

Abstract
Targeted Meitner-Auger Therapy (TMAT) has potential for personalized treatment thanks to its subcellular dosimetric selectivity, which is distinct from the dosimetry of β− and α particle emission based Targeted Radionuclide Therapy (TRT). To date, most clinical and preclinical TMAT studies have used commercially available radionuclides. These studies showed promising results despite using radionuclides with theoretically suboptimal photon to electron ratios, decay kinetics, and electron emission spectra. Studies using radionuclides whose decay characteristics are considered more optimal are therefore important for evaluation of the full potential of Meitner-Auger therapy; 119Sb is among the best such candidates. In the present work, we develop radiochemical purification of 120Sb from irradiated natural tin targets for TMAT studies with 119Sb.
Keywords: Antimony-119, Targeted Meitner-Auger Therapy, natural tin, proton irradiation, liquid-liquid extraction
Graphical Abstract:
1. Introduction
Targeted radionuclide therapy (TRT) has garnered attention in basic research and commercial sectors, accelerating the search for radionuclides with optimal therapeutic decay properties. TRT uses selective delivery systems (e.g. peptides, antibodies or other bio vectors) in combination with therapeutic radionuclides (β−, α and Meitner-Auger electron emitters) for personalized and localized treatment [1,2]. While in recent years β− and α emitters have gained much clinical popularity, notably 177Lu and 225Ac respectively, Meitner-Auger electron (MAE) emitters have been often overlooked as therapeutic candidates despite potential to offer the most selective treatment on the subcellular level [3–5].
MAE decay follows capture (EC) or isomeric transition. The energy released by this de-excitation of a higher electron energy state filling an inner electron shell vacancy is reinvested into the emission of an electron from the same atom, causing an additional electron vacancy. The process repeats, causing an electron cascade [3]. The kinetic energy of each ejected electron corresponds to the difference in energy between the energy released during electron shell vacancy filling and the ionization energy of the electron. Similarly, conversion electrons are emitted through an analogous process (called internal conversion) during nuclear de-excitations following radioactive decay. As the source of energy for electron emission is the (relatively small) difference in electron/nucleon shell energies, resultant kinetic energy for MAE’s and CE’s range from tens of eV to tens of keV [6] compared to β− (0.1–2.2 MeV) and α emission (5–8 MeV). While the low energy makes it unlikely that membrane targeted MAEs will reach target cell DNA, MAE therapy does not suffer from dose range effects [7]. Due to high particle energy and low linear energy transfer (LET), β− therapy is plagued with dose range effects, resulting in energy absorption within a large area (0.5–10 mm) surrounding decay location [8]. Alpha-emitting radionuclides have a much shorter particle range (40–100 μm), with the high particle energy accompanied by very high LET. Unfortunately, large-scale production of α emitters is unestablished and requires reactors or high-energy cyclotrons for production, which are uncommon and in high demand. Available generators can produce only limited quantities and often require decades to produce [9–12]. The advantage of MAE therapy over β− and α therapy is not only production capacity but also particle range and formation of multiple electrons (cascade) per decay. MAEs typically have electron ranges less than the length of one cell (<10 μm) [8], potentially enabling them to treat single cell metastases.
Current literature in MAE therapy primarily discusses application of commercially available candidates such as 111In, 67Ga, 125I, and 99mTc, which have clinical precedence. While these γ-emitters do undergo MAE emission, their other properties (e.g. other emissions or half-life) may limit their application for TMAT. Despite these properties, studies yield encouraging therapeutic results, demonstrating antitumor efficacy and induction of double strand DNA breaks [13–17].
Antimony-119 (119Sb, t1/2 = 38.19 h, EC = 100%) is one of the most potent radionuclides for TMAT [3,18,19]. The half-life of 119Sb is convenient for radiopharmaceutical preparation and administration. Decaying to stable 119Sn, 119Sb releases a high number (24) of Meitner-Auger and conversion (<50 keV) electrons, and importantly, the decay has a low photon-to-electron energy ratio of 0.9, expanding the therapeutic window [3].
It was shown previously that production of clinically relevant quantities of 119Sb is efficiently possible with low-energy cyclotrons via 119Sn(p,n)119Sb [21,22]. Despite being a promising TMAT candidate, few 119Sb research reports have focused on production, purification, and complexation [18]. Separation of a desired radionuclide from bulk target material often involves exploitation of chemical differences between the two species and can be achieved through a wide variety of techniques including solid-phase chromatography, liquid-liquid extraction, precipitation, complexation, and thermal diffusion, among others [23–27]. While each approach has advantages and downfalls, a robust separation technique requires reproducibility. Radioantimony/tin separations reported in the literature highlight consistency challenges. Sadeghi et al. report a technique [28] based on silica-gel column chromatography with HCl eluent; however, not only does this presumably lead to a relatively high concentration of silica in the final solution [29], but the technique is not reproducible [22]. Another study reported a more concrete method based on previously established anion-exchange chromatography [30]. Despite the successful separation, reported chromatograms are drastically different from those of the original paper, perhaps as a result of different anion-exchange solid phases [21,22]. It was also noted that timing between target dissolution and breakthrough have a “critical impact on the success of the separation” with increased time increasing tin breakthrough, likely as a result of changing of oxidation states or hydrolysis [21,22]. These consistency issues motivate development of a reliable separation method. Given historic success with liquid-liquid extraction [31,32], this method was selected for further study.
In the present work, we report irradiation of natSn to produce a broad array of antimony radioisotopes to study radiochemical separation. Anticipating biological use, liquid-liquid extraction was developed to purify radioantimony from bulk tin target material. Furthering this separation technique can be adopted for production of isotopically pure 119Sb by irradiation of enriched 119Sn.
2. Materials and Methods
2.1. General
All solvents, reagents, and resins were purchased from commercial suppliers (Sigma-Aldrich, Fisher Scientific, TCI America, Alfa Aesar, AK Scientific, Fluka) and used as received. Water used was ultrapure (18.2 MΩ cm−1 at 298 K, Milli-Q, Millipore, Billerica, MA). Irradiations were performed using the University of Wisconsin-Madison GE PET trace (Madison, Wisconsin, USA) and the TR13 cyclotron at TRIUMF (Vancouver, Canada). At the University of Wisconsin-Madison, high purity germanium (HPGe) gamma spectrometry was conducted using an aluminum-windowed detector (Ortec, Knoxville, Tennessee) with Canberra (Concord, Ontario) Model 2025 research amplifier and multichannel analyzer. This system was energy and efficiency calibrated using 241Am, 133Ba, 152Eu, 137Cs, and 60Co sources (Amersham PLC, Little Chalfont, UK). The full width at half maximum resolution is 1.8 keV at 1333 keV. At TRIUMF, an N-type co-axial HPGe gamma spectrometer from Canberra was fitted with a 0.5 mm beryllium window as previously described [33]. Detector energy, width, and efficiency calibrations were performed using a 152Eu and 133Ba source. Inductively coupled plasma mass spectrometry (ICP-MS) analysis was conducted at TRIUMF using an Agilent 8900 Triple Quadrupole ICP-MS. All measurements were acquired in helium mode.
Caution!
Antimony and Sn radioisotopes are produced by proton irradiation of natSn targets as listed in Table 1. All radionuclides are gamma emitters. Proper radiation safety procedures and shielding were implemented when handling these radionuclides in laboratories approved for radioactive material.
Table 1.
Abundance of natural tin isotopes and their produced (p, n) and (p, p*) radionuclides.
| Isotope | Natural Abundance (%)a | Radionuclide Produced via proton induced nuclear reactions |
|---|---|---|
|
| ||
| 117Sn | 7.68 | 117mSn (t1/2 = 14.0 d) |
| 118Sn | 24.22 | 118Sb (t1/2 = 3.6 min) |
| 119Sn | 8.59 | 119Sb (t1/2 = 38.2 h) |
| 120Sn | 32.58 | 120mSb (t1/2 = 5.7 d) |
| 122Sn | 4.63 | 122Sb (t1/2 = 2.7 d) |
Ref [34].
2.2. Irradiation of Tin Target
Natural tin targets (3.0 cm diameter, 0.127 mm thick, acquired from Sigma-Aldrich) were irradiated with protons (entrance energy 16 MeV for GE PETtrace and 12.8 MeV for TR13) for 1 h with a current of 5 μA.
The foil was water cooled (GE PET trace) or cooled by a double helium-cooling window configuration (TR13) with one helium jet cooling the 25 μm thick aluminum foil separating the target from the cyclotron vacuum, and the other one the front of the tin target. Irradiated targets were left in the cyclotron target area for 2 hours following irradiation. The target was then removed from the cyclotron and transported to the shielded fume hood for further radiochemical processing. The foil was trimmed to remove non-irradiated portions and reduce tin mass.
2.3. Liquid-Liquid Extraction
Target foils (between 400–480 mg after trimming) were dissolved in 12 mL of HCl (12 M) and allowed to stand in a covered (not sealed) 16 mL borosilicate KIMAX tube at room temperature overnight. 4 mL of the target solution was set aside, while 8 mL was transferred into a 50 mL Falcon centrifuge tube. A small volume of HCl (12 M, 800 μL) was used to rinse the KIMAX tube prior to adding 8 mL of the target solution. H2O2 (30% w/w, contains inhibitor, 800 μL) was added to the target solution to oxidize Sb(III) and Sn(II) to Sb(V) and Sn(IV). The solution was allowed to sit for approximately 30 minutes. Dibutyl ether (10 mL) was added to the target solution (8.8 mL), and the biphasic solution vortexed for 15 min. After the solution settled, phases were separated by pipetting organic phase from the Falcon centrifuge tube into a new 50 mL Falcon tube. Of the initial 10 mL dibutyl ether, 9 mL were collected to ensure no aqueous contamination. Next, two washing steps of the organic phase were conducted by adding 9 mL of HCl (10 M) to the dibutyl ether and vortexing the solution for 10 min before separating as previously described. For the first wash, no dibutyl ether was sacrificed (i.e., 9 mL were recovered upon separation). After the second wash, 1 mL of dibutyl ether was sacrificed, leaving 8 mL of dibutyl ether solution. Finally, back-extraction was completed by adding 8 mL of sodium citrate solution (0.1 M, pH 5.5) to the organic phase and vortexing for 30 min. Phases were separated as described above, obtaining 7 mL of citrate solution.
Gamma spectroscopy on six samples was carried out after separation (target solution, extracted target solution, HCl wash #1, HCl wash #2, extracted ether solution and final back-extracted solution). One milliliter of each solution was added to separate 2 mL glass vials to ensure counting geometry was compatible with calibration. Solutions were measured with gamma spectrometer positioned 15 cm away from detector perpendicularly in the measurement chamber, and counts were acquired until activity error reached below 10%.
ICP-MS analysis of six non-irradiated tin targets was performed on samples collected throughout the liquid-liquid extraction process. Targets were dissolved and treated as described above, without setting 4 mL of the target solution aside, starting with 12 mL of the target solution, reserving one milliliter of the target solution after each separation step, resulting in 9 mL of the citrate solution recovered. The samples were dried down overnight and converted to a 2% nitric acid matrix for analysis.
3. Results and Discussion
3.1. Target Irradiation
Though ideal for targeted radionuclide therapy application, the low energy photon emissions of 119Sb are difficult to quantify via standard HPGe detector γ-spectroscopy. From a financial perspective, radionuclidically pure 119Sb via (p,n) nuclear reaction requires expensive isotopically enriched 119Sn (~8.6% natural abundance—necessitating material recovery beyond the scope of this work). Because of these reasons, reported experiments used radionuclides produced via proton bombardment of a natural tin target (Figure 1) which is ideally suited for developing radiochemical procedure and chelation.
Figure 1.
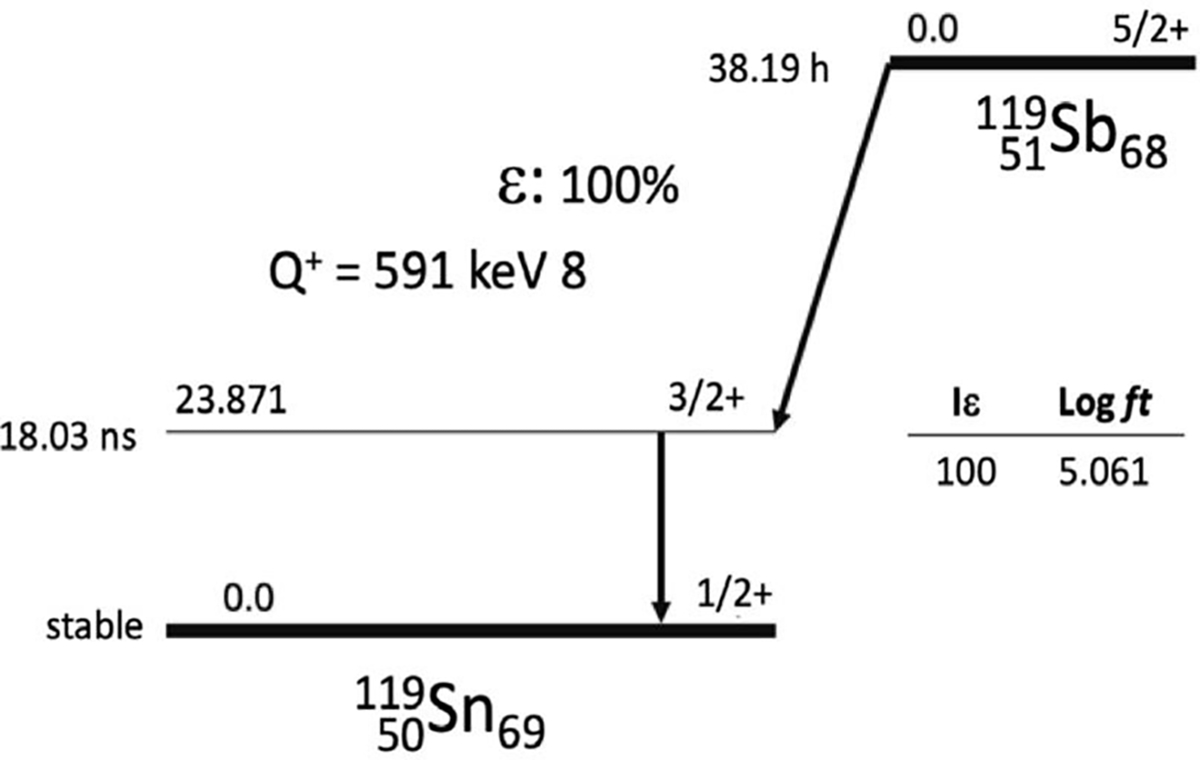
Decay scheme of 119Sb [20].
According to SRIM calculations, the beam deposits 1.7 MeV into the foil (for TR13). The 5 μA beam increases the temperature of the target to no more than 160°C, far away from the tin melting point of 232°C. Due to the diverse isotopic makeup of natural tin and their moderate (p,x) cross-sections for low energy protons,[28] several radionuclides are produced (Table 1). Following a suitable cooling period (>28 h), 120mSb (t1/2 5.76 d) and 117mSn (t1/2 14.0 d) activity remains to be used as tracers. Decay corrected to EOB for 12.8 MeV incident beam energy, (1.42 ± 0.08) MBq of 120mSb and (28.5 ± 1.1) kBq of 117mSn were produced (n = 3), and were slightly higher than TENDL calculation – 1.11 MBq of 120mSb and 25.0 kBq of 117mSn [35]. The presence of these two radiotracers allows monitoring of radiochemical separation using non-destructive γ-spectroscopy.
3.2. Purification of 120mSb by Liquid-Liquid Extraction
Figure 2 illustrates the technique used to separate radioantimony from bulk tin target material. It is crucial that H2O2 is added to the target solution shorty before performing the separation (as opposed to during target dissolution) otherwise the yield will be drastically reduced. We hypothesize this is necessary to ensure all antimony is oxidized to the pentavalent oxidation state. Once this has occurred, [120mSb]Sb(V) can be selectively extracted from the aqueous solution with dibutyl ether - only 10 minutes of vortexing is required for (near) quantitative extraction. In strong HCl, the species present in solution are [SnCl6]2− and [SbCl6]−; thus selective uptake of the latter into ether is likely a result of more diffuse charge distribution, resulting in a more hydrophobic species that prefers organic solvation [36]. Following separation of phases, two washing steps with 10 M HCl (not shown in diagram) help remove minute amounts of Sn(IV) that may have partitioned into the organic phase. Lastly, since radiolabeling is most desirably performed from aqueous solution, the Sb(V) is back-extracted into a 0.1 M sodium citrate solution, whose pH (5.5) is essential. The citrate is not only required for successful back-extraction but also mitigates Sb(V) hydrolysis, as is done for Sb(III) [37]. The success of this procedure is quantified in Table 2 and can be seen in Figure 3, where γ-ray spectra of the target solution, extracted target solution and back extracted solution are shown. The target solution most notably shows 120mSb (Eγ = 90 keV, 197 keV) and 117mSn (Eγ = 159 keV) with activities 1.42 MBq and 28.5 kBq, respectively. The extracted target solution has retained only a small amount of 120mSb (2.4%) and vast majority of 117mSn (>95%), as can be qualitatively noted by the relative intensities of peaks. Finally, the back extracted solution contains no observable 117mSn. The elemental concentration of stable tin in the final solution was determined to be lower than 18 ppm (<170 μg in total), showing that more than 99.95% of the bulk tin target is removed in the separation process. Net yield of 120mSb in the final solution (relative to target solution) was (69 ± 2) %. When correcting for sacrificed volume, the yield is >90% (Table 2). The amount of stable antimony present in the final solution was determined to be <40 μg. Experiments negating sacrificial volume were conducted providing radiochemical yield (94.6 ± 4.0) % (N=3) with 117mSn activity below limit of detection 480 Bq, which sets an upper limit of <0.1% original target Sn mass remaining in final solution.
Figure 2.
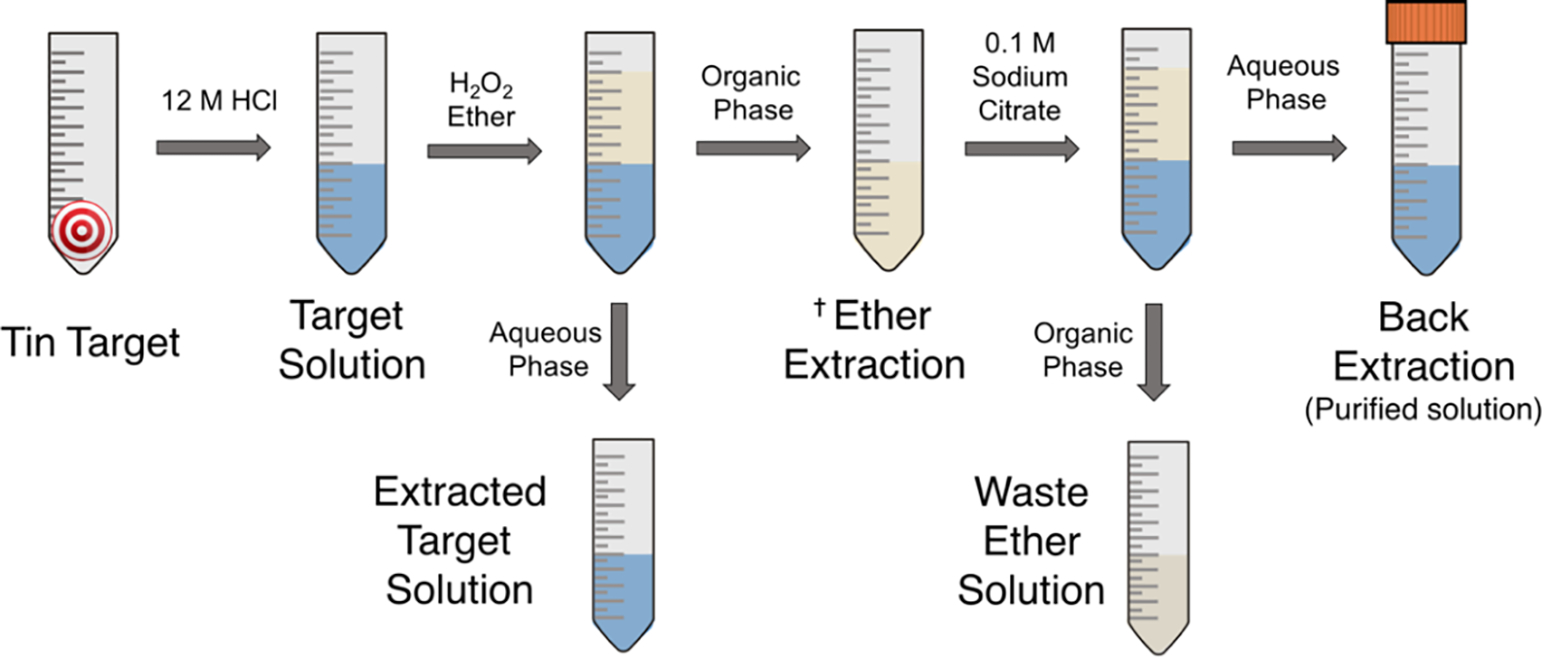
Visual representation of radioantimony purification.✝Wash ether with equal volume 10 M HCl (x2) prior to back extraction.
Table 2.
Activities of 120mSb and 117mSn expressed as a fraction over total target solution activity.a
| Solution | %120mSb Activity (100/initial activity) | % 117mSn Activity (100/initial activity) |
|---|---|---|
|
| ||
| Extracted Target Solution | 2.4% ± 0.4% | 98% ± 4% |
| HCl Wash #1 | 0.38% ± 0.07 | N.D |
| HCl Wash #2 | 0.28 ± 0.05 | N.D |
| Extracted Ether | 0.07%b | N.D |
| Final Citrate Solution (net total)c | 69% ± 2% | N.D |
| Final Citrate Solution (corrected)d | 95% ± 2% | N.D |
Values calculated using activity concentration (Bq/mL) and volume, without correcting for sacrificed volume unless otherwise specified. Activity then divided by initial activity of purified target solution. Reported error is standard deviation (n = 3). N.D. = not detected;
Due to N.D. in two trials, n = 1.
Percent isolated activity over starting activity;
Percent activity corrected for sacrificed volume over starting activity.
Figure 3.
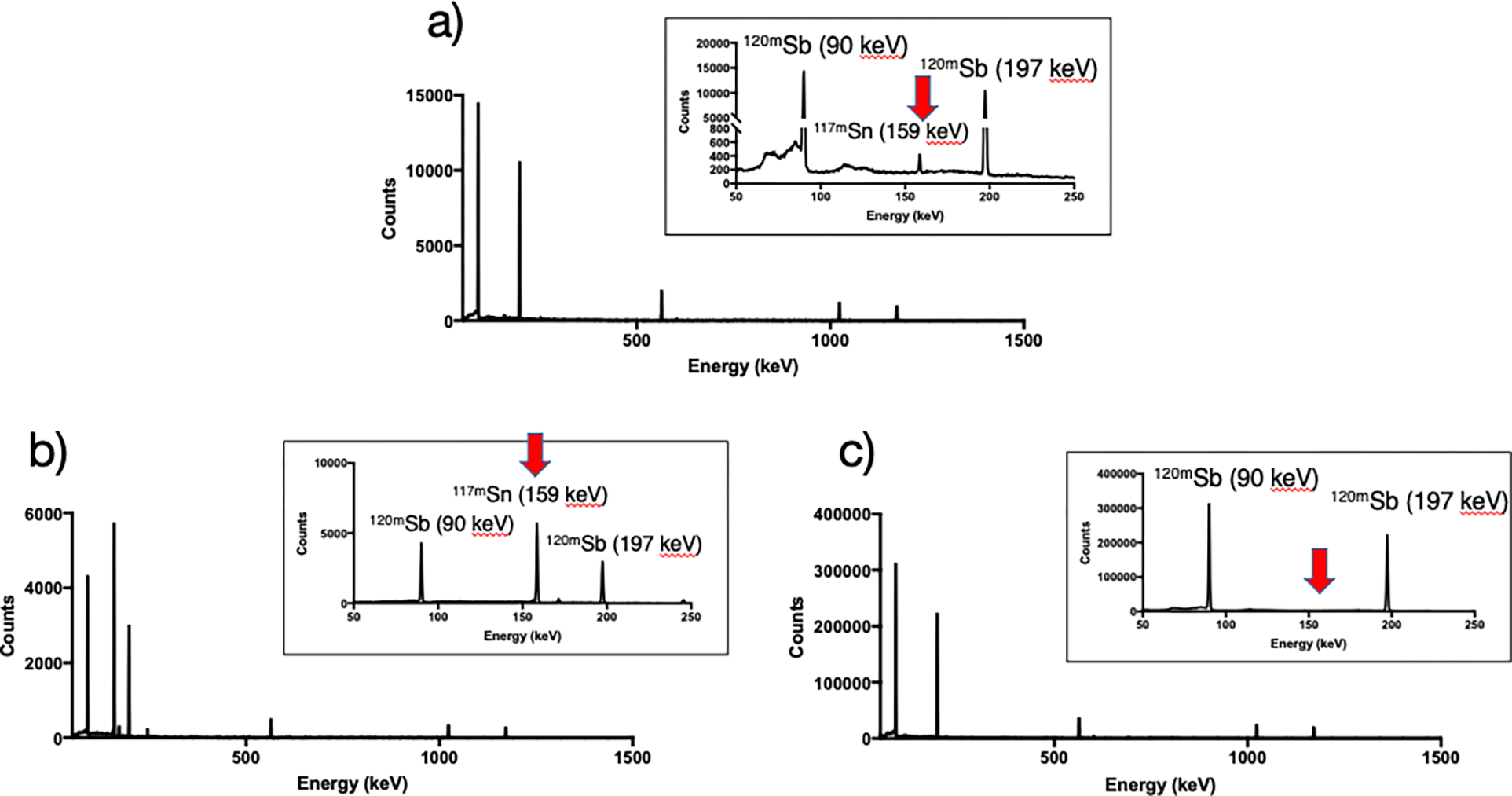
γ-ray spectra of a) target solution b) extracted target solution c) back extracted solution.
Conclusions
A natural tin target was irradiated with 12.8 MeV or 16 MeV protons to produce a variety of antimony radioisotopes. Following a two-day waiting period, the primary radionuclides of interest were 120mSb and 117mSn. A radiochemical purification method to separate radioantimony from tin using liquid-liquid extraction was developed and provided >90% radiochemical yield (corrected for sacrified volume) with separation factor of >1700. Proposed separation method can be further applied for separation of 119Sb from proton irradiated 119Sn target.
Acknowledgments
This work was supported by Canadian Institute for Health Research (CIHR) via research project (GR021373) and the Natural Sciences and Engineering Research Council (NSERC) of Canada via Discovery Grants RGPIN-2018–04997 (VR), RGPIN-2018–04772 (CO), RGPIN-2019–07207(CR) and RGPIN-2022–03388 (CH), plus CGS-M/CGS-D scholarships and a CREATE IsoSiM at TRIUMF stipend (TIK). TRIUMF receives funding via a contribution agreement with the National Research Council of Canada. We would also like to thank the TR13 Cyclotron Operations Group consisting of Toni Epp, Ryley Morgan, Spencer Staiger, and led by David Prevost for regular irradiations of tin targets. APO gratefully acknowledges support from the USA NIH T32 NRSA Institutional Predoctoral Training Fellowship 2T32CA009206–41 and NIH F31 Ruth L. Kirschstein Predoctoral Individual National Research Service Award F31CA239617. JWE gratefully acknowledges support from the NIH NCI P01CA250972. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- [1].Sgouros G, Bodei L, McDevitt MR, Nedrow JR. Radiopharmaceutical therapy in cancer: clinical advances and challenges. Nat Rev Drug Discov 2020;19:589–608. doi: 10.1038/s41573-020-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kostelnik TI, Orvig C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem Rev 2019;119:902–56. doi: 10.1021/acs.chemrev.8b00294. [DOI] [PubMed] [Google Scholar]
- [3].Filosofov D, Kurakina E, Radchenko V. Potent candidates for Targeted Auger Therapy: Production and radiochemical considerations. Nucl Med Biol 2021;94–95:1–19. doi: 10.1016/j.nucmedbio.2020.12.001. [DOI] [PubMed] [Google Scholar]
- [4].Ku A, Facca VJ, Cai Z, Reilly RM. Auger electrons for cancer therapy – a review. EJNMMI Radiopharm Chem 2019;4. doi: 10.1186/s41181-019-0075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cornelissen B A Vallis K. Targeting the Nucleus: An Overview of Auger-Electron Radionuclide Therapy. Curr Drug Discov Technol 2010;7:263–79. doi: 10.2174/157016310793360657. [DOI] [PubMed] [Google Scholar]
- [6].Eckerman K, Endo A. ICRP Publication 107. Nuclear decay data for dosimetric calculations. Ann ICRP 2008;38:7–96. doi: 10.1016/j.icrp.2008.10.004. [DOI] [PubMed] [Google Scholar]
- [7].Buchegger F, Perillo-Adamer F, Dupertuis YM, Bischof Delaloye A. Auger radiation targeted into DNA: a therapy perspective. Eur J Nucl Med Mol Imaging 2006;33:1352–63. doi: 10.1007/s00259-006-0187-2. [DOI] [PubMed] [Google Scholar]
- [8].Ramogida CF, Orvig C. Tumour targeting with radiometals for diagnosis and therapy. Chem Commun 2013;49:4720–39. doi: 10.1039/c3cc41554f. [DOI] [PubMed] [Google Scholar]
- [9].Radchenko V, Morgenstern A, Jalilian A, Ramogida C, Cutler CS, Duchemin C, et al. Production and supply of alpha particles emitting radionuclides for Targeted Alpha Therapy (TAT). J Nucl Med 2021:jnumed.120.261016. doi: 10.2967/jnumed.120.261016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ferrier MG, Radchenko V, Wilbur DS. Radiochemical aspects of alpha emitting radionuclides for medical application. Radiochim Acta 2019;107:1065–85. doi:doi: 10.1515/ract-2019-0005. [DOI] [Google Scholar]
- [11].Makvandi M, Dupis E, Engle JW, Nortier FM, Fassbender ME, Simon S, et al. Alpha-Emitters and Targeted Alpha Therapy in Oncology: from Basic Science to Clinical Investigations. Target Oncol 2018;13:189–203. doi: 10.1007/s11523-018-0550-9. [DOI] [PubMed] [Google Scholar]
- [12].Morgenstern A, Abbas K, Bruchertseifer F, Apostolidis C. Production of Alpha Emitters for Targeted Alpha Therapy. Curr Radiopharm 2008;1:135–43. doi: 10.2174/1874471010801030135. [DOI] [Google Scholar]
- [13].Morgenroth A, Dinger C, Zlatopolskiy BD, Al-Momani E, Glatting G, Mottaghy FM, et al. Auger electron emitter against multiple myeloma — targeted endo-radio-therapy with 125I-labeled thymidine analogue 5-iodo-4′-thio-2′-deoxyuridine. Nucl Med Biol 2011;38:1067–77. doi: 10.1016/j.nucmedbio.2011.02.018. [DOI] [PubMed] [Google Scholar]
- [14].Rosenkranz AA, Slastnikova TA, Khramtsov YV., Karyagina TS, Georgiev GP, Sobolev AS. Antitumor efficacy of Auger electron emitter 111In delivered by modular nanotransporter into the nuclei of cells with folate receptor overexpression. Dokl Biochem Biophys 2017;473:85–7. doi: 10.1134/S1607672917020016. [DOI] [PubMed] [Google Scholar]
- [15].Koumarianou E, Slastnikova TA, Pruszynski M, Rosenkranz AA, Vaidyanathan G, Sobolev AS, et al. Radiolabeling and in vitro evaluation of 67Ga-NOTA-modular nanotransporter – A potential Auger electron emitting EGFR-targeted radiotherapeutic. Nucl Med Biol 2014;41:441–9. doi: 10.1016/j.nucmedbio.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Balagurumoorthy P, Xu X, Wang K, Adelstein SJ, Kassis AI. Effect of distance between decaying 125I and DNA on Auger-electron induced double-strand break yield. Int J Radiat Biol 2012;88:998–1008. doi: 10.3109/09553002.2012.706360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tavares AAS, Tavares JMRS. 99mTc Auger electrons for targeted tumour therapy: A review. Int J Radiat Biol 2010;86:261–70. doi: 10.3109/09553000903564083. [DOI] [PubMed] [Google Scholar]
- [18].Randhawa P, Olson AP, Chen S, Gower-Fry KL, Hoehr C, Engle JW, et al. Meitner-Auger Electron Emitters for Targeted Radionuclide Therapy: Mercury-197m/g and Antimony-119. Curr Radiopharm 2021. doi: 10.2174/1874471014999210111201630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bernhardt P, Forssell-Aronsson E, Jacobsson L, Skarnemark G. Low-energy electron emitters for targeted radiotherapy of small tumours. Acta Oncol 2001;40:602–8. [DOI] [PubMed] [Google Scholar]
- [20].Symochko DM, Browne E, Tuli JK. Nuclear Data Sheets for A = 119. Nucl Data Sheets 2009;110:2945–3105. doi: 10.1016/j.nds.2009.10.003. [DOI] [Google Scholar]
- [21].Thisgaard H, Jensen M. Production of the Auger emitter 119Sb for targeted radionuclide therapy using a small PET-cyclotron. Appl Radiat Isot 2009;67:34–8. doi: 10.1016/j.apradiso.2008.09.003. [DOI] [PubMed] [Google Scholar]
- [22].Thisgaard H Accelerator based Production of Auger-Electron-emitting isotopes for Radionuclide Therapy. Technical University of Denmark, 2008. [Google Scholar]
- [23].Aardaneh K, Shirazi B. Radiochemical separation of 67Ga from Zn and Cu using the adsorbent resin Amberlite XAD-7. J Radioanal Nucl Chem 2005;265:47–51. doi: 10.1007/s10967-005-0787-5. [DOI] [Google Scholar]
- [24].Cyclotron Produced Radionuclides : Physical Characteristics and Production Methods. Vienna: An International Atomic Energy Agency Publication; 2009. [Google Scholar]
- [25].Sadeghi M, Mokhtari L. Rapid separation of 67,68Ga from 68Zn target using precipitation technique. J Radioanal Nucl Chem 2010;284:471–3. doi: 10.1007/s10967-010-0500-1. [DOI] [Google Scholar]
- [26].Dumortier R, Weber ME, Vera JH. Removal and recovery of gallium from aqueous solutions by complexation with sodium di-(n-octyl) phosphinate. Hydrometallurgy 2005;76:207–15. doi: 10.1016/j.hydromet.2004.11.004. [DOI] [Google Scholar]
- [27].Andrade Martins P de, Osso JA. Thermal diffusion of 67Ga from irradiated Zn targets. Appl Radiat Isot 2013;82:279–82. doi: 10.1016/j.apradiso.2013.08.012. [DOI] [PubMed] [Google Scholar]
- [28].Sadeghi M, Aboudzadeh Rovais MR, Enferadi M, Sarabadani P. Targetry and Radiochemistry for no-carrier-added production of 117,118m,119,120m,112Sb. Nukleonika 2011;56:9–15. [Google Scholar]
- [29].Lenher V, Merrill HB. The solubility of silica. J Am Chem Soc 1917;39:2630–8. doi: 10.1021/ja02257a013. [DOI] [Google Scholar]
- [30].Baluev A V, Mityakhina VS, Krasnikov LV., Galkin BY, Besnosyuk VI. Recovery of antimony-125 from tin-124 irradiated by neutrons. Czechoslov J Phys 2003;53:A417–23. doi: 10.1007/s10582-003-0054-3. [DOI] [Google Scholar]
- [31].White CE, Rose HJ. Separation of Antimony by Solvent Extraction. Anal Chem 1953;25:351–3. doi: 10.1021/ac60074a041. [DOI] [Google Scholar]
- [32].Khorasani SSMA, Khundkar. Solvent extraction of antimony(V) with ethyl acetate. Anal Chim Acta 1959;21:24–8. doi: 10.1016/0003-2670(59)80129-X. [DOI] [Google Scholar]
- [33].Robertson AKH, McNeil BL, Yang H, Gendron D, Perron R, Radchenko V, et al. 232Th-Spallation-Produced 225Ac with Reduced 227Ac Content. Inorg Chem 2020;59:12156–65. doi: 10.1021/acs.inorgchem.0c01081. [DOI] [PubMed] [Google Scholar]
- [34].Livechart - Table of Nuclides -Nuclear Structure and Decay Data n.d.
- [35].Koning AJ, Rochman D, Sublet J-C, Dzysiuk N, Fleming M, van der Marck S. TENDL: Complete Nuclear Data Library for Innovative Nuclear Science and Technology. Nucl Data Sheets 2019;155:1–55. doi: 10.1016/j.nds.2019.01.002. [DOI] [Google Scholar]
- [36].Bonner NA. The Exchange Reaction between Antimony(III) and Antimony (V) in Hydrochloric Acid Solutions. J Am Chem Soc 1949;71:3909–14. doi: 10.1021/ja01180a009. [DOI] [Google Scholar]
- [37].Özer UY, Bogucki RF. Equilibrium studies of antimony(III) chelates in aqueous solution. J Inorg Nucl Chem 1971;33:4143–53. doi: 10.1016/0022-1902(71)80514-6. [DOI] [Google Scholar]