

Executive summary
All over the world, people with sickle cell disease (an inherited condition) have premature deaths and preventable severe chronic complications, which considerably affect their quality of life, career progression, and financial status. In addition, these people are often affected by stigmatisation or structural racism, which can contribute to stress and poor mental health. Inequalities affecting people with sickle cell disease are also reflected in the distribution of the disease—mainly in sub-Saharan Africa, India, and the Caribbean—whereas interventions, clinical trials, and funding are mostly available in North America, Europe, and the Middle East. Although some of these characteristics also affect people with other genetic diseases, the fate of people with sickle cell disease seems to be particularly unfair. Simple, effective interventions to reduce the mortality and morbidity associated with sickle cell disease are available. The main obstacle preventing better outcomes in this condition, which is a neglected disease, is associated with inequalities impacting the patient populations. The aim of this Commission is to highlight the problems associated with sickle cell disease and to identify achievable goals to improve outcomes both in the short and long term.
The ambition for the management of people with sickle cell disease is that curative treatments become available to every person with the condition. Although this would have seemed unrealistic a decade ago, developments in gene therapy make this potentially achievable, albeit in the distant future. Until these curative technologies are fully developed and become widely available, health-care professionals (with the support of policy makers, funders, etc) should make sure that a minimum standard of care (including screening, prophylaxis against infection, acute medical care, safe blood transfusion, and hydroxyurea) is available to all patients.
In considering what needs to be achieved to reduce the global burden of sickle cell disease and improve the quality of life of patients, this Commission focuses on five key areas: the epidemiology of sickle cell disease (Section 1); screening and prevention (Section 2); established and emerging treatments for the management of the disease (Section 3); cellular therapies with curative potential (Section 4); and training and education needs (Section 5). As clinicians, researchers, and patients, our objective to reduce the global burden of sickle cell disease aligns with wider public health aims to reduce inequalities, improve health for all, and develop personalised treatment options. We have observed in the past few years some long-awaited momentum following the development of innovative point-of-care testing devices, new approved drugs, and emerging curative options. Reducing the burden of sickle cell disease will require substantial financial and political commitment, but it will impact the lives of millions of patients and families worldwide and the lessons learned in achieving this goal would unarguably benefit society as a whole.
Introduction
Sickle cell disease is probably the most common, serious inherited disease in the world, and one of the top 50 most common causes of non-communicable death globally, with most of these deaths occurring in African countries.1 Furthermore, it has recently been shown to be the most common contributor to mortality globally in the 5–14 year age group.1 Whereas the majority of common causes of death are decreasing, the number of deaths due to sickle cell disease are increasing globally.1 Despite this, there are fewer than five effective disease-modifying agents available and most patients in the world do not have access to any of these.2 Although curative treatment is possible, by haematopoietic stem-cell transplantation (HSCT) and emerging gene therapies, only a tiny minority of patients are currently treated with these because of the need for infrastructure, the cost, and associated adverse effects.3 In the context of increasing global health inequalities, partly driven by racism, previous calls for action on sickle cell disease have been largely ineffective. There is an urgent need for the development of research, policies, and public health strategies to reduce the morbidity and mortality associated with this long-neglected condition.4
The first paper on sickle cell disease was published in 1910 by James Herrick in Chicago (IL, USA), who described the appearance of atypical, sickle-shaped red cells in a Black dental student from Grenada, who presented with respiratory symptoms.5 Following this publication, more clinical and pathophysiological observations emerged, including the identification of haemolysis as a cause of anaemia and jaundice and vaso-occlusion as a cause of pain and ischaemic tissue damage.6 In 1948, Janet Watson published the key observation that babies did not develop symptoms of sickle cell disease in the first few months of life and attributed this to high concentrations of fetal haemoglobin (HbF).7 This realisation led to a stream of therapeutic attempts to increase HbF concentrations to recreate the asymptomatic neonatal state, which resulted in the emergence of hydroxyurea (also known as hydroxycarbamide) as the most effective and widely used drug in sickle cell disease.8 HbF is also currently the main focus of gene editing trials.9 In 1949, Linus Pauling published a paper showing that sickle cell disease was associated with atypical electrophoretic mobility of haemoglobin S and described it as a molecular disease,10 with subsequent studies showing the amino acid change responsible for the atypical haemoglobin (HbS), and the propensity of HbS to form destructive polymers when deoxygenated.6 It has also gradually emerged that there are more than 15 different genotypes that can cause sickle cell disease. The most common and severe form occurs when HbS (HbSS) is inherited from both parents, with the compound heterozygous states HbSC and HbS/β thalassaemia making up most of the remainder in most populations. The term sickle cell anaemia refers specifically to the homozyzgous condition HbSS (sometimes also including HbS/β0 thalassaemia), whereas sickle cell disease includes all genotypes causing the syndrome (appendix p 4). As molecular biology developed, the transcription of globin genes was increasingly understood and served as an example for other inherited and acquired genetic disorders.11 These observations form the backbone of our understanding of the pathophysiology of sickle cell disease: haemoglobin S is inherited, which polymerises when deoxygenated and damages the red cell; the damaged red cell occludes blood vessels causing ischaemic tissue damage, and also lyses prematurely, releasing the toxic erythrocyte contents into the circulation.12 There has been a large amount of research focusing on the downstream consequences of HbS polymerisation, showing abnormalities in nearly every measurable biological process, including inflammation, oxidative stress, blood coagulability, vascular endothelium function, nitric oxide metabolism, expression of adhesion molecules, and immune function.6 Additionally, there is progressive damage to many organs resulting in hyposplenism, renal impairment, cerebrovascular disease, avascular necrosis of bones and joints, cardiopulmonary disease, retinopathy, hepatopathy, and priapism.6
In parallel with progress in molecular and clinical understanding of sickle cell disease, it became apparent that the disease is distributed very unevenly across different populations, with the majority of patients being of African and African-Caribbean origin. Although most babies born with sickle cell disease occur in Nigeria, the Democratic Republic of Congo, and India,13 sickle cell disease is relatively common in the Indigenous Peoples of the Middle East and southern Europe, with substantial numbers of people affected in an increasing number of countries in the world because of population movements.14 The reason for this uneven distribution was suggested by John Burdon Sanderson Haldane in the 1940s, who hypothesised that carriers of sickle haemoglobin (also called sickle cell trait) had relative protection against death from malaria, which resulted in the disease becoming much more common in countries where malaria was or is an important cause of premature death. This malaria hypothesis (which also applies to other inherited red cell disorders such as α-thalassaemia and β-thalassaemia and G6PD deficiency) has largely been substantiated by further epidemiological studies, although the exact mechanism of protection is still not known.15
Although the molecular and cellular aspects of sickle cell disease are well understood and have often been used as models to understand other molecular conditions, clinical aspects have been relatively neglected. In addition to the progressive organ damage mentioned earlier, sickle cell disease is characterised by acute episodes of illness related to vaso-occlusion and infection, causing problems such as acute pain, acute chest syndrome (ACS), strokes, and—in severe cases—multiorgan failure and death.6 The majority of patients are still thought to die in childhood,16 apart from the relatively small number of patients living in some countries with better outcomes, such as many high-income countries (HICs) and Jamaica, where mean life expectancy is reduced by about 20 years compared to the non-sickle population.17 In 2013, sickle cell disease was the fourth most common cause of death in Nigeria (after malaria, lower respiratory infections, and AIDS) and the 36th most common globally.1 In addition to shortened life expectancy, sickle cell disease is also associated with substantial morbidity in all age groups, resulting from unpredictable episodes of acute illness, accumulating organ damage, neuropsychological problems, and iatrogenic complications. For example, in 2017, sickle cell disease was one of the leading non-communicable disease causes of disability-adjusted life-years in children younger than 5 years in African countries (769·4 disability-adjusted life-years per 100 000 years; 95% uncertainty interval 432·9–1076·4).1 As previously mentioned, without intervention, this global burden is expected to increase over the coming decades.
Even though sickle cell disease is a common and severe condition, there is little evidence on what constitutes effective clinical management and few treatment options, especially when compared with rarer conditions such as haemophilia A and cystic fibrosis. For example, it is unclear how simple supportive measures (eg, intravenous fluids and oxygen) should be used during acute complications. There is good evidence to support the use of phenoxymethylpenicillin prophylaxis18 and hydroxyurea,19 but until about 5 years ago these were the only drugs available to try and prevent complications. With increased commercial interest in orphan diseases and financial incentives to develop drugs for their treatment, more drugs have emerged over the past decade, with crizanlizumab,20 voxelotor,21 and L-glutamine22 variably licenced for use in different countries. Although these are expensive and non-curative drugs, they are indicative of an increasing academic and commercial interest in sickle cell disease, as suggested by the increasing number of publications, and there are currently a large number of drugs in commercial development that could offer further important benefits.23 Additionally, there is rapid progress in the development of stem cell treatments, with advances in HSCT and gene therapy,24 with the potential to make curative treatments available to all patients; however, widespread use is still a long way from clinical reality everywhere, but particularly so in low-income and middle-income countries (LMICs). Importantly, there are also ongoing advances in methods for screening and diagnosis of patients with sickle cell disease. In particular, emerging point-of-care testing (POCT) devices could facilitate the earlier identification of patients in many LMICs that do not have an established network of laboratories able to perform these tests.
It is difficult to know exactly why sickle cell disease has been so neglected, particularly when compared with other less common inherited disorders. To some extent, sickle cell has been seen as a disease mainly affecting African countries, and in those countries its relative importance was diminished by the devastation caused by infectious diseases, including malaria, diarrhoea, and AIDS. In HICs, this disease has occurred predominantly in low-income populations, who have little political influence, and has been prioritised by few health-care systems around the world. Equally, the pharmaceutical industry previously had little or no interest in the condition, although this has changed in the past decade with financial incentives to develop drugs for orphan diseases and the apparent profitability of selling such drugs. WHO has passed two resolutions on sickle cell disease: in 2006 they called for affected countries to strengthen their response to sickle cell disease and thalassaemia and in 2010 they made various statements on the prevention and management of sickle cell disease and other inherited conditions, encouraging the promotion of awareness, improved access to health-care services, and support for research. In August, 2022, the WHO regional office for Africa, African health ministers, and some other interested parties launched a campaign to reduce the burden of sickle cell disease in African countries.25 Other influential global health organisations, including the UN and the Bill & Melinda Gates Foundation, have offered some statements and support for sickle cell disease, although this is limited compared to other global health problems, such as HIV and malaria. Although this high-level support is encouraging, there has been very little progress in actual management of sickle cell disease in most parts of the world.
To sustainably achieve good health and wellbeing for all people with sickle cell disease, identifying and promoting appropriate policies that consider the priority challenges of the 21st century (eg, growing inequalities, poverty, and climate change) are needed. The aim of this Commission is to describe the current situation for people with sickle cell disease and to identify the changes that need to be made to the organisation and provision of health care to develop sustainable policies to substantially reduce morbidity and mortality over the coming decades. Although other strategies have been produced previously, most notably the US National Academy of Sciences, Engineering, and Medicine Strategic Plan and Blueprint for Action, Addressing Sickle Cell Disease,26 these have mostly been focused on particular countries, and the aim of this Commission is to take an international perspective, with representative multinational involvement, and broad recommendations relevant to both high-income and low-income regions. The main areas of focus are understanding the epidemiology, the development of screening programmes, providing better access to established treatments, ensuring that people in all countries can benefit from emerging therapies, and training health-care workers and empowering them to better manage sickle cell disease.
Section 1: epidemiology
Data quality and data quantity
Epidemiological data on disease distribution, prevalence, mortality, and morbidity are essential to monitor progress towards reducing the global burden of disease. Public health authorities require precise, reliable, comparable, and multidimensional data on the burden of disease to setup, plan, implement, and evaluate policies and break through one-size-fits-all health interventions. Currently, in most countries, many gaps in epidemiological data remain; this affects funding allocation and the development of adequate public health policies. In addition, there are substantial disparities in health-care prevention, coordination, and management programmes between HICs and LMICs, as well as within countries. Reducing these disparities would both decrease the burden caused by sickle cell disease and improve the quality of life of patients. A first step towards this goal is to agree on minimum accurate standard epidemiological data to be collected.
Databases, mapping, and networking
Although a substantial body of data has been assembled on the distribution of sickle cell disease,27–29 reliable, precise, and representative data are still scarce for the prevalence and distribution of the condition in most countries. As reflected in discrepancies between existing estimates, including those from the Global Burden of Diseases Study,1 there remains a lot of uncertainty in prevalence and burden estimates in many parts of the world. These discrepancies, combined with a long-term neglect of sickle cell disease, affect progress towards improving the management of the disease. These estimates are important to help develop national policies and often highlight the need to use standardised protocols in epidemiological studies. Furthermore, monitoring spatial and temporal changes in the frequencies of sickle cell disease, and other malaria-related red blood cell genetic disorders (eg, thalassaemia and G6PD deficiency), is essential for public health authorities to develop adequate long-term policies and interventions.
Estimates tend to be based on newborn screening and life expectancy data or rely on regional and national registries, natural history studies, clinical and administrative data, and surveys and cross-sectional reports in sample cohorts whose data are generalised.30 Many of these data are collected on a voluntary basis or are part of short-term funded research projects. Very few of these data collections are sustained in the long-term with adequate staff or funding.
The development of various international collaborative networks, such as the Global Sickle Cell Disease Network, the Sickle Pan African Research Consortium, and the Réseau d’Etudes de la Drépanocytose en Afrique Centrale (appendix p 5), has the potential to fill some of these data limitations and to promote the use of standardised protocols. These networks have all shown the benefits of collaborative work. The development of a centralised electronic portal (eg, the Sickle Cell Disease Ontology) further extended these collaborations to provide tracking systems for recruitment, data quality assurance, and reporting of research studies. Similarly, in Europe, collaborations between national registries (eg, through EuroBloodNet) have increased to standardise approaches and better monitor changes. In the USA, the Centres for Disease Control and Prevention’s Sickle Cell Data Collection programme developed a population-based longitudinal surveillance system to identify people living with sickle cell disease by centralising data from several sources. Other collaborations, such as the PhenXToolkit, have focused on the development of common data collection. The next steps are to enlarge and join up these collaborative efforts (eg, through more coordinated efforts between anglophone and francophone sub-Saharan African countries); to promote interoperability between data sources; to integrate epidemiological data with emerging omics and real-life data; and to further join up and expand all these national, regional, and global efforts to provide near real-time, consistent, and high-quality epidemiological data. Although machine learning algorithms could help fill some of the knowledge gaps, their outputs will largely depend on the quality of input data used.
Data on prevalence and mortality of sickle cell disease
Apart from a few countries with a universal newborn screening programme (eg, the UK and USA; figure 1), precise data on the annual number of children born with sickle cell disease are non-existent. A review of the burden of sickle cell disease in children younger than 5 years only identified 52 studies containing relevant data on incidence and prevalence and 15 with mortality data, further highlighting the need for better epidemiological data on sickle cell disease.31 Although epidemiological data are not necessary to set up policies and interventions, they are essential to assess changes and measure the effect of public health decisions taken.
Figure 1: Level of newborn screening programmes in each country.
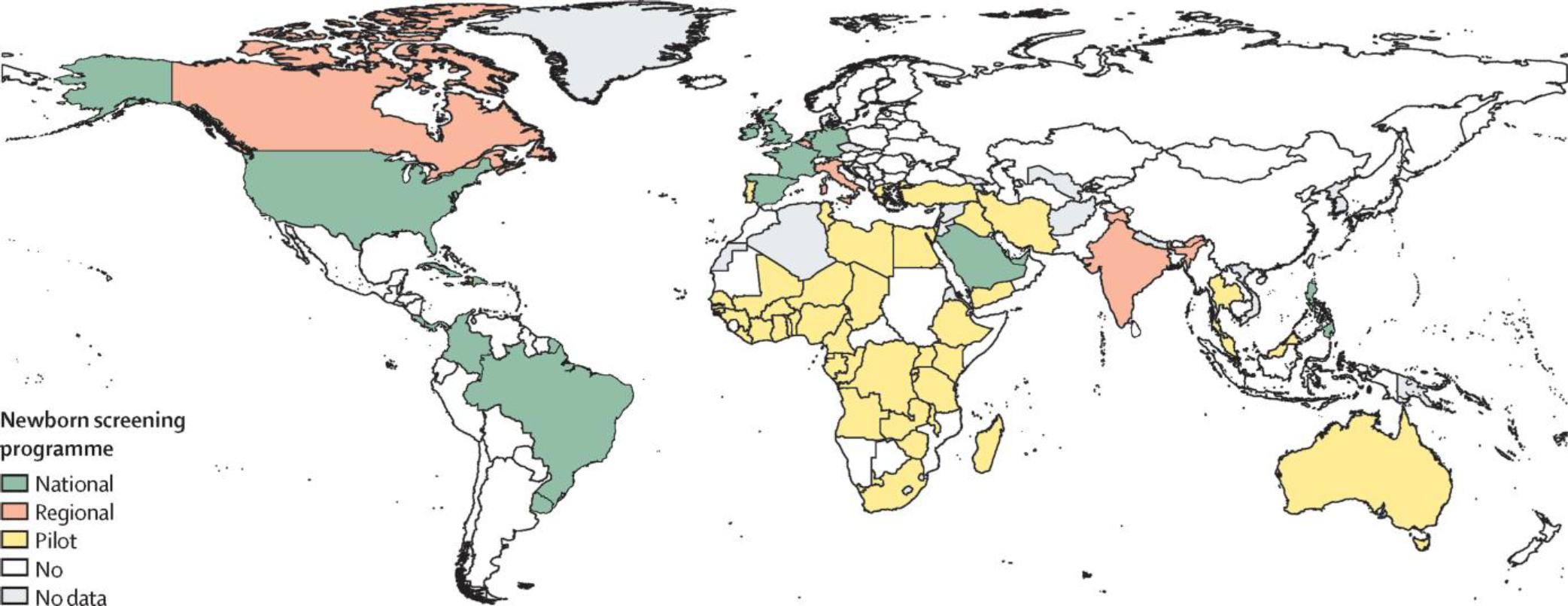
Overview of newborn screening programmes for sickle cell disease across countries, based on the following categories: national—a newborn screening programme, either universal or targeted, is in place across the entire country, can be the result of progressive implementation over years in various parts of the country (eg, the USA); regional—there is no national-level programme, but specific parts of the country (eg, provinces) have an ongoing newborn screening programme in place; pilot—there is no national or regional newborn screening programme, but there are either ongoing local newborn screening efforts (eg, Kumasi, Ghana) or one-off pilot studies to support wider programmes; none—we could not find any evidence of any newborn screening programme in these countries; no data—we did not find any information for these countries. References used to compile the data presented are listed in the appendix (pp 7–9).
Estimates suggest that around 75% of the more than 300 000 annual global children born with sickle cell anaemia occur in sub-Saharan Africa (figure 2). Data on allele frequencies of HbC and β-thalassaemia are scarce, making it difficult to estimate the numbers of births with HbSC disease and HbS/β-thalassaemia. Up-to-date and more meaningful data are required on the frequencies of expected births and outcomes of people with sickle cell disease, particularly in Africa and India.4 The generation of accurate data requires better screening and diagnostic facilities, with improved staff training. The influence of ethnic, religious, and social heterogeneities has so far been poorly studied and data on consanguinity are particularly scarce. New large-scale data collection efforts, such as the Statewide Screening Data Interface in Chhattisgarh (India), and refined modelling work on sickle cell disease accounting for these heterogeneities (eg, Scheduled and non-Scheduled populations in India)32 can have a substantial impact on estimates generated. Data on the prevalence of sickle cell disease in adults are very scarce in all countries worldwide. As a result, there is no reliable estimate of patient numbers either globally or for any country. Even in countries with the best data quality, estimates of the number of people with sickle cell disease remain very crude.33,34 Nevertheless, new data from the Global Burden of Disease 2021 study estimated that 7·74 million people (95% uncertainty interval 6·51–9·20) lived with sickle cell disease globally in 2021.1 Population and fertility projections suggest that this number will increase over the coming decades, particularly in sub-Saharan Africa. The implementation of simple measures, such as appropriate prophylaxis, immunisation, and universal screening, could lead to decreased mortality for millions of babies with sickle cell disease.35 High-quality monitoring epidemiological data would allow current estimates to be validated and updated.
Figure 2: Global distribution of sickle cell anaemia in 2023, illustrated by country-level annual births with sickle cell anaemia.
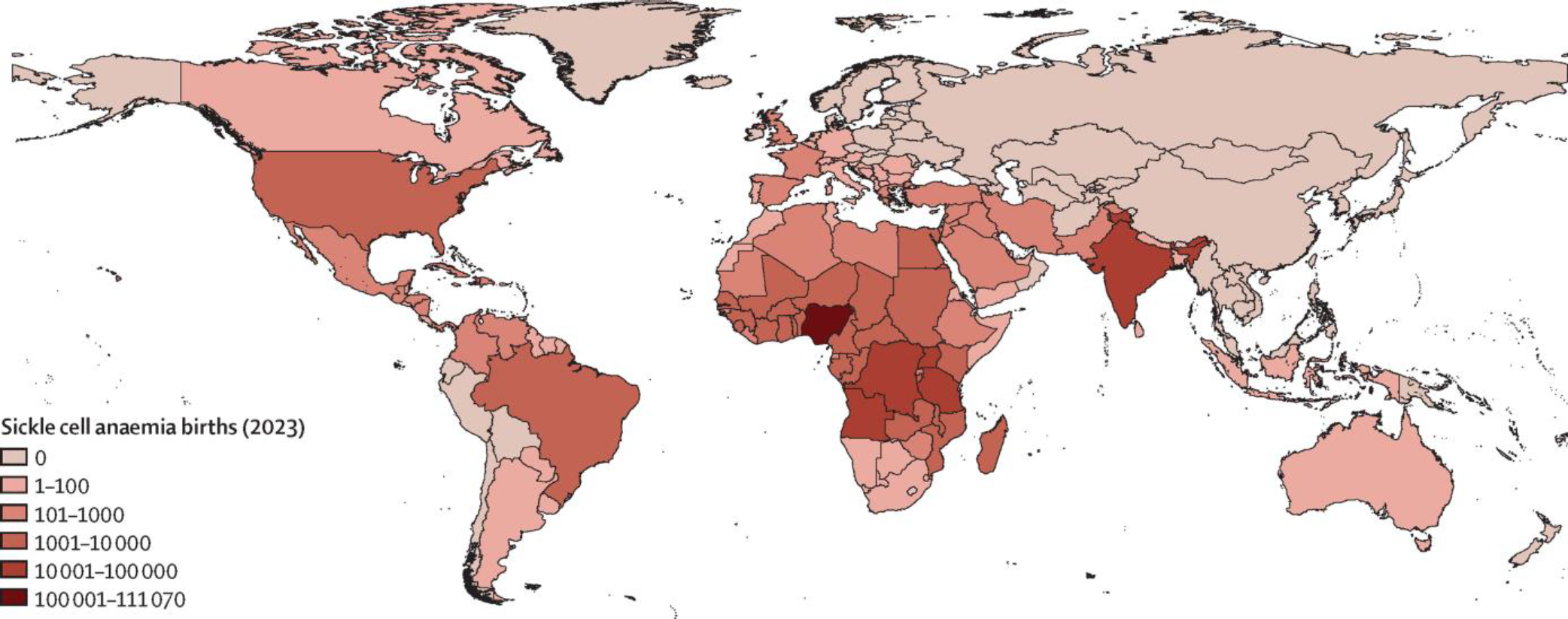
The estimates are derived from median HbS allele frequencies published in Piel and colleagues13 and probabilistic birth projections by country for 2023 from the UN World Population Prospects 2022.
The survival of adults with sickle cell disease has mainly been investigated in specific centres in North America and Europe.35,36 Although survival data from well equipped urban centres in Africa have been published,37–39 there is still a lot of uncertainty around mortality rates of people with sickle cell disease in other parts of these countries.40 Data collected as part of demographic health surveys could offer opportunities to collect representative data, as trialled in Nigeria.41 Prospective studies are required, especially in LMICs, to provide up-to-date data on risk factors for mortality.
Data on newborn screening and morbidity
Studies in HICs and Jamaica have shown the importance of newborn screening with early comprehensive care and education of the parents to substantially reduce early morbidity and mortality from sickle cell disease.42–44 Setting up appropriate follow-up of infants with sickle cell disease remains a major challenge. Furthermore, data on the use of health care, delivery of services, and costs of care are scarce in most countries, including across Europe.45 Survival of people with sickle cell disease beyond the age of 18 years in Europe and North America is common, yet their life expectancy in these countries is still 20 to 30 years lower than in the general population.
Although nationwide universal newborn screening programmes were launched in 2001 in Brazil and in 2020 in Ghana, there is still very little data available on the natural history of sickle cell disease in most areas of high prevalence. The national newborn screening programme in Ghana screened more than 200 000 babies over the course of 10 years at nine public institutions and 15 private clinics, identifying over 3000 children with sickle cell disease and providing care in local clinics.46 In Uganda, the US3 study showed the feasibility of a large-scale epidemiological study of sickle cell disease and sickle cell trait in infants of mothers who were HIV positive through the use of residual dried blood spots.47 In Tanzania, regional and district data are now being generated by analysing repurposed dried blood spots, which were part of the HIV infant diagnosis programme, with existing infrastructure.48 In India, there is negligible data on life expectancy of individuals with sickle cell disease. Although newborn screening programmes have been initiated in a few Indian states, only two studies from the states of Gujarat and Maharashtra have reported clinical outcomes;49,50 the causes of death could not be determined in most children and the follow-up rate was only around 70% in both studies.
Public–private partnerships could be helpful in implementing early diagnosis and sustained care to reduce mortality and improve the quality of data.51,52 A village-based model of care using local volunteers has shown the importance of community participation, and the possibility of assessing premature mortality rates and causes of mortality in a remote tribal village in south India has also been shown.53 One of the main challenges to improve the management of people with sickle cell disease in high-prevalence countries is reaching rural communities and making sure that people have access to ongoing care. Inexpensive POCTs used in conjunction with immunisation programmes can be a good way to start data collection, to map out and study differences in morbidity, and to setup improved health care.
Prenatal diagnosis is acceptable in many countries but requires a lot of infrastructure and therefore, data on this is scarce.54 Various preventive strategies are needed for the management and control of sickle cell disease, including antenatal screening and prenatal diagnosis early in pregnancy to offer parents informed reproductive choices. Antenatal screening is undertaken in some countries, including Australia, Bahrain, Canada, Cuba, India, Saudia Arabia, and the UK, and variably in other countries based around individual hospitals or centres. The acceptability of prenatal diagnosis and termination of pregnancies varies substantially across the world, reflecting cultural and religious beliefs. In England, although antenatal screening is widely accepted, reproductive choices depend on the timing of prenatal diagnosis, as termination of pregnancies is more acceptable with early diagnosis.55 In India, prenatal diagnosis and termination of pregnancies for sickle cell disease is acceptable even among marginalised communities. A single-centre study over 30 years showed that 527 couples opted for prenatal diagnosis of sickle cell disease and 30% of them came prospectively.54 In Cameroon, physicians considered termination in only 36% of affected pregnancies, whereas 90% of prospective mothers would opt for termination.56 In southwestern Nigeria, only 37% of doctors would recommend prenatal diagnosis for sickle cell disease, with only 17% of mothers being willing to undergo testing due to the high cost and fear of the procedure.57 One difficulty of prenatal diagnosis in sickle cell disease is that it is a variable condition and it is hard to predict the clinical severity accurately, making it difficult for parents to make informed choices; increased use of genetic modifiers of sickle cell disease, like those associated with higher HbF levels and co-inheritance of α-thalassaemia, could help to predict the clinical phenotype.56 Non-invasive prenatal diagnosis is another option to avoid the small risk of fetal loss associated with invasive methods. Although there is little data on noninvasive prenatal diagnosis for sickle cell disease, one of the largest studies on this method showed that 37 out 44 sample results were concordant with established methods.58
Economic costs
The economic burden of sickle cell disease is substantial and increasing in most countries. Irrespective of where people live and their economic situation, the cost of this disease imposes a large financial burden to them, their family, and society. In the USA, the inpatient costs, outpatient costs, and out-of-pocket costs surpass US$3 billion per year for about 100 000 patients. Median yearly costs are approximately $28 000 per patient and lifetime care cost $460 000 per patient.59 The indirect economic burden of pain events has also been recognised in the USA,60 where estimates averaged an additional $15 000 per patient per year in terms of productivity loss and around $20 000 of unpaid work lost per caregiver. In the Netherlands and the UK, some studies have looked at resource use and costs related to sickle cell disease health care.61 In LMICs, there is very little data on the economic burden, although sickle cell disease can add substantial financial pressure on patients and their families. Such information is essential to advise governments and public health authorities on the future health burden of sickle cell disease. A cost-effectiveness analysis of newborn screening and a few prophylactic interventions conducted across 47 sub-Saharan African countries could be used to guide policies and pave the way for similar studies in other regions of high prevalence. Further health economics studies, including the costings of a large-scale implementation of key interventions to reduce the burden of sickle cell disease, are needed.
Changing distribution and patient numbers
As witnessed by many haematologists, sickle cell disease is the fastest growing inherited condition in many countries worldwide.14,62,63 This increase is due to several different factors that also lead to changing demographics of the patients’ cohorts and to a more global distribution of patients in areas of the world were sickle cell disease was rare, less known, or less reported.
Evidence of increase in patients’ numbers in various settings and worldwide
Increasing numbers of people with sickle cell disease started to occur over the past few decades in Europe, North America, and Australia.64–66 Observed increases in prevalence were mostly the result of gains in life expectancy (due to reduced childhood mortality) and immigration. For example, the UK National Hemoglobinopathy Register reported 1367 sickle cell disease cases in 1979; 7800 in 2013; and 12 000 in 2019. In France, there were 1000–3000 people with sickle cell disease reported in 1992,67 whereas the total number of people with sickle cell disease in 2016 was estimated between 19 800 and 32 400.68 Population movements are also responsible for increased prevalence—for example, in Germany and Sweden.63,69
Increasing numbers of people with sickle cell disease have also been reported in LMICs in Africa, South America, and Asia and are the result of increased diagnostic and reporting capacity. In Brazil, 45 000 people with sickle cell disease were estimated in 1985,70 whereas (according to reports of the Health Ministry of Brazil) this increased to 60 000–100 000 people with sickle cell disease in 2021.
Causes of increasing numbers: increased diagnostic capacity
The implementation of sickle cell disease national newborn screening programmes has allowed the reporting of birth prevalence and shown an increase in the number of people with sickle cell disease in some HICs, including the USA42 and in many European countries.30 Birth prevalence from large studies based in HICs are summarised in the appendix (p 6). In other HICs experiencing immigration, such as Italy71 and Canada,72 only birth prevalence data from pilot newborn screening studies are available but data from long-term national sustainable programmes implemented in the health-care system are still non-existent. In African countries, birth prevalence data have also started to become available. The integration of newborn screening into existing primary health-care immunisation programmes or HIV screening programmes has been shown to be feasible and were rapidly implemented at low costs in countries such as Nigeria and Uganda.73,74 There have been few efforts so far to regularly compile prevalence data to update estimates required by public health authorities.
Partnerships between HICs and LMICs and between research, public or private bodies, and regional networking led to the implementation of pilot newborn screening programmes or screening in LMICs (eg, Antigua), diagnostic efforts in remote rural areas of Nepal and Brazil,75,76 and strengthened local efforts in the Caribbean.77 In south Asia, countrywide data and statistics are difficult to gather and there is very little information available outside India.78 Although sickle cell disease has been reported from Pakistan, Sri Lanka, Nepal, Bangladesh, and the Maldives, very little is known about the nature and the burden of the disease in these countries. Improved data are likely to start to emerge in the near future with the increased use POCT devices in LMICs.
Causes of increasing numbers: increased reporting
In several countries, the increase in patients’ numbers can be estimated owing to the increased reporting through the publication of surveys, clinical cases, research and natural history study results, or enrolment in clinical trials.79 Although all this published information does not allow the determination of precise data on birth prevalence, prevalence, and incidence, it does show an increase in patients’ numbers and growing scientific interest towards a neglected public health problem. The past decade also saw reporting from areas that previously had no data or estimates, such as Nepal and rural parts of India.
The implementation of standardised data collection is still limited geographically to a few European countries, Brazil, and organised networks. Various factors (including the scarcity of early diagnoses, data entry on a voluntary basis into the registries or databases, and the absence of longitudinal registries) have restricted systematic data collection so far. This leads to underestimating the real prevalence and to failures to track robust incidence data. A pronounced move towards widespread complete coverage of the population at risk should be one of the priorities as the number of people with sickle cell disease continues to increase.
Causes of changing distribution and demographics
The increased prevalence of sickle cell disease worldwide is coupled with a change in the distribution of the disease and a change in the demographics of the sickle cell disease population. Migration continues to increase the prevalence of sickle cell disease in many countries, with movements of people from high prevalence areas into countries where sickle cell disease was relatively rare or absent. This process started a few decades ago in Europe and Australia, with diasporas from Africa and the Middle East. This process is also ongoing in the USA, where individuals from sub-Saharan Africa represented the fastest growing immigrant population between 2010 and 2018.80 Population movements are also causing a growing ethnic diversity, increased complexity of the mutations, and increased complexity of disease genotype and phenotype, which in turn can make diagnosis and reporting more complex. The interpopulation variation and the mixture with β-thalassaemia mutations are a recognised factor of interpopulation variation and difficult interpretation in south Asia and increasingly the rest of the world. Traditional approaches to laboratory diagnosis need to be modified in the face of increased population admixture and greater variability in the genotypes underlying sickle cell disease, requiring training of laboratory staff and increased use of DNA analysis.
Reduced paediatric mortality also contributed to the increased prevalence and will determine further changes in the demographics of sickle cell disease, with increasing numbers of adults with sickle cell disease.81 The development of more complex multimorbidities is associated with older ages.
Changing life expectancy
Improved survival and comorbidities in HICs
The impact of sickle cell disease on survival was first suggested in reports (mostly from the USA) in the 1950s about the apparent low prevalence of severe sickle cell disease in older age groups. Diggs estimated a median overall survival of 14·3 years for people with sickle cell disease, with a third of the deaths occurring before the age of 5 years and half occurring between 5 and 30 years of age.82
Subsequently, the largest source of prospective data on survival came from the US Cooperative Study of Sickle Cell Disease (CSSCD; 1978–98). The CSSCD reported a median overall survival of 42 years for male participants and 48 years for female participants with sickle cell anaemia and 60 years for male participants and 68 years for female participants with HbSC disease. Compared with the general African American population, the probability of death dropped drastically after the age of 20 years. These survival estimates supported the lower life expectancy associated with severe sickle cell disease. The CSSCD was not able to establish the exact causes of death in most cases. Of the 209 deaths recorded, 171 (82%) occurred in people with no clinically diagnosed organ failure but in the circumstances of an acute event (including uncomplicated pain, pain with ACS, stroke, perioperative complications, infection, and ACS alone).83
The CSSCD gave investigators the opportunity to document and carefully determine the causes of the high rates of mortality in children described in earlier reports. After 14 670 person-years of follow-up from 1979 to 1987, the CSSCD data showed that the overall mortality was 0·5 deaths per 100 person-years, with the highest rates occurring in individuals aged less than 1 year. The cause of death was known in 54 (74%) of the 73 deaths and, among those 54 deaths, 28 (52%) people died from bacterial infection.
The Penicillin Prophylaxis Study (PROPS1, 1983–85) was conducted among young children in the CSSCD.18 Penicillin prophylaxis might have had a small ameliorative effect on mortality for individuals who were randomised to receive phenoxymethylpenicillin and a larger group that might have been placed on phenoxymethylpenicillin after the study was halted, when phenoxymethylpenicillin proved efficacious in reducing incidence of, and mortality due to, pneumococcal bacteraemia.
Following wide public health adoption of newborn screening for sickle cell disease and penicillin prophylaxis in the context of comprehensive clinical management, several HICs and Jamaica have reported drastic improvements in survival of children with sickle cell disease, increasing survival past the age of 18 years to rates above 90%, with survival of 93·9% (95% CI 90·3–96·2) from one study in the USA.84
With the proven effectiveness of newborn screening and early interventions in reducing childhood mortality in people with sickle cell disease, the expectation would be that overall survival for people with sickle cell disease in HICs would improve steadily. However, the fact that most sickle cell disease deaths had been related to acute illness events served as a cautionary note that improved childhood survival might not necessarily reduce mortality in individuals older than 5 years who die unpredictably with no clinically diagnosed organ failure. A review of mortality rates for children and adults with sickle cell disease from 1979 to 2005 using the US National Center for Health Statistics’ multiple cause of death files showed that the median age at death in 2005 was 42 years for female patients and 38 years for male patients.85 Although there was an observed annual decrease in paediatric mortality by 3%, adult mortality rate increased by 1% each year during the 16 years studied.85
The median overall survival data from Lanzkron and colleagues86 showed no improvement over those from the 1994 report from the CSSCD.It became clear that saving the lives of people with sickle cell disease during childhood might not be sufficient to improve their overall life expectancy. Interventions to reduce both organ failure and improve acute illness management were identified as a need. The switch from decreasing mortality with age in children to increasing mortality with age from 19 years of age corresponded to the switch from paediatric to adult care services in high-income health-care systems.
The most applied disease modifying therapy to address this has been hydroxyurea. The broad positive effect of hydroxyurea on the clinical course of sickle cell disease in both children and adults, and possibly on the survival of adults, suggested the need for intensified efforts to promote hydroxyurea therapy in adults with sickle cell disease.86 Despite the promise of curative interventions, HSCT and gene therapy, it is too early to assess their effect on the overall survival of people with sickle cell disease, even in high-income, well-funded health-care systems.
Mortality reduction and sustainable strategies in high-prevalence countries
In sub-Saharan Africa, where sickle cell disease has been in existence for thousands of years, traditional folklore about sickle cell disease typically describes a disease of recurrent pain attacks, small stature, increased mortality, and familial childhood death.
In 1950, Raper recorded positive sickling test results as high as 45% in one community in Uganda but not a single case of sickle cell disease.87 Comparing those findings with data from the USA, Raper suggested that sickle cell anaemia affects African Americans more frequently than it does Africans, and that this difference could be related to population admixture in the USA.87 The true solution to the apparent paradox was offered in 1952 by the Belgian Lambotte-Legrand paediatrician couple with the advice that “Prevalence of sickle cell anaemia would be found to be high if very young children in Africa were studied.”88 Even nowadays, without newborn screening, the impact of sickle cell disease on early childhood mortality often escapes public health attention.
The high mortality rate in children with sickle cell disease in sub-Saharan Africa was clearly shown by the Garki Malaria Study (1970–74) conducted in northern Nigeria.89 By screening 2742 individuals in the general population and 534 newborn babies to determine the prevalence of haemoglobin variants and genotypes, the study found only one child with sickle cell anaemia in the 1–4 years age group (instead of the six individuals expected) and only one child with sickle cell anaemia of the children aged 5 years and older (instead of the expected 53), suggesting that 98% of the people born with sickle cell anaemia had died by age 5 years.
Since the 2006 WHO report on the impact of sickle cell disease on childhood mortality, and the 50–90% early-life mortality among children born in Africa with sickle cell anaemia estimated by Grosse and colleagues,40 there has been little evidence of large-scale public health activities that could be expected to lead to substantial reduction in childhood mortality in sickle cell disease in sub-Saharan Africa. Although pilot and small-scale newborn screening projects, funded largely by foreign grants, could provide reliable birth incidence rates, mortality data remains largely unavailable. Furthermore, such poor efforts are not expected to alter the dire statistics.
The causes of the high mortality in young children with sickle cell disease in HICs are probably not fundamentally different from those in low-income, high-prevalence countries. In much of Africa, signs of infection (especially fever) are synonymous with malaria and anti-malaria therapy is often administered without previous diagnosis. However, pneumococcus is as much a major pathogen in malaria-endemic regions as it is in countries without malaria. In a large study on bacteraemia in children conducted in Kenya (1998–2008), the organisms most commonly isolated from children with sickle cell anaemia were Streptococcus pneumoniae (41%), non-typhi Salmonella spp (18%), and Haemophilus influenzae type b (12%). With no penicillin prophylaxis, 23% of children with sickle cell anaemia died.90
In the US PROPS1 study,18 13 (12%) of 110 children on placebo developed pneumococcal bacteraemia and 3 (23%) of the 13 died; 2 (2%) of 105 children on twice daily phenoxymethylpenicillin developed pneumococcal bacteraemia and both survived. The three deaths occurred within less than 9 h from onset of fever. However, penicillin prophylaxis for young children with sickle cell disease, even when they are diagnosed with sickle cell disease following an acute illness, is not uniformly practiced in many health-care institutions in Africa.
At the end of the first 10 years of the pilot newborn screening project conducted in Ghana with support from the US National Institutes of Health, 2914 of the eligible 3334 newborn babies with sickle cell disease had been enrolled for care in Kumasi and 134 (4·6%) were known to have died after enrolment. All enrolled children received twice daily penicillin prophylaxis and were followed up for comprehensive clinical care (Ohene-Frempong, unpublished). Ghana has been unable to scale up newborn screening following the pilot study and is currently screening less than 4% of newborn babies annually, mostly with foreign grants. This example illustrates the magnitude of the challenges faced by countries of high sickle cell disease prevalence.
Genetic, sociodemographic, and environmental risk factors
Although sickle cell disease is one of the most common severe monogenic diseases worldwide, and a disease that has been studied for more than a century, our capacity to define and predict disease severity is relatively low and our understanding of risk factors often remains insufficient to guide the management of people with sickle cell disease. Because sickle cell disease is a multisystem disease that can affect all the organs of the body, defining disease severity is in itself a challenge and remains a controversial topic in the field. The phenotypic variability observed is probably due to a combination of genetic, environmental, and socioeconomic factors—the respective contribution of each factor remaining poorly understood. Being able to predict the clinical course of people with sickle cell disease and to classify them has long been an important aim for better prognosis and, now, personalised treatment. Digital epidemiology initiatives based on at-home study designs, wearable devices, pervasive sensors, or engaging digital health interventions to capture real-life data (eg, knowledge, attitude, self-care practices, lifestyle behaviours, or exposure to infection) can support the data collection throughout patients’ day-to-day life, and machine learning approaches could help infer on various omics data to propose personalised recommendations supporting decision making.91
Genetics risk factors
Objective and quantifiable biomarkers of disease severity in sickle cell disease are currently scarce and this has substantially affected drug development and clinical care worldwide.92 Predicting which people are at high risk for complications with only genetic data remains difficult. Biomarkers can help both to classify people into subgroups at the population level and to predict disease severity and progression for individual patients (ie, personalised medicine). Although the five β-globin-like gene cluster haplotypes—Bantu or Central-Africa Republic (CAR), Benin (BEN), Cameroon (CAM), Senegal (SEN), and Arab-Indian (ARAB)—originally described might have identified important differences in disease severity at the population level, there is substantial overlap between the characteristics of these haplotypes. For example, data from central India has shown that severe disease can be commonly observed in people with the Arab-India haplotype, which is usually considered to be milder than the African haplotypes. Measuring ethnic variability within African haplotypes is starting to be made possible with large national newborn screening datasets and international data collected through multicentre collaborative projects (eg, Sickle In Africa and BioCADRE [NCT03352986]).
Over the past couple of decades, genetic association studies have attempted to define disease subphenotypes on the basis of markers of anaemia, haemolysis, and vascular complications.93 More than 100 blood and urine biomarkers have been correlated to at least one of the complications of sickle cell disease94 but they often explain only a small part of the overall disease variability. Cluster analysis, based on large well studied cohorts (eg, CSSCD), have further identified biomarker signatures that could aid the treatment and management of the disease.95 Although these advances have to some extent guided therapeutic developments and the clinical management of people with sickle cell disease, they have not led to generic biomarkers widely adopted in clinical settings. In the future, omics studies, accounting for genetic and non-genetic factors, might contribute to further improve our understanding of the phenotype–genotype relationships in sickle cell disease.
The most established biomarkers of survival and clinical complications in sickle cell disease are the concentrations of HbF and the coinheritance of α-thalassaemia, which both reduce the polymerisation of HbS and therefore results in a milder clinical course. Increasing HbF concentrations has been underlying the growing use of hydroxyurea to prevent complications in sickle cell disease and the development of new drugs based on endothelial cell activation, cellular adhesion, chronic inflammation, intravascular haemolysis, and nitric oxide scavenging (see Section 3).
In HICs, the quest for reliable biomarkers is likely to become more complicated with the emergence of comorbidities seen in older people with sickle cell disease.96 In LMICs, most of the current population and burden estimates and projections are based on Hardy-Weinberg Equilibrium assumptions. Better data on key factors underlying these assumptions, consanguinity in particular, will be fundamental to improve such estimates.
Environmental risk factors
A range of environmental factors, including climatic and meteorological variables (temperature, humidity, wind speed, or rainfall); air quality (indoors and outdoors pollutants, including particulate matter with a diameter of <2·5 μm or <10 μm, NO2, CO, and O3); altitude; and malaria endemicity, can potentially influence the natural history of sickle cell disease. It is very difficult to establish a clear link between specific environmental factors and pathophysiological events, and the evidence for these effects is confusing and often contradictory.97 Similarly, the US Consensus Study Report on sickle cell disease concluded that “the exact roles that such [environmental] factors play in influencing symptoms and complications [in sickle cell disease] are not well understood.98 Most of the evidence generated so far comes from Europe and the USA, although studies are starting to emerge from countries with a high prevalence of sickle cell disease.99 Studies of the general population and of other subgroups affected by other diseases (eg, cardiovascular disease, respiratory diseases, and cancers) have clearly shown the serious health effects of poor air quality, even at low rates of exposure. Despite recommendations by WHO, nine of ten people are exposed to poor air quality and the Global Burden of Disease Study estimated that air pollution contributed to 213 million disability-adjusted life-years (95% uncertainty interval 189–240) and 6·67 million deaths (5·90–7·49) in 2019.100 As it is highly probable that poor air quality would have adverse effects on individuals with sickle cell disease, quantifying risks for each pollutant would help mitigate complications and manage patients. In HICs, the absence of strong associations found so far has probably reduced opportunities for further investigations. In LMICs, ongoing efforts to collect high-resolution data on pollutant concentrations with satellite images or measurements in India, Brazil, and African countries represent great opportunities to investigate associations and health risks in sickle cell disease.
As the global population becomes increasingly urban and the impact of climate changes becomes apparent, it will be essential to better understand the effect of environmental factors on people with sickle cell disease. Estimates from the UN World Urbanization Prospects suggests that, by 2050, two-thirds of the world population will live in urban areas. These changes will be predominantly driven by changes in sub-Saharan Africa and the Indian subcontinent,101 which coincide with regions of high prevalence for sickle cell disease. Cities represent specific ecosystems in which populations can potentially benefit from easier access to health infrastructures but also have poor air quality and drastic inequalities in terms of socioeconomic status, as already observed in many megacities (see the section on sociodemographic risk factors).
In parallel, as highlighted in the 6th Assessment Report of the Intergovernmental Panel on Climate Change, millions of people will have to adapt to changing conditions of their local environment or to migrate due to the effects of more severe and more frequent events triggered by climate change (eg, droughts, storms, wildfires, and heatwaves). These local changes will mostly affect the poorest and the most vulnerable, including people with sickle cell disease and their families. For example, good hydration is part of the management of sickle cell disease, as cell dehydration promotes polymerisation and sickling. Local changes will also lead to large numbers of individuals moving within their home country, contributing to urbanisation, or internationally either to neighbouring countries or to countries less affected or with more resources. These large population movements could have an important influence on the global distribution of sickle cell disease.
These changes are likely to add to the challenges already faced by people with sickle cell disease and their families over the coming decades, making access to quality health care more difficult and affecting their mental health. There is little data on the mental health of people with sickle cell disease. A report jointly led by the UK Sickle Cell and Thalassaemia All-Party Parliamentary Group and the UK Sickle Cell Society102 stated that, during the COVID-19 pandemic, about 75% people with sickle cell disease and their carers struggled with their mental health. Providing appropriate psychological support to people with sickle cell disease should therefore be more prominent on the policy agenda in both HICs and LMICs.
Sociodemographic risk factors
The COVID-19 pandemic has further highlighted growing socioeconomic (eg, access to health care and vaccines) and racial (eg, occupation) disparities as important determinants of health. As in many other diseases, poverty can have a substantial effect on the health of people with sickle cell disease. Although a large pro portion of sickle cell disease complications would be avoidable or treatable with existing medicines (eg, hydroxyurea or penicillin prophylaxis) or interventions (eg, newborn screening), the vast majority of people with sickle cell disease worldwide live in LMICs and have little access to proper sanitation, health education, and health facilities, combined with poor nutrition, prevalent infectious diseases (eg, malaria, tuberculosis, and HIV), and exposure to toxic pollutants (eg, air pollution and heavy metals), highlighting the need for a holistic multidisciplinary approach, building on ongoing efforts73 to improve the management of sickle cell disease and reduce health inequalities.
There is some evidence that a higher proportion of people with sickle cell disease live in the lowest socioeconomic areas than the general population. In the UK, 58% of patients admitted to hospital with a primary or secondary diagnosis of sickle cell disease with or without vaso-occlusive crisis (VOC) in 2005–06 lived in areas belonging to the most deprived quintile of the English Index of Multiple Deprivation.103 In Saudi Arabia, Khan and colleagues104 found a higher percentage of children in the lowest socioeconomic class and a higher frequency of VOC and adverse events than people living in higher socioeconomic classes. In India, sickle cell disease is predominantly found among Scheduled tribes and Scheduled castes, which constitute the most socioeconomically disadvantaged population subgroups in the country.32
In the UK, people with sickle cell disease from the most socioeconomically deprived areas and with co morbidities were at highest risk of both sickle cell disease re-admissions and in-hospital mortality.103 Similarly, in the USA, financial insecurity was associated with three times more hospital admissions and re-admissions in adults with sickle cell disease.105 In children, Panepinto and colleagues106 found that sickle cell disease led to a significantly impaired health-related quality of life, even after considering the potential detrimental effect of family income on health-related quality of life. Missing school or work for patients and their relatives can have an effect on their education or career progression, further contributing to the long-term disadvantage faced by people with sickle cell disease.
Racism and stigmatisation can aggravate the effect of deprivation on people with sickle cell disease as illustrated by difficulties in accessing opioids,107 longer waiting times than for other health complications,108 and the availability of fewer financial resources than for other diseases (eg, cystic fibrosis).109 These inequalities need to be addressed. Additional funding and research for sickle cell disease and other diseases that disproportionately affect economically disadvantaged groups could help to reduce health-care disparities.
In LMICs, although high-quality care is available in well resourced centres of excellence in cities (eg, Kilifi [Kenya]38 and Dar-es-Salaam [Tanzania]37), the vast majority of people with sickle cell disease do not have access to basic health care. In Nigeria, the SPRING trial110 suggested that poverty was associated with severe anaemia in children with sickle cell disease. A cross-sectional descriptive study performed at referral centres for the treatment of haematological diseases in the northeast of Brazil identified difficulties in obtaining medications prescribed by physicians and in transportation. 111 In addition, deficiencies in nutrients associated with malnutrition and undernutrition have been shown to be linked to disease severity and health-related quality of life in both adults and children with sickle cell disease.112
Finally, migrants and refugees can face additional difficulties in relation to legal status, linguistic barriers, and cultural differences. Resulting delays in screening and adequate management can lead to severe health complications and to substantial long-term additional health-care costs.113
Epidemiology recommendations
Although the sickle cell disease community faces many challenges, there needs to be an agreement on what standard epidemiological data are needed, and these data should be collected to track progress towards set milestones, aligned with the UN’s Good Health and Well-Being Sustainable Development Goal, and to assess the effect of interventions. Proven cost-effective interventions have the potential to identify individuals with sickle cell disease early, to reduce mortality, and to prevent severe chronic complications. These interventions need to be scaled up to reflect the true global burden of sickle cell disease.
Section 2: screening
Screening for sickle cell disease
Detection programmes for any disorder depend on public health systems and life-course approaches that include screening, diagnosis, education, treatment, and comprehensive care. Screening can occur at any point throughout life but is particularly relevant in the neonatal period, premaritally and preconceptually, and as part of antenatal care (table 1).114 Appropriately trained genetic or nurse counsellors have an important part in any screening programme, particularly in the antenatal period, to discuss the possibility of prenatal diagnosis and reproductive choices.115
Table 1:
Approaches to screening for sickle cell disease
| Advantages | Disadvantages | |
|---|---|---|
|
| ||
| Preconceptual | ||
| Testing parents (or prospective parents) | ||
| Blood tests | Widely available on a voluntary basis; evidence of benefit in Saudi Arabia and Bahrain; reliable and able to distinguish most types of sickle cell disease, including compound heterozygotes | Quality assurance processes not routine in LMICs, with results varying from lab to lab |
| Conventional electrophoresis | Cheap; low maintenance equipment | Low sensitivity when compared with HPLC or capillary electrophoresis |
| HPLC | Sensitive, automated systems developed specifically for haemoglobinopathy screening | High cost of machines, reagents, and maintenance; service contracts not available in many countries |
| Capillary electrophoresis | Sensitive; separates HbE from HbA2 | Mostly used in HICs; expensive with high maintenance costs |
| POCTs | Rapid, sensitive, and low cost compared with HPLC and capillary electrophoresis; in WHO Essential Diagnostics List 3 | ·· |
| Prenatal or antenatal | ||
| Testing pregnant people | ||
| Blood tests | Integral part of the UK and Cuban newborn baby and antenatal screening programme; used for guided newborn screening targeting in India and Benin | Educational and counselling programmes not well established in many countries |
| Conventional electrophoresis | Cheap; low maintenance equipment | Low sensitivity when compared with HPLC or capillary electrophoresis |
| HPLC | Sensitive, automated systems developed specifically for haemoglobinopathy screening | High cost of machines, reagents, and maintenance; service contracts not available in many countries |
| Capillary electrophoresis | Sensitive; separates HbE from HbA2 | Mostly used in HICs; expensive with high maintenance costs |
| POCTs | Used in the Republic of Congo, Democratic Republic of the Congo, Guinea, Nigeria, Liberia, and Kenya for primary screening | ·· |
| Testing fetuses | ||
| DNA technology | Available in HICs and India | High cost; ethical issues; might be unacceptable or prohibited by law in some countries |
| Chorionic villus sampling (10–12 weeks of pregnancy) | Part of the newborn screening programme in Cuba | Invasive |
| Amniocentesis (15–20 weeks of pregnancy) | Technically easier than other fetus testing methods | Invasive |
| Analysis of circulating foetal DNA in mother’s blood | Non-invasive; no fetal risk during sampling | Still in evaluation phase |
| Testing embryos | ||
| Preimplantation genetic diagnosis with in-vitro fertilisation | Alternative to prenatal diagnosis and offer of pregnancy termination in case of an affected fetus | Very high cost; considerable physical and psychological burden for the parents |
| Postnatal | ||
| Newborn screening | ||
| Blood tests from heel-stick test | Widely implemented; extensive experience; benefit shown | ·· |
| Isoelectric focusing | Sensitive and specific; mostly used in LMICs; low cost | Labour intensive; extensive expertise needed |
| HPLC | Sensitive and specific; mostly used in HICs | High cost; requires skilled technicians |
| Capillary electrophoresis | Sensitive and specific; mostly used in HICs | High cost of equipment and maintenance; requires skilled technicians |
| Mass Spectrometry | Used in the UK and France; high throughput | Very high cost of equipment; requires skilled technicians |
| POCTs | Successfully implemented in Nigeria; evidence of benefit in Haiti, Côte d’Ivoire, Ghana, and Martinique (France); easy to use, low cost, and does not require electricity | ·· |
HICs=high-income countries. HPLC=high-performance liquid chromatography. LMICs=low-income and middle-income countries. POCTs=point-of-care tests.
Screening before conception allows the detection of the carrier status of the mother and father. With respect to sickle cell disease, during the antenatal period, the mother and fetus can be screened by various tests such as chorionic villus sampling and amniocentesis to find out if the offspring will have the disease or be a carrier.114 These tests are usually conducted at or after 12 weeks of pregnancy and are considered invasive, increasing the risk of miscarriage by up to 1%. New methodologies with non-invasive techniques employ testing of cell-free fetal DNA in the maternal circulation from 8 weeks gestation.
Postnatally, newborn babies with sickle cell disease are identified through newborn screening, which is sometimes part of broader programmes for conditions such as phenylketonuria, congenital hypothyroidism, and cystic fibrosis. It is crucially important that adequate health care is available for babies identified as having any of these conditions. For sickle cell disease, these infants need to be prescribed penicillin from the age of about 3 months and to be followed up in specialist clinics.
Screening programmes also need to consider the important stresses that often affect a family’s ability to cope with sickle cell disease, including the economic and educational consequences of time lost from work and school and the effect of chronic illness on typical family functioning and unaffected siblings. Families live with the knowledge that unpredictable acute illnesses can interrupt daily life, and there are often feelings of powerlessness, frustration, and even anger. A general scarcity of community awareness about sickle cell disease and fear of stigmatisation can restrict the support available from extended family, friends, and the community at large. Previous experience with health-care providers who lack knowledge, sensitivity, and compassion can contribute to delays in seeking appropriate health care and might engender adversarial relationships between families and providers. Failure to appreciate ethnic and cultural differences between providers and patients and families, the effect of stigmatisation, and absence of societal education about sickle cell disease could also contribute to misunderstanding and lack of trust. Thus, it is imperative that in the design of any screening programme for sickle cell disease, providers take time to listen to the concerns of patients and families, are sensitive to psychosocial and medical needs, and that they assist families in accessing available resources as needed.
Data infrastructure and surveillance systems for newborn screening
Where newborn screening programmes are established, they are cost effective and their efficiency and effectiveness depend on the smooth integration of sample collection, laboratory testing, follow-up, diagnosis, and treatment (figure 3).116–120 These programmes require information system infrastructure for follow-up and quality assurance. Inequalities between HICs and LMICs for this infrastructure are apparent. In LMICs, the absence of data collection and infrastructure makes it very difficult to assess the clinical outcomes for patients and to monitor the effectiveness of screening programmes. In addition, training is required in all aspects of establishing and running newborn screening programmes, with external quality assurance and provision of genetic counselling, follow-up of screen positive babies, and provision of comprehensive care.121
Figure 3: The network of services associated with newborn screening and the related links between the five core sections of this Commission.
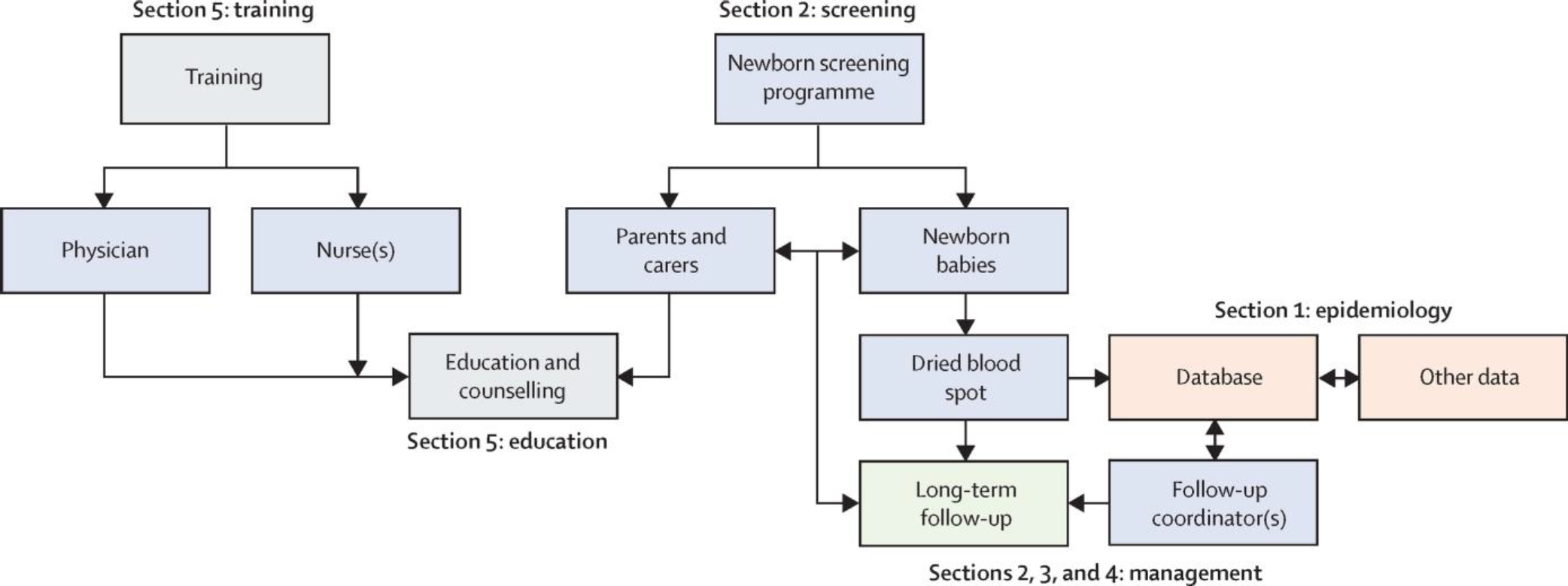
Epidemiology is in orange, screening in blue, management in green, and training and education in grey.
Screening tests
Newborn screening programmes for sickle cell disease have been in place in several regions of the world for more than 40 years (table 2). The first analytical, and still most used, techniques for screening (and diagnosis) are based upon the separation of the various haemoglobin variants in the newborn baby’s blood by their difference in electric charge.122 These techniques include isoelectric focusing (IEF), cation-exchange high-performance liquid chromatography (HPLC) and capillary electrophoresis (CE). Classic haemoglobin electrophoresis with cellulose acetate electrophoresis at alkaline pH is still a useful technique to screen for and diagnose sickle cell disease in older children and adults and is still used in many countries. However, this screening method is not usually recommended for newborn screening programmes because it might be too insensitive to reliably detect the small amounts of adult haemoglobins present in newborn babies. New technologies being developed use differences in molecular mass (through mass spectrometry) or antigenic properties (through immunoassays). Mass spectrometry is used now in some national neonatal screening programmes but relies on the use of equipment that is expensive and requires careful maintenance. Immunoassays are used in particular for POCTs but do not have an established role in screening programmes yet.
Table 2:
Newborn baby screening programmes in different regions of the world
| Year of programme initiation | Year of full national programme coverage | Comment | |
|---|---|---|---|
|
| |||
| Africa | |||
| Regional or pilot programme | |||
| Benin | 1993 | ·· | Pioneer projects; supported by NGOs, HIC agencies, and public-private initiatives |
| Ghana | 1993 | ·· | Pioneer projects; supported by NGOs, HIC agencies, and public-private initiatives |
| Cameroon, DR Congo, Gabon, Ghana, Guinea, Mali, Senegal, Tanzania, Uganda, Zambia | 1990–2020 | ·· | Regional projects partly supported by government funding |
| Burkina Faso, Kenya, Liberia, Niger, Nigeria | 1990–2020 | ·· | Regional projects partly supported by government funding |
| Europe | |||
| Universal national programmes | |||
| UK | 2002 | 2014 | Coupled newborn and antenatal screening; coverage >99%, survival at age 16 years 99·0% (95% CI 93·2–99·9) |
| Spain | 2003 | 2015 | ·· |
| Netherlands | 2007 | 2007 | ·· |
| Malta | 2017 | 2017 | ·· |
| Germany | 2020 | 2020 | ·· |
| Targeted national programmes | |||
| France | 1995 | 2000 | Targets geographical ancestry of the mother; coverage >99%, survival at age 16 years 97·1% (95% CI 95·2–98·3) |
| Regional or pilot programmes | |||
| Ireland* | 2003 | ·· | ·· |
| Belgium† | 1994 | ·· | ·· |
| Italy | 2007 | ·· | 3 of 20 regions‡ |
| Middle East | |||
| Universal national programmes | |||
| United Arab Emirates | 2002 | 2005 | ·· |
| Bahrain | 2007 | 2007 | ·· |
| Qatar | 2007 | 2007 | ·· |
| Regional or pilot programmes | |||
| Oman | 2005 | ·· | ·· |
| India | |||
| Regional programmes | |||
| 6 of 31 states and Union Territories§ | 2010 | ·· | ·· |
| North America | |||
| Universal national programmes | |||
| USA | 1973 | 2006 | Survival at age 18 years is 93·9% (95% CI 90·3–96·2) |
| Universal regional programmes | |||
| Canada | 1988–2006 | ·· | 8 of 13 provinces or territories¶ |
| Latin America | |||
| Universal national programmes | |||
| French Guiana (France) | 1995 | 1995 | Coverage >99% |
| Brazil | 2010 | 2014 | Coverage 83% |
| Regional pilot programmes | |||
| Colombia | 2000 | ·· | ·· |
| Costa Rica | 2013 | ·· | ·· |
| Uruguay | 2013 | ·· | ·· |
| West Indies | |||
| Universal national programmes | |||
| Cuba | 1983 | 1983 | Coupled antenatal screening and prenatal testing |
| French West Indies (Guadeloupe, Martinque, Saint Martin, and Saint Barthélemy) | 1984 | 1984 | Coverage >98% |
| Jamaica | 1995 | 2015 | Coverage >98% |
| Puerto Rico and the Virgin Islands (USA) | 1977 | 1987 | ·· |
| Dutch Caribbean | 2015 | 2015 | ·· |
| Regional pilot programmes | |||
| Tobago, Grenada, Saint Lucia, Saint Vincent and the Grenadines, Antigua and Barbuda, Haiti | 1990–2020 | ·· | ·· |
HIC=high-income country. NGO=non-governmental organisation.
NGO funded.
Regions of Brussels and Liège.
Regions of Veneto and Sicilia (universal), and Emilia-Romagna (targeted on ethnicity).
States of Gujarat, Maharashtra, Madhya Pradesh, Chhattisgarh, Odisha, Tamil Nadu; depending on the programme, screening is untargeted or targeted to tribal newborn babies and to newborn babies of HbAS mothers, or both.
Provinces of Ontario, British Colombia, Yukon, Quebec, New Brunswick, Nova Scotia, Prince Edward Island, and Alberta.
The main technical challenge for newborn screening is that the major haemoglobin constituent in newborn babies’ blood is HbF and that the adult haemoglobins (HbA, HbS, HbC, HbDPunjab, HbE, and HbOArab) are minor constituents. Furthermore, it is not only important to detect these haemoglobins’ presence but also to be certain if they are absent, particularly in the case of HbA. For more information on laboratory methods see the appendix (pp 2–3).
POCTs
Conventional screening programmes as established in North America, Europe, Brazil, and some Caribbean countries116–120 have not yet been established in LMICs due to the high costs, scarcity of skilled staff, inadequate electricity supplies, and absence of other basic infrastructure. Several POCTs have been developed to overcome these barriers through various approaches, including erythrocyte density, differential mobility of HbS and HbA through filter paper, and antibody-based immunoassays (qualitative lateral flow immunoassays or competitive enzyme-linked immunosorbent assays). These POCTs are inexpensive, reliable, require only a pin prick of blood, and show high specificity and sensitivity in the discrimination of the different haemoglobin phenotypes in the presence of high HbF concentrations in newborn babies. POCTs can easily be administered in rural villages with minimum training and as such have the potential to reduce health-care inequity that has made newborn screening unattainable in high-burden countries.123 Use of POCTs could increase access to universal newborn screening, although many problems remain in implementing such schemes and in following up babies found to have the condition. POCTs have been included in WHO’s Essential Diagnostics List 3.
A particular problem with POCTs is identifying some of the less common genotypes that can cause sickle cell disease, such as HbS/β+thalassaemia, which can be confused with sickle cell carriers. Screening programmes inevitably do not accurately diagnose every case, but very sensitive and highly specific approaches should be used to minimise the number of patients missed, to not falsely identify babies as affected, and to prevent unnecessary psychosocial harm to families.
Most techniques detect carriers of haemoglobin variants as a byproduct. Some sickle cell disease newborn screening programmes use this information to provide families with reproductive knowledge and informed decision making regarding sickle cell disease. Some other programmes have made the choice not to use this information for feasibility or regulatory reasons.
The status of newborn screening
Newborn screening for sickle cell disease was initiated in the USA in 1973 but it took more than 30 years to become a national programme, covering all the states of the country in 2006.117 In Europe, pilot programmes were launched in the early 1980s in France and the UK and on the French island of Guadeloupe in the Caribbean.118 In high-burden countries, the pioneering newborn screening pilot programmes were those simultaneously initiated in Benin and Ghana in 1993.124 In India, pilot programmes have been initiated since 2010 in central and southern Indian states where the incidence of sickle cell disease is the highest.49,51
Overall, the situation remains extremely heterogeneous from one country to another, from national programmes covering the whole country (USA, four European countries, Brazil, and three countries in the Middle East) to patchy pilot programmes in sub-Saharan Africa and India (table 2).
In a survey of the implementation of the WHO strategy in high-burden countries in the WHO African Region, it was found that newborn screening for sickle cell disease was being practised in 13 countries (Benin, Nigeria, Uganda, Democratic Republic of the Congo, Mali, Senegal, Ghana, Liberia, Tanzania, Kenya, Zambia, Burkina Faso, and Cameroon). Although substantial progress is being made in Ghana, none of these countries yet have a national programme because of a scarcity of funding and political commitment.
An examplar of newborn screening programme in sub-Saharan Africa: Ghana
Ghana is probably the best example of an African country with a well planned newborn screening programme for sickle cell disease.125 The pilot newborn screening programme started in 1993 and was supported in 1996 with three 5-year cycles of funding from the US National Institutes of Health (NIH) to the Komfo Anokye Teaching Hospital and Kwame Nkrumah University of Science and Technology (Kumasi) in collaboration with the Minister of Health and the Ghana Health Service.
Sample collections are done at the place of birth before discharge or at immunisation sites. Data collection, collation, and handling of samples are done by nursing staff of the screening health-care facilities. The structured programme has facilitators and coordinators at site, district, and regional levels, overseeing sample collection, communication, and shipment to the centralised screening laboratory and with a Nurse Coordinator supervising all the newborn screening activities of the coordinators of the programme. Data from the programme have been managed since inception in a backed-up database with secure access to staff only. Bet ween 1993 and 1996, 115 (85%) of 135 babies diagnosed with sickle cell disease have been located and enrolled into comprehensive care and follow-up.
In 2018, the programme transitioned to mobile data collection with the specially developed Ghana newborn screening App for all newborn screening-related data at screening sites and at the National Laboratory at Noguchi to enhance accuracy in data collection, and easier and faster management of newborn screening data. Workshops were conducted to train genetic counsellors. The Government and public health insurance supports newborn screening at sites in Kumasi and surrounding communities, but not sufficiently to allow scaling up to a national programme. Over the years, newborn screening has been sustained by external funding through international collaboration with the Government of Brazil, non-governmental organisations, the American Society of Hematology (ASH), and pharmaceutical companies (including a formal public–private agreement with the Government of Ghana). A public–private partnership from 2020 aims to scale up the programme cover to the whole country, but this has not been achieved yet.
An examplar of newborn screening programme in Europe: the UK
The history of newborn screening for sickle cell disease in Europe goes back almost 40 years, from the first pioneering pockets of screening in France and England to full implementation of newborn screening, covering the whole country in 2000 for France (targeted) and 2014 for the UK (universal).118 In both countries, it is now part of national screening programmes integrated into a comprehensive free-of-charge system to ensure a complete medical support of prevention and care.
The chosen strategy in the UK is that of a linked antenatal and neonatal screening programme. In addition, women and couples with known carrier status before pregnancy are offered genetic counselling to consider options as soon as they know they are pregnant. The antenatal programme offers screening to all pregnant people by 10 weeks of gestation. All the partners of carrier women are fast-tracked and offered information and testing. Genetic counselling is provided, and prenatal diagnosis is offered to carrier couples early enough to complete prenatal diagnosis by 12 weeks and 6 days (12⁺⁶ weeks) gestation, if the option is chosen. The same procedure is offered to pregnant people who are carriers when the biological father is unavailable.
Independently from the results of antenatal screening, all newborn babies in the UK are tested at 5 days of age via the national blood spot screening programme for nine conditions, including sickle cell disease. For sickle cell disease, testing of the blood spot is performed in 13 accredited laboratories by at least two analytical methods, including HPLC, CE, IEF and tandem mass spectrometry. The overall efficiency and quality of the programme are regularly evaluated to ensure that the main objective of the programme is met: prompt identification of babies born with sickle cell disease and timely transition into clinical care.
Advantages of coupling antenatal and neonatal programmes are to support parents at risk to make informed choices during pregnancy (and before conception); to prepare them for their newborn screening result; and to check, as a part of the quality control, that the antenatal and postnatal results are congruent. Evaluation shows that the programme is well accepted by professionals and by the public. Still, timeliness of the antenatal phase must be improved to meet the standards for antenatal maternal screening to be offered by 10 weeks gestation. Coverage of the newborn screening was 97·8% in 2018 and 2019. Survival of people with sickle cell disease at age 16 years in the UK is 99%.126
An examplar of newborn screening programme in the Middle East: Bahrain
In the Middle East, three countries are running newborn screening programmes for sickle cell disease: universal national programmes in the United Arab Emirates and Bahrain and a pilot programme in Muscat, the capital city of Oman. Newborn screening in Bahrain was included in 2007 in their national programme to control genetic diseases, which was the first to be initiated in a Middle East country (in 1984) with the support from religious scholars and the parliament.127
The screening is done on cord blood samples collected for all deliveries in all the public maternity hospitals of the country. A leaflet detailing the purpose, process, and outcomes of the screening is provided to the parents before screening and the possibility to opt out of testing is mentioned. Information regarding demographic characteristics, parental age group, and consanguinity is also recorded. Cord blood analysis is performed by HPLC in a centralised reference laboratory.
By raising awareness in the population about genetic diseases (and haemoglobinopathies in particular), the number of babies born with sickle cell disease has declined by 75% since the initiation of the national programme in 1984. Addition of newborn screening to the programme has ensured prompt identification of the affected babies and early transition into clinical care. Clinical care is provided by a network of primary care centres, with a specialist sickle cell centre at Salmaniya Medical Complex.
Differences among different regions of the world
There are marked differences in the nature of the screened populations in different parts of the world, particularly the prevalence of sickle cell disease, which determines the most effective approach to screening. Although in most programmes screening is universal, meaning that all newborn babies are tested with some form of haemoglobin analysis, some programmes aim to do blood tests only on babies deemed to be at high risk of having sickle cell disease. Targeting can be more selective by testing only newborn babies of carrier parents, as in Benin128 and in some local programmes in India49,51 and Cuba.116 Alternatively, targeting addresses populations deemed at risk based on geographical origin or ethnicity, as in France, Ireland, or one of the Italian programmes. Some programmes in India target tribal populations.32
Sampling can be done at birth or soon after at all levels of health care and by various health-care workers, mostly nurses and midwives and sometimes by laboratory personnel and doctors. In Africa, the services are generally provided only in tertiary health facilities—except in parts of Mali, Democratic Republic of the Congo, Uganda, and Ghana where samples for newborn screening are collected at all levels of the health system and transported to tertiary facilities for analysis. Sometimes sampling is performed at the first immunisation visit because it is integrated into immunisation programmes (eg, in Nigeria and Ghana). In some countries, sampling is linked to reproductive, maternal, newborn baby, and child health programmes (eg, Democratic Republic of the Congo, Gabon, Ghana, Guinea, Tanzania, and Uganda) or into HIV screening programmes (eg, Burkina Faso and Uganda). Most often, sample analysis is performed in dedicated newborn screening laboratories, with mostly IEF as the primary method in Africa; HPLC in the USA, Europe, and India; and mass spectrometry in England and France. However, this situation is changing as some countries have reported the use of POCTs for primary screening (eg, Republic of Congo, Democratic Republic of the Congo, Guinea, Liberia, and Kenya). In countries where a national programme is in place, newborn screening is fully covered, most often by public funding and sometimes by insurance companies (eg, USA, Europe, Middle East, Cuba, and Brazil). Elsewhere, the tests are paid for by a variable combination of the government, non-governmental organisations, and patients themselves in many countries. In Uganda, the Government pays for the test, whereas in Kenya, the payment source is a combination of the Government, insurance companies, and patients.
Effectiveness of the different programmes is difficult to appreciate as data are scarce and highly heterogeneous. The coverage of national programmes is high (>98% in the UK, France, the Netherlands, and USA and 85% in Brazil).118,120 Because of imprecise addresses and ever-changing telephone numbers in Africa, a major challenge of many programmes is to locate affected newborn babies and to enrol them in follow-up and care. But follow-up rates depend very much on the setting, the chosen strategy (including the point of sampling), and awareness about the disease. Linkages to HIV or reproductive, maternal, newborn baby, child, and adolescent health programmes improve early detection, enrolment into follow-up, and management of sickle cell disease as they deliver a package of educational and counselling services.
Data are scarce concerning the general effect of newborn screening on the first-year mortality of affected babies or of affected children younger than 5 years. In Angola, the first-year mortality for babies with sickle cell disease compares favourably to the national infant mortality rate (6·8% vs 9·8%).129 In Benin, the mortality rate of children with sickle cell disease younger than 5 years was 15·5 per 1000 n (ie, 10 times lower than the mortality rate of children younger than 5 years of the general population).128 In HICs with newborn screening programmes (eg, UK, France, USA), survival into adulthood is higher than 98%.84,126,130 Data concerning cost-efficiency of the various programmes are scarce. In Angola, the estimated cost per healthy life-years (HLYs) gained was US$ 1380–3565—ie, less than the gross domestic product per capita.131
Barriers to screening
Cost is one of the main barriers to screening, with other barriers existing at policy, health system, institutional, and personal levels. Conventional screening requires leadership, detailed planning, expensive equipment and reagents, technical personnel, stable power sources, logistic support (supply chain management), and education and buy-in of stakeholders and communities. Without enabling policies, budgetary allocation, and a functional public health system, newborn screening is often unsustainable (even when started by well intentioned foreign non-governmental organisations). This unsustainability is the reason for the failure to progress beyond pilot programmes in many high burden countries. Equally important barriers can be the lack of awareness and acceptability of the programme. Raising the awareness of the target population is key to a successful screening programme.132 Myths and misconceptions about sickle cell disease must be addressed while ensuring the availability of adequate screening tests kits, trained staff, and follow-up services.
The effectiveness of newborn screening and early clinical interventions
The effectiveness of newborn screening and early clinical interventions has been evident from HICs,84,126 with some rare examples of improved outcomes in LMICs, most notably Jamaica.133 However, the six-part system that includes education, screening, short-term follow-up, diagnosis, management, and evaluation134 has not been widely implemented in high-burden countries due to little government capacity and funding.135
Examples of best practices
Best practice in screening requires that all the people with sickle cell disease in each population are identified as early as possible, along with people who carry the trait. The design of any screening programme will depend on the resources available, the incidence of sickle cell disease, and the existence of other health infrastructure, such as screening programmes for other conditions. The full details about where and when the samples will be collected are important. This collection can be at birth, in the 2–6 weeks after birth, or at immunisation clinics (either at first immunisations, immunisations at birth, or later). A protocol outlining the type of tests to be used for screening and confirmation, the screening laboratory, what happens to the result, the person responsible for parental follow-up, the scheduling of clinical visits, and the comprehensive care centres should be established. The data management workflow and the personnel charged of this, as well as quality control, should be designated. Although this is best done at or soon after birth in newborn screening, every contact with health-care centres and educational institutions in high burden countries should be used to achieve universal coverage. POCTs could be particularly suited to universal screening in countries with few resources.
Consortium on newborn screening in Africa
An example of an initiative aimed to develop and expand newborn screening in Africa is the Consortium on Newborn Screening in Africa, a collaboration between the ASH and haematologists across Africa that aims to increase the region’s capacity for newborn screening and to show the benefits of screening and early therapeutic interventions for babies with sickle cell disease, working in partnership with local governments to ensure the long-term sustainability of these efforts.136 When establishing the consortium, careful preparation was made to develop governance structure and protocols, and site inspection visits were carried out to assess Consortium members’ readiness. The Consortium negotiated low equipment and reagent costs for newborn screening with IEF. Seven countries have so far been admitted into the Consortium (Ghana, Tanzania, Nigeria, Liberia, Uganda, Kenya, and Zambia). Babies with sickle cell disease detected in the programme will be followed up for 5 years to document the effectiveness of newborn screening to encourage African governments to implement universal newborn screening. The protocol, checklist, and forms developed by the Consortium could be a valuable resource for establishing other newborn screening programmes.137
Prevention of sickle cell disease
Primary prevention in the context of sickle cell disease refers to action aimed at reducing the number of babies born with sickle cell disease. This includes premarital testing and genetic counselling, with the potential to choose partners partly on the basis of their haemoglobino pathy status. Prenatal diagnosis with the termination of pregnancy is not widely practiced in most African countries, where abortion is legally restricted and cultural and ethical views are anti-abortion. However, the selective termination of affected fetuses is an integral part of screening programmes for sickle cell disease in some countries, such as the UK. In-vitro fertilisation with preimplantation genetic diagnosis, which is available in some HICs, is not feasible in most African countries.
Genetic counselling for people who are at high risk of having a child with sickle cell disease can be considered a method of primary prevention. For people who are heterozygous for the sickle cell allele, education should be provided regarding the probability of having a child with sickle cell disease. This can be implemented in the screening of adolescents within the school system.
That sickle cell disease is inherited as an autosomal recessive disease, which is potentially preventable, has been known for decades by the scientific community. However, this knowledge has not been translated into action in many high-burden countries, resulting in low rates of awareness of sickle cell disease and its implications, little availability of genetic counselling, and the absence of effective policies for universal screening and public health measures for primary prevention.
Genetic counselling for sickle cell disease
Genetic counselling is an educational procedure, the nature of which varies with the objectives. Genetic counselling in sickle cell disease is a cost-effective strategy in reducing the burden of the disease; it is a communicative process that can help to decrease the overall incidence of sickle cell disease.
The goals of genetic counselling in sickle cell disease are to provide an understanding of the inheritance of sickle cell disease and provide the people counselled with the information needed to make family planning decisions. Reproductive genetic counselling in sickle cell disease can be predominantly divided in to premarital, prenatal, and neonatal. Counselling is designed to assist in decision making; it provides objective information to individuals at risk of having a child with sickle cell disease. This information enables the individuals to make informed decisions. The counselling should also include specific information on the natural history of the type of sickle cell disease that could affect the offspring and the resources that will be required to care for an affected child. Counselling should be nondirective and objective. Counsellors should not introduce personal biases or offer specific recommendations. Counselling and education of parents of a child with sickle cell disease should be done in a kind and sensitive manner, as the parents frequently have feelings of grief, guilt, anxiety, or anger. Counselling sessions should stress the importance of health-care maintenance, compliance with prescribed prophylactic penicillin, and prompt medical evaluation of infants at the time of acute illness.
Use and awareness of genetic counselling and testing for sickle cell disease is variable across countries and is unacceptably low in countries with a high disease burden, where such services should be greatest. This scarcity of counselling will not only continue to adversely fuel the already high HbS carrier rate in these countries but will also add to the high morbidity and mortality in individuals born with the disease.
Sickle cell trait
Counselling of individuals with sickle cell trait relates predominantly to the risks of having a child with sickle cell disease. Individuals are largely fit and healthy and, hence, such counselling is necessary to increase the general public’s awareness of sickle cell trait to the level that people with sickle cell trait can make informed reproductive choices about pregnancies.
Screening recommendations
The discrepancy in access to newborn screening and early intervention between HICs and LMICs needs to be reduced. Screening for sickle cell disease is technically possible in nearly all health-care settings and is becoming more viable with POCT devices. On the basis of the evidence discussed previously, we recommend that by 2025, policies, resources, and facilities are in place to allow all babies worldwide to be tested for sickle cell disease, so that they can enter into clinical care and avoid premature death or organ damage. We also recommend that all populations at increased risk of sickle cell disease are given culturally appropriate information and counselling regarding their reproductive choices.138
Section 3: disease management—established and emerging treatments
Treatment and prevention of complications
Acute clinical events
Acute complications are common in sickle cell disease and have a characteristically abrupt onset. Whether presenting with fever, pain, dyspnoea, or other specific symptoms such as priapism or hemiplegia, people with acute clinical events warrant urgent intervention and treatment. Current medical management is frequently based on clinical experience and expert opinion, highlighting the need for prospective research to generate high quality evidence. Although appropriate treatment of acute events is an important part of sickle cell disease management, the prevention of these events is an equally important goal. In this section, three common acute clinical complications are discussed: fever, VOC, and ACS. Organ specific damage, such as stroke or splenic sequestration, are also acute events but are typically managed by blood transfusions, so are discussed separately.
Acute clinical events: infection
Fever must be systematically evaluated as bacteraemia and sepsis are the leading causes of death in children with sickle cell disease, because of functional hyposplenia or asplenia. Although there is little high-quality evidence to guide recommendations, especially in adults and LMICs, fever is typically managed aggressively in all people with sickle cell disease. Penicillin prophylaxis and immunisations greatly reduce the risk of infection with encapsulated bacteria and especially invasive pneumococcal diseases, but non-vaccine serotypes are emerging.139 Ideally, all patients with temperature greater than 38·5°C should be urgently investigated with a full blood count, blood and other relevant cultures, chest x-ray for any respiratory symptoms, and empirical antibiotics covering pneumococcus. Osteomyelitis, typically from staphylococcal or salmonella bacteria, have an increased frequency in people with sickle cell disease and can be difficult to distinguish from local vaso-occlusive events, especially since leukocytosis and inflammatory markers (eg, elevated C-reactive protein) occur in both settings. Malaria must be suspected in febrile patients living in endemic regions or after travelling to these regions.
Acute clinical events: VOCs
VOCs are the hallmark complication of sickle cell disease that manifests as severe acute pain, typically in the back, trunk, or limbs, and last several hours to several days. Diagnosis relies on the patient’s self-report as there are no specific laboratory biomarkers or imaging abnormalities. The absence of objective markers unfortunately often leads to delayed and inadequate management, and many patients are wrongfully stigmatised as drug-seeking opioid addicts.140 When available, national guidelines appropriately recommend that patients presenting for emergency care should be treated with respect and offered rapid and effective analgesia.141 Hospital personnel must be trained in pain monitoring and treatment, and should have access to local protocols for managing VOCs. Despite the event’s frequency and importance, acute pain management remains unsatisfactory for many patients, and preventive VOC treatment should be a priority.
Optimal management of VOCs is primarily for symptomatic pain control and is based mainly on expert consensus due to a scarcity of available evidence. Mild to moderate pain can be managed at home with paracetamol, non-steroidal anti-inflammatory drugs, and weak oral opioids.142 When pain cannot be relieved at home, appropriate analgesia should be rapidly administered at a health-care facility or hospital. The choice of drug, dose, and administration route is guided by the severity of the pain, the history of use of analgesia for this episode of pain, previous experience with analgesic efficacy, and adverse effects. Ideally, an individualised prescribing and monitoring protocol, established by the patient’s sickle cell disease provider, should be available.143 For severe pain, parenteral opioid treatment is usually necessary, typically intravenously or subcutaneously; however, alternative routes (including transmucosal and intranasal analgesia) can be used effectively, especially when intravenous access is delayed or difficult.144 Pain must be reassessed and treated frequently until pain relief is achieved. Regular opioid administration by patient-controlled analgesia or frequent scheduled doses is then recommended, rather than as-requested administration. Continuous opioid infusions and long-acting oral opioids can provide good pain control, but due to irregular pain intensity, some patients can become over-sedated with risk of hypoventilation and these should be administered with close monitoring. Frequent assessment of sedation, respiration, and oxygen saturation, as well as pruritus, nausea, and constipation are recommended.
Due in part to the scarcity of evidence-based data, existing guidelines have different recommendations, and several aspects of VOC management remain controversial. For example, normal saline bolus therapy for initial pain treatment is common, yet might be deleterious due to cellular dehydration that promotes sickling.145 Similarly, although patients should receive paracetamol for synergistic analgesic effects with opioids, non-steroidal anti-inflammatory drug therapy is recommended by US NIH and UK guidelines but discouraged by Brazilian and French experts for adults, fearing long-term renal toxicity.146 Corticosteroids are typically not recommended due to the potential for rebound pain, whereas incentive spirometry is important to help prevent hypoventilation and development of ACS.147
Adjunctive non-pharmacological approaches to treat pain could be useful, such as local heat, massage, distraction, meditation, exercise, and encouraging oral fluids. Nasal oxygen, intravenous fluids, oral alkaline water, and preventive anticoagulation have theoretical justification but need prospective data to strengthen the evidence. Blood transfusion is not recommended for acute pain management unless there are other clinical indications, yet many patients receive blood during hospitalisations for VOCs. After hospital discharge, weaning of opioids is also controversial, as US and UK guidelines recommend converting to oral long-acting and short-acting opioid prescriptions, whereas home-based morphine is strongly discouraged in France. National guidelines have been published in several English-speaking countries in Africa, including Uganda, Kenya, and Tanzania. However, these guidelines were based on adaptations of British guidelines and not really adapted for LMICs, and hence are more aspirational than practical.
Acute clinical events: ACS
The acute onset of lower respiratory tract signs and symptoms (ie, cough, shortness of breath, tachypnoea, retractions, or wheezing) requires evaluation and management for ACS. Minimal investigation includes chest x-ray and serial measurement of oxygen saturation by pulse oximetry, although this might not be possible in many low-income settings. By general consensus, people with signs of ACS should be hospitalised to allow close monitoring and avoid progression to respiratory failure and even death. Microbiological documentation (including blood and sputum cultures and nasopharyngeal aspirate for viral testing) is recommended.141 Supplemental oxygen is recommended to maintain oxygen saturations greater than 92–95%, depending on the patient’s baseline value, and close clinical monitoring is required for hypoxemia and bronchospasm. Bronchodilators are commonly used to help relieve reactive airway disease, but corticosteroids have been associated with late-onset clinical rebound and hospital re-admission.148
Pulmonary embolism, fat embolism, fluid overload, opioid narcosis, and hypoventilation can cause or worsen ACS. Pulmonary infection with Chlamydia pneumoniae, Mycoplasma pneumoniae, Streptococcus pneumoniae, Staphylococcus aureus, and Haemophilus influenzae have been documented in ACS,149 hence most national guidelines recommend an intravenous cephalosporin and oral macrolide antibiotic.141 However, bacterial infection is documented in a minority of cases and in France antibiotic use is relatively restricted unless fever and x-ray consolidation are present. Blood transfusion for ACS, discussed later, can help prevent rapid clinical deterioration and thus be lifesaving. For respiratory failure, high-flow humidified oxygen is recommended to reduce the need for mechanical ventilation.
Acute clinical events: chronic organ damage
Acute complications in sickle cell disease are the most frequent reason for clinical evaluation and intervention, particularly early in life, but the development of progressive damage to internal organs remains a leading cause of morbidity and mortality among adults. Repeated sickling events with tissue ischaemia, as well as chronic haemolysis and inflammation, inexorably lead to chronic organ damage. Over time, older people with sickle cell disease commonly develop kidney disease, retinopathy, pulmonary hypertension, cerebral vasculopathy, leg ulcers, avascular necrosis, and hepatobiliary complications. Management involves organ-specific screening and treatment (table 3).151,152 As described later, early use of transfusions or hydroxyurea might reduce organ damage.153,154
Table 3:
Summary of consensual expert and national guidelines in the USA, UK, and France for various complications of sickle cell disease
| Consensual expert guidelines | UK national guidelines (2nd edition, 2018)150 | French national guidelines (2nd edition, 2015)151 | US national guidelines (NIH 2014, ASH 2020)152 | Level of evidence* | |
|---|---|---|---|---|---|
|
| |||||
| Retinopathy | |||||
| Monitoring | Retinal examination after age 10 years | Every year in asymptomatic patients with sickle cell disease and on desferoxamine and deferasirox; more often in symptomatic patients or patients with a history of ophthalmopathy; every 2 or 3 years for other patients | At least every year for all patients plus slit lamp, colour vision, and electroretinogram if treatment with desferoxamine or deferasirox | Every 1–2 years if no retinopathy | Very low |
| Prevention | Laser photocoagulation if proliferative retinopathy | No intervention required for small, asymptomatic neovascular fronds; consider treatment for large, elevated sea fans or rapid growth of retinal neovascularisation | Photocoagulation of ischaemic zones | Photocoagulation in stage III retinopathy | Moderate |
| Treatment | Vitreoretinal surgery in case of vitreous haemorrhage or retinal detachment | N/A | Surgery under local anaesthesia; oxygenotherapy; no acetazolamide; no sympathomimetic; discuss transfusion | N/A | Low |
| Osteonecrosis | |||||
| Monitoring | Imaging only if intermittent or chronic hip pain; start with plain radiographs; perform MRI if normal x-ray and persistent symptoms or before surgery | N/A | N/A | N/A | Low |
| Treatment—early stages (I or II stage) | Conservative management: physiotherapy or walking aid | Injections of local anaesthetic into the joint | Consider HSCT in the femoral head | N/A | Very low |
| Treatment—advanced stages | Joint replacement surgery | Prefer the use of cementless prosthetic devices; prophylaxis of post-operative infection and thrombosis | N/A | N/A | High |
| Priapism (recurrent or stuttering) | |||||
| Prevention | Hydration | N/A | N/A | Avoid tobacco and hashish, night oxygen if nocturnal desaturation | Very low |
| Treatment | Hydration; exercise and urination; painkillers; alpha-adrenergic agonists and anti-androgens | N/A | Hydration; exercise and urination; painkillers; alpha-adrenergic agonists and anti-androgens and consult a urologist | Bloodletting to maintain Hb <10 g/dL in sickle cell anaemia, 11 g/dL in HbSC disease; referral to a psychologist | Very low |
| Leg ulcers | |||||
| Monitoring | Clinical; look for veinous insufficiency and infection | N/A | N/A | N/A | Very low |
| Prevention | Eat a nutritious and well balanced diet; local moisturisers; wear socks and well fitted shoes, insect repellents; avoid injury; treat minor trauma quickly; avoid blood test or IV lines in the lower limbs; wear compression stocking | Eat a nutritious and well balanced diet; local moisturisers; wear socks and well fitted shoes, insect repellents; avoid injury; treat minor trauma quickly; avoid blood test or IV lines in the lower limbs; wear compression stocking | Eat a nutritious and well balanced diet; local moisturisers; wear socks and well fitted shoes, insect repellents; avoid injury; treat minor trauma quickly; avoid blood test or IV lines in the lower limbs; wear compression stocking | Eat a nutritious and well balanced diet; local moisturisers; wear socks and well fitted shoes, insect repellents; avoid injury; treat minor trauma quickly; avoid blood test or IV lines in the lower limbs; wear compression stocking | Very low |
| Local treatment | Debridement; keeping the wound surface moist | N/A | Alginate phase cleaning, then vaselinated bandages then hydrocellular dressings; discussion of negative pressure (vacuum-assisted closing) with surgeons | N/A | Low |
| Treatment of infection | Antibiotics | N/A | Reserved for culture-proven bacterial infections | Reserved for culture-proven bacterial infections | Very low |
| Other treatments | Pain management; compression; bed rest | Pentoxifylline for large venous ulcers; zinc supplementation if zinc deficiency, do not stop hydroxurea | Zinc sulfate can be tried; discuss a reduction of hydroxyurea dose; try local injection of granulocyte-macrophage colony-stimulating factor | N/A | Very low |
| Transfusion | Trial of blood transfusion if intractable ulcer | N/A | N/A | N/A | Very low |
| Surgery | Microsurgery with free flap transfer and skin grafting for some intractable ulcers | N/A | N/A | N/A | Very low (high risk of failure) |
| Pulmonary hypertension | |||||
| Monitoring | Cardiac echography with measure of TRV | At least once at 18 years then every 3–5 years if normal or every year if TRV >2·5 m/s | At least once at 18 years then if evocative clinical signs | If suggestive clinical signs (ie, exercise intolerance, chest pain, fatigue, or oedema) | Low |
| Monitoring | If TRV >2·5 m/s | Refer to pulmonary hypertension centre if > 2·9m/s or if >2·5 m/s plus raised NT-proBNP or reduced 6 min walk test | To be repeated after 6 months if TRV >2·5 m/s; right heart catheterisation if >2·8 m/s at 6 months | Echography confirmed by right heart catheterisation | Moderate |
| Work out | Only for symptomatic patients | ECG; look for chronic lung disease (eg, chest radiography, pulmonary function test, 6 min walk test, arterial blood gas); thromboembolism, HIV; sleep disordered breathing; autoimmune disease; hypertension; renal and liver function | ECG; look for chronic lung disease (eg, chest radiography, pulmonary function test, 6 min walk test, arterial blood gas); thromboembolism, HIV; sleep disordered breathing; autoimmune disease; hypertension; renal and liver function | ECG; look for chronic lung disease (eg, chest radiography, pulmonary function test, 6 min walk test, arterial blood gas); thromboembolism, HIV; sleep disordered breathing; autoimmune disease; hypertension; renal and liver function | Moderate |
| Treatment | ·· | Consider disease modifying therapy with hydroxyurea or blood transfusion; vasodilator therapy for some patients with precapillary pulmonary hypertension under the supervision of a pulmonary hypertension specialist | Consider blood exchanges or hydroxycarbamide; refer to a pulmonary arterial hypertension specialist; avoid sildenafil (triggers VOCs); anticoagulation if no Moyamoya disease | Refer to a pulmonary arterial hypertension specialist | Low |
| Nephropathy | |||||
| Monitoring | Microalbuminuria plus proteinuria testing every year | N/A | N/A | N/A | Low |
| Prevention of chronic kidney disease (general measures) | High fluid intake, especially if hyposthenuria; avoid NSAIDs | NSAIDs should be avoided in patients with stage 3–5 chronic kidney disease; blood pressure target of <130/80 mmHg if the albumin to creatinine ratio is >3·5 mg/mmol | Avoid NSAIDs in all adults; systolic blood pressure target of <130 mmHg, add anticalcic if necessary | N/A | Low |
| Prevention of chronic kidney disease | ACE inhibitors or angiotensin receptor blockers | If urine protein to creatinine ratio >50 mg/mmol then add hydroxyurea | If macroalbuminuria or proteinuria, then avoid NSAIDs and iodide | In adults, if microalbuminuria or urine protein to creatinine ratio >50 mg/mmol then add ACE inhibitors or angiotensin II receptor blockers | Moderate |
| Treatment of chronic kidney disease | Usual treatment of chronic kidney disease; renal replacement if needed | N/A | No contraindication for renal transplantation unless another organ failure reduces life expectation <2 years | N/A | Low |
| Haematuria | High fluid intake, refer to urologist | Look for renal (medullary) carcinoma, calculi, or non-sickle cell disease related glomerular disease | Look for renal cancer or renal venous thrombosis; consider desmopressin if massive | N/A | Very low |
| Cerebral vasculopathy or stroke | |||||
| Monitoring | Annual transcranial Doppler velocity in children with sickle cell disease and at least one brain MRI in adults with sickle cell anaemia | Inadequate evidence to recommend routine screening by pulmonary hypertension or brain MRI to predict stroke risk in adults | Brain MRI with angiography or CT at least once at age 18 years | Monitor with angiography at least once before and after age 18 years | Moderate for pulmonary hypertension in SS or Sbeta0 patients, low for other genotypes, low for imaging |
| Primary prevention in case of atypical pulmonary hypertension | Long-term exchange transfusion therapy in children with atypical pulmonary hypertension (objective is HbS <30%) | Hydroxyurea after normalisation of pulmonary hypertension even in adults; management of risk factors for stroke (ie, hypertension, anaemia, chronic lung disease, avascular necrosis, and retinopathy) | Long-term exchange transfusion programme until age 18 years, unknown duration at adult age; no antiaggregant or anticoagulant in case of Moyamoya disease | Maintain the haemoglobin concentration >9·0 g/dL; hydroxyurea if long term exchange transfusion therapy not possible or after 1 year | High for SS/Sbeta0; low for other genotypes |
| Secondary prevention after a stroke | Long term exchange transfusion therapy (objective is HbS <30%) | To be continued in adulthood; NSAIDs as per patients without sickle cell disease; in adults with silent infarction or overt stroke, offer interval MRI scanning: consider intervention if there is progressive ischaemia or discuss stopping exchanges if no progression; consider hydroxyurea when transfusion is not possible or acceptable | Long term exchange transfusion programme in children, unknown duration in adults; no antiaggregant or anticoagulant in case of Moyamoya disease | Exchange transfusion monthly (haemoglobin >9 g/dL at all times) or hydroxyurea if regular exchanges are not possible; multidisciplinary evaluation for revascularisation surgery in addition | Moderate for exchange transfusion; very low for revascularisation |
| Cognitive impairment | |||||
| Monitoring | Surveillance in children and adults | Use simplified signalling questions; cognitive and medical evaluation to diagnose any related disorders and to identify modifiable risk factors for developmental delays or cognitive impairments | Not mentioned | Refer the patient to a specialist who might better evaluate the magnitude of the cognitive impairments and provide rehabilitative approaches | Low |
| Chronic pain | |||||
| Pharmacological treatment | ·· | Use opioids with caution, avoid escalating doses, identify key prescriptors, elaborate a prescribing plan with chronic pain team and general doctor; atypical analgesics are useful (eg, gabapentin, amitriptyline, pregabalin, and duloxetine) | Not mentioned; opioid use strongly discouraged at home | No specificity for sickle cell disease: follow the general guidelines for chronic pain | Low |
| Other therapies | ·· | Cognitive behavioural therapies or acceptance-based approaches (need further evaluation) | N/A | N/A | Very low |
| Psychotherapy | |||||
| Psychological support | Psychologists should be an integral part of multidisciplinary teams for the management of sickle cell disease | Routinely assess a patient’s desire for psychological intervention or support; encourage patients to practise cognitive behavioural therapy techniques such as relaxation; specialist staff should be aware of the importance of psychosocial issues and have access to training, support, consultation, or supervision from a psychologist | Propose psychological assessment during hospitalisation or in case of recurrent VOCs or severe morbidity; cultural issues must be taken into account since many children are migrants or children of migrants | N/A | Very low |
ASH=American Society of Hematology. ECG=electrocardiogram. HSCT=haematopoietic stem-cell transplantation. N/A=not available. NIH=US National Institutes of Health. NSAIDs=non-steroidal anti-inflammatory drugs. TRV=tricuspid regurgitant velocity. UPCR=urine protein creatinine ratio. VOC=vaso-occlusive crisis.
Gradation of evidence quality with the same methods as in Yawn and colleagues (2014).141
Standard Interventions
Blood transfusion
Red blood cell transfusions are an important and life-saving treatment for sickle cell disease, so access to safe, sufficient, and affordable blood represents a key element to the optimal management of sickle cell disease. About 90% of adults with sickle cell disease should receive at least one red blood cell transfusion in their lifetime;155 many receive sporadic transfusions for acute clinical complications, whereas some receive regular transfusions for chronic organ damage, such as stroke.
All individuals with sickle cell disease have chronic haemolytic anaemia, yet most adapt to their steady-state low haemoglobin concentration such that anaemia per se is not an indication for blood transfusion. Acute exacerbation of chronic anaemia, however, can occur in the settings of acute splenic sequestration, transient red blood cell aplasia associated with parvovirus B19 infection, and increased haemolysis, notably during severe infection such as bacterial sepsis or malaria. In these settings, a simple transfusion is indicated to increase oxygen-carrying capacity. In all cases, the post-transfusion haemoglobin concentration should not usually exceed the baseline value by more than approximately 2 g/dL or exceed 10 g/dL to avoid hyperviscosity.141 For children who have recurrent splenic sequestration, repeated transfusions can be helpful but if life-threatening anaemia develops, surgical splenectomy provides a better outcome. The optimal age for splenectomy in this setting is debated, but surgery should be delayed until all vaccinations are completed to reduce the post-operative infectious risks.
Red blood cell transfusion also reduces the percentage of erythrocytes containing HbS, which reduces the percentage of cells sickling and improves blood flow. Whenever feasible, exchange transfusion should be performed instead of simple transfusion in the setting of severe acute organ damage (eg, stroke, ACS, acute intrahepatic cholestasis, and multi-organ failure) because exchange transfusion reliably decreases the HbS to less than 30% and thereby reduces in-vivo sickling.156 Exchange transfusion can be performed either manually or as an automated erythrocytapheresis procedure where facilities permit.
The US National Heart, Lung, and Blood Institute guideline panel has recommended transfusion before surgery that requires general anaesthesia and lasts more than 1 h in people with HbSS or HbS/β0 thalassaemia, with individualised discussion according to type of surgery, known organ damage, or other medical comorbidities.141 Long-term regular transfusions to maintain HbS concentrations lower than 30% have shown efficacy in reducing the risk of a first stroke in children with atypical transcranial Doppler.153 Initially, this transfusion regimen was recommended to be lifelong,157 but hydroxyurea has been shown to be non-inferior to transfusion for primary stroke prevention in children without severe vasculopathy who received transfusion for at least a year.158 Lifelong chronic transfusion should be offered to people who have had a previous clinical ischaemic stroke, although HLA-matched HSCT offers an alternative. Regular transfusions also prevent stroke in children with silent cerebral infarcts, but the benefits of long-term transfusion in this setting must be weighed against their risks. Although randomised studies are scarce, chronic transfusion can be effective for people with recurrent ACS or recurrent VOCs but should be considered a second-line therapy after hydroxyurea.156 When available, automated exchange transfusion is usually preferable to simple transfusion because of the efficiency of the procedure to remove sickled erythrocytes and reduce the risk of iron overload.155
Long-term complications of transfusions vary in frequency and severity according to the individual country. In low-resource countries, blood is not always screened adequately so can transmit infections such as HIV, hepatitis B and C, syphilis, and malaria.159 In high-resource countries, blood supplies are readily available and routinely screened for blood-borne pathogens, so carry a very low risk of transmitting infections. Instead, the main complications are erythrocyte alloimmunisation and iron overload. Scarce venous access can also be very problematic and require insertion of indwelling catheters, which carry risks of infection and thrombosis for patients.
Erythrocyte antigens are highly polymorphic and antigenic mismatches are common between blood donors and recipients; differences in Rh and Kell antigens are particularly immunogenic and frequently lead to alloantibody formation. Alloimmunisation causes premature immune-mediated clearance of transfused erythrocytes and can lead to clinical complications, including immediate or delayed haemolytic transfusion reactions. Once present, alloimmunisation makes finding matched blood difficult and can limit the ability to transfuse safely, particularly in countries with less developed blood transfusion services. In some cases, delayed haemolytic transfusion reactions can be very severe and cause a life threatening fall in haemoglobin below its pretransfusion level, which is sometimes referred to as hyperhaemolysis. Management of this sort of very severe reaction requires a high level of expertise, since additional transfusions should be avoided if possible (even in highly anaemic people) and immunomodulatory agents might be necessary.160 To prevent alloimmunisation, a proper transfusion history is needed and ideally an extended red blood cell antigen profile by serology or genotyping, with prophylactic red blood cell antigen matching for Rh and Kell antigens performed in all patients.155
Transfusional iron overload is common among patients who have had multiple transfusions, although erythrocytapheresis limits this risk. In contrast to thalassaemia, heart function in sickle cell disease is usually preserved, although the liver can be seriously damaged. Serum ferritin concentrations are not specific or accurate to measure the iron burden (since ferritin is increased in sickle cell disease secondary to chronic inflammation) but can provide a general assessment in low-resource settings. The US National Heart, Lung, and Blood Institute guideline panel suggests iron overload screening by MRI for liver iron every 1–2 years in regularly transfused patients.141 Iron chelation is needed for patients on regular transfusions; the level of intrahepatic iron typically guides the chelation dose.
Hydroxyurea
Treatment with hydroxyurea currently constitutes the standard of care for prevention of painful VOCs in both paediatric and adult patients and is available in many parts of the world. Hydroxyurea is a cytostatic agent that induces HbF production via inhibition of ribonucleotide reductase, prolonging the S-phase of the cell cycle. Daily oral administration of hydroxyurea results in dose-related marrow suppression with stress erythropoiesis,161 which could be a crucial mechanism for HbF induction due to the proliferation and release of early erythroid progenitor cells that synthesise more HbF. Sustained increases in HbF reduce red blood cell sickling and improve red blood cell hydration, with consequent clinical benefit.
Hydroxyurea also has some HbF-independent benefits. Treatment-associated macrocytosis improves erythrocyte deformability and reduces blood viscosity.154 Leukocytes, especially granulocytes, participate in the initiation of vaso-occlusion by adhering to activated endothelium and producing inflammatory molecules that propagate vessel occlusion.162 The reduction in leukocytosis and inflammation by hydroxyurea is therefore likely to be beneficial, although excessive myelosuppression should be avoided. Furthermore, hydroxyurea might exert additional benefits by virtue of in-vivo nitric oxide donor properties, thereby activating intracellular soluble guanylyl cyclase.163 In-vivo evidence of hydroxyurea’s immediate anti-vasoocclusive actions suggests that this agent could even be exploited in acute settings, including for patients hospitalised for VOCs.164
The clinical benefits of hydroxyurea therapy for people with sickle cell disease are numerous and well established. In 1995, the randomised controlled Multicenter Study of Hydroxyurea165 documented a decreased incidence of painful VOCs and ACS, as well as reduced blood transfusions and hospitalisations. Long-term follow-up of these patients revealed a significant reduction in mortality,166 which has been observed in other adult and paediatric populations.167,168 Among infants, the BABY HUG169 trial showed reduced numbers of VOCs, ACS, and transfusions, even with a low fixed dose of 20 mg/kg per day.
National guidelines provide evidence-based recommendations for initiating and monitoring the use of hydroxy urea therapy in sickle cell disease and consensus treatment protocols for its implementation.141,170 Indications for the initiation of hydroxyurea vary among countries, typically based on the frequency and severity of vaso-occlusive complications. All guidelines emphasise the importance of informed joint decision making between patients and care providers regarding the risks and benefits of treatment. For children, hydroxyurea has the additional potential to prevent or improve future organ damage and chronic complications, hence the current consensus is to offer hydroxyurea to all children with sickle cell disease.141 However, there is not yet strong evidence that long-term hydroxyurea therapy, given either during childhood or later in life, can prevent organ damage during adulthood. Treatment guidelines in LMICs are emerging despite limited drug access and affordability.171
The HbF response to hydroxyurea depends on the average daily dose, with optimal effects observed when the dose is escalated to produce mild marrow suppression, typically an absolute neutrophil count of approximately 2·0 × 10⁹ cells per L. This type of escalation has historically been referred to as the maximum tolerated dose, although optimal dosing might be a more accurate designation. Although HbF induction is the main treatment effect, the amount of induction is heterogeneous and unpredictable, probably reflecting the numerous genetic modifiers of HbF (see the section on genetic risk factors).172
Administered orally as a single daily dose, hydroxyurea should be escalated every 4–12 weeks. An initiation dose of 15–20 mg/kg per day is well tolerated in both adults and children with sickle cell disease, and the optimal dose is typically 25–30 mg/kg per day, with a dose of 30 mg/kg per day shown to be superior for all measured outcomes (ie, reductions in acute pain episodes, ACS, transfusions, hospitalisations) to 20 mg/kg per day in a study published in 2019.19 Importantly, dose escalation does not aim to achieve a specific percentage HbF goal, but rather to achieve an acceptable amount of mild myelosuppression; patients who reach the greatest myelosuppression often have the highest HbF concentrations, which correlates with improved clinical status and survival.173 Once a stable hydroxyurea dose is established, monitoring with full blood counts (including differential leukocyte counts and reticulocytes) should be performed at periodic inter vals.141,170 Interruption of treatment is indicated only if clinically significant neutropenia, thrombocytopenia, or reticulocytopenia is detected.
Hydroxyurea treatment has almost exclusively focused on individuals with severe sickle cell disease (HbSS or HbS/β0thalassaemia), but there is some evidence to support the safety and benefits of hydroxyurea therapy for reducing pain and hospitalisation in other genotypes, including HbSC disease. A retrospective analysis of 133 adult and paediatric patients with HbSC treated with hydroxyurea reported a statistically significant decrease in painful events, especially among adults.174 However, prospective controlled trials of hydroxyurea on people with HbSC are warranted.
Concerns have been raised regarding the adverse effects of hydroxyurea on fertility for all sexes, but an analysis of testicular biopsies of 30 male children on hydroxy urea revealed no differences in spermatogonial cells.175 Potential for teratogenicity has also been proposed, but data from the USA176 and Europe177 have documented hundreds of exposed offspring without congenital anomalies, and a large review concluded that hydroxyurea at pharmacological dosing does not have in-vivo mutagenicity or carcinogenicity. 178 In addition, a prospective study concluded that hydroxyurea exposure through lactation was safe for infants, and thus breastfeeding should not be contraindicated for women with sickle cell disease on hydroxyurea therapy.179
Newer disease-modifying therapies
With improved understanding of sickling, vaso-occlusion, and other pathophysiological anomalies of sickle cell disease, an unprecedented biopharmaceutical interest has emerged that has largely focused on agents that modify cellular adhesion, oxygen affinity, and inflammation. Given the reduction in morbidity and mortality with hydroxyurea, there is also interest in identifying novel agents that induce HbF with potentially greater potency and less myelosuppression.
Induction of HbF
Elevated HbF expression in sickle cell disease, whether from inherited persistence or hydroxyurea-related reactivation, improves clinical manifestations, limits organ damage, and improves survival. Although decades of evidence have consistently documented the efficacy of hydroxyurea, its widespread use is limited, especially among adults. In addition, despite the absence of hydroxyurea-related mutagenicity or carcinogenicity, ongoing concerns exist about long-term treatment side-effects and the recommended avoidance during pre-conception, pregnancy, and lactation, which affect drug acceptance and use. There are several HbF inducers in clinical development that might overcome some of these limitations.
Tovinontrine (IMR-687) is an oral highly selective inhibitor of PDE9, which increases intracellular cyclic GMP concentrations, boosting HbF and reducing markers of haemolysis.180 An added benefit of PDE9 inhibition is that this enzyme is almost exclusively expressed in haematopoietic cells (in addition to the brain), thereby providing semi-targeted therapy for HbF induction with some anti-adhesive benefits. Although earlier trials in adults with sickle cell disease showed that IMR-687 was generally well tolerated as a monotherapy and in combination with hydroxyurea, interim results from the Ardent phase 2b clinical trial (NCT04474314) showed no statistical or clinically significant improvement in vaso-occlusive crises or HbF in a high-dose treatment group versus placebo-treated population.
HDAC1 and HDAC2 inhibition can induce HbF in sickle cell disease.181 A small study on vorinostat showed that it was well tolerated but caused no significant HbF increases. Panobinostat, an oral pan-HDAC inhibitor, is currently under investigation in adults with sickle cell disease (NCT01245179).
DNMT1 is an enzyme that epigenetically silences HbF. Decitabine, a US Food and Drug Administration (FDA)-approved DNMT1 inhibitor used to treat myelodysplastic syndrome, can increase HbF in adults with sickle cell disease, including people without clinically significant HbF responses to hydroxyurea.182 Decitabine’s intravenous administration and short biological half-life are limitations, but oral decitabine with tetrahydrouridine to block enzymatic degradation appears safe and can boost both haemoglobin and HbF.183 A large global randomised clinical trial has begun enrolment to test the efficacy of oral decitabine combined with tetrahydrouridine (NCT05405114).
Anti-sickling compounds
Since the initial demonstration almost 50 years ago that polymerisation of deoxy HbS is the first and essential step in sickle cell disease pathophysiology, the search for therapeutic agents with anti-sickling activity has been largely unsuccessful. However, several new mechanisms and compounds have become available that offer that promise to interrupt this crucial early step of erythrocyte sickling.
Voxelotor is a once daily oral small molecule approved by the FDA and European Medicines Agency (EMA) that reversibly binds to haemoglobin to increase its oxygen affinity and stabilise the oxygenated haemoglobin state. In a global, double-blind, randomised, placebo-controlled trial, voxelotor met the primary endpoint of an absolute haemoglobin increase of at least 1·0 g/dL (59% increase in the 1500mg voxelotor group vs 9% in the placebo group).184 Common adverse effects with voxelotor include headache, diarrhoea, and abdominal pain. Safety and efficacy on haemoglobin concentrations were also shown in children aged 4–11 years. Trials aimed at defining a clinical benefit, such as arterial cerebral blood flow (NCT04218084) and delayed progression of nephropathy (NCT04335721), are currently underway. Additionally, GBT021601, a highly potent, second generation HbS polymerisation inhibitor, shown in sickle cell disease mouse models to normalise haemoglobin, has entered early stage clinical trials. One important concern with all these types of drug is that a left-shifted oxygen dissociation curve could inhibit oxygen delivery to tissues.185
Haemoglobin polymerisation and red cell stability is modified by erythrocyte metabolites ATP and 2,3-diphosphyglycerate. Activation of erythrocyte PKLR lowers 2,3-diphosphyglycerate concentrations, which increases haemoglobin oxygen affinity, and inhibits HbS polymerisation. Additionally, increased PKLR activity leads to higher erythrocyte ATP concentrations, improving many aspects of erythrocyte health and potentially reducing haemolysis. Two oral small molecule PKLR activators, FT-4202186 and mitapivat,187 are currently under investigation for safety, tolerability, and efficacy in adolescents and adults with sickle cell disease (NCT04624659 and NCT04000165). PKLR activation might also reduce ineffective erythropoiesis, which could provide additional benefits for people with sickle cell disease.188
Additional therapeutic approaches
The inhibition of increased adhesiveness between different blood cells and vascular endothelium has been explored as a therapeutic option in sickle cell disease. Adhesive interactions among leukocytes, reticulocytes, endothelial cells, and platelets all contribute to sickle cell disease-related vaso-occlusion. Evidence supports a key role for membrane-bound selectins in mediating adhesion. Accordingly, selectin inhibition has been a major therapeutic focus for both prevention and treatment of acute vaso-occlusive pain. Rivipansel, a monoclonal pan-selectin inhibitor, failed to show statistically significant improvement over placebo in reducing the length of an acute VOC in hospitalised patients. However, phase 3 clinical trial indicated that the timing of rivipansel administration after the onset of pain might be crucial for accelerating acute VOC resolution (NCT02187003; NCT02433158). Currently, there is no evidence that rivipansel is an effective treatment in sickle cell disease and it should not be used unless as part of a clinical trial. In contrast, prophylactic monthly treatment with crizanlizumab, a monoclonal anti-P-selectin antibody, significantly reduced VOC pain compared with placebo, which led to FDA and EMA approvals. Studies at higher doses (NCT03814746), in children (NCT03474965), and for the prevention of priapism (NCT03938454) and progression of renal disease (NCT04053764) are ongoing. Inclacumab, a fully humanised and potentially more potent monoclonal anti-P-selectin antibody, is under investigation in adults with a 3-month dosing interval for VOC prevention (NCT04935879) and a one-time dose following an admission for acute VOC to reduce rehospitalisation (NCT04927247).
Platelet inhibition and anticoagulation have also been explored as a therapeutic option, as sickle cell disease is known to be a hypercoagulable state with widespread inflammation, which contributes to vaso-occlusion and vasculopathy through in-vivo fibrin formation. However, meaningful clinical trials have been limited by concerns over bleeding risks, especially intracranial bleeding. Sevuparin, a low molecular weight heparin, was shown to have no benefit for reducing the duration of acute VOC episodes in hospitalised patients.189 Prasugrel, an oral anti-platelet agent, did not show benefit in reducing the rate of VOCs in children aged 2–17 years with sickle cell disease.190 Whether other similar agents such as clopidogrel or even rivaroxaban are useful in sickle cell disease will require further investigation.
A wide variety of other therapeutic compounds have also been explored as potentially beneficial in sickle cell disease. Hemopexin (CL889), canakinumab, intravenous immunoglobulin therapy, simvastatin, omega-3 fatty acid supplementation, the endothelin-receptor antagonist ambrisentan, and complement inhibitor crovalimab are under consideration for clinical trials. An oral ferroportin inhibitor (vamifeport) has some reported benefits in a murine model.191 Vitamin D has various effects and has been purported to reduce infections and VOCs.192
L-glutamine was approved by the FDA in 2017 to reduce acute complications of sickle cell disease in adults and children aged 5 years and older. This approval was based on a randomised, double-blinded, placebo-controlled trial that showed a reduction in median annual VOC rates (3 vs 4, p=0·0005), time to first VOC (84 [95% CI 62–109] days vs 54 [31–73] days; HR 0·69 [0·52–0·93], p=0·02), and time to second VOC (212 [95% CI 153–250) days vs 133 [115–179] days; HR 0·68 [0·49–0·96], p=0·03).22 Adverse events were mostly gastrointestinal, probably due to the powder formulation of the medication and limited medication adherence. On the basis of this trial, L-glutamine was approved by the FDA to reduce acute complications of sickle cell disease. However, the molecule was rejected for approval by the EMA in 2019 and no additional data on the clinical benefits of L-glutamine have been published since the original study.
Considerations for low-resource settings
LMICs have both the highest number of children born with sickle cell disease and the least access to disease-modifying treatments, especially the most common—transfusions and hydroxyurea. In sub-Saharan Africa, where the prevalence of sickle cell disease is particularly high, these treatments are essential and lifesaving. Transfusions are especially important for the management of acute clinical complications that require emergency care and hospitalisation, whereas hydroxyurea can reduce the incidence of acute complications, including painful VOCs, ACS, infection, stroke, and death, while also preserving organ function.
Due to high demand, low supply, and blood safety issues, all countries in sub-Saharan Africa fail to collect enough blood to meet their transfusion needs and have frequent shortages. Individuals with sickle cell disease are especially affected by such shortages, since available units are used in other life-threatening emergencies related to obstetrics, trauma, and malaria, especially for people younger than 5 years.193 In a questionnaire-based study of 31 sickle cell centres in Nigeria, 78% of hospitals were unable to transfuse patients with sickle cell disease regularly due to blood scarcity.194 Several modifiable factors contribute to the inadequate blood supply for people with sickle cell disease in many African countries. For logistical and cultural reasons, a very low percentage of people living in LMICs donate blood; according to 2015 WHO data, the median blood donation rate in LMICs is 3–10 times lower than in HICs.193 Donation can also be limited by nutritional deficiencies (mainly iron deficiency) in potential blood donors. Blood donations in LMICs often come from paid donors and replacement donations (family members donate blood to replace that used by their relative), but less frequently from voluntary, unpaid donors.
In addition to blood scarcity, there are serious concerns about the quality of blood and related monitoring systems. Most blood collected in LMICs undergoes laboratory testing only to identify the ABO group and RhD type, without any screening of serum to identify antibodies that might affect the transfusion.194,195 In addition, transfused blood units are often selected by ABO and RhD typing alone, without a formal crossmatch to ensure accuracy in testing and labelling. Moreover, in some low-resource countries, whole blood transfusions are still performed, further increasing the risk of alloimmunisation and transfusion reactions. Although the risk of alloimmunisation might be reduced in some LMICs compared to HICs because the donor and recipient populations are of similar ethnic origin, better phenotyping, screening, and crossmatching are still needed to decrease the risk of developing acute and delayed haemolytic transfusion reactions.
Transfusion safety must also be addressed because (especially in sub-Saharan Africa and India) blood is not always tested for blood-borne pathogens such as HIV, hepatitis B and C, and syphilis. Rapid diagnostic tests are sometimes available but have variable reliability, and consequently transfusion-transmitted infections remain a substantial concern for families and people with sickle cell disease. Moreover, safe blood transfusions are often expensive and not affordable for many families. Finally, national sickle cell disease guidelines need to be developed to include the appropriate indications for transfusion in a particular country, which might well be different from guidelines currently used in Europe and North America. Treatment recommendations should consider the scarce and potentially unsafe blood supply; previous transfusion thresholds and volumes have been challenged.196
Given the various challenges toward ensuring a safe and reliable blood supply for sickle cell disease globally, wider use of hydroxyurea is an attractive treatment alternative. Decades of consistent and compelling data support its safety, efficacy, and effectiveness in adults and children with sickle cell disease. After suitable dose escalation to achieve mild myelosuppression, hydroxyurea can significantly reduce rates of VOC, ACS, hospitalisation, and transfusion, even in low-resource settings.171 Reports from Africa are now also documenting hydroxyurea’s benefits for primary and secondary stroke prevention.197 Hydroxyurea is a relatively cheap drug, and has been listed by WHO as an essential medicine for over a decade and is recommended for both children and adults with sickle cell disease.
Despite this recommendation, hydroxyurea is hardly used in many LMICs. Challenges for the effective introduction and use of hydroxyurea have been described but primarily relate to the scarcity of drug availability and affordability in low-resource settings.198 The absence of drug registration with national drug authorities is another potentially limiting factor, although hydroxyurea is already registered in many countries for the treatment of chronic myeloid leukaemia. The absence of national treatment guidelines is also limiting, although several countries are now generating their own guidelines that are based on published treatment guidelines from the USA and Europe. Lack of prescriber familiarity with hydroxyurea, including the need for monitoring blood counts and dose adjustments, is another important factor but one that is amenable to training. The cost of the drug itself and associated laboratory monitoring is also prohibitive for many families in LMICs, but efforts by pharmaceutical companies to lower prices might bring treatment closer to the affordable range. Pharmaceutical companies’ donation of hydroxyurea, possibly via intermediaries such as The Global Fund, offer one among several options to increase the availability of hydroxyurea in LMICs. However, charitable donations of hydroxyurea are unlikely to provide a sustainable solution for hydroxyurea treatment in the countries where the majority of individuals live with sickle cell disease. Additional strategies include hydroxyurea being produced in Africa, as has happened in Nigeria for over 15 years (costing about US$ 0·15 per 500mg capsule), or the development of more pharmaceutical companies producing generic formulations which could be inexpensively sold in LMICs.
Hydroxyurea has become the standard of care for sickle cell disease in HICs. To work toward health equity for sickle cell disease, hydroxyurea must be introduced widely in LMICs, especially sub-Saharan Africa. To achieve this goal, urgent efforts should be made to address the absence of treatment guidelines, inadequate clinical infrastructure and training, and drug accessibility and affordability.198 As an example worthy of mention, the public health-care system of Brazil, a middle-income country, now offers universal newborn screening for sickle cell disease, and transfusions and hydroxyurea are available free of charge in several specialised centres across the country. Before new expensive treatments can be considered for sickle cell disease in low-resource settings, including potentially curative treatment options, hydroxyurea must first become widely available.
Recommendations on established and emerging treatments
Currently, effective treatment requires access to prophylaxis against and treatment of infections, hydroxyurea, and safe blood transfusions. Table 3 summarises recommendations for basic care, based on the consensus guidelines from the USA, UK, and France. Although some LMICs (eg, Uganda, Tanzania, or Kenya) do have national guidelines, these tend to be aspirational and based on the recommendations summarised in this table.199 Although precise figures are not available, the majority of people in the world do not have access to this basic level of care. We therefore recommend that policies and resources focus on providing access to these treatments for all people across the world, with a particular goal that affordable hydroxyurea is available to all people by 2030. It is important that further new treatments are developed, and that they are tested in all appropriate health-care settings and are made affordable to all people and health-care systems throughout the world.
Section 4: disease management—cellular therapy
Allogeneic HSCT
HLA matched sibling HSCT
HSCT for sickle cell disease with an HLA matched sibling donor was first described in 1984 in a child with acute myeloid leukaemia and sickle cell disease. Since then, the approach has become the most common and arguably most effective cure for sickle cell disease. Investigations are now focused on reducing its toxicity without affecting its curative potential.200 Sibling transplantations can be performed with either myeloablative or non-myeloablative conditioning, with the latter approach being particularly useful in adults.
HSCT based on myeloablative conditioning with busulfan and cyclophosphamide was developed as a treatment for children and became the predominant approach in the late 1990s and early 2000s. Outcomes were promising in these early studies with event-free survival around 84–86% and an overall survival of 93–94%.201 A number of refinements led to an increase in event-free survival to 98% in patients younger than 30 years and 100% survival in individuals younger than 5 years. Now considered a standard of care at many centres worldwide, myeloablative matched sibling donor HSCT provides a high chance of cure, with immunological complications like severe graft-versus-host disease (GVHD) occurring in about 5% of patients and less than 5% of patients need long-term systemic immune sup pression.202
Generally, the short-term toxicities of HSCT are reversible, but the morbidity associated with myeloablative conditioning can negatively affect patients’ quality of life during the peri-transplantation period. Although infrequent, long-term risks of myeloablative-conditioning-based HSCT could include increased risk of secondary cancer, infertility, exacerbation of sickle cell disease-induced organ dysfunction, and metabolic syndrome.3
To minimise the toxicity associated with matched sibling donor HSCT, several less intense regimens have been developed. The well tolerated immunosuppressant fludarabine has been incorporated into conditioning regimens to reduce the need for alkylating drugs that are responsible for the toxicity of myeloablative regimens. A commonly used reduced intensity conditioning regimen is a combination of fludarabine, the lymphodepleting antibody alemtuzumab, and the alkylator melphalan. This low-toxicity regimen has a similar disease-free survival compared with busulfan-based myeloablative regimens. There is some preliminary data suggesting that fertility might be preserved in female patients because this reduced toxicity regimen reduces alkylator exposure.3
Non-myeloablative regimens are sufficiently low intensity that, following conditioning and in the absence of a donor stem-cell infusion, autologous blood count recovery occurs universally. Adults with sickle cell disease have cumulative organ injury, making the use of high-intensity conditioning regimens untenable due to the risk of further organ damage during HSCT. However, the non-myeloablative alemtuzumab and low-dose total body irradiation-based approach has now been shown in a primarily adult cohort of 122 patients to achieve an event-free survival of 88% (95% CI 82·0–93·7) at 1 year and 85% (77·6–92·1) at 5 years.203 Similarly encouraging results have been reported in 16 paediatric patients, with an overall survival of 100% and event-free survival of 100%, and is being studied further in a phase 2 trial (NCT03587272).204 Although toxicity is minimal and there have been no cases of severe GVHD reported, the long-term effects are still to be examined and are of particular importance in the paediatric population.
Alternative donor HSCT
As the chance of a patient with sickle cell disease finding a matched sibling donor is approximately 15%, alternative donor HSCT has been investigated to increase the donor pool. To date, none of the alternative donor HSCT approaches are comparable to the current success of matched sibling donor HSCT, but substantial improvements have been made in the past decade. Investigations into adapting conditioning and GVHD prevention continue. There are three possible types of alternative donors that can be used in HSCT: unrelated bone marrow donors, unrelated cord blood donors, and haploidentical donors.
The use of bone marrow-derived grafts from unrelated donors with 8/8 matched HLA alleles was investigated in the BMTCTN 0601 trial.205 The observed 2-year event-free survival of 69% (95% CI 42–82) in this cohort of 29 paediatric patients was lower than the contemporary matched sibling donor HSCT and was attributed to the high rates of GVHD (62% [95% CI 41–77], of which 38% were classified as extensive). 7/8 HLA-matched donors have been investigated to address the challenges of finding an 8/8 HLA-matched donor. To address the risk of GVHD, the co-stimulatory blockade agent abatacept is being evaluated as a part of the GVHD prophylaxis regimen in several studies, with promising results.206
Historically, graft failure was an issue in unrelated cord blood transplantations for people with sickle cell disease, occurring in about 50% of patients. However, in a phase 1 study of nine patients, the addition of thiotepa to an alemtuzumab–fludarabine–melphalan-based regimen imp roved the sustained engraftment rate to 78%.3 Cell dose requirements restrict the use of cord blood HSCT to younger patients, usually to those younger than around 4 years of age. To address this limitation, the ex-vivo culture expansion of cord units for transplantation has been investigated in clinical trials. In a study of cord units expanded with a nicotinamide-derivative, the median age of HSCT recipients was 13 years and an 85% (95% CI 51–96) event-free survival was observed in patients receiving an expanded plus unexpanded double cord transplantation.207 Further investigation is required to assess the applicability of this approach for older patient populations.
One of the major limitations with HSCT to treat people with sickle cell disease is the availability of suitable donors. The advantage of HLA-half-matched related or haploidentical donors is that most patients have such a donor, in the form of a parent. However, the inherent risks of severe GVHD and rejection with HLA-mismatched donors have hampered early studies. Currently two approaches for HSCT with haploidentical donors are being employed and the results are encouraging.3
Initial results with post-transplantation cyclophosphamide with a non-myeloablative conditioning regimen resulted in only about 50% of patients being disease free post-HSCT.208 With the addition of thiotepa to a reduced intensity regimen, 93% event-free survival and 100% overall survival was seen in 15 patients, with only one patient developing mild chronic GVHD. A multi-centre study is ongoing to verify these results (NCT03263559).
Historically, ex-vivo graft manipulation was affected by both severe GVHD and graft rejection, with only half of patients being cured. Two studies published in the past few years that used a CD34⁺ cell selection have shown overall survival of 84–90% and event-free survival of 69–90%.209,210 Future investigations will likely use more modern manipulation methods that better maintain cells in the graft and are important to engraftment and immune recovery.
Conclusions on HSCT
Matched sibling donor HSCT with myeloablative conditioning is now a standard curative approach requiring a small number of essential medications (panel 1), and future investigations are expected to focus on lessening toxicities by reducing the intensity of conditioning regimens. Alternative donors are, however, needed for most people with sickle cell disease seeking cure and new approaches to make these transplantations safe is an area of active research.
Panel 1: Essential medications for haematopoietic stem-cell transplantation.
Filgrastim
Magnesium sulfate
Ceftazidime
Levetiracetam
Cefepime
Amlodipine
Vancomycin
Hydralazine
Fluconazole
Labetalol
Aciclovir
Co-trimoxazole
Benzylpenicillin
Gene addition therapy
Despite improving outcomes, allogeneic HSCT might still not be a feasible option for many people with sickle cell disease, so genetic modification of autologous haematopoietic stem and progenitor cells (HSPCs) has become an area of rapidly expanding interest for the haemoglobinopathy community. Autologous genetic therapies offer two major benefits over allogeneic HSCT: eliminating the need to find a suitable donor and eliminating the risk of GVHD and immune-mediated graft rejection. Ex-vivo, viral-based gene therapy has now been used to treat various genetic disorders, including immunodeficiencies, neurodegenerative disorders, and bone marrow failure syndromes.211 Early research with gammaretroviral and early lentiviral vectors showed proof of principle by effectively supplying the gene that was missing in people with ADA deficiency and X-linked severe combined immunodeficiency,212,213 but the field suffered a setback when insertional mutagenesis led to acute leukaemia in multiple patients.214 Alterations were made to vector design, and a new generation of self-inactivating lentiviral vectors has proven well tolerated and effective, thus far without evidence of insertional oncogenesis. More than 300 patients have now been treated with HSPC gene therapy in clinical trials.215 Haemoglobinopathies are particularly well suited to correction by gene therapy because even a partial correction of the defect can lead to major clinical benefit. A gene therapy product using the addition of a modified β-globin gene to treat transfusion-dependent β-thalassaemia was among the first to receive regulatory approval by the EMA and the FDA.216
Strategies of lentiviral gene therapy for sickle cell disease include either the addition of an exogenous globin gene that will pair with α-globin to produce a non-sickling haemoglobin, or the addition of a regulatory construct that will act on the cell’s endogenous globin genes to switch expression from the mutated β-globin to γ-globin, thus inducing production of HbF. Regardless of the strategy, the metrics of success in a gene therapy trial include: features of the medicinal product, such as number of cells transduced and vector copy number per cell; quantification of the non-sickling haemoglobin; clinical outcomes; durability; and avoidance of adverse events.
The gene therapy treatment process is similar in all open clinical trials. First, autologous HSPCs must be collected from the patient. After early attempts to collect HSPCs directly via bone marrow harvest resulted in poor cell yield, several studies showed that the CXC-R4 antagonist plerixafor could effectively be used as a single-drug stem-cell mobilising agent followed by collection with apheresis in people with sickle cell disease.217–219 After collection and selection of CD34+ cells, transduction with the lentiviral vector is performed. After clinical release criteria are met (including, among other features, sufficient vector copy number in the product and adequate cell number), the modified cells are infused in the patient after preparation with an alkylator-based (generally busulfan) conditioning regimen.
Clinical trials that have reported initial results after treating people with sickle cell disease with lentiviral gene therapy are listed in the rest of this section and all sickle cell disease gene therapy trials registered as enrolling are shown in table 4.
Table 4:
Overview of ongoing or recently completed gene addition therapy studies
| Modality | Lead group | Status | Clinical trial number | Results | |
|---|---|---|---|---|---|
|
| |||||
| Globin gene addition | Addition of modified β-globin gene (bT87Q; LentiGlobin=lovotibeglogene autotemcel; bb1111) | bluebird bio | Phase 3; recruiting | NCT02140554 and NCT04293185 | 35 Group C patients infused by July, 2021, with a median duration of follow-up 20·9 months (range 8·5–28·5); 28 (97%) of 29 evaluable patients were free of severe VOCs |
| Globin gene addition | Addition of γ-globin gene (ARU-1801) | Aruvant | Phase 1/2; not recruiting | NCT02186418 | As of November, 2021, four patients have been infused; three (75%) patients with >12 months follow-up had reduced or absent VOCs |
| Globin gene addition | Addition of modified β-globin gene, (bAS3; DREPAGLOBE) | Assistance Publique—Hôpitaux de Paris | Phase 1/2; not recruiting | NCT03964792 | No clinical trial results posted yet |
| Globin gene addition | Addition of modified β-globin gene (bAS3-FB) | University of California, Los Angeles | Phase 1; recruiting | NCT02247843 | No clinical trial results posted yet |
| HbF induction | shRNA knockdown of BCL11A gene (BCH-BB694) | Boston Children’s Hospital | Phase 2; recruiting | NCT03282656 and NCT05353647 | Ten patients infused by November, 2022, with a median duration of follow-up of 30·5 months (range 2–50); nine (90%) of ten patients had reduced or absent VOCs |
VOCs=vaso-occlusive crises. shRNA=short hairpin RNA.
LentiGlobin—the addition of a modified β-globin gene
In 2008, the first patient with haemoglobinopathy was treated with gene therapy with a vector called HPV569 that delivered a modified β-globin gene (βA(T⁸⁷Q)) designed to inhibit HbS polymerisation.220,221 Subsequent modifications to this vector resulted in the vector LentiGlobin BB305.222 This vector is first being tested in people with sickle cell disease in the phase 1/2 HGB-206 trial (NCT02140554), with interim results presented at major haematology meetings in the past 10 years.223 In an initial cohort of seven patients (referred to as Group A), the individuals all engrafted but CD34+ cell yield and peripheral vector copy number per cell were low.224 Study modifications were put into place for later cohorts, including addition of a transfusion regimen before collection of autologous cells; alteration of the transduction protocol to increase vector copy number; and use of peripheral stem-cell mobilisation instead of bone marrow harvest for collection of CD34+ cells. A publication of the latest cohort in this trial (referred to as Group C) noted that 25 patients had been treated. Of 16 patients with at least six months of follow-up, total haemoglobin was 11·5 (range 9·6–16·2) g/dL with median HbS less than (or equal to) 60% of the total haemoglobin. 14 patients with at least 6 months of follow-up had 4·0 (2·0–14·0) episodes per year of VOCs or ACS during the 2 years before treatment, and these patients had no ACS or serious VOCs after treatment.225,226
BCH-BB694—the knockdown of BCL-11A to increase HbF
The protein BCL-11A is an important developmental regulator of both the initial haemoglobin switch from γ-globin to β-globin expression in infants and young children and of the persistent silencing of γ-globin expression in adults. BCL-11A, a zinc-finger protein encoded by a gene on chromosome 2p15, was not a known regulator of globin expression. In 2007–08, genome-wide association studies reported that variation at the BCL11A locus is responsible for a statistically significant degree of the variation in HbF concentrations.227,228 Preclinical studies showed that downregulation of BCL11A expression in adult human erythroid precursor cells,229 and inactivation of BCL-11A in a sickle cell mouse model,230 resulted in HbF induction and correction of the sickle cell disease defect. These data made clear that inactivating BCL-11A in people with β-haemoglobinopathies would hold promise as an effective way to increase concentrations of HbF and thus to decrease disease severity. A current clinical trial (NCT03282656) is using lentiviral gene therapy to downregulate BCL11A in haematopoietic stem cells (HSC)-derived erythroid precursors via short hairpin RNA knockdown. The vector in this study, BCH-BB694, is driven by β-globin promotor and regulatory sequences to ensure that expression is limited to the erythroid lineage. The vector contains a short hairpin RNA targeting BCL11A embedded within an endogenous microRNA scaffold. Data from the first six patients (aged 7–25 years) with a follow-up of 7–29 months showed that total HbF as a component of the total haemoglobin (HbF/[HbF+HbS]) was 21–42% at the last reported follow-up in 2021, and HbF was broadly distributed on the basis of F-cell percentages of 59–94%. Treatment was well tolerated without safety concerns beyond the expected adverse effects of conditioning therapy.231
ARU-1801—the addition of a γ-globin gene
In 2018, two patients were treated with autologous cells containing a modified γ-globin transgene. Data presented in 2020 showed HbF concentrations of 22% in the first two patients at 30 months after treatment but with only 31% F-cells. A third patient has been treated after modifying the manufacturing process, with improvement in cell number and vector copy number in the product and early suggestion of improved transgene production.232
Long-term safety
Long-term safety is one of the most crucial questions that must be investigated for all novel genetic therapies. With the new generation of lentiviral vectors, insertional oncogenesis has not been reported; however, careful monitoring of clinical status and insertion site analysis is required, typically for 15 years after treatment. Still, vector-mediated insertional mutagenesis is not the only cause of malignancy risk in trials of autologous genetic therapy. In 2020, a patient was reported to have developed myelodysplastic syndrome 3 years after infusion of the genetically modified cells.233 However, the absence of vector within the blasts indicated that vector insertion was not implicated in leukaemogenesis and this was presumed to be secondary to busulfan-related genotoxicity. In February, 2021, the LentiGlobin lentiviral sickle cell disease gene therapy trial (NCT02140554) was temporarily halted due to a reported suspected unexpected serious adverse reaction of acute myeloid leukaemia. This second patient developed acute myeloid leukaemia 5·5 years after gene therapy. In this case, the blasts did contain the vector and transgene, but there was no evidence that this integration was a driver of the malignancy. Therefore, additional investigation is needed to fully understand why at least two (4%) of 47 patients with sickle cell disease treated234 with lentiviral gene therapy have developed acute myeloid leukaemia, including the possibility of an underlying increased malignancy risk in sickle cell disease and whether there are ways to mitigate this risk.
Gene editing
Editing the genetic code to correct disease-causing mutations has been the ultimate goal of molecular genetics. Monogenic disorders, especially those due to single point mutation such as sickle cell disease, are the most amendable to genetic code correction. Several groups had previously attempted, and many successfully achieved, correction of the sickle cell disease mutation in primary human CD34+ cells or induced pluripotent stem cells with zinc-finger nucleases or transcription-activator-like effector nucleases.235 However, the advent of the precise and efficient, programmable CRISPR-associated protein 9 (Cas9) system has accelerated genome editing to treat sickle cell disease from the laboratory bench to the bedside.236,237 Novel genome editing techniques, along with the newfound understanding of the genetic regulation of the globin-like genes, have launched an era of therapeutic genetic manipulation to treat sickle cell disease. There are several target sites and gene editing methods that are currently being investigated to either increase HbF or change the sickle mutation,235,236 and many new editing tool-target site combinations are being constantly described in the literature.
Although the direct correction of the mutant sickle cell disease codon (GTG, coding for valine), either to the typical (GAG, coding for glutamic acid) or to another benign, non-sickling variant is most desirable, it has some inherent technical challenges.236 However, reversing the haemoglobin switch has been a rather simplistic and achievable alternative. Induction of HbF as a therapeutic strategy for sickle cell disease is based on the observation that individuals with sickle cell disease who co-inherit hereditary persistence of fetal haemoglobin (a naturally occurring benign genetic condition that results in persistently elevated HbF concentrations) exhibit few or no sickle cell disease effects.238 HbF inhibits HbS polymerisation.239 Additionally, induction of γ-globin genes (ie, HBG1 and HBG2) leads to competition with the β-globin gene (HBB) locus for the locus control region, a globin gene enhancer, which results in sup pression of the mutant HBBS gene expression.240,241 Several genome editing methods have been developed that induce HbF to concentrations sufficient to improve sickle cell disease pathologies. These genome editing methods that induce HbF are also less likely to induce globin chain imbalance, due to balanced production of β-like chains, in comparison with the lentiviral gene addition approaches, wherein the transgenic globin molecule is produced independently of the βS globin allele production. Systematic evaluation of patients under going treatment with gene addition methods and genome editing techniques should assess for chain imbalance and resultant ineffective erythropoiesis from unmatched globin chain aggregate deposition.
There are four main genome editing-based approaches, in various preclinical and clinical stages of development to treat sickle cell disease. These are listed in the order of those that are most advanced in clinical development to those that only have proof of concept preclinical data available.
Conventional gene editing
Conventional gene editing approaches involve making a double-stranded break in the genome at a precise location. The double-stranded break is then re-ligated by the DNA repair machinery of the cell, most frequently via the non-homologous end joining pathway and occasionally, in the presence of a template DNA, by homology-directed repair. The non-homologous end joining repair pathway leads to stochastic insertions or deletions (indels) which can disrupt either the protein coding exon sequences or functional non-coding regulatory elements in the intronic DNA. This approach has been used to disrupt the regulatory elements essential to globin switching from γ-globin (HBG1 and HBG2), which is expressed during the fetal life, to β-globin (HBB), which is expressed during the adult life, and has focused on two main targets: BCL11A on chromosome 2 and the extended β globin locus on chromosome 11
After genome-wide association studies led to the discovery of sequence variants in the gene BCL11A that are associated with elevated HbF concentrations,242 BCL-11A was identified as a potent repressor of HbF in humans.229 Early genome editing attempts used zinc-finger nuclease to disrupt exon 2 of BCL11A by introducing biallelic frameshift mutations, which led to complete inactivation of BCL11A.243 However, global knockdown of BCL-11A not only adversely affected erythroid maturation but also affected haematopoietic stem-cell expansion and engraftment.243,244 Later, pooled CRISPR screens for fine-mapping of the BCL11A intronic region identified a developmental stage-specific, erythroid lineage-restricted enhancer.245 Canver and colleagues246 further identified discrete vulnerabilities of these erythroid-specific enhancers that could be disrupted to selectively decrease BCL-11A transcription in the erythroid lineage while mitigating the HSC growth disadvantage resulting from complete BCL-11A loss. Given the specificity of BCL-11A restriction by editing of the erythroid specific enhancer and the plethora of data on these enhancers that has been generated over the past decade,247 this strategy has become the most widely adopted genome editing method and several people with sickle cell disease have been treated with autologous CD34+ cells edited at this locus (table 5). This approach is the most clinically developed and is in phase 3 clinical trials currently. Several independent groups are using this approach to disrupt the erythroid specific enhancer of BCL11A using CRISPR-Cas9248 or zinc-finger nuclease methods.249,250 Both of these nucleases have produced comparable preliminary data.
Table 5:
Overview of ongoing gene editing studies
| Modality (drug name) | Lead group (study name) | Status | Clinical trial number | Results | |
|---|---|---|---|---|---|
|
| |||||
| HbF induction | Cas9-mediated NHEJ; disruption of erythroid-specific enhancer in BCL11A gene (CTX001 or Exagamglogene autotemcel) | Vertex Pharmaceuticals/CRISPR Therapeutics (CLIMB SCD 121 Study) | Phase 1/2/3 (study closed to accrual, licensing application pending) | NCT03745287 and NCT05329649 | 31 patients with sickle cell disease (median age 23 years [range 12–34]) infused; all patients were VOC-free with a median duration of follow-up 9·6 months (range 2·0–32·3) after infusion |
| HbF induction | Cas9-mediated NHEJ, disruption of a regulatory element in the HBG1 and HBG2 promoters (OTQ923) | Novartis Pharmaceuticals | Phase 1/2 | NCT04443907 | Two patients with sickle cell disease (aged 22 and 21 years) infused; both patients were VOC-free with a follow-up of 6 and 12 months after infusion |
| HbF induction | ZFN-mediated NHEJ; disruption of erythroid-specific enhancer in BCL11A gene (BIVV003) | Sangamo Therapeutics (PRECIZN-1 Study) | Phase 1/2 | NCT03653247 | Four adult patients with sickle cell disease infused; all patients were VOC-free with a follow-up of 13–65 weeks after infusion |
| HbF induction | Cas12a (Cpf1)-mediated editing of the HBG1 and HBG2 promoters (EDIT-301) | Editas Medicine (RUBY Trial) | Phase 1/2 | NCT04853576 | No clinical trial results posted yet |
| HbF induction | Base editing of the HBG1 and HBG2 promoters (BEAM-101) | Beam Therapeutics (BEACON Trial) | Phase 1/2 | NCT05456880 | No clinical trial results posted yet |
| Correction of sickle mutation | Cas9-mediated HDR (CRISPR_SCD001) | UCSF Benioff Children’s Hospital, Oakland; University of California, Los Angeles; University of California, Berkeley | Phase 1/2 | NCT04774536 | No clinical trial results posted yet |
| Correction of sickle mutation | Cas9-mediated HDR (GPH101 or nulabeglogene autogedtemcel) | Graphite Bio (CEDAR Trial) | Phase 1/2 (study paused) | NCT04819841 | No clinical trial results posted yet; trial paused due to serious adverse event in first patient |
Data in the table updated as of Jan 11, 2023, from ClinicalTrials.gov. HDR=homology directed repair. NHEJ=non-homologous end joining. VOC=vaso-occlusive crisis.
Several cisregulatory elements have been identified within the extended globin locus on chromosome 11 (specifically in the HBG1 and HBG2 promoters, where various repressors of HbF such as BCL-11A251,252 and ZBTB7A253,254 bind), leading to haemoglobin switching from HBG1 and HBG2 to HBB.255 Disrupting these transcription factor binding motifs using CRISPR-Cas9 has been considered as another potential therapeutic strategy for HbF induction.256 Although the efficacy and safety of the conventional gene editing approaches at the BCL-11A erythroid specific enhancer and the globin locus have not yet been systematically compared, there are some key differences between these two genomic targets. Disruption of the regulatory elements in the HBG1 and HBG2 promoters represents a more targeted approach than eliminating BCL-11A expression in erythroid progenitors, which could potentially have deleterious consequences. Preliminary data suggest that disruption of the BCL11A erythroid enhancer in HSPCs could impair erythropoiesis.257 BCL11A erythroid-enhancer edited cells were found to have an erythroid differentiation defect in a mouse model and increased apoptosis during erythroid differentiation in ex-vivo erythroid cultures.257 Additionally, editing at the globin locus recapitulates some mutations that cause hereditary persistence of fetal haemoglobin, which is a naturally occurring benign condition. Individuals with hereditary persistence of fetal haemoglobin are not known to have any long-term adverse consequences affecting erythropoiesis. However, editing the HBG1 and HBG2 promoters presents a challenge that optimal HbF induction might require efficient editing at four separate loci (two HBG1 alleles and two HBG2 alleles) compared with editing of just two (or maybe even just one) alleles of the BCL11A locus. These challenges might not result in any clinically significant differences or adverse outcomes, but future clinical trials with correlative mechanistic studies to closely examine these different approaches are highly recommended.
There are at least two ongoing clinical trials, one with Streptococcus pyogenes Cas9 (NCT04443907) protein and another with Acidaminococcus sp Cas12a protein (NCT04853576) to create indels at the HBG1 and HBG2 promoters. Preliminary results from the clinical trial using the S pyogenes Cas9 for editing at the HBG1 and HBG2 locus suggest clinically relevant HbF induction and symptom control in the two patients who have undergone treatment so far.258 This approach could be an alternative to the BCL11A erythroid-specific enhancer editing.
Precise correction of sickle mutations: homology directed repair
Delivery of an extrachromosomal wild-type donor DNA sequence along with the nuclease allows the double-stranded break to be precisely corrected via the homologydirected repair pathway.259 This approach aims at replacing the sickle mutation with the wild-type sequence.260–264 Although it is indeed desirable to revert the genotype of the edited cells to the typical genotype rather than to induce HbF, there are some inherent challenges associated with this method. A high degree of sickle cell disease mutation correction by homology-directed-repair-based methods is challenging in HSCs given their quiescent nature.263,265 Concomitant formation of indels during the nuclease-mediated double-stranded break might disrupt the HBB reading frame, leading to a thalassaemia phenotype.263,266 The delivery of an exogenous DNA template for homology-directed repair is not only challenging but also potentially cytotoxic to the HSCs.267 Furthermore, given the sequence homology between different globin-like genes, there is a high risk of off-target editing.237 At the same time, monoallelic correction might be sufficient to convert sickle cell disease to the sickle cell trait phenotype.237 However, conversion to the wild-type sequence with subsequent production of adult haemoglobin would be the most desirable and natural resolution of sickle cell disease. Although current nucleases are fairly precise in their targeting of the DNA to introduce break points, repair efficiency with homology-directed repair remains low and off-target breaks remain a concern. Reported attempts at homology-directed repair correction have reported modest success in xenotransplantation and non-human primate experiments.261,268 Perhaps insufficient to produce clinical benefit as they currently stand, these methods are likely to continue to improve as methods to enhance homology-directed repair frequency or selection of HSCs with the editing of interest are developed.256 An ongoing clinical trial is attempting this approach in humans with no clinical results available yet (NCT04819841); this trial has been voluntarily paused by the sponsor because of a serious adverse event in the first patient that was thought to be related to the treatment.
Precise correction of sickle mutations: base editing
Base editors are the next generation of gene-editing nucleases that directly introduce base changes without double-stranded breaks, thus bypassing the low-efficiency homology-directed repair and non-homologous end joining mediated indels and off-target effects.269,270 Nuclease-induced double-stranded breaks activate the p53 damage-response pathway,271 which can result in prolonged cell-cycle arrest, compromise HSC viability, reduce repopulating potential, induce apoptosis, and sometimes lead to TP53 gene loss (which could promote malignant transformation of the edited cells).272 Single base edits disrupting the GATA1 motif within the BCL11A erythroid enhancer,273 as well as editing of the HBG1 and HBG2 promoter disrupting the BCL-11A binding motif in the HBG1 and HBG2 promoters,274 led to potent induction of HbF similar to indels produced by conventional CRISPR-Cas9 editing at those respective sites.275 A clinical trial exploring base editing of the HBG1 and HBG2 promoters is ongoing and recruiting participants (NCT05456880).
Although base editors are currently unable to execute a base change from T to A (required for correcting the sickle mutation to the typical genotype), adenine base editors have been shown to effectively convert sickle cell disease codon (GTG) to a variant haemoglobin G-Makassar (Hb G-Makassar, codon GCG).276 Haemoglobin G-Makassar is a rare, naturally occurring, benign, non-sickling variant found in people in southeast Asia. Individuals who are heterozygotes for haemoglobin G-Makassar exhibit normal red blood cell indices. In a xenotransplantation model, this mutation successfully rescued the sickling phenotype of erythroid progeny of edited human CD34+ cells derived from donors with sickle cell disease.276 This approach provides an efficient alternative to the HbF induction strategies but without introduction of double-stranded breaks.
However, given the novelty of base-editing techniques, little data exist on the guide RNA dependent and independent off-target activity of these nucleases, hence comprehensive off-target analysis needs to be done before base editing moves to clinical application.
Precise correction of sickle mutations: prime editing
Prime editors are the latest generation of gene editors, which can directly copy sequence information from a prime editing guide RNA into a target DNA locus, thus introducing precise changes.277 Prime editing has been shown to convert the sickle cell disease mutation to the typical genotype at relatively high efficiencies in HEK293 cells but requires further optimisation for high frequency targeted modification of human HSCs.277
In-vivo gene therapy
In-vivo gene therapy is currently the least mature platform for genetic interventions for haemoglobinopathies, yet it has the greatest promise, particularly as it relates to delivering more definitive targeted treatment to individuals living in LMICs or to people who are otherwise without access to highly specialised academic medical centres.
A collaboration was formed between the NIH and the Bill & Melinda Gates Foundation in 2019 to support the development of safe and effective in-vivo gene therapies for HIV and sickle cell disease that could be scalable and sustainable in parts of the world such as sub-Saharan Africa, where the burden of these conditions is high and access to therapies is limited.278 Lessons learned from ex-vivo gene therapy have informed several key considerations for the development of in-vivo approaches.279
Today, there are several notable impediments to in-vivo gene therapy for sickle cell disease. True HSCs are a remarkably small subpopulation of marrow-based progenitor cells and are typically non-dividing. Lentiviral vectors used in current ex-vivo gene therapy approaches use the lentiviral vesicular stomatitis virus G (VSVG) protein to promote fusion with CD34⁺ stem cells in the more quiescent G0 state through a low-density lipoprotein receptor in culture during the transduction process.280 In-vivo approaches will therefore need to overcome this resistance of HSCs in the resting state to the uptake of viral vectors or nanoparticles without the ability for cell selection or stimulation in culture, perhaps by employing new methods for targeting stem cells in situ. The use of myeloablation for ex-vivo gene therapy has ostensibly been needed to free up stem-cell niches in an otherwise hypercellular marrow and to reduce the absolute numbers of unmodified stem cells competing with genetically modified stem cells for these spaces.281 Ideally, in-vivo gene therapy would not require conditioning so the design approach must consider how effective disease amelioration can be accomplished if a small percentage of stem cells are altered. Gene-editing approaches that rely on gene disruption of targeted sequences typically use non-homologous end joining for DNA repair; however, other approaches for direct gene correction through homology-directed repair are cell cycle dependent, given that homology-directed repair occurs at the S/G2 transition, not the usual G0/G1 phase of HSCs.282 Driving HSCs into cell cycle, or otherwise activating HSCs, and then maintaining them for an extended period of time to achieve the correction would be needed to use homology-directed repair-mediated gene editing in vivo.
Specific parameters that have been deemed essential in manufacturing vectors for ex-vivo gene therapy might require reconsideration for in-vivo applications.283 One example is the target vector copy number and the general range for multiplicity of infection needed to achieve this vector copy number that have been optimised in current ex-vivo gene therapy approaches. If in-vivo gene therapy approaches involve viral vectors that are delivered by direct intravenous infusion, some (likely most) of the sample will be trapped in the liver; thus, it is not clear if the multiplicity of infection used to achieve current target vector copy number in the laboratory will be sufficient. For ex-vivo gene therapy, the percentage of CD34⁺ cells transduced with lentiviral vectors is another crucial feature of success. The inter-relationship of the efficiency of vector copy number and the percentage of CD34⁺ cells transduced with lentiviral vectors will need to be reconsidered for in-vivo approaches.
In-vivo gene therapy for sickle cell disease will likely require development of delivery systems currently not used in ex-vivo studies. Studies with the HDAd5/35++ vector system have yielded promising results in animal models that target HSCs, which are mobilised into the bloodstream before intravenous infusion of the vector that either delivers the γ-globin transgene or enables CRISPR-Cas9 gene editing of a γ-globin repressor binding site and targets the CD46 receptor of primitive haematopoietic cells.284
Studies must continue to advance understanding of the HSC microenvironment, to increase homing efficiency of modified HSCs and to optimise mobilisation to facilitate in-vivo editing of HSCs. The HSC-specific surface markers could differ between species and are altered in mobilised HSCs versus the marrow niche. More studies in various mouse models and larger animal species will be crucial before human studies.
Lipid nanoparticles have emerged as a new non-viral strategy for in-vivo delivery of CRISPR-Cas9 gene editing tools that can target HSCs.285 Nucleoside modification of mRNA that has been shown to increase translation combined with highly efficient transfection will be needed. Lipid nanoparticles that target specific cell types can deliver mRNA for gene editing reagents. Delivery of the components relative to the appropriate cell cycle phase of target cells will be important for different gene-editing platforms that seek to use homology-directed repair or non-homologous end joining-mediated DNA repair. Although it is expected that in-vivo gene therapy approaches will prevent the need for frequent, expensive hospital treatments, it is not clear if the overall costs of in-vivo gene therapy will be less than such inpatient hospital treatments. Manufacturing reagents and facilities for the preparation of viral vectors and nanoparticles are expensive. It is possible to achieve reduced costs over time, which should be pursued vigorously as these tools are being developed, so that ultimately broad access to gene therapy is achieved.
Considerations for low-resource settings
Even in HICs, individuals with sickle cell disease face barriers that prevent most people from realising the full benefits of scientific advances. Some of the conditions that contribute to overall disparities in outcomes in sickle cell disease also limit the use of HSCT.286 These include lack of awareness among patients and their care providers of the availability of curative options, lack of access to specialised care centres, and financial restrictions imposed variously by governments, health-care systems, and health insurance companies. Potential solutions include greater direct engagement of patients, creation and dissemination of multimedia, open-access educational materials describing current approaches, and advocacy for innovation in payment models.
Curative therapies for SCD currently available in clinical practice and research trials require highly complex services and facilities not likely to be available in many LMICs where the majority of the global sickle cell population live. However substantial and increasing numbers of HSCTs for sickle cell disease have been performed in some LMICs, including Brazil, Jordan, India, Iran, Egypt, South Africa, Ghana, and Nigeria, with an emerging programme in Tanzania. Some of these programmes, such as many of those in India, are focused primarily on treating private health-care patients, sometimes from overseas, who can pay for their own treatment. Collaborations between academic institutions, private industry and governments should be encouraged.278,287 Key considerations include prioritising technologies that allow for single infusions with durable effects, ensuring that the local health-care systems have adopted early detection of sickle cell disease and provision of evidence-informed early interventions, and finally devising financial and workflow models to democratise access to curative options.
Recommendations on cellular therapies
Although this Commission endorses population-based public health priorities (eg, newborn screening and early intervention), the ultimate aim is to provide access to definitive or curative therapies for every patient with sickle cell disease, irrespective of where they live, and it seems probable that this will involve some form of gene therapy. However, the widespread use of curative gene therapy will require planning and investment that will need to begin in the near future. We firmly recommend prioritising ongoing scientific and infrastructure investment to achieve effective, safe, and accessible cures globally by 2040.
Autologous curative approaches are maturing and will probably play an important role in the future given that most individuals with sickle cell disease seeking potentially curative options will still not have an HLA-matched sibling donor. We believe that most approaches currently in late-phase clinical trials have acceptable safety and efficacy; however, modifications in future trials or development of treatment centres must evolve to improve safety further, and consider cost containment to insure more global access.
Finally, it should be noted that this Commission does not address the use of germline stem-cell modifications as there is not currently international consensus on the safety and ethics of these procedures. However, given the global burden of sickle cell disease, in the future a case could be made to consider germline or heritable gene therapy in order to reduce the population at risk.
Section 5: training and educating health-care providers to improve evidence-based medical and nursing care
The increasing global burden of sickle cell disease is now well established. Despite the enhanced development of evidenced-based strategies for decreasing morbidity and mortality, few individuals with sickle cell disease in LMICs receive evidence-based medical and nursing care, resulting in thousands of preventable deaths every year. The gap in health-care practice for the prevention of death and lifelong morbidities, such as sickle cell disease-related strokes in children and young adults, is in part related to the mismatch between evidenced-based strategies for disease management and the clinical practice in LMICs. Sickle cell disease has evolved from a life-threatening illness of childhood with a shortened lifespan to a chronic disease where most children are expected to live to be at least 40 years old.1,2 As the life-course of children with sickle cell disease has changed, so have the patterns of the mortality and comorbidities associated with the disease.
The changing global demographics of sickle cell disease present unique challenges in developing a pipeline of knowledgeable medical and nursing providers. In Africa, where more than 70% of the children with sickle cell disease are born, few formal medical and nursing haematology programmes exist to ensure an evidence-based practice for sickle cell disease. In many locations in Africa, most children and adults with sickle cell disease receive nursing and medical care from health-care providers without any formal sickle cell disease training. In some cases, patients receive sickle cell disease care from nurses alone or nurses and medical officers (ie, new medical school graduates without any specialty training) with little training in evidence-based strategies for primary care and sickle cell disease practices. Countries with a traditionally low sickle cell disease prevalence, such as most European and Asian countries, have had a substantial influx of African and Middle Eastern people with sickle cell disease or people with sickle cell trait who are at risk of having offspring with sickle cell disease. The focus of the global panel of medical and nursing experts in section 5 is to: define gaps in implementing evidence-based guidelines for sickle cell disease management across individuals’ lifespan; identify best strategies for educating a multidisciplinary sickle cell disease health-care workforce in LMICs and HICs; and identify novel health-care delivery models for implementing evidence-based guidelines for sickle cell disease for nurses, neurologists, obstetricians, haematologists, and primary care physicians (the subspecialities that have pre-existing evidence based guidelines for management of sickle cell disease). We acknowledge that people with sickle cell disease, their families, and allied health-care professionals, all need education about sickle cell disease to provide comprehensive care. For this Commission, we are focusing on the education and training of health-care providers.
Education on the inheritance of sickle cell disease and non-directive genetic counselling
Medical and nursing students are typically not exposed to a formal curriculum on how to provide non-directive genetic counselling for sickle cell disease. We are unaware of any actively implemented medical school and nursing school curriculum to educate students about the inheritance pattern of sickle cell disease that includes non-directive genetic counselling learning objectives. The absence of a universal curriculum that includes genetic counselling is a missed opportunity to ensure appropriate reproductive counselling for individuals at risk of having a child with sickle cell disease. Furthermore, the lack of genetic counsellors combined with the prevalence of sickle cell trait (particularly in people of African and Asian descent, ranging from one in four individuals in West Africa to one in 12 African Americans living in the USA, and one in 50 in some populations in India) highlights the need for the development of a specific curriculum to both expand the knowledge of non-directive genetic counselling techniques and increase the number of providers certified to deliver genetic counselling. The Saudi Ministry of Health has developed training modules for genetic counselling materials used for training health-care professionals in urban and rural primary care centres. This training must have a common core content or syllabus, taking into account the religious and cultural sensitivities unique to the populations and regions where the counselling is being employed.
Existing sickle cell disease education curricula
Medical students
Most medical schools provide one to two lectures on sickle cell disease or haemoglobinopathies. We are aware of a few current sickle cell disease curricula with learning objectives adjusted for educational training (pregraduate vs postgraduate) that would assess a minimum core competency required for acute and chronic management of sickle cell disease. As an exception, faculty in Senegal and the University of Paris developed French curricula focused on these topics (panel 2).
Panel 2: Curriculum overview for training primary care providers about sickle cell disease*.
Primary care provider management
Genetics and pathophysiology of the disease; psychosocial factors; poverty and other socioeconomic factors; how primary care providers fit into the care model
The sickle cell disease perspective—individuals with sickle cell disease or caregivers
Vaccines and screening (sickle cell disease related: eye screening; heart, lung, and kidney screening; neurological screening; and sickle cell disease-specific vaccines [NHLBI guidelines141] and non-sickle cell disease related: USPSTF guidelines; pheoxymethylpenicillin [NHLBI guidelines]; depression, women’s health; hypertension; diabetes; vitamin D and bone health; sleep; acute issues)
Resources for the primary care provider: guidelines (2014 NHLBI141, 2019 ASH), checklists
Patient resources: the patient portal, apps
Medical home model and the role of the nurse or care coordinator in the coordination of care throughout the procedure
Acute and chronic pain
Their perspectives of their disease; the positives of the care they receive; the challenges of their care (eg, wait times in the emergency room and providers in care); what they would hope their primary care physicians would offer; what is most important to them as individuals with sickle cell disease and their caregivers
Acute complications
Acute pain episodes and chronic pain, including avascular necrosis and musculoskeletal issues, includes pathophysiology and treatment and the NHLBI and ASH guidelines.288
Chronic complications
Acute chest syndrome, fever and infection, priapism, splenic sequestration, abdominal pain, vision changes, venous thromboembolism (NHLBI and ASH guidelines)
Wellness
Care coordination
Preventing life-threatening infections
Avoiding sickle cell pain triggers
Managing stress
Staying active, having a healthy diet, and taking care of teeth
Safe sex, tobacco use, alcohol, and drug use
Disease-modifying therapies
Medications (hydroxyurea [hydroxycarbamide], L-glutamine, voxelotor, crizanlizumab) and chronic transfusion therapy; recognising indications and exclusions, complications, efficacies, dosing, and side effects or toxicities
Transfusion support, including red blood cell transfusions, requires extra matching (specifically at the Rh and Kell loci; ASH guidelines);290
How to manage hydroxyurea as the primary care provider (NHLBI guidelines)
Curative therapies
How to talk to patients about curative therapies
HSCT (ASH guidelines),291 post-HSCT knowledge for the primary care provider
Developments in curative treatments including clinical trials and gene therapy
ASH=American Society of Hematology. NHLBI=National Heart, Lung, and Blood Institute. USPSTF=United States Preventive Services Taskforce.
These are didactic and case-based sessions in person or done remotely through educational platforms; the sessions are primarily geared towards how a primary-care provider manages sickle cell disease in an outpatient setting.
A major gap in developing curricula for sickle cell disease education is the absence of a minimum set of teaching objectives or content knowledge required to ensure a uniform knowledge base from year to year. We identified one systematic guideline for developing a minimum knowledge base for primary care providers to become proficient in the acute and chronic management of sickle cell disease in primary care settings in Africa.292
In Lebanon, at the American University of Beirut, the first-year medical students have one lecture on sickle cell disease as part of the haematology course and all third year medical students attend the comprehensive sickle cell clinic once a week for 8 weeks. This rotation is complemented with reading materials and now includes genetic counselling for all people with haemoglobinopathies. A newly established National Committee on sickle cell disease will evaluate and recommend introducing a module on sickle cell disease to all medical school curricula in Lebanon.
In Nigeria, where the greatest number of children and adults with sickle cell disease live, medical schools offer on average 3 h of lectures on sickle cell disease and thalassaemia; however, in the country where approximately 150 000 babies with sickle cell disease are born annually, there has been no systematic approach to assess core competency in sickle cell disease care at the postgraduate, residency programme level, or in nursing schools. At the federal level, a national guideline for sickle cell disease management was developed by an expert committee and published by the Nigeria’s Minister of Health on Nov 28, 2014.293 Effective strategies are required to increase the dissemination and adoption of the national guideline into the medical and nursing curricula. A task shifting approach based on the national guideline for sickle cell disease management should be recommended to train the Community Health Extension Workers, who are responsible for the vast majority of primary care provision in all rural and underserved communities of Nigeria.
Nursing students
Comprehensive care for people with sickle cell disease relies on the chronic care model,294 and nurses are crucial members of these teams. Nursing education programmes emphasise the role of nurses as health educators and care coordinators. Prelicensure nursing education includes generalist education in the genetics of sickle cell disease. Education about the genetics of sickle cell disease in nursing school could allow nurses to provide genetic counselling to parents of children with sickle cell disease and preconception counselling to adolescents and young adults with sickle cell disease. Nurses also manage chronic complications of pain and the comorbidities of asthma by educating families on symptom management.
A precious human resource is the small cadre of nurse educators and scientists committed to the next generation of sickle cell disease nurse providers, educators, and investigators. We believe that foundations and government agencies would drastically impact the care of individuals, particularly maternal and child health, if more resources are placed in creating regional sickle cell disease nursing centres of excellence or, at the very least, providing a nurse-focused sickle cell disease curriculum.
Internal medicine and paediatric trainees
The absence of a universally accepted curriculum to provide expected sickle cell disease primary care management, despite formal evidence-based guidelines specifically developed for primary care providers,141 is a missed opportunity for building a knowledgeable sickle cell disease health-care workforce. An easy and efficient approach is to develop a consensus health-care curriculum with learning objectives for nursing and primary care providers.
Primary care providers are an under-recognised yet crucial part of the medical care for people with sickle cell disease. Individuals with sickle cell disease who have primary care providers have lower hospitalisations, readmissions, and better hydroxyurea adherence than people who do not have primary care providers.295–297 Globally, sickle cell disease care largely relies on access to primary care providers and primary care nurses. Several health-care issues typically screened and treated in the primary care setting can substantially impact people with sickle cell disease. Mental health, women’s health (including contraception), hypertension, diabetes, cancer, smoking, asthma, and vaccinations are among these topics.
A global curriculum that educates primary care providers and primary care nurses about sickle cell disease and the nuances of caring for this population is crucial. Due to the missed opportunity for educating medical and nursing students and primary care doctors mentioned previously, curriculums that target post-residency primary care providers are another potential opportunity to bridge the gap. Two of the authors of this section, a haematologist and a primary care provider (MRD and RMC), developed a curriculum to help primary care providers overcome established barriers, learn evidence-based guidelines for sickle cell disease care, and embrace the care of children and adults with sickle cell disease. The curriculum, in English, takes about 24–32 h on average and includes approximately ten sessions that address the specific components of sickle cell disease care (panel 2).298 The curriculum has evidence-based methods to overcome barriers to care and could be used to educate primary care providers across the USA. The team developed a booklet of the 2014 US National Heart, Lung, and Blood Institute guidelines, adapted for the education of individuals with sickle cell disease, their families, and primary care providers to augment this curriculum.299
Other initiatives in this area include developing a core curriculum for practitioners charged with the management of children and adults with sickle cell disease. An online course300 in French addressed the management of children and adults with sickle cell disease. The course, developed in 2013, includes topics such as diagnosis, treatment, and psychological and social aspects (70 h) and offers validated training in therapeutic education (40 h). Approximately 200 health-care providers from France, Africa, and the French West Indies have already completed the course. The University of Paris Cité acknowledges the graduates.
Although in many situations, evidence-based treatments and appropriate resources need to be established before education can be fully effective, there are also many areas where education of health-care professionals can make a very big difference. Children and adults with sickle cell disease have one of the highest incidence rates of acute and chronic neurological morbidities of any monogenic disease. Without the establishment of primary stroke prevention programmes with transcranial Doppler screening and regular blood transfusions for atypical velocities, approximately 11% of children with sickle cell anaemia will develop overt strokes.301 After introducing stroke prevention programmes in HICs, the stroke incidence rate dropped by at least 80% in a population-based study302 and decreased by 10 times in a single-centre study.303 Since 2006, and subsequently in 2011 and 2019, the American Heart Association has published evidence-based guidelines for stroke prevention and management that include sickle cell disease.301,304–306 Additionally, since 2020, the ASH has published evidence-based guidelines for the treatment and prevention of neurological morbidity in sickle cell disease.152 Despite the presence of these guidelines and the high prevalence of stroke-related morbidity and mortality in sickle cell disease, to our knowledge, a formal curriculum with learning objectives has not been established for neurologists-in-training regarding acute and chronic neurological complications of sickle cell disease across the lifespan, which is a crucial need. Further, neurologists are not always readily available. Thus, all health-care providers who care for children or adults with sickle cell disease need to be educated about the neurological complications of sickle cell disease, how to detect them by history and neurological examination, and potential treatments. A northern Nigeria research and clinical care team worked to educate nurses and non-neurologist physicians on how to perform a standardised neurological examination for stroke detection. Ultimately, this group formed stroke prevention teams.307 A programme to train nurses to perform transcranial Doppler studies has also recently been established in Tanzania.308 This low-cost model to improve prevention and treatment of strokes should be possible to implement in other LMICs. Such a strategy is required because of the scarcity of haematologists, neurologists, and radiologists to establish a systematic approach to primary stroke prevention and provide acute and chronic medical management.
Obstetric–gynaecological doctors
Informed reproductive decision making is crucial in family planning of a monogenic disease. The American College of Obstetricians and Gynaecologists recommends carrier screening for haemoglobinopathies to prepare women for the chance of delivering a child with sickle cell trait or disease.309 However, past studies show that some obstetric–gynaecological health-care providers have used sickle-dex tests for screening instead of haemoglobin analysis tests.310 As a result, other traits such as HbC and thalassaemia are missed. Prenatal counselling and follow-up represent an essential part of care and must be included in obstetric–gynaecological training. We are unaware of any formal learning objectives for obstetric–gynaecological doctors. However, women with sickle cell disease have statistically significantly increased mortality and morbidity during pregnancy when compared with women without sickle cell disease. In a large administrative dataset in the USA of 4262 births to women with sickle cell disease, the maternal mortality rate was at least 10 times greater among women with sickle cell disease (1·6 per 1000 deliveries) compared with women without sickle cell disease (0·1 per 1000 deliveries).311 In LMICs, where most children with sickle cell disease are born, the maternal mortality rate is between 7000 and 12 000 per 100 000 livebirths312–314 in comparison with the maternal mortality rate of 17 and 10 per 100 000 livebirths in the USA315 and the UK,316 respectively.
Even with the high rate of maternal–fetal mortality in women with sickle cell disease, few formal health-care delivery strategies have been developed to address this issue. At Korle Bu Teaching Hospital in Accra (Ghana), the baseline maternal mortality rate was 10 000 per 100 000 livebirths in women with sickle cell disease, compared with 1000 per 100 000 livebirths in women without sickle cell disease.317 After implementing a multidisciplinary team, the maternal mortality rate decreased from 10 791 (15 of 139) per 100 000 live births to 1176 (1 of 85) per 100 000 live births (risk ratio 0·103; 95% CI 0·014–0·779), giving an 89·1% risk reduction (p= 0·007) that was sustained 3 years after the intervention.318 The primary interventions were all low budget, including adding standard clinical care management strategies; routine assessment of haemoglobin oxygen saturation measurements at baseline and during admissions, with clinical action if there was more than a 3% drop from baseline; standardising nursing care guidelines; and ongoing quality control assessment of care with initially weekly meetings. These results clearly show that improvement in obstetrician and nursing care knowledge could result in a drastic decrease in maternal mortality rates for women with sickle cell disease. The comprehensive sickle cell disease obstetric care programme at the Korle Bu Teaching Hospital in Ghana published its management protocol319 in 2021, which can serve as an important guideline for the training of midwives, medical students, doctors, and maternal–fetal medicine fellows across Ghana and in other LMICs.
Despite the high incidence of sickle cell disease related morbidity during pregnancy, we are unaware of formal curricula to educate obstetric doctors and nurses about the prevention and treatment of complications of sickle cell disease during pregnancy outside of the Ghana experience. Two evidenced-based guidelines for the management of pregnancy in women with sickle cell disease, from the USA and the UK, could be used to establish the expected learning objective.320–322
Health-care professionals
There are some examples of the development of curricula specifically focusing on sickle cell disease, with clear learning objectives for health-care providers. One author (MRA) was actively involved with the Thalassemia International Federation in developing an online course on sickle cell disease for health-care providers. The course is available, free of charge, since April, 2021, and has modules on different aspects of sickle cell disease, including clinical complications (infections, pain, neurological complications, pregnancy, etc), treatment, and care organisation. The European Hematology Association has also developed a haematology curriculum that includes a specific and detailed description of the knowledge necessary to care for people with sickle cell disease, mostly from a European perspective. We identified one institutional collaboration between those living in HICs and those living in LMICs: the curricula by the University of Paris Cité and health-care providers in Senegal.323
Additionally, the West African Genetic Medicine Center (a World Bank Center of Excellence located at the University of Ghana) has started a genetic counselling MSc programme to train genetic counsellors for the region. Trainees are drawn from wide biomedical and clinical backgrounds including physicians, nurses, biomedical scientists, and psychologists; the programme offers appropriate detail related to inherited blood disorders, incorporating practical counselling courses focused on sickle cell disease.
It is particularly important that training programmes in each country or region reflect the availability of resources. For example, the use of blood transfusions to treat cerebrovascular disease is not possible in most LMICs and evidence-based clinical practice from Europe and the USA would not be appropriate. Training programmes need to be developed with the best available evidence adapted to local conditions. A good example of this is the way in which the Jamaica Sickle Cell Unit Clinical Care Guidelines were developed by adapting North American and UK guidelines to local needs; these guidelines have subsequently provided a basis for training throughout the English-speaking Caribbean.
Overall, we believe the absence of a core set of expected sickle cell disease competencies for haematology professionals globally substantially impairs implementing and disseminating evidenced-based practice for sickle cell disease. Further, the absence of sickle cell disease learning objectives and evaluation during annual competency service examinations explicitly lowers expectations of haematology training programme directors and trainees about the importance of sickle cell disease in their current and future clinical practice.
Improving the pipeline of investigators to advance discoveries in sickle cell disease
Although precise figures are not available, there is little research funding reserved specifically to investigate sickle cell disease, particularly when compared with many other conditions, such as cystic fibrosis and haemophilia. This scarcity of funding leads to shortages of trained investigators in sickle cell disease and is a disincentive for new researchers to become interested in the condition. Private foundations have targeted grant funding for haematologists and multidisciplinary research teams focused on sickle cell disease related problems. In the USA, the Doris Duke Charitable Foundation offered research funding opportunities specifically for sickle cell disease for 7 years (2009–15), totalling $17 million or approximately $2·4 million per year. In contrast, the US Cystic Fibrosis Foundation spends more than $100 million per year. A total of 28 new grants were provided, $450 000 for direct costs over 3 years, and seven grants were renewed. Subsequently, on the basis of this pilot funding, the Doris Duke awardees received $55·6 million from the NIH.324 The ASH also supports junior investigators with 2–3 year awards for research and training activities, and trainees focused on sickle cell disease have been among the recipients.325 This relative scarcity of funding for trainees in sickle cell disease compared to other diseases in the USA reflects the situation throughout the world, with very few funding schemes focused specifically on sickle cell disease. To our knowledge, there is no current foundation devoted to specific funding of fellows or new faculty focused on a research career in sickle cell disease anywhere in the world, in contrast to many other much rarer diseases.
Evidence-based medicine to improve and expand the pipeline of medical and nursing care in sickle cell disease
Evidence-based medicine is the purposeful use of current best evidence in making decisions about the care of individual patients. Evidence-based medicine integrates provider clinical expertise with the best available external clinical evidence from systematic research and patient preferences. In 2020, the ASH produced evidence-based guidelines in sickle cell disease for the management of heart, lung, neurological, and kidney disease;152,289 the use of blood transfusion;290 and acute and chronic pain management.288 The ASH guidelines serve as a starting point for implementing and adopting regional-specific evidenced-based guidelines for sickle cell disease. However, bridging the gap between optimal management and bedside patient management requires a paradigm shift in training physicians and nurses in low-to-middle-income and high-income settings, to improve access to treatment. Further, the value of such guidelines is undermined by the scarcity of high-grade evidence to support them, particularly when the guidelines refer to LMICs.
Opportunities for training in sickle cell disease
There is a shortage of haematologists and medical providers trained in sickle cell disease. Rural USA lacks providers for a large portion of adults with sickle cell disease. In sub-Saharan Africa, where 75% of all children with sickle cell disease are born, the demand for sickle cell disease health-care providers far exceeds available personnel. As a result, some organisations have invested in training programmes to increase the number of fellowship-trained haematologists, primary care providers, and advanced practice providers to deliver evidence-based care to children and adults with sickle cell disease.
In the USA, the Health Resources and Services Administration funded five regional Sickle Cell Disease Treatment and Demonstration Program awards to support the education of providers to learn more about evidence-based care of people with sickle cell disease. A learning collaborative is an initiative in which peers come together to study and apply quality improvement methods to a focused topic area. Learning collaboratives have also been funded to support communities learning about sickle cell disease. In the Treatment and Demonstration Program awards, supported by the Health Resources and Services Administration, the regions also formed learning collaboratives across the participating states. The Health Resources and Services Administration tasked awardees to work on quality improvement activities to increase the uptake of evidence-based guidelines, such as transcranial Doppler screening of children aged 2–16 years with sickle cell anaemia, prescribing of hydroxyurea, vaccinating, and the use of child-to-adult sickle cell disease care transition planning tools. Another learning collaborative focused on sickle cell disease care has been created by ASH in 2021. Several organisations have invested in learning modules to create sickle cell disease education programmes available to individuals living in LMICs (table 6).
Table 6:
Sample of available training modules for sickle cell disease education programmes through various organisations
| Title | Objectives | Target audience | Language | |
|---|---|---|---|---|
|
| ||||
| American Society of Hematology | Benign Hematology Curriculum: Sickle Cell Disease | Pathophysiology and clinical management of people with sickle cell disease; increase knowledge and awareness; 13 modules | Trainees in benign haematology | English |
| European Union’s Horizon 2020 Marie Skłodowska-Curie programme for education and training | ARISE: African Research and Innovative Initiative for Sickle Cell Education: Improving Research Capacity for Service Improvement | Sickle cell disease prevalence; genotypes and phenotypes; existing best practices for newborn baby screening and early diagnosis; engagement with patients, families, and policy makers to determine barriers to newborn baby screening; lab diagnosis and quality assurance systems for population screening; treatment protocols for the management of common sickle cell disease complications and transition from paediatric to adult care, health promotion strategies, and nutrition | Doctors, nurses, laboratory staff, and other health-care professionals caring for people with sickle cell disease | English |
| University of Paris | Diplôme Universitaire Enseignement on line sur la Drépanocytose | Diagnosis, treatment, and psychological and social aspects of paediatric and adult patients with sickle cell disease (70 h); offers validated training in therapeutic education (40 h) | Doctors, nurses, physiotherapists, and psychologists | French |
| Senegal and University of Paris | Diplôme Universitaire International Franco-Africain | Pathophysiology and clinical management of people with sickle cell disease with a special focus on the facilities available in Africa | Doctors, nurses, physiotherapists, and psychologists | French |
| Thalassemia International Foundation | SCD e-Course for Healthcare Professionals | Clinical complications (infections, pain, neurological complications, pregnancy, etc); treatment; care organisation | All health-care professionals | English |
| Saudi Ministry of Health | Sickle Cell Disease in KSA | Genetic counselling and modules on clinical care to be used in primary care and remote centres | Doctors, nurses, and genetic counsellors; particularly primary care settings | English |
Although opportunities for training in sickle cell disease are few in the USA, the situation is probably worse in most other countries, particularly in Africa and India where the disease is most prevalent. There is also a substantial problem in many European countries where there has been a marked increase in the numbers of people with sickle cell disease, without the means to train the medical and other staff to manage the condition.
The gap in dissemination and implementation of research for evidence-based practice
After curricula for practitioners in sickle cell disease are established, the dissemination and implementation ofthe objectives are required. In the past 10 years, implementation science has evolved as a much-needed set of tools to increase the uptake of evidence-based clinical guide lines.326–328 The same approaches of identifying the evidence-based practice or learning objectives, assessing the organisation’s culture or setting, and working through strategies to deliver the education or training are relevant to implementing curricula.329,330
In implementation research, the RE-AIM is a framework commonly used in public health to reduce the gap between evidence-based practice and actual activities.331 The reach and effectiveness of the intervention, adoption, and implementation (individual and organisational), and maintenance of the intervention is assessed after planning and intervening. A modification of the RE-AIM framework332 published in 2020 purposely brought the concept of equity into the maintenance to enhance sustainability. Given the inequities of the population of patients, their daily settings, and the settings of their medical care, this adapted version of RE-AIM332 could be considered in future dissemination and implementation of training curricula.
Recommendations on training and education
Across HICs and LMICs, we could not identify an ideal model for training and educating physicians, nurses, and other health-care professionals to look after people with sickle cell disease. There is an urgent humanitarian need to educate health-care professionals and the general public about sickle cell disease across the world, to prevent death and decrease lifelong sequelae of sickle cell disease, particularly in Africa, where 70% of newborn babies with sickle cell disease are born. However, all global regions lack an adequate approach for preparing the next cadre of leaders focused on sickle cell disease medical and nursing care and research. Small investments in training and education can lead to marked improvement in health outcomes, as shown by exemplar programmes, such as those to prevent the deaths of pregnant women with sickle cell disease in Ghana and to diagnose strokes in young children with sickle cell disease living in Nigeria and Tanzania. These programmes should be highlighted to allow replication across the settings most in need of investment (low-to-middle resource settings and settings with historically low incidence of sickle cell disease) to facilitate improvements in care.
A comprehensive partnership between private foundations, professional medical and nursing societies, and government agencies is required to tilt the global scale toward medical justice for children and adults with sickle cell disease.
Future challenges and opportunities
There have been many warnings in the past about the neglect of sickle cell disease and frequent calls to reduce its global burden and to better manage and treat people with this disease. So far, these calls, including those from WHO, have had little effect. Given growing inequalities within most countries and internationally, and substantial financial pressures to improve the health of a growing and ageing population, it will require substantial commitments to achieve the recommendations outlined in this Commission.
The number of people with sickle cell disease globally is expected to increase, bringing new challenges—eg, in terms of multimorbidity in HICs and developing adequate health-care strategies for the numbers of patients in LMICs considerably larger than those currently managed in HICs.
The development of innovative POCT devices, new drugs, and promising therapies in the past 5 years all offer great opportunities to scale up the availability and accessibility of existing tools in areas of high prevalence and to pursue further developments through ethical and equal collaborations. As illustrated by a couple of multibillion dollar acquisitions of drug companies interested in sickle cell disease treatments, there is growing awareness about sickle cell disease and the development of a cure for sickle cell disease currently has a high profile. The main challenge going forward will be to maximise the effect of investments for the benefit of patients and society overall.
To guide future progress, this Commission makes 12 key necessary and realistic recommendations for reducing the global burden of sickle cell disease (panel 3), which are further expanded in table 7.
Panel 3: Key recommendations to improve the outcomes of people with sickle cell disease.
Enable routine collection of comparable epidemiological data across all countries by 2025, requiring action by governments, national health authorities, and research funding bodies
Make national governments accountable through monitoring of public health intervention implementation for sickle cell disease care and progress, requiring action by governments, health authorities, and WHO
Ensure that all babies worldwide can be tested by 2025 to prevent long-term complications, requiring action by governments
Inform people affected by sickle cell disease about reproductive risks and choices, tailored to cultures and religions, requiring action by governments, national health authorities, public health organisations, and patient support organisations
Provide access to minimum health care specific to sickle cell disease to all people affected no matter where they live, requiring action by governments, and national health authorities.
Make hydroxyurea (hydroxycarbamide) accessible and affordable (<US$0η1 per 500mg capsule) to all people with sickle cell disease by 2030, requiring action by governments and pharmaceutical companies
Improve blood transfusion supply for people with sickle cell disease and safety (including guidelines for proper use and improvement of haemovigilance) by 2030, requiring action by governments, health authorities, blood transfusion services, and national and regional haematology societies
Accelerate the development of effective and affordable curative therapies, including prioritising scientific and infrastructure investment to achieve safe and accessible cures globally by 2040, requiring action by governments, pharmaceutic companies, and research funding bodies
Balance participation of low-income and middle-income countries versus high-income countries in clinical trials for new therapies by 2030, requiring action by drug licensing bodies, pharmaceutical companies, research funding bodies, and researchers
Educate health-care professionals and the general public about sickle cell disease and its management, requiring action from governments, health authorities, public health organisations, universities, and patient support organisations
Prioritise sickle cell disease research through dedicated funding, requiring action by governments and research funding bodies
Halve current estimates of the global burden of sickle cell disease by 2050, requiring action by WHO, governments, health authorities, public health organisations, pharmaceutical companies, research funding bodies, universities, and researchers
Table 7:
Overall Commission recommendations for each concern related to sickle cell disease
| Setting | Short-term solutions (1–3 years) | Long-term solutions (more than 3 years) | |
|---|---|---|---|
|
| |||
| Section 1: epidemiology | |||
| Limited availability of standardised epidemiological data | Worldwide | Develop standards for minimum epidemiological data needed to monitor changes and progress; build on existing infrastructures, software, and networks used for other diseases (eg, immunisation programmes); enable data sharing across datasets and registries | Support the implementation of robust, standardised electronic data collection systems, particularly in LMICs; monitor progress to reduce the burden of sickle cell disease through an online interactive portal; inform policies through reliable, regularly updated global, regional, and national burden estimates |
| Neglected large numbers of patients with sickle cell disease | High-prevalence countries | Develop tailored public health strategies, based on a better understanding of the natural history of sickle cell disease | To show the societal and economic benefits of implementing sustainable public health policies for sickle cell disease |
| Section 2: screening and prevention | |||
| Scarcity of newborn screening policies and guidelines | High-prevalence countries | Develop policies on newborn screening with budgetary allocation for implementation | Establish universal newborn screening as a public health programme with adequate provision for follow-up of babies affected |
| Absence of stable consistent and sufficient funding | High-prevalence countries | Allocate specific budgets for newborn screening | Develop a global funding mechanism for newborn screening and other sickle cell disease prevention and management interventions |
| Scarcity of organised national newborn screening programmes | LMICs | Provide tailored preconceptual programmes and genetic counselling; prioritise the implementation of newborn screening programmes | Establish national preconceptual screening programmes as part of a standard of care; establish newborn screening programmes in birthing hospitals; develop Centres of Excellence with multidisciplinary providers |
| Section 3: Disease management—established and emerging treatments | |||
| Need for comprehensive national sickle cell disease programmes | Worldwide | Write guidelines that are geographically and socioeconomically appropriate | Arrange low-cost distribution of vaccines, antibiotics, analgesics, and hydroxyurea (hydroxycarbamide) |
| Poor management of VOCs and other acute events | Worldwide | Set up facilities with transcranial Doppler screening; create local protocols for managing pain; provide adequate opioid analgesics; encourage incentive spirometry; treat fever with antibiotics and antimalarials as appropriate | Increase awareness of psychosocial issues; create practical tools for evaluation and improvement of treatment adherence |
| Poor access to safe blood transfusions | LMICs | Write guidelines for proper use of blood transfusions, limiting paid donors; screen serum for red blood cell alloantibodies; crossmatch blood before releasing units | Establish a national transfusion programme; develop tools for recording historical red blood cell alloantibodies; improve haemovigilance and blood safety |
| Poor access to safe blood transfusions | Worldwide | Increase blood supply for patients with sickle cell disease patients | ·· |
| Poor access to affordable hydroxyurea | LMICs | Register hydroxyurea nationally for the treatment of sickle cell disease; write treatment guidelines adapted to the socioeconomic setting | Improve clinical infrastructure for drug distribution and treatment; increase accessibility to drug supply and avoid stock-outs; improve affordability to less than US$1 per week |
| Section 4: management—cellular therapies | |||
| Little awareness of curative therapies | Worldwide | Enhance education and advocacy of governmental agencies | Create educational materials that are multimedia and in appropriate languages |
| Little access to allogeneic HSCT | Worldwide | Secure essential medications for HSCT; improve coverage in national plans or through insurance payors | Increase accessibility to drug supply; improve affordability |
| Little access to allogeneic HSCT | HICs | Provide safe, universal HLA typing of all children with sickle cell disease, covered by insurance where applicable; enhance blood banking capabilities for safe collection and storage of components, especially platelets; establish radiotherapy services to deliver irradiation for conditioning (total body) and to irradiate blood products | ·· |
| Little access to allogeneic HSCT | LMICs | Develop capacity for transplantation and access to clinical trials; develop patient selection criteria on the basis of local considerations | Identify additional centres with sufficient infrastructure to perform safe and effective allogeneic HSCT |
| Little access to autologous HSCT for gene therapy | HICs | Support late-stage clinical trials that will lead to registration of commercial gene therapy; encourage additional early phase studies of innovative approaches that could be safer, more effective, and less costly | ·· |
| Little access to autologous HSCT for gene therapy | LMICs | ·· | To identify additional centres for autologous HSCT |
| Little investment in development of in-vivo gene therapy | Worldwide | Encourage and support ongoing research and innovation; prioritise investments in systems that support in-vivo delivery of genetic therapies | Explore technology solutions, such as closed systems for point of care manufacturing, that will reduce dependence on specialised centres; support ongoing studies on non-genotoxic preparative regimen to eliminate the need for radiation or chemotherapy |
| Section 5: training and educating health-care providers to improve evidence-based medicine and care | |||
| Limited global curriculum for training and certification of sickle cell disease care providers | Worldwide | Develop a global curriculum and adapt it to local circumstances; facilitate advocacy for inclusion in health-care worker training by local practitioners; create regional sickle cell disease nursing Centres of Excellence or, at the very least, provide a nurse-focused sickle cell disease curriculum; develop educational resources for health-care professionals, such as social workers, genetic counsellors, physiotherapists, occupational therapists, speech and language therapists, and psychologists; target curriculum development for primary care physicians, obstetricians, and midwives for improved identification and counselling of carriers and couples | Foster an international multidisciplinary educational meeting (IT solution) for sickle cell disease clinicians; update curriculum every 2 years |
| Absence of centres offering training on sickle cell disease | Worldwide (lead by centres of excellence; support provided to groups of similar providers; should be regionalised and aligned to care settings | Capitalise on expansion of online training and telemedicine to standardise and disseminate training | Set up secondary and tertiary consultations to facilitate evidence-based care and support local practitioners; develop sanctioned educational and clinical support partnerships: Centres of Excellence linked with geographical zones of LMICs (eg, quarterly succinct online meetings with short updates, new learning, or cases with learning moderated by Centres of Excellence) |
| Scarcity of funding | Worldwide | Foundation funding for fellows or new faculty to focus a career of research in SCD | ·· |
HICs=high-income countries. HSCT=haematopoietic stem-cell transplantation. LMICs=low-income and middle-income countries. VOC=vaso-occlusive crisis.
Conclusions
Overall, improvements for people with sickle cell disease worldwide have been very limited over the past few decades. Until recently, hydroxyurea was the only available drug for the treatment of sickle cell disease, and most individuals with sickle cell disease in high prevalence areas remain undiagnosed or untreated. Furthermore, throughout their lives, most patients do not receive adequate health care and are victims of racism and stigmatisation.
The beginning of the journey would seem to be starting newborn screening programmes in every country with large numbers of people with sickle cell disease; although the technology to do this is increasingly available, with POCTs and widespread use of mobile phones, it is still a huge undertaking to establish even the most basic programme in most African countries. The end of the journey is also imaginable, in which every patient in the world is offered curative treatment—possibly through the emerging field of in-vivo gene editing, although this requires major technical, logistical, and economic advances, and it would take substantial commitment to make this a reality within the next three decades.
Education has often been shown to be a driver of societal, economic, and health improvements. Awareness and understanding of the disease within the general population, training, and knowledge of sickle cell disease within health-care professionals, and familiarity with the challenges faced by public health services among decision makers would all contribute to improved life and quality of care for people with sickle cell disease and their families.
Although there remains many unknowns concerning the natural history of the disease and uncertainties in relation to the exact burden of sickle cell disease, a series of inexpensive and effective tools to identify people with sickle cell disease, to reduce early mortality, and to prevent severe chronic complications are readily available to be rolled out at scale. There is strong evidence for their benefits and cost-effectiveness, and strategies building on existing infrastructure to maximise investments and effects.
This Commission brings together the enthusiasm and ambitious aspirations of a diverse and multidisciplinary group of international experts in sickle cell disease—complemented by input from advisory board members from academia, advocacy groups, policy institutions, and funders—to seize current opportunities to make the world a better place for all people with sickle cell disease by defining priorities and setting short-term, mid-term, and long-term recommendations to guide public health policies and investments. Our ambition is to monitor and document progress towards these recommendations and to evaluate achievements and shortfalls within the next decade, while curative treatments continue to be developed in parallel.
Supplementary Material
Search strategy and selection criteria.
Separate literature searches were performed by the authors of the different sections of this Commission. References were identified through searches of PubMed from its inception to January 31, 2023, with the search terms “sickle”, “sickle cell disease”, “sickle cell anaemia”, “HbSC disease” or “HbS/beta thalassaemia” in combination with particular procedures, treatments, and complications including “epidemiology”, “screening”, “diagnosis”, “treatment”, “vaccination”, “penicillin prophylaxis”, “hydroxyurea”, “hydroxycarbamide”, “blood transfusion”, “acute pain”, “acute chest syndrome”, “stroke”, “haematopoietic stem cell transplantation”, “bone marrow transplantation”, “gene therapy”, “gene editing”, “training”, and “education”. ClinicalTrials.gov was searched for evidence of unreported and ongoing clinical trials from its inception to Jan 11, 2023. Articles and abstracts were also identified through searches of the authors’ own files. The final reference list was generated on the basis of originality and relevance to the broad scope of this Commission. References were also selected for their importance and ease of access.
Acknowledgments
Akshay Sharma acknowledges support from the American Society of Hematology in the form of the Scholar Award and from the Doris Duke Charitable Foundation. We thank the members of the Advisory Group who provided insights and feedback on the report: Robert Brodsky (American Society of Hematology); Antonio Almeida (European Hematology Association); Karin Butler (The National Institute for Health and Care Excellence); Lwimba Kasongo (Global Alliance of Sickle Cell Disease Organizations); Pascale Jeanot Tuseo (Action Solidarité Madagascar); Matshidiso Moeti (WHO Regional Office for Africa); Nicola Skyers (Jamaican Ministry of Health & Wellness); José Valverde (DG Sante, European Commission); Sindy Escobar Alvarez (Doris Duke Charitable Foundation); Béatrice Garrette (Fondation Pierre Fabre); Christophe Przybylski (Fondation Pierre Fabre).
Footnotes
Declaration of interests
MRA has received grants or contracts with Novartis, Pfizer, Global Blood Therapeutics, and Forma Therapeutics; and participated in Data Safety Monitoring Boards or Advisory Boards for Vertex, GBT, Forma, Novartis, and Aggios. BA has participated in Data Safety Monitoring Boards or Advisory Boards for Agios, Aruvant, bluebird bio, CRISPR, Accordant (CVS), Emmaus, Forma Therapeutics, Fulcrum, Global Blood Therapeutics, Hemanext, Novartis, NovoNordisk, and Vertex. RF has received grants from the European Horizon 2020 and Horizon Europe schemes and a contract with Global Blood Therapeutics; received consulting fees from Novartis and Global Blood Therapeutics; received honoraria for a Physicians’ Education Resource and from Global Blood Therapeutics; participated in Data Safety Monitoring Boards or Advisory Boards for Vertex, Addmedica and Novonordisk; and a leadership or fiduciary role as a member of the Steering Committee and Coordinator of the Red Cell Disorder Working Group of the Italian Association of Pediatric Hematology Oncology. NC has received a research grant for Novartis Precision Medicine. MdM has received honoraria for presentations by Addmedica and Novartis and support from Addmedica for attending a conference; and participated in a Data Safety Monitoring Board or Advisory Board for Addmedica, Vertex, and Novartis. JE has received grants from the European Horizon 2020 scheme and participated in the Data Safety Monitoring Board or Advisory Board of the Fondation Pierre Fabre. EE has received consulting fees from bluebird bio. ALG has an honorary advisory role on the Australian Sickle Cell Advisory Board. IMI has received a grant from the American Society of Hematology; consulting fees from Agios Pharmaceuticals; and honoraria from the American Society of Hematology. LCJ received royalties for a book chapter. AAK has received support from the Health resources and Services Administration. MLP has received consulting fees from the American College of Medical Genetics and Genomics; and participated in a Data Safety Monitoring Board or Advisory Board for the American College of Medical Genetics and Genomics and the American Academy of Pediatrics. ON has received travel support from the American Society of Hematology to attend a conference. DCR has received consulting fees from Vertex, Forma, Agios, and Vifor; received honoraria from Vertex; and participated in a Data Safety Monitoring Board or Advisory Board for TauRx and Mitsubishi. AS has received a scholar award from the American Society of Hematology, an advancing cures research award from the Doris Duke Charitable Foundation, and a Working group award from the Center for ELSI Resources and Analysis; consulting fees from Spotlight Therapeutics, Medexus, Vertex, Editas Medicine, and Sangamo Therapeutics; has received honoraria from Vindico Medical Education; and has a leadership or fiduciary role in the American Society for Transplantation and Cellular Therapy Content Committee, the Scientific Executive Committee, Sickle Cell Transplant Advocacy & Research Alliance, and the Pediatric Transplantation and Cellular Therapy Consortium Supportive Care Committee; and has financial interests with CRISPR Therapeutics, Vertex Pharmaceuticals, Novartis Pharmaceuticals, Magenta Therapeutics, and Beam Therapeutics. AAT has received consulting fees from GlaxoSmithKline and Global Blood Therapeutics; and honoraria from Novartis. REW has received consulting fees from Nova Laboratories; participated in a Data Safety Monitoring Board for Novartis and Editas; and received donations and support for clinical trials from Bristol Myers Squib, Addmedica, and Hemex Health. DJW has received a grant or contract from the US National Institutes for Health; received payment for expert testimony from Fredslough Law Firm; participated in a Data Safety Monitoring Board or Advisory board for Northwestern University; and is the founder and chairman of eNursing.
Contributor Information
Frédéric B Piel, Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London, London, UK.
David C Rees, Department of Paediatric Haematology, King’s College London, King’s College Hospital, London, UK.
Michael R DeBaun, Department of Pediatrics, Vanderbilt-Meharry Center of Excellence for Sickle Cell Disease, Vanderbilt University Medical Center, Nashville, TN, USA.
Obiageli Nnodu, Department of Haematology and Blood Transfusion, College of Health Sciences and Centre of Excellence for Sickle Cell Disease Research and Training, University of Abuja, Abuja, Nigeria.
Brigitte Ranque, Department of Internal Medicine, Georges Pompidou European Hospital, Assistance Publique—Hopitaux de Paris Centre, University of Paris Cité, Paris, France.
Alexis A Thompson, Division of Hematology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Russell E Ware, Division of Hematology and Global Health Center, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA.
Miguel R Abboud, Department of Pediatrics and Adolescent Medicine, and Sickle Cell Program, American University of Beirut, Beirut, Lebanon.
Allistair Abraham, Division of Blood and Marrow Transplantation, Children’s National Hospital, Washington, DC, USA.
Emmanuela E Ambrose, Department of Paediatrics and Child Health, Bugando Medical Centre, Mwanza, Tanzania.
Biree Andemariam, New England Sickle Cell Institute, University of Connecticut Health, Connecticut, USA.
Roshan Colah, Department of Haematogenetics, Indian Council of Medical Research National Institute of Immunohaematology, Mumbai, India.
Raffaella Colombatti, Pediatric Oncology Hematology Unit, Department of Women’s and Children’s Health, University of Padua, Padua, Italy.
Nicola Conran, Department of Clinical Medicine, School of Medical Sciences, Center of Hematology and Hemotherapy (Hemocentro), University of Campinas—UNICAMP, Campinas, Brazil.
Fernando F Costa, Department of Clinical Medicine, School of Medical Sciences, Center of Hematology and Hemotherapy (Hemocentro), University of Campinas—UNICAMP, Campinas, Brazil.
Robert M Cronin, Department of Internal Medicine, The Ohio State University, Columbus, OH, USA.
Mariane de Montalembert, Department of Pediatrics, Necker-Enfants Malades Hospital, Assistance Publique—Hopitaux de Paris Centre, Paris, France.
Jacques Elion, Paris Cité University and University of the Antilles, Inserm, BIGR, Paris, France.
Erica Esrick, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Harvard Medical School, Boston, MA, USA.
Anthea L Greenway, Department Clinical Haematology, Royal Children’s Hospital, Parkville and Department Haematology, Monash Health, Clayton, VIC, Australia.
Ibrahim M Idris, Department of Hematology, Aminu Kano Teaching Hospital/Bayero University Kano, Kano, Nigeria.
David-Zacharie Issom, Department of Business Information Systems, School of Management, HES-SO University of Applied Sciences and Arts of Western Switzerland, Geneva, Switzerland.
Dipty Jain, Department of Paediatrics, Government Medical College, Nagpur, India.
Lori C Jordan, Department of Pediatrics, Division of Pediatric Neurology, Vanderbilt University Medical Center, Nashville, TN, USA.
Zane S Kaplan, Department of Clinical Haematology, Monash Health and Monash University, Melbourne, VIC, Australia.
Allison A King, Departments of Pediatrics and Internal Medicine, Divisions of Pediatric Hematology and Oncology and Hematology, Washington University School of Medicine, St Louis, MO, USA.
Michele Lloyd-Puryear, Eunice Kennedy Shriver National Institute of Child Health and uman Development, National Institutes of Health, Bethesda, MD, USA.
Samuel A Oppong, Department of Obstetrics and Gynecology, University of Ghana Medical School, Accra, Ghana.
Akshay Sharma, Department of Bone Marrow Transplantation and Cellular Therapy, St Jude Children’s Research Hospital, Memphis, TN, USA.
Lillian Sung, Division of Haematology/Oncology, The Hospital for Sick Children, Toronto, ON, Canada.
Leon Tshilolo, Institute of Biomedical Research/CEFA Monkole Hospital Centre and Official University of Mbuji-Mayi, Mbuji-Mayi, Democratic Republic of the Congo.
Diana J Wilkie, Department of Biobehavioral Nursing Science, College of Nursing, University of Florida, Gainesville, FL, USA.
Kwaku Ohene-Frempong, Division of Hematology, Children’s Hospital of Philadelphia, Pennsylvania, USA; Sickle Cell Foundation of Ghana, Kumasi, Ghana.
References
- 1.GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol 2023; published online June 15. 10.1016/S2352-3026(23)00118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers 2018; 4: 18010. [DOI] [PubMed] [Google Scholar]
- 3.de la Fuente J, Gluckman E, Makani J, et al. The role of haematopoietic stem cell transplantation for sickle cell disease in the era of targeted disease-modifying therapies and gene editing. Lancet Haematol 2020; 7: e902–11. [DOI] [PubMed] [Google Scholar]
- 4.Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Arch Dis Child 2015; 100: 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910. Yale J Biol Med 2001; 74: 179–84. [PMC free article] [PubMed] [Google Scholar]
- 6.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010; 376: 2018–31. [DOI] [PubMed] [Google Scholar]
- 7.Watson J, Starman AW, Bilello FP. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948; 215: 419–23. [DOI] [PubMed] [Google Scholar]
- 8.Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992; 79: 2555–65. [PubMed] [Google Scholar]
- 9.Mussolino C, Strouboulis J. Recent approaches for manipulating globin gene expression in treating hemoglobinopathies. Front Genome Ed 2021; 3: 618111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science 1949; 110: 543–48. [DOI] [PubMed] [Google Scholar]
- 11.Vinjamur DS, Bauer DE, Orkin SH. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br J Haematol 2018; 180: 630–43. [DOI] [PubMed] [Google Scholar]
- 12.Hebbel RP. Reconstructing sickle cell disease: a data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am J Hematol 2011; 86: 123–54. [DOI] [PubMed] [Google Scholar]
- 13.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 2013; 381: 142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piel FB, Tatem AJ, Huang Z, Gupta S, Williams TN, Weatherall DJ. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health 2014; 2: e80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weatherall DJ. Genetic variation and susceptibility to infection: the red cell and malaria. Br J Haematol 2008; 141: 276–86. [DOI] [PubMed] [Google Scholar]
- 16.Ranque B, Kitenge R, Ndiaye DD, et al. Estimating the risk of child mortality attributable to sickle cell anaemia in sub-Saharan Africa: a retrospective, multicentre, case-control study. Lancet Haematol 2022; 9: e208–16. [DOI] [PubMed] [Google Scholar]
- 17.Gardner K, Bell C, Bartram JL, et al. Outcome of adults with sickle cell disease admitted to critical care—experience of a single institution in the UK. Br J Haematol 2010; 150: 610–13. [DOI] [PubMed] [Google Scholar]
- 18.Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986; 314: 1593–99. [DOI] [PubMed] [Google Scholar]
- 19.John CC, Opoka RO, Latham TS, et al. Hydroxyurea dose escalation for sickle cell anemia in Sub-Saharan Africa. N Engl J Med 2020; 382: 2524–33. [DOI] [PubMed] [Google Scholar]
- 20.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 2017; 376: 429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howard J, Ataga KI, Brown RC, et al. Voxelotor in adolescents and adults with sickle cell disease (HOPE): long-term follow-up results of an international, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol 2021; 8: e323–33. [DOI] [PubMed] [Google Scholar]
- 22.Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 2018; 379: 226–35. [DOI] [PubMed] [Google Scholar]
- 23.Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov 2019; 18: 139–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosanwo TO, Bauer DE. Editing outside the body: ex vivo gene-modification for β-hemoglobinopathy cellular therapy. Mol Ther 2021; 29: 3163–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.WHO. African health ministers launch drive to curb sickle cell disease toll. 2022. https://www.afro.who.int/news/african-health-ministers-launch-drive-curb-sickle-cell-disease-toll (accessed Jan 11, 2023).
- 26.National Academies of Sciences, Engineering, Medicine. Addressing sickle cell disease: a strategic plan and blueprint for action. Washington (DC): National Academies Press, 2020. [PubMed] [Google Scholar]
- 27.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 2013; 10: e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livingstone FB. Frequencies of hemoglobin variants. Oxford: Oxford University Press, 1985. [Google Scholar]
- 29.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008; 86: 480–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lobitz S, Telfer P, Cela E, et al. Newborn screening for sickle cell disease in Europe: recommendations from a Pan-European Consensus Conference. Br J Haematol 2018; 183: 648–60. [DOI] [PubMed] [Google Scholar]
- 31.Wastnedge E, Waters D, Patel S, et al. The global burden of sickle cell disease in children under five years of age: a systematic review and meta-analysis. J Glob Health 2018; 8: 021103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hockham C, Bhatt S, Colah R, et al. The spatial epidemiology of sickle-cell anaemia in India. Sci Rep 2018; 8: 17685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassell KL. Population estimates of sickle cell disease in the US. Am J Prev Med 2010; 38 (suppl): S512–21. [DOI] [PubMed] [Google Scholar]
- 34.Dormandy E, James J, Inusa B, Rees D. How many people have sickle cell disease in the UK? J Public Health 2018; 40: e291–95. [DOI] [PubMed] [Google Scholar]
- 35.Lee A, Thomas P, Cupidore L, Serjeant B, Serjeant G. Improved survival in homozygous sickle cell disease: lessons from a cohort study. BMJ 1995; 311: 1600–02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maitra P, Caughey M, Robinson L, et al. Risk factors for mortality in adult patients with sickle cell disease: a meta-analysis of studies in North America and Europe. Haematologica 2017; 102: 626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makani J, Cox SE, Soka D, et al. Mortality in sickle cell anemia in Africa: a prospective cohort study in Tanzania. PLoS One 2011; 6: e14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uyoga S, Macharia AW, Mochamah G, et al. The epidemiology of sickle cell disease in children recruited in infancy in Kilifi, Kenya: a prospective cohort study. Lancet Glob Health 2019; 7: e1458–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mbiya BM, Kalombo DK, Mukendi YN, et al. Improvement of SCD morbimortality in children: experience in a remote area of an African country. BMC Health Serv Res 2021; 21: 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med 2011; 41 (suppl 4): S398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nnodu OE, Oron AP, Sopekan A, Akaba GO, Piel FB, Chao DL. Child mortality from sickle cell disease in Nigeria: a model-estimated, population-level analysis of data from the 2018 Demographic and Health Survey. Lancet Haematol 2021; 8: e723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Therrell BL Jr, Lloyd-Puryear MA, Eckman JR, Mann MY. Newborn screening for sickle cell diseases in the United States: a review of data spanning 2 decades. Semin Perinatol 2015; 39: 238–51. [DOI] [PubMed] [Google Scholar]
- 43.Streetly A, Sisodia R, Dick M, Latinovic R, Hounsell K, Dormandy E. Evaluation of newborn sickle cell screening programme in England: 2010–2016. Arch Dis Child 2018; 103: 648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mason K, Gibson F, Gardner RA, Serjeant B, Serjeant GR. Prevention of sickle cell disease: observations on females with the sickle cell trait from the Manchester project, Jamaica. J Community Genet 2016; 7: 127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colombatti RSL, Sainati L. Management of children with sickle cell disease in Europe: current situation and future perspectives. EMJ Hematol 2016; 4: 129–35. [Google Scholar]
- 46.Ohene-Frempong K, Oduro J, Tetteh H, Nkrumah F. Screening newborns for sickle cell disease in Ghana. Pediatrics 2008; 121 (suppl 2): S120–21. [Google Scholar]
- 47.Ndeezi G, Kiyaga C, Hernandez AG, et al. Burden of sickle cell trait and disease in the Uganda Sickle Surveillance Study (US3): a cross-sectional study. Lancet Glob Health 2016; 4: e195–200. [DOI] [PubMed] [Google Scholar]
- 48.Ambrose EE, Smart LR, Charles M, et al. Surveillance for sickle cell disease, United Republic of Tanzania. Bull World Health Organ 2020; 98: 859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Italia Y, Krishnamurti L, Mehta V, et al. Feasibility of a newborn screening and follow-up programme for sickle cell disease among South Gujarat (India) tribal populations. J Med Screen 2015; 22: 1–7. [DOI] [PubMed] [Google Scholar]
- 50.Upadhye DS, Jain DL, Trivedi YL, Nadkarni AH, Ghosh K, Colah RB. Neonatal screening and the clinical outcome in children with sickle cell disease in central India. PLoS One 2016; 11: e0147081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colah RB, Mehta P, Mukherjee MB. Newborn screening for sickle cell disease: Indian experience. Int J Neonatal Screen 2018; 4: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Odame I, Jain D. Sickle cell disease: progress made & challenges ahead. Indian J Med Res 2020; 151: 505–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raman V, Seshadri T, Joice SV, N Srinivas P. Sickle cell disease in India: a scoping review from a health systems perspective to identify an agenda for research and action. BMJ Glob Health 2021; 6: e004322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colah RB, Nadkarni AH, Gorakshakar AC, et al. The changing trends in prenatal diagnosis of hemoglobinopathies in India: the quest of a single center to reduce the burden of disease over three decades. Hemoglobin 2021; 45: 112–18. [DOI] [PubMed] [Google Scholar]
- 55.Weil LG, Charlton MR, Coppinger C, Daniel Y, Streetly A. Sickle cell disease and thalassaemia antenatal screening programme in England over 10 years: a review from 2007/2008 to 2016/2017. J Clin Pathol 2020; 73: 183–90. [DOI] [PubMed] [Google Scholar]
- 56.Wonkam A, Ngo Bitoungui VJ, Ngogang J. Perspectives in genetics and sickle cell disease prevention in Africa: beyond the preliminary data from Cameroon. Public Health Genomics 2015; 18: 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olatunya OS, Babatola AO, Ogundare EO, et al. Perceptions and practice of early diagnosis of sickle cell disease by parents and physicians in a southwestern state of Nigeria. ScientificWorldJournal 2020; 2020: 4801087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daniel Y, Van Campen J, Silcock L, et al. Non-invasive prenatal diagnosis (NIPD) of sickle-cell disease by massively parallel sequencing of cell-free fetal DNA in maternal serum. Blood 2019; 134 (suppl 1): 2085 (abstr). [Google Scholar]
- 59.Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol 2009; 84: 323–27. [DOI] [PubMed] [Google Scholar]
- 60.Holdford D, Vendetti N, Sop DM, Johnson S, Smith WR. Indirect economic burden of sickle cell disease. Value Health 2021; 24: 1095–101. [DOI] [PubMed] [Google Scholar]
- 61.Thielen FW, Houwing ME, Cnossen MH, et al. Cost of health care for paediatric patients with sickle cell disease: an analysis of resource use and costs in a European country. Pediatr Blood Cancer 2020; 67: e28588. [DOI] [PubMed] [Google Scholar]
- 62.Colombatti R, Dalla Pozza LV, Mazzucato M, Sainati L, Pierobon M, Facchin P. Hospitalization of children with sickle cell disease in a region with increasing immigration rates. Haematologica 2008; 93: 463–64. [DOI] [PubMed] [Google Scholar]
- 63.Kunz JB, Cario H, Grosse R, Jarisch A, Lobitz S, Kulozik AE. The epidemiology of sickle cell disease in Germany following recent large-scale immigration. Pediatr Blood Cancer 2017; 64: e26550. [DOI] [PubMed] [Google Scholar]
- 64.Davis LR, Huehns ER, White JM. Survey of sickle-cell disease in England and Wales. Br Med J (Clin Res Ed) 1981; 283: 1519–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corriveau-Bourque C, Bruce AA. The changing epidemiology of pediatric hemoglobinopathy patients in northern Alberta, Canada. J Pediatr Hematol Oncol 2015; 37: 595–99. [DOI] [PubMed] [Google Scholar]
- 66.Crighton G, Wood E, Scarborough R, Ho PJ, Bowden D. Haemoglobin disorders in Australia: where are we now and where will we be in the future? Intern Med J 2016; 46: 770–79. [DOI] [PubMed] [Google Scholar]
- 67.Lena-Russo D, North ML, Girot R. Epidemiology of genetic hemoglobin diseases in metropolitan France. Rev Prat 1992; 42: 1867–72 (in French). [PubMed] [Google Scholar]
- 68.Leleu H, Arlet JB, Habibi A, et al. Epidemiology and disease burden of sickle cell disease in France: a descriptive study based on a French nationwide claim database. PLoS One 2021; 16: e0253986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hemminki K, Li X, Försti A, Sundquist J, Sundquist K. Thalassemia and sickle cell anemia in Swedish immigrants: genetic diseases have become global. SAGE Open Med 2015; 3: 2050312115613097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salzano FM. Incidence, effects, and management of sickle cell disease in Brazil. Am J Pediatr Hematol Oncol 1985; 7: 240–44. [DOI] [PubMed] [Google Scholar]
- 71.Colombatti R, Martella M, Cattaneo L, et al. Results of a multicenter universal newborn screening program for sickle cell disease in Italy: a call to action. Pediatr Blood Cancer 2019; 66: e27657. [DOI] [PubMed] [Google Scholar]
- 72.Yorke D, Mitchell J, Clow C, et al. Newborn screening for sickle cell and other hemoglobinopathies: a Canadian pilot study. Clin Invest Med 1992; 15: 376–83. [PubMed] [Google Scholar]
- 73.Nnodu OE, Sopekan A, Nnebe-Agumadu U, et al. Implementing newborn screening for sickle cell disease as part of immunisation programmes in Nigeria: a feasibility study. Lancet Haematol 2020; 7: e534–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hernandez AG, Kiyaga C, Howard TA, et al. Trends in sickle cell trait and disease screening in the Republic of Uganda, 2014–2019. Trop Med Int Health 2021; 26: 23–32. [DOI] [PubMed] [Google Scholar]
- 75.Marchand M, Gill C, Malhotra AK, et al. The assessment and sustainable management of sickle cell disease in the Indigenous Tharu population of Nepal. Hemoglobin 2017; 41: 278–82. [DOI] [PubMed] [Google Scholar]
- 76.Souza RC, Miranda Neto PAD, Santos JRN, et al. Sickle cell anaemia prevalence among newborns in the Brazilian Amazon–savanna transition region. Int J Environ Res Public Health 2019; 16: 1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Antoine M, Lee K, Donald T, et al. Prevalence of sickle cell disease among Grenadian newborns. J Med Screen 2018; 25: 49–50. [DOI] [PubMed] [Google Scholar]
- 78.Darshana T, Rees D, Premawardhena A. Hydroxyurea and blood transfusion therapy for sickle cell disease in South Asia: inconsistent treatment of a neglected disease. Orphanet J Rare Dis 2021; 16: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibbons C, Geoghegan R, Conroy H, et al. Sickle cell disease: time for a targeted neonatal screening programme. Ir Med J 2015; 108: 43–45. [PubMed] [Google Scholar]
- 80.Cintron-Garcia J, Guddati AK. Effect of immigration on mortality trends in sickle cell patients. Am J Blood Res 2020; 10: 172–78. [PMC free article] [PubMed] [Google Scholar]
- 81.Thein SL, Howard J. How I treat the older adult with sickle cell disease. Blood 2018; 132: 1750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diggs LM. Anatomic lesions in sickle cell disease. In: Abramson H, Bertles JF, Wethers DL, eds. Sickle cell disease: diagnosis, management, education, and research. St Louis (MO): CV Mosby, 1973: 189–229. [Google Scholar]
- 83.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639–44. [DOI] [PubMed] [Google Scholar]
- 84.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010; 115: 3447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: US, 1979–2005. Public Health Rep 2013; 128: 110–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med 2008; 148: 939–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Raper A Sickle-cell disease in Africa and America—a comparison. J Trop Med Hyg 1950; 53: 49–53. [Google Scholar]
- 88.Lambotte-Legrand J, Lambotte-Legrand C. Sickle cell anemia in the native child of Belgian Congo. Ann Soc Belg Med Trop (1920) 1951; 31: 207–34 (in French). [PubMed] [Google Scholar]
- 89.Fleming AF, Storey J, Molineaux L, Iroko EA, Attai ED. Abnormal haemoglobins in the Sudan savanna of Nigeria. I. Prevalence of haemoglobins and relationships between sickle cell trait, malaria and survival. Ann Trop Med Parasitol 1979; 73: 161–72. [DOI] [PubMed] [Google Scholar]
- 90.Williams TN, Uyoga S, Macharia A, et al. Bacteraemia in Kenyan children with sickle-cell anaemia: a retrospective cohort and case-control study. Lancet 2009; 374: 1364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Issom DZ, Hardy-Dessources MD, Romana M, Hartvigsen G, Lovis C. Toward a conversational agent to support the self-management of adults and young adults with sickle cell disease: usability and usefulness study. Front Digit Health 2021; 3: 600333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalpatthi R, Novelli EM. Measuring success: utility of biomarkers in sickle cell disease clinical trials and care. Hematology (Am Soc Hematol Educ Program) 2018; 2018: 482–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dubert M, Elion J, Tolo A, et al. Degree of anemia, indirect markers of hemolysis, and vascular complications of sickle cell disease in Africa. Blood 2017; 130: 2215–23. [DOI] [PubMed] [Google Scholar]
- 94.Rees DC, Gibson JS. Biomarkers in sickle cell disease. Br J Haematol 2012; 156: 433–45. [DOI] [PubMed] [Google Scholar]
- 95.Du M, Van Ness S, Gordeuk V, et al. Biomarker signatures of sickle cell disease severity. Blood Cells Mol Dis 2018; 72: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sandhu MK, Cohen A. Aging in sickle cell disease: co-morbidities and new issues in management. Hemoglobin 2015; 39: 221–24. [DOI] [PubMed] [Google Scholar]
- 97.Tewari S, Brousse V, Piel FB, Menzel S, Rees DC. Environmental determinants of severity in sickle cell disease. Haematologica 2015; 100: 1108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.US National Academies of Sciences, Engineering, Medicine. Addressing sickle cell disease: a strategic plan and blueprint for action. 2020. https://www.nationalacademies.org/our-work/addressing-sickle-cell-disease-a-strategic-plan-and-blueprint-for-action (accessed Jan 11, 2023). [PubMed]
- 99.Asnani MR, Knight Madden J, Reid M, Greene L-G, Lyew-Ayee P. Socio-environmental exposures and health outcomes among persons with sickle cell disease. PLoS One 2017; 12: e0175260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020; 396: 1223–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Our World in Data. Share of people living in urban areas, 2020. https://ourworldindata.org/grapher/share-of-population-urban (accessed Jan 11, 2023).
- 102.The Sickle Cell and Thalassaemia All-Party Parliamentary Group. Cast aside and forgotten: a report into the experiences of those living with sickle cell or caring for someone with sickle cell during the COVID-19 pandemic. 2021. https://www.sicklecellsociety.org/wp-content/uploads/2021/01/Cast-Aside-and-Forgotten-SCTAPPG-Report-Final.pdf (accessed Jan 11, 2023).
- 103.Aljuburi G, Laverty AA, Green SA, Phekoo KJ, Bell D, Majeed A. Socio-economic deprivation and risk of emergency readmission and inpatient mortality in people with sickle cell disease in England: observational study. J Public Health 2013; 35: 510–17. [DOI] [PubMed] [Google Scholar]
- 104.Khan SA, AlSiny F, Makki A, Ali A, AlAnsari I, Khan S. Socioeconomic status dependent medical complexities in children with sickle cell disease in Saudi Arabia. Saudi J Biol Sci 2020; 27: 1781–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cronin RM, Hankins JS, Byrd J, et al. Risk factors for hospitalizations and readmissions among individuals with sickle cell disease: results of a US survey study. Hematology 2019; 24: 189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Panepinto JA, Pajewski NM, Foerster LM, Sabnis S, Hoffmann RG. Impact of family income and sickle cell disease on the health-related quality of life of children. Qual Life Res 2009; 18: 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fearon A, Marsh A, Kim J, Treadwell M. Pediatric residents’ perceived barriers to opioid use in sickle cell disease pain management. Pediatr Blood Cancer 2019; 66: e27535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Haywood C Jr, Tanabe P, Naik R, Beach MC, Lanzkron S. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013; 31: 651–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Farooq F, Mogayzel PJ, Lanzkron S, Haywood C, Strouse JJ. Comparison of US federal and foundation funding of research for sickle cell disease and cystic fibrosis and factors associated with research productivity. JAMA Netw Open 2020; 3: e201737. [DOI] [PubMed] [Google Scholar]
- 110.Bello-Manga H, Galadanci AA, Abdullahi S, et al. Low educational level of head of household, as a proxy for poverty, is associated with severe anaemia among children with sickle cell disease living in a low-resource setting: evidence from the SPRING trial. Br J Haematol 2020; 190: 939–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fernandes TA, Medeiros TM, Alves JJ, et al. Socioeconomic and demographic characteristics of sickle cell disease patients from a low-income region of northeastern Brazil. Rev Bras Hematol Hemoter 2015; 37: 172–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kamal S, Naghib MM, Al Zahrani J, Hassan H, Moawad K, Arrahman O. Influence of nutrition on disease severity and health-related quality of life in adults with sickle cell disease: a prospective study. Mediterr J Hematol Infect Dis 2021; 13: e2021007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Canatan D, Vives Corrons JL, Piacentini G, et al. Immigration and screening programs for hemoglobinopathies in Italy, Spain and Turkey. Acta Biomed 2021; 92: e2021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghosh K, Ghosh K, Agrawal R, Nadkarni AH. Recent advances in screening and diagnosis of hemoglobinopathy. Expert Rev Hematol 2020; 13: 13–21. [DOI] [PubMed] [Google Scholar]
- 115.Chakravorty S, Dick MC. Antenatal screening for haemoglobinopathies: current status, barriers and ethics. Br J Haematol 2019; 187: 431–40. [DOI] [PubMed] [Google Scholar]
- 116.Marcheco-Teruel B Sickle cell anemia in Cuba: prevention and management, 1982–2018. MEDICC Rev 2019; 21: 34–38. [DOI] [PubMed] [Google Scholar]
- 117.El-Haj N, Hoppe CC. Newborn screening for SCD in the USA and Canada. Int J Neonatal Screen 2018; 4: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Daniel Y, Elion J, Allaf B, et al. Newborn screening for sickle cell disease in Europe. Int J Neonatal Screen 2019; 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Knight-Madden J, Lee K, Elana G, et al. Newborn screening for sickle cell disease in the Caribbean: an update of the present situation and of the disease prevalence. Int J Neonatal Screen 2019; 5: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Silva-Pinto AC, Alencar de Queiroz MC, Antoniazzo Zamaro PJ, Arruda M, Pimentel Dos Santos H. The neonatal screening program in Brazil, focus on sickle cell disease (SCD). Int J Neonatal Screen 2019; 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Diallo DA, Guindo A. Sickle cell disease in sub-Saharan Africa: stakes and strategies for control of the disease. Curr Opin Hematol 2014; 21: 210–14. [DOI] [PubMed] [Google Scholar]
- 122.Frömmel C Newborn screening for sickle cell disease and other hemoglobinopathies: a short review on classical laboratory methods-isoelectric focusing, HPLC, and capillary electrophoresis. Int J Neonatal Screen 2018; 4: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Steele C, Sinski A, Asibey J, et al. Point-of-care screening for sickle cell disease in low-resource settings: a multi-center evaluation of HemoTypeSC, a novel rapid test. Am J Hematol 2019; 94: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rahimy MC. Early detection and medical management of sickle cell anemia: five years of experience in Cotonou. Arch Pediatr 1999; 6 (suppl 2): S343–44 (in French). [DOI] [PubMed] [Google Scholar]
- 125.Segbefia CI, Goka B, Welbeck J, et al. Implementing newborn screening for sickle cell disease in Korle Bu Teaching Hospital, Accra: results and lessons learned. Pediatr Blood Cancer 2021; 68: e29068. [DOI] [PubMed] [Google Scholar]
- 126.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in east London. Haematologica 2007; 92: 905–12. [DOI] [PubMed] [Google Scholar]
- 127.Al Arrayed S, Al Hajeri A. Newborn screening services in Bahrain between 1985 and 2010. Adv Hematol 2012; 2012: 903219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rahimy MC, Gangbo A, Ahouignan G, Alihonou E. Newborn screening for sickle cell disease in the Republic of Benin. J Clin Pathol 2009; 62: 46–48. [DOI] [PubMed] [Google Scholar]
- 129.McGann PT, Ferris MG, Ramamurthy U, et al. A prospective newborn screening and treatment program for sickle cell anemia in Luanda, Angola. Am J Hematol 2013; 88: 984–89. [DOI] [PubMed] [Google Scholar]
- 130.Couque N, Girard D, Ducrocq R, et al. Improvement of medical care in a cohort of newborns with sickle-cell disease in north Paris: impact of national guidelines. Br J Haematol 2016; 173: 927–37. [DOI] [PubMed] [Google Scholar]
- 131.McGann PT, Grosse SD, Santos B, et al. A cost-effectiveness analysis of a pilot neonatal screening program for sickle cell anemia in the Republic of Angola. J Pediatr 2015; 167: 1314–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.James J, Dormandy E. Improving screening programmes for sickle cell disorders and other haemoglobinopathies in Europe: the role of patient organisations. Int J Neonatal Screen 2019; 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.King LG, Bortolusso-Ali S, Cunningham-Myrie CA, Reid ME. Impact of a comprehensive sickle cell center on early childhood mortality in a developing country: the Jamaican experience. J Pediatr 2015; 167: 702–5.e1. [DOI] [PubMed] [Google Scholar]
- 134.Brousse V, Arnaud C, Lesprit E, et al. Evaluation of outcomes and quality of care in children with sickle cell disease diagnosed by newborn screening: a real-world nation-wide study in France. J Clin Med 2019; 8: 1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.WHO Regional Committee for Africa. Progress in the implementation of the African Region sickle-cell strategy 2010–2020: information document. Brazzaville: World Health Organization Regional Office for Africa, 2020. [Google Scholar]
- 136.Therrell BL Jr. US newborn screening policy dilemmas for the twenty-first century. Mol Genet Metab 2001; 74: 64–74. [DOI] [PubMed] [Google Scholar]
- 137.Green NS, Zapfel A, Nnodu OE, et al. The Consortium on Newborn Screening in Africa for sickle cell disease: study rationale and methodology. Blood Adv 2022; 6: 6187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Okeke CO, Chianumba RI, Isa H, Asala S, Nnodu OE. Using dried blood spot on HemoTypeSC™, a new frontier for newborn screening for sickle cell disease in Nigeria. Front Genet 2022; 13: 1013858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oligbu G, Fallaha M, Pay L, Ladhani S. Risk of invasive pneumococcal disease in children with sickle cell disease in the era of conjugate vaccines: a systematic review of the literature. Br J Haematol 2019; 185: 743–51. [DOI] [PubMed] [Google Scholar]
- 140.Ballas SK. Opioids and sickle cell disease: from opium to the opioid epidemic. J Clin Med 2021; 10: 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014; 312: 1033–48. [DOI] [PubMed] [Google Scholar]
- 142.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet 2017; 390: 311–23. [DOI] [PubMed] [Google Scholar]
- 143.Tanabe P, Silva S, Bosworth HB, et al. A randomized controlled trial comparing two vaso-occlusive episode (VOE) protocols in sickle cell disease (SCD). Am J Hematol 2018; 93: 159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Telfer P, Bahal N, Lo A, Challands J. Management of the acute painful crisis in sickle cell disease—a re-evaluation of the use of opioids in adult patients. Br J Haematol 2014; 166: 157–64. [DOI] [PubMed] [Google Scholar]
- 145.Carden MA, Patil P, Ahmad ME, Lam WA, Joiner CH, Morris CR. Variations in pediatric emergency medicine physician practices for intravenous fluid management in children with sickle cell disease and vaso-occlusive pain: a single institution experience. Pediatr Blood Cancer 2018; 65: e26742. [DOI] [PubMed] [Google Scholar]
- 146.Bartolucci P, El Murr T, Roudot-Thoraval F, et al. A randomized, controlled clinical trial of ketoprofen for sickle-cell disease vaso-occlusive crises in adults. Blood 2009; 114: 3742–47. [DOI] [PubMed] [Google Scholar]
- 147.Bellet PS, Kalinyak KA, Shukla R, Gelfand MJ, Rucknagel DL. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N Engl J Med 1995; 333: 699–703. [DOI] [PubMed] [Google Scholar]
- 148.Sobota A, Graham DA, Heeney MM, Neufeld EJ. Corticosteroids for acute chest syndrome in children with sickle cell disease: variation in use and association with length of stay and readmission. Am J Hematol 2010; 85: 24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med 2000; 342: 1855–65. [DOI] [PubMed] [Google Scholar]
- 150.Sickle Cell Society. Standards for the clinical care of adults with sickle cell disease in the UK. London: Sickle Cell Society, 2018. [Google Scholar]
- 151.Habibi A, Arlet JB, Stankovic K, et al. Recommandations françaises de prise en charge de la drépanocytose de l’adulte: actualisation 2015. Rev Méd Interne 2015; 36 (suppl 1): 5S3–84. [DOI] [PubMed] [Google Scholar]
- 152.DeBaun MR, Jordan LC, King AA, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv 2020; 4: 1554–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 1998; 339: 5–11. [DOI] [PubMed] [Google Scholar]
- 154.Ware RE. Optimizing hydroxyurea therapy for sickle cell anemia. Hematology (Am Soc Hematol Educ Program) 2015; 2015: 436–43. [DOI] [PubMed] [Google Scholar]
- 155.Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematology (Am Soc Hematol Educ Program) 2013; 2013: 439–46. [DOI] [PubMed] [Google Scholar]
- 156.Davis BA, Allard S, Qureshi A, et al. Guidelines on red cell transfusion in sickle cell disease part II: indications for transfusion. Br J Haematol 2017; 176: 192–209. [DOI] [PubMed] [Google Scholar]
- 157.Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med 2005; 353: 2769–78. [DOI] [PubMed] [Google Scholar]
- 158.Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia—TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet 2016; 387: 661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Candotti D, Tagny-Tayou C, Laperche S. Challenges in transfusion-transmitted infection screening in Sub-Saharan Africa. Transfus Clin Biol 2021; 28: 163–70. [DOI] [PubMed] [Google Scholar]
- 160.Pirenne F, Yazdanbakhsh K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood 2018; 131: 2773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Socolovsky M Molecular insights into stress erythropoiesis. Curr Opin Hematol 2007; 14: 215–24. [DOI] [PubMed] [Google Scholar]
- 162.Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc 2018; 68: 263–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cokic VP, Andric SA, Stojilkovic SS, Noguchi CT, Schechter AN. Hydroxyurea nitrosylates and activates soluble guanylyl cyclase in human erythroid cells. Blood 2008; 111: 1117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Conran N, de Alvarenga Maximo C, Oliveira T, Fertrin KY, Lobo C, Costa FF. Safe use of hydroxycarbamide in sickle cell disease patients hospitalized for painful vaso-occlusive episodes during the randomized, open-label HELPS study. Br J Haematol 2022; 199: 153–57. [DOI] [PubMed] [Google Scholar]
- 165.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med 1995; 332: 1317–22. [DOI] [PubMed] [Google Scholar]
- 166.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol 2010; 85: 403–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood 2010; 115: 2354–63. [DOI] [PubMed] [Google Scholar]
- 168.Lobo CL, Pinto JF, Nascimento EM, Moura PG, Cardoso GP, Hankins JS. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br J Haematol 2013; 161: 852–60. [DOI] [PubMed] [Google Scholar]
- 169.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011; 377: 1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Qureshi A, Kaya B, Pancham S, et al. Guidelines for the use of hydroxycarbamide in children and adults with sickle cell disease: a British Society for Haematology guideline. Br J Haematol 2018; 181: 460–75. [DOI] [PubMed] [Google Scholar]
- 171.Tshilolo L, Tomlinson G, Williams TN, et al. Hydroxyurea for children with sickle cell anemia in Sub-Saharan Africa. N Engl J Med 2019; 380: 121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Menzel S, Thein SL. Genetic modifiers of fetal haemoglobin in sickle cell disease. Mol Diagn Ther 2019; 23: 235–44. [DOI] [PubMed] [Google Scholar]
- 173.Fitzhugh CD, Hsieh MM, Allen D, et al. Hydroxyurea-increased fetal hemoglobin is associated with less organ damage and longer survival in adults with sickle cell anemia. PLoS One 2015; 10: e0141706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Luchtman-Jones L, Pressel S, Hilliard L, et al. Effects of hydroxyurea treatment for patients with hemoglobin SC disease. Am J Hematol 2016; 91: 238–42. [DOI] [PubMed] [Google Scholar]
- 175.Gille AS, Pondarré C, Dalle JH, et al. Hydroxyurea does not affect the spermatogonial pool in prepubertal patients with sickle cell disease. Blood 2021; 137: 856–59. [DOI] [PubMed] [Google Scholar]
- 176.Ballas SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. J Natl Med Assoc 2009; 101: 1046–51. [DOI] [PubMed] [Google Scholar]
- 177.de Montalembert M, Voskaridou E, Oevermann L, et al. Real-life experience with hydroxyurea in patients with sickle cell disease: results from the prospective ESCORT-HU cohort study. Am J Hematol 2021; 96: 1223–31. [DOI] [PubMed] [Google Scholar]
- 178.Ware RE, Dertinger SD. Absence of hydroxyurea-induced mutational effects supports higher utilisation for the treatment of sickle cell anaemia. Br J Haematol 2021; 194: 252–66. [DOI] [PubMed] [Google Scholar]
- 179.Ware RE, Marahatta A, Ware JL, McElhinney K, Dong M, Vinks AA. Hydroxyurea exposure in lactation: a pharmacokinetics study (HELPS). J Pediatr 2020; 222: 236–39. [DOI] [PubMed] [Google Scholar]
- 180.McArthur JG, Svenstrup N, Chen C, et al. A novel, highly potent and selective phosphodiesterase-9 inhibitor for the treatment of sickle cell disease. Haematologica 2020; 105: 623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Perrine SP, Ginder GD, Faller DV, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N Engl J Med 1993; 328: 81–86. [DOI] [PubMed] [Google Scholar]
- 182.Koshy M, Dorn L, Bressler L, et al. 2-deoxy 5-azacytidine and fetal hemoglobin induction in sickle cell anemia. Blood 2000; 96: 2379–84. [PubMed] [Google Scholar]
- 183.Molokie R, Lavelle D, Gowhari M, et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med 2017; 14: e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med 2019; 381: 509–19. [DOI] [PubMed] [Google Scholar]
- 185.Quinn CT, Ware RE. Increased oxygen affinity: to have and to hold. Blood 2021; 138: 1094–95. [DOI] [PubMed] [Google Scholar]
- 186.Shrestha A, Chi M, Wagner K, et al. FT-4202, an oral PKR activator, has potent antisickling effects and improves RBC survival and Hb levels in SCA mice. Blood Adv 2021; 5: 2385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Matte A, Federti E, Kung C, et al. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a β-thalassemia mouse model. J Clin Invest 2021; 131: e144206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.El Nemer W, Godard A, El Hoss S. Ineffective erythropoiesis in sickle cell disease: new insights and future implications. Curr Opin Hematol 2021; 28: 171–76. [DOI] [PubMed] [Google Scholar]
- 189.Biemond BJ, Tombak A, Kilinc Y, et al. Sevuparin for the treatment of acute pain crisis in patients with sickle cell disease: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Haematol 2021; 8: e334–43. [DOI] [PubMed] [Google Scholar]
- 190.Heeney MM, Hoppe CC, Abboud MR, et al. A multinational trial of prasugrel for sickle cell vaso-occlusive events. N Engl J Med 2016; 374: 625–35. [DOI] [PubMed] [Google Scholar]
- 191.Nyffenegger N, Zennadi R, Kalleda N, et al. The oral ferroportin inhibitor vamifeport improves hemodynamics in a mouse model of sickle cell disease. Blood 2022; 140: 769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee MT, Kattan M, Fennoy I, et al. Randomized phase 2 trial of monthly vitamin D to prevent respiratory complications in children with sickle cell disease. Blood Adv 2018; 2: 969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.WHO. Blood safety and availability. Geneva: World Health Organization, 2020. [Google Scholar]
- 194.Diaku-Akinwumi IN, Abubakar SB, Adegoke SA, et al. Blood transfusion services for patients with sickle cell disease in Nigeria. Int Health 2016; 8: 330–35. [DOI] [PubMed] [Google Scholar]
- 195.Diop S, Pirenne F. Transfusion and sickle cell anemia in Africa. Transfus Clin Biol 2021; 28: 143–45. [DOI] [PubMed] [Google Scholar]
- 196.Maitland K, Kiguli S, Olupot-Olupot P, et al. Immediate transfusion in African children with uncomplicated severe anemia. N Engl J Med 2019; 381: 407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lagunju I, Brown BJ, Oyinlade AO, et al. Annual stroke incidence in Nigerian children with sickle cell disease and elevated TCD velocities treated with hydroxyurea. Pediatr Blood Cancer 2019; 66: e27252. [DOI] [PubMed] [Google Scholar]
- 198.Power-Hays A, Ware RE. Effective use of hydroxyurea for sickle cell anemia in low-resource countries. Curr Opin Hematol 2020; 27: 172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Paintsil V, Ally M, Isa H, et al. Development of multi-level standards of care recommendations for sickle cell disease: experience from SickleInAfrica. Front Genet 2023; 13: 1052179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Abraham AA, Tisdale JF. Gene therapy for sickle cell disease: moving from the bench to the bedside. Blood 2021; 138: 932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017; 129: 1548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Eapen M, Brazauskas R, Walters MC, et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol 2019; 6: e585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Alzahrani M, Damlaj M, Jeffries N, et al. Non-myeloablative human leukocyte antigen-matched related donor transplantation in sickle cell disease: outcomes from three independent centres. Br J Haematol 2021; 192: 761–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guilcher GMT, Monagel DA, Nettel-Aguirre A, et al. Nonmyeloablative matched sibling donor hematopoietic cell transplantation in children and adolescents with sickle cell disease. Biol Blood Marrow Transplant 2019; 25: 1179–86. [DOI] [PubMed] [Google Scholar]
- 205.Shenoy S, Eapen M, Panepinto JA, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood 2016; 128: 2561–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ngwube A, Shah N, Godder K, Jacobsohn D, Hulbert ML, Shenoy S. Abatacept is effective as GVHD prophylaxis in unrelated donor stem cell transplantation for children with severe sickle cell disease. Blood Adv 2020; 4: 3894–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Parikh S, Brochstein JA, Galamidi E, Schwarzbach A, Kurtzberg J. Allogeneic stem cell transplantation with omidubicel in sickle cell disease. Blood Adv 2021; 5: 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv 2017; 1: 652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gaziev J, Isgrò A, Sodani P, et al. Haploidentical HSCT for hemoglobinopathies: improved outcomes with TCRαβ+/CD19+-depleted grafts. Blood Adv 2018; 2: 263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Foell J, Schulte JH, Pfirstinger B, et al. Haploidentical CD3 or α/β T-cell depleted HSCT in advanced stage sickle cell disease. Bone Marrow Transplant 2019; 54: 1859–67. [DOI] [PubMed] [Google Scholar]
- 211.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science 2018; 359: eaan4672. [DOI] [PubMed] [Google Scholar]
- 212.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000; 288: 669–72. [DOI] [PubMed] [Google Scholar]
- 213.Blaese RM, Culver KW, Miller AD, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science 1995; 270: 475–80. [DOI] [PubMed] [Google Scholar]
- 214.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003; 302: 415–19. [DOI] [PubMed] [Google Scholar]
- 215.Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet 2021; 22: 216–34. [DOI] [PubMed] [Google Scholar]
- 216.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med 2018; 378: 1479–93. [DOI] [PubMed] [Google Scholar]
- 217.Tisdale JF, Pierciey FJ Jr, Bonner M, et al. Safety and feasibility of hematopoietic progenitor stem cell collection by mobilization with plerixafor followed by apheresis vs bone marrow harvest in patients with sickle cell disease in the multi-center HGB-206 trial. Am J Hematol 2020; 95: e239–42. [DOI] [PubMed] [Google Scholar]
- 218.Esrick EB, Manis JP, Daley H, et al. Successful hematopoietic stem cell mobilization and apheresis collection using plerixafor alone in sickle cell patients. Blood Adv 2018; 2: 2505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lagresle-Peyrou C, Lefrère F, Magrin E, et al. Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica 2018; 103: 778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010; 467: 318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 2001; 294: 2368–71. [DOI] [PubMed] [Google Scholar]
- 222.Negre O, Eggimann AV, Beuzard Y, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(a(t87q))-globin gene. Hum Gene Ther 2016; 27: 148–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kanter J, Tisdale JF, Mapara MY, et al. Resolution of sickle cell disease manifestations in patients treated with LentiGlobin gene therapy: updated results from the phase 1/2 hgb-206 group c study. Blood 2019; 134 (suppl 1): 990 (abstr). [Google Scholar]
- 224.Kanter J, Walters MC, Hsieh M, et al. interim results from a phase 1/2 clinical study of LentiGlobin gene therapy for severe sickle cell disease. Blood 2017; 130 (suppl): 527 (abstr).28611024 [Google Scholar]
- 225.Kanter J, Walters MC, Krishnamurti L, et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med 2022; 386: 617–28. [DOI] [PubMed] [Google Scholar]
- 226.Thompson AA, Walters MC, Mapara MY, et al. Resolution of serious vaso-occlusive pain crises and reduction in patient-reported pain intensity: results from the ongoing phase 1/2 hgb-206 group c study of lentiglobin for sickle cell disease (bb1111) gene therapy. Blood 2020; 136 (suppl 1): 16–17 (abstr). [Google Scholar]
- 227.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 2007; 39: 1197–99. [DOI] [PubMed] [Google Scholar]
- 228.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA 2008; 105: 11869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008; 322: 1839–42. [DOI] [PubMed] [Google Scholar]
- 230.Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 2011; 334: 993–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Esrick EB, Lehmann LE, Biffi A, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med 2021; 384: 205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Grimley M, Asnani M, Shrestha A, et al. Early results from a phase 1/2 study of aru-1801 gene therapy for sickle cell disease (SCD): manufacturing process enhancements improve efficacy of a modified gamma globin lentivirus vector and reduced intensity conditioning transplant. Blood 2020; 136 (suppl 1): 20–21 (abstr). [Google Scholar]
- 233.Hsieh MM, Bonner M, Pierciey FJ Jr, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv 2020; 4: 2058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Leonard A, Tisdale JF. A pause in gene therapy: reflecting on the unique challenges of sickle cell disease. Mol Ther 2021; 29: 1355–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bhoopalan SV, Yen JS, Levine RM, Sharma A. Editing human hematopoietic stem cells: advances and challenges. Cytotherapy 2023; 25: 261–69. [DOI] [PubMed] [Google Scholar]
- 236.Doerfler PA, Sharma A, Porter JS, Zheng Y, Tisdale JF, Weiss MJ. Genetic therapies for the first molecular disease. J Clin Invest 2021; 131: e146394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Drysdale CM, Nassehi T, Gamer J, Yapundich M, Tisdale JF, Uchida N. Hematopoietic-stem-cell-targeted gene-addition and gene-editing strategies for β-hemoglobinopathies. Cell Stem Cell 2021; 28: 191–208. [DOI] [PubMed] [Google Scholar]
- 238.Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci 1998; 850: 38–44. [DOI] [PubMed] [Google Scholar]
- 239.Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood 2017; 129: 2719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bartman CR, Hsu SC, Hsiung CC, Raj A, Blobel GA. Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell 2016; 62: 237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet 2003; 35: 190–94. [DOI] [PubMed] [Google Scholar]
- 242.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA 2008; 105: 1620–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chang KH, Smith SE, Sullivan T, et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev 2017; 4: 137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Psatha N, Reik A, Phelps S, et al. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with β-thalassemia major. Mol Ther Methods Clin Dev 2018; 10: 313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013; 342: 253–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Canver MC, Smith EC, Sher F, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015; 527: 192–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wu Y, Zeng J, Roscoe BP, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 2019; 25: 776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med 2021; 384: 252–60. [DOI] [PubMed] [Google Scholar]
- 249.Lessard S, Rimmele P, Ling H, et al. Zinc finger nuclease-mediated disruption of the BCL11A erythroid enhancer results in enriched biallelic editing, increased fetal hemoglobin, and reduced sickling in erythroid cells derived from sickle cell disease patients. Blood 2019; 134 (suppl 1): 974 (abstr). [Google Scholar]
- 250.Moran K, Ling H, Lessard S, et al. Ex vivo gene-edited cell therapy for sickle cell disease: disruption of the BCL11A erythroid enhancer with zinc finger nucleases increases fetal hemoglobin in plerixafor mobilized human CD34+ cells. Blood 2018; 132 (suppl 1): 2190 (abstr). [Google Scholar]
- 251.Métais JY, Doerfler PA, Mayuranathan T, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv 2019; 3: 3379–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Traxler EA, Yao Y, Wang YD, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016; 22: 987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Weber L, Frati G, Felix T, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv 2020; 6: eaay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Antoniani C, Meneghini V, Lattanzi A, et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 2018; 131: 1960–73. [DOI] [PubMed] [Google Scholar]
- 255.Masuda T, Wang X, Maeda M, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016; 351: 285–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Humbert O, Radtke S, Samuelson C, et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med 2019; 11: eaaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Chang K-H, Sanchez M, Heath J, et al. Comparative studies reveal robust HbF induction by editing of HBG1/2 promoters or BCL11A erythroid-enhancer in human CD34+ cells but that BCL11A erythroid-enhancer editing is associated with selective reduction in erythroid lineage reconstitution in a xenotransplantation model. Blood 2018; 132 (suppl 1): 409 (abstr). [Google Scholar]
- 258.Sharma A, Boelens JJ, Cancio MI, et al. Treatment of individuals with severe sickle cell disease with OTQ923, an autologous, ex vivo, CRISPR/Cas9-edited, CD34+ hematopoietic stem and progenitor cell product, leads to durable engraftment and fetal hemoglobin induction. Blood 2022; 140 (suppl 1): 1906–08 (abstr). [Google Scholar]
- 259.Urnov FD, Miller JC, Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005; 435: 646–51. [DOI] [PubMed] [Google Scholar]
- 260.Pattabhi S, Lotti SN, Berger MP, et al. In vivo outcome of homology-directed repair at the HBB gene in HSC using alternative donor template delivery methods. Mol Ther Nucleic Acids 2019; 17: 277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wilkinson AC, Dever DP, Baik R, et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat Commun 2021; 12: 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cai L, Bai H, Mahairaki V, et al. A universal approach to correct various HBB gene mutations in human stem cells for gene therapy of beta-thalassemia and sickle cell disease. Stem Cells Transl Med 2018; 7: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Hoban MD, Cost GJ, Mendel MC, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015; 125: 2597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Hoban MD, Lumaquin D, Kuo CY, et al. CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol Ther 2016; 24: 1561–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Park SH, Lee CM, Dever DP, et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res 2019; 47: 7955–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016; 539: 384–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Yang H, Ren S, Yu S, et al. Methods favoring homology-directed repair choice in response to CRISPR/Cas9 induced-double strand breaks. Int J Mol Sci 2020; 21: 6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Uchida N, Li L, Nassehi T, et al. Preclinical evaluation for engraftment of CD34+ cells gene-edited at the sickle cell disease locus in xenograft mouse and non-human primate models. Cell Rep Med 2021; 2: 100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017; 551: 464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016; 533: 420–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 2018; 24: 927–30. [DOI] [PubMed] [Google Scholar]
- 272.Leibowitz ML, Papathanasiou S, Doerfler PA, et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat Genet 2021; 53: 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zeng J, Wu Y, Ren C, et al. Therapeutic base editing of human hematopoietic stem cells. Nat Med 2020; 26: 535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wang L, Li L, Ma Y, et al. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Res 2020; 30: 276–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Li C, Georgakopoulou A, Mishra A, et al. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood Adv 2021; 5: 1122–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Newby GA, Yen JS, Woodard KJ, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021; 595: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019; 576: 149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Cohen J, Kaiser J. Gates and NIH join forces on HIV and sickle cell diseases. Science 2019; 366: 558–59. [DOI] [PubMed] [Google Scholar]
- 279.Cannon P, Asokan A, Czechowicz A, et al. Safe and effective in vivo targeting and gene editing in hematopoietic stem cells: strategies for accelerating development. Hum Gene Ther 2021; 32: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Amirache F, Lévy C, Costa C, et al. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 2014; 123: 1422–24. [DOI] [PubMed] [Google Scholar]
- 281.Ikawa Y, Miccio A, Magrin E, Kwiatkowski JL, Rivella S, Cavazzana M. Gene therapy of hemoglobinopathies: progress and future challenges. Hum Mol Genet 2019; 28: R24–30. [DOI] [PubMed] [Google Scholar]
- 282.Kesavan G Innovations in CRISPR-based therapies. Mol Biotechnol 2023; 65: 138–45. [DOI] [PubMed] [Google Scholar]
- 283.Urbinati F, Campo Fernandez B, Masiuk KE, et al. Gene therapy for sickle cell disease: a lentiviral vector comparison study. Hum Gene Ther 2018; 29: 1153–66. [DOI] [PubMed] [Google Scholar]
- 284.Li C, Wang H, Georgakopoulou A, Gil S, Yannaki E, Lieber A. In vivo HSC gene therapy using a bi-modular HDAd5/35++ vector cures sickle cell disease in a mouse model. Mol Ther 2021; 29: 822–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Cullis PR, Hope MJ. Lipid nanoparticle systems for enabling gene therapies. Mol Ther 2017; 25: 1467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lee L, Smith-Whitley K, Banks S, Puckrein G. Reducing health care disparities in sickle cell disease: a review. Public Health Rep 2019; 134: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.McCune JM, Stevenson SC, Doehle BP, Trenor CC 3rd, Turner EH, Spector JM. Collaborative science to advance gene therapies in resource-limited parts of the world. Mol Ther 2021; 29: 3101–02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Brandow AM, Carroll CP, Creary S, et al. American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv 2020; 4: 2656–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Liem RI, Lanzkron S, D Coates T, et al. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv 2019; 3: 3867–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv 2020; 4: 327–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kanter J, Liem RI, Bernaudin F, et al. American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv 2021; 5: 3668–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.African Centre of Excellence for Maternal and Child Health. Sickle cell disease in Africa. http://ceasamef.sn/wp-content/uploads/2019/09/Guide-La-Drepanocytose-en-Afrique-senegal-.pdf (accessed Jan 11, 2023).
- 293.Olatunya OS, Adekile AD. What every physician should know about the national guidelines for the control and management of sickle cell disease and the parent handbook for sickle cell disease in Nigeria. Niger J Clin Pract 2017; 20: 123–25. [DOI] [PubMed] [Google Scholar]
- 294.Wagner EH, Austin BT, Von Korff M. Organizing care for patients with chronic illness. Milbank Q 1996; 74: 511–44. [PubMed] [Google Scholar]
- 295.Mainous AG 3rd, Rooks B, Tanner RJ, Carek PJ, Black V, Coates TD. Shared care for adults with sickle cell disease: an analysis of care from eight health systems. J Clin Med 2019; 8: 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Brodsky MA, Rodeghier M, Sanger M, et al. Risk factors for 30-day readmission in adults with sickle cell disease. Am J Med 2017; 130: 601.e9–15. [DOI] [PubMed] [Google Scholar]
- 297.Crego N, Douglas C, Bonnabeau E, et al. Sickle-cell disease co-management, health care utilization, and hydroxyurea use. J Am Board Fam Med 2020; 33: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.National Institute for Children’s Health Quality. Improving transitions in care saves lives. https://nichq.org/insight/improving-transitions-care-saves-lives (accessed June 8, 2023).
- 299.Cronin RM, Mayo-Gamble TL, Stimpson SJ, et al. Adapting medical guidelines to be patient-centered using a patient-driven process for individuals with sickle cell disease and their caregivers. BMC Hematol 2018; 18: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.University of Paris Cité. Online university diploma on sickle cell disease. https://odf.u-paris.fr/fr/offre-de-formation/diplome-d-universite-1/sciences-technologies-sante-STS/du-enseignement-on-line-sur-la-drepanocytose-I73G5WVE.html (in French; accessed Jan 11, 2023).
- 301.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998; 91: 288–94. [PubMed] [Google Scholar]
- 302.Fullerton HJ, Adams RJ, Zhao S, Johnston SC. Declining stroke rates in Californian children with sickle cell disease. Blood 2004; 104: 336–39. [DOI] [PubMed] [Google Scholar]
- 303.Enninful-Eghan H, Moore RH, Ichord R, Smith-Whitley K, Kwiatkowski JL. Transcranial Doppler ultrasonography and prophylactic transfusion program is effective in preventing overt stroke in children with sickle cell disease. J Pediatr 2010; 157: 479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Sacco RL, Adams R, Albers G, et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Stroke 2006; 37: 577–617. [DOI] [PubMed] [Google Scholar]
- 305.Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011; 42: 227–76. [DOI] [PubMed] [Google Scholar]
- 306.Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2019; 50: e344–418. [DOI] [PubMed] [Google Scholar]
- 307.Ghafuri DL, Abdullahi SU, Dambatta AH, et al. Establishing sickle cell disease stroke prevention teams in Africa is feasible: program evaluation using the RE-AIM framework. J Pediatr Hematol Oncol 2022; 44: e56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Ambrose EE, Latham TS, Songoro P, et al. Hydroxyurea with dose escalation for primary stroke risk reduction in children with sickle cell anaemia in Tanzania (SPHERE): an open-label, phase 2 trial. Lancet Haematol 2023; 10: e261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Adamski C, Terhaar C, DaRe J, Fernandes P, Settler C, Dohany L. Hemoglobinopathy evaluation in routine carrier testing: results to inform guidelines and protocols [5O]. Obstet Gynecol 2019; 133: 163.30531566 [Google Scholar]
- 310.Moerdler S, Fraint E, Silver E, et al. Prenatal hemoglobinopathy screening practices: areas for improvement. Blood 2019; 134 (suppl 1): 2298 (abstr). [Google Scholar]
- 311.Alayed N, Kezouh A, Oddy L, Abenhaim HA. Sickle cell disease and pregnancy outcomes: population-based study on 8.8 million births. J Perinat Med 2014; 42: 487–92. [DOI] [PubMed] [Google Scholar]
- 312.Odum CU, Anorlu RI, Dim SI, Oyekan TO. Pregnancy outcome in HbSS-sickle cell disease in Lagos, Nigeria. West Afr J Med 2002; 21: 19–23. [PubMed] [Google Scholar]
- 313.Wilson NO, Ceesay FK, Hibbert JM, et al. Pregnancy outcomes among patients with sickle cell disease at Korle-Bu Teaching Hospital, Accra, Ghana: retrospective cohort study. Am J Trop Med Hyg 2012; 86: 936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Muganyizi PS, Kidanto H. Sickle cell disease in pregnancy: trend and pregnancy outcomes at a tertiary hospital in Tanzania. PLoS One 2013; 8: e56541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Hoyert DL, Miniño AM. Maternal mortality in the United States: changes in coding, publication, and data release, 2018. Natl Vital Stat Rep 2020; 69: 1–18. [PubMed] [Google Scholar]
- 316.Knight M, Bunch K, Kenyon S, Tuffnell D, Kurinczuk JJ. A national population-based cohort study to investigate inequalities in maternal mortality in the United Kingdom, 2009–17. Paediatr Perinat Epidemiol 2020; 34: 392–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Oppong SA, Asare EV, Olayemi E, et al. Multidisciplinary care results in similar maternal and perinatal mortality rates for women with and without SCD in a low-resource setting. Am J Hematol 2019; 94: 223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Asare EV, Olayemi E, Boafor T, et al. Implementation of multidisciplinary care reduces maternal mortality in women with sickle cell disease living in low-resource setting. Am J Hematol 2017; 92: 872–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Oppong SA, Stewart J-A, DeBaun MR. Management of pregnant women and newborns with sickle cell disease. In: Gladwin M, Kato GJ, Novelli EM, eds. Sickle Cell Disease. Beijing: McGraw Hill, 2021: 195–211. [Google Scholar]
- 320.Royal College of Obstetricians and Gynaecologists. No G. management of sickle cell disease in pregnancy. London: Royal College of Obstetricians and Gynaecologists, 2011. [Google Scholar]
- 321.Amer YS, Sabr Y, ElGohary GM, et al. Quality assessment of evidence-based clinical practice guidelines for the management of pregnant women with sickle cell disease using the AGREE II instrument: a systematic review. BMC Pregnancy Childbirth 2020; 20: 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.ACOG Committee on Obstetrics. ACOG practice bulletin no. 78: hemoglobinopathies in pregnancy. Obstet Gynecol 2007; 109: 229–37. [DOI] [PubMed] [Google Scholar]
- 323.University of Paris Cité. International francoafrican interuniversity diploma—controlling sickle cell disease. https://odf.u-paris.fr/fr/offre-de-formation/diplome-d-universite-1/sciences-technologies-sante-STS/du-enseignement-on-line-sur-la-drepanocytose-I73G5WVE.html (in French; accessed Jan 11, 2023).
- 324.Escobar Alvarez SN, Myers ER. Impact of a grant program to spur advances in sickle cell disease research. Blood Adv 2021; 5: 3855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Scholarships Broxmeyer H. and awards ensure the future of hematology. Hematologist 2010; published online May 1. 10.1182/hem.V7.3.6301. [DOI] [Google Scholar]
- 326.Luo L, King AA, Carroll Y, et al. Electronic health record-embedded individualized pain plans for emergency department treatment of vaso-occlusive episodes in adults with sickle cell disease: protocol for a preimplementation and postimplementation study. JMIR Res Protoc 2021; 10: e24818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Hankins JS, Shah N, DiMartino L, et al. Integration of mobile health into sickle cell disease care to increase hydroxyurea utilization: protocol for an efficacy and implementation study. JMIR Res Protoc 2020; 9: e16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Duckworth L, Black LV, Ezmigna D, et al. Spirometry use in patients with sickle cell disease with and without asthma and acute chest syndrome: a Multicenter Study. eJHaem 2020; 1: 239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Price DW, Wagner DP, Krane NK, et al. What are the implications of implementation science for medical education? Med Educ Online 2015; 20: 27003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.McGaghie WC, Barsuk JH, Cohen ER, Kristopaitis T, Wayne DB. Dissemination of an innovative mastery learning curriculum grounded in implementation science principles: a case study. Acad Med 2015; 90: 1487–94. [DOI] [PubMed] [Google Scholar]
- 331.Glasgow RE, Vogt TM, Boles SM. Evaluating the public health impact of health promotion interventions: the RE-AIM framework. Am J Public Health 1999; 89: 1322–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Shelton RC, Chambers DA, Glasgow RE. An extension of RE-AIM to enhance sustainability: addressing dynamic context and promoting health equity over time. Front Public Health 2020; 8: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.