

Abstract
Objective:
Alcohol use in Metabolic dysfunction-associated steatohepatitis (MASH) is associated with increased risk of fibrosis and liver-related death. Here, we aimed to identify mechanism through which repeated alcohol binges exacerbate liver injury in a high fat-cholesterol-sugar diet (MASH diet) induced model of MASH.
Design:
C57BL/6 mice received either chow or MASH diet for 3 months with or without weekly alcohol binges. Neutrophil infiltration, neutrophil extracellular traps (NETs), and fibrosis were evaluated.
Results:
We found that alcohol binges in MASH increase liver injury and fibrosis. Liver transcriptomic profiling revealed differential expression of genes involved in extracellular matrix reorganization, neutrophil activation, and inflammation compared to alcohol or MASH diet alone. Alcohol binges specifically increased NETs formation in MASH livers in mice and NETs were also increased in human livers with MASH plus alcohol use. We discovered that cell-free NETs are sensed via NLRP3. Furthermore, we show that cell-free NETs in vitro induce a pro-fibrotic phenotype in hepatic stellate cells (HSCs) and pro-inflammatory monocytes. In vivo, neutrophil depletion using anti-Ly6G antibody or NETs disruption with DNase treatment abrogated monocyte and HSCs activation and ameliorated liver damage and fibrosis. In vivo, inhibition of NLRP3 using MCC950 or NLRP3 deficiency attenuated NETs formation, liver injury and fibrosis in MASH plus alcohol diet-fed mice (graphical abstract).
Conclusion:
Alcohol binges promote liver fibrosis via NET-induced activation of hepatic stellate cells and monocytes in MASH. Our study highlights the potential of inhibition of NETs and/or NLRP3, as novel therapeutic strategies to combat the pro-fibrotic effects of alcohol in MASH.
Keywords: NETs, MetALD, ALD, stellate cell, neutrophils, fibrosis, macrophage, inflammasome, inflammation
INTRODUCTION
Alcohol-associated liver disease (ALD) and Metabolic dysfunction-associated Steatotic liver disease (MASLD) account for the majority of chronic liver diseases worldwide and represent therapeutic challenges.(1) Obesity and associated metabolic syndrome often coexist with alcohol consumption and is now recognized as a new disease phenotype, named as MetALD.(2–4) MetALD represents a separate group of patients with MASLD who consume alcohol in the amount of 140–350 g/week for females and 210–420 g/week for males and recognizes that MetALD can be either MASLD or ALD predominant.(4) Recent epidemiology studies found that excessive alcohol use in MASLD exacerbates the risk of liver fibrosis and hepatocellular carcinoma (HCC) (5) and can serve as prognostic cofactors for long-term morbidity and mortality.(5–7)
ALD and MASH share common pathological and histological stages that range from steatosis, hepatitis, to fibrosis/cirrhosis, and increased risk of HCC.(8, 9) The distinguishing histological features of alcoholic hepatitis (AH) include the abundance of infiltrating neutrophils surrounding ballooned hepatocytes and Mallory-Denk bodies.(8, 10) Despite sufficient information about the respective pathogenesis of MASH and ALD, the mechanisms governing the combined insult of MASH and ALD remain unclear.
Increased neutrophil infiltration in the liver is one of the hallmarks of ALD, which contributes to liver damage.(10, 11) Damaged hepatocytes and activated Kupffer cells secrete chemokines such as CXC motif ligand (CXCL)1, which recruit neutrophils at the site of injury.(11) Neutrophils also release neutrophil extracellular traps (NETs) that are composed of condensed chromatin and granular proteins such as citrullinated histone (cit-H3) and neutrophil elastase (NE), respectively.(11) Previously, we have shown that binge alcohol consumption leads to the spontaneous formation of NETs in the liver and promotes liver damage and inflammation.(12, 13)
Nod-like receptor protein 3 (NLRP3) inflammasome complex, including caspase 1 and IL-1β, was shown to be involved in both ALD and MASH.(14) NLRP3 is expressed in various cell types in the liver, including hepatocytes, hepatic stellate cells (HSCs), macrophages, and neutrophils.(14, 15) NLRP3 overexpression in HSCs, the major cell type contributing to liver fibrosis, leads to activation and differentiation of quiescent HSCs into myofibroblasts,(16, 17) extracellular matrix deposition and fibrosis.(17)
Here, we report that repeated alcohol binges in a mouse model of MASH accelerate liver damage, inflammation, and fibrosis. Alcohol binges in MASH diet-fed mice result in transcriptomic signatures of extracellular matrix reorganization and inflammation. Notably, alcohol binges increase neutrophil infiltration in MASH liver. We discovered that cell-free NETs, sensed by NLRP3, lead to the activation of HSCs and promote pro-inflammatory monocyte phenotypes. Our study demonstrates that in vivo disruption of NETs by DNase, or depletion of neutrophils using anti-Ly6G antibody ameliorates alcohol-induced liver fibrosis in MASH.
RESULTS
1. Alcohol binges augment liver injury in MASLD/MASH mice
To study the pathomechanism of the combined insult of MASH plus alcohol on the liver, we administered weekly alcohol binges in mice fed a MASH diet for 3 months (Fig1A, S1A). The MASH diet resulted in a significant weight gain compared to chow- or alcohol groups; mice on MASH diet plus alcohol binges had lower body weight compared to MASH diet alone (Fig1B-C). Blood alcohol levels were comparable between mice that received alcohol binges with or without the MASH diet (Fig S1B).
Fig. 1: Alcohol binges accelerate liver injury in MASH diet fed mice.
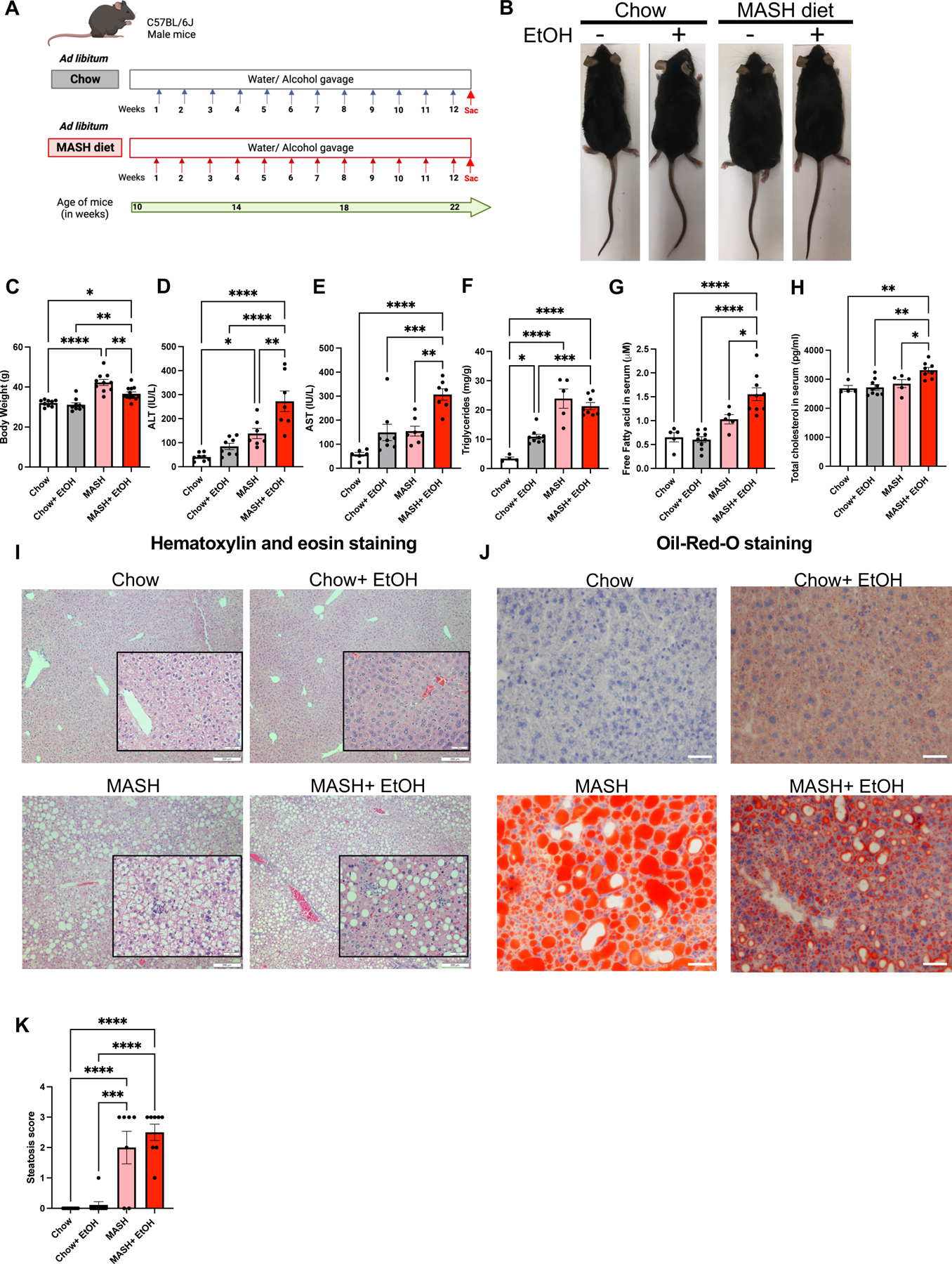
(A) Feeding schematics for combined liver injury. C57BL/6 wild type (WT) mice (n=10) were fed on MASH diet plus alcohol binges with a single diet as a control for 12 weeks. (B-C) Representative images of mice and the body weight of mice are shown as a bar graph. (D-E) ALT & AST levels were measured from serum. (F) Triglyceride levels were measured from the liver. (G-H) Free fatty acid and total cholesterol was measured from the serum. Formalin-fixed liver sections were stained with (I) Hematoxylin and eosin, scale bar=200 μm (Insets scale bar=50 μm) (J) Oil-red-O stain, scale bar=50 μm, and representative slides are shown. (K) Steatosis score is shown as graph * p≤0.05, **p<0.005, ***p<0.0005, ****p<0.00005. n=4–8 mice/group.
Liver injury in MASH diet-fed mice was indicated by increased ALT and AST as compared to chow controls (Fig1D-E). Alcohol binges further increased transaminase levels in MASH mice compared to those with either the MASH diet or alcohol binges alone (Fig1D-E).
Hepatic steatosis was evident by increased triglyceride levels (Fig1F), free fatty acid (Fig 1G), total cholesterol (Fig1H), histologic section on H&E (Fig1I) and Oil-Red-O staining (Fig1J) in MASH and MASH plus alcohol groups (Fig1F-K and Fig S1C). There was elevation in fasting glucose levels in mice with MASH diet with or without alcohol binges compared to their respective controls (FigS1D), while only MASH mice developed significant insulin resistance when compared to the chow diet-fed mice (FigS1E).
2. Alcohol binges exacerbate fibrosis in MASH diet fed mice
Because both ALD and MASH progress to liver fibrosis,(17) we hypothesized that combined liver injury by alcohol and MASH diet accelerates fibrosis and promotes disease progression. In support of this hypothesis, Sirius red staining demonstrated increased fibrosis in MASH plus alcohol-administered mice compared to the MASH-diet or alcohol alone groups (Fig2A-B).
Fig 2. Alcohol binges exacerbate fibrosis in MASH mice.
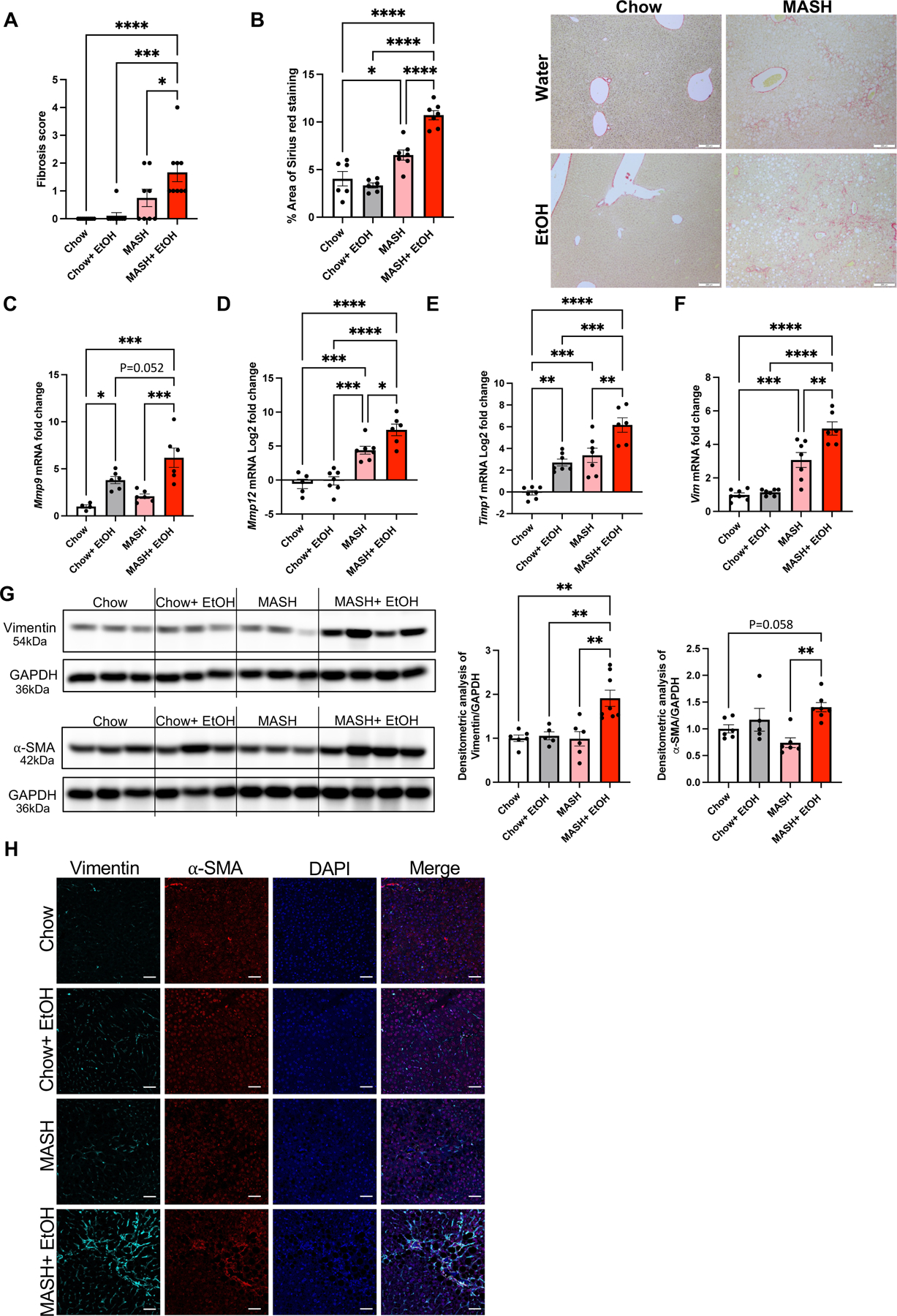
(A) Histological scoring of fibrosis. (B) Formalin-fixed liver sections were stained with Sirius red stain and representative slides are shown. (n=6–7 mice/group and average of 3–5 images per mice). Scale bar=200 μm. The percentage area of Sirius red staining is quantified using Image J. Liver RNA was used to determine Mmp9 (C), Mmp12 (D), Timp1 (E), and Vim (F) mRNA levels by qPCR (n=6–7 mice/group). 18s was used to normalize Cq values. (G) Liver lysates were analyzed by western blotting for α-SMA and vimentin, using GAPDH as a loading control. The densitometric analysis of α-SMA and vimentin. (n=5–8 mice/group). (H) Co-immunofluorescence staining with vimentin and α-SMA in mouse liver. Scale bar=50 μm. * p≤0.05, **p<0.005, ***p<0.0005, ****p<0.00005
Matrix metalloproteinases (MMPs) and metallopeptidase inhibitors (TIMP1) are involved in matrix remodeling and contribute to liver fibrosis.(17) The mRNA levels of Mmp9, Mmp12, Timp1, and Vimentin were significantly upregulated in mice with MASH plus alcohol compared to their respective controls (Fig2C-F). Profibrotic markers such as α-SMA and vimentin, as assessed by Western blots (Fig2G and FigS2A-B) and immunofluorescence (Fig2H), were significantly upregulated in the MASH plus alcohol group compared to all other groups. These results suggest that alcohol binges exacerbate liver fibrosis in diet-induced MASH.
3. Transcriptomic analysis shows differential expression of signaling pathways in combined liver injury.
Next, we evaluated liver mRNA on the nCounter fibrosis panel. Heatmap showed clear segregation of the differentially expressed genes (DEGs) in MASH plus alcohol compared to their respective controls (Fig3A). Indeed, we found 280 genes, with p≤0.05, that were differentially expressed in the MASH plus alcohol group as compared to chow (Supplementary File 2). In the alcohol- and MASH-only groups, 260 and 215 genes were differentially expressed, respectively (Supplementary File 2). Furthermore, approximately 11% of DEGs in the MASH plus alcohol group were unique to this group, as shown in the Venn diagram (Fig3B). There was a substantial number of upregulated genes in the MASH plus alcohol as compared to MASH alone or alcohol alone, as evident from the volcano plot (Fig3C, S3A-3B, Supplementary File 2). GO term analysis, as depicted by the bubble plot, highlighted the different biological processes that are upregulated in the MASH plus alcohol group and in respective controls (Fig3D, S3C-S3D). Functional analysis of DEGs revealed upregulation of pathways associated with extracellular matrix reorganization and inflammation in the MASH plus alcohol compared to alcohol or MASH alone (Fig3E-F). Interestingly, genes associated with neutrophils and their activation, such as neutrophil Fcγ receptor genes (Fcer), Cxcl1, Bst2 were upregulated in MASH plus alcohol as compared to chow (Fig3G). In alcohol alone group, neutrophil-associated genes, Cxcl1, Arg1, and Cd14 were the only upregulated genes (FigS3D). These data indicated that alcohol promotes disease pathogenesis in MASH by modulating signaling pathways related to neutrophil infiltration and extracellular-matrix remodeling.
Fig 3. Differential expression of signaling pathways in MASH plus alcohol binges.
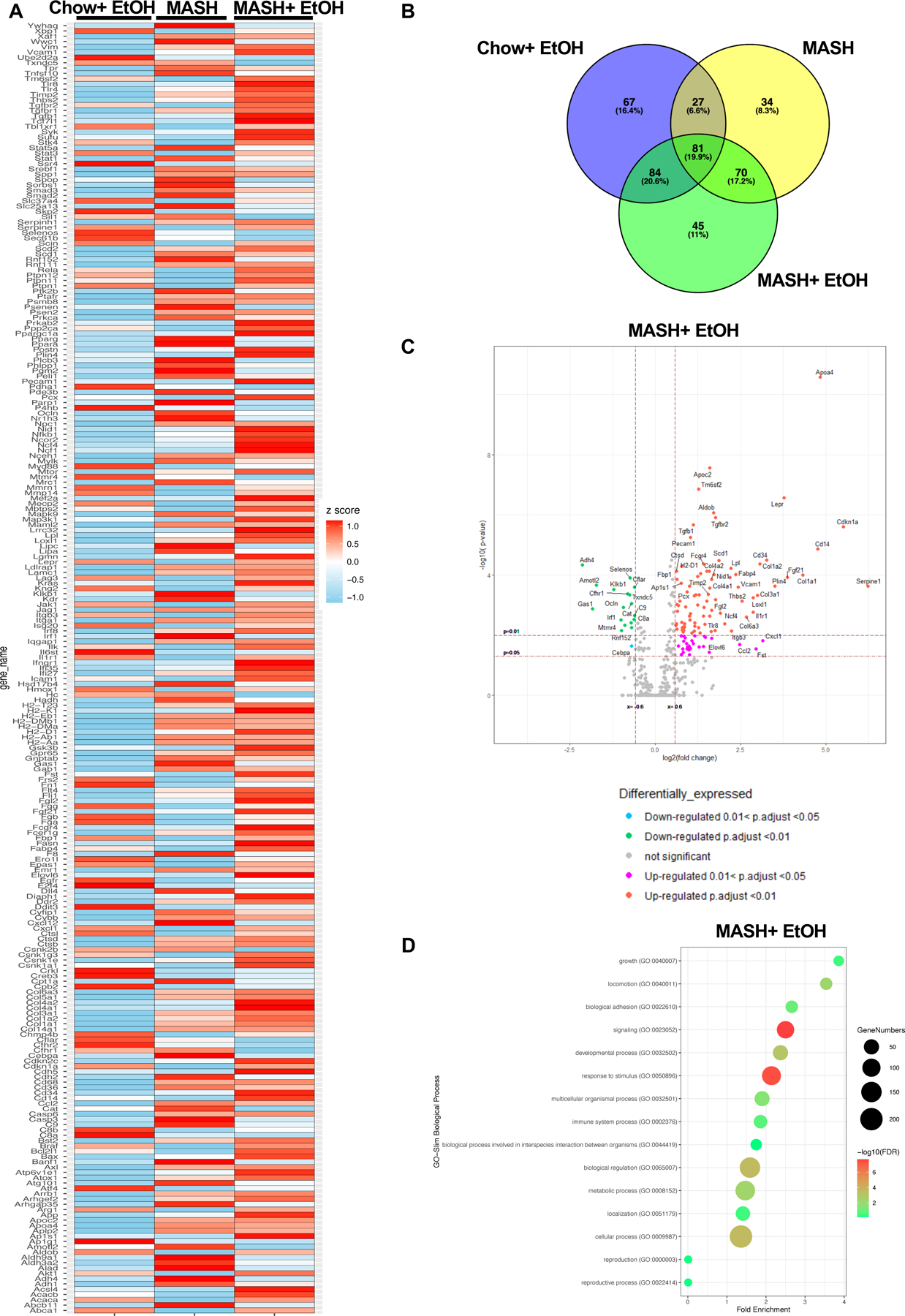
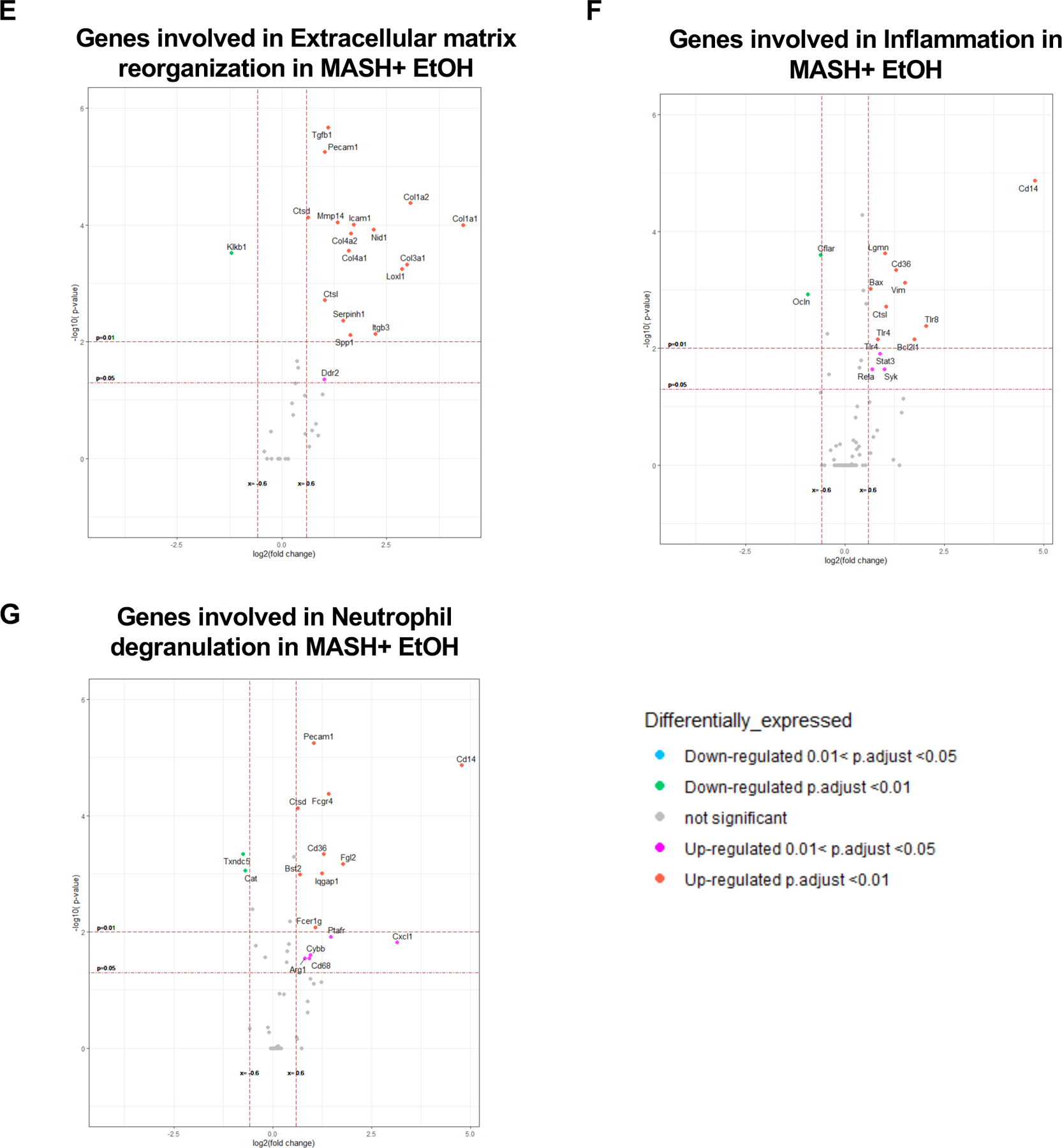
(A) Heatmap of differentially expressed genes (DEGs) in alcohol only, MASH only, and MASH plus alcohol with p≤0.05, and normalized to chow. (n=6mice/group). (B) Venn diagram showing the unique DEGs in each group as compared to chow. (C) Volcano plot depicting DEGs in MASH plus alcohol. (D) Bubble plot depicting GO term analysis of the biological process. Volcano plot depicting DEGs involved in extracellular matrix reorganization (E), inflammation (F), and neutrophil degranulation (G) in MASH plus alcohol as compared to chow.
4. Alcohol binges in MASH specifically promote neutrophil infiltration and Neutrophil Extracellular Traps (NETs) formation in the liver
Prominent neutrophil infiltration in the liver is a hallmark of ALD.(11) In our MASH model alcohol binges resulted in a significant increase in genes involved in neutrophil infiltration (Fig3D). Consistent with this, we found a significant increase in Ly6G+ cells in the livers of mice that received both alcohol and MASH diet, compared to alcohol or MASH diet alone (Fig4A-B).
Fig 4. Alcohol binges promote neutrophil infiltration and neutrophil extracellular trap formation in MASH mice.
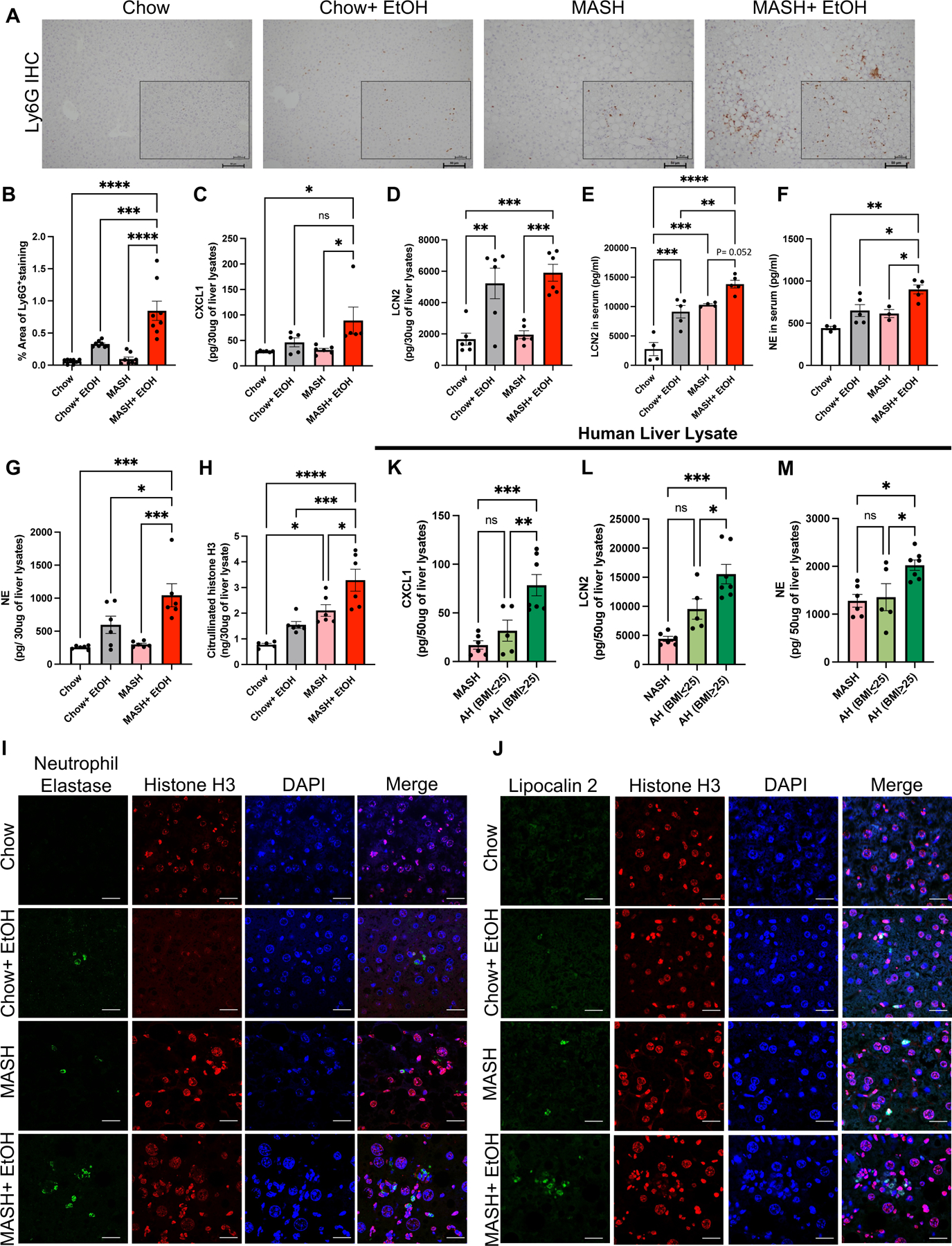
(A) Representative Ly6G immunohistochemistry images from liver sections. Scale bar=50 μm (n=8 mice/group and average of 3–5 images per mice). (B) Quantification of percentage area of Ly6G+ positive cells using Image J. Whole-cell liver lysates were used to detect CXCL1(C), LCN2 (D), NE (G) and Cit-H3 (H) by ELISA. (n=3–8 mice/group). (E-F) Levels of LCN2 and NE in serum as measured by ELISA (n=3–5mice/group). (I-J) Co-immunofluorescence staining with NE and Histone H3 (I) and with LCN2 and Histone H3 (J) to visualize NETs production in mouse liver. scale bar=50 μm. Whole-cell human liver lysates were used to detect CXCL1(K), LCN2 (L), and NE (M) by ELISA. (n=6 patients/MASH, n=5 patients/AH (BMI≤25) and n=7 patients/AH (BMI≥25)). * p<0.05, **p<0.005, ***p<0.0005, ****p<0.0000
Neutrophil recruitment can be triggered by chemokines released from damaged cells in the liver; therefore, we hypothesized that increased neutrophil infiltration was due to the increased expression of chemokines in liver. Consistent with this hypothesis, we observed a significant increase in CXCL1 and lipocalin2 (LCN2) levels in the liver lysates of mice fed on MASH diet plus alcohol compared to the MASH diet alone (Fig4C-E). Alcohol alone increased LCN2 expression in the serum and liver lysate which was further exacerbated in liver lysates and serum of MASH plus alcohol mice (Fig4D-E). These results suggest that alcohol binges in MASH increase chemokine production that correlate with neutrophil infiltration.
To explore the mechanism by which alcohol-induced neutrophil infiltration contributes to liver injury, we evaluated neutrophil extracellular trap (NETs) formation. Neutrophil elastase (NE) and citrullinated-histone 3 (cit-H3) are used as markers of NETs formation (also known as NETosis).(13, 18, 19) We found a significant increase in the levels of NE in serum and liver in the MASH plus alcohol group (Fig4F-G). Furthermore, cit-H3 levels were also increased in the liver, suggesting enhanced NETs formation in MASH diet plus alcohol mice compared to MASH diet or alcohol alone (Fig4H). Notably, we detected increased NE and LCN2 immunofluorescence in the liver sections of the MASH diet plus alcohol mice confirming NETs (Fig4I-J).
To delineate mechanisms by which alcohol induces NETs, we inhibited aldehyde dehydrogenase using disulfiram or treated neutrophils with antioxidant N-acetylcysteine (NAC) that both prevented alcohol-induced NETs formation (FigS4A). Furthermore, acetaldehyde, a metabolite of alcohol, directly induced NETs formation (FigS4A), suggesting NETs formation is induced by reactive oxygen species, alcohol and its metabolites.
To validate findings in mice, we next assessed livers from patients with ALD. Notably, in ALD patients with BMI ≥ 25, CXCL1, LCN2 expression and NE were significantly higher compared to patients with BMI≤25 or NASH with no history of alcohol use (Fig4K-M) indicating greater neutrophil activation in ALD the presence of increased BMI.
Altogether, these results suggest that alcohol binges induce increased neutrophil infiltration and NETs release in MASH, in the liver of mice and in AH patients with BMI≥25.
5. NETs directly activate hepatic stellate cells via NLRP3 sensing
The fibrillar structure of NETs is composed of DNA and proteins such as High mobility group box 1 (HMGB1), and histones which can be recognized as Damage-associated molecular patterns (DAMPs); however, the exact mechanism for NETs recognition is not fully explored.(20) As we found a significant increase in NETs formation in the MASH plus alcohol group, we hypothesized that NETs could act as DAMPs to activate stellate cells and thereby promote fibrosis.
To test this hypothesis, human blood neutrophils were isolated and NETs were induced in vitro using alcohol or palmitic acid (PA) (Fig5A). NETosis was quantified by measuring NET-associated neutrophil elastase and by western blot of cit-H3. Both alcohol and PA stimulation resulted in increased NETs formation (FigS4B and S4C).
Fig 5. In vitro neutrophil extracellular traps activate stellate cells activation via NLRP3.
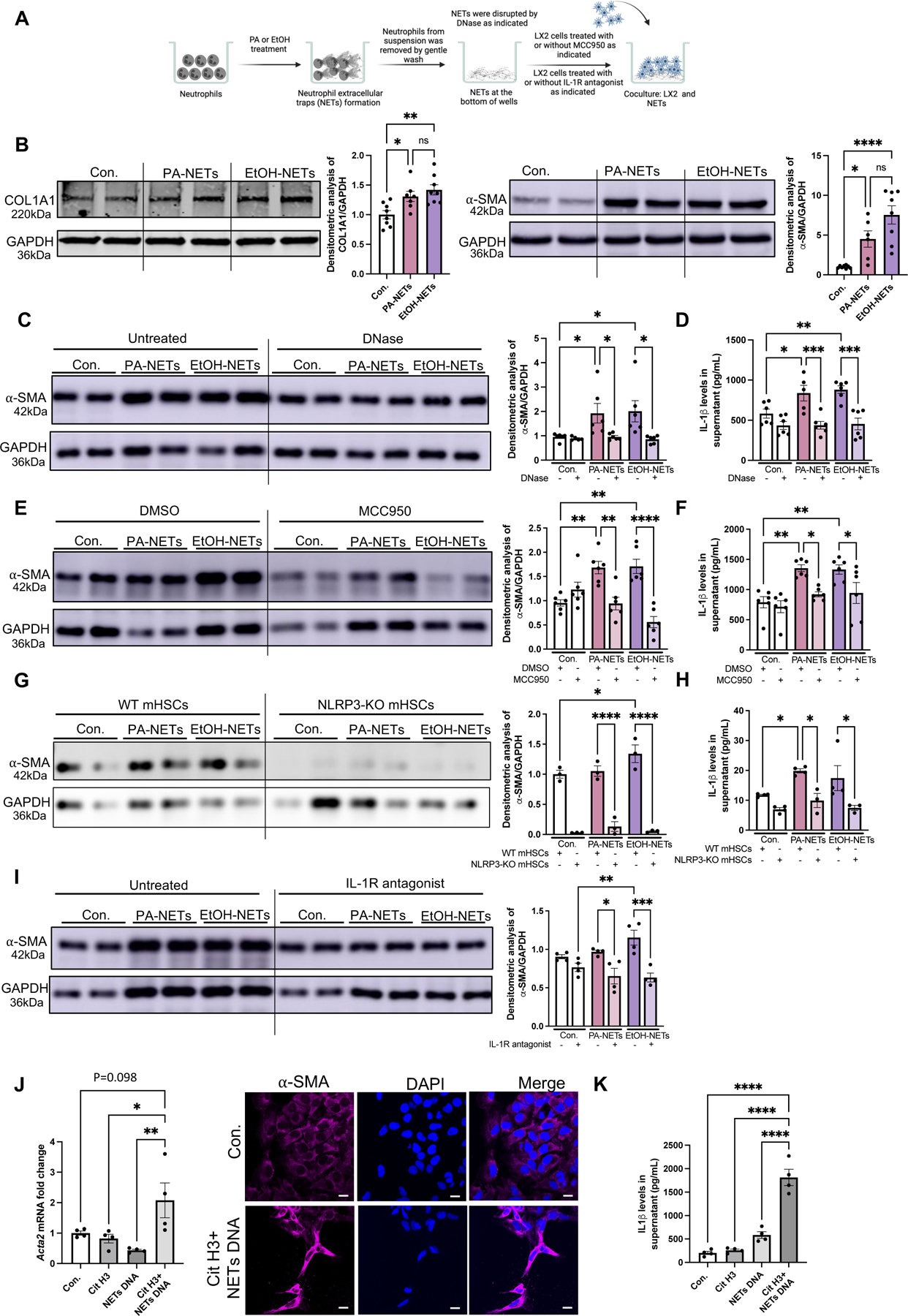
(A) Human neutrophils were treated with PA or EtOH for NETs formation, followed by coculturing NETs with LX2 cells. (B) LX2-lysates were used to detect α-SMA and COL1A1 by Western blot. The densitometry analysis is shown as bar graph (n=8). (C) LX2 lysates after coculturing with DNase treated NETs were used to detect α-SMA by Western blot. The densitometry analysis is shown as bar graph (n=6). (D) LX2 supernatant after coculturing with DNase treated NETs were used to detect IL-1β by ELISA (n=6). (E) EtOH/PA-induced NETs were cocultured with MCC950-treated LX2 cells. LX2 cell lysates were used to detect α-SMA by western blot. The densitometry analysis is shown as bar graph (n=6). (F) MCC950/DMSO-treated LX2 supernatant after coculturing with EtOH/PA NETs were used to detect IL-1β by ELISA (n=6). (G) EtOH/PA-induced NETs from WT neutrophils were cocultured with WT mouse HSCs or NLRP3-KO HSCs. The densitometry analysis is shown as bar graph (n=3). (H) WT mouse HSCs or NLRP3-KO HSCs supernatants after coculturing with EtOH/PA NETs were used to detect IL-1β by ELISA. The densitometry analysis is shown as bar graph (n=3). (I) EtOH/PA-induced NETs were cocultured with IL-1 receptor antagonist-treated LX2 cells. LX2 cell lysates were used to detect α-SMA by western blot. The densitometry analysis is shown as bar graph (n=4). (J) LX2 cells RNA after culturing with recombinant cit-H3, NETs DNA or combination of cit-H3 and NETs DNA (n=4). Acta2 mRNA levels was determined by qPCR. 18s was used to normalize Cq values. Immunofluorescence staining with α-SMA and DAPI after culturing LX2 cells with recombinant cit-H3, and NETs DNA, scale bar=50 μm. (K) LX2 supernatant after treating LX2 cells with cit-H3, NETs DNA or combination of citH3 and NETs DNA were used to detect IL-1β by ELISA (n=4). * p<0.05, **p<0.005, ***p<0.0005, ****p<0.00005
To evaluate the functional role of NETs, neutrophils in the supernatant were removed, and cell-free NETs were cocultured with LX2 cells for 24h (Fig5A). We found that both PA- and ethanol-induced cell-free NETs increased expression of α-SMA and COL1A1 (Fig5B) as well as IL-1β release from LX2 cells (Fig5D). To assess the specificity of cell-free NETs-induced HSCs activation, we treated NETs with Deoxyribonuclease I (DNase) before co-culturing with LX2 cells. We found that disruption of the DNA structure with DNase partially abrogated the NET-induced increase in α-SMA levels and IL-1β release in LX2 cells (Fig5C and 5D).
The NLRP3 inflammasome can be activated by intermediate filaments such as vimentin and by the fibrillar structure of β-amyloid.(21, 22) Therefore, we hypothesized that activation of HSCs by cell-free NETs, that have a fibrillar structure, might be mediated by the NLRP3 inflammasome. To test this hypothesis, we first evaluated NLRP3 expression in LX2 cells after treating them with PA, ethanol or TGF-β as a positive control.(23) We observed a significant increase in the NLRP3 protein expression as shown FigS5A. Thereafter, we cocultured NETs with LX2 cells in the presence or absence of MCC950, an NLRP3 inflammasome inhibitor. Upon NETs exposure, LX2 cells treated with MCC950 showed a significant decrease in α-SMA production and IL-1β release as compared to untreated cells (Fig5E and 5F). To further assess the direct role of NLRP3 in HSCs activation, we isolated HSCs from WT mice and cocultured them with alcohol or PA induced NETs, which showed an increase in the expression of α-SMA and IL-1β release. In contrast, HSCs isolated from NLRP3-KO mice showed no increase in α-SMA levels or IL-1β release after stimulation with NETs, suggesting that NLRP3 plays an indispensable role in HSCs activation in response to NETs (Fig 5G and 5H).
Because we observed an increase in IL-1β release after coculturing with NETs, we next evaluated whether activation of LX2 cells was dependent on IL-1β. We treated the LX2 cells with an IL-1 receptor antagonist (Anakinra) and cocultured them with PA- or alcohol-induced NETs. In the presence of IL-1 receptor antagonist we found decreased levels of α-SMA, suggesting an IL-1β dependent mechanism of NET-induced activation in LX2 cells (Fig5I).
Next, to understand how NLRP3 interacts with NETs, we performed Glutathione S-transferase (GST)-pull down assay using GST-tagged NLRP3 and CitH3 (FigS5B). With the increasing concentration of Cit-H3 we found an increase in the binding of NLRP3-GST with Cit-H3, indicating a direct binding of NLRP3 with CitH3 (FigS5B).
To further assess mechanisms of NET-induced HSCs activation, we reconstituted NETs in vitro using recombinant cit-H3 and NET-DNA. Alcohol-induced NET-DNA was isolated as described previously,(20) and described in the Materials/Methods. LX2 cells stimulation with the combination of cit-H3 plus NET-DNA induced significant induction of Acta2 mRNA levels as compared to NET-DNA or cit-H3 alone (Fig5J-left panel). Immunofluorescence revealed increased induction of α-SMA in LX2 cells treated with cit-H3 plus NET-DNA, as compared to untreated cells, indicating that the combination of DNA and protein components of NETs can effectively induce HSCs activation (Fig5J-right panel). Furthermore, IL-1β production in HSC was also significantly increased by the combined stimulation of cit-H3 plus NET-DNA but not - by cit-H3 or NET-DNA alone (Fig5K). These results demonstrate that cell-free NETs activate HSCs and are sensed by NLRP3.
6. Cell-free NETs induce proinflammatory cytokine production in monocytes via NLRP3 sensing
Recruitment of monocyte-derived macrophages and Kupffer cell activation are major contributors to MASH and ALD pathogenesis.(24) When NETs generated as described in Fig 6A were cocultured with human monocyte cells (THP1), we found significant increases in IL-1β, Tumor necrosis factor-α (TNFα), and Monocyte chemoattractant protein-1 (MCP1) production (Fig6B-D). Furthermore, dual stimulation of monocytes with NETs plus LPS resulted in significantly higher induction of IL-1β, TNFα, and MCP1 than just NETs or Lipopolysaccharide (LPS) alone (Fig6B-D). Based on this robust monocyte activation by the dual signals of LPS and fibrillar NETs, we speculated that NLRP3 can be the mechanism by which monocyte sense cell-free NETs. In support of this hypothesis, we found a significant attenuation in IL-1β production, in presence of NLRP3 inhibition with MCC950, indicating a role of NLRP3 inflammasome in NETs-plus LPS-induced activation of monocytes (Fig6B). However, none of these cytokines were inhibited by NLRP3 inhibition in LPS alone stimulated cells suggesting that MCC950 inhibited the NET-induced monocyte activation. Furthermore, DNase treatment, significantly reduced NET-induced IL-1β production in monocytes (Fig6E).
Fig 6. Neutrophil extracellular traps activate monocytes via NLRP3 inflammasomes.
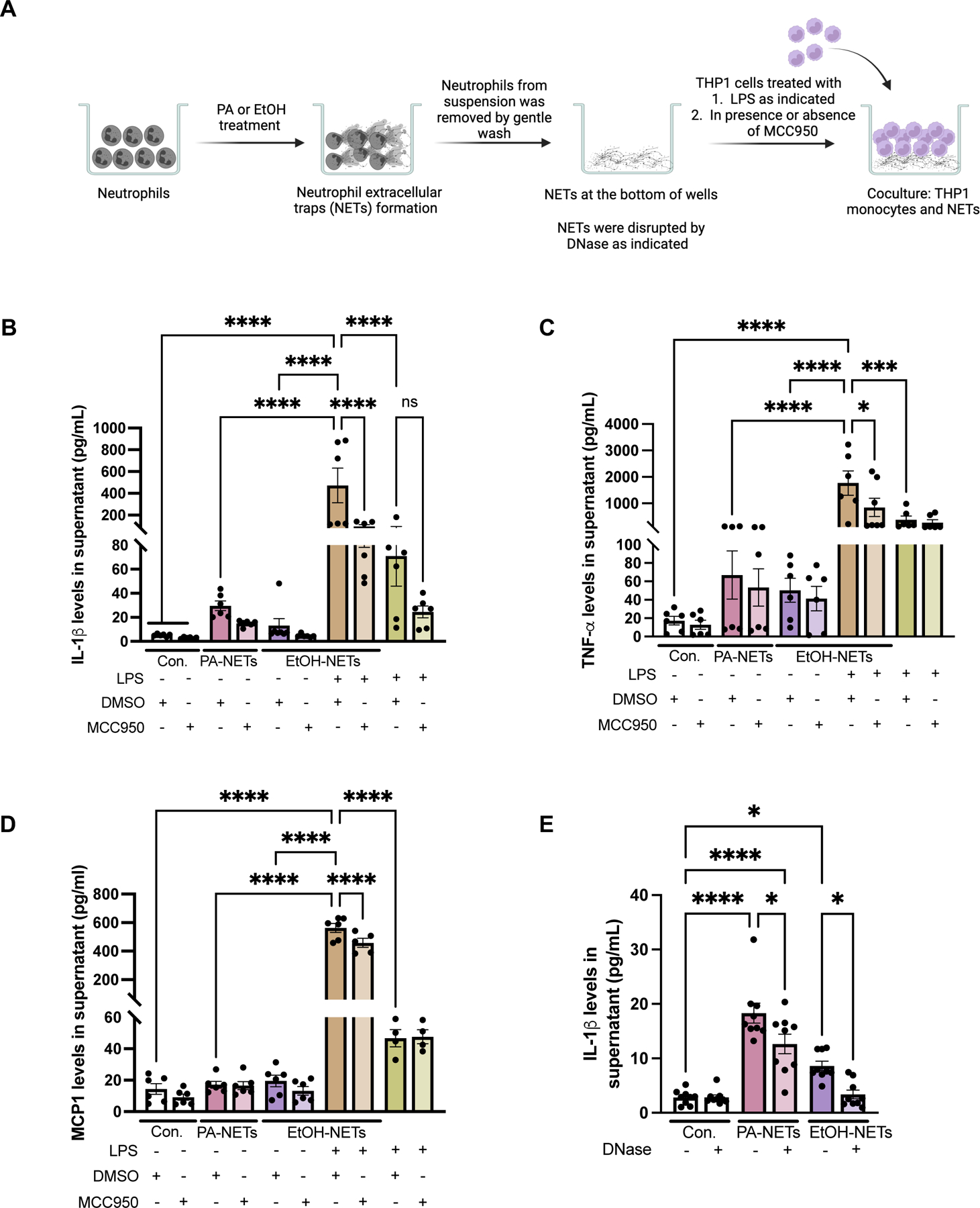
(A) Human neutrophils were treated with PA or EtOH for NETs formation, followed by coculturing NETs with THP1 cells pretreated with MCC950 or DMSO. For dual stimulation, THP1 cells cocultured with PA or EtOH-induced NETs were treated with 10ng/ml of LPS, in presence of MCC950 or DMSO. THP1 supernatants after culturing with NETs were used to detect the level of IL-Iβ (B), TNF-α (C) and MCP-1(D) by ELISA (n=6). (E) THP1 supernatant after coculturing with DNase treated NETs were used to detect IL-1β by ELISA (n=9). * p<0.05, **p<0.005, ***p<0.0005, ****p<0.00005
7. Neutrophil depletion or DNase treatment in vivo ameliorate liver damage and fibrosis in combined liver injury by alcohol binges and MASH
Given our observation that NETs activate stellate cells and induce pro-inflammatory phenotype in monocytes, we next evaluated the therapeutic role of neutrophil depletion and disruption of NETs in vivo. First, we used anti-Ly6G antibody or DNase, twice a week, for the last six weeks of alcohol and MASH diet (Fig7A). We evaluated circulating neutrophils (CD45+CD11b+Ly6G+) by flow cytometry and observed a 2-fold increase in MASH plus alcohol binge mice, which was significantly attenuated in anti-Ly6G antibody treated MASH plus alcohol mice (Fig7B). There was no significant change in CD45+CD11b+Ly6G+ neutrophils in MASH plus alcohol mice treated with DNase (Fig7B). The significant increase in transaminase levels (ALT and AST) after alcohol binges in MASH diet was strikingly attenuated in the presence of anti-Ly6G antibody or DNase treatment, respectively, indicating a decrease in liver damage (Fig7C-D). The histologic H&E liver sections (Fig7E) in MASH plus alcohol group in the presence of either anti-Ly6G antibody or DNase treatment showed reduction in micro- and macrovesicular steatosis (Fig7E).
Fig 7. Depletion of neutrophils and disruption of NETs by DNase ameliorate liver damage and fibrosis in combined liver injury by alcohol binges and MASH.
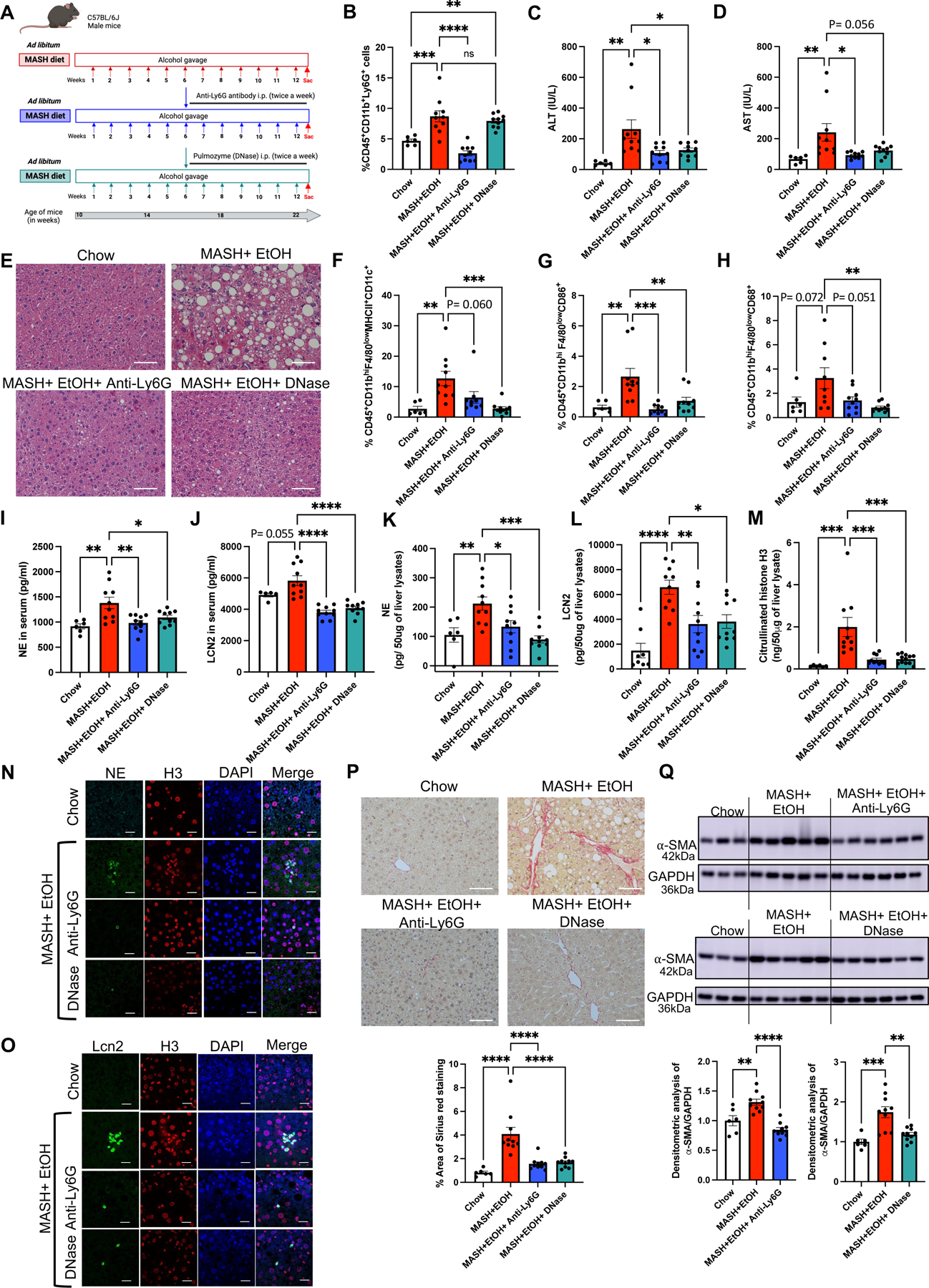
(A) Feeding schematics for combined liver injury with a therapeutic intervention of anti-Ly6G antibody and DNase treatment as described in Methods section. (B) Flow cytometry analysis of neutrophils (CD45+CD11b+Ly6G+) in liver immune cells. (C-D). ALT & AST levels were measured from serum. (E) Formalin-fixed liver sections were stained with Hematoxylin and eosin representative slides are shown, scale bar =50 μm. (F-H) Flow cytometry analysis of monocyte-derived macrophages, (CD45+CD11bhiF4/80lowMHCII+CD11c+, CD45+CD11bhiF4/80lowCD86+, CD45+CD11bhiF4/80lowCD68+). Levels of NE (I), and LCN2 (J), in serum as measured by ELISA. Whole-cell liver lysates were used to detect NE (K), LCN2 (L), and Cit-H3 (M) by ELISA. Co-immunofluorescence staining with NE and Histone H3 (N) and with LCN2 and Histone H3 (O) to visualize NETs production in mouse liver. scale bar=50 μm. (P) Formalin-fixed liver sections were stained with Sirius red stain, and representative slides are shown. (n=6–10 mice/group and average of 3–5 images per mice). Scale bar=50 μm. The percentage area of Sirius red staining is quantified using Image J. (Q) Liver lysates were analyzed by western blotting for α-SMA, using GAPDH as a loading control. The densitometric analysis of α-SMA is shown as bar graph. * p<0.05, **p<0.005, ***p<0.0005, ****p<0.00005 (n=6mice/chow group and n=10mice/all other group).
Furthermore, the increase in infiltrating monocyte-derived macrophages, CD45+CD11bhiF4/80lowMHCII+CD11c+, CD45+CD11bhiF4/80lowCD86+, and CD45+CD11bhiF4/80lowCD68+ in MASH plus alcohol mice was significantly reduced or showed a decreasing trend after DNase t and anti-Ly6G treatment, respectively (Fig 7F-H). These results suggest that inhibition of NETs formation or neutrophil depletion attenuate monocyte-derived macrophages infiltration in the liver after alcohol binge and MASH insults.
Next, we evaluated markers of NETs in the serum and liver. MASH plus alcohol-related increases in serum and liver NE (Fig7I & 7K), and Cit-H3 (Fig7M) were prevented by either anti-Ly6G antibody or DNase treatment (Fig7I, 7K, 7M). The significant increase in LCN2 in MASH plus alcohol mice in serum and liver lysates was also attenuated by anti-Ly6G antibody or DNase treatment (Fig7J and 7L). Immunofluorescence staining of NE and LCN2 revealed decreased NETs in the liver sections of the mice treated with either anti-Ly6G antibody or DNase compared to untreated MASH plus alcohol mice (Fig7N-O).
In addition to inhibition of NETs, anti-Ly6G antibody or DNase treatment significantly attenuated liver fibrosis in MASH plus alcohol fed mice as indicated by reduction in the profibrotic markers, collagen and α-SMA on Sirius Red staining (Fig2B & 7P) and Western blots (Fig2G & 7Q), respectively. These results indicated a therapeutic benefit of NETs disruption or neutrophil depletion to ameliorate liver damage and fibrosis in a combined liver injury from MASH diet and alcohol binges.
8. Alcohol binges significantly upregulate NLRP3 inflammasome activation in MASH and NLRP3 inhibition attenuates NETs and fibrosis in a combined liver injury model
Since previous studies have shown that MASH diet and alcohol administration, individually, can lead to inflammasome activation in both macrophages and hepatocytes and that the NLRP3 inflammasome is involved in fibrosis,(14, 25, 26) we next evaluated whether the NLRP3 inflammasome was activated by alcohol binge in MASH.
The initial signal in NLRP3 activation priming from Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) mediated increase in transcript levels of inflammasome components.(14) We found a significant increase in the mRNA levels of Nlrp3 (Fig8A), Pro-caspase-1 (Casp1) (Fig8B) and Il1b (Fig8C) in MASH plus alcohol compared to either alcohol or MASH alone. We also found that other inflammasomes such as Nlrc4 inflammasome, but not Aim2, was significantly increased in MASH plus alcohol compared to controls (FigS6A-B). Functional activation of the NLRP3 inflammasome was indicated by a significant increase in cleaved caspase-1 and cleaved IL-Iβ in MASH plus alcohol group compared to MASH alone (Fig8E-H).
Fig 8. Alcohol binges plus MASH diet activates NLRP3 inflammasome and inhibition of NLRP3 attenuates neutrophil infiltration and NETs formation in mice.
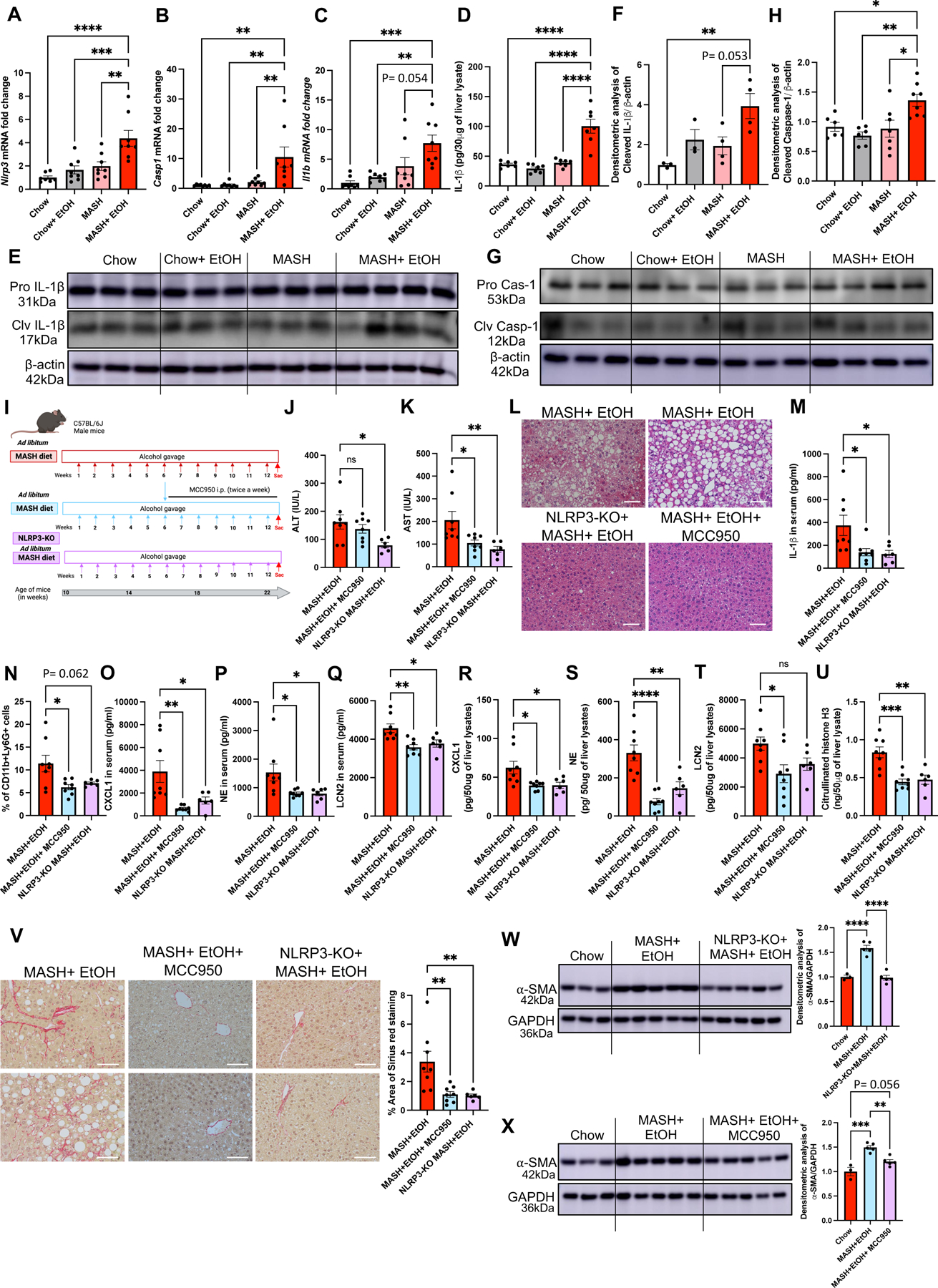
Liver RNA was used to determine Nlrp3 (A), Casp1 (B) and Il1b (C) mRNA levels by qPCR. 18s was used to normalize Cq values (n=6–8/group). Liver lysates were used to detect IL-1β by ELISA (D) and by western blot (E-F). Liver lysates were used to detect Cleaved Caspase-1 by western blot (G-H) (n=6–8/group). (I) Feeding schematics for combined liver injury with a therapeutic intervention of MCC950 or NLRP3-KO mice fed on MASH diet plus alcohol as described in Methods. (J-K) ALT & AST levels were measured from serum. (L) Formalin-fixed liver sections were stained with Hematoxylin and eosin representative slides are shown, scale bar =50 μm. (M) IL-1β level in the mice serum was detected by ELISA. (N) Flow cytometry analysis of neutrophils (CD45+CD11b+Ly6G+) in liver immune cells. Levels of CXCL1 (O), NE (P), and LCN2 (Q), in serum as measured by ELISA. Whole-cell liver lysates were used to detect CXCL1 (R), NE (S), and LCN2 (T), and Cit-H3 (U) by ELISA. (V) Formalin-fixed liver sections were stained with Sirius red stain, and representative slides are shown. (n=6–8 mice/group and average of 3–5 images per mice). Scale bar=50 μm. (W-X) Liver lysates were analyzed by western blotting for α-SMA, using GAPDH as a loading control. The densitometric analysis of α-SMA is shown as bar graph. * p<0.05, **p<0.005, ***p<0.0005, ****p<0.00005 (n=6mice/NLRP3-KO group and n=8 mice/all other group).
Next, we validated the involvement of NLRP3 inflammasome in the MASH plus alcohol binge model by either inhibiting NLRP3 inflammasome with MCC950, for the last six weeks of alcohol and MASH diet in WT mice or feeding whole body NLRP3-KO mice MASH diet plus weekly alcohol binges for three months (Fig8I). Liver damage (as assessed by ALT and AST levels) was significantly attenuated in NLRP3-KO mice, whereas only AST was significantly decreased in mice that received MCC950 (Fig8J-K). The histologic H&E liver sections in NLRP3-KO mice or MCC950 administered mice showed reduction in steatosis (Fig8L). MASH plus alcohol-induced increase in serum IL-1β levels (Fig8M) was significantly attenuated by MCC950 administration and in NLRP3-KO mice, suggesting that NLRP3 inhibition can prevent the liver damage and inflammation caused by the combined liver injury.
Our results suggested the involvement of NLRP3 in sensing cell-free NETs in HSCs and monocytes. Therefore, we next questioned if NLRP3 was involved in promoting neutrophil infiltration, and NETs formation in liver by MASH plus alcohol. Using NLRP3-KO mice or MCC950 administration in MASH plus alcohol model for three months, we found a significant reduction in infiltrating neutrophils (CD45+CD11b+Ly6G+) in the liver of MASH plus alcohol-treated mice, as compared to WT mice (Fig8N). This reduction in neutrophils correlated with significant attenuation of CXCL1 (Fig8O & 8R), and NE (Fig8P & 8S), both in serum and liver, in MCC950 administered and NLRP3-KO mice as compared to MASH plus alcohol WT mice. LCN2 was also significantly decreased in serum and liver after MCC950 administration and in the serum of NLRP3-KO mice (Fig8Q & 8T). In liver lysates, Cit-H3 was reduced in NLRP3-KO and MCC950 administrated mice (Fig8U) further strengthening our observation that NLRP3 plays an indispensable role in NETs formation
Importantly, Sirius Red staining (Fig8V) and α-SMA western blots (Fig8W and 8X) showed a significant decrease in fibrosis in the MASH plus alcohol mice treated with MCC950 as well as in NLRP3-KO mice as compared to WT mice on MASH diet plus alcohol alone, suggesting NLRP3 inhibition attenuates liver fibrosis.
In summary, our results indicate that NLRP3 is involved alcohol-induced recruitment of neutrophils and plays a regulatory role in NETs formation. Therefore, alcohol-induced NETs formation and NET-induced inflammasome activation point to a dual role of NLRP3 in orchestrating liver injury and fibrosis in the context of alcohol binges in MASH.
DISCUSSION
Alcohol consumption in individuals with MASLD is often ignored. However, recent studies suggest that even moderate alcohol consumption (2–4 drinks/day) accelerates hepatic steatosis, facilitates its progression of fibrosis, and increases the risk of liver-related death and HCC.(2, 5) Therefore, a separate group of patients with MASLD that consume 140–350 g/week for females and 210–420 g/week for males of alcohol is named as MetALD.(4)
In this study, we report that alcohol binges in combination with MASH synergistically promote liver fibrosis. We found that alcohol binges in MASH diet-fed mice resulted in a dramatic increase in neutrophil infiltration and NETs formation, which was regulated by the NLRP3 inflammasome (Fig 9A and graphical abstract). We discovered that alcohol-induced cell-free NETs trigger a pro-fibrotic HSCs phenotype and promote pro-inflammatory cytokine production from monocytes. We identified a unique role of the NLRP3 inflammasome in sensing cell-free NETs by directly interacting with cit-H3, both in HSCs and monocytes. We also demonstrate the therapeutic benefits of neutrophil depletion by anti-Ly6G antibody, NETs disruption by DNase treatment or NLRP3 inhibition using MCC950 administration in a preclinical model of alcohol binges in MASH (Fig 9A).
Fig 9. Schematic diagram depicting the mechanistic details of exacerbated liver fibrosis by MASH diet plus alcohol binge.
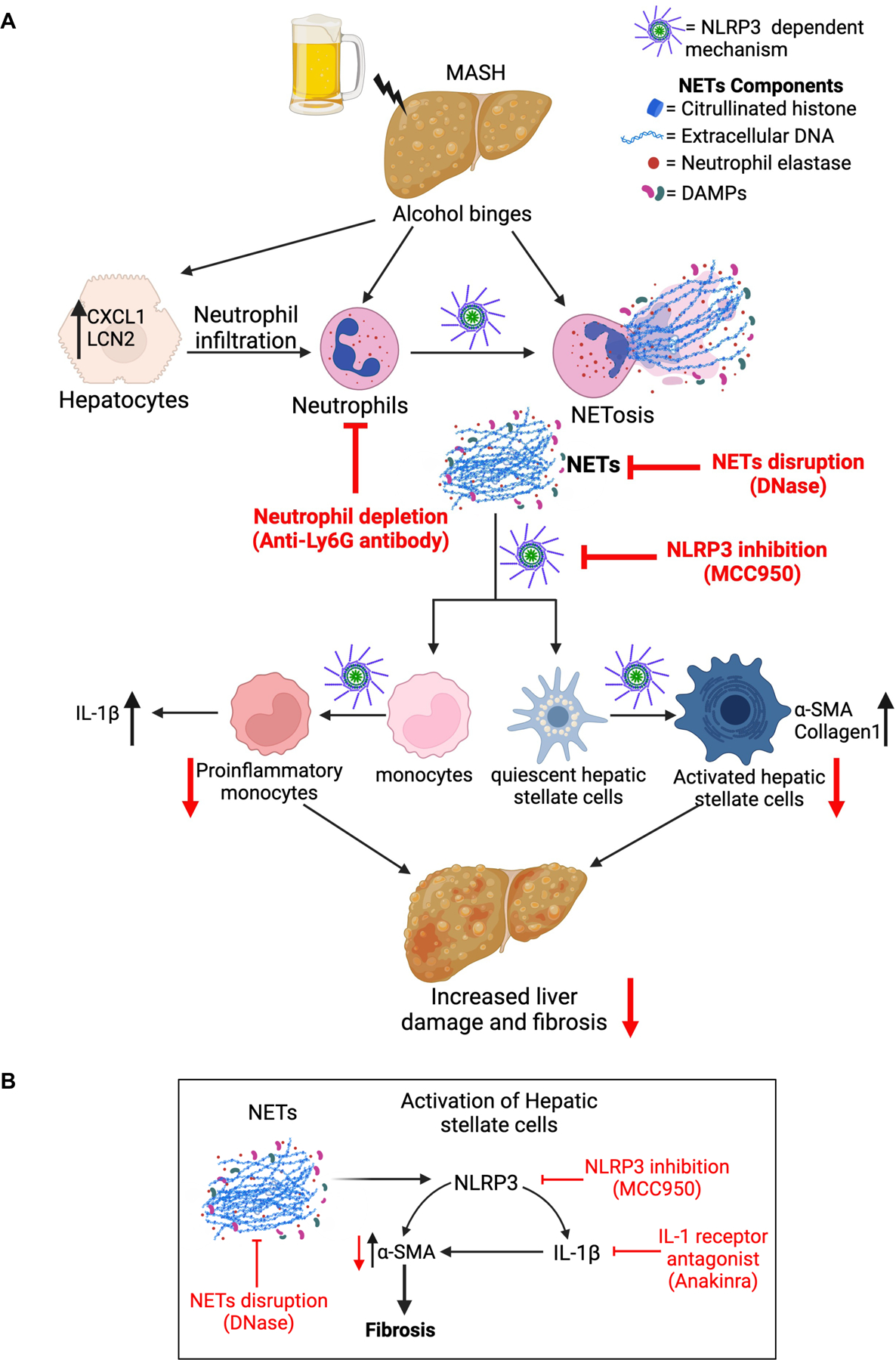
(A) Briefly, alcohol binges in MASH diet fed mice lead to increased neutrophil infiltration and NETs formation in NLRP3-depedent manner. NETs are sensed by monocytes and hepatic stellate cells via NLRP3 which promotes IL-1β production from monocytes and increases α-SMA and Collagen1 production from hepatic stellate cells. Activation of hepatic stellate cells and pro-inflammatory cytokine production from monocytes promotes liver damage and fibrosis. Neutrophil depletion by anti-Ly6G antibody, NETs disruption by DNase, and NLRP3 inhibition by MCC950, ameliorates liver damage and fibrosis. (B) NETs are sensed by hepatic stellate cells by NLRP3, which promotes IL-1β production from stellate cells. NLRP3 and IL-1β increase α-SMA production from hepatic stellate cells. Activation of hepatic stellate cells promotes fibrosis. Disrupting NETs by DNase, inhibiting NLRP3 by MCC950, or blocking IL-1β by IL-1 receptor antagonist attenuates α-SMA production from hepatic stellate cells and reduces fibrosis.
Both binge drinking and obesity show an increasing trend globally.(2, 3) Our experimental murine model mimics human alcohol binge drinking and a diet rich in high fat-cholesterol-sucrose which displays accelerated fibrosis in 12 weeks as compared to MASH diet alone. We previously showed that the MASH diet alone (high fat-cholesterol-sucrose diet) results in progression from MASLD to MASH (but no fibrosis at 12 weeks) and early HCC (48 weeks) similar to the natural history of MASH in human disease.(27) Murine models of MASH, based on defined diets such as Gubra Amylin NASH (GAN) diet or non-trans-fat Western diets (WD-NTF) develop fibrosis after 24–30 weeks.(28, 29) Our results are consistent with previous studies where in our MASH alone mice showed mild fibrosis (pathology score 0–1) and no significant increase in the molecular markers of fibrosis such as α-SMA and vimentin while MASH plus alcohol significantly increased fibrosis. Previous studies with high-fat diet (HFD) and alcohol, where alcohol was either provided as single binge or multiple binges for one month after establishing MASH induced oxidative, Endoplasmic Reticulum (ER), and mitochondrial stresses along with steatohepatitis.(30, 31) Our model recapitulates not only the binge drinking pattern commonly seen in obesity but importantly highlights novel neutrophil-related mechanisms involved in liver disease progression and exacerbated fibrosis in response to simultaneous injury by alcohol and MASH diet. Therefore, this paradigm of alcohol and MASH diet with extended feeding time and/or chronic alcohol consumption will provide a preclinical model to recapitulate the entire spectrum of MetALD pathologies.
We discovered that in setting of MASH, alcohol binge induces significant neutrophil infiltration and activation in the liver. While alcohol binge alone increased neutrophil numbers in chow-fed mice, intriguingly, both transcriptomic profiling and immunohistochemistry revealed significantly higher Ly6G+ neutrophil infiltration in the liver of mice fed on MASH diet plus alcohol. These neutrophils resulted in a dramatic increase in NETosis, as observed by high LCN2, NE, and cit-H3, in an alcohol plus MASH diet. A recent study showed that alcohol and ROS can directly induce NETs.(13, 32) Interestingly, a study on animal model of SARS-CoV-2 animal model showed that FDA-approved drug for alcohol use disorder, disulfiram (an inhibitor of aldehyde dehydrogenase) inhibits NETs formation.(33) Our in vitro results also showed the induction of NETs by alcohol in human neutrophil was reduced by anti-oxidant N-Acetyl Cysteine and disulfiram. Further, NETs formation is increased in acetaldehyde treated neutrophils suggesting that alcohol and its metabolites can regulate NETs formation.
Our transcriptomic data showed that mice with alcohol plus MASH diet exhibited the most robust transcriptomic profile of fibrosis and tissue remodeling, which directly correlated with histological findings of accelerated fibrosis compared to alcohol or MASH diet alone. Previous reports have shown a close relationship between neutrophils and fibrosis.(31) NETs comprise of DAMP molecules such as histone, DNA and HMGB1, and inhibition of NETs formation by PAD4 inhibitors or DNase1, was previously shown to attenuate HMGB1 and histone-mediated ischemic reperfusion liver injury.(13, 34) Here we demonstrate for the first time that alcohol- or PA-induced cell-free NETs directly activate stellate cells and induce IL-1β as well as α-SMA and collagen production (Fig9B). This is mediated by NETs that are found in the liver tissue. The potential of cell-free NETs in modulating cell functions in tissues has been shown in other sterile inflammatory conditions.(35) Interestingly, no secondary stimulus was required for robust activation of HSCs. We speculate, however, that the NET-induced HSCs activation is accelerated by repeated alcohol binges in MASH. Here, we demonstrate that in vivo disruption of NETs by DNase or depleting neutrophils by anti-Ly6G antibody ameliorated liver fibrosis caused by MASH and alcohol binges. Interestingly, few previous studies described the presence of NETs in MASH alone, and in a recent study, we reported a significantly increased presence of NETs in the liver and circulation of patients with alcohol-associated hepatitis.(13, 36) In this study we found significantly higher neutrophil elastase and lipocalin expression in livers of AH patients with BMI >25 compared to AH patients with normal BMI or with MASH only patients, indicating NETs activation. It has been suggested that activated neutrophils stay in the liver for a prolonged time.(13) This is also possible because activated HSCs secrete granulocyte-macrophage colony-stimulating factor and interleukin-15 to prolong the survival of neutrophils, and this interaction likely promotes fibrosis.(31)
We previously reported that NET-producing neutrophils (or NETosis neutrophils) induce monocyte differentiation to CD14+CD16+ monocytes.(13) Here, we deciphered that alcohol-induced NETs directly activate monocytes and induce a significantly greater increase in IL-1β, TNF-α and MCP-1 production combined with LPS as a second stimulus. Furthermore, we found that disruption of the fibrillar structure of NETs by DNase partially attenuated IL-1β release from monocytes. These results were in accordance with a recent study that highlighted the combination of histone and DNA, and not individual components, promoted cytokine production in macrophages in atherosclerosis.(20) We show that in vivo disruption of NETs by DNase or depletion of neutrophils by anti-Ly6G antibody, reduced CD45+CD11b+Ly6Chi pro-inflammatory monocytes and decreased the number of liver infiltrating monocyte-derived macrophages. Consistent with our observation, a recent study showed that DNase treatment in diabetic mice led to atherosclerosis resolution by reducing CD68+ macrophages.(37) Our results strongly highlight a novel role of NETs in sustaining inflammation through monocyte activation.
NLRP3 serves as a sensor of various sterile danger signals, including various crystals, intermediate filaments, and fibrillar DAMPs such as cholesterol, uric acid, vimentin and β-amyloid.(21, 22, 38, 39) Previous studies have implicated the activation of NLRP3 inflammasome in liver injury and fibrosis.(14, 25, 26) We speculated that the fibrillar structure of cell-free NETs might activate NLRP3 in HSCs and monocytes. In support of this hypothesis, we discovered that inhibiting NLRP3 in LX2 cells or HSCs from NLRP3-KO mice dampened NET-induced α-SMA and IL-β production (Fig9B). We show that disruption of the fibrillar structure of NETs with DNase in vitro partially attenuated the NET-induced HSCs activation (Fig9B). In vitro reconstitution of NETs with cit-H3 and NET-DNA further suggested that only fully assembled NETs (both histone and DNA) activate HSC. We further demonstrated that NLRP3 senses NETs by directly interacting with cit-H3. Previous studies showed direct interaction of NLRP3 with oxidized mitochondrial DNA(40, 41) and given that alcohol-induced NETs DNA comprise of both nuclear and mitochondrial DNA,13 we cannot rule out whether NLRP3 can directly interact with NETs DNA, or the role of other patten recognition receptors such as TLR9, TLR4 in sensing the nucleic acid components of NETs.
NLRP3 inflammasome activation leads to cleavage of IL-1β and release.(14) We found that inhibiting the NLRP3 with MCC950 attenuated the NET-induced release of IL-1β and production of α-SMA from HSCs (Fig9B). Therefore, we speculated that a NLRP3-IL-1β dependent pathway might be required for NET-mediated HSCs activation. In support with this hypothesis, we found a decrease in α-SMA in LX2 cells in the presence of IL-1 receptor antagonist. The effect of IL-1β on promoting HSCs activation was shown before; therefore, we speculate that NET-induced IL-1β is a secondary mechanism in HSCs activation by NETs (Fig9B). Importantly, inhibiting NLRP3 with MCC950 or whole body NLRP3-KO mice displayed reduced liver injury and inflammation. These results suggest a NLRP3-dependent propagation of inflammation and fibrosis after alcohol binges in MASH.
A potential relationship between NLRP3 and neutrophil infiltration was reported earlier in the murine model of gout and hepatic ischemia liver injury.(35, 42) Our findings also indicate that NLRP3 is involved in neutrophil infiltration in the liver. We discovered that alcohol-induced NETs in MASH plus alcohol was dependent on NLRP3, as NLRP3-KO mice or NLRP3 inhibition by MCC950 administration attenuated neutrophil infiltration and NETs formation.
A previous study has shown that chronic alcohol feeding in ob/ob mice induced both micro- and macrovesicular steatosis.(43) Similarly, cross-sectional studies with NASH patients’ liver biopsies showed positive correlation of microvesicular steatosis with hepatocyte ballooning and Mallory-Denk bodies.(44) Interestingly, in our MASH plus alcohol mice, we observed a similar pattern of both macro- and micro-vesicular steatosis and increase in free fatty acids and total cholesterol. Furthermore, depletion of neutrophils or NLRP3 inhibition, improved steatosis in alcohol plus MASH diet-fed mice. We speculate that this could be related to improved insulin sensitivity, reduced lipid accumulation and reduced infiltration of inflammatory immune cells, as reported by previous studies.(45–47)
In conclusion, our results highlight the crucial role of binge alcohol-induced neutrophil liver infiltration and NETs production in accelerating liver injury, inflammation, and fibrosis in MASH. We show for the first time that alcohol-induced cell-free NETs activate HSCs and monocytes (Fig 9), thereby contributing to the acceleration of inflammation and fibrosis in MASH (Fig 9). We conclude that neutrophil depletion and NETs disruption can prevent liver damage and fibrosis. Our data suggest that NLRP3 inflammasome inhibition prevents alcohol-induced neutrophil liver infiltration, NETs-mediated liver damage, and HSCs activation in the alcohol binges plus MASH mouse model. Our study highlights the previously unrecognized effect of alcohol binges in MASLD/MASH by accelerating the progression of liver disease.
MATERIALS/METHODS
Feeding regimen and MASH diet plus alcohol binge model used in this study
Mice were fed on a high fat-cholesterol-sucrose diet (MASH diet) (from Research Diets, Inc. New Jersey, USA-D09061704) or standard laboratory chow diet ad libitum for 3 months. We have previously reported that administration of a MASH diet results in features of the metabolic syndrome, that progresses from MASH to fibrosis mimicking the progression of human MASH(27). MASH diet and chow-fed mice were randomly divided into two groups and either received 5g/kg alcohol gavage or water gavage once every week for 3 months. Survival was > 75% in mice that received alcohol binges plus MASH diet and 90% in alcohol binges alone (FigS1A). All the mice were euthanized 9h after the last alcohol gavage. After experimental treatments, blood and liver samples were collected, processed immediately, and stored at −80 °C for further analysis. Insulin tolerance test was performed as described previously and described in supplementary Material and Methods.(48) Administration of anti-Ly6G, DNase and MCC950 is included in supplementary Material and Methods. All animal procedures were approved by the BIDMC institutional animal care and use committee (IACUC, protocol #019–2019, #30–2022). Animal experiments were reported using ARRIVE reporting guidelines.(49)
Statistical analysis
Statistical significance was determined using one-way ANOVA, or unpaired t-test. Each experiment was repeated at least three times to determine the biological significance. Data are shown as mean ± SEM and were considered statistically significant at P ≤ 0.05. GraphPad Prism software (version 9; GraphPad Software Inc.) was used for analysis.
Additional Methods
Please see the supplementary material and method section for additional materials and methodology used in this study.
Data availability statement
All the data relevant to the study are included in the article or uploaded as supplementary information
Study Approval
All animal procedures were approved by the UMASS and BIDMC institutional animal care and use committee (IACUC, protocol #019–2019, #30–2022).
Peripheral blood was collected from healthy subjects, which was approved by the BIDMC IRB (Protocol# 2019P000521) full committee review. All participants were informed about blood sampling for clinical research and signed at the consent form.
Supplementary Material
What is already known on this topic
Clinical studies, suggest that heavy alcohol use accelerates progression of Metabolic dysfunction-associated steatohepatitis (MASH) to fibrosis and cirrhosis.
What this study adds
Using transcriptomic profiling and mechanistic approaches, our study shows alcohol binges accelerates fibrosis in MASH by neutrophil recruitment to the liver and NETs release, which directly activate hepatic stellate cells and promote pro-inflammatory cytokine production in monocytes via NLRP3, in vitro and in vivo.
How this study might affect research, practice or policy
This study identifies a unique role for NLRP3 in sensing alcohol-induced NETs, leading to HSCs and monocyte activation and shows that inhibiting NETs and/or NLRP3 attenuate the pro-fibrotic effects of alcohol in MASH.
ACKNOWLEDGMENT
We acknowledge the Microscopy and Histopathology Core B of the Harvard Digestive Disease Center at the Beth Israel Deaconess Medical Center and NIH grant P30DK034854 for resources, technology, and expertise, including Dr. Lay-Hong Ang for immunostaining and assistance with confocal microscopy and Suzanne White for paraffin sectioning and special stains. The authors thank Dr. Ruchi Chauhan for assistance in preparing the manuscript.
FUNDING
Research reported in this publication was supported by National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under award number R01AA017729, R01AA011576, and U01AA026933 to GS.
Microscopy and Histopathology Core B of the Harvard Digestive Disease Center at the Beth Israel Deaconess Medical Center received National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health-grant P30DK034854.
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviation
- AH
Alcoholic Hepatitis/Alcohol-associated Hepatitis
- AIM2
Absent in Melanoma 2
- ALD
Alcoholic Liver Disease/Alcohol-associated Liver Disease
- ALT
Alanine aminotransferase
- ARG1
Arginase 1
- AST
Aspartate aminotransferase
- BST2
Bone Marrow Stromal Cell Antigen 2
- Cit-H3
Citrullinated Histone 3
- CXCL1
Chemokine (C-X-C motif) ligand 1
- DNase
Deoxyribonuclease I
- DAMPs
Damage-associated molecular patterns
- DEG
Differentially expressed gene
- Fcer
Fcγ receptor genes
- HCC
Hepatocellular carcinoma
- HSCs
Hepatic stellate cells
- IL-1β
Interleukin 1 beta
- IL-18
Interleukin 18
- LCN2
Lipocalin 2
- LMNCs
Liver mononuclear cells
- LPS
Lipopolysaccharides
- MFI
Mean fluorescent intensity
- MMP
Matrix metalloproteinases
- MASLD
Metabolic Dysfunction Associated Steatotic Liver Disease
- MASH
Metabolic Dysfunction Associated Steatohepatitis
- NE
Neutrophil Elastase
- NET(s)
Neutrophil extracellular trap(s)
- NLRP3
Nod-like receptor protein 3
- NLRC4
NLR Family CARD Domain Containing 4
- PECAM
Platelet endothelial cell adhesion molecule
- RBCs
Red blood cells
- SSC
Side scatter
- TIMP
Tissue Inhibitor of Metalloproteinase
- TNF-α
Tumor necrosis factor alpha
- TLR4
Toll-like receptor 4
Footnotes
CONFLICT OF INTEREST
G.S. reports being a paid consult for Durect Corporation, Cyta Therapeutics, Generon, Terra Firma, Quest Diagnostics, Pandion Therapeutics, Surrozen, Merck, Novartis, Pfizer, Lab Corp, Intercept and Takeda. She has stock options in Glympse Bio, Satellite Bio and Ventyx.
References
- 1.Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70(1):151–71. [DOI] [PubMed] [Google Scholar]
- 2.Boyle M, Masson S, Anstee QM. The bidirectional impacts of alcohol consumption and the metabolic syndrome: Cofactors for progressive fatty liver disease. J Hepatol 2018;68(2):251–67. [DOI] [PubMed] [Google Scholar]
- 3.Ntandja Wandji LC, Gnemmi V, Mathurin P, Louvet A. Combined alcoholic and non-alcoholic steatohepatitis. JHEP Rep 2020;2(3):100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023. [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM, Stepanova M, Ong J, Yilmaz Y, Duseja A, Eguchi Y, et al. Effects of Alcohol Consumption and Metabolic Syndrome on Mortality in Patients With Nonalcoholic and Alcohol-Related Fatty Liver Disease. Clin Gastroenterol Hepatol 2019;17(8):1625–33 e1. [DOI] [PubMed] [Google Scholar]
- 6.Decraecker M, Dutartre D, Hiriart JB, Irles-Depe M, Marraud des Grottes H, Chermak F, et al. Long-term prognosis of patients with alcohol-related liver disease or non-alcoholic fatty liver disease according to metabolic syndrome or alcohol use. Liver Int 2022;42(2):350–62. [DOI] [PubMed] [Google Scholar]
- 7.Kuntsche E, Kuntsche S, Thrul J, Gmel G. Binge drinking: Health impact, prevalence, correlates and interventions. Psychol Health. 2017;32(8):976–1017. [DOI] [PubMed] [Google Scholar]
- 8.Hosseini N, Shor J, Szabo G. Alcoholic Hepatitis: A Review. Alcohol Alcohol 2019;54(4):408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stengel JZ, Harrison SA. Nonalcoholic Steatohepatitis: Clinical Presentation, Diagnosis, and Treatment. Gastroenterol Hepatol (N Y). 2006;2(6):440–9. [PMC free article] [PubMed] [Google Scholar]
- 10.Toshikuni N, Tsutsumi M, Arisawa T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(26):8393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho Y, Szabo G. Two Faces of Neutrophils in Liver Disease Development and Progression. Hepatology. 2021;74(1):503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bukong TN, Cho Y, Iracheta-Vellve A, Saha B, Lowe P, Adejumo A, et al. Abnormal neutrophil traps and impaired efferocytosis contribute to liver injury and sepsis severity after binge alcohol use. J Hepatol 2018;69(5):1145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcoholic hepatitis. J Hepatol 2022. [DOI] [PubMed] [Google Scholar]
- 14.de Carvalho Ribeiro M, Szabo G. Role of the Inflammasome in Liver Disease. Annu Rev Pathol 2022;17:345–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrasek J, Iracheta-Vellve A, Saha B, Satishchandran A, Kodys K, Fitzgerald KA, et al. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J Leukoc Biol 2015;98(2):249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG, et al. NLR Family Pyrin Domain-Containing 3 Inflammasome Activation in Hepatic Stellate Cells Induces Liver Fibrosis in Mice. Hepatology. 2019;69(2):845–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 2021;18(3):151–66. [DOI] [PubMed] [Google Scholar]
- 18.Stoimenou M, Tzoros G, Skendros P, Chrysanthopoulou A. Methods for the Assessment of NET Formation: From Neutrophil Biology to Translational Research. Int J Mol Sci 2022;23(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawabata K, Hagio T, Matsuoka S. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol 2002;451(1):1–10. [DOI] [PubMed] [Google Scholar]
- 20.Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep 2020;31(5):107602. [DOI] [PubMed] [Google Scholar]
- 21.dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun 2015;6:6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friker LL, Scheiblich H, Hochheiser IV, Brinkschulte R, Riedel D, Latz E, et al. beta-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep 2020;30(11):3743–54 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang H, Seo E, Oh YS, Jun HS. TGF-beta activates NLRP3 inflammasome by an autocrine production of TGF-beta in LX-2 human hepatic stellate cells. Mol Cell Biochem 2022;477(5):1329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol 2013;3(2):785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2016;64(3):955–65. [DOI] [PubMed] [Google Scholar]
- 26.Calcagno DM, Chu A, Gaul S, Taghdiri N, Toomu A, Leszczynska A, et al. NOD-like receptor protein 3 activation causes spontaneous inflammation and fibrosis that mimics human NASH. Hepatology. 2022;76(3):727–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganz M, Bukong TN, Csak T, Saha B, Park JK, Ambade A, et al. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J Transl Med 2015;13:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallage S, Avila JEB, Ramadori P, Focaccia E, Rahbari M, Ali A, et al. A researcher’s guide to preclinical mouse NASH models. Nat Metab 2022;4(12):1632–49. [DOI] [PubMed] [Google Scholar]
- 29.Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Animal Models of Fibrosis in Nonalcoholic Steatohepatitis: Do They Reflect Human Disease? Adv Nutr 2020;11(6):1696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology. 2015;62(4):1070–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Z, Xu MJ, Cai Y, Wang W, Jiang JX, Varga ZV, et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell Mol Gastroenterol Hepatol 2018;5(3):399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azzouz D, Khan MA, Palaniyar N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov 2021;7(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adrover JM, Carrau L, Dassler-Plenker J, Bram Y, Chandar V, Houghton S, et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight 2022;7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaral FA, Costa VV, Tavares LD, Sachs D, Coelho FM, Fagundes CT, et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum 2012;64(2):474–84. [DOI] [PubMed] [Google Scholar]
- 36.van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. 2018;68(4):1347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight 2020;5(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iracheta-Vellve A, Petrasek J, Satishchandran A, Gyongyosi B, Saha B, Kodys K, et al. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J Hepatol 2015;63(5):1147–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cabral A, Cabral JE, Wang A, Zhang Y, Liang H, Nikbakht D, et al. Differential Binding of NLRP3 to non-oxidized and Ox-mtDNA mediates NLRP3 Inflammasome Activation. Commun Biol 2023;6(1):578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012;36(3):401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue Y, Shirasuna K, Kimura H, Usui F, Kawashima A, Karasawa T, et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J Immunol 2014;192(9):4342–51. [DOI] [PubMed] [Google Scholar]
- 43.Romics L Jr., Mandrekar P, Kodys K, Velayudham A, Drechsler Y, Dolganiuc A, et al. Increased lipopolysaccharide sensitivity in alcoholic fatty livers is independent of leptin deficiency and toll-like receptor 4 (TLR4) or TLR2 mRNA expression. Alcohol Clin Exp Res 2005;29(6):1018–26. [DOI] [PubMed] [Google Scholar]
- 44.Tandra S, Yeh MM, Brunt EM, Vuppalanchi R, Cummings OW, Unalp-Arida A, et al. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol 2011;55(3):654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips BE, Lantier L, Engman C, Garciafigueroa Y, Singhi A, Trucco M, et al. Improvement in insulin sensitivity and prevention of high fat diet-induced liver pathology using a CXCR2 antagonist. Cardiovasc Diabetol 2022;21(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Sun K, Zhou Y, Wang H, Zhou Y, Liu S, et al. NLRP3 inflammasome inhibitor CY-09 reduces hepatic steatosis in experimental NAFLD mice. Biochem Biophys Res Commun 2021;534:734–9. [DOI] [PubMed] [Google Scholar]
- 47.Chen F, Liu Y, Li Q, Wang F. Inhibition of hepatic NLRP3 inflammasome ameliorates non-alcoholic steatohepatitis/hepatitis B - induced hepatic injury. Clin Res Hepatol Gastroenterol 2023;47(1):102056. [DOI] [PubMed] [Google Scholar]
- 48.Vinue A, Gonzalez-Navarro H. Glucose and Insulin Tolerance Tests in the Mouse. Methods Mol Biol 2015;1339:247–54. [DOI] [PubMed] [Google Scholar]
- 49.Omary MB, Cohen DE, El-Omar E, Jalan R, Low M, Nathanson M, et al. Not All Mice Are the Same: Standardization of Animal Research Data Presentation. Gut 2016;65(6):894–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data relevant to the study are included in the article or uploaded as supplementary information