

Abstract
Anti‐HER2 therapy is indicated for erb‐b2 receptor tyrosine kinase 2 (ERBB2)‐amplified/overexpressing endometrial carcinoma (EC). Mutations constitute another mode of ERBB2 activation, but only rare ERBB2‐mutated ECs have been reported. We sought to characterize the clinicopathologic and genetic features of ERBB2‐mutated EC. From an institutional cohort of 2638 ECs subjected to clinical tumor‐normal panel sequencing, 69 (2.6%) with pathogenic ERBB2 mutation(s) were identified, of which 11 were also ERBB2‐amplified. The most frequent ERBB2 hotspot mutations were V842I (38%) and R678Q (25%). ERBB2 mutations were clonal in 87% of evaluable cases. Immunohistochemistry revealed low HER2 protein expression in most ERBB2‐mutated ECs (0/1+ in 66%, 2+ in 27%); all 3+ tumors (7.3%) were also ERBB2‐amplified. Compared to ERBB2‐wildtype ECs (with or without ERBB2 amplification), ERBB2‐mutated/non‐amplified ECs were enriched for the microsatellite instability‐high (MSI‐H) and, to a lesser extent, DNA polymerase epsilon, catalytic subunit (POLE) molecular subtypes, and associated with high tumor mutational burden and low chromosomal instability. Survival outcomes were similar between patients with ERBB2‐mutated/non‐amplified versus wildtype EC, whereas ERBB2 amplification was associated with worse prognosis on univariate, but not multivariate, analyses. In conclusion, ERBB2 mutation defines a rare subgroup of ECs that is pathogenically distinct from ERBB2‐wildtype and ERBB2‐amplified ECs.
Keywords: endometrial cancer, ERBB2, HER2, microsatellite instability, mutation
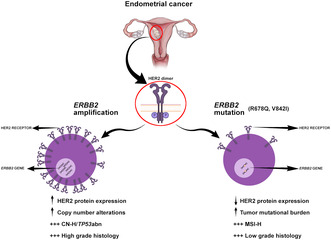
ERBB2 amplification is a known driver of copy number‐high/TP53‐abnormal high‐grade endometrial carcinoma and is associated with HER2 overexpression. In this retrospective cohort of clinically annotated endometrial carcinomas, ERBB2 pathogenic mutations define a distinct and non‐overlapping subgroup of tumors, typically with low levels of HER2 expression, high tumor mutational burden, microsatellite instability‐high status, and low‐grade endometrioid histology.
Abbreviations
- ADC
antibody‐drug conjugate
- amp
amplified
- CI
confidence interval
- CN‐H
copy number‐high
- CN‐L
copy number‐low
- EC
endometrial carcinoma
- ERBB2
erb‐b2 receptor tyrosine kinase 2
- FGA
fraction of genome altered
- FIGO
International Federation of Gynecology and Obstetrics
- HR
hazard ratio
- IHC
immunohistochemistry
- IQR
interquartile range
- MMR
mismatch repair
- MSI‐H
microsatellite instability‐high
- MSK‐IMPACT
Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets
- mut
mutated
- non‐amp
non‐amplified
- NOS
not otherwise specified
- NSMP
no specific molecular profile
- OS
overall survival
- PFS
progression‐free survival
- POLE
DNA polymerase epsilon, catalytic subunit
- SNV
single nucleotide variant
- TCGA
The Cancer Genome Atlas
- TMB
tumor mutational burden
- TP53abn
TP53 abnormal
- wt
wildtype
1. Introduction
Endometrial carcinoma (EC) is the most common gynecologic malignancy, with endometrioid, serous, and clear cell carcinomas comprising the major histologic subtypes. Complementing the traditional histologic classification, The Cancer Genome Atlas (TCGA) study of EC identified four molecular subtypes [1]: (1) DNA polymerase epsilon, catalytic subunit (POLE), ultra‐mutated; (2) microsatellite instability‐high (MSI‐H), hypermutated; (3) copy number‐high (CN‐H), serous‐like; and (4) copy number‐low (CN‐L), endometrioid. These molecular subtype classes are associated with distinct outcomes, with POLE having the most favorable outcome, MSI‐H and CN‐L intermediate outcomes, and CN‐H ECs having the worst outcomes.
There is recent interest in the tyrosine kinase receptor HER2, encoded by the erb‐b2 receptor tyrosine kinase 2 (ERBB2) oncogene, as a therapeutic target for high‐grade ECs. ERBB2 amplification leads to HER2 protein overexpression and is correlated with poor prognosis in several tumor types, including breast, gastroesophageal, and ECs [2, 3, 4]. Anti‐HER2 therapies, including the monoclonal antibody, trastuzumab, constitute an important therapeutic option for HER2‐positive breast and gastroesophageal tumors [5]. Trastuzumab has been incorporated into National Comprehensive Cancer Network guidelines for treatment of advanced and recurrent serous EC with HER2 overexpression/ERBB2 amplification [6], based on a randomized phase II study demonstrating improved survival outcomes in this patient population [7, 8].
ERBB2 amplification in EC is primarily restricted to those of CN‐H/TP53‐abnormal (TP53abn) molecular subtype [9, 10], and ERBB2‐amplified serous/high‐grade carcinomas likely represent only a subset of all HER2‐driven ECs. In addition to amplification, ERBB2 may also be altered by somatic mutations, which has been described in other tumor types, including breast, bladder, gastrointestinal, and lung cancers [11, 12, 13, 14, 15]. Pan‐cancer sequencing studies have revealed ERBB2 mutations to be most common in bladder/urinary tract cancers (7–8%), followed by stomach (4–5%) and bile duct (4–5%) cancers [11, 12, 13, 14, 15]. Mutations involving the tyrosine kinase domain, encompassing exons 19, 20, and 21 (amino acids 720–987) are most prevalent overall, however, specific mutations vary in frequency between different tumor types. For example, in non‐small cell lung cancer, the most common mutation is p.Y772_A775dup, while biliary tract and breast cancers more commonly harbor S310F/Y and L755 mutations, respectively [11, 12].
In vitro overexpression systems have shown most mutations ultimately increase kinase activity, resulting in HER2 phosphorylation and activation of downstream signaling, accompanied by cellular transformation [11, 16]. ERBB2 mutations are generally considered to confer resistance to trastuzumab, through constitutive activation of kinase activity, despite receptor blockade, or by interfering with drug binding [17]. However, neratinib, an irreversible pan‐HER tyrosine kinase inhibitor, demonstrated promising pre‐clinical activity across different types of ERBB2 mutations, which led to a Phase II basket trial, SUMMIT (NCT01953926), evaluating neratinib in advanced pre‐treated ERBB2‐mutant solid tumors [18]. Clinical responses were variable and dependent on cancer type (with clinical efficacy observed primarily in breast, biliary and cervical cancers), the specific ERBB2 mutation, and presence of other co‐existing mutations. New opportunities for targeting ERBB2 mutations have also emerged with the development of antibody‐drug conjugates (ADCs), including trastuzumab emtansine [19, 20] and trastuzumab deruxtecan [21], which have shown clinical activity in patients with non‐small cell lung cancer with ERBB2 mutations.
Unlike other cancer types, the prevalence and spectrum of ERBB2 mutations in EC, as well as their clinicopathologic associations have not been well characterized. This knowledge could potentially pave the way towards exploring novel therapies to target ERBB2 mutations in this tumor type. Therefore, in this study, we sought to characterize the clinical, histopathologic and genetic features of ECs harboring pathogenic ERBB2 mutations.
2. Materials and methods
2.1. Case selection
The study methodology conforms to the standards set by the Declaration of Helsinki. The study was approved by Memorial Sloan Kettering Cancer Center Institutional Review Board and written informed consent for molecular profiling was obtained from all patients (IRB #12‐245). Of consented EC patients who underwent clinical FDA‐authorized tumor‐normal sequencing using Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT) [22], between 1/2014 and 03/2022 (n = 2638), ECs with pathogenic ERBB2 mutations were identified [23]. Demographic and clinicopathologic data, including age at diagnosis, International Federation of Gynecologic and Obstetrics (FIGO) 2009 stage, clinical follow‐up, as well as information on anti‐HER2 therapy and radiologic response, if applicable, were extracted from electronic medical records. For comparison, 1790 ERBB2 wildtype ECs (including those with ERBB2 amplification, n = 99), annotated with clinical and molecular subtype information (see Section 2.3), were identified from a previously published dataset (1/2014–12/2020) [24]. For analysis of data from the Cancer Genome Atlas (TCGA) study of EC [1], information on ERBB2 mutation, tumor histology and molecular subtype were extracted from the cBioPortal for Cancer Genomics website (http://www.cbioportal.org).
2.2. Sequencing analysis
All ECs included underwent clinical FDA‐authorized tumor‐normal MSK‐IMPACT panel sequencing targeting 341–505 genes, as previously reported [25]. Somatic mutations and tumor mutational burden were extracted from MSK‐IMPACT. ERBB2 somatic mutations were considered pathogenic based on OncoKB [23]. Copy number alterations and loss of heterozygosity (LOH) were defined using facets [26], as previously described [27, 28]. The cancer cell fractions of somatic mutations were computed using absolute (v1.0.6) [29], and a mutation was classified as clonal if its probability of being clonal was > 50% or if the lower bound of the 95% confidence interval of its cancer cell fraction was > 90%, as previously described [27, 28].
2.3. EC molecular subtype classification
Molecular subtyping was performed using our previously described integrated molecular – immunohistochemistry (IHC)‐based approach [24]. In brief, ECs were classified as (1) POLE molecular subtype based on the presence of a POLE hotspot exonuclease domain mutation [30], (2) MSI‐H molecular subtype if the MSK‐IMPACT‐based MSIsensor score [31] was ≥ 10 and/or DNA mismatch repair‐deficient (MMR)‐deficient based on IHC, (3) CN‐H/TP53abn molecular subtype based on the presence of a pathogenic TP53 genetic alteration, or (4) CN‐L/no specific molecular profile (NSMP) if any of the defining features of the other subtypes were lacking.
2.4. Histopathologic review and immunohistochemical analysis
All available diagnostic slides from ERBB2‐mutated (ERBB2‐mut) ECs were re‐reviewed by a gynecologic pathologist (M.H.C.) for confirmation of histological subtype and grade, according to WHO 2020 criteria [32]. HER2 IHC was performed (clone 4B5; Ventana, Tucson, AZ, USA) on all available cases, on the same tissue block used for MSK‐IMPACT sequencing. The percentage of tumor cells with absent, weak, moderate, or strong membranous staining was estimated, and HER2 IHC score was assigned, using newly proposed EC‐specific guidelines, based on criteria used in the clinical trial by Fader et al. [7, 8], endorsed by the College of American Pathologists [33].
2.5. Statistical analysis
Correlative analyses between ERBB2 mutation status and clinicopathologic variables were performed using Wilcoxon rank sum test and Fisher's exact test, for continuous and categorical variables, respectively, with multiple comparisons adjusted using the Benjamini and Hochberg method. For survival analyses, only patients who received their primary treatment at MSK and had MSK‐IMPACT sequencing performed on primary tumors were included [n = 1012, including ERBB2‐mut/non‐amplified (mut/non‐amp), n = 34, ERBB2‐wildtype/non‐amplified (wt/non‐amp), n = 936, ERBB2‐wildtype/amplified (wt/amp), n = 39, ERBB2‐mut/amplified (mut/amp), n = 3]. Progression‐free survival (PFS) was defined from the time of pathologic diagnosis of EC to first recurrence or progression, by imaging or pathologic confirmation, death or last follow‐up date, whichever came first. Overall survival (OS) was defined as the time from diagnosis to death or last follow‐up. Non‐events were censored at the last follow‐up date. Left truncation methodology was applied to address selection bias as patients needed to be selected after the date of MSK‐IMPACT, as previously described [34]. Survival curves were generated using the Kaplan–Meier survival method, and hazard ratios (HR) with 95% confidence intervals (CI) and P‐values were obtained by the Cox proportional hazard model, accounting for left truncation. All tests were two‐sided and a P‐value < 0.05 was considered statistically significant. All analyses were performed using r version 4.1.2 (https://www.R‐project.org/).
3. Results
3.1. ERBB2 mutations in EC
From a cohort of 2638 ECs across histologic types, 69 (2.6%) had known pathogenic ERBB2 mutations, of which 11 (16%) also had concurrent ERBB2 amplification, and 8 (12%) ECs harbored multiple pathogenic ERBB2 mutations. The most frequent ERBB2 hotspot mutations were V842I (26/69, 38%), located in the kinase domain, and R678Q (16/69, 23%), situated in the juxtamembrane domain (Fig. 1A). Other recurrent mutations included kinase domain mutations (L755S, n = 5; D769H, n = 3; T862A, n = 3; V777M, n = 2), and mutations involving the furin‐like extracellular domain (S310F/Y, n = 6) and juxtamembrane domain (V697L, n = 2). Of 61 evaluable cases with sufficient tumor purity, assessment of the cancer cell fractions revealed that ERBB2 mutations were clonal in 87% (n = 53) of cases.
Fig. 1.
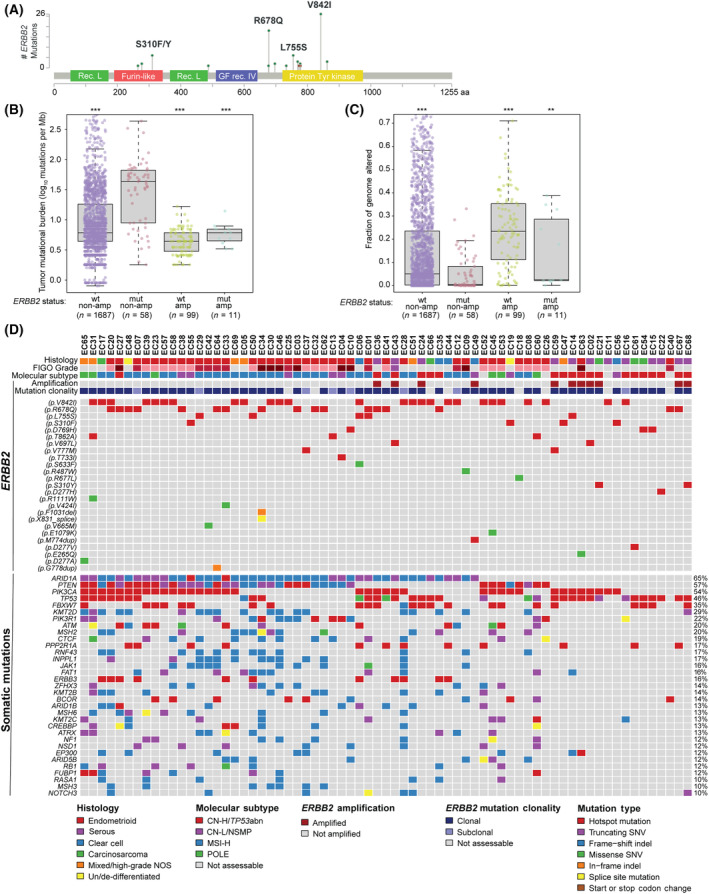
Genomic landscape of 69 ERBB2‐mutated endometrial carcinomas. (A) Lollipop plot showing frequencies of specific ERBB2 activating mutations. (B, C) Targeted panel sequencing‐based tumor mutational burden (B) and fraction of genome altered (C), stratified by ERBB2 mutation (wt, wildtype; mut, mutated) and copy number (non‐amp, non‐amplified; amp, amplified) status. Boxplots show median with interquartile range (IQR), with boundaries of whiskers at 1.5 times IQR. **P < 0.01, ***P < 0.001, Wilcoxon rank sum test. (D) Oncoplot displaying ERBB2 mutations and recurrent somatic mutations in ERBB2‐mutated endometrial cancers. Mutation types, histologic subtype, molecular subtype, ERBB2 amplification status, and clonality of ERBB2 mutations are annotated according to the legend. CN‐H/TP53abn, copy number‐high/TP53 abnormal; CN‐L/NSMP, copy number‐low/no specific molecular profile; high‐grade EC‐NOS, high‐grade endometrial carcinoma, not otherwise specified; Indel, insertion/deletion; MSI‐H, microsatellite instability‐high; SNV, single nucleotide variant.
3.2. Somatic genetic landscape of ERBB2‐mutated ECs
The global genomic landscape of ERBB2‐mut/non‐amp ECs was characterized by a significantly higher tumor mutational burden (TMB; median 43.2 mutations per Mb, range: 1.8–436.2), relative to ECs lacking ERBB2 mutation or amplification (ERBB2‐wt/non‐amp: median 6.1 mutations per Mb, range: 0.8–667.9, P < 0.001) and ECs with ERBB2 amplification, but no mutation (ERBB2‐wt/amp: median 4.4 mutations per Mb, range: 1.8–16.7, P < 0.001, Fig. 1B). Chromosomal instability, inferred from the fraction of genome altered (FGA), was low in ERBB2‐non‐amp ECs, particularly those with ERBB2 mutation (ERBB2‐mut/non‐amp: 0.5%, range: 0–33.1% vs ERBB2‐wt/non‐amp: 5.1%, range: 0–95.7%, P < 0.001), in contrast to ERBB2‐wt/amp ECs, which typically showed high FGAs (23.4%, range: 0–71.0%, P < 0.001; Fig. 1C). Overall, the rare ECs with both ERBB2 mutation and amplification (ERBB2‐mut/amp) had relatively low TMB (6.1 mutations per Mb, range: 3.3–14) and FGA (2.5%, range: 0.08–38.8%), though definitive conclusions cannot be drawn due to the limited numbers.
Assessment of cancer gene alterations revealed that the most frequent co‐existing mutations in ERBB2‐mut ECs involved ARID1A (65%), PTEN (57%) and PIK3CA (54%), which are characteristic of endometrioid and clear cell carcinomas [1, 24] (Fig. 1D). Genetic alterations typical of high‐grade ECs, namely, TP53 (46%), FBXW7 (35%) and PPP2R1A (17%), were also observed [1, 35].
For comparison, among the publicly available TCGA cohort of 529 ECs that underwent whole‐exome sequencing, 15 (2.8%) cases harbored ERBB2 pathogenic mutations, of which one case harbored two distinct ERBB2 mutations (V842I and L755S) [1]. Consistent with the results from our cohort, V842I (n = 4) and R678Q (n = 5) were the most common mutations, and the only other recurrent mutation was L755S (n = 3).
3.3. Clinicopathologic features and associations with molecular subtype
The spectrum of EC histologic subtypes was represented among ERBB2‐mut/non‐amp ECs (Fig. 2A, Table 1), including endometrioid (66%; of which 79% were Grades 1 or 2), serous (6.9%) and clear cell (6.9%) carcinomas, carcinosarcoma (6.9%), mixed EC/high‐grade EC, not otherwise specified (10%), and undifferentiated/de‐differentiated EC (3.4%). Similar frequencies were observed across ERBB2‐wt/non‐amp ECs. However, significant differences became apparent when stratifying by molecular subtype [24]. Consistent with the high TMB observed in ERBB2‐mut/non‐amp ECs, these tumors were enriched for MSI‐H (59%) and POLE (11%) molecular subtypes (compared to 24% MSI‐H and 5.6% POLE in ERBB2‐wt/non‐amp ECs, P < 0.001). Of note, TMB was consistently higher in ERBB2‐mut/non‐amp compared to ERBB2‐wt/non‐amp ECs, even within MSI‐H (ERBB2‐mut/non‐amp: median 50.3 mutations per Mb, range: 4.4–88.6, vs ERBB2‐wt/non‐amp: median 29.8 mutations per Mb, range: 0.9–397.9; P < 0.001) and microsatellite‐stable (MSS; ERBB2‐mut/non‐amp: median 10.1 mutations per Mb, range: 3.3–436.2, vs ERBB2‐wt/non‐amp: median 5.3 mutations per Mb, range: 0.8–667.9; P < 0.001) subgroups, whilst no significant differences in FGA values were observed (Fig. S1A,B). Patients with ERBB2‐mut/non‐amp and ERBB2‐wt/non‐amp ECs had a similar age distribution (median 60 vs 63, P = 0.041) and did not significantly differ with respect to body mass index (BMI, median 28.0 vs 29.7 kg·m−2, P = 0.08) or stage at presentation (P = 0.22). Consistent with our findings, in the TCGA cohort [1], most ERBB2‐mut ECs (11/15, 73%) were endometrioid and of MSI‐H molecular subtype (n = 11, 73%).
Fig. 2.
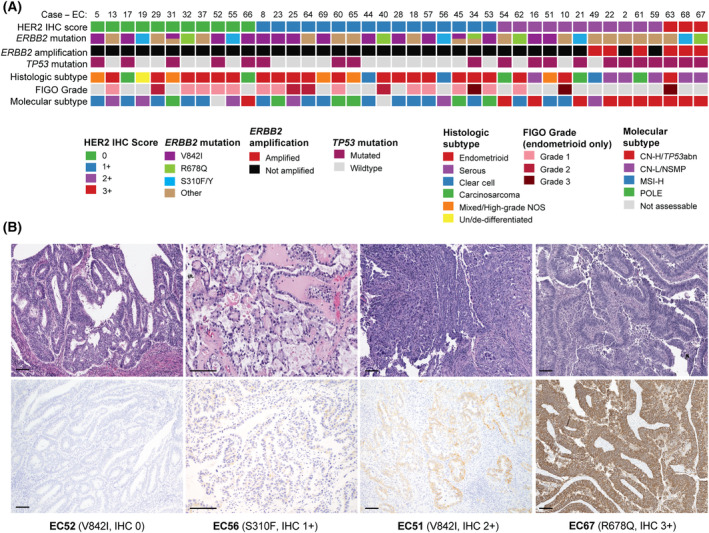
Histologic features and HER2 immunohistochemical analysis of ERBB2‐mutated endometrial carcinomas. (A) Stratification of ERBB2‐mutated endometrial carcinomas by HER2 immunohistochemistry (IHC) score (n = 41). HER2 IHC scores, and other tumor characteristics are color‐coded according to the legend. CN‐H/TP53abn, copy number‐high/TP53 abnormal; CN‐L/NSMP, copy number‐low/no specific molecular profile; high‐grade EC‐NOS, high‐grade endometrial carcinoma, not otherwise specified; MSI‐H, microsatellite instability‐high. (B) Photomicrographs of representative cases (H&E and HER2 IHC): EC52, endometrioid carcinoma, Grade 1 (100× magnification); EC56, clear cell carcinoma (200× magnification); EC51, high‐grade EC‐NOS (100× magnification); EC67, serous carcinoma (ERBB2‐mutated and amplified, 100× magnification). Scale bar represents 100 μm.
Table 1.
Demographic and clinicopathologic features of endometrial cancers stratified by ERBB2 genetic alteration status. The number of cases in each category do not always sum up to the total number of cases in the cohort due to missing data values. amp, amplified; CN‐H/TP53abn, copy number‐high/TP53 abnormal; CN‐L/NSMP, copy number‐low/no specific molecular profile; FIGO, International Federation of Gynecology and Obstetrics; High‐grade EC‐NOS, high‐grade endometrial carcinoma, not otherwise specified; MSI‐H, microsatellite instability‐high; mut, mutated; non‐amp, non‐amplified; wt, wildtype.
| Characteristic | ERBB2 mutation/copy number status, N = 1859 | P‐value a | ||||
|---|---|---|---|---|---|---|
| wt/non‐amp, N = 1691, n (%) | mut/non‐amp, N = 58, n (%) | wt/amp, N = 99, n (%) | mut/amp, N = 11, n (%) | mut/non‐amp vs wt/non‐amp | mut/non‐amp vs wt/amp | |
| Age at diagnosis [median, years (range)] | 63 (21–96) | 60 (31–83) | 66 (54–86) | 70 (54–75) | 0.041 | < 0.001 |
| BMI [median, kg·m−2, (range)] | 29.7 (14.9–67.6) | 28.0 (16.9–48.4) | 28.3 (19.3–48.2) | 29.3 (21.6–52.7) | 0.08 | 0.95 |
| Stage (FIGO 2009) | ||||||
| I | 906 (59) | 40 (71) | 25 (29) | 3 (33) | 0.22 | < 0.001 |
| II | 65 (4.2) | 3 (5.4) | 4 (4.7) | 1 (11) | ||
| III | 309 (20) | 9 (16) | 24 (28) | 2 (22) | ||
| IV | 251 (16) | 4 (7.1) | 32 (38) | 3 (33) | ||
| Histologic type | ||||||
| Endometrioid | 948 (56) | 38 (66) | 7 (7.1) | 1 (9.1) | 0.12 | < 0.001 |
| Serous | 233 (14) | 4 (6.9) | 34 (34) | 8 (73) | ||
| Clear cell | 47 (2.8) | 4 (6.9) | 5 (5.1) | 1 (9.1) | ||
| Carcinosarcoma | 196 (12) | 4 (6.9) | 25 (25) | 1 (9.1) | ||
| Mixed/high‐grade EC‐NOS | 140 (8.3) | 6 (10) | 23 (23) | 0 (0) | ||
| Undifferentiated/de‐differentiated | 36 (2.1) | 2 (3.4) | 1 (1.0) | 0 (0) | ||
| Unclassifiable | 91 (5.4) | 0 (0) | 4 (4.0) | 0 (0) | ||
| FIGO grade (for endometrioid only) | ||||||
| 1 or 2 | 741 (81) | 30 (79) | 5 (71) | 0 (0) | 0.84 | 0.64 |
| 3 | 175 (19) | 8 (21) | 2 (29) | 1 (100) | ||
| Molecular subtype | ||||||
| POLE | 95 (5.6%) | 6 (11%) | 0 | 0 | < 0.001 | < 0.001 |
| MSI‐H | 404 (24%) | 32 (59%) | 0 | 0 | ||
| CN‐L/NSMP | 561 (33%) | 7 (13%) | 8 (8.1%) | 1 (9.1%) | ||
| CN‐H/TP53abn | 631 (37%) | 9 (17%) | 91 (92%) | 10 (91%) | ||
Fisher exact test, two‐tailed.
Significant differences between ERBB2‐mut/non‐amp versus ERBB2‐wt/amp ECs were observed with respect to age (P < 0.001), stage (P < 0.001), and distribution of histologic (P < 0.001) and molecular subtypes (P < 0.001; Table 1). Specifically, patients with ECs with ERBB2 amplification were significantly older, more frequently presented at advanced stage, with enrichment of high‐grade histologic types (serous/mixed carcinomas and carcinosarcoma), and CN‐H/TP53abn molecular subtype. Of the 11 ERBB2‐mut/amp ECs, 10/11 were CN‐H/TP53abn (serous, n = 8, grade 3 endometrioid, n = 1, carcinosarcoma, n = 1), and the remaining case was an MSI‐H clear cell carcinoma.
3.4. Genetic features of MSI‐H ERBB2‐mutated ECs
Across ERBB2‐mut ECs, those of MSI‐H molecular subtype were particularly enriched for V842I and R678Q hotspot mutations (25/32, 78%, of MSI‐H, vs 14/33, 42%, of other molecular subtypes, P = 0.005). Of the 32 ERBB2‐mut EC of MSI‐H molecular subtype, the mechanism of MMR‐deficiency/MSI varied, with 9 (28%) being associated with MLH1 promoter hypermethylation. Of those negative for MLH1 promoter hypermethylation, available germline testing results revealed 9/21 (43%) cases were associated with Lynch syndrome, with an underlying pathogenic germline mutation in one of the MMR genes, including MLH1 (n = 1), MSH2 (n = 4) or MSH6 (n = 4). In addition, 1 (4.8%) had an MUTYH germline mutation (along with MSH2 somatic mutations). Somatic MMR gene mutations were present in 12/21 (57%) cases. For the two patients that were negative for MLH1 promoter hypermethylation and of unknown germline status, one had isolated MSH6 loss by IHC without any MMR gene mutations, and the other had loss of MSH2 and MSH6 expression and co‐existing MSH2 tumor mutations.
3.5. Immunohistochemical analysis of HER2 protein expression in ERBB2‐mutated ECs
HER2 IHC was performed (Fig. 2A,B) on 41 ERBB2‐mut ECs with available tissue and demonstrated that the majority had low levels of HER2 expression, with the following distribution of HER2 IHC scores: 0, 27% (n = 11); 1+, 39% (n = 16); 2+, 27% (n = 11); and 3+, 7.3% (n = 3). All tumors with IHC 3+ and 3/11 (27%) with IHC 2+ harbored both ERBB2 mutation and amplification. There was a significant association between the presence of a TP53 mutation and higher levels of HER2 protein expression, with TP53 mutation observed in 11/14 (79%) ECs with HER2 IHC scores of 2+/3+, compared to 9/27 (33%) cases with HER2 IHC 0/1+ scores (P = 0.009). Similarly, the CN‐H/TP53abn molecular subtype (which excludes MSI‐H ECs with TP53 mutation), was associated with increased HER2 protein expression (10/14, 71%, of HER2 IHC 2+/3+, vs 1/27, 3.7%, of HER2 IHC 0/1+, P < 0.001). The most common mutations (V842I, R678Q, S310F/Y) were observed in ECs across the range of HER2 IHC scores and there was no apparent relationship between specific mutation and HER2 expression level.
3.6. Clinical outcomes and response to trastuzumab therapy
The median follow‐up for the 1012 EC patients who met criteria for survival analysis (see Section 2; ERBB2‐wt/non‐amp, n = 936; ERBB2‐mut/non‐amp, n = 37; ERBB2‐wt/amp, n = 39; ERBB2‐mut/amp, n = 3) was 21.8 months (range 0.6–214.5 months) and there were 83 deaths. Median PFS was not reached for non‐amplified ERBB2‐wt and ERBB2‐mut ECs (mut/non‐amp vs wt/non‐amp: HR 0.51, 95% CI 0.19–1.36) and was 12.8 (95% CI 7.8–18.4) months for ERBB2‐wt/amp ECs (wt/amp vs wt/non‐amp: HR 4.32, 95% CI 2.84–6.57, P < 0.001; Fig. 3A, Table S1). Median OS was not reached for ERBB2‐wt/non‐amp and ERBB2‐mut/non‐amp ECs (mut/non‐amp vs wt/non‐amp: HR 0.39, 95% CI 0.05–2.78) and was 31.9 (95% CI 24.8‐NE) months for ERBB2‐wt/amp ECs (wt/amp vs wt/non‐amp: HR 4.35, 95% CI 2.16–8.76, P < 0.001; Fig. 3B). PFS and OS were un‐estimable for ERBB2‐mut/amp ECs, due to limited sample size. ERBB2 genetic alteration status was no longer significant on multivariate analysis after adjusting for age, stage and molecular subtype (Table 2). Among ERBB2‐mut ECs, no survival differences were observed between cases harboring V842I or R678Q ERBB2 mutations compared to other pathogenic ERBB2 mutations.
Fig. 3.
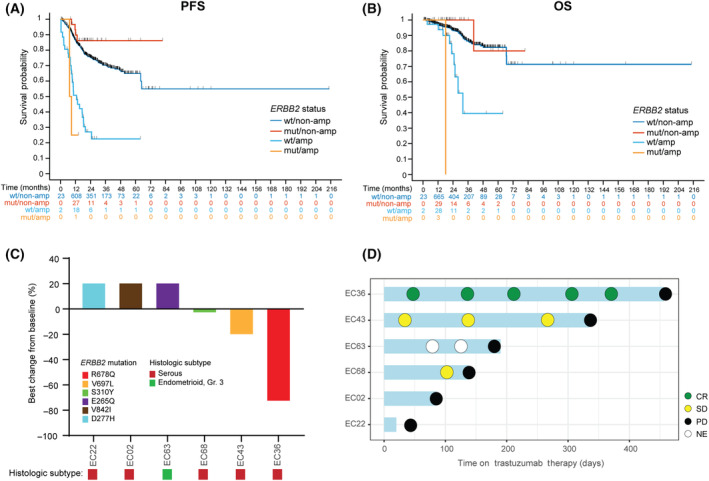
Clinical outcomes of ERBB2‐mutated endometrial carcinomas. (A) Progression‐free (PFS) and (B) overall survival (OS) in endometrial carcinomas stratified by ERBB2 mutation (wt, wildtype; mut, mutated) and copy number (non‐amp, non‐amplified; amp, amplified) status. (C) Waterfall plot showing treatment responses to trastuzumab combined with chemotherapy. In EC02, EC22, and EC63, bars showing a 21% increase denotes appearance of new non‐target lesions at first evaluable computed tomography scan performed while on treatment. Specific ERBB2 mutations and histologic subtype are color‐coded as indicated in the legend. (D) Swimmer's plot showing best response to trastuzumab, time on treatment, and time to disease progression. CR, complete response; NE, not evaluable (non‐CR, non‐PD); PD, progression of disease; PR, partial response.
Table 2.
Multivariate analysis for progression‐free and overall survival. CI, confidence interval; CN‐H/TP53abn, copy number‐high/TP53 abnormal; CN‐L/NSMP, copy number‐low/no specific molecular profile; FIGO, International Federation of Gynecology and Obstetrics; HR, hazard ratio; MSI‐H, microsatellite instability‐high.
| Characteristic | Progression‐free survival | Overall survival | ||
|---|---|---|---|---|
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | |
| Age | 1.02 (1.00–1.03) | 0.029 | 1.04 (1.01–1.06) | 0.006 |
| Molecular subtype | ||||
| POLE/MSI‐H | – | < 0.001 | – | < 0.001 |
| CN‐L/NSMP | 1.01 (0.64–1.59) | 1.47 (0.64–3.38) | ||
| CN‐H/TP53abn | 2.86 (1.98–4.13) | 3.91 (1.94–7.86) | ||
| Stage (FIGO 2009) | ||||
| I/II | – | < 0.001 | – | < 0.001 |
| III | 3.96 (2.82–5.55) | 3.78 (2.07–6.90) | ||
| IV | 7.18 (5.09–10.1) | 7.86 (4.52–13.7) | ||
| ERBB2 status | ||||
| Wildtype | – | 0.52 | – | 0.69 |
| Mutated | 0.89 (0.33–2.43) | 0.54 (0.07–3.89) | ||
| Amplified | 1.28 (0.83–1.99) | 1.24 (0.60–2.56) | ||
Six patients with ERBB2‐mut ECs received anti‐HER2 therapy (Fig. 3C,D). All patients were treated with trastuzumab combined with chemotherapy in the recurrent setting. By MSK‐IMPACT, 5 also harbored concurrent ERBB2 amplification; in the remaining case (EC22), low level ERBB2 amplification was detected by fluorescence in situ hybridization only (ERBB2/CEP17 ratio: 2.1, ERBB2 copy number: 3.5). Trastuzumab was administered once every 3 weeks and number of doses received ranged from 1 to 19 (median 8), with treatment lasting until disease progression. Clinical responses with associated specific ERBB2 mutations were as follows: complete response, n = 1 (R678Q), stable disease, n = 2 (V697L, S310Y), and progressive disease, n = 3 (V842I, D277H, E265Q). Median time from treatment initiation to disease progression was 164 days (range 20–456 days).
4. Discussion
Across epithelial malignancies, oncogenic activation of ERBB2 occurs predominantly by gene amplification and less commonly by mutation. In EC, the prevalence of ERBB2 amplification has been reported to be 3.8% across all histologic subtypes [9]. In the current study, we show that a comparable proportion of ECs (2.6%) harbor ERBB2 mutations. While ERBB2 amplification is essentially exclusive to high‐grade histologic types, including serous ECs and carcinosarcomas, of CN‐H/TP53‐altered molecular subtype [9, 24, 35], ERBB2 mutations occur predominantly in an MSI‐H background, and most are low‐grade endometrioid carcinomas. Furthermore, while ERBB2 amplification leads to protein overexpression, the majority of ERBB2‐mut ECs have low or undetectable levels of HER2 expression by IHC. Our results indicate that ERBB2 mutations and amplification, although involving the same gene, define distinct pathologic subgroups of EC, and may necessitate distinct therapeutic approaches.
The observation that ERBB2‐mut ECs have high TMB and associated with MSI‐H and to a lesser extent, POLE molecular subgroups, suggests that ERBB2 mutation originated as part of a “mutator phenotype.” ERBB2 mutations have also been reported at higher frequency in MSI‐H compared to microsatellite‐stable colorectal cancers. In most of the EC cases, ERBB2 mutations were clonal, however, indicating that they likely occurred early in carcinogenesis, followed by selective clonal expansion. Furthermore, our cohort was restricted to ERBB2 variants that were annotated as mutational hotspots and/or “pathogenic.” Our analyses thus provide compelling evidence that ERBB2 mutations are true pathogenic drivers in EC, rather than mere passenger mutations, even when occurring in the context of a high TMB/MSI‐H background.
Despite MLH1 promoter hypermethylation being the more prevalent cause for MMR‐deficiency in EC (~ 70% of MMR‐deficient ECs) [36], MSI‐H ECs with ERBB2 mutations were enriched for germline or somatic MMR mutations (up to 72%). These results complement previous work that reported ERBB2 mutations in only 3% of ECs with MLH1 promoter hypermethylation, compared to 29% and 13% of ECs with MMR germline and somatic mutations, respectively [36]. The biological explanation for this observation is unclear. However, ECs with MLH1 promoter hypermethylation showed lower TMB compared to those harboring MMR germline/somatic mutations [36], which further suggests that ERBB2 activation may selectively promote tumor cell survival in the setting of high TMB.
The prognostic impact of ERBB2 mutations varies across other tumor types. In breast, ERBB2 mutations are associated with poorer OS in invasive lobular, but not ductal, carcinomas [13]. Furthermore, higher rates of complete response to chemotherapy were achieved in ERBB2‐mut compared to ERBB2‐wt bladder cancers [14], while no associations with clinical outcomes were observed in lung cancer [15]. Similarly, ERBB2 mutation status was not associated with prognosis in our EC cohort.
The V842I and V678Q hotspot mutations, in the kinase and juxtamembrane domains respectively, are by far, the most frequent pathogenic variants in EC, together making up 61% of cases, followed by S310F/Y (8.7%). Interestingly, these are also the 3 most common ERBB2 mutations observed in colorectal cancer [37]. Future work is necessary to determine whether these specific mutations preferentially drive carcinogenesis in intestinal and endometrial epithelial cells, particularly in the context of an MSI‐H/high mutation burden genetic background.
Pre‐clinical studies have shown variability with respect to kinase activity, phosphorylation of downstream signaling proteins, transformation potential and drug sensitivity between different mutations [11, 16, 38, 39]. Concerning the most prevalent mutations in our EC cohort, V842I confers in vitro resistance to trastuzumab and a reversible kinase inhibitor, lapatinib, while conflicting data to neratinib was reported across studies [11, 16, 40]. In contrast, R678Q is associated with sensitivity to trastuzumab, lapatinib and neratinib [38, 39]. There is more evidence supporting S310F/Y to be sensitive to anti‐HER2 therapy. A patient‐derived xenograft model of S310Y‐mutated colorectal cancer was sensitive to trastuzumab, lapatinib and neratinib, with the highest activity observed in trastuzumab combined with neratinib [40]. Cabel et al. [41] reported two patients, one with cervical and the other with EC, both harboring ERBB2 S310Y mutation, who achieved partial responses after treatment with a combination regimen of paclitaxel, trastuzumab, and everolimus (due to co‐existing mTOR pathway alteration). In the SUMMIT trial, 3 of 12 patients with cervical cancer treated with neratinib achieved partial responses, and all 3 had tumors with S310Y/F mutations, and one had a V842I mutation [18, 42].
In our cohort, only six patients received trastuzumab therapy, due to concurrent HER2 overexpression/amplification. Interestingly, the only two patients with objective responses had tumors with ERBB2 mutations at positions R678Q and S310Y, respectively, whilst a patient with an ERBB2‐V842I‐mutated EC progressed on therapy, consistent with pre‐clinical functional characterization of these mutations. Co‐existing ERBB2 mutation and amplification is rare and observed in < 5% of solid tumors [43]. Given that most ERBB2 mutations, particularly those in the kinase domain, are associated with resistance to trastuzumab, prior work has shown that in ERBB2‐amp metastatic breast cancer patients who received trastuzumab combined with chemotherapy as first‐line treatment, those with concurrent ERBB2 mutations had shorter PFS compared to the ERBB2‐wt group (median PFS 4.7 vs 11.0 months) [44].
In the SUMMIT basket trial, of the seven patients with ERBB2‐mut EC treated with neratinib, the best response was stable disease in four patients, with ERBB2 mutations at S310Y (n = 2), V777L (n = 1) and R678Q (n = 1), and disease progression in three patients, with ERBB2 mutations at V842I, V697L, and P761del [18]. While the numbers are small, neratinib alone does not appear to be particularly effective for ERBB2‐mut EC. Combination therapy with other therapeutic agents or alternative HER2‐directed therapies should be explored for this patient population. Given the association between ERBB2‐mutation and MSI‐H status, many of these patients would be eligible for immunotherapy [45]; hence combining anti‐HER2 therapy with an immune checkpoint inhibitor may represent a potential strategy.
Emerging HER2 ADCs, which have demonstrated clinical efficacy in HER2‐low/negative tumors may be particularly promising for ERBB2‐mut ECs, most of which, have low or undetectable HER2 expression by IHC. Recent work has demonstrated that ERBB2 mutations enhance internalization of receptor‐bound trastuzumab emtansine, and objective responses trastuzumab emtansine were observed in patients with ERBB2‐mut lung cancer patients, including those with low or undetectable (IHC score 0/1+) HER2 expression [19, 20]. Trastuzumab deruxtecan is a next‐generation HER2‐targeting ADC, which has previously demonstrated remarkable efficacy in HER2‐low breast cancer, attributed, to its potent cytotoxic payload with high drug‐to‐antibody ratio (8 : 1) and its bystander killing effect of neighboring HER2‐non‐expressing tumor cells [46]. In a Phase 2 trial of trastuzumab deruxtecan in patients with ERBB2‐mut lung cancer, most of whom lacked co‐existing ERBB2 amplification, the objective response rate was 55% and durable responses were observed independent of HER2 expression level [21]. As the mechanism of action involves internalization of the receptor‐ADC complex to deliver the cytotoxic payload, rather than inhibition of downstream signaling, ADCs are efficacious across ERBB2 mutations, involving extracellular or kinase domains.
The present study has some limitations inherent to its retrospective nature. The number of ERBB2‐mut ECs that were appropriate to be included in the survival analysis was small, being restricted to those who received their entire treatment course and follow‐up at our institution. Only 6 patients received trastuzumab therapy and all had ERBB2 amplification/HER2 overexpression; with this small sample size, definitive conclusions cannot be drawn concerning the impact of specific mutations on treatment response. Nevertheless, consistent with previous reports [18, 38, 39, 40, 41], clinical benefit was observed in the ECs with ERBB2‐R678Q and S310Y mutations, while the ERBB2‐V842I‐mutated EC was among those resistant to treatment.
5. Conclusions
This retrospective cohort study characterizes the clinicopathologic features and molecular genetic landscape of ECs harboring pathogenic ERBB2 mutations, thereby defining a rare subgroup of ECs, which is enriched for MSI‐H molecular subtype and pathogenically distinct from ERBB2‐wt and ERBB2‐amp ECs. Future prospective trials will be needed to assess the efficacy of other HER2 targeting agents in ERBB2‐mut EC, either as monotherapy or part of a combination regimen, which may include immunotherapy. Since ERBB2‐mut and ERBB2‐amp ECs constitute largely non‐overlapping groups, our results suggest that more ECs patients may potentially benefit from novel anti‐HER2 therapies.
Conflict of interest
BW reports grant funding by Repare Therapeutics, outside the scope of the current study. The remaining authors declare no competing interests.
Author contributions
BW and MHC contributed to conception, design, and supervision. PS contributed to bioinformatics analysis. QZ and AI contributed to statistical analysis. MNB, PS, SM, WM, CD, TB, NRA‐R, CA, LHE, BW, and MHC contributed to data collection. MNB, MHC, and BW contributed to data interpretation/analysis. All authors reviewed, edited, and approved the manuscript.
Peer review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1002/1878‐0261.13698.
Supporting information
Fig. S1. ERBB2‐mutated endometrial carcinomas are associated with increased tumor mutational burden.
Table S1. Univariate associations with progression‐free and overall survival.
Acknowledgements
We thank Irina Linkov (MSK Pathology Core Facility) for assistance with immunohistochemistry, Hunter Green for assistance with archival tissue retrieval, and Michelle Wu for assistance with retrieval of clinical data. Research reported in this publication was supported by a Foundation of Women's Cancer Grant (PI: MHC) and in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748 S5) core facility grant. MNB was supported by Fonds de recherche du Québec – Santé and the Ovarian Cancer Research Alliance. BW is supported in part by Breast Cancer Research Foundation and Cycle for Survival grants.
Britta Weigelt and M. Herman Chui are co‐senior authors.
Data accessibility
Targeted sequencing data supporting the findings of this study will be available at cBioPortal for Cancer Genomics (www.cbioportal.org) upon publication of this manuscript.
References
- 1. Levine DA, Getz G, Gabriel SB, Cibulskis K, Lander E, Sivachenko A, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. 10.1038/nature12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER‐2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J Clin Oncol. 1993;11(10):1936–1942. 10.1200/jco.1993.11.10.1936 [DOI] [PubMed] [Google Scholar]
- 3. Hechtman JF, Polydorides AD. HER2/neu gene amplification and protein overexpression in gastric and gastroesophageal junction adenocarcinoma: a review of histopathology, diagnostic testing, and clinical implications. Arch Pathol Lab Med. 2012;136(6):691–697. 10.5858/arpa.2011-0168-RS [DOI] [PubMed] [Google Scholar]
- 4. Morrison C, Zanagnolo V, Ramirez N, Cohn DE, Kelbick N, Copeland L, et al. HER‐2 is an independent prognostic factor in endometrial cancer: association with outcome in a large cohort of surgically staged patients. J Clin Oncol. 2006;24(15):2376–2385. 10.1200/jco.2005.03.4827 [DOI] [PubMed] [Google Scholar]
- 5. Oh DY, Bang YJ. HER2‐targeted therapies – a role beyond breast cancer. Nat Rev Clin Oncol. 2020;17(1):33–48. 10.1038/s41571-019-0268-3 [DOI] [PubMed] [Google Scholar]
- 6. 2020 National Comprehensive Cancer Network . Uterine neoplasms (version 1.2020). Available from: https://www.nccn.org/ [DOI] [PubMed]
- 7. Fader AN, Roque DM, Siegel E, Buza N, Hui P, Abdelghany O, et al. Randomized phase II trial of carboplatin‐paclitaxel versus carboplatin‐paclitaxel‐Trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. J Clin Oncol. 2018;36(20):2044–2051. 10.1200/JCO.2017.76.5966 [DOI] [PubMed] [Google Scholar]
- 8. Fader AN, Roque DM, Siegel E, Buza N, Hui P, Abdelghany O, et al. Randomized phase II trial of carboplatin‐paclitaxel compared with carboplatin‐paclitaxel‐trastuzumab in advanced (stage III–IV) or recurrent uterine serous carcinomas that overexpress Her2/Neu (NCT01367002): updated overall survival analysis. Clin Cancer Res. 2020;26(15):3928–3935. 10.1158/1078-0432.Ccr-20-0953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ross DS, Devereaux KA, Jin C, Lin DY, Zhang Y, Marra A, et al. Histopathologic features and molecular genetic landscape of HER2‐amplified endometrial carcinomas. Mod Pathol. 2022;35(7):962–971. 10.1038/s41379-021-00997-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vermij L, Horeweg N, Leon‐Castillo A, Rutten TA, Mileshkin LR, Mackay HJ, et al. HER2 status in high‐risk endometrial cancers (PORTEC‐3): relationship with histotype, molecular classification, and clinical outcomes. Cancers (Basel). 2020;13(1):44. 10.3390/cancers13010044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robichaux JP, Elamin YY, Vijayan RSK, Nilsson MB, Hu L, He J, et al. Pan‐cancer landscape and analysis of ERBB2 mutations identifies poziotinib as a clinically active inhibitor and enhancer of T‐DM1 activity. Cancer Cell. 2019;36(4):444–457.e7. 10.1016/j.ccell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H, Miao J, Wen Y, Xia X, Chen Y, Huang M, et al. Molecular landscape of ERBB2 alterations in 14,956 solid tumors. Pathol Oncol Res. 2022;28:1610360. 10.3389/pore.2022.1610360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kurozumi S, Alsaleem M, Monteiro CJ, Bhardwaj K, Joosten SEP, Fujii T, et al. Targetable ERBB2 mutation status is an independent marker of adverse prognosis in estrogen receptor positive, ERBB2 non‐amplified primary lobular breast carcinoma: a retrospective in silico analysis of public datasets. Breast Cancer Res. 2020;22(1):85. 10.1186/s13058-020-01324-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Groenendijk FH, de Jong J, Fransen van de Putte EE, Michaut M, Schlicker A, Peters D, et al. ERBB2 mutations characterize a subgroup of muscle‐invasive bladder cancers with excellent response to neoadjuvant chemotherapy. Eur Urol. 2016;69(3):384–388. 10.1016/j.eururo.2015.01.014 [DOI] [PubMed] [Google Scholar]
- 15. Arcila ME, Chaft JE, Nafa K, Roy‐Chowdhuri S, Lau C, Zaidinski M, et al. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 2012;18(18):4910–4918. 10.1158/1078-0432.ccr-12-0912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237. 10.1158/2159-8290.cd-12-0349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaibar M, Beltrán L, Romero‐Lorca A, Fernández‐Santander A, Novillo A. Somatic mutations in HER2 and implications for current treatment paradigms in HER2‐positive breast cancer. J Oncol. 2020;2020:6375956. 10.1155/2020/6375956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hyman DM, Piha‐Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2‐ and HER3‐mutant cancers. Nature. 2018;554(7691):189–194. 10.1038/nature25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado‐trastuzumab emtansine for patients with HER2‐mutant lung cancers: results from a phase II basket trial. J Clin Oncol. 2018;36(24):2532–2537. 10.1200/jco.2018.77.9777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li BT, Michelini F, Misale S, Cocco E, Baldino L, Cai Y, et al. HER2‐mediated internalization of cytotoxic agents in ERBB2 amplified or mutant lung cancers. Cancer Discov. 2020;10(5):674–687. 10.1158/2159-8290.Cd-20-0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, et al. Trastuzumab deruxtecan in HER2‐mutant non‐small‐cell lung cancer. N Engl J Med. 2022;386(3):241–251. 10.1056/NEJMoa2112431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering‐integrated mutation profiling of actionable cancer targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. 10.1016/j.jmoldx.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017:1–16. 10.1200/po.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rios‐Doria E, Momeni‐Boroujeni A, Friedman CF, Selenica P, Zhou Q, Wu M, et al. Integration of clinical sequencing and immunohistochemistry for the molecular classification of endometrial carcinoma. Gynecol Oncol. 2023;174:262–272. 10.1016/j.ygyno.2023.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen R, Seshan VE. FACETS: allele‐specific copy number and clonal heterogeneity analysis tool for high‐throughput DNA sequencing. Nucleic Acids Res. 2016;44(16):e131. 10.1093/nar/gkw520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst. 2018;110(9):1030–1034. 10.1093/jnci/djy028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Da Cruz Paula A, da Silva EM, Segura SE, Pareja F, Bi R, Selenica P, et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod Pathol. 2020;33(8):1606–1617. 10.1038/s41379-020-0514-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. 10.1038/nbt.2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. León‐Castillo A, Britton H, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol. 2020;250(3):323–335. 10.1002/path.5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable pan‐cancer microsatellite instability assessment by using targeted next‐generation sequencing data. JCO Precis Oncol. 2017;2017:1–17. 10.1200/PO.17.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. WHO Classification of Tumours 5th edition – Female Genital Tumours. WHO Classification of Tumours Editorial Board, editor. Lyon: IARC Press; 2020.
- 33. 2023 College of American Pathologists . Template for reporting results of biomarker testing of specimens from patients with carcinoma of gynecologic origin (version 1.1.0.0). [cited 2024 May 1]. Available from: https://documents.cap.org/documents/Gynecologic.Bmk_1.1.0.0.REL_CAPCP.pdf
- 34. Praiss AM, Marra A, Zhou Q, Rios‐Doria E, Momeni‐Boroujeni A, Iasonos A, et al. TERT promoter mutations and gene amplification in endometrial cancer. Gynecol Oncol. 2023;179:16–23. 10.1016/j.ygyno.2023.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Momeni‐Boroujeni A, Dahoud W, Vanderbilt CM, Chiang S, Murali R, Rios‐Doria EV, et al. Clinicopathologic and genomic analysis of. Clin Cancer Res. 2021;27(9):2613–2623. 10.1158/1078-0432.CCR-20-4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manning‐Geist BL, Liu YL, Devereaux KA, Paula ADC, Zhou QC, Ma W, et al. Microsatellite instability‐high endometrial cancers with MLH1 promoter hypermethylation have distinct molecular and clinical profiles. Clin Cancer Res. 2022;28(19):4302–4311. 10.1158/1078-0432.Ccr-22-0713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ross JS, Fakih M, Ali SM, Elvin JA, Schrock AB, Suh J, et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer. 2018;124(7):1358–1373. 10.1002/cncr.31125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagano M, Kohsaka S, Ueno T, Kojima S, Saka K, Iwase H, et al. High‐throughput functional evaluation of variants of unknown significance in ERBB2. Clin Cancer Res. 2018;24(20):5112–5122. 10.1158/1078-0432.Ccr-18-0991 [DOI] [PubMed] [Google Scholar]
- 39. Pahuja KB, Nguyen TT, Jaiswal BS, Prabhash K, Thaker TM, Senger K, et al. Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell. 2018;34(5):792–806.e5. 10.1016/j.ccell.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5(8):832–841. 10.1158/2159-8290.Cd-14-1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cabel L, Fuerea A, Lacroix L, Baldini C, Martin P, Hollebecque A, et al. Efficacy of histology‐agnostic and molecularly‐driven HER2 inhibitors for refractory cancers. Oncotarget. 2018;9(11):9741–9750. 10.18632/oncotarget.24188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oaknin A, Friedman CF, Roman LD, D'Souza A, Brana I, Bidard FC, et al. Neratinib in patients with HER2‐mutant, metastatic cervical cancer: findings from the phase 2 SUMMIT basket trial. Gynecol Oncol. 2020;159(1):150–156. 10.1016/j.ygyno.2020.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chmielecki J, Ross JS, Wang K, Frampton GM, Palmer GA, Ali SM, et al. Oncogenic alterations in ERBB2/HER2 represent potential therapeutic targets across tumors from diverse anatomic sites of origin. Oncologist. 2015;20(1):7–12. 10.1634/theoncologist.2014-0234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yi Z, Rong G, Guan Y, Li J, Chang L, Li H, et al. Molecular landscape and efficacy of HER2‐targeted therapy in patients with HER2‐mutated metastatic breast cancer. NPJ Breast Cancer. 2020;6:59. 10.1038/s41523-020-00201-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ott PA, Bang YJ, Berton‐Rigaud D, Elez E, Pishvaian MJ, Rugo HS, et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1‐positive endometrial cancer: results from the KEYNOTE‐028 study. J Clin Oncol. 2017;35(22):2535–2541. 10.1200/JCO.2017.72.5952 [DOI] [PubMed] [Google Scholar]
- 46. Ferraro E, Drago JZ, Modi S. Implementing antibody‐drug conjugates (ADCs) in HER2‐positive breast cancer: state of the art and future directions. Breast Cancer Res. 2021;23(1):84. 10.1186/s13058-021-01459-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ERBB2‐mutated endometrial carcinomas are associated with increased tumor mutational burden.
Table S1. Univariate associations with progression‐free and overall survival.
Data Availability Statement
Targeted sequencing data supporting the findings of this study will be available at cBioPortal for Cancer Genomics (www.cbioportal.org) upon publication of this manuscript.