirm that AAV-cre infusion reduced HCN1 expression in the AIC (cre: 0.65 ± 0.2 vs. GFP: 1.07 ± 0.4, Fig. 11C). Next, using electrical stimulation and recording in the DLS, we found that the ablation of HCN1 expression in the AIC reduced the magnitude ofMOR-LTD in AAV-cre-injected mice, relative to AAV-GFP-injected mice (cre: 91 ± 12% vs. GFP: 77 ± 10%; Fig. 11D-H). Next, we tested whether the reduction of presynaptic HCN1 channels produced changes in basal striatal glutamatergic transmission. We found that the decrease of AIC HCN1 channels did not affect any synaptic parameters (Fig. 11I-M). These data suggest that HCN1 channels specifically on AIC inputs are necessary for MOR-LTD expression in the DLS.
Figure 11. Genetic ablation of HCN1 channels in the AIC reduces MOR-mediated LTD expression in the DLS.
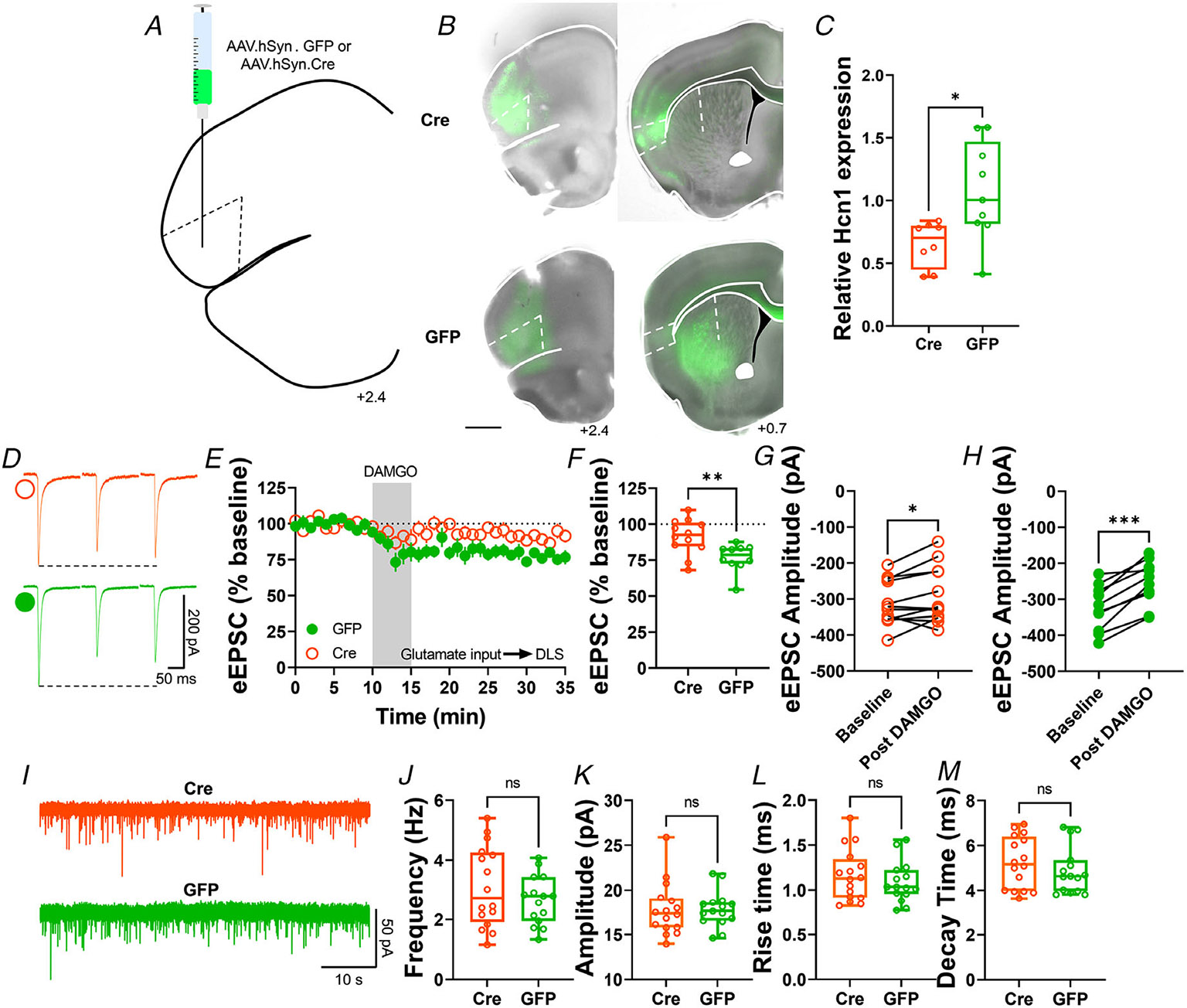
A, schematic figure of coronal brain slice showing injection paradigm enabling knock out of HCN1 channel expression in AIC in HCN1-flox mice. An AAV vector encoding for either cre-recombinase (AAV.hSyn.cre) or eGFP (AAV.hSyn.eGFP) was injected at least 4 weeks prior to recordings. B, coronal brain slice showing the AAV-cre or AAV-GFP infection of AIC and dorsal striatal terminal expression (bar scale = 1000 μm). C, AAV.cre reduced the expression of HCN1 in the AIC after 4 weeks post-injection (cre: 0.65 ± 0.06 vs. GFP: 1.07 ± 0.13, unpaired t test, P = 0.0139, t15 = 2.785, n = 8 cre-injected mice and n = 8 GFP-injected mice). D, representative eEPSC traces showing the effects of DAMGO (0.3 μM, 5 min) application in brain slices of AAV-injected HCN1-flox mice. E–F, the conditional decrease of HCN1 channel from AIC, blunted MOR-LTD in DLS (cre: 91 ± 4% vs. GFP: 77 ± 3%, unpaired t test, P = 0.00634, t20 = 3.05, n = 12 neurons from six cre-injected mice and n = 10 neurons from four GFP-injected mice). G, HCN1 reduction did not fully disrupt MOR-LTD, because there was still a small, but significant reduction of eEPSC amplitude after DAMGO application (0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.0415, t11 = 2.31, n = 12 neurons from six mice). H, control GFP-injected mice showed normal decrease of eEPSC amplitude after DAMGO application 0–10 min baseline vs. final 10 min of recording; paired t test, P = 0.000101, t9 = 6.55, n = 10 neurons from four mice). I, representative synaptic current traces from HCN1-flox mice showing the spontaneous excitatory postsynaptic currents (sEPSCs) in AAV-cre (orange trace) and AAV-GFP (green trace) injected mice. J–M, HCN1 channel ablation from AIC synapses did not affect frequency (cre: 3.1 ± 0.3 Hz vs. GFP: 2.7 ± 0.2 Hz, unpaired t test, P = 0.277, t29 = 1.11, n = 16 neurons from four cre-injected mice and n = 15 neurons from three GFP-injected mice), amplitude (cre: 17.8 ± 0.7 pA vs. GFP: 17.8 ± 0.5 pA, unpaired t test, P = 0.980, t29 = 0.025, n = 16 neurons from four cre-injected mice and n = 15 neurons from three GFP-injected mice), rise time (cre: 1.16 ± 0.07 ms vs. GFP: 1.09 ± 0.06 ms, unpaired t test, P = 0.415, t29 = 0.822, n = 16 neurons from four cre-injected mice and n = 15 neurons from three GFP-injected mice) and decay time (cre: 5.24 ± 0.29 ms vs. GFP: 4.9 ± 0.27 ms, unpaired t test, P = 0.395, t29 = 0.396, n = 16 neurons from four cre-injected mice and n = 15 neurons from three GFP-injected mice) of the sEPSC events in DLS. Time course data represent means ± SEM. Box plots show average of the final 10 min of recording and represent median and interquartile ranges. ns = not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
The current study demonstrates that the mechanism of MOR-mediated LTD is similar to the mechanism of induction of glutamatergic CB-LTD in that it involves cAMP-PKA-protein translation signalling and both occur at AIC-DLS synapses (Atwood, Lovinger et al., 2014; Lonart et al., 2003; Mato et al., 2008; Yasuda et al., 2008; Yin et al., 2006; Younts et al., 2016). However, we found notable differences such as a lack of dependence on mTOR signalling (Fig. 8) and a novel mechanism involving HCN1 signalling for MOR-LTD (Figs 10 and 11). Figure 13 summarizes the findings of the paper and illustrates the differential mechanisms involved in the expression of MOR-mediated LTD. Furthermore, the MOR-LTD mechanism is different from that of MOR-STD (Fig. 13), suggesting that MOR-mediated plasticity does not utilize a common pathway at all glutamatergic synapses.
Figure 13. Summary of the proposed presynaptic signalling pathways that are involved in MOR-mediated LTD and STD.
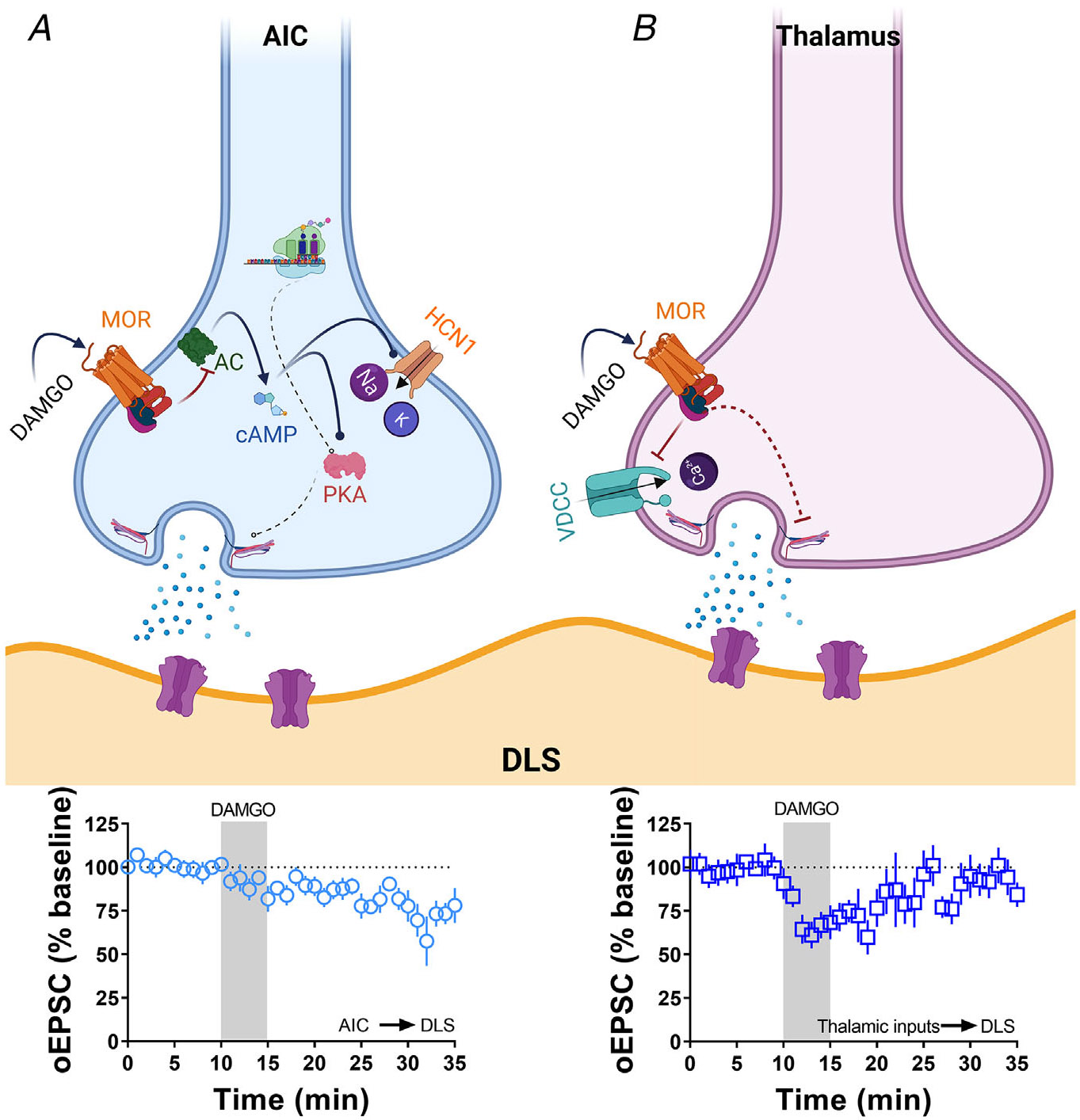
A, the activation of MOR at AIC inputs in the DLS induces a decrease in the production of cAMP (via adenylyl cyclase (AC) inhibition), which impacts the activation of HCN1 channels and the function of presynaptic PKA. Furthermore, the presence of local protein synthesis is critical for the maintaining of the glutamatergic LTD shown below. B, the activation of MOR at thalamic inputs in the DLS does not utilize the mechanisms illustrated in (A). Therefore, the mechanism is conceptualized to involve MOR-driven Gβγ block of voltage-dependent calcium channels producing a transitory reduction in the release of glutamate in the DLS as shown below. Schematic figure was created with BioRender.com.
Our previous report demonstrates that MORs expressed in AIC are the sole source of MOR-mediated LTD at glutamate inputs to the DLS (Munoz et al., 2018). However, in the DMS, MORs on the medial prefrontal cortex, anterior cingulate cortex and basolateral amygdala inputs are capable of inducing LTD (Muñoz et al., 2020). MOR-LTD also occurs locally at cholinergic interneuron synapses that co-release glutamate on to MSNs (Munoz et al., 2018; Muñoz et al., 2020). However, alcohol only disrupts MOR-LTD at AIC inputs to DLS and at medial prefrontal and anterior cingulate cortex inputs to DMS leaving MOR plasticity at thalamostriatal, amygdalostriatal and cholinergic interneurons unaffected (Munoz et al., 2018; Muñoz et al., 2020). We also found that the opioid oxycodone disrupts MOR-LTD in the DLS (Atwood, Kupferschmidt et al., 2014), and given that this is mediated by AIC-DLS MORs (Munoz et al., 2018), it follows that alcohol and opioids have similar actions on MOR plasticity at this synapse. Given that alcohol (and presumably oxycodone and other opioids) does not affect MOR plasticity at these other non-corticostriatal synapses, differential mechanisms that underlie MOR plasticity at each type of synapse may confer susceptibility to the deleterious effects of these drugs and could account for their mechanisms of action in the development of habitual or compulsive use of the drugs.
Previous work has shown that the inhibition of PKA disrupts CB-LTD in the nucleus accumbens (Mato et al., 2008) and that presynaptic PKA is required to induce C-LTD in the cerebellum (Lonart et al., 2003) and cannabinoid synaptic plasticity in the hippocampus (Yasuda et al., 2008). Moreover, previous reports had shown that the activation of AC by forskolin enhances corticostriatal EPSC amplitudes (Cho et al., 2008) and blocked delta opioid receptor-mediated LTD of inhibitory postsynaptic currents (Patton et al., 2016). cAMP/PKA signalling has also been implicated in presynaptic type 2/3 metabotropic glutamate receptor- and type 1b serotonin receptor-mediated LTD at various synapses in the brain (Atwood, Kupferschmidt et al., 2014). As previously reported (Duffy & Nguyen, 2003), we confirmed that the activation of postsynaptic PKA is necessary for the induction of LTP in CA1 pyramidal neurons from the hippocampus, but not for MOR-LTD in the DLS (Fig. 5). Until now, the role of the cAMP/PKA signalling pathway in MOR-mediated plasticity had not been addressed. Our results demonstrate that presynaptic PKA inhibition blocked AIC-DLS MOR-LTD (Figs 4 and 13) but not thalamostriatal MOR-STD (Figs 9D-F and 13). Interestingly, the magnitudes and time courses of eEPSCs and oEPSCs did not differ between measures (Fig. 4A, D, H, K, O and R), suggesting that PKA is not required for an acute MOR-mediated inhibition of glutamate release, but it is required for the maintenance of MOR-LTD. On the other hand, we found that AC activation disrupted both acute MOR-mediated inhibition (in fact turning it into a transient potentiation) and MOR-LTD (Figs 6 and 13), but had no effect on MOR-STD (Figs 9G-I, M and 13). Altogether these data suggest that the cAMP/PKA pathway may be a general mechanism whereby GPCRs that signal through Gαi may induce LTD, but a receptor’s ability to couple to Gαi does not necessarily mean that it will induce cAMP/PKA-dependent LTD upon activation.
Protein translation is necessary for striatal CB-LTD (Yin et al., 2006) and is involved in GPCR-mediated LTD at some synapses, but not others (Atwood, Kupferschmidt et al., 2014; Younts et al., 2016), suggesting that protein translation can be an important component of glutamatergic LTD, depending on the synapse. Indeed, local protein translation can take place in the presynaptic region, where the activation of CB1 receptors enhances protein translation, being critical for the induction and not the maintenance of inhibitory LTD (Younts et al., 2016). Here we demonstrated that protein translation is necessary to produce MOR-LTD (Figs 7 and 13), but not MOR-STD (Figs 9J-M and 13). Our data are consistent with the previous reports on protein translation-dependent CB-LTD, and suggest that it is presynaptic protein translation that is involved in MOR-LTD expression. However, cycloheximide has been reported to also activate p38/MAPK signalling (Lockhead et al., 2020) and several studies have shown a critical role of p38 kinases in the induction of LTD and LTP (Asih et al., 2020). Therefore, more data are needed to determine whether this is definitively protein translation-mediated in the DLS.
Although we have previously reported that MOR- and CB-LTD are mutually occlusive in the DLS (Atwood, Kupferschmidt et al., 2014), and here we found that there were similarities between the MOR-LTD and CB-LTD mechanisms, we found some differences which make both types of LTD distinct. In the DLS, MOR-LTD of glutamate input is restricted to the AIC (Munoz et al., 2018), which differs from the more broadly expressed (and overlapping at AIC-DLS synapses, Fig. 3) CB-LTD (Gremel et al., 2016; Wu et al., 2015), suggesting that these two forms of LTD may occur in distinct synaptic environments that could utilize differing mechanisms. It is likely that even in synapses where both receptors are expressed (e.g. in AIC terminals) that each receptor shares partially overlapping, but not identical signalling pathways.
A clear example of distinct signalling mechanisms identified here is in the role of the mTOR pathway in CB-LTD and MOR-LTD. The CB1 receptor can signal via mTOR in the hippocampus, where the increase of protein translation induced by CB1 receptor activation is mTOR-dependent, producing inhibitory CB-LTD (Younts et al., 2016). We predicted that we would see the same effect in the DLS for both CB-LTD and for MOR-LTD. Partially correct in our prediction, we found that CB-LTD is blocked after the inhibition of mTOR in the DLS (Fig. 8A-D), but the mechanism of MOR-LTD did not utilize mTOR (Fig. 8E-H). These novel data regarding CB1-mTOR signalling in the DLS provide a rationale for future experiments addressing the role of CB1-mTOR signalling in dorsal striatal-related behaviours.
Presynaptic cations such as Na+ and K+ are important for the propagation of action potentials and subsequent glutamate release (Chen & Lui, 2021). HCN channels are permeable to Na+ and K+ and modulated by cAMP (Sartiani et al., 2017), and they can decrease neurotransmission by restricting presynaptic Ca2+ influx (Huang et al., 2011). We did identify a critical role for the HCN1 channel in MOR-LTD expression (Figs 10, 11 and 13). It has been reported that HCN1 channels can be found at presynaptic sites in the globus pallidus, where the inhibition by ZD7288 produced an increase of miniature inhibitory postsynaptic current frequency, with no changes in amplitude (Boyes et al., 2007). The block or deletion of HCN1 causes an increase in the frequency of miniature EPSCs in entorhinal cortical pyramidal neurons (Huang et al., 2011). Here we showed that a broad blockade of HCN channels alters glutamatergic synaptic transmission release (Fig. 10E-I). However, when we deleted just HCN1 from AIC inputs to the DLS (Fig. 11C), we did not observe any changes in basal glutamate transmission (Fig. 11I-M). The differences in the effects of the broad blockade and synapse-specific deletion may be attributed to the overall contribution of HCN1s at AIC inputs relative to the population of glutamate synapses. Genetic deletion did not completely ablate MOR-LTD expression (Fig. 11G). This could be due to an incomplete AAV vector transduction (most recordings had a complete loss of LTD) or perhaps that HCN1 contributes to LTD differently at AIC inputs to different subtypes of MSN. Therefore, in the future, it will also be important to investigate the role of HCN1 in MOR-LTD at AIC inputs to D1- or D2-expressing MSNs (direct or indirect pathway respectively). It has recently been reported that HCN1 contributes to alcohol preference (Salling & Harrison, 2020). Given that alcohol disrupts MOR-LTD in DLS, this mechanism of MOR-HCN1 signalling may be a target of alcohol’s effects and contribute to alcohol-related, dorsal striatal-dependent behaviours such as habitual and compulsive alcohol drinking. As the opioid oxycodone also disrupts MOR-LTD it will be intriguing to determine the impact of both oxycodone and alcohol on AIC-DLS HCN1 function. HCN1 may therefore have therapeutic potential in alcohol and opioid use disorders.
In conclusion, these findings indicate that glutamatergic synapses in the DLS have distinct mechanisms of plasticity induction mediated by the same types of receptors. The specific signalling processes that result in these different forms of plasticity might explain why different types of plasticity are differentially sensitive to drugs of abuse and alcohol (Fig. 13) (Atwood, Lovinger et al., 2014; Munoz et al., 2018; Muñoz et al., 2020). However, further work is required to resolve what components of these mechanisms render some synapses susceptible to the deleterious effects of these drugs while others are resistant. It will also be important to decipher the behavioural relevance of AIC-DLS synapse HCN1 channels and CB1-mTOR signalling in the DLS.
Supplementary Material
Key points.
Mu opioid receptor-mediated long-term depression at anterior insular cortex inputs to dorsolateral striatum involves presynaptic cAMP/PKA signalling and protein translation, similar to known mechanisms of cannabinoid long-term depression.
Dorsal striatal cannabinoid long-term depression also occurs at anterior insular cortex inputs to the dorsolateral striatum. Dorsal striatal cannabinoid long-term depression requires mTOR signalling, similar to hippocampal cannabinoid long-term depression, but dorsal striatal mu opioid long-term depression does not require mTOR signalling.
Mu opioid long-term depression requires presynaptic HCN1 channels at anterior insular cortex inputs to dorsolateral striatum.
Funding
This work was supported by NIH/NIAAA grant R01 AA027214.
Biography

Braulio Munoz graduated with a PhD in Molecular & Cell Biology from the University of Concepcion, Chile, during which he studied the role of glycine receptors on nucleus accumbens. He undertook his postdoctoral training in Dr Brady Atwood’s laboratory on mu opioid-mediated and cannabinoid-mediated glutamatergic plasticity. Dr Munoz is currently an Assistant Research Professor in Dr Atwood’s laboratory in the Department of Pharmacology and Toxicology at Indiana University School of Medicine (IUSM). His main focus is studying glutamatergic and inhibitory (GABA, glycine) synaptic plasticity with in corticostriatal neurocircuits. Specifically, on the effect of alcohol on synaptic plasticity in the neurocircuits that mediate alcohol drinking behaviours.
Footnotes
The peer review history is available in the Supporting information section of this article (https://doi.org/10.1113/JP283513#support-information-section).
Competing interests
None declared.
Supporting information
Additional supporting information can be found online in the Supporting Information section at the end of the HTML view of the article. Supporting information files available:
Statistical Summary Document
Peer Review History
Data availability statement
All data in this study are available upon reasonable request to the corresponding author. All reagents and other materials used for this work are commercially available.
References
- Asih PR, Prikas E, Stefanoska K, Tan ARP, Ahel HI, & Ittner A (2020). Functions of p38 MAP kinases in the central nervous system. Frontiers in Molecular Neuroscience, 13, 570586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, & Lovinger DM (2014). Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nature Neuroscience, 17(4), 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM, & Mathur BN(2014). Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends in Neuroscience (Tins), 37(11), 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G, Kang L, Li H, Li Y, Pu L, Xia P, Ma L, & Pei G(2007). Morphine and heroin differentially modulate in vivo hippocampal LTP in opiate-dependent rat. Neuropsychopharmacology, 32(8), 1738–1749. [DOI] [PubMed] [Google Scholar]
- Boyes J, Bolam JP, Shigemoto R, & Stanford IM(2007). Functional presynaptic HCN channels in the rat globus pallidus. European Journal of Neuroscience, 25(7), 2081–2092. [DOI] [PubMed] [Google Scholar]
- Burton AC, Nakamura K, & Roesch MR (2015). From ventral-medial to dorsal-lateral striatum: Neural correlates of reward-guided decision-making. Neurobiology of Learning and Memory, 117, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, & Lui F (2021). Neuroanatomy, neuron action potential. In StatPearls. StatPearls Publishing. Copyright © 2021, StatPearls Publishing LLC., Treasure Island (FL). [PubMed] [Google Scholar]
- Cho HS, Lee HH, Choi SJ, Kim KJ, Jeun SH, Li Q-Z, & Sung K-W (2008). Forskolin enhances synaptic transmission in rat dorsal striatum through NMDA receptors and PKA in different phases. Kjpp, 12, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit LH, & Janak PH(2016). Habitual alcohol seeking: Neural Bases and possible relations to alcohol use disorders. Alcoholism: Clinical and Experimental Research, 40(7), 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M, & Nugent FS(2011a). Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology, 61(7), 1166–1171. [DOI] [PubMed] [Google Scholar]
- Dacher M, & Nugent FS(2011b). Opiates and plasticity. Neuropharmacology, 61(7), 1088–1096. [DOI] [PubMed] [Google Scholar]
- DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, Pickens CL, Cinar R, Saksida LM, Kunos G, Lovinger DM, Bussey TJ, Camp MC, & Holmes A(2013). Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. PNAS, 110(36), 14783–14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Chavkin C, & Milner TA(2007). Opioid systems in the dentate gyrus. Progress in Brain Research, 163, 245–263. [DOI] [PubMed] [Google Scholar]
- Duffy SN, & Nguyen PV (2003). Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. The Journal of Neuroscience, 23(4), 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, & Robbins TW (2016). Drug addiction: Updating actions to habits to compulsions ten years on. Annual Review of Psychology, 67(1), 23–50. [DOI] [PubMed] [Google Scholar]
- Fritz BM, Munoz B, & Atwood BK(2019). Genetic selection for alcohol preference in mice alters dorsal striatum neurotransmission. Alcoholism, Clinical and Experimental Research, 43(11), 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz BM, Munoz B, Yin F, Bauchle C, & Atwood BK(2018). A high-fat, high-sugar ‘western’ diet alters dorsal striatal glutamate, opioid, and dopamine transmission in mice. Neuroscience, 372, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, Chancey JH, Atwood BK, Luo G, Neve R, Ramakrishnan C, Deisseroth K, Lovinger DM, & Costa RM(2016). Endocannabinoid modulation of orbitostriatal circuits gates habit formation. Neuron, 90(6), 1312–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes SL, Salinas AG, Lovinger DM, & Blackwell KT(2017). Long-term plasticity of corticostriatal synapses is modulated by pathway-specific co-release of opioids through κ-opioid receptors. Journal of Physiology, 595(16), 5637–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Chen F, Li B, & Hu Z(2014). Neurophysiology of HCN channels: From cellular functions to multiple regulations. Progress in Neurobiology, 112, 1–23. [DOI] [PubMed] [Google Scholar]
- Hilário MRF, & Costa RM(2008). High on habits. Frontiers in Neuroscience, 2(2), 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, & Lesscher HMB(2014). Rodent models for compulsive alcohol intake. Alcohol, 48(3), 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Li G, Aguado C, Lujan R, & Shah MM(2017). HCN1 channels reduce the rate of exocytosis from a subset of cortical synaptic terminals. Science Reports, 7(1), 40257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC, & Shah MM(2011). Presynaptic HCN1 channels regulate CaV3.2 activity and neurotransmission at select cortical synapses. Nature Neuroscience, 14(4), 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iremonger KJ, & Bains JS(2009). Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. Journal of Neuroscience, 29(22), 7349–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Becker JAJ, Befort K, & Kieffer BL (2009). Reward processing by the opioid system in the brain. Physiological Reviews, 89(4), 1379–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhead S, Moskaleva A, Kamenz J, Chen Y, Kang M, Reddy AR, Santos SDM, & Ferrell JE, Jr(2020). The apparent requirement for protein synthesis during G2 phase is due to checkpoint activation. Cell Reports, 32(2), 107901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, & Linden DJ(2003). Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell, 115(1), 49–60. [DOI] [PubMed] [Google Scholar]
- Lovinger DM(2010). Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology, 58(7), 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato S, Lafourcade M, Robbe D, Bakiri Y, & Manzoni OJ(2008). Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca++ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology, 54(1), 87–94. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, & Ludwig A(1999). Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biological Chemistry, 380(7–8), 975–980. [DOI] [PubMed] [Google Scholar]
- Munoz B, Fritz BM, Yin F, & Atwood BK(2018). Alcohol exposure disrupts mu opioid receptor-mediated long-term depression at insular cortex inputs to dorsolateral striatum. Nature Communication, 9(1), 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz B, Haggerty DL, & Atwood BK(2020). Synapse-specific expression of mu opioid receptor long-term depression in the dorsomedial striatum. Scientific Reports, 10(1), 7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazzaro C, Greco B, Cerovic M, Baxter P, Rubino T, Trusel M, Parolaro D, Tkatch T, Benfenati F, Pedarzani P, & Tonini R(2012). SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nature Neuroscience, 15(2), 284–293. [DOI] [PubMed] [Google Scholar]
- O’Tousa D, & Grahame N (2014). Habit formation: Implications for alcoholism research. Alcohol, 48(4), 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton MH, Roberts BM, Lovinger DM, & Mathur BN(2016). Ethanol disinhibits dorsolateral striatal medium spiny neurons through activation of a presynaptic delta opioid receptor. Neuropsychopharmacology, 41(7), 1831–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postea O, & Biel M(2011). Exploring HCN channels as novel drug targets. Nature Reviews Drug Discovery, 10(12), 903–914. [DOI] [PubMed] [Google Scholar]
- Reeves KC, Shah N, Muñoz B, & Atwood BK(2022). Opioid receptor-mediated regulation of neurotransmission in the brain. Frontiers in Molecular Neuroscience, 15, 919773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renteria R, Cazares C, & Gremel CM(2020). Habitual ethanol seeking and licking microstructure of enhanced ethanol self-administration in ethanol-dependent mice. Alcoholism, Clinical and Experimental Research, 44(4), 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salling MC, & Harrison NL(2020). Constitutive genetic deletion of Hcn1 increases alcohol preference during adolescence. Brain Sciences, 10(11), 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartiani L, Mannaioni G, Masi A, Novella Romanelli M, & Cerbai E (2017). The hyperpolarization-activated cyclic nucleotide–gated channels: From biophysics to pharmacology of a unique family of ion channels. Pharmacological Reviews, 69(4), 354–395. [DOI] [PubMed] [Google Scholar]
- Shen F, Jin S, Duan Y, Liang J, Zhang M, Jiang F, & Sui N (2018). Distinctive changes of L-type calcium channels and dopamine receptors in the dorsomedial and dorsolateral striatum after the expression of habitual cocaine-seeking behavior in rats. Neuroscience, 370, 139–147. [DOI] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, & Ding JB(2015). Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Reports, 10(1), 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Huang Y, & Tsumoto T(2008). Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proceedings of the National Academy of Sciences, 105(8), 3106–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Davis MI, Ronesi JA, & Lovinger DM(2006). The role of protein synthesis in striatal long-term depression. The Journal of Neuroscience, 26(46), 11811–11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younts TJ, Monday HR, Dudok B, Klein ME, Jordan BA, Katona I, & Castillo PE(2016). Presynaptic protein synthesis is required for long-term plasticity of GABA release. Neuron, 92(2), 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in this study are available upon reasonable request to the corresponding author. All reagents and other materials used for this work are commercially available.