

Abstract
The Sec61 complex forms a protein-conducting channel in the endoplasmic reticulum membrane that is required for secretion of soluble proteins and production of many membrane proteins. Several natural and synthetic small molecules specifically inhibit Sec61, generating cellular effects that are useful for therapeutic purposes, but their inhibitory mechanisms remain unclear. Here we present near-atomic-resolution structures of human Sec61 inhibited by a comprehensive panel of structurally distinct small molecules—cotransin, decatransin, apratoxin, ipomoeassin, mycolactone, cyclotriazadisulfonamide, and eeyarestatin. All inhibitors bind to a common lipid-exposed pocket formed by the partially open lateral gate and plug domain of Sec61. Mutations conferring resistance to the inhibitors are clustered at this binding pocket. The structures indicate that Sec61 inhibitors stabilize the plug domain in a closed state, thereby preventing the protein-translocation pore from opening. Our study provides the atomic details of Sec61–inhibitor interactions and the structural framework for further pharmacological studies and drug design.
Introduction
The universally conserved heterotrimeric Sec61 complex (SecY in prokaryotes) plays essential roles in biosynthesis of more than one third of proteins in all species (for recent review, see ref.1). In eukaryotes, secretory proteins are first translocated into the endoplasmic reticulum (ER) by the Sec61 complex before reaching the cell surface by vesicular trafficking. The Sec61 complex also mediates membrane integration of many proteins, including most cell surface receptors and cell adhesion molecules. The Sec61/SecY channel has an hourglass-like structure with a pore constriction (termed the pore ring) halfway across the membrane, which is gated by a movement of a plug-like ER-lumenal (or extracellular in SecY) domain of the channel2. In addition, the channel has a seam (lateral gate) in the wall that can open laterally in the plane of the membrane to release transmembrane segments (TMs) of membrane protein clients into the lipid phase. Concerted opening of the lumenal and lateral gates is also required for initial insertion of the client protein’s hydrophobic signal sequence or uncleavable signal anchor into the channel (Fig. 1a).
Figure 1. Cryo-EM structures of the human Sec61 complex inhibited by various small-molecule inhibitors.
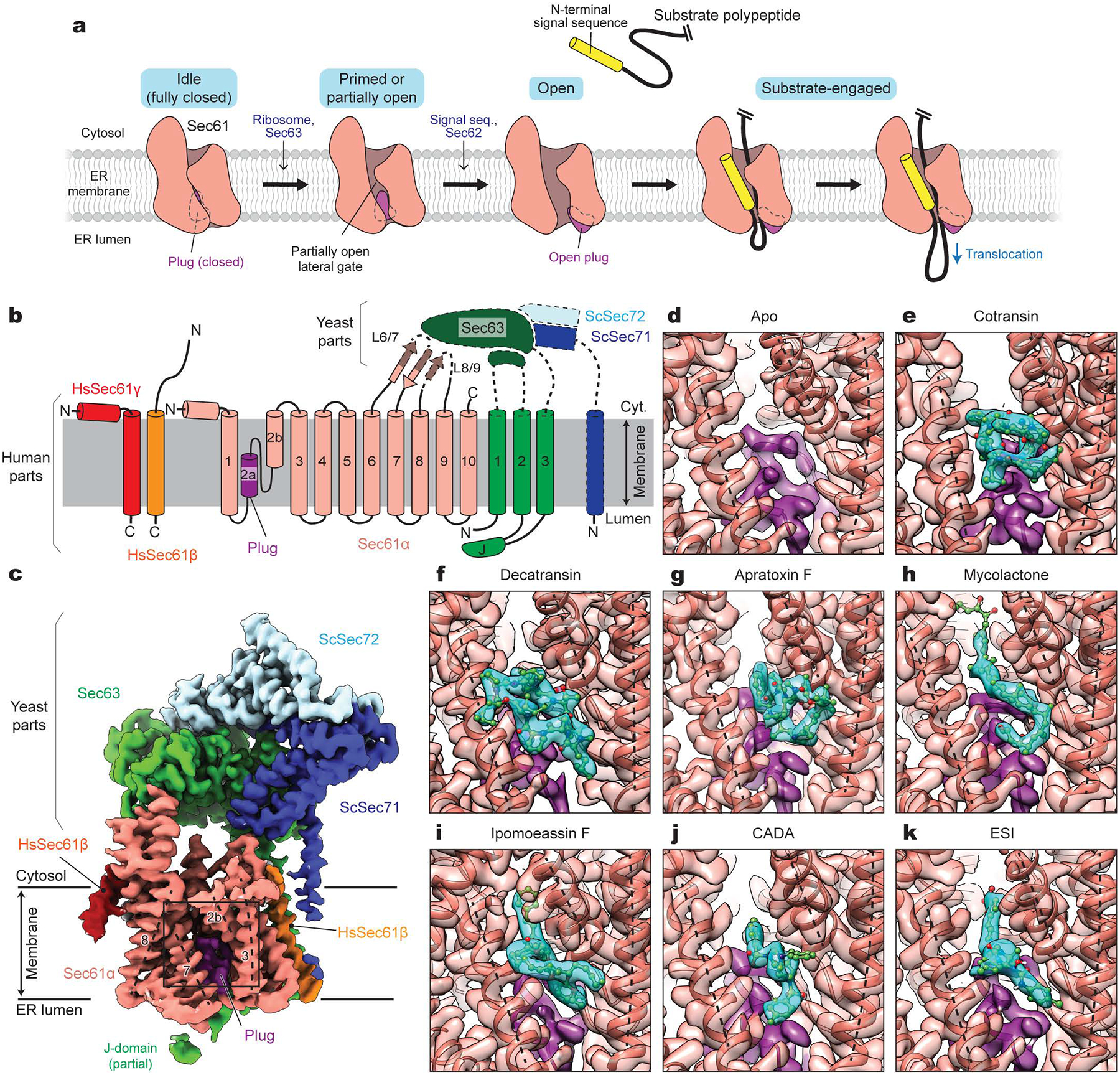
a, Architecture of the Sec61 channel and overall model for gating and substrate engagement. b, Design of a human-yeast chimeric Sec complex. Parts derived from human and yeast proteins are outlined with solid and dashed lines, respectively. Note that except for the cytosolic L6/7 and L8/9 loops, Sec61α is from the human SEC61A1 protein sequence. Hs, Homo sapiens; Sc, Saccharomyces cerevisiae; J, J-domain. c, 2.7-Å-resolution cryo-EM map of the chimeric Sec complex in an apo state (Class 1, unsharpened map). The lateral gate helices are indicated by dashed lines and TM numbers. The region outlined by a rectangle indicates the inhibitor-binding site (also see d–k). d–k, Views into the inhibitor-binding site of Sec61α of apo and inhibitor-bound structures. Cryo-EM maps (semi-transparent surface) and atomic models were overlaid. Inhibitor and plug densities are shown in cyan and purple, respectively. Dashed lines indicate lateral gate helices TMs 2b, 3, and 7 as in c.
The Sec61/SecY channel translocates polypeptides either co-translationally by docking a translating ribosome or post-translationally by engaging a fully synthesized polypeptide client. In eukaryotes, the post-translational mode is enabled by association of the channel with two additional membrane proteins Sec63 and Sec62 (ref.3,4). X-ray crystallography and cryo-electron microscopy (cryo-EM) have visualized structures of the Sec61/SecY channel in different functional states and revealed how it is gated and engages with client proteins2,5–14. The current model posits that association of a ribosome or Sec63 slightly perturbs (“primes”) or partially opens the lateral gate7,11,12 (Fig. 1a). Insertion of the client polypeptide needs further widening of the lateral opening and a displacement of the plug away from the pore, which occur in a cooperative manner. In cotranslational translocation, these conformational changes are presumed to be induced by an interaction between the channel and the signal sequence/anchor7,9, whereas in post-translational translocation, they seem to be mediated by Sec6213.
Several natural and synthetic small molecules bind to Sec61 and inhibit protein translocation (for review, see ref.15,16). These inhibitors have been investigated as potential anticancer, antiviral, and/or immunosuppressive agents17–20. Inhibition of Sec61 leads to downregulation of disease-related and clinically-relevant proteins, such as cytokines, cell surface receptors, and viral membrane proteins. Indeed, one such Sec61 inhibitor is currently being tested in a phase-I clinical trial for treatment of solid tumor malignancies21. A founding class of Sec61 inhibitors is a group of fungal-derived cyclic heptadepsipeptides named cotransins22–24. Other naturally occurring inhibitors discovered to date are decatransin, mycolactone, apratoxins, coibamide A, and ipomoeassin F, which are produced by certain fungal, bacterial, and plant species25–30. In addition, two synthetic compounds cyclotriazadisulfonamide (CADA) and eeyarestatin I (ESI) have also been shown to inhibit the Sec61 channel31,32. These inhibitors are structurally unrelated to each other, but several of them have been suggested to bind to an overlapping site in the Sec61 channel based on their abilities to compete for Sec61 binding. Remarkably, cotransin and CADA inhibit Sec61 in a client-specific manner22,23,33, whereas other inhibitors act more broadly independent of clients. Biochemical data suggest that cotransin likely interacts with the lateral gate and/or the plug of Sec61 (ref.34). However, key information regarding the actions of these inhibitors remains unavailable, including molecular details about Sec61-inhibitor interactions, which specific steps along the translocation process are inhibited, and what underlies client-specific versus broad-spectrum inhibition. This has limited our capability to design or discover additional therapeutically promising small-molecule agents that target Sec61.
Results
Cryo-EM analysis of inhibitor-bound Sec61
To understand the mechanism of Sec61 inhibition, we sought to determine high-resolution structures of inhibitor-bound Sec61 using cryo-EM. To date, all mammalian Sec61 structures have been obtained from ribosome-bound cotranslational complexes7,8. However, due to the flexibility of Sec61 with respect to the ribosome, this approach limits the resolution of Sec61 to only ~5 Å, a resolution that is impractical to model protein side chains and small ligands7. This limitation is also apparent in the recent cryo-EM structures of the Sec61-ribosome complex treated with mycolactone35, a cotransin derivative (KZR-8445)36, or a CADA derivative (CK147)37. By contrast, we previously attained 3.1–3.7-Å resolution structures of the Sec61 channel from fungal post-translational translocation complexes11,13 (termed the Sec complex), which contained Sec62, Sec63 and fungal-specific nonessential Sec71 and Sec72 in addition to the three (α, β, and γ) subunits of the Sec61 complex. Thus, we reasoned that use of the Sec complex would be an effective approach to study Sec61 inhibitors.
To enable high-resolution cryo-EM analysis of inhibitor-bound human Sec61, we designed a chimeric Sec complex, whose transmembrane and cytosolic domains are derived from the human and yeast proteins, respectively (Fig. 1b). Our initial efforts employing the entirely yeast or human Sec complex were unsuccessful. The yeast Sec complex incubated with cotransin ‘compound 2’ (hereafter referred to as cotransin CP2)25 did not show any cotransin-like feature in the cryo-EM map (Extended Data Fig. 1 a and b). This could be due to a lower binding affinity of cotransin CP2 towards yeast Sec61 compared to mammalian Sec6125, the presence of detergent in the sample, or both. We could see a putative cotransin CP2 density in a cryo-EM structure of the human Sec complex lacking Sec62, but the resolution could not be improved beyond ~5 Å, probably due to high flexibility of the cytosolic domain of human Sec63 (Extended Data Fig. 1 c–f). We hypothesized that the resolution could be improved by replacing the cytosolic domain of Sec63 in the human complex with the yeast counterpart, as yeast Sec63, along with additional Sec71–Sec72 proteins, consistently showed well-defined features in our previous cryo-EM studies11,13. While this chimeric construct would not be functional for post-translational translocation without a matching chimeric Sec62 subunit that can interact with both human Sec61 and yeast Sec63, we expected that inhibitors would still bind efficiently to the channel as the Sec61 sequence is mostly human.
The human-yeast chimeric Sec complex reconstituted into a peptidisc38 indeed yielded dramatically improved structures at overall 2.5 to 2.9-Å resolution with most side-chain densities well defined (Fig 1c, and Extended Data Figs. 2–4 and Supplementary Table 1). In the absence of inhibitors, particle images could be sorted into two three-dimensional (3-D) classes with minor differences (Extended Data Fig. 2 b–h). In both classes, the Sec61 channel adopts a similar conformation, including a partially open lateral gate and a closed plug, as expected for a complex lacking Sec62 (ref.13). However, the two classes showed slightly different arrangements of Sec61 with respect to Sec63-Sec71-Sec72 due to a loose contact between the engineered L6/7 loop of Sec61α and the FN3 domain of yeast Sec63 in Class 2 (Extended Data Fig. 2 g and h).
For inhibitor-bound structures, we used five naturally occurring inhibitors, cotransin CP2, decatransin, apratoxin F, ipomoeassin F, and mycolactone; and two designed synthetic compounds CADA and ESI. Focused refinement masking out the cytosolic domains of Sec63-Sec71-Sec72 further improved the map of the Sec61 complex (at overall resolution of 2.6 to 3.2 Å) showing clear, well-defined density features for the added inhibitor (Fig. 1 d–k, Extended Data Figs. 3 and 4, and Supplementary Table 2). Local resolution around the inhibitor-binding region was on par with or better than the overall resolution owing to relatively uniform resolution distributions (Extended Data Fig. 3f). Reliable atomic models of inhibitor molecules could be built into the densities of inhibitors based on their two-dimensional (2-D) chemical structures (Fig. 1 d–k). However, we note that positions and orientations of certain atoms and bonds may deviate from their true structures as our structures do not resolve individual atoms. When we compared the cotransin CP2-bound Sec61 structures from the human and chimeric Sec complexes, the two structures were essentially superimposable (Extended Data Fig. 5). This suggests that the Sec61 channel in the chimeric complex can adopt the conformations that are compatible with inhibitor binding observed in the human Sec complex.
Inhibitor-binding site
Despite their diverse chemical structures, all analyzed inhibitors are found to bind essentially to the same site in the Sec61 channel (Figs. 1 and 2 a–c, and Extended Data Fig. 6). The pocket is formed at the partially open lateral gate, approximately halfway across the membrane. The inhibitors commonly interact with lateral gate helices TMs 2b, 3, and 7 of the Sec61α subunit. However, it should be noted that the actual structure of the pocket substantially varies depending on the bound inhibitor because the lateral gate adopts different degrees of opening (Fig. 2d, and Extended Data Fig. 6). The width of the lateral gate opening is widest in the cotransin CP2-bound structure and narrowest in the ipomoeassin F-bound structure. During protein translocation, the lateral gate of the Sec61/SecY channel dynamically adopts closed or variable open states by a relative motion between the N- and C- terminal halves of the α subunit2,5–14. Our structures show that inhibitors bind to the lateral gate in one of these partially open states facilitated by the conformational flexibility of Sec61 and form a tight fit with the pocket. A notable example of such flexibility can be seen in the ipomoeassin F-bound structure, where the inhibitor binding even caused partial dissociation of Sec61 from Sec63 to accommodate a near-closed conformation of the lateral gate (Extended Data Fig. 7). Compared to natural inhibitors, the interfaces of CADA and ESI to Sec61 seem less extensive, possibly explaining the lower (micromolar-range) affinities of these synthetic inhibitors (Extended Data Fig. 6).
Figure 2. Structural plasticity of the inhibitor-binding pocket.
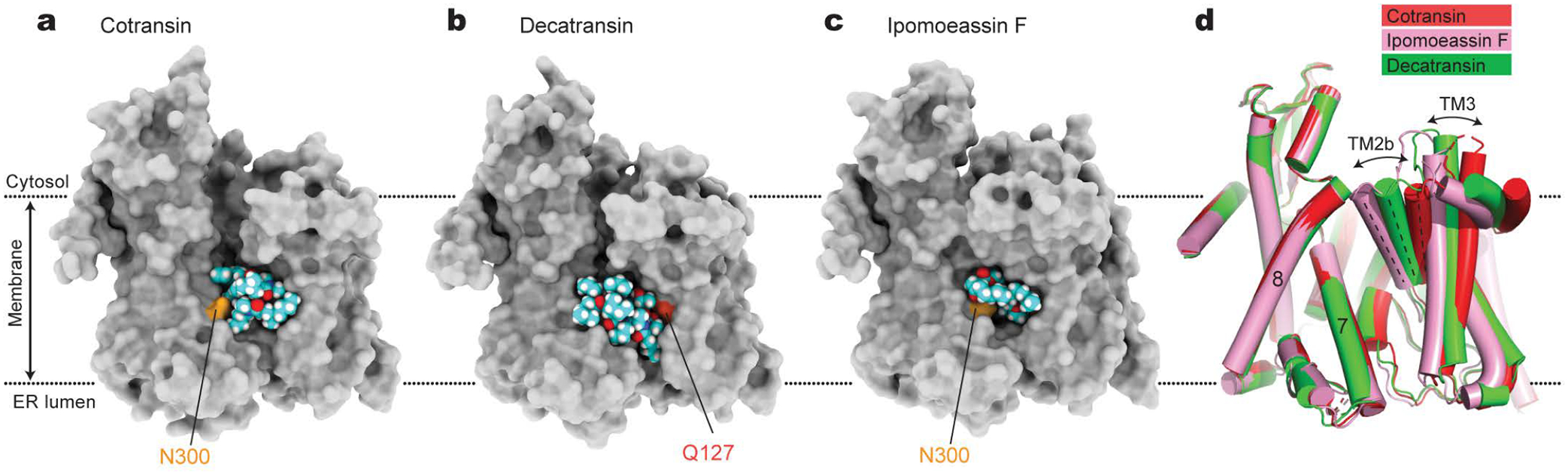
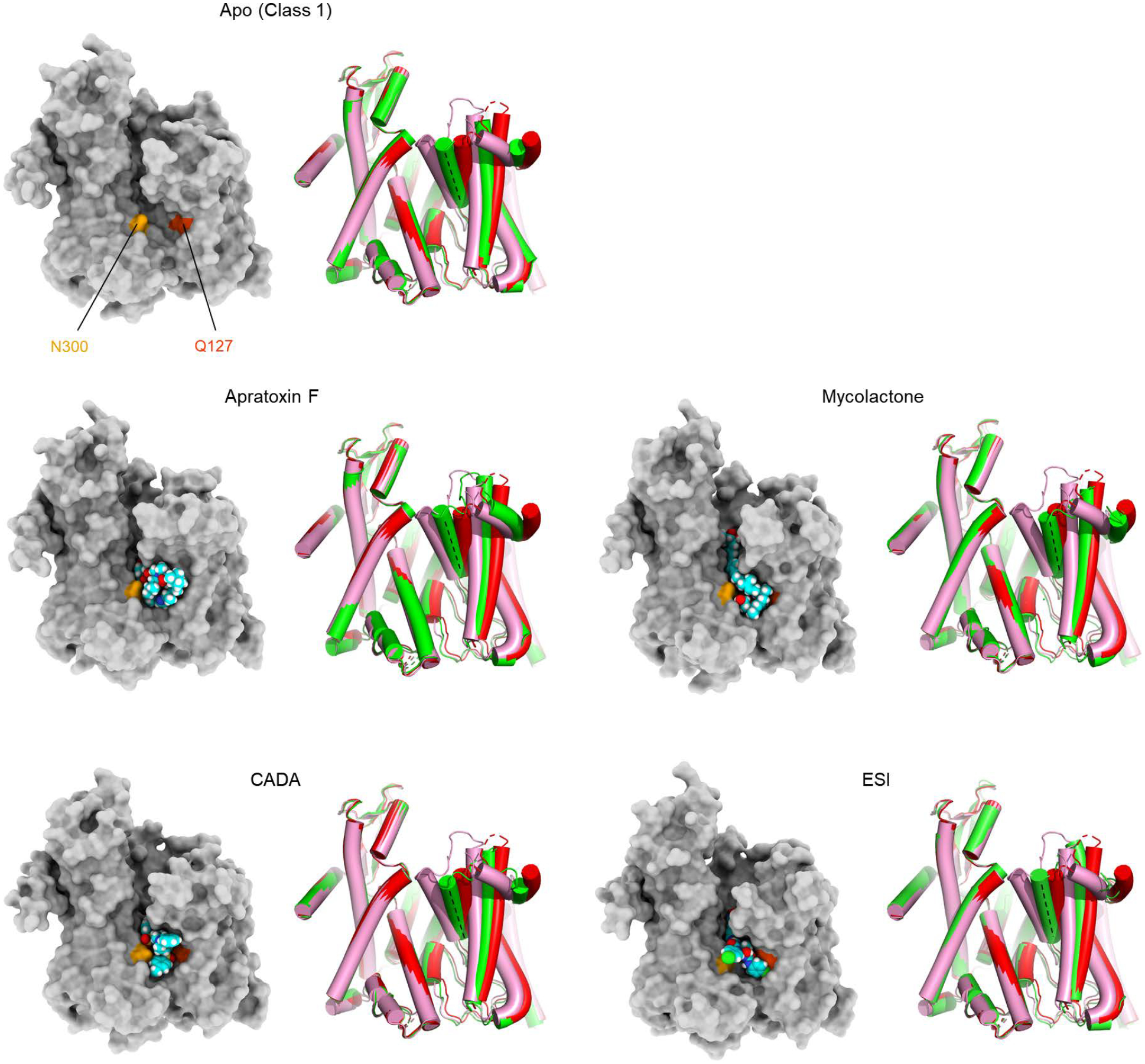
a–c, The inhibitor-binding pocket of Sec61 and bound inhibitors are shown in surface (protein) and sphere (inhibitors) representations. Shown are for cotransin CP2 (a), decatransin (b), and ipomoeassin F (c). For other inhibitors, see Extended Data Fig. 6. Conserved polar amino acids N300 and Q127 at the inhibitor binding site (also see Fig. 3) are indicated in light and dark orange, respectively. Note that part (cinnamate moiety) of ipomoeassin is deeply buried inside the channel and invisible in this representation. d, Superposition of the Sec61 structures bound to cotransin CP2 (red), decatransin (green), and ipomoeassin F (pink). Note differences in the lateral gate opening due to the varying position of the N-terminal half of Sec61α, particularly TMs 2b and 3.
In addition to the lateral gate, the plug and pore ring critically participate in binding of all inhibitors. The partially open lateral gate of inhibited Sec61 is reminiscent of conformations observed with substrate-engaged Sec61. In fact, the inhibitor binding site largely coincides with where a signal sequence docks upon the insertion of a substrate protein into the channel9,10. However, one crucial difference exists between polypeptide substrates and inhibitors: unlike the signal sequence, all inhibitors also form a direct contact with the plug in a closed position through hydrophobic moieties (Figs. 1 and 3). Many inhibitors even further intercalate into the dilated, crescent-shaped pore ring and interact with pore-ring residues (Ile81, Val85, Ile179, Ile183, Ile292, and/or Ile449). In the cases of mycolactone and ESI, their extended chain penetrates deeply into the channel interior and occupies a substantial space of the channel’s cytosolic funnel (Fig. 3 and Extended Data Figs. 6 and 8). These parts of mycolactone and ESI are known to be critical for their inhibitory activity17,32.
Figure 3. Maps for interactions between Sec61 and inhibitors.
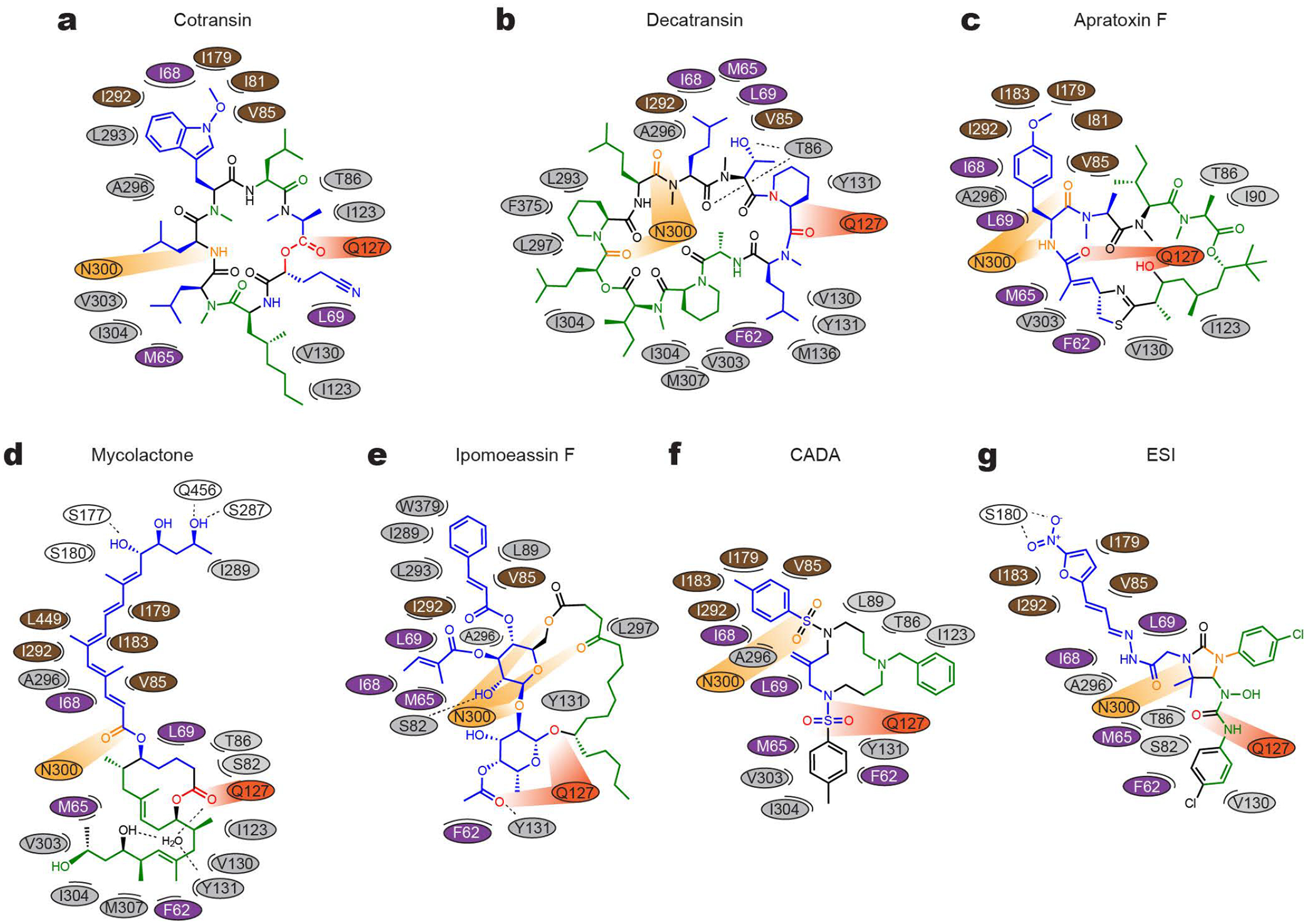
a, The chemical structure of cotransin CP2 and the positions of amino acids (ovals) of Sec61 in the immediate vicinity are drawn in a two-dimensional representation. Different colors were used for ovals to indicate regions in Sec61α: purple–plug, brown–pore ring, gray–lateral gate, light and dark oranges–polar cluster Q127/N300, and white–others. In the cotransin CP2 chemical structure, main lipid-exposed parts are in green whereas channel-facing parts are in blue. Moieties in orange and red interact with N300 and Q127 respectively. b–g, As in a, but drawn for decatransin (b), apratoxin F (c), mycolactone (d), ipomoeassin F (e), CADA (f), and ESI (g). Dashed lines indicate putative hydrogen bonds. Note that in the mycolactone-bound structure, a water molecule coordinated by Sec61 and mycolactone was observed in the pocket. For 3D structures, see Extended Data Fig. 8.
Structures of inhibitors and their interactions with Sec61
Except for cotransin and apratoxin, the structures of which were determined in organic solvents by NMR spectroscopy or X-ray crystallography39,40, 3D structures of most Sec61 inhibitors were unknown. Our cryo-EM structures now reveal their 3D structures in association with the Sec61 channel. Notably, conformations of cotransin CP2 and apratoxin F in our cryo-EM structures are highly similar to those structures determined in organic solvent39,40. This might be because the inhibitor-binding site in Sec61 forms a markedly hydrophobic environment. Particularly, the pocket is open towards the lipid phase (Figs. 1 and 2), and thus, all inhibitors are expected to interact with hydrocarbon tails of membrane lipids. The lipid-exposed parts of inhibitors are predominantly hydrophobic (Fig. 3). Similarly, the parts of inhibitors that face the Sec61 channel are mostly hydrophobic as they form contacts with hydrophobic side chains from the lateral gate, plug, and pore ring of Sec61α.
While van der Waals interactions between apolar groups of inhibitors and Sec61 seem to be dominant contributors to inhibitor binding, our cryo-EM structures also show a recurring pattern of polar interactions between Sec61 and inhibitors. In the closed channel, the lateral gate contains a conserved polar cluster halfway across the membrane, formed mainly by the side chain amide groups of Gln127 (Q127) in TM3 and Asn300 (N300) in TM7. Mutations in this polar cluster has been shown to affect the energetics of channel gating41. In the inhibitor-bound structures, Q127 and N300 are separated by lateral gate opening, but instead they do form polar interactions with certain oxygen and nitrogen atoms in the backbones of the inhibitors. Given that these prong-like polar interactions are present in a predominantly hydrophobic milieu, it is likely that they substantially strengthen inhibitor binding at the pocket (see below).
Mutations in Sec61 conferring resistance to inhibitors
Several point mutations in Sec61α have been found to confer resistance to Sec61 inhibitors25,26,28–30,34,35. These mutations are mostly located in the plug and the lateral gate. Given the direct interactions between inhibitors and these parts, disruption of the inhibitor binding surface could be a mechanism for these mutations. However, it has also been proposed that mutations might work indirectly through altering the conformation of the channel35. Extensive biochemical studies of the Sec61/SecY complexes have well established that mutations in the lateral gate, plug, and pore ring often change the gating behavior of the channel41–43. The best-known examples are prl mutations that give rise to relaxed client selectivity through increased propensity of channel opening. Thus, this phenotypic complexity has obscured how Sec61 mutations confer resistance to inhibitors. Moreover, positions of the identified mutations were often redundant and sparse, limiting detailed investigation of their mechanisms.
To biochemically probe inhibitor-binding sites in the Sec61 complex, we conducted a comprehensive mutational analysis fully blinded from our cryo-EM study. We focused on two inhibitors cotransin CP2 and ipomoeassin F, which were readily available to us. In addition to anti-proliferation activities on mammalian cancer cell lines, these compounds also cause growth retardation of yeast cells in a Sec61α (Sec61p)-specific manner25. Therefore, we tested 84 point mutations on 34 amino acid positions in yeast Sec61α for their half-maximal growth inhibitory concentration (IC50) (Supplementary Table 3). Positions were mainly chosen from the cytosolic funnel and lateral gate as they were likely candidates to bind inhibitors (each site was typically mutated to either Asp or Trp). This led us to identify 19 and 14 new resistance-conferring positions for cotransin CP2 and ipomoeassin F, respectively.
We then mapped the mutation positions onto the cryo-EM structures. The results clearly show that most resistance mutations are clustered around bound cotransin CP2 or ipomoeassin F (Figure 4 a and b), suggesting that their primary mechanism is through directly impairing the inhibitor-binding surface. However, some mutations (e.g., mutations equivalent to R66I/G and E78K in human Sec61α) are located at distal sites in the plug, and they may act through a conformational change in the plug domain. The plug makes a substantial contact with all tested inhibitors and is one of the most mobile parts of Sec61. Thus, altered structure or dynamics of the plug may explain the weakened inhibitor binding.
Figure 4. Inhibitor-resistant mutations.
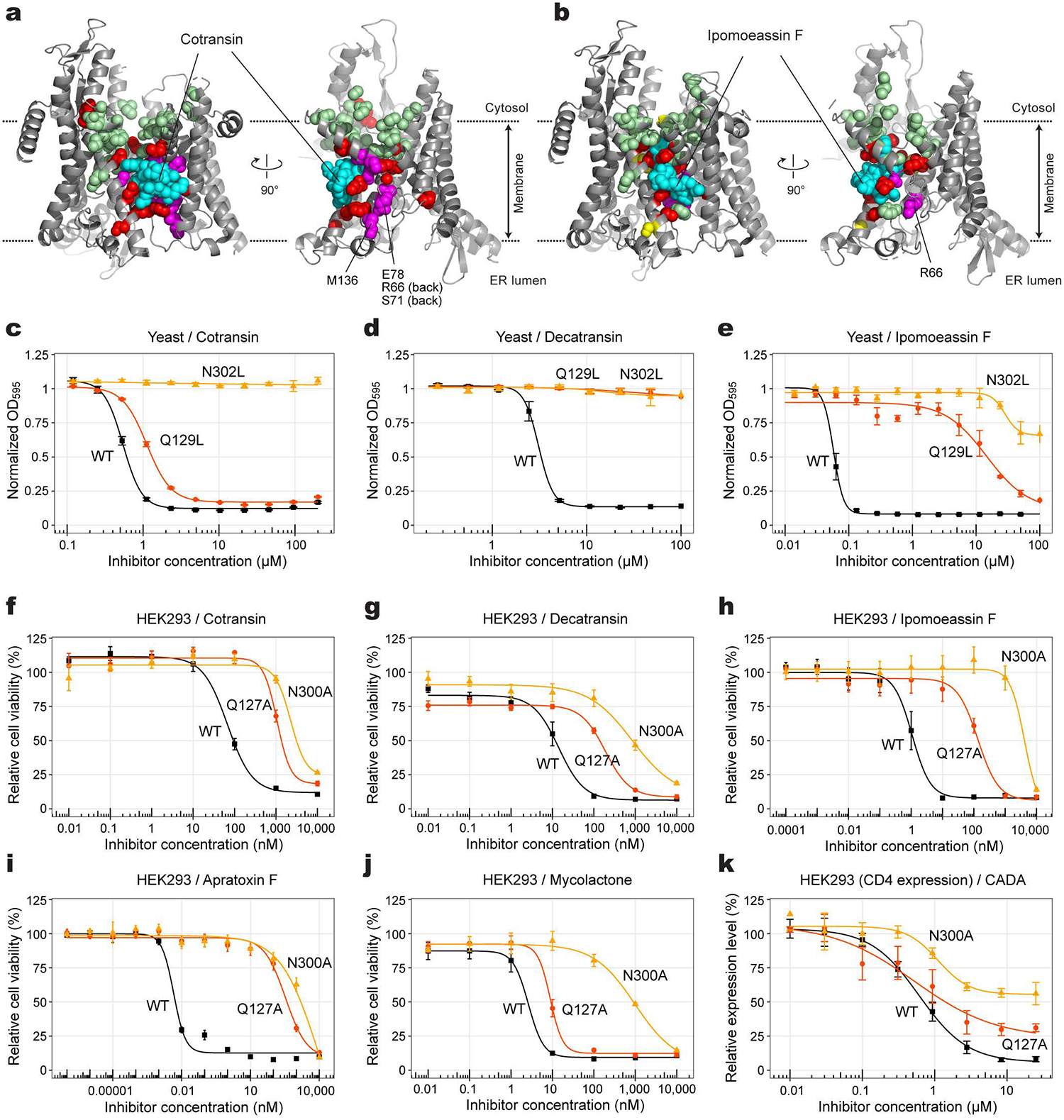
a, Positions of mutations tested with yeast Sec61 were mapped onto the cotransin CP2-bound structure (also see Supplementary Table 3). Left, front view; right, cutaway side view. Cotransin CP2 (cyan) and amino acid side chains are shown as spheres. Red and pale green spheres indicate positions in which mutation to Asp or Trp develops high and no cotransin CP2 resistance, respective. Magenta, positions of other resistant mutations previously reported25,34. b, As in a, but with ipomoeassin-F-resistant mutations. Yellow spheres additionally show positions that give rise to moderate ipomoeassin F resistance. c–e, Effects of Sec61 lateral gate polar amino acid mutations on yeast growth inhibition by cotransin CP2, decatransin, and ipomoeassin F (residue numbers are according to yeast Sec61). Shown are means, s.e.m., and fitted curves (n=3 independent experiments for cotransin CP2 and decatransin; n=4 independent experiments for ipomoeassin F). f–j, Dose-response curves for indicated inhibitors from viability assays of cultured human (HEK293) cells expressing the indicated Sec61α variant (residue numbers are according to human SEC61A1; means and s.e.m., n=4 independent experiments). k, Inhibition of expression of CD4 in HEK293 by CADA (means and s.e.m., n=4 independent experiments).
Lastly, we investigated the importance of polar interactions at the binding site by mutational analysis. In the yeast growth assay, we found that an N302L mutation in yeast Sec61α (equivalent to N300L in human Sec61α) confers strong resistance to cotransin CP2, decatransin, and ipomoeassin F (Fig. 4 c–e). A Q129L mutant (equivalent to Q127L in human Sec61α) showed strong to intermediate resistance to decatransin and ipomoeassin F and mild effects on cotransin CP2. We further tested the effects of Q127 and N300 mutations on inhibition using human cells because several inhibitors exerted little or no effects on yeast growth even at high concentrations (200 μM for mycolactone and apratoxin F; and 1 mM for CADA and ESI). We generated stable HEK293 cell lines that overexpress Sec61α (Extended Data Fig. 9). All natural-product inhibitors potently inhibited cell viability at nano- or subnano- molar concentrations in wild-type expressing cells (Fig. 4 f–j). Similar to the yeast-based assay, expression of Q127A or N300A mutant Sec61α markedly shifted dose-response curves to higher inhibitor concentrations, suggesting that the mutations substantially decrease inhibitor-binding affinities. Unlike natural inhibitors, we could not observe Sec61-dependent cytotoxicity in the cell-based assay from synthetic designed inhibitors CADA and ESI. However, CADA inhibited expression of human CD4 with an IC50 of 0.6 μM, similar to previous report31, and the Q127A and N300A mutations substantially reduced the inhibition by CADA (Fig. 4k and Extended Data Fig. 9). In all these experiments, the N300 mutations generally led to stronger resistance than the Q127 mutations. This might be explained in part by the structural observation that the side-chain amide of N300 more directly faces toward the inhibitors compared to Q127 (Extended Data Fig. 8).
Discussion
Our study reveals how Sec61 inhibitors interact with the channel and block the protein translocation. Remarkably, all seven tested inhibitors were found to bind to the same site in the channel formed by a partially open lateral gate and the fully closed plug domain, suggesting that this mode of interaction provides possibly the most effective mechanism for small molecules to inhibit the Sec61 channel. Among all known major Sec61 inhibitors to date, coibamide A is the only compound that was not included in the present study. However, given the previous observations that it competes with apratoxin A and mycolactone for Sec61 binding and that its resistant mutation could be found also in the plug30, coibamide A is likely to bind to the same or an overlapping site. We note that the mycolactone and CADA derivative CK147 models proposed in the recent medium-resolution cryo-EM studies35,37 using ribosome-bound Sec61 fundamentally differ in both position and conformation from those we found in our study (Extended Data Fig. 10). Although the mycolactone (PDB 6Z3T) and CK147 (PDB 8B6C) models in these studies might represent alternative binding modes, understanding of these discrepancies would require further investigation, such as high-resolution cryo-EM analysis of inhibited cotranslational complexes. We also note the KZR-8445-bound Sec61 structure36 and our cotransin CP2-bound structure show some differences. While the overall conformation of the channel and the location of the binding pocket seem consistent, the orientation of the KZR-8445 model is different from that of cotransin CP2 in our study. This discrepancy is more likely due to a limited map resolution of the ribosome–Sec61–KZR-8445 structure, although we cannot rule out a possibility that it may originate from minor structural differences between the two cotransin compounds.
Despite distinct chemical structures of the inhibitors, some common features among them could be inferred from our results. First, the inhibitors have two major clusters of hydrophobic moieties, one arranged to interact with the plug and the lateral gate, and the other with membrane lipids. The Sec61-facing sides are characterized by strong surface complementarity for the binding pocket, while the lipophilicity of the other parts would also contribute to efficient binding as the pocket exists within the plane of the membrane. Second, all inhibitors form polar interactions between their backbone and the side chains of the lateral gate (mainly N300 and Q127 of Sec61α). We found that this is crucial for Sec61 binding affinity. These polar groups of inhibitors would also provide some water solubility of the compounds. Third, certain inhibitors, such as mycolactone and ESI, further penetrate the cytosolic funnel of Sec61 forming additional polar and hydrophobic interactions therein. These interactions likely contribute to the binding energy of the inhibitor and their broad-spectrum activity.
Our data indicate that all known Sec61 inhibitors block the protein translocation process commonly by locking both lateral and lumenal gates of Sec61 into translocation-incompetent conformations (Fig. 5a). Although the lateral gate stays partially open, it does not provide sufficient space for a signal sequence/anchor to pass. Importantly, the lumenal gate, i.e., the plug, remains fully closed such that the client polypeptide cannot insert into the pore. Overall, all three key gating elements—the lateral gate, plug, and pore ring—are cemented together by the inhibitor at their interface, thereby prohibiting their concerted opening required for the client protein insertion.
Figure 5. Proposed model for Sec61 inhibition.
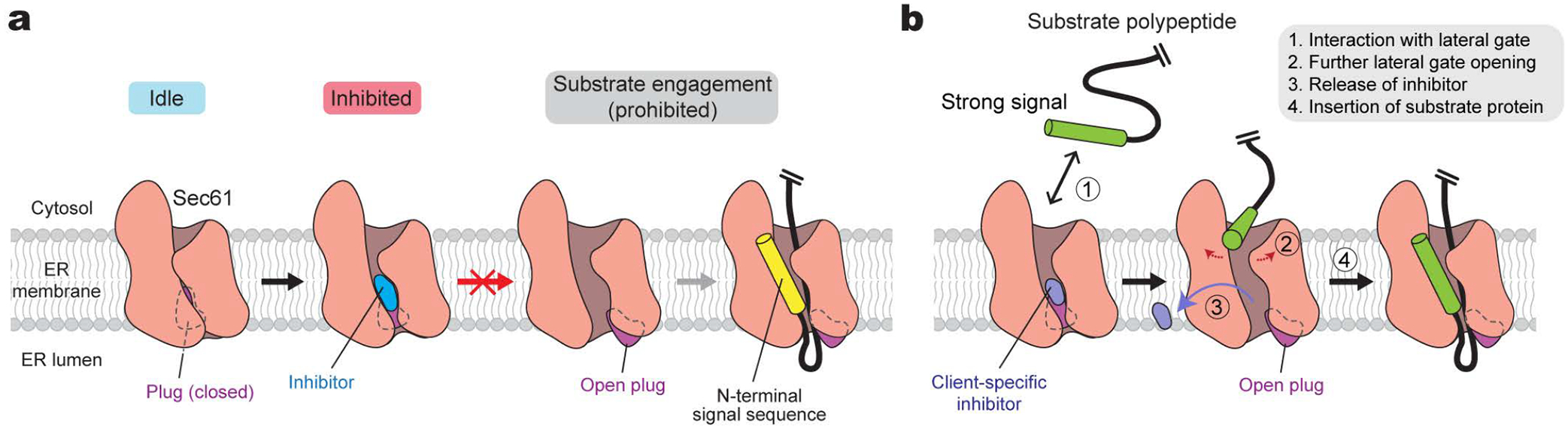
a, General model for the mechanism of Sec61 inhibitors. Inhibitors bind to Sec61 in a partially open conformation and preclude the plug from opening. This prevents substrate polypeptide insertion. b, A proposed model for client-specific inhibition. Certain client-specific inhibitors may allow an interaction between strong signals (e.g., TM signal anchors) and the channel such that the signal sequence/anchor is wedged into the partially open lateral gate. This would further open the lateral gate to cause release of the inhibitor. Inhibitors forming less interactions with the pore and plug, rendering the lateral gate into a more open conformation, and/or displaying a weaker overall affinity are likely to be overcome by this manner.
Although further investigations would be necessary, our comparative analysis also hints at why certain inhibitors exhibit client-dependent translocation inhibition. Cotransin and CADA have been shown to be less effective in blocking translocation of client proteins containing a stronger targeting signal, such as a signal sequence with higher hydrophobicity or a TM signal anchor44,45. Our structures show that in the cotransin CP2-bound structure, the lateral gate adopts a relatively more open conformation on the cytosolic side (Fig. 2). This may allow certain hydrophobic interactions between the lateral gate and the incoming signal sequence/anchor (Fig. 5b). A stronger interaction exerted by a stronger targeting signal probably tends to further pry open the lateral gate, promoting the inhibitor to be released. Although the lateral gate of the CADA-bound structure is not as wide as that of cotransin CP2-bound Sec61, its relatively low binding affinity (~0.2–2 μM) might facilitate certain hydrophobic signals to overcome inhibition. On the other hand, those inhibitors that deeply insert into the pore and cytosolic funnel of the channel, such as mycolactone and ESI, may tend to exert broad-spectrum inhibition by additionally impeding client insertion into the pore.
It remains unclear whether binding of an inhibitor requires prior opening of the Sec61 channel. In our chimeric complex, the lateral gate is partially opened by Sec63. In co-translational translocation, it has been generally thought that the ribosome docking alone does not open the lateral gate to a considerable extent7, which seems necessary for inhibitor binding. However, a transient breathing motion of the channel might allow inhibitors to bind. Single-molecule fluorescence studies of the bacterial SecY channel have indicated that the lateral gate spontaneously fluctuates between closed and open states without any binding partner46,47. Thus, it is possible that inhibitor binding may not require priming or partial opening of the channel induced allosterically by the ribosome or Sec63.
It is also unclear how inhibitors affect other functions of the Sec61 channel beyond its role in protein translocation. Previous studies have reported that the Sec61 channel is responsible for passive calcium leakage from the ER lumen and that certain Sec61 inhibitors, such as mycolactone and ESI, enhance this leak48,49. Although our current data do not provide structural insight into calcium permeation by Sec61, it is possible that some relevant conformations for this activity were not captured in our analysis. It is also possible that the calcium leakage involves other translocon components that are absent in our study. Given the importance of calcium in the physiology of metazoan cells, these outstanding issues warrant further study.
Lastly, the rich structural and mechanistic knowledge we provide here can facilitate structure-guided design of Sec61 inhibitors. The Sec61 channel has been considered as a promising target for therapeutic intervention due to its essential role in production of many cytokines, surface receptors, and cell adhesion molecules that are clinically relevant. Nevertheless, currently available Sec61 inhibitors would need further structural optimizations to improve their effectiveness and pharmacological properties while reducing undesired cytotoxicity. Our new approach enabling high-resolution structural analysis of human Sec61 and bound ligands would accelerate efforts to understand the mechanisms of new Sec61 inhibitors and optimize previously identified molecules.
Methods
Sec61 Inhibitors
Isolation of cotransin CP2 and decatransin from fungal species have been described previously25. For apratoxin F, ipomoeassin F, and mycolactone, we used synthetic versions. Synthesis of apratoxin F (ref.50), ipomoeassin F (ref.51), mycolactone (ref.52) has been as described previously. We note that apratoxin F and its more commonly studied analog apratoxin A have only a minor structural difference and both are known to exhibit comparable IC50 values on mammalian cancer cell lines50. We also note that the used synthetic mycolactone is a 4:1 mixture of two epimers at C12 in favor of the natural configuration. CADA and ESI were purchased from Calbiochem (cat# 534337 and 324521, respectively). Inhibitors were dissolved in dimethyl sulfoxide (DMSO) at 10 mM (for decatransin, ipomoeassin F, and mycolactone), 20 mM (for cotransin CP2, apratoxin F, and CADA), or 50 mM (for ESI) before use.
Plasmid constructs for cryo-EM studies
The plasmids and yeast strain to express the S. cerevisiae Sec complex have been described previously11,13. To express the human Sec complex in Spodoptera frugiperda (Sf9) cells, we modified a Bac-to-Bac baculovirus expression vector (Invitrogen) adapting the multigene-expression approach from MoClo Yeast ToolKit (YTK)53 as follows. First, we generated part plasmids for a baculovirus polyhedrin (PH) promoter and a SV40 polyA signal, and an accepter plasmid (pBTK1) consisting of the backbone of pFastBac-1 (including a Tn7L element, an ampicillin resistance gene, a pUC E. coli origin of replication, a Tn7R element and a gentamycin resistance gene) and a BsmBI–superfolder GFP (sfGFP)–BsmBI acceptor cassette from pYTK096 (ref.53). Gene fragments encoding human Sec subunits were chemically synthesized and individually cloned into the entry plasmid pYTK001 as coding sequence (CDS) parts. Amino acids sequences of human (denoted by “Hs”) Sec61, Sec62, and Sec63 subunits are from the following entries in UniProt: P61619 (S61A1_HUMAN) for HsSec61α, P60468 (SC61B_HUMAN) for HsSec61β, P60059 (SC61G_HUMAN) for HsSec61γ, Q99442 (SEC62_HUMAN) for HsSec62, and Q9UGP8 (SEC63_HUMAN) for HsSec63. For the pYTK001-HsSec63 plasmid, a DNA segment encoding a human rhinovirus (HRV) 3C-cleavable linker (amino acid sequence: GAGSNSLEVLFQGPTAAAA; italic, HRV 3C cleavage site) and an enhanced green fluorescence protein (eGFP) were inserted immediately before the stop codon of HsSec63. To generate single Sec gene expression cassettes, each Sec subunit CDS was assembled with connectors (from pYTK003–007 and pYTK067–072), the PH promoter, and the SV40 terminator into pYTK095 using BsaI Golden Gate cloning. Then, all Sec subunit expression cassettes were assembled into pBTK1 using BsmBI Golden Gate cloning. In this multigene plasmid, the expression cassettes were arranged in the following order: PH-HsSec61α-SV40 | PH-HsSec61γ-SV40 | PH-HsSec61β-SV40 | PH-HsSec63–3C-eGFP-SV40 | PH-HsSec62-SV40.
The plasmid expressing the human-yeast chimeric Sec complex was made similarly to the human Sec complex plasmid with modifications of pYTK95 HsSec61α and HsSec63 expression constructs as follows. To modify Sec61α, two substitution mutations were introduced in cytosolic loops of HsSec61α using PCR to replace (1) amino acid residues 263–278 (VDLPIKSARYRGQYNT) with the corresponding yeast sequence (residues 265–280; YELPIRSTKVRGQIGI) and (2) amino acid residues 394–411 (LKEQQMVMRGHRETSMVH) with amino acids 395–412 of ScSec61 (FKDQGMVINGKRETSIYR; “Sc” denotes Saccharomyces cerevisiae). The substitutions in HsSec63 were introduced using Gibson assembly by first substituting amino acid residues 30–96 (ATY…VKK) with amino acids 29–93 of ScSec63 (MTL…RRN), followed by substitution of residues 215 to the C-terminus (SIR…stop) with the corresponding sequence from ScSec63 (residues 246–stop; TQS…stop). Fragments of ScSec63 were amplified from genomic DNA of yeast strain BY4741. In the multigene pBTK1 construct of the chimeric Sec complex, HsSec62 cassette was omitted, and instead, the cassettes for ScSec71 (PH-ScSec71-SV40) and ScSec72 (PH-ScSec72-SV40) were added. The CDS fragments of ScSec71 and ScSec72 were amplified by PCR from genomic DNA of yeast strain BY4741 and cloned into pYTK001. Like other single subunit expression plasmids, ScSec71 and ScSec72 CDSs were assembled into pYTK095 together with the PH promoter and the SV40 polyA signal before use for the BsmBI assembly.
Protein Expression
Baculovirus bacmids encoding the human or chimeric Sec complex were generated by transforming the respective pBTK1 plasmid into the DH10Bac E. coli cells (Invitrogen) according to the manufacturer’s instructions. Bacmids were isolated using a DNA midiprep kit (Epoch Life Science). 40 mL of a Sf9 suspension culture were prepared in ESF921 medium (Expression Systems) to a density of ~1.5 M/mL. 40 μg bacmid DNA were mixed with 80 μg PEI Max transfection reagent (PolySciences) in 4 mL Dulbecco’s phosphate-buffered saline (DPBS). After incubating at 22°C for 20–30 minutes, the DNA:PEI mixture was added to the culture. Supernatant containing P1 virus was harvested ~4 days post transfection and stored at 4°C after supplementing 5% FBS (Gibco). Expression of the Sec complex was carried out by adding 0.5 mL P1 virus to 0.7 L of Sf9 cells at density of ~1.5 M/ml that were prepared in a 2-L baffled flask. Cells were harvested by centrifugation typically two to three days post-infection upon verifying uniform expression of green fluorescence under microscope. Cell pellets were frozen in liquid nitrogen and stored at −80°C until use.
Protein Purification
The yeast Sec complex was purified from yeast strain ySI8 (ref.13). This strain expresses a “pore mutant (PM)” version of ScSec61, the pore ring residues of which were mutated to amino acids corresponding to HsSec61α (M90L/T185I/M294I/M450L). The yeast Sec complex was purified as described previously11,13. After Superose 6 (GE Life Sciences) size-exclusion chromatography, the purified protein was concentrated to ~4 mg/mL in 20 mM Tris pH 7.5, 100 mM NaCl, 1mM EDTA, 2 mM DTT, and 0.02% glycol-diosgenin (GDN; Anatrace) and mixed with 100 μM cotransin CP2 for 0.5–1 h before preparing cryo-EM grids.
To purify the human Sec complexes, Sf9 cell pellets were first resuspended in lysis buffer containing 50 mM Tris-HCl pH 7.5, 200 mM NaCl, 2 mM dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic acid (ETDA) supplemented with protease inhibitors (5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin A, and 1.2 mM PMSF). All subsequent steps were carried out in ice or at 4°C. The cells were broken with a glass Dounce homogenizer using ~100 strokes. After removing large debris by brief centrifugation (4,000 g, 10 min), membranes were pelleted by ultracentrifugation for 1.5 h (125,000 g, Beckman Type 45 Ti). The membrane pellet was resuspended in ~10 pellet volumes of lysis buffer supplemented with 5 μM cotransin CP2. Membranes were solubilized by an addition of 1% lauryl maltose neopentyl glycol (LMNG; Anatrace) and 0.2% cholesteryl hemisuccinate (CHS; Anatrace) for 2 h. Then, the lysate was clarified by ultracentrifugation at 125,000 g for 1 h. The clarified lysate was then supplemented with 2 μg Serratia marcescens nuclease and incubated with home-made anti-GFP nanobody Sepharose beads for 1.5 h. Beads were washed with wash buffer containing 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM DTT, 1 mM ETDA, 0.02% GDN, and 5 μM cotransin CP2 (hereafter, 5 μM cotransin CP2 was included in all buffers). The complex was eluted by incubating beads with the HRV 3C protease overnight. The eluate was collected and concentrated to ~10 mg/ml by Amicon Ultra (cutoff 100 kDa). The sample was then injected to a Superose 6 increase column (GE Life Sciences) equilibrated with the wash buffer. Peak fractions were pooled and concentrated to ~6 mg/mg, before preparing cryo-EM grids.
The chimeric Sec complex was purified similarly using the method to purify the human Sec complex but with minor modifications. First, the Sec complex was purified without supplementing Sec61 inhibitors during purification (inhibitors were added to the purified Sec complex before cryo-EM grid preparation). Second, to solubilize membranes, 1% n-dodecyl-β-D-maltopyranoside (DDM; Anatrace) and 0.2% CHS was used instead of LMNG/CHS. For column wash, the buffer contained 0.02% DDM and 0.004% CHS instead of GDN. Third, the Sec complex was reconstituted into a peptidisc38 as follows. After concentrating the eluate from GFP-nanobody beads to ~10 mg/ml, the Sec complex was mixed with the peptidisc protein (Peptidisc Lab) at a weight ratio of 1.5:1 (peptidisc to Sec). After incubating for 1 h, the mixture was injected into a Superose 6 Increase column equilibrated with 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM DTT and 1 mM ETDA. Peak factions were pooled and concentrated to ~10 mg/ml, and one of the Sec61 inhibitors was added for ~1 h before preparing cryo-EM grids. The inhibitor concentrations used were: 100 μM for cotransin CP2, 100 μM for decatransin, 100 μM for apratoxin F, 100 μM for ipomoeassin F, 100 μM for mycolactone, 200 μM for CADA, and 500 μM for ESI. These concentrations, except for that of ESI, correspond to a 2–4-fold molar excess to the protein concentration (~52 μM) to ensure saturated binding. A higher concentration was used for ESI based on a relatively low (70 μM) IC50 reported in an in-vitro experiment32.
Cryo-EM data acquisition
Immediately prior to preparing cryo-EM grids, 3 μM Fos-Choline-8 (Anatrace) was added to the protein sample. The sample was then applied to a gold Quantifoil R 1.2/1.3 holey carbon grid (Quantifoil) that was glow discharged for 35 sec using PELCO easiGlow glow discharge cleaner. The grid was blotted for 3–4 sec using Whatman No. 1 filter paper and plunge frozen using Vitrobot Mark IV (FEI) set at 4°C and 100% humidity.
The yeast Sec complex dataset (1,578 movies) was acquired on FEI Talos Arctica electron microscope operated at an acceleration voltage of 200 kV, with Gatan K2 Summit direct electron detector. A magnification of 36,000x under super resolution mode (with the physical pixel size of 1.14 Å) was used with a nominal defocus range that was set between −0.8 to −2.2 μm. Each micrograph was composed of 42 frames with total exposure of 50 e−/pixel.
The human Sec complex dataset (3,499 movies) was collected on FEI Titan Krios G2 microscope operating at an acceleration voltage of 300 kV and equipped with a Gatan Quantum Image Filter (slit width of 20 eV) and a Gatan K3 direct electron detector. A magnification of 64,000x under the super-resolution mode (with physical pixel size of 0.91 Å) was used at a defocus range that was set between −0.8 and −2.0. Each micrograph was composed of 42 frames with total exposure of 50 e−/pixel. Exposures were performed with beam shifts onto 9 holes (3 by 3) per stage movement.
All chimeric Sec complex datasets were acquired on an FEI Titan Krios G3i electron microscope operating at an acceleration voltage of 300 kV, with a Gatan K3 Summit direct electron detector and a Gatan Quantum Image Filter (with 20 eV slit width). A magnification of 81,000x under the super-resolution mode (with physical pixel size of 1.05 Å) was used at a defocus range that was set between −0.8 and −2.0. Each micrograph was composed of 50 frames with a total exposure of 50 e−/pixel. Exposures were performed with beam shifts onto 9 holes (3 by 3) per stage movement (often acquiring movies for two non-overlapping areas per hole). All datasets were acquired using SerialEM software54.
Cryo-EM image analysis
Preprocessing of the movies and particle image extraction were done using Warp55. Motion correction and CTF estimation were performed on images divided to 7 × 5 tiles, and particles (256 × 256 pixels) were picked by the BoxNet algorithm in Warp. All subsequent image processing procedures were perform using cryoSPARC v3.3 (ref.56).
Cotransin CP2-treated pore mutant ScSec complex: A data processing flowchart diagram is shown in Extended Data Fig. 1a. A dataset of 528,128 auto-picked particles was classified into fifty 2-D class averages. Using visual inspection of the output, classes that represented empty micelles or poor-quality classes were removed and particles grouped into well-resolved classes corresponding to a single copy of the full Sec complex were selected (385,686 particles). Three ab-initio 3-D maps were then generated in cryoSPARC using the selected particles, followed by heterogeneous refinement. One 3-D class with 274,913 particles refined to a density map exhibiting defined Sec complex features. Non-uniform refinement of the particles in this class yielded a consensus map with 3.9-Å overall resolution. The particles were further separated into two 3-D classes using a heterogeneous refinement with inputs of the consensus map and the consensus map with manually erased Sec62. After a subsequent round of non-uniform refinement 174,058 particles yielded a map of ScScSec complex with Sec62 at 4.0-Å overall resolution, and 100,855 particles yielded a map of ScSec complex without Sec62 at 4.2-Å overall resolution.
Cotransin CP2-bound wildtype HsSec complex: A data processing flowchart diagram is shown in Extended Data Fig. 1d. A dataset of 601,465 auto-picked particles was classified into seventy 2-D class averages. Selected classes yielded 330,005 particles that were then reconstructed into three 3-D classes using ab-initio reconstruction followed by heterogeneous refinement. One major class, with 202,946 particles, was selected for further refinement. Non-uniform refinement of this class resulted in a reconstruction only at 7.4-Å resolution due to poor image alignment. Thus, for the final map, we used the ab-initio reconstruction method (without splitting the particle sets for half maps) with the maximal refinement resolution manually set to 5.0-Å (Extended Data Fig. 1f).
Apo chimeric Sec complex: A data processing flowchart diagram is shown in Extended Data Fig. 2b. Using 2-D classifications starting with 616,121 auto-picked particles, we selected 363,027 particles for 3-D reconstruction. Following an ab-initio refinement step generating four initial maps and a heterogeneous refinement step we identified two major 3-D classes with distinguishable full Sec complex features. Each of these classes were refined using non-uniform refinement, local CTF refinement, and another round of non-uniform refinement, yielding full maps of the chimeric Sec complex at overall resolutions of 2.7 and 2.8 Å from 188,637 particles (Class 1) and 147,081 particles (Class 2), respectively. The Sec61 channel was further refined by masking out the cytosolic domains of the complex and performing local refinement, yielding overall channel resolutions of 3.0 Å (Class 1) and 3.4 Å (Class 2).
Apratoxin F-bound chimeric Sec complex: A data processing flowchart diagram is shown in Extended Data Fig. 3b. Using 2-D classifications starting with 910,463 auto-picked particles, we selected 534,411 particles for 3-D reconstruction. Following an ab-initio refinement step generating four initial maps and a heterogeneous refinement step we identified two structurally indistinguishable major 3-D classes with defined full Sec complex features. The particles from the two classes were combined and refined using non-uniform refinement, local CTF refinement, and another round of non-uniform refinement, yielding a full map of the apratoxin F bound chimeric Sec complex at an overall resolution of 2.5 Å from 497,555 particles. The Sec61 channel was further refined by masking out the cytosolic domains of the complex and performing local refinement, producing an overall channel resolution of 2.6 Å.
All other inhibitor-bound datasets were processed using a workflow described for Apratoxin F-bound structure with minor variations in the numbers of classes in 2-D and 3-D classification procedures. For details, see Supplementary Information Figs. 1 and 2. Statistics for final refined maps are shown in Extended Data Fig. 4 and Supplementary Tables 1 and 2.
Model building and refinement
Atomic model building and refinement were performed using Coot (version 0.9.8.1)57 and Phenix (version 1.19.2)58. An initial model was built by docking an ScSec complex model (PDB ID 7KAH; ref.13) into the cryo-EM map of the cotransin CP2-bound complex using UCSF chimera (version 1.16)59 and rebuilding the polypeptide chains. For building and refining of Sec61 and inhibitor models, we used maps from focused (local) refinements as they typically showed better protein side-chain and inhibitor features than full maps. The initial model was further improved by using our highest-resolution map, which was obtained from the apratoxin F-bound complex. This model was then used to build atomic models for apo and other inhibitor-bound complexes by docking the model to the map using UCSF Chimera and locally adjusting it into the map in Coot. The restraint models of inhibitors were generated from SMILES strings of inhibitors using the Grade web server (http://grade.globalphasing.org) or the eLBOW tool of Phenix. The atomic models of inhibitors were then fitted into the cryo-EM map in Coot. We note that stereochemistry of decatransin has not been experimentally determined. We assumed that all amino acid residues of decatransin are in an L or S configuration based on an observation that no epimerase was found in the biosynthetic gene cluster of decatransin. The configuration of Cα of the homoleucine-derived 2-hydroxy carboxylic acid remains ambiguous25, but we also assumed that it is in an S configuration. The resulting model could be fitted well into the cryo-EM map. The atomic models were refined with Phenix real-space refinement using maps that were sharpened with B-factors estimated based on the Guinier plots and low-pass-filtered at their overall resolution. The refinement resolution was also limited to the overall resolution of the maps in Phenix. Structural validation was performed using MolProbity included in the Phenix package. UCSF Chimera, ChimeraX (version 1.4), and PyMOL (version 2.5.1; Schrödinger) were used to prepare figures in the paper.
Mutagenesis of yeast Sec61α and IC50 measurements
Except for the experiment shown in Fig. 4c, cotransin CP2 IC50 measurements were based on the yeast strain RSY1293 (matα, ura3-1, leu2-3,−112, his3-11,−15, trp1-1, ade2-1, can1-100, sec61::HIS3, [pDQ1]) (ref.60). In strain RSY1293URA, pDQ1, i.e., YCplac111 (LEU2 CEN) containing the gene of an N-terminally His-tagged, otherwise wild-type Sec61α (Sec61p) with its own promoter, was exchanged for YCplac33 (URA3 CEN) containing the same insert. Mutations in sec61 were introduced in pDQ1 by PCR and transformed into RSY1293URA, followed by elimination of the URA3 plasmid containing wild-type using 5-fluoro-orotic acid. Finally, the presence of the mutation was confirmed by sequencing.
For experiments shown in Fig. 4 c–e and ipomoeassin IC50 measurements, we used the yeast strain BY4743Δ8a (mat a, ura3Δ0, leu2Δ0, his3Δ1, lys2Δ0, snq2::KanMX4; pdr3::KanMX4; pdr5::KanMX4; pdr1::NAT1; yap1::NAT1; pdr2Δ; yrm1Δ; yor1Δ) lacking eight genes involved in drug resistance (efflux pumps SNQ2, PDR5, and YOR1, and transcription factors PDR1, PDR2, PDR3, YAP1, and YRM1)61. This strain showed higher sensitivity to ipomoeassin F compared to RSY1293. YCplac33 containing (untagged) SEC61 with 200 bp of its own upstream and 205 bp of its own downstream sequence was transformed into BY4743Δ8a. Genomic SEC61 together with 194 bp 5’- and 204 bp 3’-noncoding sequence was replaced with a hygromycin resistance cassette using pAG32 (HphMX4) resulting in the strain BY4743Δ9aURA (sec61::HphMX4 [pSEC61-YCplac33]). Finally, pDQ1 containing the mutated sec61 versions were transformed and the wild-type SEC61 URA3 plasmid counterselected. Plasmid exchange was validated by PCR. The IC50 measurements for cotransin CP2 in the BY4743Δ9aURA background paralleled those in the RSY1293.
IC50 measurements were performed as described previously25 by testing log-phase cultures in 96-well microtiter plates in YPD medium with serial dilutions of the compound. The assay volume was 120 μl/well, start OD600 was 0.05, DMSO was normalized to 2%. Curves were calculated by taking the 19 h OD600 measurements and applying a log regression curve fit in TIBCO Spotfire (version 3.2.1).
Preparation of stable mammalian cell lines
A DNA segment encoding human SEC61A1 was synthesized and cloned into pcDNA5/FRT/TO (Life Technologies) followed by addition of a C-terminal HA-tag, an internal ribosome entry site (IRES), and enhanced green fluorescence protein (EGFP) sequences. Point mutants of SEC61A1 were generated by PCR. All cell lines were maintained in DMEM supplemented with 10% FBS, 1% Penicillin/Streptomycin (Gibco), and 15 μg/mL blasticidin S (Gold Biotechnology). In a 6-well dish, Flp-In™ T-REx™−293 cells (Invitrogen, Cat. # R78007) were plated at a density of 5 × 105 cells/well overnight at 37 °C. Plasmids pcDNA5/FRT/TO-SEC61A1-HA-IRES-GFP and pOG44 were co-transfected at a 3:1 ratio with Lipofectamine 3000 (Thermo Fisher, Cat. # L3000001). The next day, media was replaced. After another 24h, cells were transferred to a 10cm dish containing fresh DMEM with 15 μg/mL blasticidin S and 50 μg/mL Hygromycin B (Gold Biotechnology). Cells were continually monitored with media replacement until several 1mm cell colonies became visible (about 3 weeks). Cells were then dissociated and expanded into new 10-cm dishes for cell maintenance. All cell lines were sequence verified by PCR using primers (forward: AAA GTG CTG TGG ACC GCT ATC, reverse: CC AAC TGG ATA AGC ACG TGC TG) specific to the synthetic human SEC61A1 coding sequence prior to use.
HEK293 viability assay
SEC61A1-overexpressing Flp-In T-Rex-293 cells were seeded in 96-well flat-bottom plates at 5 × 103 cells/well in media containing 1 μg/mL doxycycline overnight at 37°C. The following day, serial dilutions of compounds were performed in doxycycline-supplemented DMEM media, and cells were treated with compound dilutions for a total incubation volume of 150 μL (0.11% DMSO v/v). After 72-h incubation, cell viability was measured by addition of 15 μL resazurin reagent (Biotium, Cat. #30025–1). The plate was incubated for 2 h at 37°C and fluorescence intensity read using a BMG Labtech CLARIOstar plate reader (fluorescence Intensity, excitation at 545 nm and emission at 600nm). The data were processed with Excel, and the curves were generated using R software (version 4.0.5) and ggplot2 (version 3.3.3) and drc (version 3.0–1packages.
Human CD4 expression assay
A plasmid expressing a full-length human CD4 cDNA construct under a cytomegalovirus (CMV) promoter (Horizon Discovery, Cat. #MHS6278–202801784) was purchased. An additional single Ser-Gly linker and Strep-tag was appended the C-terminus to the CD4 coding sequence, resulting in pCMV-huCD4-Strep. SEC61A1-overexpressing Flp-In T-Rex-293 cells were seeded in a 12-well plate at 4 × 105 cells/well and pretreated with 1 μg/mL doxycycline overnight at 37°C. Cells were transfected with pCMV-huCD4-Strep using Lipofectamine LTX (Invitrogen, Cat. #15338030) according to manufacturer protocol. After 6 h, the cells were treated with dilutions of CADA (0.25% DMSO v/v). At 24 h post-treatment, cells were harvested by pipetting, washed with 500μL PBS, then lysed directly on ice (50 mM Tris pH 6.8, 2% SDS, 6% glycerol, supplemented with 5 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin A, 1 mM PMSF, and 2μg/mL Benzonase nuclease). Lysates were then clarified by spinning for 10 min at 17,000g. Protein concentrations were measured by a BCA assay (Thermo Scientific, Cat. #23227) before preparation of samples in SDS-PAGE sample loading buffer. Lysates were separated on 10% Bis-Tris gels (10 μg/well), transferred to PVDF membranes. Immunoblotting was performed using antibodies against Strep-tag (Genscript, Cat. #A01732; 1:2000), HA-tag (Cell Signaling Technology, Cat. #C29F4, 1:1000), and human SEC61A1 (Cell Signaling Technology, Cat. #D7Q6V, 1:1000). Band intensities were quantified by ImageJ. The data were processed with Excel, and the curves were generated using R software and ggplot2 and drc packages.
Extended Data
Extended Data Figure 1. Cryo-EM analysis of the yeast and human Sec complexes.
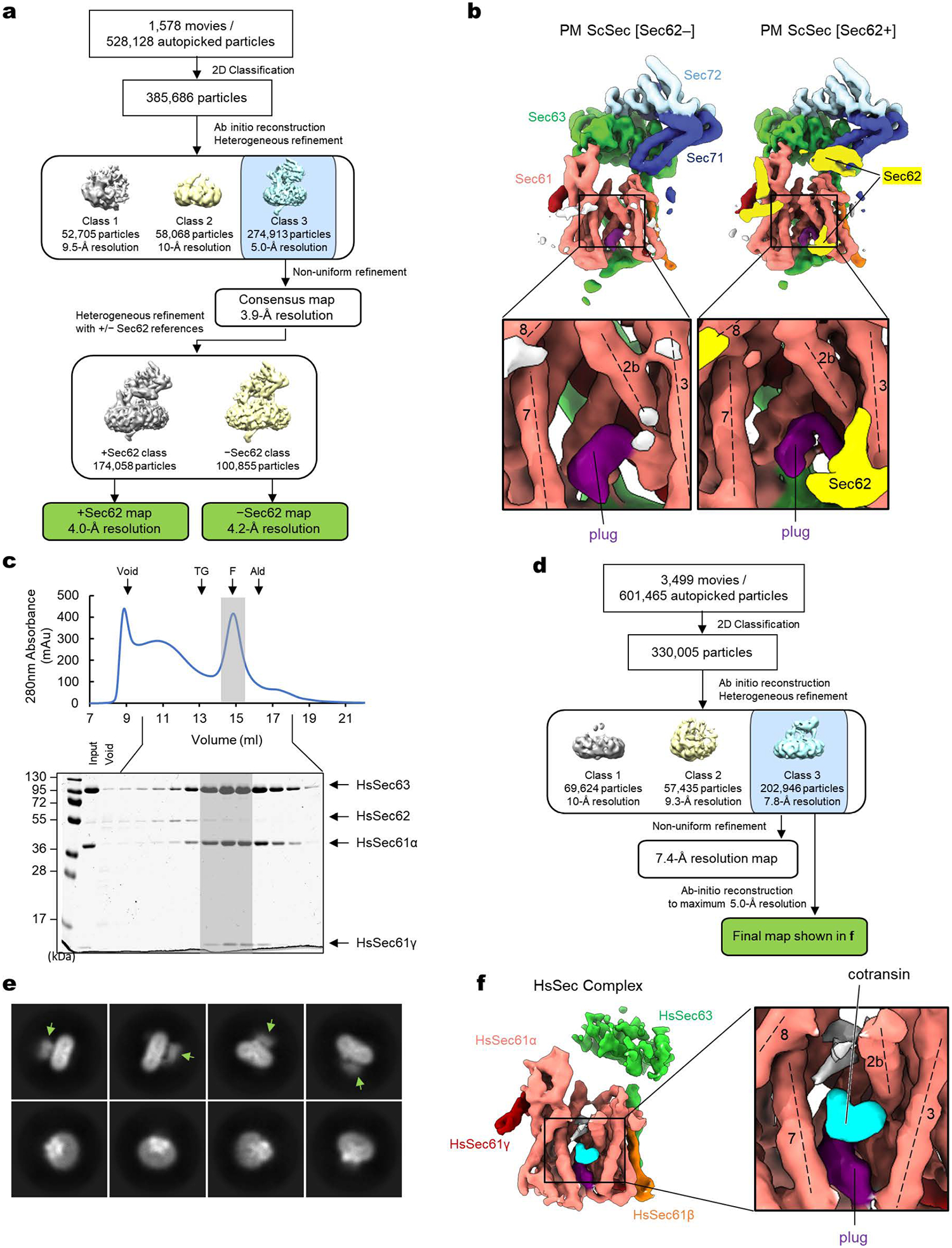
a, A schematic of the single-particle cryo-EM analysis of the yeast Sec (ScSec) complex incubated with cotransin. Note that the particles were sorted into two 3D classes, with and without Sec62, due to partial occupancy of Sec62. b, 3D reconstructions of the ScSec complex with and without ScSec62 (shown in yellow). No cotransin-like density was observed in either class. For this experiment, we used a pore ring mutant (PM; M90L/T185I/M294I/M450L) that stabilize the plug towards a closed conformation. c, Purification of the human Sec (HsSec) complex. Shown is a Superose 6 size-exclusion chromatography elution profile with fractions analyzed on a Coomassie-stained SDS gel. Note that under the used purification condition, HsSec62 does not co-purify at a stoichiometric ratio or stably comigrate with the Sec61–Sec63 complex. The fractions indicated by gray shade were used for cryo-EM. MW standards: Tg, thyroglobulin; F, ferritin; Ald, aldolase. The experiment was repeated twice independently with similar results. d, A schematic of the single-particle analysis of HsSec complex incubated with cotransin. Due to a poor refinement result from nonuniform refinement in cryoSPARC, the final reconstruction was obtained by the ab-initio refinement function of cryoSPARC (see f). e, Representative 2D classes of the HsSec complex. Diffuse cytosolic features of Sec63 (green arrowheads) suggest its flexibility or disorderedness. f, The 3D reconstruction of the HsSec complex. A putative cotransin feature (cyan) is visible at the lateral gate.
Extended Data Figure 2. Cryo-EM analysis of the chimeric Sec complex in an apo form.
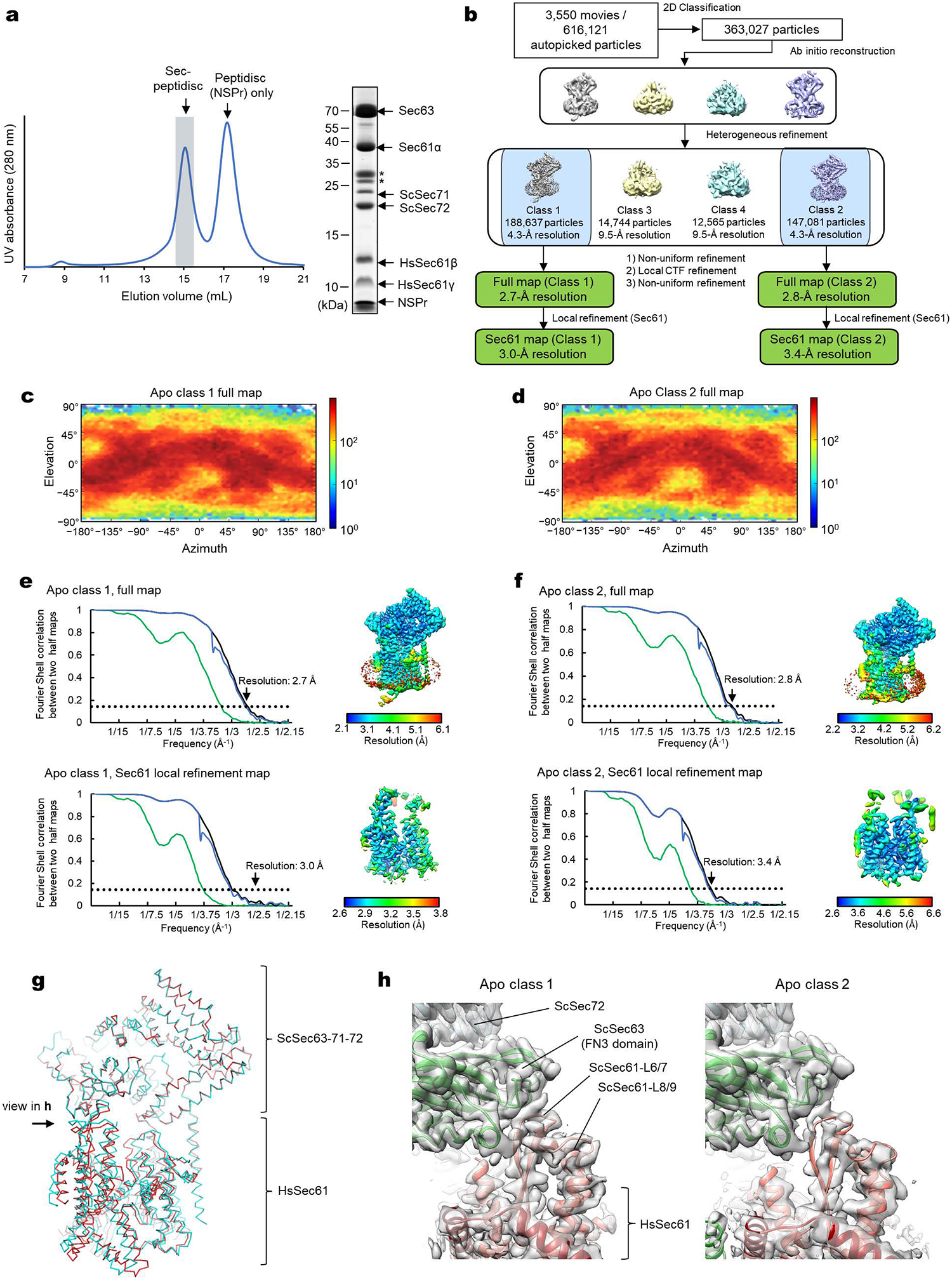
a, Purification of the chimeric Sec complex reconstituted in a peptidisc. Left, Superose 6 elution profile; right, Coomassie-stained SDS gel of the peak fraction. The fraction marked by gray shade was used for cryo-EM. Asterisks, putative species of glycosylated ScSec71. The experiment was repeated at least four times independently with similar results. b, A schematic of the cryo-EM analysis of the chimeric Sec complex in an apo state. c and d, Distributions of particle view orientations in the final reconstructions of Classes 1 (c) and 2 (d). e and f, Fourier shell correlation (FSC) curves and local resolution maps of the final reconstructions. g, Superimposition of the Class 1 and 2 atomic models (based on the cytosolic domains) shows a slight difference in relative positions between Sec63–Sec71–Sec72 and the Sec61 complex. h, Side views showing the contact between the engineered cytosolic loops of Sec61α and the FN3 domain of ScSec63. Note that in Apo Class 2, the contact is more poorly packed than Class 1.
Extended Data Figure 3. Cryo-EM analysis of the chimeric Sec complex in an inhibitor (apratoxin F)-bound form.
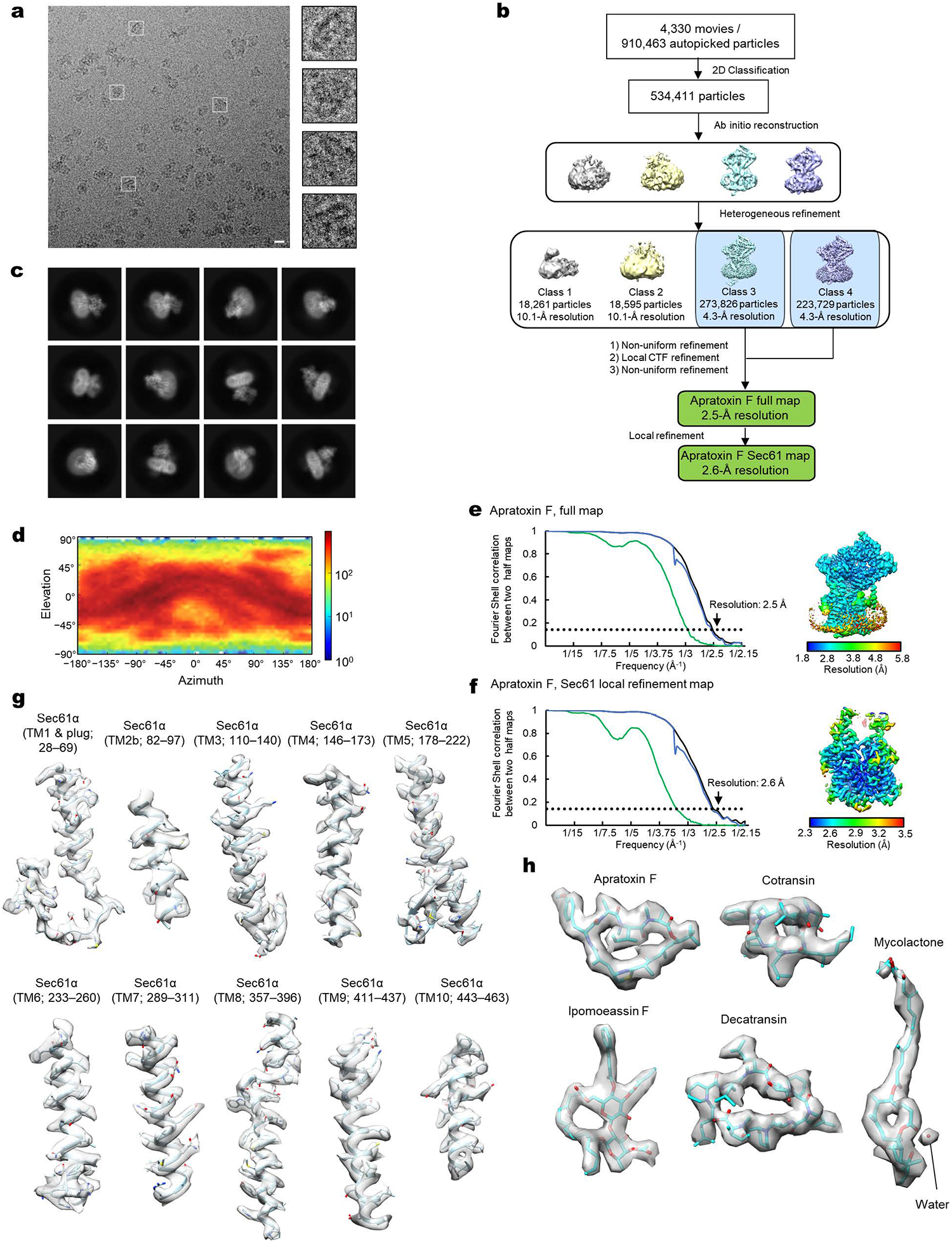
a, Images of a representative micrograph and particles of the apratoxin F-bound chimeric Sec complex. Scale bar, 10 nm. b, A schematics of the cryo-EM analysis of the apratoxin F-bound chimeric Sec complex. c, Representative 2D classes of the apratoxin F-bound Sec complex. d, Distribution of particle view orientations in the final reconstruction. e, The FSC curve and local resolution map of the final reconstruction (full Sec complex map). f, As in e, but for the map from focused (local) refinement. g, Segmented density maps of the apratoxin F-bound Sec61α subunit. h, Segmented density features of bound natural inhibitors.
Extended Data Figure 4. FSC curve and local resolution maps of inhibitor-bound Sec complexes.
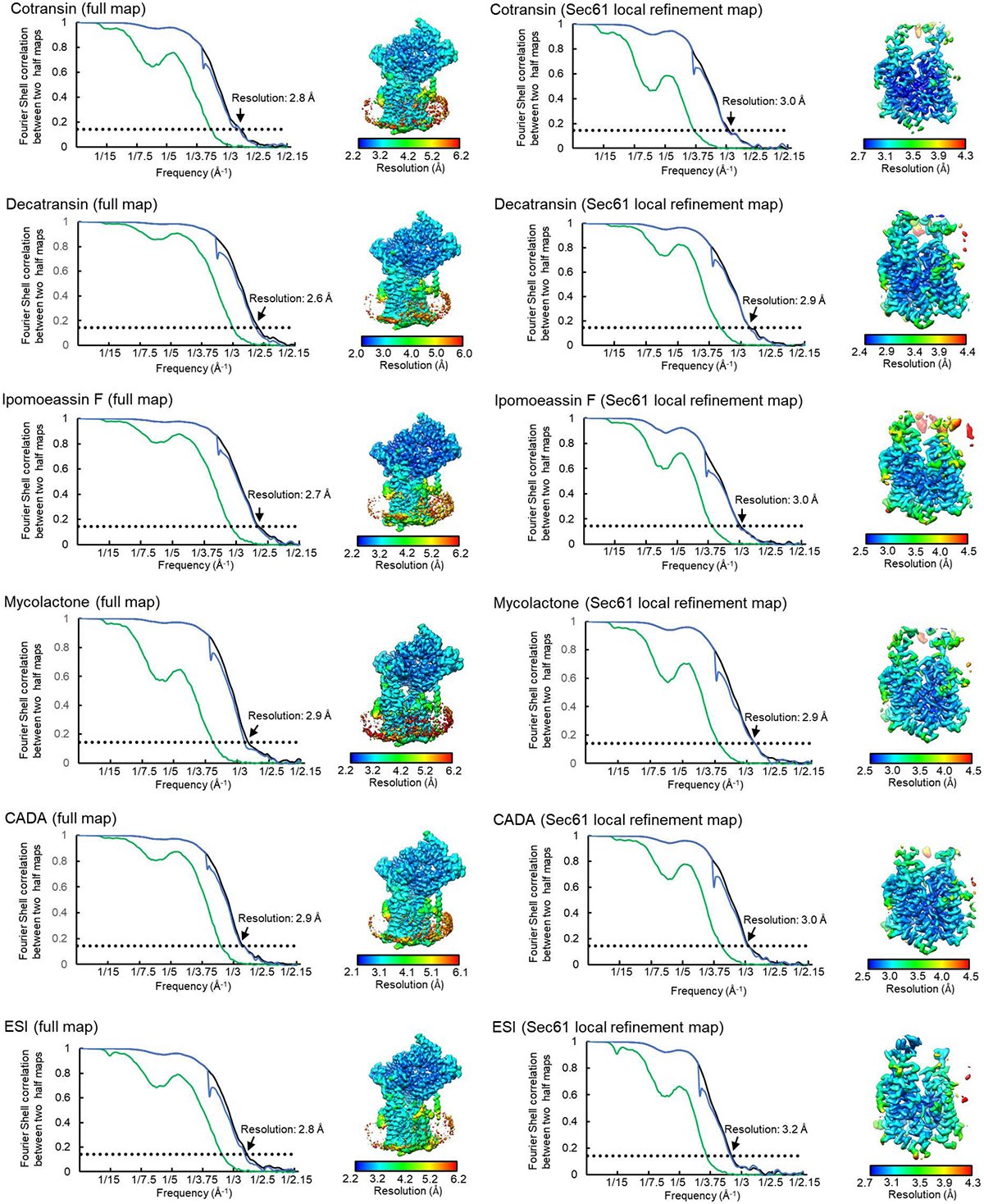
As in Extended Data Figure 3 e and f, but for all other inhibitor-bound structures.
Extended Data Figure 5. Comparison between the structures of cotransin-bound human and chimeric Sec complexes.
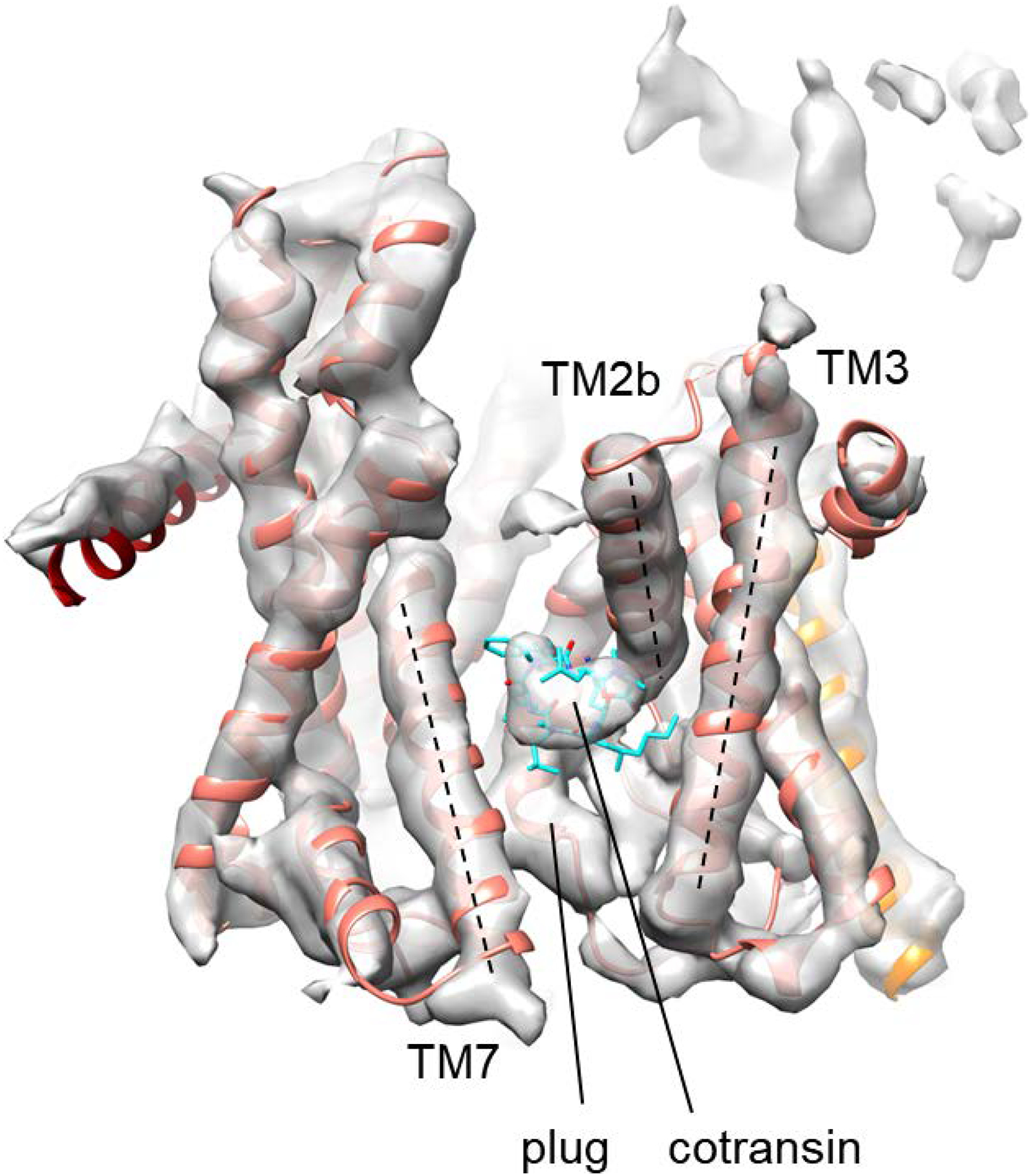
The high-resolution structure of the cotransin-bound chimeric Sec complex (ribbon representation for Sec61 and stick representation for cotransin) is docked into the low-resolution cotransin-bound human Sec complex structure (the semi-transparent gray density map; also see Extended Data Fig. 1f). The features of Sec61α and the bound cotransin are essentially superimposable between the two structures. Dashed lines, lateral gate helices (TM2b, TM3, and TM7).
Extended Data Figure 6. Variation in the extent of lateral gate opening in inhibitor-bound structures.
As in Fig. 2 a and b, but showing other inhibitor-bound structures. In all panels showing a lateral gate comparison, cylindrical representations in red and pink are the cotransin- and ipomoeassin F- bound structures, respectively, whereas the representation in green is the structure with the indicated inhibitor.
Extended Data Figure 7. Conformational flexibility of the chimeric Sec complex allows ipomoeassin F binding.
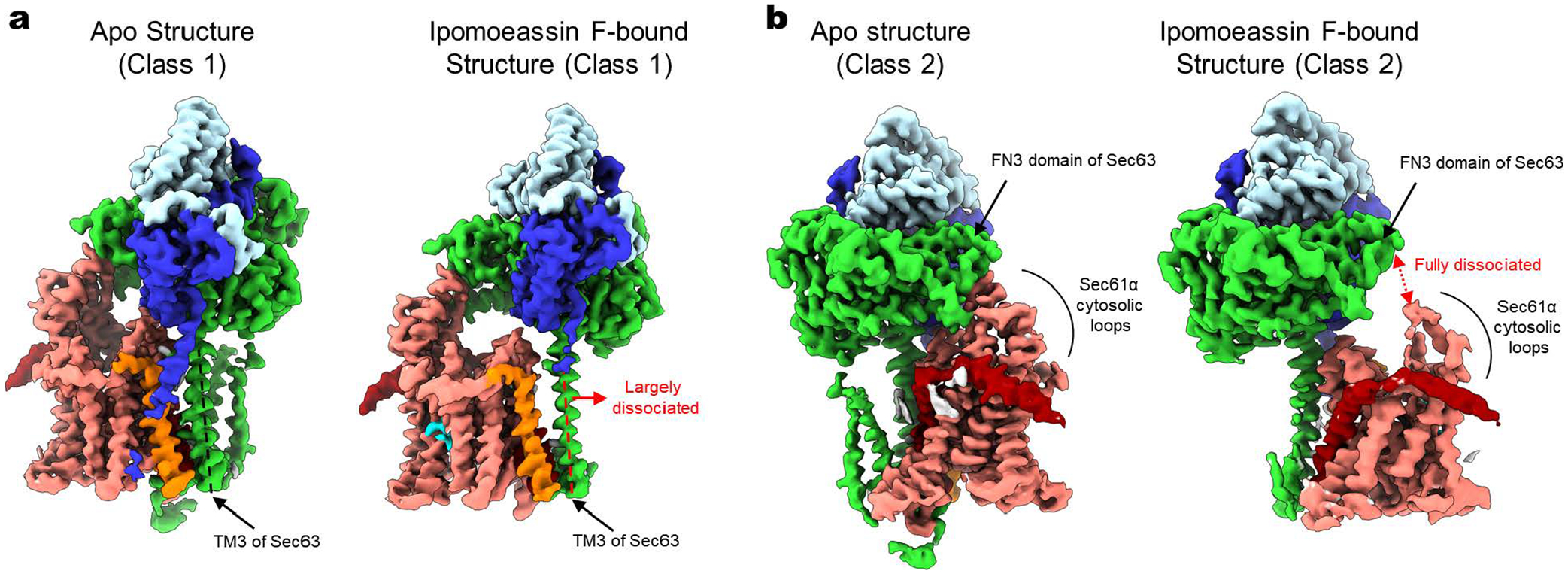
Binding of ipomoeassin F causes a narrower opening of the Sec61 lateral gate compared to the apo complex structures (also see Fig. 2), and this is enabled by disengagement of the Sec61 channel from TM3 (Class 1; panel a) or FN3 domain (Class 2; panel b) of Sec63. For comparison, the structures of the apo complex are also shown.
Extended Data Figure 8. 3D maps for interactions between Sec61 and inhibitors.
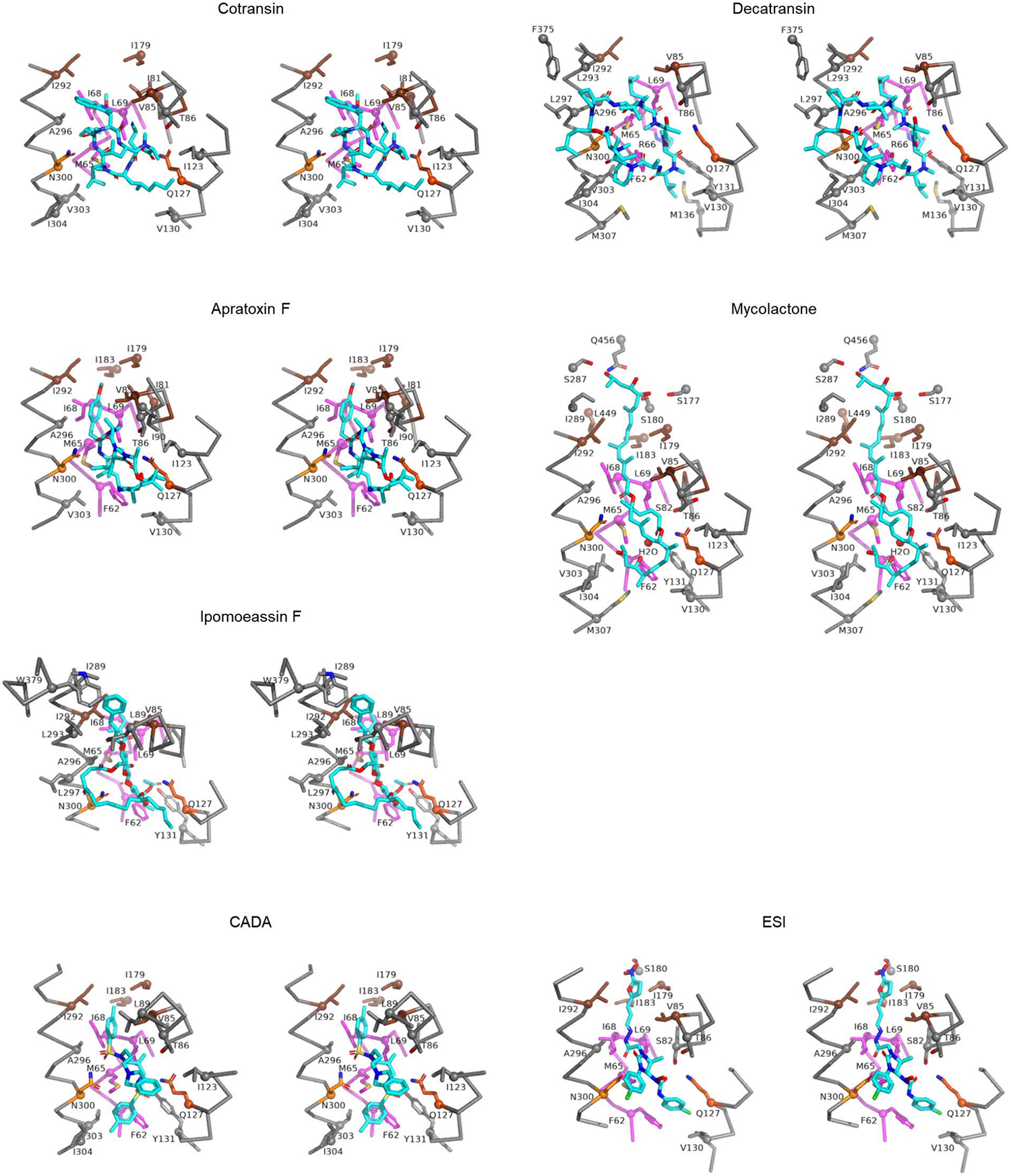
Shown are stereo-views into the inhibitor-binding site. Inhibitors and adjacent protein side chains are shown in a stick representation together with Cα traces for TM2b, TM3, TM7, and the plug. The views are roughly similar between the different structures but adjusted for each structure for more clear representations. The following colors are used to differentiate parts: brown, pore ring residues; magenta, plug; lighter orange; N300, darker orange, Q127. All inhibitors are shown in cyan with certain atom-dependent coloring (nitrogen-blue, oxygen-red, sulfur-yellow, and chlorine-green).
Extended Data Figure 9. Generation of HEK293 cell lines with expression of additional SEC61A1 and effects of CADA in CD4 expression.
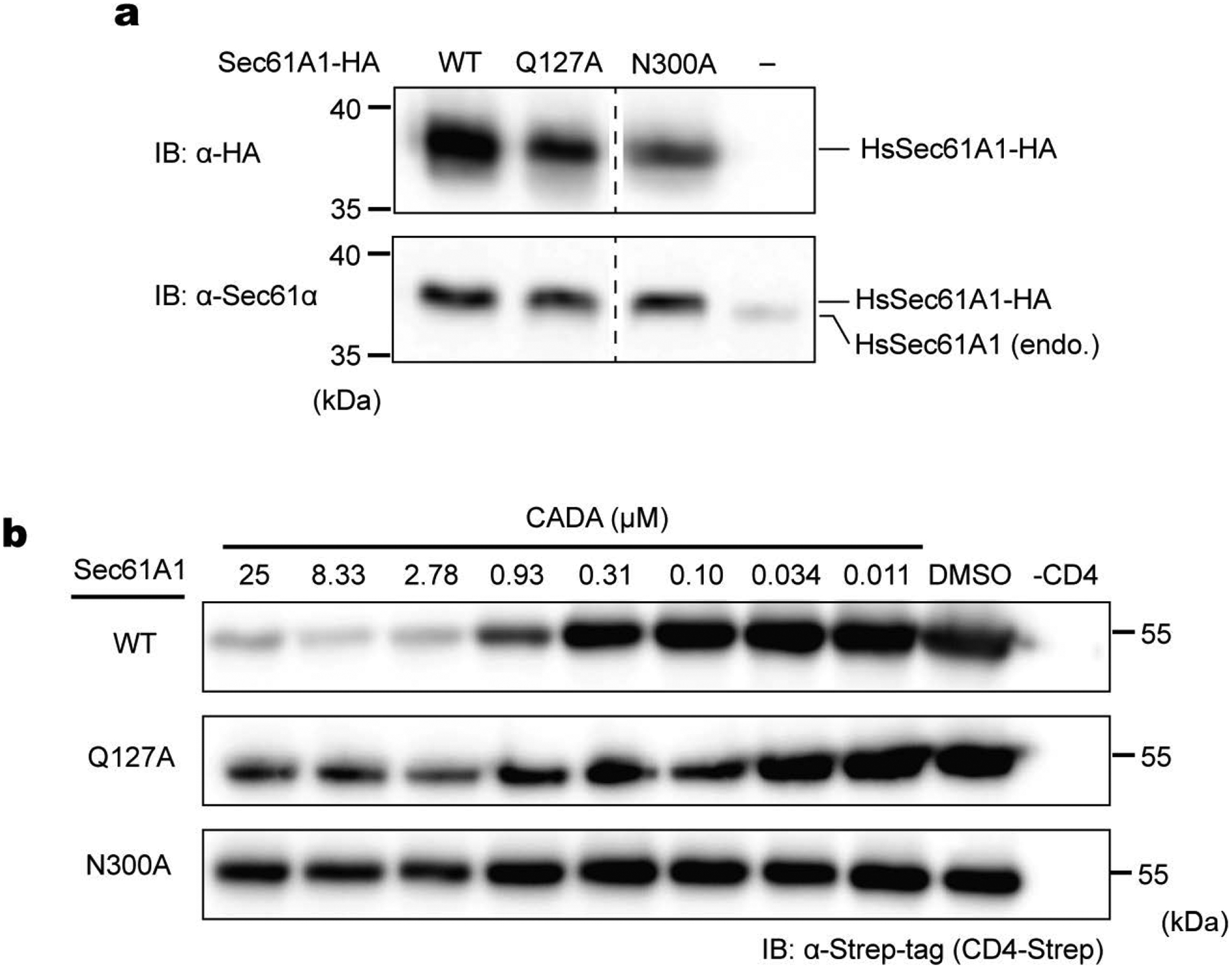
a, Expression of indicated human Sec61A1 in stable HEK293 (T-Rex-293) cells was confirmed by western-blotting with anti-HA-tag and anti-Sec61A1 antibodies. b, Human CD4 with a C-terminal Strep-tag was expressed in the indicated HEK293 cell lines by transient transfection, and the CD4 expression level after treating cells with the indicated concentrations of CADA was measured by SDS-PAGE and western-blotting. Four replicates were performed, and the dose-response curves are shown in Fig. 4k.
Extended Data Figure 10. Comparison with the mycolactone model by Gérard et al.
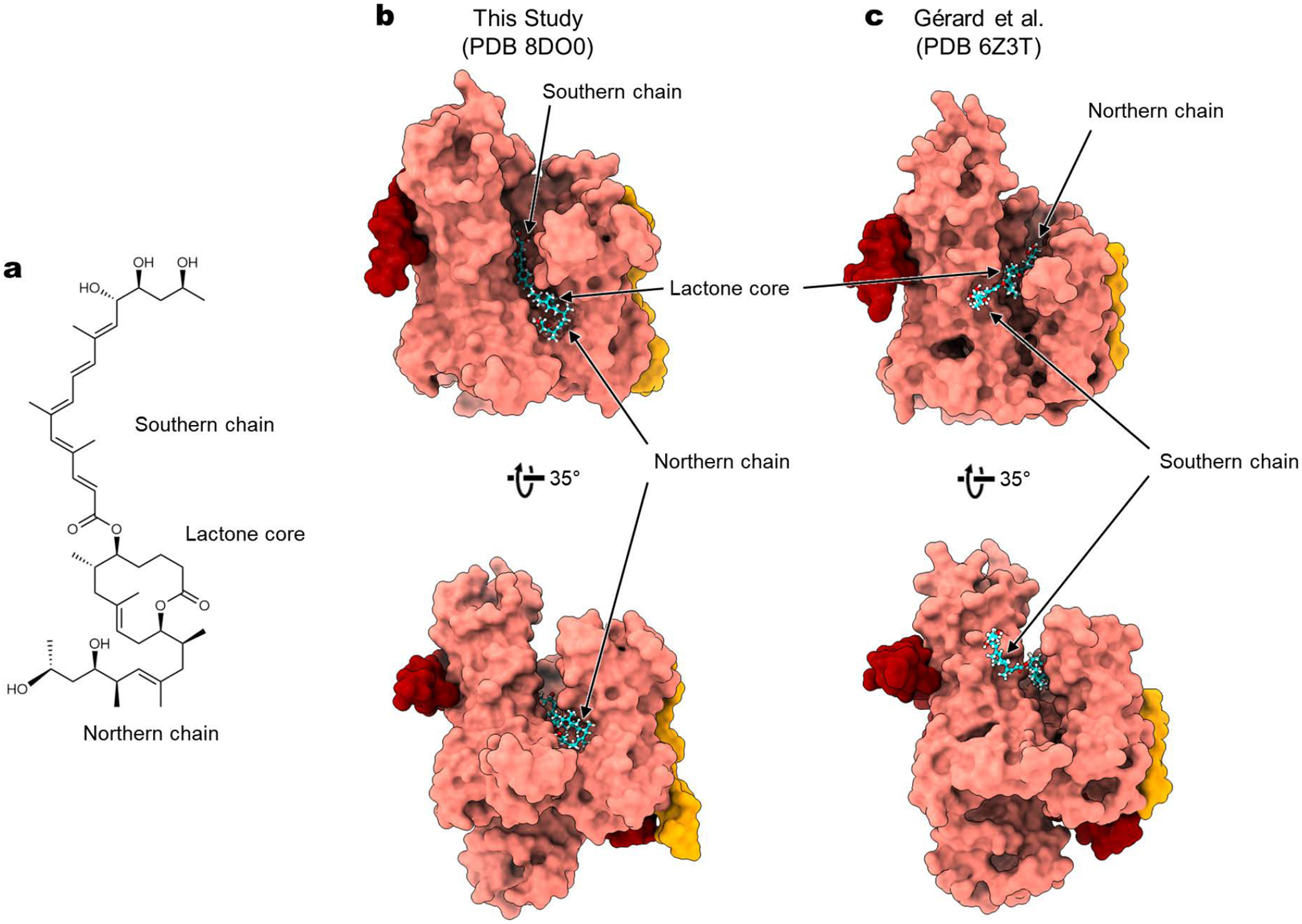
a, Chemical structure of mycolactone A/B. b, Structure of mycolactone-bound Sec61 in the current study. c, Structure of mycolactone-bound Sec61 in Gérard et al. (ref. 35). Note that the position and orientation of mycolactone are markedly different between the two structures. For example, the southern chain of mycolactone is buried into the cytosolic funnel of Sec61 in our study, whereas it is in the membrane in the study by Gérard et al. Other notable discrepancies include a one-residue shift in the helical register of the Sec61α TM7, which forms a lateral gate in Gérard et al.
Supplementary Material
Acknowledgments
We thank Dan Toso for support for electron microscope operation, Guanghui Zong for ipomoeassin F synthesis, Philippe Mathys and Ralph Riedl for help acquiring the IC50 data. E.P. was supported by the Vallee Scholars Program, Pew Biomedical Scholars Program, and Hellman Fellowship. S.I. and L.W. were supported by a National Institutes of Health training grant (5T32GM008295). M.S and T.J. were supported by the Swiss National Science Foundation (31003A-182519). N.B was supported by Fondation Raoul Follereau and Fondation Pour Le Développement De La Chimie Des Substances Naturelles Et Ses Applications. W.Q.S. (synthesis of ipomoeassin F) was supported by an AREA grant from National Institutes of Health (GM116032). C.F. and L.X. were supported by the Ohio State University.
Footnotes
Competing interests
During the revision of the manuscript, the Park lab (E.P. and L.W.) signed a sponsored research collaboration agreement with Kezar Life Sciences. The remaining authors declare no competing interests.
Data availability
EM maps and models are available through EM Data Bank (EMDB) and Protein Data Bank (PDB) under the following accession codes: EMD-27581 and PDB-8DNV for the apo class 1 structure, EMD-27582 and PDB-8DNW for the apo class 2 structure, EMD-27583 and PDB-8DNX for the cotransin CP2-bound complex, EMD-27584 and PDB-8DNY for the decatransin-bound complex, EMD-27585 and PDB-8DNZ for the apratoxin-F-bound complex, EMD-27586 and PDB-8DO0 for the mycolactone bound complex, EMD-27587 and PDB-8DO1 for ipomoeassin-F-bound complex, EMD-27588 and PDB-8DO2 for the CADA-bound complex, and EMD-27589 and PDB-8DO3 for the eeyarestatin-I-bound complex. Additional full Sec complex maps were also deposited to EMDB (see Supplementary Table 1 for accession codes).
References
- 1.Itskanov S et al. Mechanism of Protein Translocation by the Sec61 Translocon Complex. Cold Spring Harb Perspect Biol (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van den Berg B et al. X-ray structure of a protein-conducting channel. Nature 427, 36–44 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Deshaies RJ et al. Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature 349, 806–808 (1991). [DOI] [PubMed] [Google Scholar]
- 4.Panzner S et al. Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell 81, 561–570 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Egea PF et al. Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc Natl Acad Sci U S A 107, 17182–17187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park E et al. Structure of the SecY channel during initiation of protein translocation. Nature 506, 102–106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voorhees RM et al. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 157, 1632–1643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gogala M et al. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506, 107–110 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Voorhees RM et al. Structure of the Sec61 channel opened by a signal sequence. Science 351, 88–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L et al. Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature 531, 395–399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itskanov S et al. Structure of the posttranslational Sec protein-translocation channel complex from yeast. Science 363, 84–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X et al. Structure of the post-translational protein translocation machinery of the ER membrane. Nature 566, 136–139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itskanov S et al. Stepwise gating of the Sec61 protein-conducting channel by Sec63 and Sec62. Nat Struct Mol Biol 28, 162–172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weng TH et al. Architecture of the active post-translational Sec translocon. EMBO J 40, e105643 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pauwels E et al. Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway. Int J Mol Sci 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luesch H et al. Natural products as modulators of eukaryotic protein secretion. Nat Prod Rep 37, 717–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guenin-Mace L et al. Shaping mycolactone for therapeutic use against inflammatory disorders. Sci Transl Med 7, 289ra285 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Heaton NS et al. Targeting Viral Proteostasis Limits Influenza Virus, HIV, and Dengue Virus Infection. Immunity 44, 46–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vermeire K et al. CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology 302, 342–353 (2002). [DOI] [PubMed] [Google Scholar]
- 20.O’Keefe S et al. Ipomoeassin-F inhibits the in vitro biogenesis of the SARS-CoV-2 spike protein and its host cell membrane receptor. J Cell Sci 134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowe E et al. Preclinical evaluation of KZR-261, a novel small molecule inhibitor of Sec61. Journal of Clinical Oncology 38, 3582–3582 (2020). [Google Scholar]
- 22.Besemer J et al. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 436, 290–293 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Garrison JL et al. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 436, 285–289 (2005). [DOI] [PubMed] [Google Scholar]
- 24.MacKinnon AL et al. Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J Am Chem Soc 129, 14560–14561 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junne T et al. Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. J Cell Sci 128, 1217–1229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paatero AO et al. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem Biol 23, 561–566 (2016). [DOI] [PubMed] [Google Scholar]
- 27.McKenna M et al. Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J Cell Sci 129, 1404–1415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baron L et al. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J Exp Med 213, 2885–2896 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zong G et al. Ipomoeassin F Binds Sec61alpha to Inhibit Protein Translocation. J Am Chem Soc 141, 8450–8461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tranter D et al. Coibamide A Targets Sec61 to Prevent Biogenesis of Secretory and Membrane Proteins. ACS Chem Biol 15, 2125–2136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vermeire K et al. Signal peptide-binding drug as a selective inhibitor of co-translational protein translocation. PLoS Biol 12, e1002011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cross BC et al. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J Cell Sci 122, 4393–4400 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pauwels E et al. A Proteomic Study on the Membrane Protein Fraction of T Cells Confirms High Substrate Selectivity for the ER Translocation Inhibitor Cyclotriazadisulfonamide. Mol Cell Proteomics 20, 100144 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackinnon AL et al. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. Elife 3, e01483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerard SF et al. Structure of the Inhibited State of the Sec Translocon. Mol Cell 79, 406–415 e407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rehan S et al. Signal peptide mimicry primes Sec61 for client-selective inhibition. bioRxiv, 2022.2007.2003.498529 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pauwels E et al. Structural insights into TRAP association with ribosome-Sec61 complex and translocon inhibition by a CADA derivative. Sci Adv 9, eadf0797 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlson ML et al. The Peptidisc, a simple method for stabilizing membrane proteins in detergent-free solution. Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hommel U et al. The 3D-structure of a natural inhibitor of cell adhesion molecule expression. FEBS Lett 379, 69–73 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Luesch H et al. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J Am Chem Soc 123, 5418–5423 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Trueman SF et al. A gating motif in the translocation channel sets the hydrophobicity threshold for signal sequence function. J Cell Biol 199, 907–918 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith MA et al. Modeling the effects of prl mutations on the Escherichia coli SecY complex. J Bacteriol 187, 6454–6465 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Junne T et al. Mutations in the Sec61p channel affecting signal sequence recognition and membrane protein topology. J Biol Chem 282, 33201–33209 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Klein W et al. Defining a conformational consensus motif in cotransin-sensitive signal sequences: a proteomic and site-directed mutagenesis study. PLoS One 10, e0120886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Puyenbroeck V et al. Preprotein signature for full susceptibility to the co-translational translocation inhibitor cyclotriazadisulfonamide. Traffic 21, 250–264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fessl T et al. Dynamic action of the Sec machinery during initiation, protein translocation and termination. Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mercier E et al. Lateral gate dynamics of the bacterial translocon during cotranslational membrane protein insertion. Proc Natl Acad Sci U S A 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhadra P et al. Mycolactone enhances the Ca2+ leak from endoplasmic reticulum by trapping Sec61 translocons in a Ca2+ permeable state. Biochem J 478, 4005–4024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gamayun I et al. Eeyarestatin Compounds Selectively Enhance Sec61-Mediated Ca(2+) Leakage from the Endoplasmic Reticulum. Cell Chem Biol 26, 571–583 e576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao L Synthetic Apratoxin F and Novel Analogues - Molecules for Anticancer Mechanistic and Therapeutic Applications, The Ohio State University, (2017). [Google Scholar]
- 51.Zong G et al. Total Synthesis and Biological Evaluation of Ipomoeassin F and Its Unnatural 11R-Epimer. J Org Chem 80, 9279–9291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chany AC et al. A diverted total synthesis of mycolactone analogues: an insight into Buruli ulcer toxins. Chemistry 17, 14413–14419 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Lee ME et al. A Highly Characterized Yeast Toolkit for Modular, Multipart Assembly. ACS Synth Biol 4, 975–986 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Tegunov D et al. Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods 16, 1146–1152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Punjani A et al. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Emsley P et al. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Pilon M et al. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J 16, 4540–4548 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoepfner D et al. Selective and specific inhibition of the plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 11, 654–663 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
EM maps and models are available through EM Data Bank (EMDB) and Protein Data Bank (PDB) under the following accession codes: EMD-27581 and PDB-8DNV for the apo class 1 structure, EMD-27582 and PDB-8DNW for the apo class 2 structure, EMD-27583 and PDB-8DNX for the cotransin CP2-bound complex, EMD-27584 and PDB-8DNY for the decatransin-bound complex, EMD-27585 and PDB-8DNZ for the apratoxin-F-bound complex, EMD-27586 and PDB-8DO0 for the mycolactone bound complex, EMD-27587 and PDB-8DO1 for ipomoeassin-F-bound complex, EMD-27588 and PDB-8DO2 for the CADA-bound complex, and EMD-27589 and PDB-8DO3 for the eeyarestatin-I-bound complex. Additional full Sec complex maps were also deposited to EMDB (see Supplementary Table 1 for accession codes).