

Abstract
Chronic cocaine administration causes instability in extracellular glutamate in the nucleus accumbens that is thought to contribute to the vulnerability to relapse. A computational framework was developed to model glutamate in the extracellular space, including synaptic and nonsynaptic glutamate release, glutamate elimination by glutamate transporters and diffusion, and negative feedback on synaptic release via metabotropic glutamate receptors (mGluR2/3). This framework was used to optimize the geometry of the glial sheath surrounding excitatory synapses, and by inserting physiological values, accounted for known stable extracellular, extrasynaptic concentrations of glutamate measured by microdialysis and glutamatergic tone on mGluR2/3. By using experimental values for cocaine-induced reductions in cystine-glutamate exchange and mGluR2/3 signaling, and by predicting the down-regulation of glutamate transporters, the computational model successfully represented the experimentally observed increase in glutamate that is seen in rats during cocaine-seeking. This model provides a mathematical framework for describing how pharmacological or pathological conditions influence glutamate transmission measured by microdialysis.
Keywords: glutamate transporter, glial geometries, cystineglutamate exchange, mGluR2/3, non-synaptic release, microdialysis
Repeated cocaine administration causes enduring changes in glutamate transmission in the nucleus accumbens that may contribute to relapse vulnerability (Kalivas et al., 2005). These changes include alterations in glutamate release (McFarland et al., 2003), postsynaptic glutamate signaling (Conrad et al., 2008), dendritic spine morphology (Robinson and Kolb, 2004), and group II metabotropic glutamate receptors (mGluR2/3; Xi et al., 2002). The diversity of neuroadaptations has proven difficult to synthesize into a portrait of cocaine-induced pathology. While obtaining experimental measurements of glutamate transmission is critical, an alternate approach is to mathematically model an ‘archetypal’ synapse by extracting common features of the synaptic environment from a large number of synapses (Clements et al., 1992; Kleinle et al., 1996; Rusakov and Kullmann, 1998; Rusakov, 2001; Barbour, 2001; Diamond, 2005; Saftenku, 2005). These models have focused on synaptic glutamate release, diffusion out of the synapse and elimination by glutamate transporters (XAG) in an effort to understand the accessibility of synaptically released glutamate to the extracellular environment.
The mathematical models cited are based upon in vitro electrophysiological research and are appropriate for assessing concentrations of glutamate in the synaptic cleft and the near adjacent perisynaptic environment. However, in vivo extrasynaptic concentrations assessed by microdialysis reveal that the majority of glutamate outside of the synaptic cleft is not of synaptic origin (Miele et al., 1996; Timmerman and Westerink, 1997; Melendez et al., 2005). Also, extracellular glutamate in tissue slices and cell culture experiments is partly of nonsynaptic origin (Jabaudon et al., 1999; Haydon, 2001; Le Meur et al., 2007). While a number of sources of nonsynaptic extracellular glutamate have been suggested (Danbolt, 2001; Haydon, 2001; Cavelier et al., 2005), extracellular glutamate measured by microdialysis in the accumbens arises primarily from cystine-glutamate exchange (xc−; Baker et al., 2002; Xi et al., 2002). xc− is the rate-limiting step in glutathione synthesis (McBean, 2002), and glutamate derived from xc− stimulates perisynaptic mGluR2/3, and thereby inhibits synaptic glutamate release (Xi et al., 2002; Moran et al., 2005).
These data indicate that mathematical modeling of glutamate transmission should include nonsynaptic sources of glutamate. Moreover, rats withdrawn from chronic cocaine administration show dysregulation of extracellular glutamate in the nucleus accumbens due, in part, to reduced xc− and mGluR2/3 signaling (Baker et al., 2003; Madayag et al., 2007). Therefore, including extrasynaptic glutamate is required to model relevant cocaine-induced neuroplasticity. Also, while mathematical models considering only synaptically released glutamate predict that each glutamate synapse functions in relative isolation from other synapses (Kleinle et al., 1996; Barbour, 2001; Lehre and Rusakov, 2002; Sykova, 2004), microdialysis during cocaine-seeking measures significant overflow of synaptic glutamate (McFarland et al., 2003, 2004).
In order to reproduce cocaine-induced adaptations in extracellular glutamate, we modeled synaptic glutamate transmission, different glial geometries populated with XAG and xc−, and the regulation of glutamate release by mGluR2/3. Combining physiological values from the literature and empirically derived changes produced by chronic cocaine, the proposed mathematical framework was able to accurately portray both basal and cocaine altered extracellular glutamate levels as measured by microdialysis.
EXPERIMENTAL PROCEDURES
Model inputs, baseline diffusion, binding and transport parameters
Baseline physiological parameters for glutamate transmission were employed, primarily as described in previous models of glutamate transmission (Table 1). The principal mechanisms involved in transient glutamate dynamics in the perisynaptic region are glutamate diffusion out of the synapse after release, binding to transporters and uptake into glia (Danbolt, 2001), production of glutamate by the xc− located in glia (Pow, 2001; Sato et al., 2002), and activation of mGluR2/3 autoreceptors reducing synaptic release probability (Dietrich et al., 2002; Losonczy et al., 2003; Billups et al., 2005).
Table 1.
Ranges for parameter values used in model
| Parameter | Range of values (citation) | Model valuea |
|---|---|---|
|
| ||
| Diffusion coefficient (μm2/ms) | 0.05–0.41 (Rusakov and Kullmann, 1998, Saftenku, 2005) | 0.05 |
| k1 (M−1ms−1) | 104 (Lehre and Rusakov, 2002) | 104 |
| k−1 (ms−1) | 0.2 (GLAST/GLT; Lehre and Rusakov, 2002) | 0.2 |
| k2 (ms−1) | 0.1 (Lehre and Rusakov, 2002) | 0.1 |
| No. of molecules per release | 4700–80,000 (Bruns and Jahn, 1995) | 10,000 |
| Intersynaptic distance (μm) | 2–20 (Rusakov, 2001) | 2 |
| Kd for mGluR2/3 (μM) | 0.1–0.3 (Schoepp and True, 1992) | 0.187 |
| Maximum release probability | 0.1–0.5 (Trommershauser et al., 2003; Billups et al., 2005; Volynski et al., 2006) | 0.4 |
| XAG conc. (molecules/μm2)b | 550–3780 (Bergles and Jahr, 1997; Lehre and Danbolt, 1998; Colombo, 2005) | See ‘b’ below |
| xc– (mM h−1)c | 5–50 (Basal values from Warr et al., 1999; Baker et al., 2003) | 41 |
Surface density (molecules/μm2) of XAG was distributed as follows: G1a-1575, G1b-970, G2a-790, G2b-560, G3a-260, G3b-150, G4a-0, G4b-0; corresponding volume density (×10−21 mol) of XAG: G1a-1.089, G1b-1.085, G2a-1.082, G2b-1.08, G3a-0.602, G3b-0.463, G4a-0, G4b-0.
xc− was distributed uniformly in seven compartments of G4b: (i=12, j=2–8).
Synaptic release and regulation by mGluR2/3 autoreceptors.
In vivo estimates of basal firing frequency in prefrontal cortical (PFC) neurons projecting to the nucleus accumbens range from 1 to 3 Hz with the capacity for periods of burst firing up to 15 Hz (Chang et al., 1997; Peters et al., 2005; Sun and Rebec, 2006). Although the probability that an action potential will release a synaptic vesicle can range from <0.1–1 depending upon the experimental preparation (Allen and Stevens, 1994; Murthy and Sejnowski, 1997), the average synaptic release probability more typically ranges from 0.1–0.5, with estimates for cortex being at ~0.4 (Trommershauser et al., 2003; Billups et al., 2005; Volynski et al., 2006). Release probability at glutamatergic synapses is reduced by up to 50% following stimulation of presynaptic mGluR2/3 autoreceptors (Dietrich et al., 2002; Losonczy et al., 2003; Billups et al., 2005), which are located outside of the synaptic cleft (Alagarsamy et al., 2001). Using in vivo microdialysis it has been shown that blocking mGluR2/3 elevates extracellular concentrations of glutamate (Xi et al., 2002) and electrophysiological studies in tissue slices reveal that the glutamate providing this tone is derived primarily from nonsynaptic sources (Bandrowski et al., 2003; Moran et al., 2005). Given these studies indicating that partial tone exists on mGluR2/3 regulating glutamate release, the basal levels of glutamate in the vicinity of perisynaptic mGluR2/3 were adjusted in the present model to produce ~50% occupancy, based upon the range of and values reported at this receptor (0.1–0.3 μM glutamate; Schoepp and True, 1992). In the proposed model, presynaptic tone on mGluR2/3 was computed as release probability. mGluR2/3 is a glial sheath -coupled metabotropic receptor, and analysis of GTPγS binding reveals that G protein signaling by stimulating mGluR2/3 is increased as a logarithm of agonist dose (Xi et al., 2002; Bowers et al., 2004). Thus, the relationship between release probability and mGluR2/3 occupation was modeled as the logarithm of glutamate concentration, with a glutamate (Schoepp and True, 1992) and maximum release probability with no mGluR2/3 stimulation set at 0.4 (see above). Each action potential provoking glutamate release (a function of firing frequency and release probability) resulted in an instantaneous vesicular release of a fixed number of molecules into the cleft. This fixed number was selected iteratively from the range 4700–80,000 reported by Bruns and Jahn (1995) and set at 10,000 (Table 1).
Diffusion.
In a complex medium, several factors can impose constraints on diffusion, including geometry, binding, uptake, viscosity, temperature, or change in structure with time (Nicholson, 2001; Sykova, 2004; Diamond, 2005; Saftenku, 2005). Diffusion in the extracellular space is typically characterized by volume fraction (void space/total tissue volume) and tortuosity (hindrance to diffusion imposed by local boundaries or local viscosity) (Nicholson, 2001). Volume fraction in brain tissue is estimated to be around 0.2 (Nicholson and Sykova, 1998). Tortuosity varies due to constriction, wiggle and topological factors (Nicholson, 2001) and is estimated to be ~1.2–2.4 based on diffusion measurements over a range of 100–300 μm (Nicholson, 2001). Rice and Nicholson (1991) reported values for α and λ as 0.21 and 1.54, respectively, for the rat striatum. To account for the complex factors cited, diffusion coefficient values have been reported in the range from 0.05–0.41 μm2/ms (Saftenku, 2005), based on typical tortuosity estimates. Further, different cellular elements including spines, small axonal boutons, protein, glia, and microfilaments may result in additional tortuosity in the microenvironment of a synapse (Saftenku, 2005). Experimental estimates of diffusion coefficients in the perisynaptic region have not been reported for synapses with tightly packed glia. In the proposed model, with high density glia close to the synapse, we iteratively determined the diffusion coefficients to satisfy steady state and transient constraints on glutamate concentrations at three locations (, and in Fig. 1). This iterative process is described in more detail below.
Fig. 1.
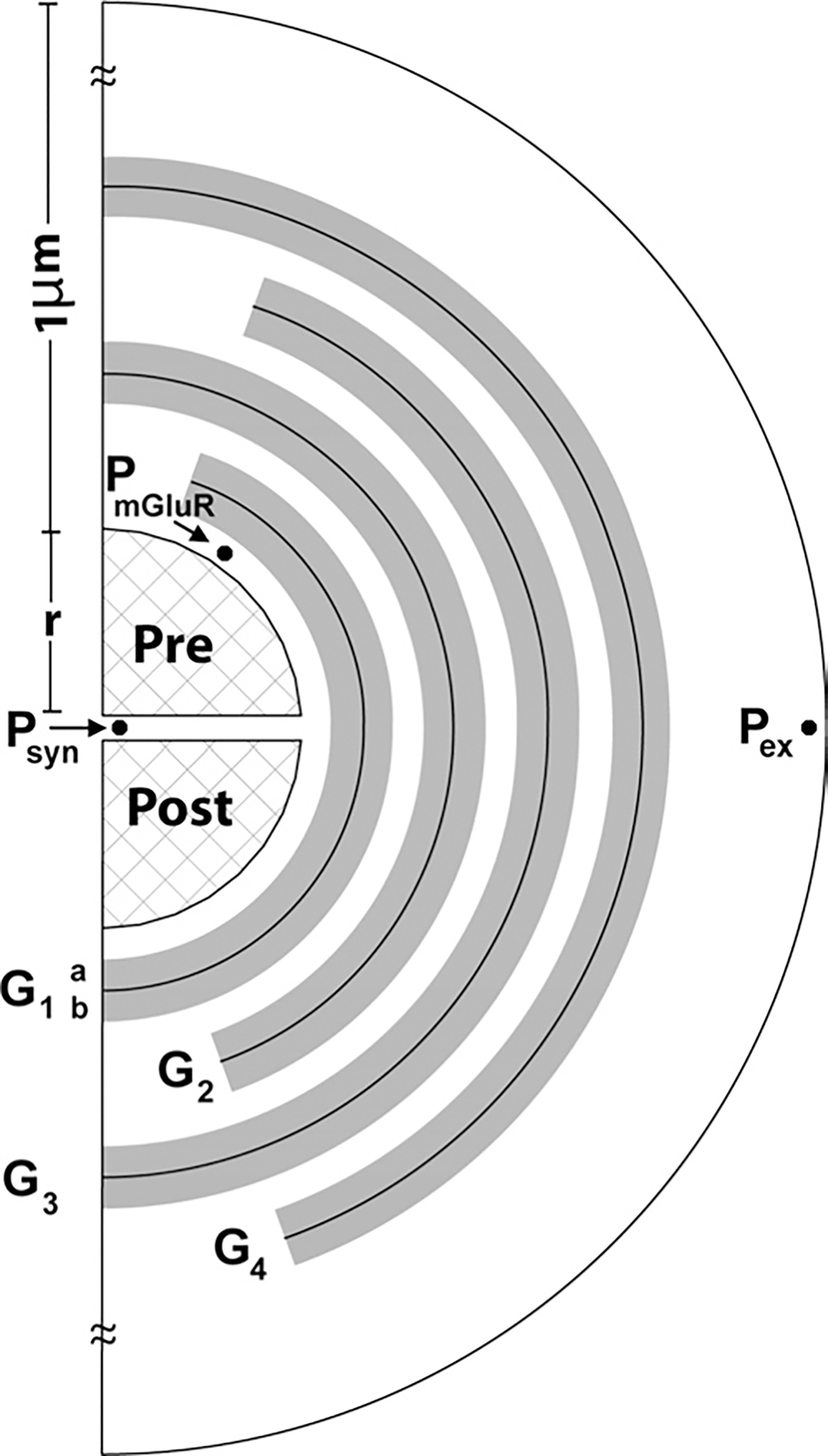
The glial configuration used to study glutamate homeostasis in the perisynaptic space around the PFC-accumbens synapse. The model depicts XAG and xc− in glial regions (shaded) in varying concentrations. The cleft () separates the two hemispheres of radius surrounded by being the closest to the synapse) with the highest density of XAG in and decreasing in radially outward sheaths. Each sheath is 50 nm thick with an impermeable surface in the middle, and with XAG volume-populated in the 25 nm thick space on either side, permitting interaction with glutamate molecules in those regions. The perisynaptic space is partitioned in radial (step ) and tangential (step ) directions as in Rusakov (2001). Binding, uptake and efflux are computed for each compartment. Glutamate concentrations were measured at three sites, within the synaptic cleft (at ), in the perisynaptic region containing presynaptic mGluR2/3 (at ), and at the site where dialysis probe measures extracellular glutamate (at ).
XAG.
Glutamate transport into glia is the primary mechanism for eliminating extracellular glutamate (Danbolt, 2001). XAG uptake rates depend on local glutamate concentration and the kinetics of transporter binding (see eqn. 3 below). The glutamatergic axon terminals from the PFC to the accumbens were assumed to be covered by a (Lehre et al., 1995). The density of XAG is non-uniform, and glial membranes that face neuropil have a higher expression of transporters than membrane surfaces facing other glia (Cholet et al., 2002). XAG are expressed with a high density in the hippocampus, with surface density ranging from 2500 to 10,800 molecules/μm2 (Bergles and Jahr, 1997; Lehre and Danbolt, 1998). Based upon glutamate uptake assays (Colombo, 2005) and transporter binding studies (Danbolt, 2001) it was estimated that surface density values for XAG in the nucleus accumbens are 22–35% (550–3780 molecules/μm2) of the value in the hippocampus and cortex. Thus, for the present model (where XAG is volume populated as described later), the equivalent surface density of XAG was determined iteratively by varying it within the range of 550–3780 molecules/μm2 (Table 1).
xc–.
Wyatt et al. (1996) estimated the maximum uptake rate for cystine to be 450 μM h−1 based on cerebellar slices. The density of xc− in the cortex is higher by a factor of 2.4 compared with the cerebellar molecular layer (1 mM h−1; Warr et al., 1999). Based on microdialysis studies, Baker et al. (2003) reported basal extracellular glutamate concentrations to be 1.1 and 5.6 μM in the prefrontal cortex and nucleus accumbens, respectively. Iterations to satisfy model constraints resulted in the consideration of a range from 5–50 mM h−1 for the density of xc− in the nucleus accumbens and a final value of 41 mM h−1 under basal conditions (Table 1).
Model inputs and cocaine-induced neuroadaptations
The parameters adjusted in the model to estimate neuroadaptive changes produced by withdrawal from chronic cocaine are outlined in Table 2. Withdrawal from daily cocaine administration elicits a 50% reduction in for [35S] cystine uptake into accumbens tissue slices (Baker et al., 2003), and recently a 40% reduction in cystine uptake was reported after withdrawal from cocaine self-administration (Madayag et al., 2007). Also, a 40% reduction in the concentration of glial transporter protein (GLT-1) has been reported in membrane homogenates from the nucleus accumbens of rats withdrawn from cocaine self-administration (Knackstedt et al., 2007). Based upon these data the concentration of xc− was reduced by 50% (Table 2). Previous studies using [35S]GTPγS binding in accumbens homogenates revealed that G protein coupling to mGluR2/3 is reduced by approximately 70% after cocaine (Xi et al., 2002). Assuming a logarithmic relationship between [35S]GTPγS binding and vesicle release probability (see above), the cocaine-induced reduction in mGluR2/3 function was modeled as a change in release probability from 0.14 (control) to 0.34 (cocaine-treated condition). Thus, a release event occurred every 2.9 action potentials in the cocaine case, instead of every 7.1 action potentials under basal conditions. Finally, cocaine-seeking behavior is associated with an increase in firing frequency of accumbens neurons, driven in part by inputs from the prefrontal cortex (Hollander and Carelli, 2007). Basal firing frequency in prefrontal pyramidal neurons can range from 1 to 15 Hz, and the neurons can burst fire at a rate of >1 burst/s and a single burst event can include >50 spikes (Sun and Rebec, 2006). The in vivo basal firing of prefrontal pyramidal cells is reduced after withdrawal from self-administered cocaine (Trantham et al., 2002; Sun and Rebec, 2006), but the firing rate during burst events is significantly elevated in response to a cocaine injection in rats trained to self-administer cocaine (Sun and Rebec, 2006). The apparent hyperexcitability of prefrontal neurons after chronic cocaine may be associated with the increased excitability observed in dissociated prefrontal pyramidal cells after chronic cocaine (Dong et al., 2005). Given this complex profile of changes in pyramidal cell firing after chronic cocaine, in the present model the behavioral transition to cocaine-seeking was modeled simply as an increase in overall firing frequency from 1 (basal) to 15 Hz (cocaine-seeking).
Table 2.
Parameters altered by chronic cocaine administration
| Parameter | Control | Cocaine | Reference |
|---|---|---|---|
|
| |||
| Glutamate concentration at Pex (μM; basal) | 5.6±1.0 | 2.89±0.34 | Baker et al., 2003; Szumlinski et al., 2006 |
| Peak glutamate in Pex (μM; during food seeking/cocaine-seeking) | 5.6±1.0 | 13.3±1.4 | McFarland et al., 2003, 2004 |
| xc− (mM h−1) | 41 | 20.5a | Baker et al., 2003 |
| Release probability | 0.14 (Basal) | 0.34 (Basal)b | Xi et al., 2002 |
| Firing freq (Hz) (basal) | 2 | 1 | Sun and Rebec, 2006; Trantham et al., 2002 |
| Firing freq (Hz) (drug-seeking) | N/A | 3–15 | Chang et al., 1997; Sun and Rebec, 2006 |
Based upon increase in Kmfor cystine from 2.1±0.2–4.2±0.2 μM; 28.3±7.9% reduction in catalytic subunit of xc− (xCT).
Based upon 70% reduction in mGluR2/3-induced GTPγS binding.
Modeling the synapse and glial geometry
Upon release at the center of the synapse, glutamate molecules diffuse through the porous cleft into the perisynaptic space (Barbour and Hausser, 1997), where XAG dense astrocytes reduce glutamate spillover to near zero (Diamond and Jahr, 2000; Danbolt 2001). The configuration of the (Fig. 1) is akin to that previously reported (Rusakov, 2001), but distinct in that in the present model we include xc−. Also, as an approximation of glial folds, the glial membranes were modeled in the form of multiple impermeable sheaths (the dark line at the center of each sheath in Fig. 1 represents an impermeable surface, i.e. flux=0 across this surface) with porous space in between them. XAG was volume populated on both sides of each (permeable to glutamate up to 25 nm thickness on each side of the impermeable center surface of the 50 nm thick ). Glutamate concentration at mGluR2/3 receptors was monitored in the model at the presynaptic location in Fig. 1 (compartment , starting at ).
The extracellular space is thus modeled as a porous medium with four whose centerlines were 75 nm apart (close to the range of 38–64 nm reported in the extracellular space of the rat neocortex in vivo by Thorne and Nicholson, 2006). Of this 75 nm, 50 nm is volume populated with XAG and/or xc−, as described above. This permits the glutamate molecules to move up to 75 nm between the impermeable surfaces of each sheath. Based upon studies indicating that the highest densities of XAG are closer to the synapse (Lehre and Danbolt, 1998; Danbolt, 2001; Cholet et al., 2002), had the highest density of XAG and the density decreased radially outwards to . xc− were modeled as being located on the outer surface of the glial membranes of regions (Table 1). Beyond the last , the extracellular space contained only glutamate without XAG or xc−. The experimentally defined concentrations of extracellular glutamate reported by in vivo microdialysis (Table 2) were modeled as being at point in Fig. 1, outside glial region .
Mathematical details.
In the configuration of Fig. 1, the two synaptic hemispheres were assumed rigid permitting no diffusion (i.e. flux=0 along the periphery), with synaptic radius from the center, and a separation of (synaptic cleft) (Rusakov and Kullmann, 1998; Rusakov, 2001; Diamond, 2005). Around this synapse are 40 concentric 25 nm thick shell compartments resulting in the outer boundary of the perisynaptic region modeled being at a distance of 1 μm from the edge of the synapse. Each of these shells was divided into nine compartments (20° angle increments, ) circumferentially, permitting XAG and xc− concentrations to be assigned individually to each compartment of any shell.
The synaptic cleft volume was discretized into cylindrical segments where was the outer radius of each of the cylindrical segment with being the thickness. The width of each cylinders is , such that the volume of each concentric cylinder is , with the contact surface between adjacent elements being . The extracellular space were discretized into concentric spherical elements each of thickness , and each spherical element was divided into annular sections where was determined by . In the model for the cleft, , and , and for the spherical shells, and rad.
The specific mathematical equations used are described next. These standard conservation and flux equations (see Rusakov, 2001 for a comprehensive description including derivations) were used to analyze the effect of the proposed glial geometry. A mass balance for extracellular glutamate in each compartment (with XAG and/or xc−, as appropriate) yields eqn. 1 (Rusakov, 2001),
| (1) |
where was the time step, was the surface area between adjacent volume elements in the radial direction, and was the surface area shared by adjacent volume elements in the tangential direction, with . The radial and tangential fluxes into the compartment were denoted by and , respectively. Each compartment had a volume of . The term accounted for the production of glutamate by xc− and unbinding of glutamate from the transporters , where is the constant production rate of glutamate by xc− for compartment , while the term accounted for the reduction in glutamate due to transporter binding. For compartments that are not populated with XAG or xc−, the corresponding terms in eqn. 1 are omitted. Also, eqn. 1 is appropriately modified for the compartments in the synaptic cleft, to exclude XAG, xc−, and the tangential flux, and include synaptic release.
The glutamate flux between any two adjacent volume compartments and was computed by eqn. 2,
| (2) |
where was the spatial distance between compartment centroids and the diffusion coefficient. For each compartment, this flux was calculated considering two others connected to it radially, and two connected in the tangential direction. Within any glial compartment, binding of glutamate with transporters is governed by eqn. 3 (Rusakov and Kullmann, 1998),
| (3) |
where [], [], and [] represent the compartmental concentrations of glutamate, transporter, and the bound complex, respectively, and represents uptake rate of glutamate by XAG.
The discrete form of the differential equation for this kinetic equation is given by eqn. set 4 (Rusakov, 2001):
| (4) |
The kinetics for XAG were taken from Rusakov (2001) and Lehre and Rusakov (2002) who based it on experiments reported in the literature (Wadiche et al., 1995; Bergles and Jahr, 1998), , , and . For the outermost shell, e.g. , the boundary condition of flux=0 was imposed at the outer edge of all compartments, to simulate identical neighboring synapses. That is, no flux enters or leaves the outer boundary of this shell.
Iterative evaluation.
The computational model was developed using C++ software, Microsoft Visual Studio 2005 package (Microsoft Corporation, Washington), and an integration time step of 0.5 μs was used. The concentration of glutamate was considered uniform in each compartment and this concentration was updated (eqns. 1–4) at each integration interval based on diffusion, uptake by XAG, and production rates for glutamate, as appropriate. Conservation of molecules was confirmed at each time step by computing the numbers of free, bound and transported glutamate molecules. To check for numerical accuracy, we decreased the integration time step by a factor of 10 and found no significant change in concentration estimates. Similarly, insignificant changes in the same estimates were found with variation of spatial resolution of compartments by 50%.
To implement a volume fraction of (Nicholson and Sykova, 1998) in the model shown in Fig. 1 (which was also iteratively derived; details not shown), we approximated shells to be representing cellular obstacles (i.e. space that glutamate cannot flow into), with an effective extracellular space from for glutamate overflow. This implies that is now measured in shell 20. The model showed that in the space outside the (i.e. outside shell 12) the steady state concentration of glutamate was uniform for any number of total outside shells, and differed by less than 0.01 μM for all cases considered. This observation justifies selection of anywhere in the space outside for measurement purposes.
As cited earlier, diffusion coefficients close to the synapse have not been reported for synapses with tight glial coverage. With the porous glial geometry in Fig. 1, we considered three apparent diffusion coefficients, one in the synapse ( near ), one in the sheath region ( in the region that has ) and one outside the region ( in the region that has ). We noticed that the flow dynamics was governed solely by , with insignificant effects due to variations in and within the range of 0.05–0.41 μm2/ms (data not shown). Accordingly, we used a uniform value of D (from the same range cited above) for all the regions in the model, without loss of accuracy. It should be noted that the added geometric tortuosity in the model.
The model was adjusted by changing the following parameters within the ranges outlined in Table 1: number of molecules/release, xc− concentration, diffusion coefficient and XAG concentration. The iterative process began with values in the lower end of the ranges for these parameters, while monitoring the concentrations of glutamate at , and (Fig. 1), for the basal control case (2 Hz). When the densities of XAG were iteratively changed in , their relative proportions were maintained, i.e. density and so on. Through this iterative process, numerous solutions were found that satisfied empirically determined concentrations at , , and for the control case at 2 Hz (Table 2).
After satisfying the requirements for the basal control case, we simulated the basal cocaine and drug-seeking situation by modeling known cocaine-induced changes to xc− and mGluR2/3 signaling (modeled as release probability, see above). Through further iterative changes we identified multiple parameter sets that satisfied some of the constraints in Table 2, and the model values listed in Table 1 constitute values that satisfied all the constraints simultaneously.
RESULTS
Geometry of the
Multiple 3-D spherical configurations were studied for glia surrounding the synapse by varying glial coverage, thickness and openings (similar to those in Rusakov, 2001; Barbour, 2001; data not shown). Table 1 shows the range of diffusion coefficients, number of molecules per release, as well as XAG and xc− concentrations in the various . These were varied iteratively to determine the configuration that brought glutamate concentration at (extracellular compartment sampled by microdialysis) into the range outlined in Table 2 at both low and high firing frequencies. At the same time, concentrations at and were constrained to be <200 nM. This process involved simultaneous variations of the parameters (see Experimental Procedures). Following this iterative process, the configuration in Fig. 1 proved most robust at sustaining glutamate concentrations within the acceptable ranges. Of note, the basal control concentration at did not exceed the range measured by microdialysis at firing frequencies of 15 Hz (Table 3, Fig. 2A). Also, by providing resistance to the flow of glutamate, this configuration established the necessary gradient to support levels of extracellular glutamate at approaching those estimated from in vitro slice physiology (Herman and Jahr, 2007) and at that are consistent with in vivo tone being present on mGluR2/3 (Xi et al., 2002). Thus, at both low and high frequency stimulation, remained between 0.1 and 0.3 μM, which approximates the for glutamate binding to mGluR2/3 (0.19 μM; Schoepp and True, 1992).
Table 3.
Model estimates at varying firing frequencies using control and chronic cocaine parameters
| Parameter | Control basal | Control biological reward seeking | Cocaine basala | Cocaine drug seekinga |
|---|---|---|---|---|
|
| ||||
| Firing freq (Hz) | 2 | 15 | 1 | 15 |
| Release probability | 0.14 | 0.12 | 0.34 | 0.30 |
| XAG (moles) | 5.4×10−21 | 5.4×10−21 | 3.24×10−21 | 3.24×10−21 |
| xc− (mM h−1) | 41 | 41 | 20.5 | 20.5 |
| Estimates of steady state Glu concentrations at three locations | ||||
| Psyn (μM) | 0.16 | 0.19 | 0.24 | 1.05 |
| PmGluR (μM) | 0.195 | 0.28 | 0.27 | 1.19 |
| Pex (μM) | 5.04 | 6.58 | 3.03 | 12.4 |
Cocaine-induced reduction in XAG (40%), xc− (50%) and mGluR2/3 signaling (70%; modeled as release probability).
Fig. 2.
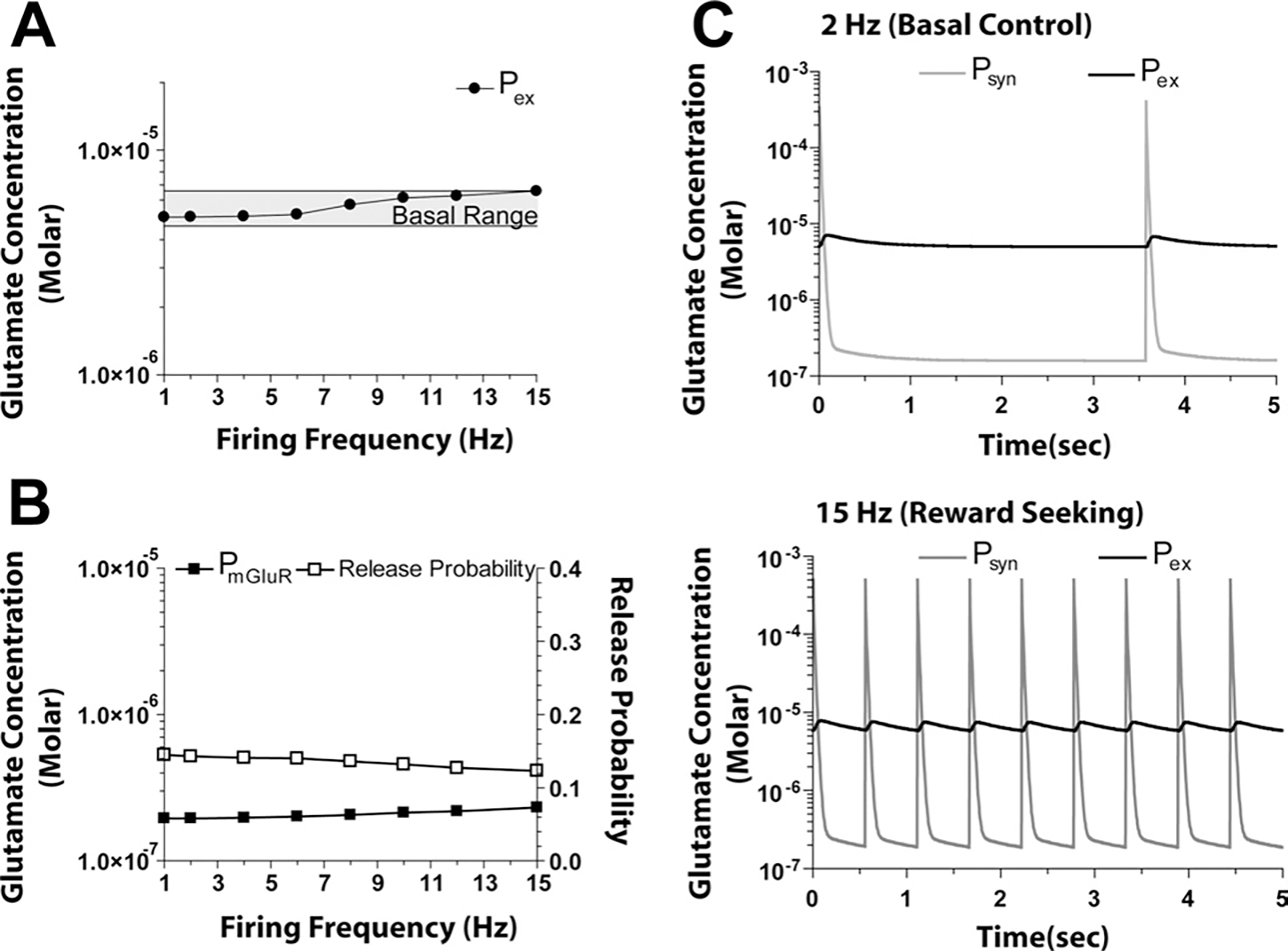
Concentrations of glutamate at different spatial locations under control conditions. (A) The increase in glutamate at remained within the basal range over the entire 1–15 Hz range of firing. (B) As firing frequency increases, the concentration of glutamate in the vicinity of perisynaptic mGluR2/3 autoreceptors (at ) increases producing a concomitant decrease in release probability. (C) Model output at 2 and 15 Hz over 5 s, illustrating the dynamic changes in synaptic (at ), and extracellular glutamate (at ).
Fig. 2B shows how the increase in associated with increased firing frequency negatively regulated release probability, i.e. as increased with increasing synaptic release, the release probability decreased from 0.14–0.12. Thus, as firing frequency ranged from 1 to 15 Hz, the peak concentration at reached as high as 10 mM, which, when averaged over 100 μs around this peak, resulted in a maximum value of 0.5 mM (Fig. 2C). As well, transient glutamate concentrations in the synapse (at ) were biphasic and within ranges reported by Clements (1996) and Bergles et al. (1999). The resting concentration at between release events ranged from 0.16–0.19 μM (Table 3). These levels are somewhat higher than recent published estimates which range from 25 to 100 nM using tonic activity at N-methyl-D-aspartic acid (NMDA) receptors in tissue culture (Herman and Jahr, 2007; Le Meur et al., 2007), and could reflect a lack of neuronal glutamate uptake in the present model. Fig. 2C also shows the effect of seeking natural rewards (e.g. food that was modeled as an increase in firing frequency to 15 Hz). In this control situation, the level of extracellular glutamate at agreed with measurements and was not significantly different from basal (i.e. remained in the range of 4.6–6.6 μM; McFarland et al., 2003).
Effect of withdrawal from chronic cocaine
Table 2 illustrates the alterations made in parameters by incorporating experimentally determined values for reduced xc− and mGluR2/3 desensitization after chronic cocaine (Xi et al., 2002; Baker et al., 2003). In addition, concentrations at approximated the basal values determined by microdialysis in the accumbens after withdrawal from chronic cocaine, as well as peak values elicited after inducing cocaine-seeking.
The model constraints for the basal extracellular concentration measured by dialysis in after cocaine was in the range of 2.55–3.23 μM (Baker et al., 2003; Szumlinski et al., 2006), and Fig. 3 shows that when the chronic cocaine-induced changes in xc−, mGluR2/3 and basal firing frequency were introduced, the model accurately depicted the reduced basal levels. However, when cocaine-seeking was introduced into the model (i.e. 15 Hz firing frequency) the extracellular concentration of glutamate () was expected to be in the range of 11.9–14.7 μM (McFarland et al., 2003, 2004; Szumlinski et al., 2006), and levels attained only 6.3 μm (see Fig. 3, 0% reduction in XAG).
Fig. 3.
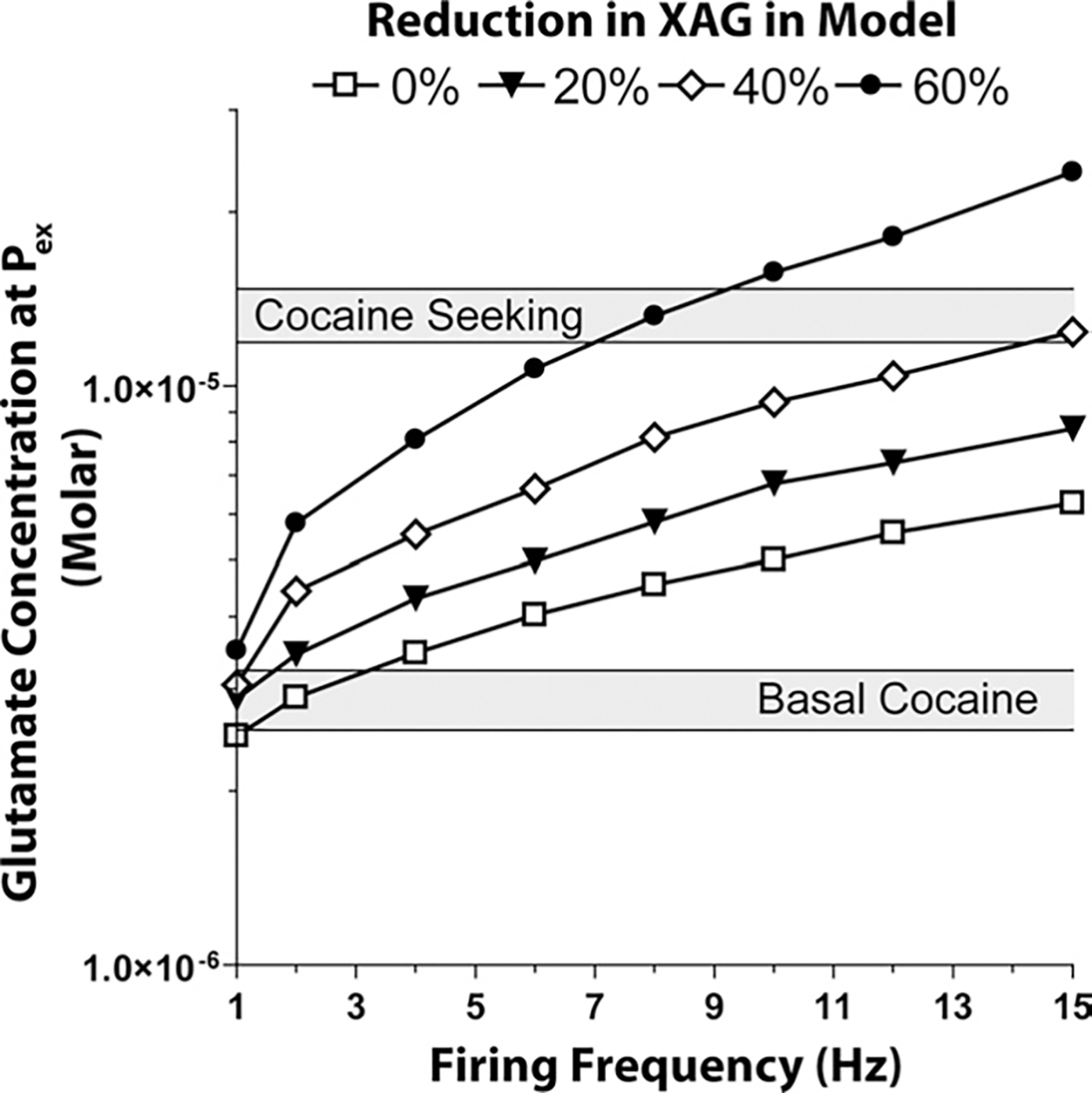
Effect of reducing XAG on the concentration of extracellular glutamate at , in cocaine-treated rats. To model the cocaine condition, the function of xc− and mGluR2/3 was reduced by 50% and 70%, respectively. Iterations of the model were then run at different percent decreases in the concentration of XAG over a firing frequency range of 1–15 Hz.
Down-regulation of XAG predicts chronic cocaine concentrations
Since the computational model based upon established cocaine-induced neuroadaptations failed to reproduce observed extracellular glutamate concentrations elicited during cocaine-induced drug seeking, other parameters were considered, such as XAG, molecules per release event, volume fraction, ECS width, and apparent diffusion coefficient. Multiple iterations of changes in these parameters revealed that down-regulating XAG produced changes in extracellular glutamate that mimicked those elicited in vivo, while changing the other parameters had lesser or no impact. Fig. 3 shows the iterative analysis of changing XAG. When XAG was reduced by 40%, increasing firing frequency to 15 Hz produced extracellular glutamate levels in the range observed by microdialysis during cocaine seeking (11.9–14.7 μM).
Fig. 4 illustrate the outcome for concentrations at and after introducing the cocaine-altered parameters for xc− and mGluR2/3 (modeled as release probability, see Experimental Procedures), a 40% reduction in XAG based upon the data in Fig. 3, and stimulating synaptic transmission at 1–15 Hz. Over a firing frequency of 1–15 Hz, the change in concentration at was similar to that at (Table 3). Note that release probability did not change appreciably even though increased as a function of increased firing frequency due to the fact that mGluR2/3 signaling is reduced by 70% after chronic cocaine (Xi et al., 2002). By including a 40% reduction in XAG along with the cocaine-induced reductions in xc− and mGluR2/3 signaling, values were in the expected range under both basal (1 Hz) and cocaine seeking (15 Hz) conditions.
Fig. 4.
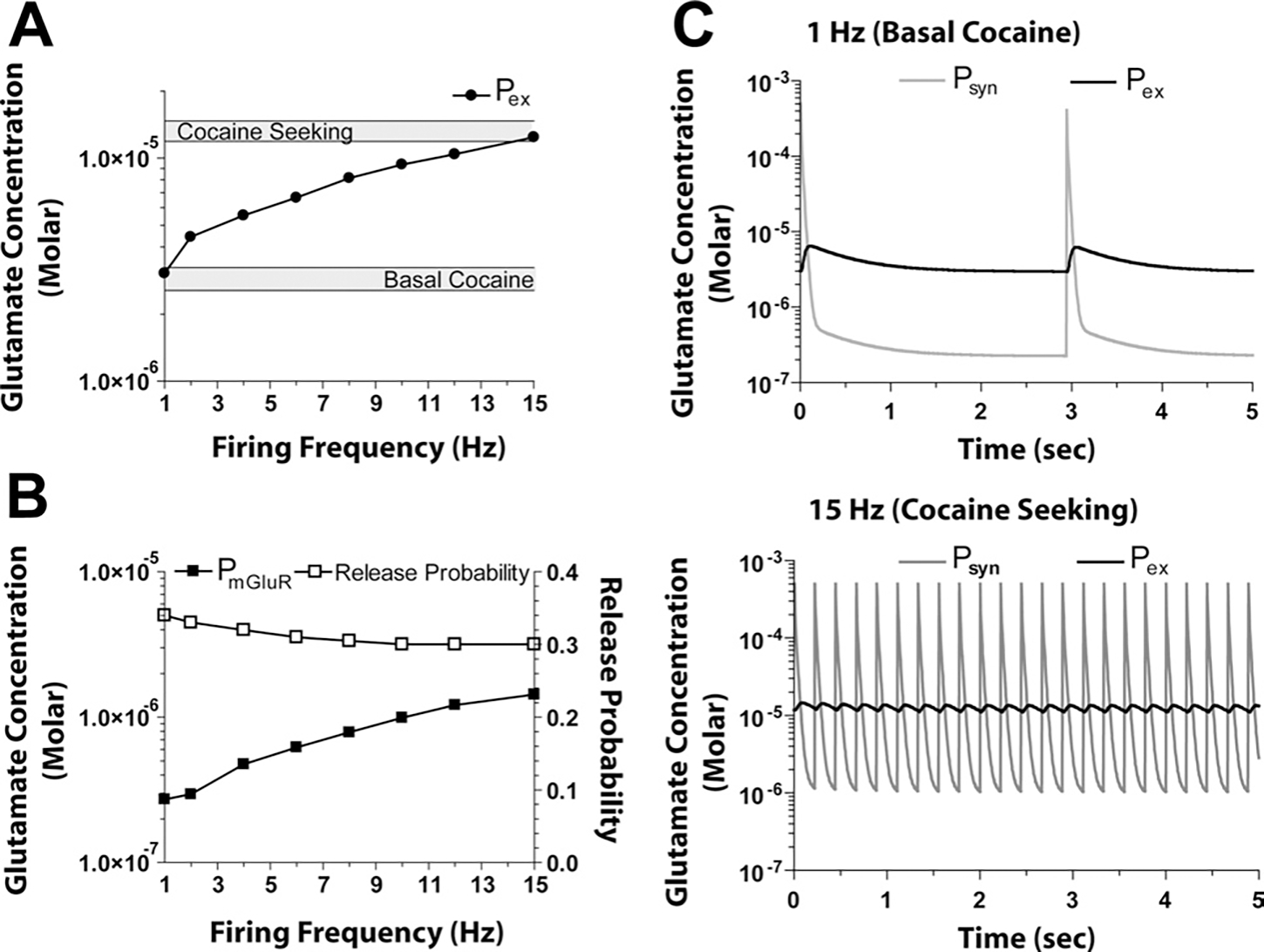
Concentrations of glutamate at three spatial locations under cocaine conditions (i.e. xc− reduced 50%, mGluR2/3 signaling reduced 70%, XAG reduced 40%). (A) The increase in glutamate at was within the basal range at 1 Hz and increases to the cocaine-seeking range at 15 Hz firing frequency. (B) As firing frequency increased, the concentration of glutamate in the vicinity of perisynaptic mGluR2/3 autoreceptors (at ) increased with a concomitant decrease in release probability. (C) Model output at 1 and 15 Hz over 5 s, illustrating the dynamic changes in synaptic (at ), and extracellular concentration (at ).
DISCUSSION
A computational modeling framework for studying glutamate homeostasis in prefrontal glutamatergic synapses onto nucleus accumbens spiny cells is reported that reproduced extracellular glutamate concentrations as measured by in vivo microdialysis. The parameters used include those previously employed in computational models of excitatory neurotransmission, such as synaptic release, diffusion from the synaptic cleft and glutamate uptake, as well as parameters not typically modeled, including xc− and negative feedback on synaptic release by perisynaptic mGluR2/3. These latter parameters were included to model changes in extracellular glutamate concentrations produced by chronic cocaine administration that are hypothesized to result at least in part from cocaine-induced reductions in xc− and mGluR2/3 signaling (Xi et al., 2002; Baker et al., 2003; Moran et al., 2005). The computational model successfully reproduced extracellular concentrations at different firing frequencies in control accumbens. Although incorporating cocaine-induced reductions in xc− and mGluR2/3 signaling reproduced the reduction in concentrations at at low firing frequencies, it was necessary to incorporate a reduction in XAG to predict the large increase at that occurs at the higher firing frequencies achieved during cocaine-seeking. Importantly, recent reports indicate that XAG is reduced in the accumbens after withdrawal from self-administered cocaine, including lower levels of the primary glial transporter protein, GLT-1, and a decrease in3[H]-glutamate uptake (Knackstedt et al., 2007).
Effect of chronic cocaine on glutamatergic transmission
Withdrawal from repeated cocaine administration results in two changes in extracellular glutamate measured by microdialysis: 1) reduced basal concentrations, and 2) increased levels of glutamate after an acute injection of cocaine that induces cocaine-seeking or sensitized motor activity (Pierce et al., 1996; Reid and Berger, 1996; Hotsenpiller et al., 2001; Baker et al., 2003; McFarland et al., 2003; Madayag et al., 2007). Under basal conditions, glutamate measured by microdialysis is almost entirely of nonsynaptic origin (Miele et al., 1996; Timmerman and Westerink, 1997; Melendez et al., 2005), while the increase following a cocaine injection in chronic cocaine-treated animals is of synaptic origin (i.e. blocked by tetrodotoxin (TTX) or inhibiting prefrontal glutamatergic inputs to the accumbens; Pierce et al., 1996; McFarland et al., 2003). Importantly, an increase in extracellular glutamate (either synaptic or nonsynaptic) does not accompany an acute injection of cocaine or operant responding in animals trained to seek natural rewards such as food (Pierce et al., 1996; Hotsenpiller et al., 2001; McFarland et al., 2003). Thus, in the accumbens of animals chronically pretreated with cocaine, synaptic glutamate transmission appears to escape from the immediate synaptic environment and is measured in significant amounts outside of the synaptic region. The overflow of synaptic glutamate in animals withdrawn from cocaine is in contrast to the lack of diffusion by significant amounts of synaptic glutamate to adjacent synapses predicted under physiological conditions by previous mathematical models (Barbour, 2001; Lehre and Rusakov, 2002; Sykova, 2004) or empirically derived using in vivo microdialysis (Miele et al., 1996; Timmerman and Westerink, 1997; Melendez et al., 2005). Thus, it is possible that the cocaine-induced glutamate overflow may be a critical event in addiction. However, stress induces overflow of glutamate in the striatum or prefrontal cortex that is inhibited by TTX (Moghaddam, 2002), indicating that at least some biological stimuli can also induce release of synaptic glutamate measurable by dialysis.
The concentrations of glutamate estimated by the model at and at are presumably capable of stimulating perisynaptic and synaptic glutamate receptors in adjacent synapses, since at 15 Hz firing frequency (e.g. during drug-seeking), the model predicted that the concentration of glutamate at and at is 1.1 and 1.2 μM, respectively, and the estimated values for mGluR2 and NMDA receptors are in the range of 200 nM and 2 μM, respectively (Patneau and Mayer, 1990; Schoepp and True, 1992). Moreover, this concentration of glutamate would be expected to partially desensitize NMDA receptors (Cavelier et al., 2005), and could contribute to the increase in AMPA/NMDA current ratio (Kourrich et al., 2007) and AMPA receptor membrane insertion seen after chronic cocaine (Conrad et al., 2008).
Limitations of the proposed mathematical model
Two general limitations exist in the proposed model. The first limitation is the simplicity of the model relative to the known physiology and cocaine-induced changes in glutamate transmission. Notably, only occupancy of mGluR2/3 is considered, but occupancy of mGluR1 or mGluR5 can be expected to change glutamate release and synaptic scaling (Malenka and Bear, 2004; Kreitzer and Malenka, 2005), and mGluR1/5 content and/or function is altered by chronic cocaine administration (Swanson et al., 2001; Szumlinski et al., 2006). In addition to xc−, there are other sources of nonsynaptic glutamate release that may tonically stimulate glutamate receptors, such as calcium-dependent release from astroglia and release from junction hemi-channels (Danbolt, 2001; Cavelier et al., 2005). Also, multiple parameter variation studies (e.g. simultaneous variations in volume fraction and down-regulation of XAG) should be performed to investigate if they reliably predict the observed changes in extracellular glutamate concentration. Finally, while the glial geometry used in the framework is a reflection of endogenous tortuosity, it oversimplifies the more varied in vivo structural geometry. Thus, future models need to consider additional dynamic cellular processes that accompany alterations in firing frequency, as well as more complicated morphological geometries. In addition, it will be important to examine the model in situations known to inhibit drug-seeking in vivo, such as by activating xc− using N-acetylcysteine (Baker et al., 2003; Madayag et al., 2007).
A second important consideration is that in contrast to the standard mathematical models using postsynaptic currents to empirically validate synaptic concentrations of extracellular glutamate, the present model employed in vivo microdialysis measures. Although the strengths of microdialysis are that estimates are made in vivo and nonsynaptic release is readily determined, microdialysis induces damage artifacts that are distinct from the damage artifacts produced by dissecting tissue for in vitro measurements. Two distinctions between estimates of extracellular glutamate made in vitro versus with in vivo microdialysis are particularly relevant. The first is that previous microdialysis estimates of extraction fraction (i.e. the slope of the line in the no net flux experiment; Bungay et al., 2003) that are used to determine the elimination rate of glutamate in brain tissue by passing different concentrations of glutamate through the probe, found no apparent change in uptake (Baker et al., 2003). In contrast, both [3H]-glutamate uptake and membrane protein content of GLT-1 are reduced ~40% in the accumbens (Knackstedt et al., 2007). Recent modeling of microdialysis concludes that the extraction fraction may not be a reliable estimate of transmitter uptake (Bungay et al., 2003; Chen, 2006). The reasons for this are twofold. 1) The presence of a tissue trauma layer changes the tissue resistance and volume in the vicinity of the dialysis probe. While this markedly affects the estimates of extraction fraction, it does not impact the no net flux estimate of basal transmitter concentration. 2) The distribution of XAG within the present model is based upon data indicating that uptake sites are concentrated in the vicinity of the synaptic cleft (Lehre and Danbolt, 1998; Danbolt, 2001), while nonsynaptic glutamate release via xc− was inversely distributed with the highest concentration of xc− being found away from the synapse (Sato et al., 2002). This distribution of XAG and xc− can contribute to both the lack of TTX sensitivity in basal glutamate levels and the relatively poor capacity to detect uptake-dependent changes in the extraction fraction (Bungay et al., 2003).
The second distinction raised by modeling glutamate transmission based upon microdialysis measurements relative to in vitro measures is revealed by estimates of extracellular glutamate using NMDA currents in tissue slices being one to three orders of magnitude less than dialysis measurements (Cavelier et al., 2005; Herman and Jahr, 2007). However, this fact is largely incorporated into the proposed model that contains a steep gradient of glutamate concentrations between the synapse ( where the electrophysiological measures are obtained) and the site where the dialysis measurements occur ().
CONCLUSIONS
A computational framework of glutamate transmission is presented that incorporates both synaptic and nonsynaptic glutamate release and homeostatic regulation of synaptic release via stimulation of mGluR2/3 autoreceptors. This model accurately reproduced the basal levels of extracellular glutamate measured by microdialysis, as well as the levels of glutamate in the vicinity of mGluR2/3 that provide inhibitory tone on synaptic release. However, in order to achieve changes in extracellular glutamate observed during cocaine seeking, the model required a 40% downregulation of XAG. Thus, this model provides a general mathematical framework for describing how pharmacological or pathological conditions influence glutamate transmission, and for predicting potential cocaine-induced neuroadaptations (e.g. reduced XAG) that may be important to experimentally evaluate.
Acknowledgments—
This research was supported in part by USPHS grants DA015369, DA03906 (P.W.K.), and subcontract from DA015369 to University of Missouri (S.S.N.).
Abbreviations:
- Dsyn,Dsh and Dex
diffusion coefficient in the synapse, between the sheath and extracellular space
- Gi
glial sheath
- GLT-1
glutamate transporter protein
- mGluR2/3
metabotropic glutamate receptors
- NMDA
N-methyl-d-aspartic acid
- PFC
prefrontal cortex
- Psyn,PmGluR, and Pex
glutamate concentrations at synapse, mGluR and extracellular space
- TTX
tetrodotoxin
- XAG
glutamate transporters
- xc
cystine-glutamate exchange
REFERENCES
- Alagarsamy S, Sorensen SD, Conn PJ (2001) Coordinate regulation of metabotropic glutamate receptors. Cur Opin Neurobiol 11:357–362. [DOI] [PubMed] [Google Scholar]
- Allen C, Stevens CF (1994) An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci USA 91:10380–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW (2002) The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 22:9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW (2003) Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6:743–749. [DOI] [PubMed] [Google Scholar]
- Bandrowski AE, Huguenard JR, Prince DA (2003) Baseline glutamate levels affect group I and II mGluRs in layer V pyramidal neurons of rat sensorimotor cortex. J Neurophysiol 89:1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B (2001) An evaluation of synapse independence. J Neurosci 21:7969–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Hausser M (1997) Intersynaptic diffusion of neurotransmitter. Trends Neurosci 20:377–384. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE (1997) Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19:1297–1308. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE (1998) Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in hippocampus. J Neurosci 18:7709–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE (1999) Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol 9:293–298. [DOI] [PubMed] [Google Scholar]
- Billups B, Graham BP, Wong AY, Forsythe ID (2005) Unmasking group III metabotropic glutamate autoreceptor function at excitatory synapses in the rat CNS. J Physiol (Lond) 565:885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, Lanier SM, Kalivas PW (2004) Activator of G-protein signaling 3: a gatekeeper of cocaine sensitization and drug-seeking. Neuron 42:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D, Jahn R (1995) Real-time measurement of transmitter release from single synaptic vesicles. Nature 377:62–65. [DOI] [PubMed] [Google Scholar]
- Bungay PM, Newton-Vinson P, Isele W, Garris PA, Justice JB Jr (2003) Microdialysis of dopamine interpreted with quantitative model incorporating probe implantation trauma. J Neurochem 86:932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Hamann M, Rossi D, Mobbs P, Attwell D (2005) Tonic excitation and inhibition of neurons: ambient transmitter sources and computational consequences. Prog Biophys Mol Biol 87:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Zhang L, Janak PH, Woodward DJ (1997) Neuronal responses in prefrontal cortex and nucleus accumbens during heroin self-administration in freely moving rats. Brain Res 754:12–20. [DOI] [PubMed] [Google Scholar]
- Chen KC (2006) Effects of tissue trauma on the characteristics of microdialysis zero-net-flux method sampling neurotransmitters. J Theor Biol 238:863–881. [DOI] [PubMed] [Google Scholar]
- Cholet N, Pellerin L, Magistretti PJ, Hamel E (2002) Similar perisynaptic glial localization for Na+, K+-ATPase alpha 2 subunit and the glutamate transporters GLAST and GLT-1 in the somatosensory cortex. Cereb Cortex 12:515–525. [DOI] [PubMed] [Google Scholar]
- Clements JD, Robin A, Lester J, Tong G, Jahr CE, Westbrook GL (1992) The time course of glutamate in the synaptic cleft. Science 258:1498–1501. [DOI] [PubMed] [Google Scholar]
- Clements JD (1996) Transmitter time course in synaptic cleft: its role in central synaptic function. Trends Neurosci 19:163–171. [DOI] [PubMed] [Google Scholar]
- Colombo JA (2005) Glutamate uptake by rat brain astroglia incubated in human cerebrospinal fluid. Med Sci Monitor 11:BR13–BR17. [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Hen LJ, Shaham Y, Marinelli M, Wolf ML (2008) Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine seeking. Nature 454(7200):118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105. [DOI] [PubMed] [Google Scholar]
- Diamond JS (2005) Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci 25:2906–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (2000) Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol 83:2835–2843. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Kral T, Clusmann H, Friedl M, Schramm J (2002) Presynaptic group II metabotropic glutamate receptors reduce stimulated and spontaneous transmitter release in human dentate gyrus. Neuropharmacology 42:297–305. [DOI] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ (2005) Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci 25:936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon P (2001) Glia: listening and talking to the synapse. Nat Neurosci 2:185–191. [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM (2007) Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J Neurosci 27:3535–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotsenpiller G, Giorgetti M, Wolf ME (2001) Alterations in behaviour and glutamate transmission following presentation of stimuli previously associated with cocaine exposure. Eur J Neurosci 14:1843–1855. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U (1999) Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci U S A 96:8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J (2005) Unmanageable motivation in addiction: pathology in prefrontal-accumbens glutamate transmission. Neuron 45:647–650. [DOI] [PubMed] [Google Scholar]
- Kleinle J, Vogt K, Luscher H-R, Muller R, Senn W, Wyler K, Streit J (1996) Transmitter concentrations profiles in the synaptic cleft: an analytical model of release and diffusion. Biophys J 71:2413–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez R, Kalivas PW (2007) Cocaine self-administration alters the expression of proteins associated with glutamatergic transmission and homeostasis at cortico-accumbens synapses. Society for Neuroscience annual meeting, San Diego, CA: Abstract 815.8. [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007) Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27:7921–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2005) Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci 25:10537–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15:1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Rusakov D (2002) Asymmetry of glia near synapse favors presynaptic glutamate escape. Biophys J 83(1):125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Anulo MC, Audinat E (2007) Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol 580:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Somogyi P, Nusser Z (2003) Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J Neurophysiol 89:1910–1919. [DOI] [PubMed] [Google Scholar]
- Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, Grier MD, Baker DA (2007) Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci 27:13968–13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21. [DOI] [PubMed] [Google Scholar]
- McBean GJ (2002) Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci 23:299–302. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23:3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW (2004) Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci 24:1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, Vuthiganon J, Kalivas PW (2005) Regulation of extracellular glutamate in the prefrontal cortex: focus on the cystine glutamate exchanger and group I metabotropic glutamate receptors. J Pharmacol Exp Ther 314:139–147. [DOI] [PubMed] [Google Scholar]
- Miele M, Boutelle MG, Fillenz M (1996) The source of physiologically stimulated glutamate efflux from the striatum of conscious rats. J Physiol (Lond) 497:745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B (2002) Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry 51:775–787. [DOI] [PubMed] [Google Scholar]
- Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK (2005) Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci 25:6389–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ (1997) Heterogeneous release properties of visualized individual hippocampal synapses. Neuron 18:599–612. [DOI] [PubMed] [Google Scholar]
- Nicholson C (2001) Diffusion and related transport mechanism in brain tissue. Rep Prog Physics 64:815–884. [Google Scholar]
- Nicholson C, Sykova E (1998) Extracellular space structure revealed by diffusion analysis. Trends Neurosci 21:207–215. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML (1990) Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J Neurosci 10:2385–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters YM, O’Donnell P, Carelli RM (2005) Prefrontal cortical cell firing during maintenance, extinction, and reinstatement of goal-directed behavior for natural reward. Synapse 56(2):74–83. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW (1996) Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16(4):1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow DV (2001) Visualizing the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia 34:27–38. [DOI] [PubMed] [Google Scholar]
- Reid MS, Berger SP (1996) Evidence for sensitization of cocaine-induced nucleus accumbens glutamate release. Neurosci Lett 7:1325–1329. [DOI] [PubMed] [Google Scholar]
- Rice ME, Nicholson C (1991) Diffusion characteristics and extracellular volume fraction during normoxia and hypoxia in slices of rat neostriatum. J Neurophysiol 65(2):264–272. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B (2004) Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47:33–46. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM (1998) Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci 18:3158–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA (2001) The role of perisynaptic glial sheaths in glutamate spillover and extracellular Ca2+ depletion. Biophys J 81:1947–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftenku EE (2005) Modeling of slow glutamate diffusion and AMPA receptor activation in the cerebellar glomerulus. J Theor Biol 234:363–382. [DOI] [PubMed] [Google Scholar]
- Sato K, Keino-Masu K, Masu M, Bannai S (2002) Distribution of cystine/glutamate exchange transporter, system xc−, in the mouse brain. J Neurosci 22:8028–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, True RA (1992) 1S-3R-ACPD-sensitive (metabotropic) [3H] glutamate receptor binding in membranes. Neurosci Lett 145:100–104. [DOI] [PubMed] [Google Scholar]
- Sun W, Rebec GV (2006) Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci 26:8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CJ, Baker DA, Carson D, Worley PF, Kalivas PW (2001) Repeated cocaine administration attenuates group I metabotropic glutamate receptor-mediated glutamate release and behavioral activation: A potential role for Homer1b/c. J Neurosci 21:9043–9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykova E (2004) Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience 129:861–876. [DOI] [PubMed] [Google Scholar]
- Szumlinski KK, Abernathy KE, Oleson EB, Kullmann M, Lominac KD, He DY, Ron D, During M, Kalivas PW (2006) Homer isoforms differentially regulate cocaine-induced neuroplasticity. Neuropsychopharmacology 4:768–777. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Nicholson C (2006) In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci U S A 103:5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman W, Westerink BH (1997) Brain microdialysis of GABA and glutamate: what does it signify? Synapse 27:242–261. [DOI] [PubMed] [Google Scholar]
- Trantham H, Szumlinski K, McFarland K, Kalivas PW, Lavin A (2002) Repeated cocaine administration alters the electrophysiological properties of prefrontal cortical neurons. Neuroscience 113:749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommershauser J, Schneggenburger R, Zippelius A, Neher E (2003) Heterogeneous presynaptic release probabilities: functional relevance for short-term plasticity. Biophys J 84:1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volynski KE, Rusakov DA, Kullmann DM (2006) Presynaptic fluctuations and release-independent depression. Nat Neurosci 9:1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP (1995) Kinetics of a human glutamate transporter. Neuron 14:1019–1027. [DOI] [PubMed] [Google Scholar]
- Warr O, Takahashi M, Attwell D (1999) Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol 514:789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt I, Gyte A, Simpson MG, Widdowson PS, Lock EA (1996) The role of glutathione in L-2-chloropropionic acid induced cerebellar granule cell necrosis in the rat. Arch Toxicol 70(11):724–735. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Baker DA, Shen H, Carson DS, Kalivas PW (2002) Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J Pharmacol Exp Ther 300:162–171. [DOI] [PubMed] [Google Scholar]