

Abstract
Intrinsically disordered protein regions exist in a collection of dynamic interconverting conformations that lack a stable three-dimensional structure. These regions are structurally heterogeneous, ubiquitous, and found across all kingdoms of life. Despite the absence of a defined 3D structure, disordered regions are essential for cellular processes ranging from transcriptional control and cell signalling to sub-cellular organization. Through their conformational malleability and adaptability, disordered regions extend the repertoire of macromolecular interactions and are readily tunable by their structural and chemical context, making them ideal responders to regulatory cues. Recent work has led to major advances in understanding the link between protein sequence and conformational behaviour in disordered regions, yet the link between sequence and molecular function is less well-defined. Here, we consider the biochemical and biophysical foundations that underlie how and why disordered regions can engage in productive cellular functions, provide examples of emerging concepts, and discuss how protein disorder contributes to intracellular information processing and regulation of cellular function.
Introduction
Molecular interactions directly determine cellular fate and function. Proteins are the central conduits for the reception, processing, and transmission of cellular information, a collection of activities we refer to as ‘molecular communication’. Proteins often control biological function through well-structured molecular interactions mediated by folded domains. However, many proteins also possess intrinsically disordered regions (IDRs)1-5, protein domains that can mediate essential cellular interactions without long-lived (stable) structures.
IDRs are defined by an amino acid sequence that gives rise to dynamic polypeptide chains unable to acquire a stable tertiary structure3. This inability to fold often reflects an insufficient proportion of hydrophobic amino acids to form a hydrophobic core. Despite the absence of a well-defined 3D structure, IDRs are essential for cellular function. They are found across all cellular locations, from integral membrane proteins to soluble cytoplasmic proteins to chromatin-associated proteins (Fig. 1). They function in cellular processes including but not limited to transcription, translation, signalling, cell division, genome maintenance, immune surveillance, circadian biology, and cellular homeostasis6-15. On the molecular scale, IDRs can function as flexible linkers, as tunable modules for molecular recognition, as binding interfaces for simultaneous interactions with multiple partners, as cellular sensors, and as drivers of subcellular organization3,16-22. IDRs range in length from short (5-10 residue) to long (1000+ residue) regions, and can exist as tails, linkers, and loops. Along an IDR, distinct sequence properties can be concentrated in specific parts of the sequence, enabling discrete molecular functions to co-exist in a single IDR23-25. While serving a variety of functions, a common feature shared by many IDRs is their ability to enable multivalent, tunable, and malleable molecular recognition that would otherwise be challenging to mediate via folded domains. In this way, IDRs offer a route to enhance and expand molecular communication.
Figure 1: IDRs are central to cellular function.
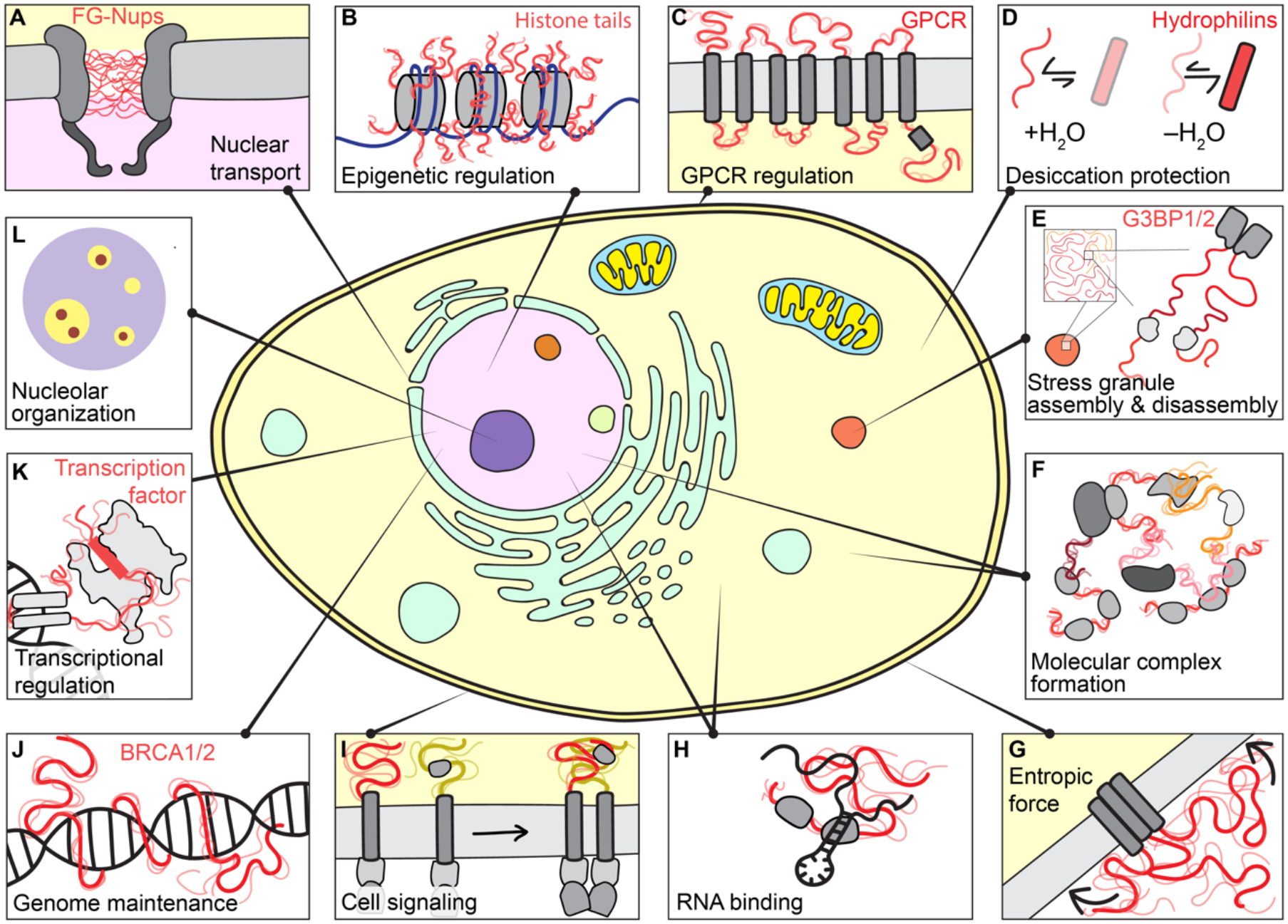
IDRs play critical cellular roles across cellular compartments. From top left clockwise. (a) The nuclear pore complex is a macromolecular portal that controls the partitioning of biomolecules between the nucleus and cytosol and regulate passage through the nuclear envelope. The central lumen of its pore is filled with a chemically-tuned meshwork of IDRs — phenylalanine-glycine (FG) repeats — from nucleoporin (Nup) proteins that enable selectivity through favourable transient interactions with nuclear transport receptors. (b) Histones are among the most abundant proteins in Eukaryotes and act as positively charged counterions to compact negative DNA into chromatin. Histone tails are IDRs that undergo extensive post-translational modification (PTM), enabling both changes to the intrinsic biophysical behaviour and the recruitment or exclusion of partner proteins to determine epigenetic state. (c) G-protein coupled receptors (GPCRs) are a large class of membrane-bound receptors that transduce extracellular stimuli into chemical information. Many GPCRs contain IDRs in their intracellular and extracellular loops and tails. These IDRs are highly variable in composition and length, suggesting they may act as evolutionary-labile sensors connected to a more conserved signal-transduction machine. (d) For many organisms, resilience to low levels of water is among the strongest selective pressures. Most identified desiccation-resistance proteins (e.g., hydrophilins, CAHS proteins etc.) are disordered when in aqueous environments, although many also acquire helicity upon desiccation. The molecular details that underlie how and why disordered proteins appear to play key roles in desiccation tolerance remains enigmatic. (e) Stress granules are an evolutionarily conserved class of cytoplasmic condensate that form in response to cellular stress. In humans, stress granule formation often depends on the largely disordered paralogous proteins G3BP1/2. More broadly, however, many core stress granule proteins contain large IDRs, potentially related to their roles in RNA binding and environmental responsiveness. (f) IDRs are often found in multidomain proteins that facilitate the formation of large dynamic macromolecular complexes. In these, they may act as flexible linkers connecting folded domains, or as molecular recognition modules that facilitate complex formation. (g) IDRs can exert entropic force, here shown in membrane proteins. Any reduction in available volume of an IDR – for example, by the presence of an adj acent membrane – results in a corresponding force proportional to the entropic cost levied by the lost volume (highlighted by arrows). (h) IDRs are often found in RNA binding proteins. They can bind RNA directly and can enhance or suppress the binding affinity of canonical RNA binding domains. Given the size mismatch between mRNA and most proteins, productive RNA recognition events may require the collective behaviour of many proteins, and IDRs may contribute to both protein–protein and protein–RNA interactions. (i) Transmembrane signalling proteins (e.g., T-cell receptors, cytokine receptors, and growth factor receptors) often contain intracellular disordered regions that contribute to signal amplification upon receptor clustering. These regions can interact with other IDRs, act as a platform upon which downstream signalling molecules can co-assemble or undergo PTMs (especially phosphorylation) to indicate signalling status. (j) Genome maintenance represents an essential set of cellular programmes conserved from yeast to humans. Many of the core proteins that drive central steps in different aspects of genome maintenance contain large IDRs with important cellular functions (e.g., p53, BRCA1, BRCA2, ATM, MLH, XPA). These IDRs may aid in the coordination of DNA repair by recruiting other proteins but may also interact directly with DNA. (k) Transcription factors are DNA-binding proteins that dictate the set of genes being expressed at any given moment. Most transcription factors contain IDRs. In addition to mediating the recruitment of appropriate partner proteins – which also typically contain IDRs – to activate or repress gene expression (often via folding-upon binding), emerging work suggests transcription factor IDRs can even guide the specific of transcription factors for DNA sequences. (l) Biomolecular condensates are membrane-less non-stochiometric assemblies that concentrate specific biomolecules and exclude others. IDRs, owing to their multivalency, can participate in phase transitions associated with biomolecular condensate formation. In particular, the nucleolar substructure observed in vitro and in vivo is coordinated at least in part by sequence features in IDRs. These observations illustrate how mesoscopic organization can emerge despite disorder at the level of individual molecules.
Protein disorder is ubiquitous across the kingdoms of life. In eukaryotic proteomes, 30-40% of residues are in IDRs, with a similar fraction in many viruses25,26. An entire protein can be disordered, in which case the protein is referred to as an intrinsically disordered protein (IDP). However, most protein disorder is found in IDRs positioned terminally (tails) or connecting two folded domains (linkers) (Fig. 2a and Box 1). Around 70% of proteins in the human proteome possess one or more IDRs of 30 residues or longer (see Box 1). While prokaryotes contain fewer IDRs (see Box 1), emerging work suggests these also play key roles27.
Figure 2: IDRs exist in ensembles dictated by protein sequence features.
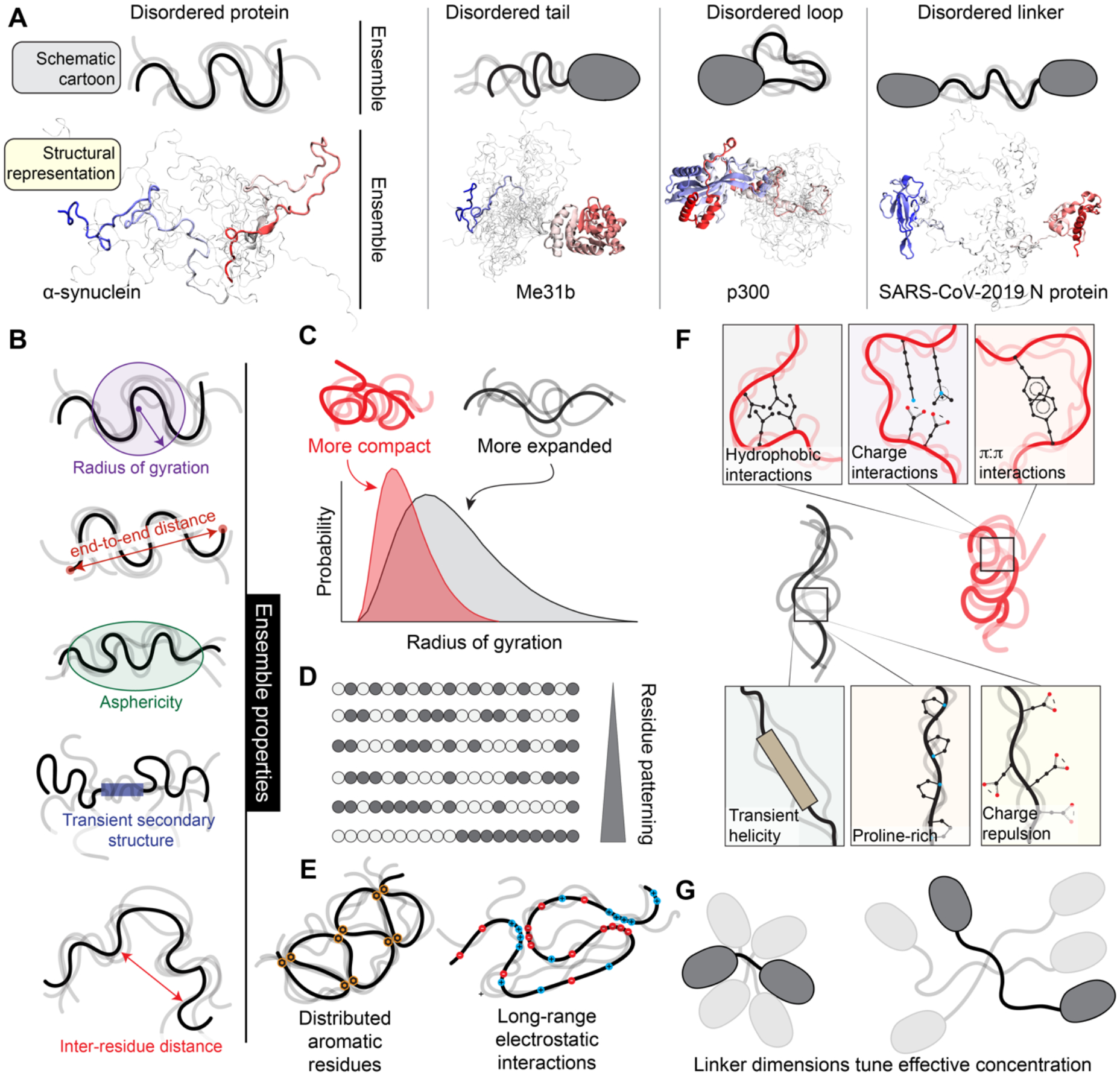
(a). IDRs exist in ensembles — a collection of dynamic conformations that are energetically accessible to a disordered region. Although folded domains also exist in ensembles, the conformations associated with a folded domain are typically structurally similar. By contrast, for IDRs, ensemble conformations are highly heterogeneous. Here we compare structural models for IDR ensembles in different molecular contexts (bottom) with schematized representations of IDR ensembles (top). Only a small number of separate conformations are shown for visual accessibility, however in reality, IDRs exchange between tens of thousands of different conformations. The four proteins depicted here are examples of IDRs from either a fully disordered protein (furthest left) or IDRs in different structural contexts. In each representation, one specific conformation is highlighted, and a collection of additional conformations are superimposed in shaded lines, with the goal of illustrating the structural heterogeneity associated with an ensemble. For a clearer demonstration of an ensemble, see Movie M1, a rendering from an all-atom simulation of the low complexity domain from the RNA binding protein hnRNPA1 (see Box 1). PDB codes for structures: left (homology model based on PDB:4CT5), centre (PDB:6GYR), right (PBD: 6YI3); note disordered regions are not visible in deposited PDB structures. (b) Because IDRs exist in ensembles, they cannot be represented by a single 3D structure. Consequently, IDR ensembles are described in terms of ensemble properties: specific metrics that can be measured, calculated, or predicted for the collection of conformations to quantify the ensemble. Commonly used ensemble properties include the radius of gyration and the end-to-end distance (measures of global ensemble dimensions), asphericity (a measure of ensemble shape), transient secondary structure (a measure of local structural acquisition) and inter-residue distances (a measure of specific ensemble dimensions). These properties can be calculated from simulations or measured experimentally (see Box 2). (c) IDR ensemble properties should ideally be described in terms of probability distributions. For example, the distribution of the radius of gyration is shown for two IDRs. One IDR (red) is compact, while the other IDR (black) is more expanded. (d) IDR ensembles often depend on residue patterning, which quantifies how segregated/clustered residues of one chemical group (here depicted as white or grey beads) are with respect to another. (e) Local sequence properties can influence IDR ensembles, such as charge patterning (left) and evenly spaced aromatic residues (right). (f) Overall, IDR ensemble properties are a consequence of the sequence-encoded physical chemistry and the context-dependence of interactions endowed by that physical chemistry. (g) Ensemble properties of IDR linkers tune the effective concentration of folded domains to one another. Two folded domains connected by a short IDR are inherently close to one another, yet if long IDRs are relatively compact, folded domains will remain close, despite the superficially “large” intervening disordered linker (see panel 2c). For two domains that interact with one another, linker properties (modulated via post-translational modifications or changes in linker sequence over evolution) can therefore tune inter-domain communication, thereby influencing local inhibition or activation or altering binding affinity for target molecules.
Box 1. Identifying IDRs.
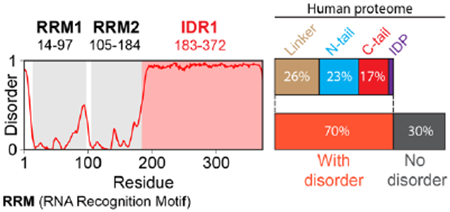
Early work on IDRs was driven by bioinformatics, with initial predictors enabling disordered and folded domains to be delineated 391-394 Over the last twenty-five years, disorder predictors have become increasingly accurate. In 2021, the first Critical Assessment of Intrinsic Disorder (CAID) competition was held, comparing different predictors in terms of accuracy and performance51. Based on results from the most recent CAID competition, the accuracy among the top ten predictors is similar, with AlphaFold2 also performing well52. Predictors have also gotten faster. For example, using one of the top-performing predictors, metapredict V2-FF, all IDRs in the human proteome can be predicted in a few minutes24,395. Disorder predictors provide a linear assessment of whether a residue falls within a disordered region or not (see figure, disorder profile for the human RNA binding protein hnRNPAl; RNA Recognition Motifs [RRMs] are folded domains). Proteome-wide analysis with metapredict (V2-FF) reveals that across the human proteome, ~40% of proteins have IDRs that are 100 residues or longer (18,074 IDRs), and ~70% of proteins possess IDRs that are 30 residues or longer (29,698 IDRs). Of those 29,698 IDRs, ~37% are linkers, ~34% are N-terminal tails, ~25% are C-terminal tails, and the remainder are fully disordered proteins. Such proteome-wide analyses have helped reveal that IDRs are common in eukaryotes and viruses while generally less common in bacteria and archaea26.
In addition to predicting IDRs, a repository of known SLiMs exist in the Eukaryotic Linear Motif Resource 264 Although the number of known SLiMs now approaches the thousands, it is estimated that up to 100,000 different SLiMs could exist255. While consensus SLiMs can be identified from sequence, whether these function as bona fide SLiMs typically requires direct experimental validation, highlighting the importance of context in licensing SLiM function.
Instead of a stable 3D structure, IDRs exist in a collection of rapidly interconverting structurally distinct conformations known as an ensemble (Fig. 2a,b, Movie M1)2,28,29. An ensemble can be considered the landscape of accessible IDR conformations. Although folded domains also exist in ensembles, these are typically much less structurally heterogeneous than those of IDRs30. Moreover, while it is convenient to discuss IDRs and folded domains as distinct entities, in reality, they exist along a continuum of structural heterogeneity31. Just as structure and folds (e.g., four-helix bundle, β-barrel) can quantitatively describe a folded domain, an IDR can be quantitatively described by its ensemble properties32-34
Ensemble properties are quantifiable parameters that describe 3D features derived from the ensemble. They include global IDR dimensions (i.e., how expanded or compact the protein conformations in an ensemble are), local transient structure (i.e., lowly populated helices and extended conformations), and inter-residue distances (Fig. 2b). IDR global dimensions are often quantified by the radius of gyration, end-to-end distance, or hydrodynamic radius. Importantly, ensemble properties are determined by molecular interactions encoded by the IDR sequence and its context (discussed below) and can be determined using experimental and computational approaches (see Box 2).
Box 2. Characterizing IDRs.
Experimental characterization of IDR ensemble properties can be achieved via a range of experimental approaches. Measuring residue-specific interactions relies on techniques that provide residue-specific information. These include NMR spectroscopy396,397, single-molecule Forster Resonance Energy Transfer (smFRET) with specific positions labeled398,399, and hydrogen-deuterium exchange mass spectrometry400. NMR and smFRET also enable global, ensemble properties to be measured401,402, as do additional techniques, including ensemble FRET120, small angle X-ray scattering (SAXS)403-405, dynamic light scattering (DLS)18, fluorescence correlation spectroscopy (FCS)406, circular dichroism (CD)407, and collision cross-section mass spectrometry (CCS-MS)408. While measuring both local (e.g. helicity, NMR chemical shifts) and global (e.g. radius of gyration, end-to-end distance) IDR ensemble properties for the same IDR can be time-consuming and challenging, integrative biophysical studies — in which several methods measure distinct properties of a single IDR — have played key roles in developing our current understanding of sequence-ensemble relationships32,57,61,64,87,246,402,409-415.
Computational characterization of IDR ensembles has been essential in understanding sequence-to-ensemble relationships416. Computational approaches can generally be classified as either top-down or bottom-up. Bottom-up approaches offer predictions of ensemble properties without experimental data. Top-down approaches take experimental data and construct ensembles consistent with those data. For bottom-up approaches, molecular simulations at a range of resolutions have proven invaluable64,112,113,117,410,411,417,418. While – historically speaking – many all-atom forcefields lead to the over-compaction of IDRs, recent efforts to address this weakness have led to major improvements419-425. In parallel, improvements in coarse-grained forcefields have also enabled rapid characterization of ensemble properties 335,426-429. In a recent preprint, ensemble properties of all IDRs in the human proteome were calculated from coarse-grained simulations60, while instantaneous predictions of global dimensions using deep learning based approaches trained on coarse-grained simulations enable ensemble properties (e.g., radius of gyration, end-to-end distance) to be predicted directly from sequence in milliseconds 24. For top-down approaches, tools including flexible-meccano430 and EOM431 for building ensembles from experimental data and various approaches for selecting an ensemble from the larger set of conformations and reweighting to optimize correspondence with the experimental data (e.g., ASTEROIDS432, Bayesian inference433,434, maximum entropy approaches435, metainference436, and deep learning437) have been applied to construct experimentally consistent ensembles at atomistic resolution438,439.
IDR ensemble properties can play key roles in biological function33. For example, transient secondary structure can predispose an IDR to bind a specific partner and play important roles in binding energetics21,35,36. In other instances, the average end-to-end distance of an IDR that connects two folded domains may position these at a functionally-relevant average distance from one another37-40. As a corollary, the modulation of ensemble properties can influence cellular function. Understanding that IDRs are defined by sequence-specific ensembles with unique physicochemical features acknowledges that ensemble properties can alter in response to molecular interactions, changes in the cellular environment, or post-translational modifications (PTMs).
Ensemble properties are best described in terms of probability distributions (Fig. 2c). Furthermore, IDR ensembles can possess well-defined structural and conformational preferences encoded by the underlying protein sequence, biasing them towards certain functionally relevant conformations or average ensemble properties. Just as folded proteins have a sequence-structure-function relationship, IDRs possess an analogous sequence-ensemble-function relationship, where that ensemble can be quantified in terms of ensemble properties (Fig. 2b, c).
The ensemble properties of an IDR depend on both the IDR sequence and its context. We define context as i) the local solution context, i.e., the proximity to other biomolecules (proteins, nucleic acids, lipids, small molecules, etc.), solution temperature, presence of osmolytes or ions, ii) the chemical context of the IDR, namely PTMs and changes in pH leading to protonation and deprotonation effects41, and iii) the structural context, i.e., the presence or absence of adjacent folded domains. Moreover, the binding of IDRs to ligands – be they other proteins, DNA, RNA, lipids, metal ions, carbohydrates, or other molecules – can influence ensemble properties and contribute to context42-45. While context can also alter folded domain ensembles, the absence of a network of stable intramolecular contacts in IDRs means they are generally more sensitive to changes in context46. Given that contexts can alter IDR ensemble properties in various ways, and changes in ensemble properties can be synergistic or antagonistic to specific functions, it stands to reason that IDR function can be tuned or even completely rewired by different combinations of ensemble-influencing perturbations. This allows IDRs to integrate complex signalling cascades and crosstalk across many cellular input pathways.
The molecular details that underlie how IDRs confer biological function are, in many cases, opaque. This knowledge gap partly stems from the need to integrate molecular biophysics and cell biology to fully interpret how function emerges, e.g., sequence-specific effects may alter IDR ensembles and hence function. In this Review, we aim to provide the conceptual tools needed to tease apart the molecular basis for IDR-mediated cellular function and regulation.
Sequence-to-Ensemble Relationships in IDRs
The relative deficiency of hydrophobic amino acids in many IDRs means their sequence composition often differs from folded proteins. It is therefore possible to assess the probability of a region being disordered from its sequence alone. Indeed, many accurate and robust disorder predictors have emerged over the years (see Box 1). Moreover, recent advances in structure prediction have provided a convenient corollary to disorder prediction; the absence of a predicted structure from tools such as AlphaFold2 (refs. 47,48) and trRosetta49 implicates a region as being disordered (although the resulting structure predicted by these tools should not be taken as a faithful prediction of the ensemble properties50). As a result, IDRs can generally be confidently identified from the amino acid sequence51,52.
Unconstrained by the requirement to fold into a 3D structure, paralogous and orthologous IDR sequences can be highly variable across evolution (see Box 3)53-55. This can make sequence alignment difficult and often misleading, necessitating alternative routes to measure conservation38,55-59. In particular, the underlying physical chemistry encoded by an IDR sequence dictates the resulting ensemble, and the properties of the ensemble can dictate function. Thus, one approach for understanding conservation and function in IDRs is by considering if and how ensemble properties might contribute to function, enabling the decoding of sequence-ensemble-function relationships24,38,60.
Box 3. The Evolution of IDRs.
IDRs often show poor sequence conservation when assessed by alignment-based metrics53,54,178,440,441. This poor conservation could be interpreted as a lack of important cellular function, yet the realization that IDRs play many critical roles in molecular and cellular biology invalidates this interpretation. An emerging paradigm suggests that conservation in IDRs can operate at the level of sequence features as opposed to on specific amino acid sequences38,55-58,72,92,174,178,442. If the conserved features include SLiMs, these may ‘diffuse’ around within an IDR, such that even if a SLiM is well-conserved, its relative or absolute position need not be 178,179. IDRs in which certain regions are highly conserved, as based on multiple sequence alignment, may reflect evolutionary coupling between that region and a folded partner, whereby the rate of change for this region has been slowed to match the partner’s surface, as shown recently for the bacterial tubulin homolog FtsZ442-444. Alternatively, variation in IDR sequences across evolutionary timescales may lead to compensatory changes in protein interaction networks, such that the overall function of a cellular programme is preserved even as individual disordered regions change445.
There are at least two related possible reasons for the limited sequence conservation observed in IDRs. First, because IDRs lack a specific 3D structure, they are not sensitive to (i) destabilizing mutations, (ii) mutations that impact folding pathways, or (iii) mutations that disrupt specific finely tuned allosteric networks. By contrast, in the case of folded domains, mutations can impact all three of these. As an example, mutations across enzymes can have a substantial impact on their function by altering stability, folding, and/or allosterically regulating function446,447. In effect, a stable 3D structure imparts a tight and cooperative coupling between sequence, fold, and function, and its absence loosens this coupling. Second, as discussed in the main text, IDR-mediated functions often depend on sequence features instead of specific sequences. Given that natural selection operates on the level of function, not on sequence, two IDRs with equivalent functionality are equally fit, regardless of how similar their sequences are. In this way, combining IDR sequence analysis with evolutionary analysis is one route to aid in identifying sequence features that may be important for molecular function55,59,92,178,263.
Amino Acid Physical Chemistry Defines Sequence-to-Ensemble Relationships
The twenty natural amino acids offer a chemically diverse set of building blocks to encode distinct ensemble properties33,34,61. The relative abundance and position of different amino acids are often called sequence features. For sequence-ensemble relationships, certain sequence features are more influential than others. The number, charge, and relative positioning – termed patterning – of charged residues are key determinants of ensemble properties in IDRs providing repulsive and attractive electrostatic interactions coupled with favourable free energies of solvation (Fig. 2d, e, f)58,62-70. Aromatic residues can engage in intramolecular interactions driven by their sidechain π:π interactions (π-electrons), cations:π interactions (with arginine, lysine, and protonated histidine), methyl:π interactions, or hydrophobic interactions (with aliphatic residues) (Fig. 2e, 2f)57,71-73. Aliphatic residues can drive intramolecular interactions via the hydrophobic effect and desolvation, whereas polar residues can engage in hydrogen bonds or dipole-dipole interactions18,74-76. Finally, due in part to steric effects, proline residues generally make chain dimensions more expanded than they would otherwise be, and, along with glycine, suppress transient helicity and β-strand formation61,63,77-80. In all cases, the clustering and patterning of these different residues can impact ensemble properties57,61,81-83. In addition to genetically-encoded sequence biases, IDRs are disproportionately post-translationally modified compared to folded domains84,85. By dynamically re-writing sequence chemistry through PTMs, IDR ensembles can be modulated in a reversible and controllable way86-90. In summary, sequence features can be quantified via recently established sequence parameters, enabling comparison between IDRs without reliance on (often impossible) sequence alignments4,25,34,55,91-93.
Sequence features – and hence ensemble properties – can be used for comparisons, evolutionary analysis, and quantitative predictions relevant to understanding IDR function55,56,92,94-96. For example, the C-terminal IDR in the Polycomb Repressive Complex 1 protein PSC is essential, poorly conserved as assessed by sequence alignments, yet highly conserved in terms of disorder and charge properties, highlighting the potential for function to be maintained with minimal sequence conservation58. More broadly, the preservation of overall charge or charge clusters in seemingly divergent IDRs has been used to explain functional conservation across evolutionary lineages or between seemingly unrelated proteins59,97-101. Finally, changes in IDR sequence features can compensate for evolutionary changes in IDR length, if ensemble properties are conserved. For example, in a linker IDR from the Adenovirus protein E1A, the fraction of proline and negatively-charged residues decreases as the linker sequence becomes longer (more residues), such that the global dimensions of the linker are conserved, a phenomenon termed conformational buffering38.
Attractive and Repulsive Intramolecular Interactions Determine Ensemble Properties
Attractive or repulsive intra-molecular interactions encoded by IDR sequence features can influence ensemble properties (Fig. 2b). These effects can be local or global and can act synergistically or antagonistically, with consequences for IDR-associated function.
Local lowly-populated (10-30%) helicity is common in IDRs, and is driven by local sequence features that stabilize the network of backbone hydrogen bonds found in α-helices17,21,102-106 Transient helicity can orientate sidechains to pre-organize binding interfaces. For example, the N-terminal disordered region of the androgen receptor can bind small molecules via two transiently-formed helical regions that arrange hydrophobic sidechains in a way that let them sandwich and bind small hydrophobic molecules107. In some systems, transient helicity appears to be evolutionarily tuned, in others, it determines molecular specificity. While the presence of transient helicity does not necessarily imply functional significance, conserved elements that form transient helices – especially those with aliphatic or aromatic residues along the helix face – often appear as functionally important elements within IDRs. The importance of transient helicity is further underscored by the fact that mutations that modulate helicity can lead to disease21,35,104,108-110.
Attractive and repulsive interactions along the IDR chain can lead to global chain compaction or expansion, driven by different chemical origins 24,33,60. Compaction here refers to a scenario in which an ensemble has a smaller global dimensions that expected by chance, whereas expansion means the ensemble is larger than expected by chance. Evenly-distributed aromatic or hydrophobic residues can drive labile attractive intramolecular interactions, as is seen in many low-complexity prion-like domains (Fig. 2e, left)18,57,72,76,111-113. Alternatively, clusters of oppositely charged residues can interact through long-range electrostatic attraction, as can aromatic and arginine residues (Fig. 2e, right)72,81-83,114,115. Finally, long repeats of some polar amino acids can lead to chain compaction via local dipole interactions and hydrogen bonding. In the case of polyglutamine (polyQ), a combination of helix formation and long-range intramolecular dipole interactions appears to govern global chain dimensions74,116-119. However, other polar tracts (e.g., glycine-serine repeats) behave as flexible chains that are neither overly compact nor expanded120,121. Chain compaction serves various functional roles, including modulating accessibility of binding motifs122 or enhancing the local concentration of adhesive interactions that can drive the formation of biomolecular condensates (discussed in the section IDRs and Biomolecular Condensates )18,57,123.
In addition to attractive interactions, some IDRs are enriched in residues that minimize intra-molecular interactions61,78,124,125. These self-avoiding IDRs serve various roles. For linker IDRs that connect folded domains, linker length and sequence features influence the interaction between the folded domains. By setting the effective concentration of the folded domains for one another, dynamic and flexible (or compact) linkers enhance inter-domain interactions in a length-dependent manner, while expanded linkers suppress inter-domain interaction (Fig. 2g)37,38,69,121,126,127. These effects can be regulated by PTMs, offering a route to tune interdomain interaction22,37,128. Changing the effective concentration of two folded domains can tune partner binding129, impact autophosphorylation22, and alter allosteric communication between folded domains40,130,131. Self-avoiding IDRs can also serve scaffolding roles, as seen for the disordered tail of the transmembrane protein LAT, onto which several SH2 domains can bind at defined distances132, or in the growth hormone receptor133. Finally, IDRs can exert an entropic force. This is an intermolecular effect, whereby a reduction in the volume accessible to an IDR-ensemble causes it to “push” against any molecular components that reduce its volume134. Given the generated entropic force is proportional to the loss of ensemble volume, IDR chain dimensions can tune the strength of the force by altering the volume occupied by the ensemble124,134,135. This entropic force can tune binding events124, sense/influence membrane curvature125,135,136, or even enable entropy-driven translocation of IDRs through bacterial cell walls137.
IDRs in Context
Many IDRs function by engaging in intermolecular interactions with other biomolecules. IDRs can interact in various ways (discussed in the following two sections). These include but are not limited to (i) sequence motifs composed of ~5 – 12 residue elements that encode sequence-specific recognition modules recurrent in many different and even unrelated proteins, known as Short Linear Motifs (SLiMs)138, (ii) multivalent interactions driven by specific sequence features (e.g., distributed aromatic residues or clusters of positively charged residues), (iii) folding-upon-binding to an appropriate partner, or (iv) some combination of these. IDRs can be highly multivalent, with several SLiMs or repeats orchestrating higher-order complexes, as seen in signalling hubs13. Moreover, IDRs may possess repetitive features that encode multivalency and lead to the formation of biomolecular condensates (discussed in the section IDRs and Biomolecular Condensates )139-141. Intra- and intermolecular interactions driven by IDRs can be suppressed or enhanced by changes in context that affect the physical chemistry of the amino acid residues (Fig. 3a-f). These changes in context can be transient or long-lived and can emerge from various origins.
Figure 3: IDR ensemble properties are context dependent.
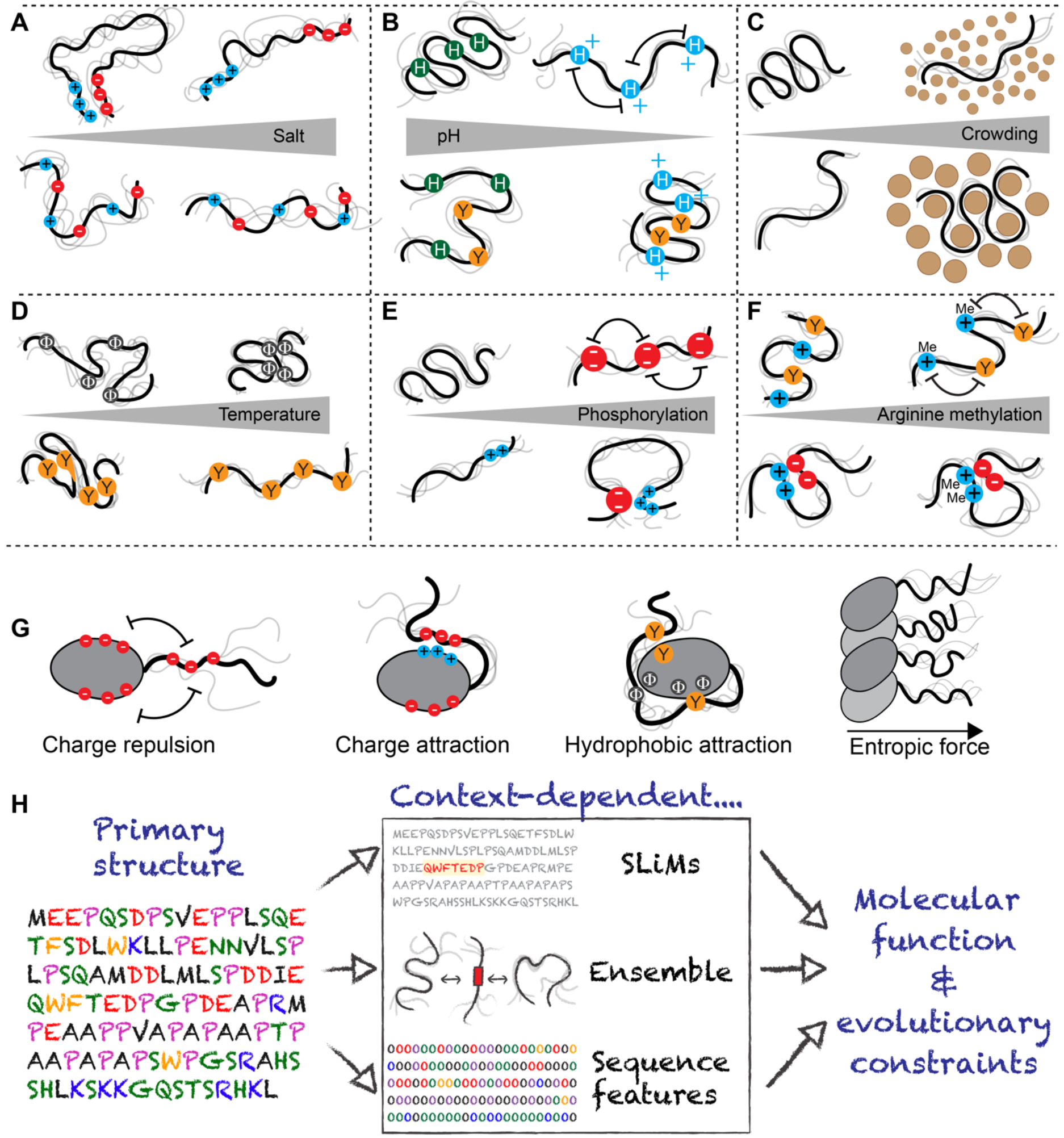
Behaviour of the IDR ensemble is highly context dependent. (a) Highly charged IDRs can be sensitive to changes in salt, although how salt influences ensemble properties depend on the IDR sequence features and the salt. If IDRs possess clusters of oppositely charged residues, these clusters can interact with one another driving chain compaction, an effect that is reduced as salt concentration is increased (top). By contrast, if charged residues are uniformly patterned, an increase in salt concentration may have a comparatively modest impact on IDR dimensions as no strong intramolecular interactions are found (bottom). Finally, divalent ions can bind to clusters of negatively charged residues with effects on local and global compaction (not shown). (b) Changes in pH can influence IDRs with amino acids that may be protonated (Asp, Glu, His) or deprotonated (Lys, Tyr, Arg, His) within physiological regimes. As a note, arginine deprotonation would seem to be almost impossible under physiological conditions. For uncharged IDRs with many histidine residues, a reduction in pH can lead to histidine protonation, driving intramolecular repulsion and leading to chain expansion (top). Conversely, if an IDR contains histidine and aromatic residues, protonation can lead to strong cations interactions between positively charged histidine and aromatic residues, driving chain compaction (bottom). (c) IDR dimensions respond to crowders differently; if crowders have weakly favourable non-specific interactions with IDRs then small crowders can drive IDR expansion while large crowders drive compaction. As a result, some IDRs may be well-poised to act as sensors of cellular crowding on specific length scales. (d) IDRs are sensitive to changes in temperature. For IDRs enriched in aliphatic hydrophobic residues (i.e., valine, leucine, isoleucine, methionine, alanine), the enhanced strength of the hydrophobic effect at higher temperatures leads to chain compaction (top). For IDRs enriched in aromatic residues, π:π interactions are enthalpically dominated, such that as temperature increases π:π interactions become weaker, and these chains become more expanded (bottom), and for IDRs in general, there is a loss of polyproline-II structures - an extended left-handed secondary structure that usually but not necessarily involves prolines - with temperature, leading to compaction. (e) Phosphorylation can have opposing effects on IDR dimensions. Phosphorylation of an uncharged region can lead to chain expansion driven by electrostatic repulsion between phosphate groups (top). However, phosphorylation of IDRs with clusters of positively charged residues can lead to chain compaction, driven by electrostatic interactions between phosphorylated residues and residues with positively charged clusters (bottom). Both effects can occur within a single IDR. Phosphorylation also impacts local structure and can stabilize and destabilize transient helices in a position dependent manner (not shown) (f) Arginine methylation weakens cation:p interactions between arginine and aromatic groups, which could lead to an increase in IDR dimensions (top). However, methylation does not neutralize arginine, such that intramolecular interactions driven by arginine-acidic residue interactions would likely be largely unaffected. (g) As solution context can influence IDR properties, folded domains adjacent to IDRs can do so too. The impact that folded domain surface features have on IDR ensemble properties depends on the chemistry of the folded domain and the IDR sequence. From left to right: Same charged residues on a folded domain surface and an IDR will repel one another, preventing intramolecular interaction and ensuring an IDR is projected into solution, away from the folded domain. Oppositely charged residues on a folded domain surface and an IDR will attract one another, driving intramolecular interaction. Hydrophobic interactions between aliphatic and/or aromatic residues on folded domain surfaces and IDRs can lead to intradomain interaction. If many IDRs are projected from a filament formed from folded domains, inter-IDR interaction and repulsion can lead to a bottle-brush architecture and a resulting entropic force. (h) Figure summarizing a current model for IDR function. IDRs are encoded by their amino acid sequence (left). That sequence determines the presence of SLiMs (middle top), the overall ensemble (middle center) and the presence of sequence features (middle bottom). All three properties and/or their functionality are influenced by IDR context. Ultimately, these context-dependent properties dictate both molecular function and the evolutionary constraints that govern IDR sequence variation over generations.
Physicochemical context
Physicochemical context can substantially alter IDR form and function46. For example, electrostatic interactions can be screened by changes in ionic strength (ionic activity), as can occur from an influx of Ca2+, Na+, K+, or by cellular sulfation gradients142-144. Interactions can be enabled or suppressed upon protonation or deprotonation of titratable groups upon pH changes, as occurs in transit from the cytosol to endosomes, during cellular stress, or in disease states with high glycolytic activity97,145,146. Macromolecular crowding can alter IDR global dimensions, e.g., upon hyper-osmotic shock or due to enhanced ribosomal production, implicating IDRs as potential mechanosensors62,147,148. Many proteins involved in desiccation tolerance are also disordered prior to desiccation, yet acquire helicity upon desiccation149-151. Finally, IDRs often show temperature-dependent changes in their molecular interactions, an effect capitalized on by IDRs that act as cellular thermosensors, as seen in the yeast heat shock response or in cellular programs that control germination in plants18,152-156.
Post-translational modifications
Post-translational modifications (PTMs) offer another way to alter IDR context. PTMs enable covalent but reversible changes in IDR sequence chemistry, which can influence intra- and inter-molecular interactions61,67,68. Given the importance of charged residues in determining IDR global dimensions, phosphorylation (gain of negative charge) and lysine acetylation (loss of positive charge) are two examples of PTM-mediated charge changes that can directly drive expansion or compaction, and hence may impact ensemble properties, depending on how these PTMs alter IDR sequence properties and where they are positioned157,158. Phosphorylation can also enable switch-like behaviour, whereby adding a phosphate moiety substantially changes an IDR’s ensemble14,159-161. For example, phosphorylation of the stress granule protein G3BP1 alters long-range intramolecular electrostatic interactions and suppresses RNA binding114, while the protein 4E-BP2 can switch from a disordered ensemble to a stable folded state upon phosphorylation162.
Structural context
Finally, the structural context of an IDR can alter ensemble behaviour and molecular function. For IDRs connected to folded domains, the steric impact of the folded regions and their chemical makeup can significantly influence IDR ensemble properties and function (Fig. 3g)163-167. This is true for IDRs in the same polypeptide chain but also for those in multiprotein assemblies, as is the case for histone tails168,169. For example, charged patches on the surface of folded domains can enable IDR interactions if complementary charged regions are found in the IDR163,166,170. Similarly, if IDRs are found adjacent to binding sites on folded domains, they can behave as locally tethered competitive inhibitors171-173. Moreover, even IDRs that do not engage in attractive interactions but are adjacent to ligand binding sites can impede ligand binding through entropic effects. Mechanistically, this can occur if ligand binding reduces the accessible volume of the IDR, as seen for the IDR of the UDP-α-D-glucose-6-dehydrogenase124,134,174 In summary, the ensemble properties of IDRs are inherently tuned by their context such that changes in context offer a complex and multifaceted route to recode and reroute IDR function.
Sequence and context are inextricably intertwined
Ultimately, IDR function depends on sequence and context (Fig. 3h). Sequence can be viewed through two complementary lenses: (i) the sequence-encoded 3D ensemble (or 4D ensemble, if the timescales of conformational re-arrangement are considered) and (ii) the 2D (d1 = residue identity, d2 = position) sequence-encoded information, such as sequence features or SLiMs. These two lenses are not independent – IDRs with certain sequence features will reliably show certain ensemble properties – yet they provide complementary views. For example, sequence changes to a motif may have no discernible impact on ensemble properties, yet these changes may entirely abrogate function. Finally, context can impact ensemble properties and sequence-encoded information and may do so to different extents. For example, phosphorylation may alter the net charge substantially but may have no major impact on global ensemble properties61,79, or it may induce or decrease local helicity dependent on the position within the helix and its sequence. 175,176.
A challenge in studying IDRs is that the functional importance of ensemble properties vs. sequence features vs. SLiMs is system-specific. A SLiM may be essential for one function, while the IDR's overall net charge may be the most important factor for another. Moreover, two IDRs may have similar ensemble properties (e.g., similar overall ensemble dimensions) even if their sequences differ in composition or length 38,61,69,79. This redundancy leads to a much looser relationship between sequence and molecular function, raising challenges and opportunities for evolutionary analysis (see Box 3). As a result, IDRs often appear less well-conserved when assessed by linear sequence alignment53,55,177,178. Exceptions here are SLiMs, which often have conserved sequence positions, although this is not a requirement179,180. Notwithstanding these challenges, an interpretable understanding of IDR function is accessible if the underlying biochemical and biophysical principles are jointly considered.
Modes of Molecular Interactions Mediated by IDRs
Molecular recognition reflects the specific, non-covalent interaction between two different molecules. The canonical model is one in which chemical and shape complementarity cooperate to enable specific binding events, as a hand fits a glove181-183. For IDRs, where one or both interacting partners exist as disordered ensembles, the models for molecular recognition require rethinking. Indeed, as described below, IDRs may comply with known interaction models, but they also extend the possible mechanisms through which molecular recognition is achieved. In this way, IDRs expand the cell's communication toolbox by offering complementary alternatives to the traditional 1:1 model of molecular specificity.
IDRs can bind other biomolecules through three main mechanisms: (i) Coupled folding and binding, where a disordered region folds to enable shape and chemistry complementarity in the bound complex102,184,185. Coupled folding and binding may involve an entire IDR, a single subregion, or two or more locally folded anchors connected by a disordered linker 186-188. (ii) As a fuzzy complex, where a finite number of structurally distinct bound-state configurations are observed and needed for function189,190. (iii) As a fully disordered bound-state complex, where both partners remain disordered.
The delineation of binding modes in this section is convenient from a didactic standpoint. However, molecular recognition involves a continuum of binding modes and principles from multiple mechanisms will likely be relevant for any given binding event. Indeed, the same IDR can bind to different partners with different mechanisms, as seen for the C-terminal tail of RNA Polymerase II111,113,191. Moreover, binding affinity192,193, specificity194-196, and even the binding mechanism can be tuned by context, as discussed above196,197. The range of potential partners bound via different mechanisms enables context-dependent crosstalk between various cellular programs and pathways. This tunability also has the potential for errors: miscommunication driven by aberrant interactions, signifying the need for negative design principles to minimize unwanted interactions.
Coupled Folding and Binding
In coupled folding and binding, either a subregion or the entire IDR folds upon binding to a folded – or disordered - partner, typically with the involvement of a conserved SLiM (Fig. 4a)198-202. In this situation, the free energy of binding must compensate for the loss of entropy experienced upon folding a disordered chain. Compared to binding a folded partner, the magnitude of this can be fairly small (on average ~2.5 kcal mol−1)203,204, but remains within a range that can determine biological functions205. Compensation may come from enthalpic contributions from inter or intra-molecular non-covalent bond formation but could also be entropic, driven by the release of solvent from hydrophobic residues and/or the release of counterions from charged side chains 36,206,207. Coupled folding and binding can follow induced fit 102, conformational selection 208, or – as is usually the case – some combination of the two, and kinetic measurements are needed for teasing these apart209-212. Coupled folding and binding can involve various interactions, including IDR–protein, IDR–DNA, and IDR–RNA199,213-215. In many ways, coupled folding and binding is analogous to intermolecular protein folding, as opposed to the intramolecular process one typically associates with protein folding in general.
Figure 4: IDRs enable a range of molecular recognition modes.
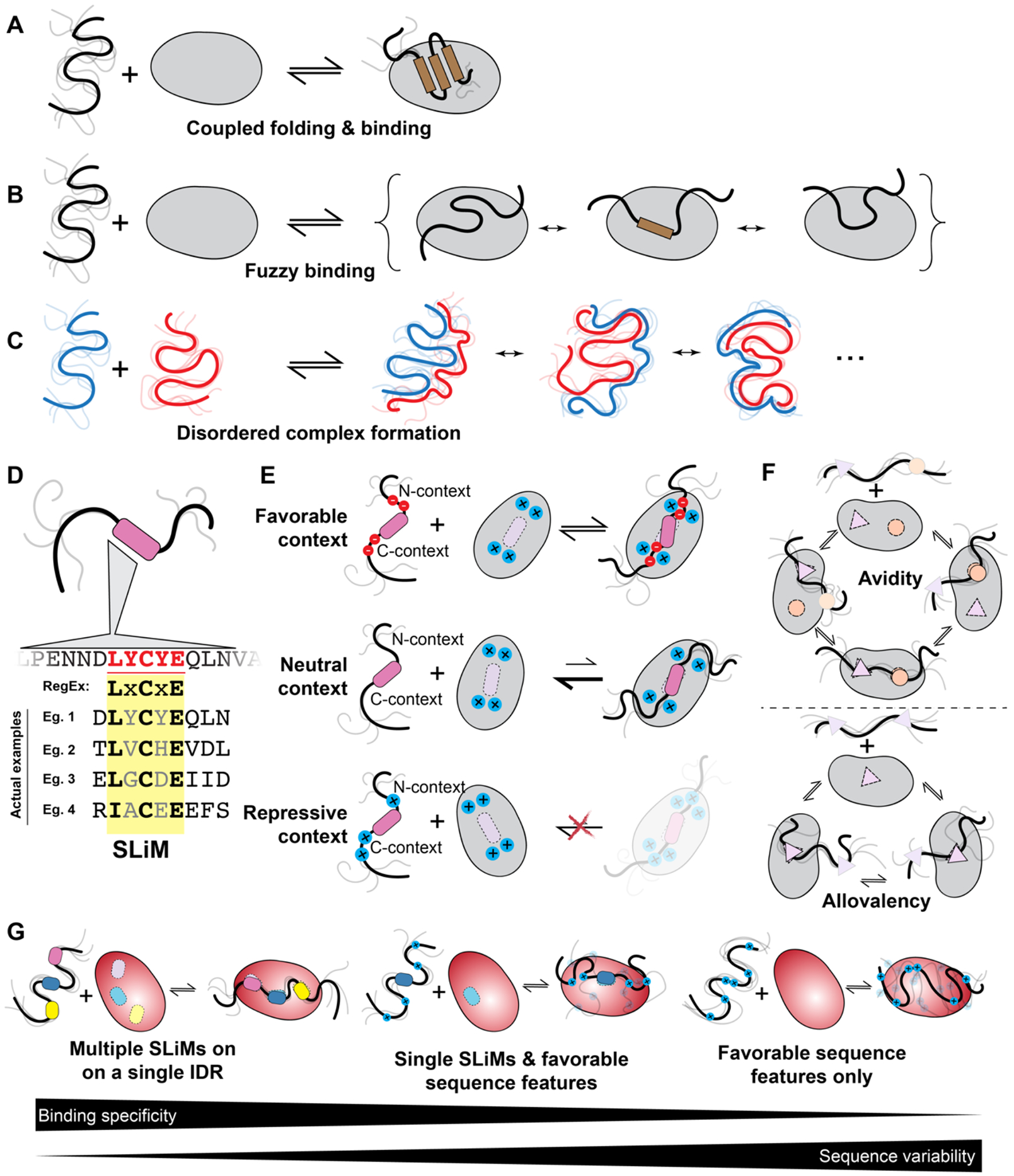
(a) IDRs can bind partners via coupled folding and binding, where an IDR (or a subregion) folds upon interaction with its partner, be it DNA, RNA, protein, or a membrane. (b) IDRs can bind partners via fuzzy interactions, whereby multiple structurally distinct bound states are relevant to function. Illustrated here is a scenario where an IDR consistently interacts with the same interface in structurally distinct bound states. However, fuzzy interactions could also involve a scenario whereby an IDR possesses several non-overlapping motifs or binding residues that exchange in binding a single interface on the surface of a folded domain. (c) IDRs can bind disordered partners to form fully disordered complexes where no persistent structure or contacts are seen in either partner in the bound state. (d) IDR molecular recognition is often facilitated by Short Linear Motifs (SLiMs). These are often well-described as a consensus motif with evolutionary conserved and invariant positions, while other positions are partially or fully redundant. As a result, SLiMs can be described in terms of “regular expressions” (RegExs), a term borrowed from computer science that describes patterning matching when a subset of positions in a sequence are under some set of constraints (e.g., the PIP box binding to PCNA (QxxLxxFF), where X is any amino acid). (e) The sequence context around SLiMs is a critical determinant of binding. The same SLiM present in different proteins may bind with high affinity or not all, depending on the complementary chemical interactions between the residues flanking a SLiM and the surface surrounding the binding site. Thus, when the features of the flanking regions match those of the binding partner, the context is favourable (top), when no determining features are present, only the SLiM is deterministic for binding (middle) and when the features of the flanking regions and those of the binding partner surface do not match, the context is repressive (bottom). (f) Binding of IDRs often involves avidity and allovalency. Avidity emerges when multiple binding sites (e.g. SLiMs) enable two molecules to interact through two or more independent binding interfaces (top). Allovalency reflects the situation in which a single binding site on one partner is complemented by multiple identical binding interfaces on another (bottom). (g) IDRs can encode binding specificity in a variety of ways. Multiple SLiMs within a single IDR offer one route to high-specificity (and high affinity) binding, whereby only a limited set of partners possess binding interfaces common to all the SLiMs present, providing specificity combinatorily via many weak motifs (left). While conceptually this may be straightforward to understand, a growing body of work suggests the existence of a continuum of multivalent binding modes, whereby a combination of SLiMs and sequence features enable a trade off between sequence conservation and binding to a specific target (middle). Finally, IDRs may interact solely via chemical specificity, whereby specific sequence features lead to favourable interactions between the IDR and a partner, such as a positively-charged IDR binding to a negatively charged partner (right). The discriminatory power available for such a simple sequence feature may be limited, and other properties such as number of charges or charge density or properties yet to be discovered may enable specific molecular recognition
The molecular details surrounding coupled folding and binding are tuned to fit the needs of the cell. A well-described example is the N-terminal IDR from the master tumour suppressor p53, which undergoes coupled folding and binding, and for which residual helicity tunes affinity and specificity in inter-molecular interactions21,35,184,200,216,217. A more recent example shows evolutionary fine-tuning of helicity and that the correlation between the amount of residual helicity in the IDR and binding affinity for a folded domain is manifested in altered bound-state lifetime218. In some systems, like the pro-apoptotic BH3-only proteins, the conformational landscape of coupled folding and binding is encoded by the IDR sequence219, as opposed to being templated by different folded partners220. In contrast, for the measles virus nucleoprotein, coupled folding and binding of the C-terminal IDR is driven by an induced folding pathway, whereby intermolecular contacts form before or in parallel with intramolecular folding102,221. As a final example, the nuclear co-activator domain NCBD from p300/CBP is a hub domain that is folded yet metastable. Upon binding one of its many partners – the disordered activation domain of the nuclear receptor coactivator ACTR – a transient electrostatically-steered complex forms, followed by an intramolecular folding reaction that stabilizes both proteins42. The formation of a stable bound-state complex from states where both partners are partially (NCBD) or fully (ACTR) disordered reflects the fact that NCBD can form distinct structured complexes with different disordered partners 217,222-225. In this way, a single partially folded domain can function as a multi-modal input receptor for cell signalling, transducing the identity and concentration of potential binding partners into distinct structural complexes.
Fuzzy Binding
In fuzzy binding, a number of structurally distinct states make up the bound complex (Fig. 4b)190,226,227. Fuzzy binding may involve static disorder, where each individual binding event yields a structurally distinct bound state that remains stable for its lifetime without exchanging to another state228. An extreme example of static disorder is the assembly of amyloid fibrils formed from disordered proteins229-231. While structurally-distinct fibres can and do form, interconversion between fibres of different structural states appears effectively impossible once formed. Alternatively, fuzzy binding may involve dynamic disorder, in which the bound state complex rearranges on timescales that are fast when compared to the timescales for dissociation. For dynamic disorder, fuzzy complexes could involve just a handful of structurally-distinct bound conformations that interconvert, or could reflect a scenario in which IDR conformational heterogeneity is similar in the bound and unbound states17,187,232. A classic example is the complex formed between the activation domain of the yeast transcription factor GCN4 and the co-activator Gal11 (refs. 233-236).
Fuzzy interactions are ubiquitous across IDR-mediated molecular recognition events. As one example, nuclear import and export rely on nuclear transport receptors, folded proteins that enable the passage of an appropriate cargo through the lumen of the nuclear pore complex237,238. The phenylalanine-glycine (FG) repeats from IDRs of nuclear pore proteins form fuzzy complexes with nuclear transport receptors239. This dynamic interaction is central to the ability of the nuclear pore to provide a chemical selectivity filter, a feature conserved across evolution240-242.
Transcription factor IDRs and their cognate co-activators can also form fuzzy complexes with some folding-upon-binding of local motifs7,187,233,243. Indeed, modulation of transcription factor interactions by competing binding partners or PTMs may enable fine-tuning of gene expression in a manner that allows different inputs to enhance or suppress transcriptional output, in effect acting as a network switch for signal integration 7,42,187,233,234,236,244. For example, the interaction between the folded TAZ1 domain of a transcription coactivator and IDRs from two transcription factors (HIF-1α and CITED2) provides a remarkable example of dynamic allosteric regulation enabled by a fuzzy complex188. When measured independently, HIF-1α binds TAZ1 and CITED2 with an equal affinity. Consequently, it might seem impossible for CITED2 to ever fully outcompete HIF-1α from binding to TAZ1. However, upon the interaction of CITED2 with the HIF-1α–TAZI complex, a transient ternary complex involving all three proteins is formed. Here, CITED2 takes over a shared binding site on TAZ1, leading to a conformational re-arrangement of TAZ1 and a subsequent reduction in affinity for HIF-1α. This complex allosteric mechanism highlights how IDRs can reshape folded domain ensembles to modulate molecular function.
Fully Disordered Complexes
The third mechanism of IDR-mediated recognition is one in which two IDRs bind one another and remain disordered in their bound state (Fig. 4c). For disordered bound-state ensembles, binding can be driven by distributed complementary chemical interactions that undergo fast timescale conformational re-arrangements, leading to a highly dynamic, heterogeneous bound-state ensemble245. These distributed chemical interactions can be driven by electrostatic interactions, aromatic interactions, or, in principle, any interaction mode whereby degenerate multivalency, i.e., the presence of many binding interfaces with approximately the same interaction strength, is encoded in an IDR.
The first rigorously characterized example of a fully disordered complex is the interaction between the negatively-charged histone chaperone prothymosin α and the positively-charged linker histone H1.0 (refs. 169,246-248). The interaction between such oppositely and highly charged proteins could be considered an extreme case of multivalency, with many short-lived and rapidly exchanging interactions between the individual charged groups. It could alternatively be considered as an average (mean field) electrostatic attraction that holds the two dynamically interconverting chains in very close proximity to one another with ultra-high affinity. Importantly, due to the electrostatic nature of this interaction, the measured affinity is exquisitely sensitive to salt, enabling binding affinities to be tuned by ionic strength in a rheostat-like manner. Dynamic, high-affinity interactions offer advantages to fast regulation of biology. Histone H1.0 also forms a high-affinity disordered complex with the nucleosome. The strength of this interaction should, in principle, impede nucleosome remodelling. However, enabled by the molecular dynamics found in H1.0 bound states, prothymosin α can dynamically outcompete the nucleosome, dislodging H1.0 by forming a transient H1.0–prothymosin α–nucleosome heterotrimer, followed by the release of H1.0, in a process of competitive substitution169,247. This ensures that nucleosomal remodelling can occur on timescales compatible with biological regulation. Similarly, disordered complexes have been observed for IDR-RNA interaction43,44. In the SARS-CoV-2 nucleocapsid protein, preprinted works shows that a short N-terminal IDR adjacent to the canonical, folded RNA binding domain enhances RNA binding ~50-fold, yet this IDR remains fully disordered in the bound complex44,174. Another example can be drawn from the nuclear pore complex. The interior of the nuclear pore provides a local chemical environment defined by tethered IDRs with FG repeats240,242,249. These disordered FG repeats interact with one another (homotypic interactions) via distributed phenylalanine residues, leading to a finely tuned chemical portal that enables efficient nucleo-cytoplasmic transport based on the surface-exposed chemistry of molecules in transit250. As a point of comparison, if those molecules in transit are folded domains (e.g., nuclear import receptors), they will interact with individual FG-rich IDRs as a fuzzy complex (as discussed above).
IDR-mediated Binding is Multifaceted
The separation of IDR-mediated binding modes into three subclasses might imply mechanistic stringency of interactions, making it possible to neatly categorize a given molecular complex. Yet, in reality, IDR-associated binding events can involve multiple modes. Fuzzy complexes often involve some degree of folding upon binding226. Folded domains are far from rigid, and IDR-associated binding may enhance or suppress molecular dynamics in folded domains251. We emphasize that this continuum of binding modes reflects the structural malleability associated with IDRs, and that it is generally worth considering all the types of interactions when understanding how an IDR may interact with a partner.
Molecular Specificity in IDR-mediated Interactions
Given the many different binding modes available, IDRs may appear poised to be promiscuous and adaptable. However, lack of specificity is not a general trait, and it may not be obvious if and how IDRs can encode specific molecular recognition. Specificity is defined in terms of both affinities and the availability of ligands252. The importance of affinity is obvious — if a protein/IDR binds many ligands with equal affinity, it would be considered promiscuous, such that binding one ligand with higher affinity than all others is typically how specificity is described. However, ligand availability is also key. A protein/IDR may — in principle — bind many different ligands, but if one is highly abundant, then it will behave with high specificity252,253. Thus, specificity is tunable by the cell. As a result, in a situation where affinities are low and/or many different binding-competent ligands are present, an IDR may appear promiscuous, despite that under a different scenario (a single binding-competent ligand), it may appear specific. While specificity can be encoded in canonical sequence-specific structured interfaces, emerging work suggests that specificity can also be obtained by combining several molecular interfaces on a single IDR.
SLiM-Mediated Specificity
One source of binding specificity is through SLiMs (Fig. 4d)138,186,254,255. SLiMs can bind to partner proteins in concert with the acquisition of secondary structure, as is seen for the PIP-Box motifs that bind PCNA, a trimeric DNA clamp that plays a central role in DNA replication192,256. SLiMs may also bind without taking on any specific structure, as is seen for the Disordered Ubiquitin-Binding Motif, a SLiM seen across many proteins257. Some folded binding partners can accommodate different SLiM-carrying IDRs that bind with different degrees (and kinds) of structure and disorder20,258. The converse is also true; the same SLiM can bind folded partners that differ substantially in tertiary structure, likely because of closely overlapping SLiMs259. As such, both simultaneously and competitively, one IDR can bind many folded domains, and a single folded domain can bind many IDRs, offering the opportunity for complex, context-dependent interactomes 260.
SLiMs enable specific molecular recognition, yet they often possess substantial redundancy. Redundancy here reflects the fact that for a SLiM binding to a specific partner, a subset of SLiM positions may be essential for binding, while other redundant positions can tolerate sequence changes (i.e., are partially or fully redundant) 138,261-263. This architecture means that SLiMs are frequently described in terms of so-called regular expressions, a computer science term used for pattern-matching that encompasses one or more unique sequences. For example, one such regular expression is “LxCxE”, where ‘x’ implies any residue is tolerated, whereas the leu (L), Cys (C), and Glu (E) are required138,255,264. This scenario is further complicated because this redundancy can depend on the binding partner. For example, for one partner, the appropriate regular expression might be LxCxE, while for another, it might be the more restrictive L[K|R]CxE – i.e., the second position must be positively charged. The potential for multiple constraints on SLiM variation depending on which binding partners are relevant can lead to complex patterns in sequence conservation and divergence 178,265.
Another source of complexity in SLiM-mediated binding is via overlapping SLiMs, a scenario in which several SLiMs partially overlap one another. Overlapping SLiMs enable competition-based regulation of intracellular communication (e.g., in signalling cascades). For example, the intracellular IDR from the transmembrane Growth Hormone Receptor (GHR) possesses overlapping SLiMs for two different kinases, such that direct competition between these kinases leads to distinct downstream signalling profiles depending on which kinase is bound266. Similarly, SLiMs in the N-terminal IDR of p53 bind several partners with different affinities and structures, leading to distinct downstream responses21,216,217,267-272. In short, overlapping SLiMs that define mutually exclusive binding interfaces provide a means to build biological exclusive “OR” (XOR) logic gates, where either one or another bound state can exist.
Specific mutations in SLiMs can have devastating phenotypic consequences273. Mutations in a degron SLiM embedded in the N-terminal IDR of β-catenin lead to unfettered proliferative growth in a variety of cancers274. Similarly, in cases with overlapping SLiMs, mutation can change the balance between interactors, rewiring downstream signalling138,260,275. While IDRs are often less susceptible to individual point mutations, SLiMs are an exception, where single mutations can abrogate or instigate function178,179,273,276.
The importance of understanding SLiM-mediated molecular recognition has catalyzed efforts to systematically measure SLiM binding using high-throughput methods260,277-279. Identified SLiMs are catalogued in a database of curated entries, which includes both specific instances and inferred regular expressions264. In essence, SLiMs can be thought of as short, flexible sequence-specific protein interfaces that enable molecular targeting for intracellular communication.
SLiM Context
Recent work has implicated the importance of the local sequence context into which a SLiM has evolved, or has evolved around a SLiM17,31,41,178,224,262,280,281. Rather than existing as independent binding modules, the N- and C-terminal regions flanking a SLiM can influence molecular recognition, either by ensuring a SLiM is fully accessible or by providing additional auxiliary interactions that contribute to productive binding encounters (Fig. 4e)41,224,280. A SLiM and its sequence context can cooperate synergistically to enhance the affinity and specificity of interactions of IDRs with their cellular targets. For example, a C-terminal lysine-rich region is required adjacent to the PxxPxK proline-rich motif for correct SH3 domain recognition in the HS1–HPK1 interaction282. Similarly, flanking regions and phosphorylation sites around the LxCxE motif of the human papillomavirus E7 protein tune binding affinity, controlling molecular interactions that impact cellular proliferation283. Finally, work on proteins that interact with PCNA revealed that most PCNA-binding motifs reside in IDRs, and that changes in flanking regions that increase the number of positive charges in the IDR tune affinity across four orders of magnitude192. These results implicate an emerging hierarchical model for specificity, where motifs and flanking regions cooperate to enable short-term fine-tuning via PTMs and long-term (evolutionary) fine-tuning via changes in the protein sequence.
The emerging importance of flanking regions in determining SLiM binding affinity and specificity reflects the conceptual challenge that regions around SLiMs are often poorly conserved, as assessed by linear sequence alignment. This apparent lack of conservation has given way to an appreciation that sequence features (discussed above) may be conserved despite divergence in primary structure (see Box 3) 6,55-57,59,92,178,244,284 When viewed through this lens, specificity can be dually encoded via two distinct types of interactions. If we accept that SLiMs enable sequence specificity (i.e., SLiMs cannot tolerate being shuffled; the action of randomly re-ordering the sequence without changing composition), then flanking regions essential for binding that lack bona fide SLiMs can be considered to possess sequence feature specificity (i.e., chemical specificity). Chemical specificity reflects local sequence chemistry that is complementary to a binding partner (Fig. 4e) 41,89,90,178,252. While conservation of SLiMs may require specific residues to be retained, conservation of sequence features can be achieved despite large-scale remodelling of the underlying amino acid sequence. Finally, flanking regions can overrule a SLiM by presenting incompatible features that prohibit binding. Thus, the presence of a sequence that — in principle — matches a known SLiM regular expression is not necessarily sufficient to define a bona fide SLiM (i.e., a motif that reliably binds its expected partner). For molecular communication, this hierarchical recognition that combines SLiMs with local sequence context enables specific, and in some cases high-affinity binding, with only a few conserved amino acids.
Balancing Affinity and Specificity
Although presenting a relatively limited binding interface, individual SLiMs can be highly specific. For example, TFIIS N-terminal domain (TND)-interacting motifs (TIMs) are SLiMs from transcription regulators that selectively recognize specific domains in the eukaryotic elongation machinery20. Although SLiMs can be high-affinity206,285, in many cases, the binding of individual SLiMs — especially if surrounded by sub-optimal flanking regions — can be relatively weak262. One way to enhance the binding affinity (and specificity) of an IDR is to embed multiple SLiMs that bind non-overlapping sites in a partner. If each SLiM binds a different recognition interface and only the appropriate partner possesses the full set of recognition interfaces, binding can be both high affinity (due to an avidity effect, Fig. 4f) and high specificity (due to the combinatorics) despite individually weak binding affinities associated with any single SLiM (Fig. 4g)38,110,286,287.
An alternative to carrying multiple SLiMs is for an IDR to possess a single SLiM with specific sequence features that interact via chemical specificity with a given partner or set of partners. This is similar to how SLiM context influences binding, but in this case sequence features may stretch far (10s-to-100s of residues) from the SLiM location, as opposed to simply defining a local permissive context. These sequence features may not offer the same degree of specificity that multiple SLiMs would. However, because these sequence features operate at the level of distributed chemical interactions instead of sequence-specific binding interfaces (as SLiMS can), they place a much lower burden on sequence conservation in the IDR or, indeed, sequence or structural conservation in the folded domain17,55,110,178,288. Moreover, an IDR with a single SLiM can interact specifically with many different partners that share only a single SLiM-binding interface, e.g., a PDZ-binding SLiM can bind many different proteins as long as each possesses a PDZ domain with surface chemistry complementary to the flanking sequence around the SLiM38,262,289. If intracellular communication lines depend on the fidelity of messages passed, the repertoire of molecular interfaces — from sequence-specific motifs to an appropriate net charge offer a broad toolkit for ensuring reception, transmission, and fine-tuning of those messages 18,120,146,152,154,160,162,290-292.
Combining multiple equivalent binding sites (i.e., SLiMs, repeats, or individual residues) in a single IDR can also enhance affinity through allovalency (Fig. 4f)189,275,293. Allovalency refers to a multiplicative increase in affinity brought about by a high copy number of independent binding sites that bind to the same site on a partner. For example, increasing the number of FG repeats in a nuclear pore IDR revealed that the low per-FG repeat affinity avoids high-avidity interaction between FG-nucleoporins and nuclear transport receptors (NTRs) while the many FG repeats promote frequent FG-NTR contacts, resulting in enhanced selectivity294.
The dynamic ranges of affinities, timescales, and specificities available to IDRs are no different from those observed for folded domains203. Indeed, fully disordered complexes can form with picomolar affinity246, while individual SLiMs that fold upon binding may bind with high discriminatory power yet weak affinities262. Although there are numerous examples of IDRs that fold upon binding225,295, they likely only constitute a fraction of complexes involving IDR, allowing for a much broader view of how disorder contributes to molecular communication in cells. As biophysical/biochemical studies typically examine binding between small fragments from larger IDRs and cognate partners, it raises the question of how disorder-based interaction may manifest in full-length proteins. Moving towards studying proteins in context, we are only beginning to understand where and how disordered complexes contribute to function and cellular regulation. IDRs provide a broad toolkit of distinct mechanisms of molecular recognition that can enable complex, highly tunable interactions that underlie transcriptional networks, signalling pathways, and cellular organization.
IDRs and Biomolecular Condensates
Recently, the role of IDRs in biomolecular phase transitions has captured increasing attention (Fig. 5a, b). Assemblies formed via phase transitions are often called biomolecular condensates, a catch-all term defining membrane-less non-stochiometric assemblies that concentrate specific biomolecules and exclude others296. Condensates can range in diameter from a few nanometers (e.g., transcriptional condensates) to micrometers (e.g., membraneless organelles such as nucleoli or P-granules)19,297-299. Condensates can also possess different material properties, with some behaving like liquids and others like solids300. While all droplets formed by phase separation are condensates, not all condensates form via phase separation296. The physical principles underlying phase transitions in biology have been reviewed extensively elsewhere140,301-305, as has the form and function of biomolecular condensates296,300,306,307. As such, our focus here is on the roles IDRs can play in biomolecular condensates but not on the underlying physical principles.
Figure 5: IDRs can undergo phase separation and contribute to biomolecular condensate formation.
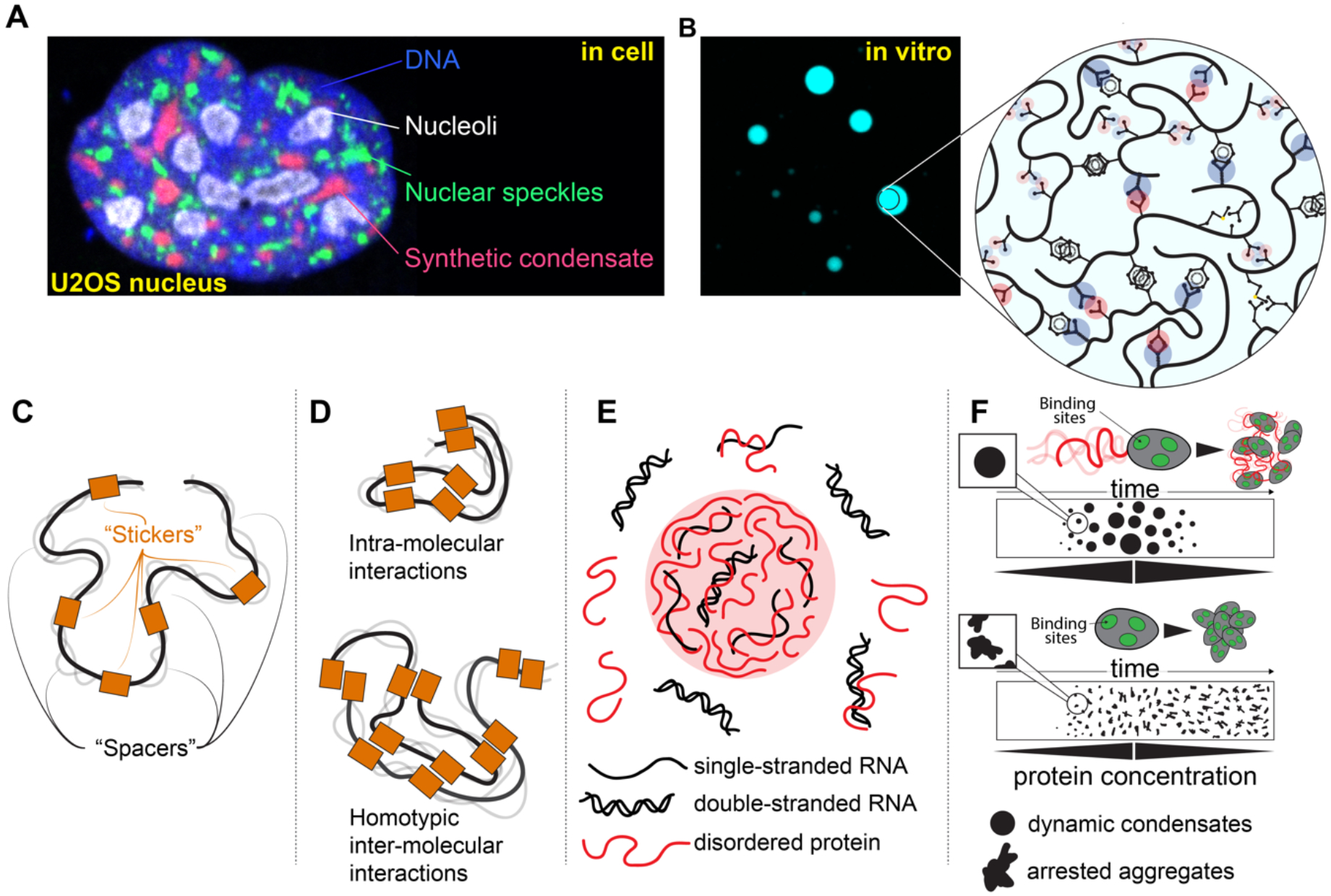
(a) Biomolecular condensates are non-stoichiometric assemblies that concentrate specific biomolecules while excluding others. In cells, many condensates can co-exist, as shown here where nucleoli, nuclear speckles, and synthetic condensates generated using the PopTag oligomerization domain coexist in the same U2OS cell nucleus. (b) Condensates formed in vitro and in vivo through phase separation are often stabilized by IDRs, with a variety of distinct chemical interactions tuning condensate formation, maintenance, and material state. (c) IDRs that drive phase transitions can be described in terms of stickers and spacers, where stickers reflect regions or residues that have an outsized role in driving attractive interactions, while spacers are regions that connect stickers. (d) For IDRs that drive homotypic phase separation where many copies of the same IDR interact, favourable multivalent intra-molecular drive chain compaction, whereas favourable multivalent inter-molecular interactions drive phase separation. (e) If intra-condensate IDR concentrations are high, the high concentration of sidechain chemistries presented by the many IDR molecules effectively provides a novel solvent environment that can destabilize e.g., nucleic acid duplexes, but could also in principle catalyze chemical reactions. (f) The presence of IDRs adjacent to folded domains can prevent the formation of arrested condensates (irreversibly formed) through IDRs acting as local molecular ‘lubricants’. If the IDR engages in many weak interactions with the surface of the folded domain, those interactions can impede strong intermolecular interactions between folded domains that would otherwise lead to arrested condensates. In this way IDRs can act to ensure the condensates are dynamic and, upon a reduction in overall protein concentration, undergo disassembly. Here, folded domains are represented with discrete binding sites that mediate interactions with other folded domains. If folded domains lack IDRs, they readily assemble via interactions between folded domains, but those condensates become trapped irreversibly on the timescale of the schematic. In contrast, if folded domains possess IDRs, the IDRs lubricate folded domain interactions, leading to dynamic and reversible condensate formation. Image in part a is a courtesy of Steven Boeynaems, Baylor College of Medicine, Houston, TX.
Molecular Basis for Phase Transitions
In general, IDRs are neither necessary nor sufficient for phase transitions308. Nevertheless, there are many specific examples where IDRs are both necessary and sufficient and many more cases where IDRs modulate phase transitions. One reason why IDRs are often found to be associated with phase transitions is the same reason that IDRs enable dynamic, tunable molecular recognition: multivalency (Fig. 5b)39,57,309. Phase separation requires multivalent interactions that enable networks. IDRs provide a convenient platform upon which SLiMs and surrounding sequence features can cooperate to enable multivalency301,308.
One framework for describing multivalent IDRs is in terms of “stickers” and “spacers”, a framework originally developed for associative polymers 39,57,301,305,310-316. Stickers are defined as regions or residues that are the primary drivers of attractive multivalent interactions, while spacers connect stickers and influence overall solubility as well as sticker-sticker cooperativity (Fig. 5c). This is a deliberate simplification when applied to biomolecules in that “spacer” regions can and do contribute crucial attractive or repulsive interactions to tune biomolecular phase transitions112,163,313,317. Nonetheless, the stickers-and-spacers offers a convenient approach to capture the most important sequence-determinants of IDR-mediated phase transitions39,57,72,318-323. If multivalent IDRs are fully flexible and interact via homotypic interactions, there exists a symmetry between the degree of chain compaction (intra-molecular interaction) and the extent of phase separation (inter-molecular interaction) (Fig. 5d)57,123,324. While multivalency is not sufficient for phase separation (i.e., multivalent molecules can form system-spanning gels rather than locally dense droplets), it is certainly necessary39,311.
Roles of IDRs in Condensates
The biophysical roles of IDRs in condensates are manifold. In some systems, IDRs can be the drivers of condensate formation, whereas in others, IDRs tune condensate formation, dictate condensate material properties (e.g., liquid-like, solid-like), prevent amorphous aggregation, or enable condensate regulation via PTMs. Within condensates, IDRs could – potentially – interact via all the possible mechanisms described in Section Modes of Molecular Interactions Mediated by IDRs. As such, in addition to influencing condensate formation, IDRs can facilitate the recruitment or exclusion of other molecular components, called clients, into condensates325. While it is often convenient to think of clients as passive bystanders, the influence that client recruitment can have – either on the chemical environment within a condensate or on the species that enable recruiting – means condensates are unavoidably responsive to changes in their composition311,323,326.
IDRs in pan-kingdom DEAD-box helicases (DDXs) are at least in some cases necessary and sufficient to drive condensates in vitro and in vivo 327-333. For human DDX4, the N-terminal IDR drives condensate formation via distributed multivalent interactions mediated by aromatic and arginine residues along with clusters of charged residues248,329,334. This particular molecular grammar has been identified in many other IDRs as mediating attractive interactions for phase transitions, as have additional sequence features, including contributions from aliphatic and polar residues57,72,99,111-113,240,241,248,310,335-339. Moreover, these features are readily altered via PTMs, which can enhance or suppress attractive interactions that drive condensate formation98,114,340-344.
Condensates formed by the DDX4 N-terminal IDR reduce the stability of duplexed nucleic acids, illustrating the ability of condensates to form unique chemical environments that facilitate specific chemistries (Fig. 5e) 345. By doing so, condensates offer the potential to enhance biological processes like RNA folding and enzyme catalysis19,326,346. In this way, condensates provide a means to define local states, augmenting molecular communication by creating filters (local regions that are only accessible to certain biomolecules), amplifiers (small changes in the intracellular environment can manifest in the formation or dissolution of entire organelles), and resistors (condensates that buffer the concentration of soluble components)311,326,345,347.
While some IDRs are essential for condensate formation, in many situations, they tune or modulate assembly153,348-350. The N-terminal IDRs in the yeast prion protein Sup35 and the fruit fly RNA binding protein Me31b prevent adjacent folded domains from forming kinetically arrested (i.e., “irreversible”) condensates, and instead facilitate the formation of reversible liquid-like assemblies (Fig. 5f) 97,330. In the yeast RNA binding protein Pab1, the major IDR is dispensable for condensate formation in vitro and in vivo, yet acts as a tunable thermosensor, where the hydrophobicity of the IDR tunes the temperature at which condensates form18. More broadly, IDRs in condensates have been implicated in environmental sensing in other contexts, including thermosensing in plants155,351, cellular crowding352, pH sensing18,97, osmotic shock353,354, and water availability355. In many of these examples, condensate behaviour is correlated with distinct biological phenotypes, including plant flowering, seed germination, cellular survival, and gene expression. Indeed, a growing body of work suggests IDRs may be poised to act as sensors of the cellular environment, with condensates offering one such mechanism46,120,356.
In addition to driving or tuning the formation of condensates, IDRs can influence condensate material properties with consequences for cellular function. These include intra-condensate viscosity, surface tension, permeability, and elasticity. Even seemingly subtle sequence changes (arginine to lysine) can change condensate viscosity by orders of magnitude357,358. While it is tempting to expect functional condensates to be liquid-like, many studies suggest variability and that condensate material properties must be tuned for condensate function359. For example, the Caulobactor crescentus protein PopZ forms a large condensate at the cell poles, where it plays key roles in asymmetric cell division360-362. Mutations that enhance or suppress PopZ condensate viscosity impact cellular fitness, yet large-scale mutations that preserve material properties have no effect on fitness, highlighting the importance of the properties of the material state349. The ability to orthogonally permutate IDR sequence features in a manner that preserves condensate properties (e.g., exchanging one set of chemical interactions that drive attractive interactions for an alternative, chemically distinct set) is one route to test the biological importance of condensates. If two chemically orthogonal types of interactions give rise to condensates with similar properties and preserved function, this is strong evidence that the condensate, not the specific chemical properties of the IDRs, are key for function.
The physics of phase transitions offer many features that could be co-opted for molecular communication and cellular function, including force generation, hypersensitivity spurred by abrupt changes, concentration buffering, molecular selectivity, and the ability to integrate disparate input signals (e.g., pH, temperature, ligands) that lead to a common output (the formation of the same condensates) 18,242,329,347,363,364 Condensates can form via many different modes of molecular interactions. While it may be tempting to ascribe molecular functions to IDR-dependent condensates, it is worth remembering that many IDRs are intrinsically multivalent. The sequence features that enable IDRs to form or modulate condensates are the same as those that drive IDR-mediated interactions in conventional molecular recognition. One possibility is that the primary function of a condensate-associated IDR is to form or modulate condensates (as illustrated for the Pab1 IDR18). An alternative explanation is that condensate formation is an unavoidable epiphenomenon associated with multivalency and that multivalent IDRs can and will form condensates regardless of whether those assemblies have biological roles. A key challenge for the field is delineating between these two possible explanations.
Conclusions and perspective
IDRs are ubiquitous and essential for normal cellular regulation, yet many questions regarding the molecular basis for their functions remain unanswered. A primary challenge in studying IDRs comes from their inherently context-dependent functions.
Interpreting the functional roles of folded domains benefits from the fundamental paradigm that form (i.e., structure) dictates function, allowing folded domains to be classified as a dehydrogenase, a kinase, an immunoglobulin domain etc. 365. In these examples, a complex biomolecule is captured (rightly or wrongly) in a way that allows us to exchange the molecular complexity of a 3D structure with a single interpretable descriptor. By contrast, IDRs are conformationally heterogeneous, and their behaviour and function are multifaceted and context-dependent. Their function depends on an often yet-to-be-deciphered combination of ensemble properties, sequence features, and motifs, where the relative importance of these three factors varies from IDR to IDR and from function to function. Consequently, simple terms that would describe an IDR as a “binding domain” or as a “proline rich domain” are at best insufficient and at worst misleading. Instead, we suggest embracing the underlying biochemistry and biophysics of IDRs is essential to make sense of sequence–ensemble–function relationships.
Based on emerging work by many groups, we propose that a core role of IDRs is in the reception, processing, and transmission of cellular information (i.e., molecular communication). The various molecular interaction modes enabled by IDRs extend the repertoire of molecular functions offered by folded domains. Importantly, the context-dependent nature of IDR-mediated interactions means that through splicing, changes in the cellular environment, changes via PTMs, and presence/absence of different binding partners, IDR function can be tuned or entirely re-defined. An important open question is if and how the cellular environment alters biochemical and biophysical conclusions drawn from in vitro or in silico work292,366-369. Moreover, while most insights into IDR functions are made from studies of proteins found within the cell, extracellular communication may well rely on IDRs in similar manners. Currently understudied is also the role of isoforms and proteoforms, two ways of regulating protein function for which IDRs are statistically enriched 84,370-373.
Although most annotated disease-causing mutations affect structured regions of proteins374, over 20% of human disease mutations occur in IDRs276,375-378. While IDRs are — in general — less sensitive to single-point mutations, there are many examples in which seemingly small changes in sequence chemistry can have substantial effects on IDR-dependent molecular recognition. For example, given their often loose determinants of specificity, SLiMs may appear or be removed seemingly out of nowhere (ex nihilo)179, as seen in the lung-cancer-related P495T mutation in the GRH IDR in which a binding site for a negative regulator is lost108, or the glucose transporter GLUT1 where the appearance of a di-leucine motif causes mis-trafficking in GLUT1 deficiency syndrome276. For IDRs that mediate intermolecular interactions, even small changes can lead to aggregation-prone proteins that drive aberrant cellular assemblies377,379-383. Finally, repeat expansions, frameshift mutations, and large-scale genetic rearrangements can all lead to novel IDR-containing proteins that drive human disease338,339,377,382-388. Despite clear examples, our understanding of how mutations in IDRs contribute to pathophysiology is in its infancy, necessitating detailed biochemical investigation to decode the principles that underlie the sequence-ensemble-dysfunction in human disease.
One common perception of IDRs is that their interactions may be “weak” or “non-specific”. As discussed, specificity by IDRs is, in many cases, enabled by multivalency, where a combination of SLiMs or sequence features can act synergistically to define specificity and affinity, linking sequence to function. While it is tempting to consider binding affinity as a proxy for the importance of a given interaction, sensitive and responsive regulation of high-affinity interactions raises many challenges. Weaker binding affinity may reflect interactions that are most easily regulated. Indeed, the importance of weak, motif-based interactions for cellular physiology is implied by the fact that many viruses rewire cellular programmes through molecular mimicry of host protein SLiMs 279,389,390. While weaker interactions are harder to measure in vitro, are more strongly influenced by their solution context, and may only be functionally important under specific conditions, their importance in determining cellular state and in enabling tunable intracellular communication is abundantly clear. As such, we propose that IDRs are poised to enable a specific class of regulatable, evolutionarily-prone interactions that allow for adaptation over short (minutes), medium (epigenetic/generational), and long (evolutionary) timescales.
Supplementary Material
M1 Rendering of all-atom simulation of the hnRNPA1 IDR to illustrate the conformational heterogeneity within an atomistic ensemble57. Conformations were generated through all-atom Monte Carlo simulations, which show good agreement with experimental characterization.
Acknowledgements
The authors wish to thank Rohit Pappu for discussions in the initial phase of writing. We also thank Gary Daughdrill, Aidan Flynn, Julie Forman-Kay, Per Jemth, Alan Moses, Johan G. Olsen, Rohit Pappu, Benjamin Schuler, Karen Skriver, and Shahar Sukenik for valuable comments and suggestions. We thank Steven Boeynaems for original microscopy images in Figure 5.
Funding
This work was supported by the Novo Nordisk Foundation challenge grant REPIN, rethinking protein interactions (NNF18OC0033926 to BBK), by the Danish Research Councils (9040-00164B to BBK), by the United States National Science Foundation (NSF) (NSF 2128068 to ASH), by the United States National Institutes of Health (DP2 CA290639-01 to ASH), and by the Human Frontiers in Science Program (HFSP) (RGP0015/2022 to ASH).
Glossary
- radius of gyration
Also written as Rg. This parameter is a measure of global ensemble dimensions and reports on the average distance between the center of mass of the IDR and the individual atoms.
- end-to-end distance
Also written as Re This parameter is a measure of global ensemble dimensions and reports on the average distance between the first and last residues in the IDR.
- hydrodynamic radius
Also written as Rh. This parameter is a measure of global ensemble dimensions and reports on the radius associated with a sphere that would diffuse through the solution at the same speed the IDR in question would, after correcting for solution viscosity.
- sequence features
Properties of an IDR amino acid sequence that are determined by the composition and patterning of different amino acids. Sequence features can – by definition – be determined directly from sequence. Several commonly-used sequence features can be calculated using the CIDER webserver.
- π:π interactions
Interactions mediated by delocalized π electron clouds, seen in amino acids with aromatic side chains.
- prion-like domains (PLD)
A class of protein domains defined by being of low complexity (many similar amino acids) and possessing enrichment for polar amino acids (especially glutamine, asparagine, glycine, and serine), often with additional aromatic residues. PLDs are defined using the PLAAC webserver with default parameters. While PLDs have been found to phase separate, their presence should not be taken as evidence that a protein will phase separate. They are named after yeast prions, in which a PLD was originally defined.
- induced fit
A mode of binding in which the IDR is templated into a specific conformation by a binding partner. Unlike conformational selection, the bound-state conformation of the IDR is never/rarely visited in the unbound ensemble, and the act of binding “induces” this bound-state conformation.
- conformational selection
A mode of binding in which the IDR binds a partner by adopting a binding-competent conformation in the unbound ensemble, which then binds without further conformational rearrangement. Unlike induced fit, the bound-state configuration of the IDR is visited in the unbound ensemble, such that the binding partner “selects” a specific conformation to bind.
- associative polymers
A class of polymer architecture in which specific regions or monomers contribute associated (attractive) interactions. See foundational work by Cate & Whitten (1986) and Semenov & Rubinstein (1998).
- molecular grammar
When used in the context of IDRs and biomolecular condensates, this refers to the grammer of sequence features that dictate the driving forces for condensate formation and the resulting material properties.
- forcefields
In molecular simulations, forcefields are the set of equations and parameters used to describe the chemical physics of the molecular system of interest. All-atom forcefields used for simulating disordered proteins include ABSINTH, amber03ws, a99SB-disp, CHARMM36m, and DES-Amber417,420-423.
- deep learning
Deep learning is a branch of machine learning concerned with models that contain large numbers of parameters. It has received substantial attention due to its ability to perform complex pattern recognition, especially for text and images. In the biological sciences, deep learning has been applied to protein structure prediction, disorder prediction, and, more recently, the prediction of ensemble properties.
Related links
- Metapredict disorder predictor: https://metapredict.net/
- CAID prediction portal: https://caid.idpcentral.org/submit
- Eukaryotic Linear Motif (ELM) resource: http://elm.eu.org/
- PLAAC Webserver for identify prion-like domains: http://plaac.wi.mit.edu/
- CIDER Webserver for calculating sequence properties: http://pappulab.wustl.edu/CIDER/
- Link to bioinformatic analysis referred to in this paper: https://github.com/holehouse-lab/supportingdata/tree/master/2023/holehouse_and_kragelund_2023
Footnotes
Competing Interest
A.S.H. is a scientific consultant with Dewpoint Therapeutics and on the Scientific Advisory Board for Prose Foods. All other authors declare no conflicts of interest.
References:
- 1. Dunker AK et al. Intrinsically disordered protein. J. Mol. Graph. Model 19, 26–59 (2001). This article (along with Wright & Dyson 1999, Uversky 2001, and Tompa 2002, refs 2,4, and 5, respectively) makes the original arguments that IDRs can and do have important roles in cellular function.
- 2.Wright PE & Dyson HJ Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol 293, 321–331 (1999). [DOI] [PubMed] [Google Scholar]
- 3.van der Lee R. et al. Classification of intrinsically disordered regions and proteins. Chem. Rev 114, 6589–6631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uversky VN Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci 27, 527–533 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Brodsky S, Jana T & Barkai N Order through disorder: The role of intrinsically disordered regions in transcription factor binding specificity. Curr. Opin. Struct. Biol 71, 110–115 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Fuxreiter M. et al. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol 4, 728–737 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Bai X-C & Chen ZJ Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 53, 43–53 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Cuylen S. et al. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 535, 308–312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelham JF, Dunlap JC & Hurley JM Intrinsic disorder is an essential characteristic of components in the conserved circadian circuit. Cell Commun. Signal 18, 181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tompa P & Csermely P The role of structural disorder in the function of RNA and protein chaperones. The FASEB Journal (2004). [DOI] [PubMed] [Google Scholar]
- 12.Altmeyer M. et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun 6, 8088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright PE & Dyson HJ Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol 16, 18–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payliss BJ et al. Phosphorylation of the DNA repair scaffold SLX4 drives folding of the SAP domain and activation of the MUS81-EME1 endonuclease. Cell Rep. 41, 111537 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Witus SR et al. BRCA1/BARD1 intrinsically disordered regions facilitate chromatin recruitment and ubiquitylation. EMBO J. 42, e113565 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanez Orozco IS et al. Identifying weak interdomain interactions that stabilize the supertertiary structure of the N-terminal tandem PDZ domains of PSD-95. Nat. Commun 9, 3724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watson M. et al. Hidden Multivalency in Phosphatase Recruitment by a Disordered AKAP Scaffold. J. Mol. Biol 434, 167682 (2022). [DOI] [PubMed] [Google Scholar]
- 18.Riback JA et al. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 168, 1028–1040.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feric M. et al. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 165, 1686–1697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cermakova K. et al. A ubiquitous disordered protein interaction module orchestrates transcription elongation. Science 374, 1113–1121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Borcherds W. et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol 10, 1000–1002 (2014). This paper offers direct evidence that ensemble properties of IDRs can directly influence cellular function.
- 22.Dyla M & Kjaergaard M Intrinsically disordered linkers control tethered kinases via effective concentration. Proc. Natl. Acad. Sci. U. S. A 117, 21413–21419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millard PS et al. IDDomainSpotter: Compositional bias reveals domains in long disordered protein regions-Insights from transcription factors. Protein Sci. 29, 169–183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotthammer JM, Ginell GM, Griffith D, Emenecker RJ & Holehouse AS Direct Prediction of Intrinsically Disordered Protein Conformational Properties From Sequence. bioRxiv 2023.05.08.539824 (2023) doi: 10.1101/2023.05.08.539824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holehouse AS, Das RK, Ahad JN, Richardson MOG & Pappu RV CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J 112, 16–21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue B, Dunker AK & Uversky VN Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn 30, 137–149 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Cohan MC & Pappu RV Making the Case for Disordered Proteins and Biomolecular Condensates in Bacteria. Trends Biochem. Sci 45, 668–680 (2020). [DOI] [PubMed] [Google Scholar]
- 28. Dyson HJ & Wright PE Equilibrium NMR studies of unfolded and partially folded proteins. Nat. Struct. Biol 5 Suppl, 499–503 (1998). This perspective – along with Wright & Dyson 1999 [ref 2] – makes the case that an ensemble-centric biophysical lens is crucial for understanding disordered proteins.
- 29.Mittag T & Forman-Kay JD Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol 17, 3–14 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Henzler-Wildman K & Kern D Dynamic personalities of proteins. Nature 450, 964–972 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Babu MM, Kriwacki RW & Pappu RV Structural biology. Versatility from protein disorder. Science 337, 1460–1461 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Lazar T. et al. PED in 2021: a major update of the protein ensemble database for intrinsically disordered proteins. Nucleic Acids Res. 49, D404–D411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das RK, Ruff KM & Pappu RV Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol 32, 102–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao AH, Lyle N & Pappu RV Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem. J 449, 307–318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crabtree MD et al. Conserved Helix-Flanking Prolines Modulate Intrinsically Disordered Protein:Target Affinity by Altering the Lifetime of the Bound Complex. Biochemistry 56, 2379–2384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wicky BIM, Shammas SL & Clarke J Affinity of IDPs to their targets is modulated by ion-specific changes in kinetics and residual structure. Proc. Natl. Acad. Sci. U. S. A 114, 9882–9887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dyla M, González Foutel NS, Otzen DE & Kjaergaard M The optimal docking strength for reversibly tethered kinases. Proc. Natl. Acad. Sci. U. S. A 119, e2203098119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. González-Foutel NS et al. Conformational buffering underlies functional selection in intrinsically disordered protein regions. Nat. Struct. Mol. Biol 29, 781–790 (2022). In this study, the authors present evidence that a viral IDR-linker is conserved with respect to ensemble dimensions despite large-scale sequence and length variation.
- 39.Harmon TS, Holehouse AS, Rosen MK & Pappu RV Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Q, Li M, Lai L & Liu Z Allostery of multidomain proteins with disordered linkers. Curr. Opin. Struct. Biol 62, 175–182 (2020). [DOI] [PubMed] [Google Scholar]
- 41. Bugge K. et al. Interactions by Disorder - A Matter of Context. Front Mol Biosci 7, 110 (2020). This review synthesizes a core and emerging idea in the field of disordered proteins; that IDR function is strongly influenced by context.
- 42.Sturzenegger F. et al. Transition path times of coupled folding and binding reveal the formation of an encounter complex. Nat. Commun 9, 4708 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmstrom ED, Liu Z, Nettels D, Best RB & Schuler B Disordered RNA chaperones can enhance nucleic acid folding via local charge screening. Nat. Commun 10, 2453 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cubuk J. et al. The disordered N-terminal tail of SARS CoV-2 Nucleocapsid protein forms a dynamic complex with RNA. bioRxiv 2023.02.10.527914 (2023) doi: 10.1101/2023.02.10.527914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stuchell-Brereton MD et al. Apolipoprotein E4 has extensive conformational heterogeneity in lipid-free and lipid-bound forms. Proc. Natl. Acad. Sci. U. S. A 120, e2215371120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moses D, Ginell GM, Holehouse AS & Sukenik S Intrinsically disordered regions are poised to act as sensors of cellular chemistry. Trends Biochem. Sci 0, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jumper J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tunyasuvunakool K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baek M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruff KM & Pappu RV AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol 433, 167208 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Necci M, Piovesan D, CAID Predictors, DisProt Curators & Tosatto SCE Critical assessment of protein intrinsic disorder prediction. Nat. Methods 18, 472–481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conte AD et al. Critical assessment of protein intrinsic disorder prediction (CAID) - Results of round 2. Proteins (2023) doi: 10.1002/prot.26582. [DOI] [PubMed] [Google Scholar]
- 53.Brown CJ, Johnson AK, Dunker AK & Daughdrill GW Evolution and disorder. Curr. Opin. Struct. Biol 21, 441–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown CJ, Johnson AK & Daughdrill GW Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol 27, 609–621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zarin T. et al. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. Elife 8, (2019). This study offers a systematic assessment of how conservation in IDRs could act at the level of sequence features instead of specific linear sequence.
- 56.Cohan MC, Shinn MK, Lalmansingh JM & Pappu RV Uncovering Non-random Binary Patterns Within Sequences of Intrinsically Disordered Proteins. J. Mol. Biol 434, 167373 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martin EW et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699 (2020). This study provides a detailed biophysical assessment of how sequence features and ensemble properties cooperate to determine the driving forces for phase separation in low-complexity disordered regions.
- 58.Beh LY, Colwell LJ & Francis NJ A core subunit of Polycomb repressive complex 1 is broadly conserved in function but not primary sequence. Proc. Natl. Acad. Sci. U. S. A 109, E1063–71 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zarin T, Tsai CN, Nguyen Ba AN & Moses AM Selection maintains signaling function of a highly diverged intrinsically disordered region. Proc. Natl. Acad. Sci. U. S. A 114, E1450–E1459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tesei G. et al. Conformational ensembles of the human intrinsically disordered proteome: Bridging chain compaction with function and sequence conservation. bioRxiv 2023.05.08.539815 (2023) doi: 10.1101/2023.05.08.539815. [DOI] [Google Scholar]
- 61.Martin EW et al. Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. J. Am. Chem. Soc 138, 15323–15335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Müller-Späth S. et al. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 107, 14609–14614 (2010). This article (along with Marsh & Forman-Kay [2010] and Mao et al. PNAS [2010], refs 63 and 64, respectively) offers direct evidence that IDRs net charge influences global dimensions.
- 63.Marsh JA & Forman-Kay JD Sequence Determinants of Compaction in Intrinsically Disordered Proteins. Biophys. J 98, 2383–2390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mao AH, Crick SL, Vitalis A, Chicoine CL & Pappu RV Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 107, 8183–8188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Das RK & Pappu RV Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U. S. A 110, 13392–13397 (2013). This study is the first to systematically show that the patterning of charged residues in IDRs can be an important feature that influences ensemble behaviour.
- 66.Sawle L & Ghosh K A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J. Chem. Phys 143, 085101 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Kulkarni P. et al. Phosphorylation-induced conformational dynamics in an intrinsically disordered protein and potential role in phenotypic heterogeneity. Proc. Natl. Acad. Sci. U. S. A 114, E2644–E2653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jin F & Gräter F How multisite phosphorylation impacts the conformations of intrinsically disordered proteins. PLoS Comput. Biol 17, e1008939 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sørensen CS & Kjaergaard M Effective concentrations enforced by intrinsically disordered linkers are governed by polymer physics. Proc. Natl. Acad. Sci. U. S. A 116, 23124–23131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zeng X, Ruff KM & Pappu RV Competing interactions give rise to two-state behavior and switch-like transitions in charge-rich intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 119, e2200559119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Portz B. et al. Structural heterogeneity in the intrinsically disordered RNA polymerase II C-terminal domain. Nat. Commun 8, 15231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bremer A. et al. Deciphering how naturally occurring sequence features impact the phase behaviours of disordered prion-like domains. Nat. Chem 14, 196–207 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plevin MJ, Bryce DL & Boisbouvier J Direct detection of CH/pi interactions in proteins. Nat. Chem 2, 466–471 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Crick SL, Jayaraman M, Frieden C, Wetzel R & Pappu RV Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. U. S. A 103, 16764–16769 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Holehouse AS, Garai K, Lyle N, Vitalis A & Pappu RV Quantitative assessments of the distinct contributions of polypeptide backbone amides versus side chain groups to chain expansion via chemical denaturation. J. Am. Chem. Soc 137, 2984–2995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S & Deniz AA A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proceedings of the National Academy of Sciences 104, 2649–2654 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Theillet F-X et al. The alphabet of intrinsic disorder: I. Act like a Pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord Proteins 1, e24360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boze H. et al. Proline-rich salivary proteins have extended conformations. Biophys. J 99, 656–665 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibbs EB et al. Phosphorylation induces sequence-specific conformational switches in the RNA polymerase II C-terminal domain. Nat. Commun 8, 15233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rauscher S, Baud S, Miao M, Keeley FW & Pomès R Proline and glycine control protein self-organization into elastomeric or amyloid fibrils. Structure 14, 1667–1676 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Beveridge R. et al. Ion Mobility Mass Spectrometry Uncovers the Impact of the Patterning of Oppositely Charged Residues on the Conformational Distributions of Intrinsically Disordered Proteins. J. Am. Chem. Soc 141, 4908–4918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Das RK, Huang Y, Phillips AH, Kriwacki RW & Pappu RV Cryptic sequence features within the disordered protein p27Kip1 regulate cell cycle signaling. Proc. Natl. Acad. Sci. U. S. A 113, 5616–5621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sherry KP, Das RK, Pappu RV & Barrick D Control of transcriptional activity by design of charge patterning in the intrinsically disordered RAM region of the Notch receptor. Proc. Natl. Acad. Sci. U. S. A 114, E9243–E9252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ginell GM, Flynn AJ & Holehouse AS SHEPHARD: a modular and extensible software architecture for analyzing and annotating large protein datasets. Bioinformatics 39, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iakoucheva LM et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choy MS, Page R & Peti W Regulation of protein phosphatase 1 by intrinsically disordered proteins. Biochem. Soc. Trans 40, 969–974 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gomes G-NW et al. Conformational Ensembles of an Intrinsically Disordered Protein Consistent with NMR, SAXS, and Single-Molecule FRET. J. Am. Chem. Soc 142, 15697–15710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nash P. et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414, 514–521 (2001). [DOI] [PubMed] [Google Scholar]
- 89.Mittag T. et al. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proceedings of the National Academy of Sciences 105, 17772–17777 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Borg M. et al. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl. Acad. Sci. U. S. A 104, 9650–9655 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ruff KM Predicting Conformational Properties of Intrinsically Disordered Proteins from Sequence. Methods Mol. Biol 2141, 347–389 (2020). [DOI] [PubMed] [Google Scholar]
- 92.Zarin T. et al. Identifying molecular features that are associated with biological function of intrinsically disordered protein regions. Elife 10, e60220 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ginell GM & Holehouse AS Analyzing the Sequences of Intrinsically Disordered Regions with CIDER and localCIDER. in Intrinsically Disordered Proteins: Methods and Protocols (eds. Kragelund BB & Skriver K) vol. 2141 103–126 (Springer US, 2020). [DOI] [PubMed] [Google Scholar]
- 94.Ghosh K, Huihui J, Phillips M & Haider A Rules of Physical Mathematics Govern Intrinsically Disordered Proteins. Annu. Rev. Biophys (2022) doi: 10.1146/annurev-biophys-120221-095357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huihui J & Ghosh K Intra-chain interaction topology can identify functionally similar Intrinsically Disordered Proteins. Biophys. J (2021) doi: 10.1016/j.bpj.2020.11.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huihui J & Ghosh K An analytical theory to describe sequence-specific inter-residue distance profiles for polyampholytes and intrinsically disordered proteins. J. Chem. Phys 152, 161102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Franzmann TM et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 359, eaao5654 (2018). [DOI] [PubMed] [Google Scholar]
- 98.Yamazaki H, Takagi M, Kosako H, Hirano T & Yoshimura SH Cell cycle-specific phase separation regulated by protein charge blockiness. Nat. Cell Biol 24, 625–632 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lyons H. et al. Functional partitioning of transcriptional regulators by patterned charge blocks. Cell 186, 327–345.e28 (2023). In this paper, the authors show that specific patterning of charged residues in IDRs enable specific molecular recruitment to transcriptional condensates.
- 100.Jankowski MS et al. The formation of a fuzzy complex in the negative arm regulates the robustness of the circadian clock. bioRxiv 2022.01.04.474980 (2022) doi: 10.1101/2022.01.04.474980. [DOI] [Google Scholar]
- 101.Brendel V & Karlin S Association of charge clusters with functional domains of cellular transcription factors. Proc. Natl. Acad. Sci. U. S. A 86, 5698–5702 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robustelli P, Piana S & Shaw DE Mechanism of Coupled Folding-upon-Binding of an Intrinsically Disordered Protein. J. Am. Chem. Soc 142, 11092–11101 (2020). [DOI] [PubMed] [Google Scholar]
- 103.Das RK, Crick SL & Pappu RV N-terminal segments modulate the α-helical propensities of the intrinsically disordered basic regions of bZIP proteins. J. Mol. Biol 416, 287–299 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Milles S. et al. An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci Adv 4, eaat7778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Daughdrill GW Disorder for Dummies: Functional Mutagenesis of Transient Helical Segments in Disordered Proteins. Methods Mol. Biol 2141, 3–20 (2020). [DOI] [PubMed] [Google Scholar]
- 106.Davey NE The functional importance of structure in unstructured protein regions. Curr. Opin. Struct. Biol 56, 155–163 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Zhu J, Salvatella X & Robustelli P Small molecules targeting the disordered transactivation domain of the androgen receptor induce the formation of collapsed helical states. Nat. Commun 13, 6390 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chhabra Y. et al. A growth hormone receptor SNP promotes lung cancer by impairment of SOCS2-mediated degradation. Oncogene 37, 489–501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Loening NM, Saravanan S, Jespersen NE, Jara K & Barbar E Interplay of Disorder and Sequence Specificity in the Formation of Stable Dynein-Dynactin Complexes. Biophys. J 119, 950–965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clark S. et al. Multivalency regulates activity in an intrinsically disordered transcription factor. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Burke KA, Janke AM, Rhine CL & Fawzi NL Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell 60, 231–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Murthy AC et al. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol 26, 637–648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murthy AC et al. Molecular interactions contributing to FUS SYGQ LC-RGG phase separation and co-partitioning with RNA polymerase II heptads. Nat. Struct. Mol. Biol 28, 923–935 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang P. et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 181, 325–345.e28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guillen-Boixet J. et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 181, 346–361.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Warner JB 4th et al. Monomeric Huntingtin Exon 1 Has Similar Overall Structural Features for Wild-Type and Pathological Polyglutamine Lengths. J. Am. Chem. Soc 139, 14456–14469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Newcombe EA et al. Tadpole-like Conformations of Huntingtin Exon 1 Are Characterized by Conformational Heterogeneity that Persists regardless of Polyglutamine Length. J. Mol. Biol 430, 1442–1458 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Urbanek A. et al. Flanking Regions Determine the Structure of the Poly-Glutamine in Huntingtin through Mechanisms Common among Glutamine-Rich Human Proteins. Structure 28, 733–746.e5 (2020). [DOI] [PubMed] [Google Scholar]
- 119.Elena-Real CA et al. The structure of pathogenic huntingtin exon 1 defines the bases of its aggregation propensity. Nat. Struct. Mol. Biol 30, 309–320 (2023). [DOI] [PubMed] [Google Scholar]
- 120. Moses D. et al. Revealing the Hidden Sensitivity of Intrinsically Disordered Proteins to their Chemical Environment. J. Phys. Chem. Lett 11, 10131–10136 (2020). ). In this work, the authors combine experiment, simulation, and theory to examine how IDRs show sequence-dependent sensitivity to changes in the solution context.
- 121.Sørensen CS & Kjaergaard M Measuring Effective Concentrations Enforced by Intrinsically Disordered Linkers. Methods Mol. Biol 2141, 505–518 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Mateos B. et al. Hyperphosphorylation of Human Osteopontin and Its Impact on Structural Dynamics and Molecular Recognition. Biochemistry (2021) doi: 10.1021/acs.biochem.1c00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dignon GL, Zheng W, Best RB, Kim YC & Mittal J Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 115, 9929–9934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Keul ND et al. The entropic force generated by intrinsically disordered segments tunes protein function. Nature 563, 584–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Busch DJ et al. Intrinsically disordered proteins drive membrane curvature. Nat. Commun 6, 7875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Borcherds W. et al. Optimal Affinity Enhancement by a Conserved Flexible Linker Controls p53 Mimicry in MdmX. Biophys. J 112, 2038–2042 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kjaergaard M. Estimation of Effective Concentrations Enforced by Complex Linker Architectures from Conformational Ensembles. Biochemistry 61, 171–182 (2022). [DOI] [PubMed] [Google Scholar]
- 128.Martin IM et al. Phosphorylation tunes elongation propensity and cohesiveness of INCENP’s intrinsically disordered region. J. Mol. Biol 434, 167387 (2022). [DOI] [PubMed] [Google Scholar]
- 129.Sherry KP, Johnson SE, Hatem CL, Majumdar A & Barrick D Effects of Linker Length and Transient Secondary Structure Elements in the Intrinsically Disordered Notch RAM Region on Notch Signaling. J. Mol. Biol 427, 3587–3597 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Motlagh HN, Wrabl JO, Li J & Hilser VJ The ensemble nature of allostery. Nature 508, 331–339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li M, Cao H, Lai L & Liu Z Disordered linkers in multidomain allosteric proteins: Entropic effect to favor the open state or enhanced local concentration to favor the closed state? Protein Sci. 27, 1600–1610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang WYC, Ditlev JA, Chiang H-K, Rosen MK & Groves JT Allosteric Modulation of Grb2 Recruitment to the Intrinsically Disordered Scaffold Protein, LAT, by Remote Site Phosphorylation. J. Am. Chem. Soc 139, 18009–18015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Seiffert P. et al. Orchestration of signaling by structural disorder in class 1 cytokine receptors. Cell Commun. Signal 18, 132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yu F & Sukenik S Structural Preferences Shape the Entropic Force of Disordered Protein Ensembles. J. Phys. Chem. B 127, 4235–4244 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zeno WF et al. Synergy between intrinsically disordered domains and structured proteins amplifies membrane curvature sensing. Nat. Commun 9, 4152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zeno WF et al. Molecular Mechanisms of Membrane Curvature Sensing by a Disordered Protein. J. Am. Chem. Soc 141, 10361–10371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Halladin DK et al. Entropy-driven translocation of disordered proteins through the Gram-positive bacterial cell wall. Nat Microbiol 6, 1055–1065 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Davey NE et al. Attributes of short linear motifs. Mol. Biosyst 8, 268–281 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Holehouse AS Chapter 7 - IDPs and IDRs in biomolecular condensates. in Intrinsically Disordered Proteins (ed. Salvi N) 209–255 (Academic Press, 2019). [Google Scholar]
- 140.Posey AE, Holehouse AS & Pappu RV Chapter One - Phase Separation of Intrinsically Disordered Proteins. in Methods in Enzymology (ed. Rhoades E) vol. 611 1–30 (Academic Press, 2018). [DOI] [PubMed] [Google Scholar]
- 141.Brangwynne CP, Tompa P & Pappu RV Polymer physics of intracellular phase transitions. Nat. Phys 11, 899–904 (2015). [Google Scholar]
- 142.Duchesne L. et al. Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol. 10, e1001361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kleinschmit A. et al. Drosophila heparan sulfate 6-O endosulfatase regulates Wingless morphogen gradient formation. Dev. Biol 345, 204–214 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yan D & Lin X Shaping morphogen gradients by proteoglycans. Cold Spring Harb. Perspect. Biol 1, a002493 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Triandafillou CG, Katanski CD, Dinner AR & Drummond DA Transient intracellular acidification regulates the core transcriptional heat shock response. Elife 9, e54880 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gutierrez JI et al. SWI/SNF senses carbon starvation with a pH-sensitive low-complexity sequence. Elife 11, e70344 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Soranno A. et al. Single-molecule spectroscopy reveals polymer effects of disordered proteins in crowded environments. Proc. Natl. Acad. Sci. U. S. A 111, 4874–4879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Delarue M. et al. mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell 174, 338–349.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Battaglia M, Olvera-Carrillo Y, Garciarrubio A, Campos F & Covarrubias AA The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 148, 6–24 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Boothby TC & Pielak GJ Intrinsically Disordered Proteins and Desiccation Tolerance: Elucidating Functional and Mechanistic Underpinnings of Anhydrobiosis. Bioessays 39, 1700119 (2017). [DOI] [PubMed] [Google Scholar]
- 151.Boothby TC et al. Tardigrades Use Intrinsically Disordered Proteins to Survive Desiccation. Mol. Cell 65, 975–984.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wuttke R. et al. Temperature-dependent solvation modulates the dimensions of disordered proteins. Proc. Natl. Acad. Sci. U. S. A 111, 5213–5218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Quiroz FG & Chilkoti A Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat. Mater 14, 1164–1171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kjaergaard M. et al. Temperature-dependent structural changes in intrinsically disordered proteins: formation of alpha-helices or loss of polyproline II? Protein Sci. 19, 1555–1564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jung J-H et al. A prion-like domain in ELF3 functions as a thermosensor in Arabidopsis. Nature 585, 256–260 (2020). [DOI] [PubMed] [Google Scholar]
- 156.Zhu P, Lister C & Dean C Cold-induced Arabidopsis FRIGIDA nuclear condensates for FLC repression. Nature 599, 657–661 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cuylen-Haering S. et al. Chromosome clustering by Ki-67 excludes cytoplasm during nuclear assembly. Nature (2020) doi: 10.1038/s41586-020-2672-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kumar S & Hoh JH Modulation of repulsive forces between neurofilaments by sidearm phosphorylation. Biochem. Biophys. Res. Commun 324, 489–496 (2004). [DOI] [PubMed] [Google Scholar]
- 159.Doncic A. et al. Compartmentalization of a bistable switch enables memory to cross a feedback-driven transition. Cell 160, 1182–1195 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Koivomagi M. et al. Multisite phosphorylation networks as signal processors for Cdk1. Nat. Struct. Mol. Biol 20, 1415–1424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhou M, Kim JK, Eng GWL, Forger DB & Virshup DM A Period2 Phosphoswitch Regulates and Temperature Compensates Circadian Period. Mol. Cell 60, 77–88 (2015). [DOI] [PubMed] [Google Scholar]
- 162. Bah A. et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519, 106–109 (2015). Here, the authors show that multisite phosphorylation of a disordered region can mediate intramolecular folding into a stable 3D structure, offering an example of phospho-conditional folding.
- 163.Mittal A, Holehouse AS, Cohan MC & Pappu RV Sequence-to-Conformation Relationships of Disordered Regions Tethered to Folded Domains of Proteins. J. Mol. Biol 430, 2403–2421 (2018). [DOI] [PubMed] [Google Scholar]
- 164.Taneja I & Holehouse AS Folded domain charge properties influence the conformational behavior of disordered tails. Curr Res Struct Biol 3, 216–228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zheng T, Galagedera SKK & Castañeda CA Previously uncharacterized interactions between the folded and intrinsically disordered domains impart asymmetric effects on UBQLN2 phase separation. Protein Sci. (2021) doi: 10.1002/pro.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Martin EW et al. Interplay of folded domains and the disordered low-complexity domain in mediating hnRNPA1 phase separation. Nucleic Acids Res. 49, 2931–2945 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bjarnason S. et al. DNA binding redistributes activation domain ensemble and accessibility in pioneer factor Sox2. bioRxiv 2023.06.16.545083 (2023) doi: 10.1101/2023.06.16.545083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Farr SE, Woods EJ, Joseph JA, Garaizar A & Collepardo-Guevara R Nucleosome plasticity is a critical element of chromatin liquid–liquid phase separation and multivalent nucleosome interactions. Nat. Commun 12, 1–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heidarsson PO et al. Release of linker histone from the nucleosome driven by polyelectrolyte competition with a disordered protein. Nat. Chem 14, 224–231 (2022). [DOI] [PubMed] [Google Scholar]
- 170.Chen Q, Yang R, Korolev N, Liu CF & Nordenskiöld L Regulation of Nucleosome Stacking and Chromatin Compaction by the Histone H4 N-Terminal Tail–H2A Acidic Patch Interaction. J. Mol. Biol 429, 2075–2092 (2017). [DOI] [PubMed] [Google Scholar]
- 171.Staby L. et al. Flanking Disorder of the Folded αα-Hub Domain from Radical Induced Cell Death1 Affects Transcription Factor Binding by Ensemble Redistribution. J. Mol. Biol 433, 167320 (2021). [DOI] [PubMed] [Google Scholar]
- 172.Staby L. et al. Disorder in a two-domain neuronal Ca2+-binding protein regulates domain stability and dynamics using ligand mimicry. Cell. Mol. Life Sci 78, 2263–2278 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Corless EI et al. The flexible N-terminus of BchL autoinhibits activity through interaction with its [4Fe-4S] cluster and released upon ATP binding. J. Biol. Chem 296, 100107 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Alston JJ, Soranno A & Holehouse AS Conserved molecular recognition by an intrinsically disordered region in the absence of sequence conservation. bioRxiv 2023.08.06.552128 (2023). [Google Scholar]
- 175.Hendus-Altenburger R. et al. A phosphorylation-motif for tuneable helix stabilisation in intrinsically disordered proteins - Lessons from the sodium proton exchanger 1 (NHE1). Cell. Signal 37, 40–51 (2017). [DOI] [PubMed] [Google Scholar]
- 176.Hendus-Altenburger R. et al. The human Na(+)/H(+) exchanger 1 is a membrane scaffold protein for extracellular signal-regulated kinase 2. BMC Biol. 14, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Brown CJ et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol 55, 104–110 (2002). [DOI] [PubMed] [Google Scholar]
- 178.Langstein-Skora I. et al. Sequence- and chemical specificity define the functional landscape of intrinsically disordered regions. bioRxiv 2022.02.10.480018 (2022) doi: 10.1101/2022.02.10.480018. [DOI] [Google Scholar]
- 179.Davey NE, Cyert MS & Moses AM Short linear motifs--ex nihilo evolution of protein regulation. Cell Commun. Signal 13, 43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sangster AG, Zarin T & Moses AM Evolution of short linear motifs and disordered proteins Topic: yeast as model system to study evolution. Curr. Opin. Genet. Dev 76, 101964 (2022). [DOI] [PubMed] [Google Scholar]
- 181.Garboczi DN et al. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature vol. 384 134–141 (1996). [DOI] [PubMed] [Google Scholar]
- 182.Rebek J Jr. Model studies in molecular recognition. Science 235, 1478–1484 (1987). [DOI] [PubMed] [Google Scholar]
- 183.Brooijmans N & Kuntz ID Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct 32, 335–373 (2003). [DOI] [PubMed] [Google Scholar]
- 184.Rogers JM, Wong CT & Clarke J Coupled folding and binding of the disordered protein PUMA does not require particular residual structure. J. Am. Chem. Soc 136, 5197–5200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen T, Song J & Chan HS Theoretical perspectives on nonnative interactions and intrinsic disorder in protein folding and binding. Curr. Opin. Struct. Biol 30C, 32–42 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Ivarsson Y & Jemth P Affinity and specificity of motif-based protein-protein interactions. Curr. Opin. Struct. Biol 54, 26–33 (2019). [DOI] [PubMed] [Google Scholar]
- 187.Staller MV et al. Directed mutational scanning reveals a balance between acidic and hydrophobic residues in strong human activation domains. Cell Syst 13, 334–345.e5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Berlow RB, Dyson HJ & Wright PE Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 543, 447–451 (2017). In this study, the authors reveal how dynamic inter-molecular competition coupled with disorder-driven allosteric changes can lead to complex regulatory behaviour in IDR binding.
- 189.Olsen JG, Teilum K & Kragelund BB Behaviour of intrinsically disordered proteins in protein–protein complexes with an emphasis on fuzziness. Cell. Mol. Life Sci 74, 3175–3183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tompa P & Fuxreiter M Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci 33, 2–8 (2008). [DOI] [PubMed] [Google Scholar]
- 191.Eick D & Geyer M The RNA polymerase II carboxy-terminal domain (CTD) code. Chem. Rev 113, 8456–8490 (2013). [DOI] [PubMed] [Google Scholar]
- 192.Prestel A. et al. The PCNA interaction motifs revisited: thinking outside the PIP-box. Cell. Mol. Life Sci (2019) doi: 10.1007/s00018-019-03150-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Follis AV et al. Regulation of apoptosis by an intrinsically disordered region of Bcl-xL. Nat. Chem. Biol 14, 458–465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.He F. et al. Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc. Natl. Acad. Sci. U. S. A 116, 8859–8868 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Krois AS, Dyson HJ & Wright PE Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. U. S. A 115, E11302–E11310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 197.Phatnani HP & Greenleaf AL Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936 (2006). [DOI] [PubMed] [Google Scholar]
- 198.Wright PE & Dyson HJ Linking folding and binding. Curr. Opin. Struct. Biol 19, 31–38 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sugase K, Dyson HJ & Wright PE Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 (2007). [DOI] [PubMed] [Google Scholar]
- 200.Rogers JM, Steward A & Clarke J Folding and binding of an intrinsically disordered protein: fast, but not “diffusion-limited.” J. Am. Chem. Soc 135, 1415–1422 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gianni S, Morrone A, Giri R & Brunori M A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem. Biophys. Res. Commun 428, 205–209 (2012). [DOI] [PubMed] [Google Scholar]
- 202.Dogan J, Schmidt T, Mu X, Engström Å & Jemth P Fast association and slow transitions in the interaction between two intrinsically disordered protein domains. J. Biol. Chem 287, 34316–34324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Teilum K, Olsen JG & Kragelund BB Globular and disordered-the non-identical twins in protein-protein interactions. Front. Mol. Biosci 2, 40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lazar T, Tantos A, Tompa P & Schad E Intrinsic protein disorder uncouples affinity from binding specificity. Protein Sci. 31, e4455 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Skriver K, Theisen FF & Kragelund BB Conformational entropy in molecular recognition of intrinsically disordered proteins. Curr. Opin. Struct. Biol (in press), (2023). [DOI] [PubMed] [Google Scholar]
- 206.O’Shea C. et al. Structures and short linear motif of disordered transcription factor regions provide clues to the interactome of the cellular hub protein radical-induced cell Death1. J. Biol. Chem 292, 512–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Theisen FF et al. αα-hub coregulator structure and flexibility determine transcription factor binding and selection in regulatory interactomes. J. Biol. Chem 298, 101963 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Arai M, Sugase K, Dyson HJ & Wright PE Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc. Natl. Acad. Sci. U. S. A 112, 9614–9619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shammas SL, Crabtree MD, Dahal L, Wicky BIM & Clarke J Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem 291, 6689–6695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gianni S, Dogan J & Jemth P Distinguishing induced fit from conformational selection. Biophys. Chem 189, 33–39 (2014). [DOI] [PubMed] [Google Scholar]
- 211.Dogan J & Jemth P Only kinetics can prove conformational selection. Biophys. J 107, 1997–1998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hammes GG, Chang Y-C & Oas TG Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. U. S. A 106, 13737–13741 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Dyson HJ & Wright PE Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol 12, 54–60 (2002). [DOI] [PubMed] [Google Scholar]
- 214.Spolar RS & Record MT Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science 263, 777–784 (1994). [DOI] [PubMed] [Google Scholar]
- 215.Suarez IP, Burdisso P, Benoit MPMH, Boisbouvier J & Rasia RM Induced folding in RNA recognition by Arabidopsis thaliana DCL1. Nucleic Acids Res. 43, 6607–6619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vise PD, Baral B, Latos AJ & Daughdrill GW NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 33, 2061–2077 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lee CW, Martinez-Yamout MA, Dyson HJ & Wright PE Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 49, 9964–9971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Elkjaer S. et al. Evolutionary fine-tuning of residual helix structure in disordered proteins manifests in complex structure and lifetime. Commun. Biol 6, 63 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Crabtree MD, Mendonça CATF, Bubb QR & Clarke J Folding and binding pathways of BH3-only proteins are encoded within their intrinsically disordered sequence, not templated by partner proteins. J. Biol. Chem 293, 9718–9723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Toto A. et al. Molecular recognition by templated folding of an intrinsically disordered protein. Sci. Rep 6, 21994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Longhi S. et al. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem 278, 18638–18648 (2003). [DOI] [PubMed] [Google Scholar]
- 222.Demarest SJ et al. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 415, 549–553 (2002). [DOI] [PubMed] [Google Scholar]
- 223.Qin BY et al. Crystal structure of IRF-3 in complex with CBP. Structure 13, 1269–1277 (2005). [DOI] [PubMed] [Google Scholar]
- 224.Karlsson E. et al. Disordered regions flanking the binding interface modulate affinity between CBP and NCOA. J. Mol. Biol 167643 (2022). [DOI] [PubMed] [Google Scholar]
- 225.Jemth P. et al. Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins. Science Advances 4, eaau4130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Fuxreiter M. Fold or not to fold upon binding—does it really matter? Curr. Opin. Struct. Biol (2019). [DOI] [PubMed] [Google Scholar]
- 227.Sharma R, Raduly Z, Miskei M & Fuxreiter M Fuzzy complexes: Specific binding without complete folding. FEBSLett. 589, 2533–2542 (2015). [DOI] [PubMed] [Google Scholar]
- 228.Jiang Y, Rossi P & Kalodimos CG Structural basis for client recognition and activity of Hsp40 chaperones. Science 365, 1313–1319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Shi Y. et al. Structure-based classification of tauopathies. Nature 598, 359–363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yang Y. et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 375, 167–172 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yang Y. et al. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature 610, 791–795 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Berlow RB, Dyson HJ & Wright PE Multivalency enables unidirectional switch-like competition between intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 119, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Tuttle LM et al. Gcn4-Mediator Specificity Is Mediated by a Large and Dynamic Fuzzy Protein-Protein Complex. Cell Rep. 22, 3251–3264 (2018). In this study, the authors provide biophysical characterization of fuzzy transcription factor–co-activator binding.
- 234.Brzovic PS et al. The acidic transcription activator Gcn4 binds the mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol. Cell 44, 942–953 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Staller MV et al. A High-Throughput Mutational Scan of an Intrinsically Disordered Acidic Transcriptional Activation Domain. CellSyst 6, 444–455.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sanborn AL et al. Simple biochemical features underlie transcriptional activation domain diversity and dynamic, fuzzy binding to Mediator. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Görlich D, Prehn S, Laskey RA & Hartmann E Isolation of a protein that is essential for the first step of nuclear protein import. Cell 79, 767–778 (1994). [DOI] [PubMed] [Google Scholar]
- 238.Görlich D & Kutay U Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol 15, 607–660 (1999). [DOI] [PubMed] [Google Scholar]
- 239.Milles S. et al. Plasticity of an ultrafast interaction between nucleoporins and nuclear transport receptors. Cell 163, 734–745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Frey S & Görlich D A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell 130, 512–523 (2007). This paper provides the first dissection of the sequence rules that determine what would now be referred to as biomolecular condensates, illustrating how IDRs can self-assemble into materials with biological activity.
- 241.Frey S, Richter RP & Görlich D FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 314, 815–817 (2006). [DOI] [PubMed] [Google Scholar]
- 242.Schmidt HB & Görlich D Nup98 FG domains from diverse species spontaneously phase-separate into particles with nuclear pore-like permselectivity. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Buholzer KJ et al. Multilayered allosteric modulation of coupled folding and binding by phosphorylation, peptidyl-prolyl cis/trans isomerization, and diversity of interaction partners. J. Chem. Phys 157, 235102 (2022). [DOI] [PubMed] [Google Scholar]
- 244.Staller MV Transcription factors perform a 2-step search of the nucleus. Genetics (2022) doi: 10.1093/genetics/iyac111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Schuler B. et al. Binding without folding - the biomolecular function of disordered polyelectrolyte complexes. Curr. Opin. Struct. Biol 60, 66–76 (2019). [DOI] [PubMed] [Google Scholar]
- 246. Borgia A. et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 555, 61–66 (2018). This paper provides conclusive evidence that disordered proteins can form high-affinity biomolecular complexes that remain dynamic without the acquisition of any secondary or tertiary structure or persistent contacts.
- 247.Sottini A. et al. Polyelectrolyte interactions enable rapid association and dissociation in high-affinity disordered protein complexes. Nat. Commun 11, 5736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Galvanetto N. et al. Extreme dynamics in a biomolecular condensate. Nature 1–8 (2023). In this work, the authors combine single-molecule studies and simulations to show that intra-condensate dynamics in IDR condensates can remain fast, despite macroscopic condensate viscosity being high.
- 249. Yu M. et al. Visualizing the disordered nuclear transport machinery in situ. Nature 617, 162–169 (2023). In this work, the authors determine the conformational behaviour of FG-repeat IDRs inside the nuclear pore complex.
- 250.Frey S. et al. Surface Properties Determining Passage Rates of Proteins through Nuclear Pores. Cell 174, 202–217.e9 (2018). [DOI] [PubMed] [Google Scholar]
- 251.Schneider R, Blackledge M & Jensen MR Elucidating binding mechanisms and dynamics of intrinsically disordered protein complexes using NMR spectroscopy. Curr. Opin. Struct. Biol 54, 10–18 (2019). [DOI] [PubMed] [Google Scholar]
- 252.Teilum K, Olsen JG & Kragelund BB On the specificity of protein-protein interactions in the context of disorder. Biochem. J 478, 2035–2050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Eaton BE, Gold L & Zichi DA Let’s get specific: the relationship between specificity and affinity. Chem. Biol 2, 633–638 (1995). [DOI] [PubMed] [Google Scholar]
- 254.Kumar M. et al. ELM—the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 48, D296–D306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Tompa P, Davey NE, Gibson TJ & Babu MM A million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 (2014). This perspective provides a clear assessment of the importance that SLiMs endow upon IDRs, and their prevalence across eukaryotic proteomes.
- 256.Bruning JB & Shamoo Y Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-delta p66 subunit and flap endonuclease-1. Structure 12, 2209–2219 (2004). [DOI] [PubMed] [Google Scholar]
- 257.Dreier JE et al. A context-dependent and disordered ubiquitin-binding motif. Cell. Mol. Life Sci 79, 484 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lee BJ et al. Rules for Nuclear Localization Sequence Recognition by Karyopher2. Cell 126, 543–558 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Oldfield CJ et al. Flexible nets: disorder and induced fit in the associations of p53 and 14–3-3 with their partners. BMC Genomics 9 Suppl 1, S1 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Davey NE, Simonetti L & Ivarsson Y The next wave of interactomics: Mapping the SLiM-based interactions of the intrinsically disordered proteome. Curr. Opin. Struct. Biol 80, 102593 (2023). [DOI] [PubMed] [Google Scholar]
- 261.Hadži S, Loris R & Lah J The sequence-ensemble relationship in fuzzy protein complexes. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Nguyen HQ et al. Quantitative mapping of protein-peptide affinity landscapes using spectrally encoded beads. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mihalič F. et al. Evolution of affinity between p53 transactivation domain and MDM2 across the animal kingdom demonstrates high plasticity of motif-mediated interactions. Protein Sci. 32, e4684 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kumar M. et al. The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res. 50, D497–D508 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Davey NE, Shields DC & Edwards RJ Masking residues using context-specific evolutionary conservation significantly improves short linear motif discovery. Bioinformatics 25, 443–450 (2009). [DOI] [PubMed] [Google Scholar]
- 266.Chhabra Y. et al. Tyrosine kinases compete for growth hormone receptor binding and regulate receptor mobility and degradation. Cell Rep. 42, 112490 (2023). [DOI] [PubMed] [Google Scholar]
- 267.Raj N & Attardi LD The Transactivation Domains of the p53 Protein. Cold Spring Harb. Perspect. Med 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bochkareva E. et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. U. S. A 102, 15412–15417 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Di Lello P. et al. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 22, 731–740 (2006). [DOI] [PubMed] [Google Scholar]
- 270.Miller Jenkins LM et al. Characterization of the p300 Taz2–p53 TAD2 Complex and Comparison with the p300 Taz2–p53 TAD1 Complex. Biochemistry 54, 2001–2010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rowell JP, Simpson KL, Stott K, Watson M & Thomas JO HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 20, 2014–2024 (2012). [DOI] [PubMed] [Google Scholar]
- 272.Lee M-S et al. Solution structure of MUL1-RING domain and its interaction with p53 transactivation domain. Biochem. Biophys. Res. Commun 516, 533–539 (2019). [DOI] [PubMed] [Google Scholar]
- 273.Mészáros B, Kumar M, Gibson TJ, Uyar B & Dosztányi Z Degrons in cancer. Sci. Signal 10, (2017). [DOI] [PubMed] [Google Scholar]
- 274.Provost E. et al. Functional correlates of mutation of the Asp32 and Gly34 residues of beta-catenin. Oncogene 24, 2667–2676 (2005). [DOI] [PubMed] [Google Scholar]
- 275.Olsen JG et al. Checkpoint activation by Spd1: a competition-based system relying oi tandem disordered PCNA binding motifs. bioRxiv 2023.05.11.540346 (2023) doi: 10.1101/2023.05.11.540346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Meyer K. et al. Mutations in Disordered Regions Can Cause Disease by Creating Dileucine Motifs. Cell 175, 239–253.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 277.Kliche J. et al. Large-scale phosphomimetic screening identifies phospho-modulated motif-based protein interactions. Mol. Syst. Biol e11164 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kassa E. et al. Evaluation of affinity-purification coupled to mass spectrometry approaches for capture of short linear motif-based interactions. Anal. Biochem 663, 115017 (2023). [DOI] [PubMed] [Google Scholar]
- 279. Mihalič F. et al. Large-scale phage-based screening reveals extensive pan-viral mimicry of host short linear motifs. Nat. Commun 14, 2409 (2023). In this study, a high-throughput analysis identified 1712 SLiM-based virus–host interactions, where viruses engage in molecular mimicry to subvert host programmes.
- 280.Stein A & Aloy P Contextual specificity in peptide-mediated protein interactions. PLoS One 3, e2524 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Karlsson E, Ottoson C, Ye W, Andersson E & Jemth P Intrinsically Disordered Flanking Regions Increase the Affinity of a Transcriptional Coactivator Interaction across Vertebrates. Biochemistry (2023) doi: 10.1021/acs.biochem.3c00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Siligardi G. et al. The SH3 domain of HS1 protein recognizes lysine-rich polyproline motifs. Amino Acids 42, 1361–1370 (2012). [DOI] [PubMed] [Google Scholar]
- 283.Palopoli N, González Foutel NS, Gibson TJ & Chemes LB Short linear motif core and flanking regions modulate retinoblastoma protein binding affinity and specificity. Protein Eng. Des. Sel 31, 69–77 (2018). [DOI] [PubMed] [Google Scholar]
- 284.Brodsky S. et al. Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol. Cell 79, 459–471.e4 (2020). [DOI] [PubMed] [Google Scholar]
- 285.Strzalka W, Oyama T, Tori K & Morikawa K Crystal structures of the Arabidopsis thaliana proliferating cell nuclear antigen 1 and 2 proteins complexed with the human p21 C-terminal segment. Protein Sci. 18, 1072–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Berlow RB, Martinez-Yamout MA, Dyson HJ & Wright PE Role of Backbone Dynamics in Modulating the Interactions of Disordered Ligands with the TAZ1 Domain of the CREB-Binding Protein. Biochemistry 58, 1354–1362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bhattacharyya RP et al. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311, 822–826 (2006). [DOI] [PubMed] [Google Scholar]
- 288.Mihalic F. et al. Conservation of affinity rather than sequence underlies a dynamic evolution of the motif-mediated p53/MDM2 interaction in teleosts. bioRxiv 2023.08.24.554616 (2023) doi: 10.1101/2023.08.24.554616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Benz C. et al. Proteome-scale mapping of binding sites in the unstructured regions of the human proteome. Mol. Syst. Biol 18, e10584 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Jephthah S, Staby L, Kragelund BB & Skepö M Temperature Dependence of Intrinsically Disordered Proteins in Simulations: What are We Missing? J. Chem. Theory Comput 15, 2672–2683 (2019). [DOI] [PubMed] [Google Scholar]
- 291.Cuevas-Velazquez CL et al. Intrinsically disordered protein biosensor tracks the physical-chemical effects of osmotic stress on cells. Nat. Commun 12, 5438 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Moses D. et al. Structural biases in disordered proteins are prevalent in the cell. bioRxiv 2021.11.24.469609 (2022) doi: 10.1101/2021.11.24.469609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Klein P, Pawson T & Tyers M Mathematical modeling suggests cooperative interactions between a disordered polyvalent ligand and a single receptor site. Curr. Biol 13, 1669–1678 (2003). [DOI] [PubMed] [Google Scholar]
- 294.Sparks S, Hayama R, Rout MP & Cowburn D Analysis of multivalent IDP interactions: Stoichiometry, affinity, and local concentration effect measurements. Methods Mol. Biol 2141, 463–475 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Rogers JM et al. Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl. Acad. Sci. U. S. A 111, 15420–15425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Cho W-K et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Chong S. et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Brangwynne CP et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 (2009). [DOI] [PubMed] [Google Scholar]
- 300.Shin Y & Brangwynne CP Liquid phase condensation in cell physiology and disease. Science 357, (2017). [DOI] [PubMed] [Google Scholar]
- 301.Choi J-M, Holehouse AS & Pappu RV Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys 49, 107–133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Dignon GL, Best RB & Mittal J Biomolecular Phase Separation: From Molecular Driving Forces to Macroscopic Properties. Annu. Rev. Phys. Chem 71, 53–75 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Berry J, Brangwynne CP & Haataja M Physical principles of intracellular organization via active and passive phase transitions. Rep. Prog. Phys 81, 046601 (2018). [DOI] [PubMed] [Google Scholar]
- 304.Mittag T & Pappu RV A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol. Cell 82, 2201–2214 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Pappu RV, Cohen SR, Dar F, Farag M & Kar M Phase Transitions of Associative Biomacromolecules. Chem. Rev (2023) doi: 10.1021/acs.chemrev.2c00814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Boeynaems S. et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 28, 420–435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Lyon AS, Peeples WB & Rosen MK A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol 1–21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Martin EW & Holehouse AS Intrinsically disordered protein regions and phase separation: sequence determinants of assembly or lack thereof. Emerg Top Life Sci 4, 307–329 (2020). [DOI] [PubMed] [Google Scholar]
- 309.Li P. et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Wang J. et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688–699.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Choi J-M, Dar F & Pappu RV LASSI: A lattice model for simulating phase transitions of multivalent proteins. PLoS Comput. Biol 15, e1007028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Choi J-M, Hyman AA & Pappu RV Generalized models for bond percolation transitions of associative polymers. Phys Rev E 102, 042403 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ginell GM & Holehouse AS An Introduction to the Stickers-and-Spacers Framework as Applied to Biomolecular Condensates. Methods Mol. Biol 2563, 95–116 (2023). [DOI] [PubMed] [Google Scholar]
- 314.Cates ME & Witten TA Chain conformation and solubility of associating polymers. Macromolecules 19, 732–739 (1986). [Google Scholar]
- 315.Semenov* AN & Rubinstein M Thermoreversible Gelation in Solutions of Associative Polymers. 1. Statics. Macromolecules 31, 1373–1385 (1998). [Google Scholar]
- 316.Rubinstein M & Semenov AN Thermoreversible Gelation in Solutions of Associating Polymers. 2. Linear Dynamics. Macromolecules 31, 1386–1397 (1998). [Google Scholar]
- 317.Rekhi S. et al. Expanding the molecular language of protein liquid-liquid phase separation. bioRxiv 2023.03.02.530853 (2023) doi: 10.1101/2023.03.02.530853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Holehouse AS, Ginell GM, Griffith D & Böke E Clustering of Aromatic Residues in Prion-like Domains Can Tune the Formation, State, and Organization of Biomolecular Condensates. Biochemistry 60, 3566–3581 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Ruff KM et al. Sequence grammar underlying the unfolding and phase separation of globular proteins. Mol. Cell 82, 3193–3208.e8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Yang Y, Jones HB, Dao TP & Castañeda CA Single Amino Acid Substitutions in Stickers, but Not Spacers, Substantially Alter UBQLN2 Phase Transitions and Dense Phase Material Properties. J. Phys. Chem. B 123, 3618–3629 (2019). [DOI] [PubMed] [Google Scholar]
- 321.Farag M. et al. Condensates formed by prion-like low-complexity domains have small-world network structures and interfaces defined by expanded conformations. Nat. Commun 13, 7722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Abbas M, Lipiński WP, Nakashima KK, Huck WTS & Spruijt E A short peptide synthon for liquid-liquid phase separation. Nat. Chem 13, 1046–1054 (2021). [DOI] [PubMed] [Google Scholar]
- 323.Ruff KM, Dar F & Pappu RV Ligand effects on phase separation of multivalent macromolecules. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lin Y-H & Chan HS Phase Separation and Single-Chain Compactness of Charged Disordered Proteins Are Strongly Correlated. Biophys. J 112, 2043–2046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Banani SF et al. Compositional Control of Phase-Separated Cellular Bodies. Cell 166, 651–663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Riback JA et al. Composition-dependent thermodynamics of intracellular phase separation. Nature 581, 209–214 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Elbaum-Garfinkle S. et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U. S. A 112, 7189–7194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Hondele M. et al. DEAD-box ATPases are global regulators of phase-separated organelles. Nature 573, 144–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Nott TJ et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 57, 936–947 (2015). This study is among the first systematic biophysical investigations into the sequence determinants of phase separation as driven by IDRs.
- 330.Sankaranarayanan M. et al. Adaptable P body physical states differentially regulate bicoid mRNA storage during early Drosophila development. Dev. Cell 56, 2886–2901.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Whitman BT, Wang Y, Murray CRA, Glover MJN & Owttrim GW Liquid-liquid phase separation of the DEAD-box cyanobacterial RNA helicase redox (CrhR) into dynamic membraneless organelles in Synechocystis sp. Strain PCC 6803. Appl. Environ. Microbiol 89, e0001523 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Li Q. et al. DEAD-box helicases modulate dicing body formation in Arabidopsis. Sci Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Iserman C. et al. Condensation of Ded1p Promotes a Translational Switch from Housekeeping to Stress Protein Production. Cell 181, 818–831.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Brady JP et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. U. S. A 114, E8194–E8203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Joseph JA et al. Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy. Nature Computational Science 1, 732–743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Pak CW et al. Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell 63, 72–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Han TW et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779 (2012). [DOI] [PubMed] [Google Scholar]
- 338.Kato M. et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Boeynaems S. et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 65, 1044–1055.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Sang D. et al. Condensed-phase signaling can expand kinase specificity and respond to macromolecular crowding. Mol. Cell 82, 3693–3711.e10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Sridharan S. et al. Systematic discovery of biomolecular condensate-specific protein phosphorylation. Nat. Chem. Biol 1–11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Milovanovic D, Wu Y, Bian X & De Camilli P A liquid phase of synapsin and lipid vesicles. Science 361, 604–607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Kim TH et al. Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829 (2019). [DOI] [PubMed] [Google Scholar]
- 344.Guo YE et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Nott TJ, Craggs TD & Baldwin AJ Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat. Chem 8, 569–575 (2016). [DOI] [PubMed] [Google Scholar]
- 346.Peeples W & Rosen MK Mechanistic dissection of increased enzymatic rate in a phase-separated compartment. Nat. Chem. Biol 17, 693–702 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Klosin A. et al. Phase separation provides a mechanism to reduce noise in cells. Science 367, 464–468 (2020). [DOI] [PubMed] [Google Scholar]
- 348.To P. et al. Intrinsically Disordered Regions Promote Protein Refoldability and Facilitate Retrieval from Biomolecular Condensates. bioRxiv 2023.06.25.546465 (2023) doi: 10.1101/2023.06.25.546465. [DOI] [Google Scholar]
- 349.Lasker K. et al. The material properties of a bacterial-derived biomolecular condensate tune biological function in natural and synthetic systems. Nat. Commun 13, 5643 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Dzuricky M, Rogers BA, Shahid A, Cremer PS & Chilkoti A De novo engineering of intracellular condensates using artificial disordered proteins. Nat. Chem 12, 814–825 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Chen D. et al. Integration of light and temperature sensing by liquid-liquid phase separation of phytochrome B. Mol. Cell 82, 3015–3029.e6 (2022). [DOI] [PubMed] [Google Scholar]
- 352.Boyd-Shiwarski CR et al. WNK kinases sense molecular crowding and rescue cell volume via phase separation. Cell 185, 4488–4506.e20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Wang B. et al. Condensation of SEUSS promotes hyperosmotic stress tolerance in Arabidopsis. Nat. Chem. Biol 18, 1361–1369 (2022). [DOI] [PubMed] [Google Scholar]
- 354.Jalihal AP et al. Multivalent Proteins Rapidly and Reversibly Phase-Separate upon Osmotic Cell Volume Change. Mol. Cell 0, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Dorone Y. et al. A prion-like protein regulator of seed germination undergoes hydration-dependent phase separation. Cell 184, 4284–4298.e27 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Yoo H, Triandafillou C & Drummond DA Cellular sensing by phase separation: Using the process, not just the products. J. Biol. Chem 294, 7151–7159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Boeynaems S. et al. Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proc. Natl. Acad. Sci. U. S. A 116, 7889–7898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Fisher RS & Elbaum-Garfinkle S Tunable multiphase dynamics of arginine and lysine liquid condensates. Nat. Commun 11, 4628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Woodruff JB, Hyman AA & Boke E Organization and Function of Non-dynamic Biomolecular Condensates. Trends Biochem. Sci 43, 81–94 (2018). [DOI] [PubMed] [Google Scholar]
- 360.Bowman GR et al. A polymeric protein anchors the chromosomal origin/ParB complex at a bacterial cell pole. Cell 134, 945–955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Ellisman MH, McAdams HH & Shapiro L Caulobacter PopZ forms a polar subdomain dictating sequential changes in pole composition and function. Molecular (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Lasker K. et al. Selective sequestration of signalling proteins in a membraneless organelle reinforces the spatial regulation of asymmetry in Caulobacter crescentus. Nat Microbiol (2020) doi: 10.1038/s41564-019-0647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Sanders DW et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 181, 306–324.e28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Bergeron-Sandoval L-P et al. Endocytic proteins with prion-like domains form viscoelastic condensates that enable membrane remodeling. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Mistry J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Plitzko JM, Schuler B & Selenko P Structural Biology outside the box-inside the cell. Curr. Opin. Struct. Biol 46, 110–121 (2017). [DOI] [PubMed] [Google Scholar]
- 367.Theillet F-X et al. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chem. Rev 114, 6661–6714 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Theillet F-X et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50 (2016). [DOI] [PubMed] [Google Scholar]
- 369.König I. et al. Single-molecule spectroscopy of protein conformational dynamics in live eukaryotic cells. Nat. Methods 12, 773–779 (2015). [DOI] [PubMed] [Google Scholar]
- 370.Buljan M. et al. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct. Biol 23, 443–450 (2013). [DOI] [PubMed] [Google Scholar]
- 371.Buljan M. et al. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Niklas KJ, Bondos SE, Dunker AK & Newman SA Rethinking gene regulatory networks in light of alternative splicing, intrinsically disordered protein domains, and post-translational modifications. Front Cell Dev Biol 3, 8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Zhou J, Zhao S & Dunker AK Intrinsically Disordered Proteins Link Alternative Splicing and Post-translational Modifications to Complex Cell Signaling and Regulation. J. Mol. Biol 430, 2342–2359 (2018). [DOI] [PubMed] [Google Scholar]
- 374.Subramanian S & Kumar S Evolutionary anatomies of positions and types of disease-associated and neutral amino acid mutations in the human genome. BMC Genomics 7, 306 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Vacic V & Iakoucheva LM Disease mutations in disordered regions—exception to the rule? Mol. Biosyst 8, 27–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Vacic V. et al. Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput. Biol 8, e1002709 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Tsang B, Pritisanac I, Scherer SW, Moses AM & Forman-Kay JD Phase Separation as a Missing Mechanism for Interpretation of Disease Mutations. Cell 183, 1742–1756 (2020). [DOI] [PubMed] [Google Scholar]
- 378.Mészáros B, Hajdu-Soltész B, Zeke A & Dosztányi Z Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules 11, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Kim HJ et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Patel A. et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015). [DOI] [PubMed] [Google Scholar]
- 381.Dao TP et al. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 69, 965–978.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Banani SF et al. Genetic variation associated with condensate dysregulation in disease. Dev. Cell 57, 1776–1788.e8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Mensah MA et al. Aberrant phase separation and nucleolar dysfunction in rare genetic diseases. Nature 614, 564–571 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Basu S. et al. Unblending of Transcriptional Condensates in Human Repeat Expansion Disease. Cell (2020) doi: 10.1016/j.cell.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Gemayel R. et al. Variable Glutamine-Rich Repeats Modulate Transcription Factor Activity. Mol. Cell 59, 615–627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Boeynaems S. et al. Aberrant phase separation is a common killing strategy of positively charged peptides in biology and human disease. bioRxiv 2023.03.09.531820 (2023) doi: 10.1101/2023.03.09.531820. [DOI] [Google Scholar]
- 387.Chandra B. et al. Phase Separation Mediates NUP98 Fusion Oncoprotein Leukemic Transformation. Cancer Discov. 12, 1152–1169 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Shirnekhi HK, Chandra B & Kriwacki RW The Role of Phase-Separated Condensates in Fusion Oncoprotein–Driven Cancers. Annu. Rev. Cancer Biol 7, 73–91 (2023). [Google Scholar]
- 389.Simonetti L, Nilsson J, Mclnerney G, Ivarsson Y & Davey NE SLiM-binding pockets: an attractive target for broad-spectrum antivirals. Trends Biochem. Sci (2023) doi: 10.1016/j.tibs.2022.12.004. [DOI] [PubMed] [Google Scholar]
- 390.Madhu P, Davey NE & Ivarsson Y How viral proteins bind short linear motifs and intrinsically disordered domains. Essays Biochem. (2022) doi: 10.1042/EBC20220047. [DOI] [PubMed] [Google Scholar]
- 391.Romero P, Obradovic Z & Dunker AK Folding minimal sequences: the lower bound for sequence complexity of globular proteins. FEBSLett. 462, 363–367 (1999). [DOI] [PubMed] [Google Scholar]
- 392.Romero Obradovic & Dunker K Sequence Data Analysis for Long Disordered Regions Prediction in the Calcineurin Family. Genome Inform. Ser. Workshop Genome Inform 8, 110–124 (1997). [PubMed] [Google Scholar]
- 393.Romero P, Obradovic Z, Kissinger C, Villafranca JE & Dunker AK Identifying disordered regions in proteins from amino acid sequence. in Proceedings of International Conference on Neural Networks (ICNN’97) vol. 1 90–95 vol.1 (1997). [Google Scholar]
- 394.Uversky VN, Gillespie JR & Fink AL Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins: Struct. Funct. Bioinf 41, 415–427 (2000). [DOI] [PubMed] [Google Scholar]
- 395.Emenecker RJ, Griffith D & Holehouse AS Metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys. J 120, 4312–4319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Gibbs EB, Cook EC & Showalter SA Application of NMR to studies of intrinsically disordered proteins. Arch. Biochem. Biophys 628, 57–70 (2017). [DOI] [PubMed] [Google Scholar]
- 397.Camacho-Zarco AR et al. NMR Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins. Chem. Rev 122, 9331–9356 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Schuler B, Soranno A, Hofmann H & Nettels D Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys 45, 207–231 (2016). [DOI] [PubMed] [Google Scholar]
- 399.Brucale M, Schuler B & Samorì B Single-molecule studies of intrinsically disordered proteins. Chem. Rev 114, 3281–3317 (2014). [DOI] [PubMed] [Google Scholar]
- 400.Balasubramaniam D & Komives EA Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochim. Biophys. Acta 1834, 1202–1209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Leeb S & Danielsson J Obtaining Hydrodynamic Radii of Intrinsically Disordered Protein Ensembles by Pulsed Field Gradient NMR Measurements. Methods Mol. Biol 2141, 285–302 (2020). [DOI] [PubMed] [Google Scholar]
- 402.Fuertes G. et al. Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Proc. Natl. Acad. Sci. U. S. A 114, E6342–E6351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Kikhney AG & Svergun DI A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. (2015). [DOI] [PubMed] [Google Scholar]
- 404.Martin EW, Hopkins JB & Mittag T Small-angle X-ray scattering experiments of monodisperse intrinsically disordered protein samples close to the solubility limit. Methods Enzymol. 646, 185–222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Riback JA et al. Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science 358, 238–241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Cubuk J, Stuchell-Brereton MD & Soranno A The biophysics of disordered proteins from the point of view of single-molecule fluorescence spectroscopy. Essays Biochem. 66, 875–890 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Chemes LB, Alonso LG, Noval MG & de Prat-Gay G Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods Mol. Biol 895, 387–404 (2012). [DOI] [PubMed] [Google Scholar]
- 408.Stuchfield D & Barran P Unique insights to intrinsically disordered proteins provided by ion mobility mass spectrometry. Curr. Opin. Chem. Biol 42, 177–185 (2018). [DOI] [PubMed] [Google Scholar]
- 409.Kassem N. et al. Order and disorder-An integrative structure of the full-length human growth hormone receptor. Sci Adv 7, eabh3805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Borgia A. et al. Consistent View of Polypeptide Chain Expansion in Chemical Denaturants from Multiple Experimental Methods. J. Am. Chem. Soc 138, 11714–11726 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Zheng W. et al. Probing the action of chemical denaturant on an intrinsically disordered protein by simulation and experiment. J. Am. Chem. Soc 138, 11702–11713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Naudi-Fabra S, Tengo M, Jensen MR, Blackledge M & Milles S Quantitative Description of Intrinsically Disordered Proteins Using Single-Molecule FRET, NMR, and SAXS. J. Am. Chem. Soc 143, 20109–20121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Clark PL, Plaxco KW & Sosnick TR Water as a Good Solvent for Unfolded Proteins: Folding and Collapse are Fundamentally Different. J. Mol. Biol 432, 2882–2889 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Best RB Emerging consensus on the collapse of unfolded and intrinsically disordered proteins in water. Curr. Opin. Struct. Biol 60, 27–38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Guseva S. et al. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci Adv 6, eaaz7095 (2020). This paper, along with Milles et al. 2018 [ref. 104], provide the biophysical basis for weak multivalent interactions that underlie how viral proteins can mediate condensate formation.
- 416.Shea J-E, Best RB & Mittal J Physics-based computational and theoretical approaches to intrinsically disordered proteins. Curr. Opin. Struct. Biol 67, 219–225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Vitalis A & Pappu RV ABSINTH: A new continuum solvation model for simulations of polypeptides in aqueous solutions. J. Comput. Chem 30, 673–699 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Zerze GH, Best RB & Mittal J Sequence- and Temperature-Dependent Properties of Unfolded and Disordered Proteins from Atomistic Simulations. J. Phys. Chem. B 119, 14622–14630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Zerze GH, Zheng W, Best RB & Mittal J Evolution of All-Atom Protein Force Fields to Improve Local and Global Properties. J. Phys. Chem. Lett 10, 2227–2234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Piana S, Robustelli P, Tan D, Chen S & Shaw DE Development of a Force Field for the Simulation of Single-Chain Proteins and Protein–Protein Complexes. J. Chem. Theory Comput 16, 2494–2507 (2020). [DOI] [PubMed] [Google Scholar]
- 421.Robustelli P, Piana S & Shaw DE Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A 115, E4758–E4766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Huang J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Best RB, Zheng W & Mittal J Balanced protein–water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Piana S, Donchev AG, Robustelli P & Shaw DE Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119, 5113–5123 (2015). [DOI] [PubMed] [Google Scholar]
- 425.Huang J & MacKerell AD Jr. Force field development and simulations of intrinsically disordered proteins. Curr. Opin. Struct. Biol 48, 40–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Tesei G, Schulze TK, Crehuet R & Lindorff-Larsen K Accurate model of liquid–liquid phase behavior of intrinsically disordered proteins from optimization of single-chain properties. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Palazzesi F, Prakash MK, Bonomi M & Barducci A Accuracy of current all-atom force-fields in modeling protein disordered states. J. Chem. Theory Comput 11, 2–7 (2015). [DOI] [PubMed] [Google Scholar]
- 428.Baul U, Chakraborty D, Mugnai ML, Straub JE & Thirumalai D Sequence effects on size, shape, and structural heterogeneity in intrinsically disordered proteins. J. Phys. Chem. B 123, 3462–3474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Dignon GL, Zheng W, Kim YC, Best RB & Mittal J Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol 14, e1005941 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Ozenne V. et al. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 28, 1463–1470 (2012). [DOI] [PubMed] [Google Scholar]
- 431.Tria G, Mertens HDT, Kachala M & Svergun DI Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Nodet G. et al. Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J. Am. Chem. Soc 131, 17908–17918 (2009). [DOI] [PubMed] [Google Scholar]
- 433.Bottaro S, Bengtsen T & Lindorff-Larsen K Integrating Molecular Simulation and Experimental Data: A Bayesian/Maximum Entropy Reweighting Approach. Methods Mol. Biol 219–240 (2018) doi: 10.1007/978-1-0716-0270-6_15. [DOI] [PubMed] [Google Scholar]
- 434.Brookes DH & Head-Gordon T Experimental Inferential Structure Determination of Ensembles for Intrinsically Disordered Proteins. J. Am. Chem. Soc 138, 4530–4538 (2016). [DOI] [PubMed] [Google Scholar]
- 435.Leung HTA et al. A Rigorous and Efficient Method To Reweight Very Large Conformational Ensembles Using Average Experimental Data and To Determine Their Relative Information Content. J. Chem. Theory Comput 12, 383–394 (2016). [DOI] [PubMed] [Google Scholar]
- 436.Bonomi M, Camilloni C, Cavalli A & Vendruscolo M Metainference: A Bayesian inference method for heterogeneous systems. Sci Adv 2, e1501177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Zhang O. et al. Learning to evolve structural ensembles of unfolded and disordered proteins using experimental solution data. J. Chem. Phys 158, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Bonomi M, Heller GT, Camilloni C & Vendruscolo M Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol 42, 106–116 (2017). [DOI] [PubMed] [Google Scholar]
- 439.Thomasen FE & Lindorff-Larsen K Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins. Biochem. Soc. Trans 50, 541–554 (2022). [DOI] [PubMed] [Google Scholar]
- 440.Chen JW, Romero P, Uversky VN & Dunker AK Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res 5, 879–887 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Chen JW, Romero P, Uversky VN & Dunker AK Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J. Proteome Res 5, 888–898 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Shinn MK et al. Connecting sequence features within the disordered C-terminal linker of Bacillus subtilis FtsZ to functions and bacterial cell division. Proc. Natl. Acad. Sci. U. S. A 119, e2211178119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Pancsa R, Zsolyomi F & Tompa P Co-evolution of intrinsically disordered proteins with folded partners witnessed by evolutionary couplings. Int. J. Mol. Sci 19, 3315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Toth-Petroczy A. et al. Structured States of Disordered Proteins from Genomic Sequences. Cell 167, 158–170.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Hsu IS et al. A functionally divergent intrinsically disordered region underlying the conservation of stochastic signaling. PLoS Genet. 17, e1009629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Markin CJ et al. Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics. Science 373, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Pincus D. et al. Engineering allosteric regulation in protein kinases. Sci. Signal. 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
M1 Rendering of all-atom simulation of the hnRNPA1 IDR to illustrate the conformational heterogeneity within an atomistic ensemble57. Conformations were generated through all-atom Monte Carlo simulations, which show good agreement with experimental characterization.