

Abstract
African swine fever (ASF) is an asymptomatic infection of warthogs and bushpigs, which has become an emergent disease of domestic pigs, characterized by hemorrhage, lymphopenia, and disseminated intravascular coagulation. It is caused by a large icosohedral double-stranded DNA virus, African swine fever virus (ASFV), with infection of macrophages well characterized in vitro and in vivo. This study shows that virulent isolates of ASFV also infect primary cultures of porcine aortic endothelial cells and bushpig endothelial cells (BPECs) in vitro. Kinetics of early and late gene expression, viral factory formation, replication, and secretion were similar in endothelial cells and macrophages. However, ASFV-infected endothelial cells died by apoptosis, detected morphologically by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling and nuclear condensation and biochemically by poly(ADP-ribose) polymerase (PARP) cleavage at 4 h postinfection (hpi). Immediate-early proinflammatory responses were inhibited, characterized by a lack of E-selectin surface expression and interleukin 6 (IL-6) and IL-8 mRNA synthesis. Moreover, ASFV actively downregulated interferon-induced major histocompatibility complex class I surface expression, a strategy by which viruses evade the immune system. Significantly, Western blot analysis showed that the 65-kDa subunit of the transcription factor NF-κB, a central regulator of the early response to viral infection, decreased by 8 hpi and disappeared by 18 hpi. Both disappearance of NF-κB p65 and cleavage of PARP were reversed by the caspase inhibitor z-VAD-fmk. Interestingly, surface expression and mRNA transcription of tissue factor, an important initiator of the coagulation cascade, increased 4 h after ASFV infection. These data suggest a central role for vascular endothelial cells in the hemorrhagic pathogenesis of the disease. Since BPECs infected with ASFV also undergo apoptosis, resistance of the natural host must involve complex pathological factors other than viral tropism.
Viral hemorrhagic fevers (VHFs) are considered newly emergent diseases. VHFs are basically distinguishable in two groups according to the occurrence or not of a disseminated intravascular coagulation (DIC) during the infection. Mechanisms leading to DIC are still poorly understood, but it is assumed that the endothelium plays a key role in its occurrence and evolution (32). In nonpathologic conditions, the endothelium maintains a barrier between tissues and blood, and it contributes actively to the control of hemostatic balance by providing a nonthrombogenic surface (50). Activation of endothelium, induced by cytokines (mainly tumor necrosis factor alpha [TNF-α], interferons [IFNs], interleukin-1 [IL-1], IL-6, and IL-8) or pathogenic agents, is characterized by the expression of a proinflammatory and/or a procoagulant phenotype. A common feature of viruses inducing VHFs is that they infect and replicate in cells from the monocyte/macrophage lineage (20). However, during VHFs, there is increasing evidence that viruses damage the endothelium directly by infecting vascular endothelial cells. For example, replication of Ebola and Marburg viruses occurs both in macrophages (15) and in primary endothelial cultures (39), with the viral glycoprotein binding specifically to endothelial cells (51). In dengue virus infection, damage to the endothelium is both by direct infection (5) and by TNF-α produced from infected macrophages (4) but also involves apoptosis (25). The pathogenesis of VHFs is not fully understood, and the interactions between these viruses or viral proteins and endothelial cells are still poorly studied.
African swine fever (ASF) is a VHF caused by a double-stranded DNA virus, African swine fever virus (ASFV). The disease pathogenicity ranges from lethal to moderately virulent or nonvirulent according to virus strains and host species. Highly virulent ASFV isolates such as Malawi are responsible for a lethal contagious VHF in domestic pigs, whereas it is a persistent infection in African warthogs (Phacochoerus aethiopicus) and bushpigs (Potamochoerus porcus) (43), considered ASFV natural reservoirs (3). Histopathology studies of ASFV-infected pig tissue revealed that the disease is characterized by DIC (47), fibrinolysis (46), and lymphopenia, due to extensive apoptosis in lymph nodes (12, 31). An important feature of the pathology for ASFV involves bystander apoptosis of uninfected lymphocytes, which interestingly has been seen for other VHFs, such as Ebola virus and classical swine fever virus (16, 38).
Although macrophages have long been considered the primary site of viral replication, there is some in vivo evidence that vascular endothelial cells can support replication of the ASFV (18, 35), resulting in their activation and loss with increased vascular permeability and fibrin deposits (19). This study was undertaken to investigate the interactions between ASFV and endothelial cells, to set up an in vitro model to correlate the underlying mechanism for this viral disease with other VHFs. Here, we present novel data that a highly virulent strain of ASFV, Malawi Lil 20/1, productively infected and replicated in primary cultures of porcine aortic endothelial cells (PAECs) and bushpig endothelial cells (BPECs). Moreover, ASFV induced apoptosis in these cells, demonstrated morphologically by nuclear condensation and terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) labeling and biochemically by poly(ADP-ribose) polymerase (PARP) cleavage and annexin V staining. ASFV infection blocked PAEC activation by inhibiting the expression of a proinflammatory phenotype and inducing a procoagulant phenotype. Moreover, infection with ASFV downregulated IFN-α-induced expression of major histocompatibility complex (MHC) class I, a mechanism to prevent antigen presentation and allow infected PAECs to escape from immune detection. Cleavage of PARP occurred by 4 h postinfection (hpi), while a second caspase substrate seen in PAECs undergoing apoptosis, the 65-kDa subunit of the transcription factor NF-κB p65 (24), decreased after 8 hpi. Proteolysis of both was inhibited by z-VAD-fmk, demonstrating that caspase activation occurred rapidly after ASFV infection. Virus production, however, was completed before cells died. We hypothesize that ASFV-induced hemorrhage may result not only from vascular damage by factors secreted from infected macrophages but also as a direct result of the infection of endothelial cells and their subsequent apoptosis.
MATERIALS AND METHODS
Cells and viruses.
Virulent ASFV Malawi Lil 20/1 was isolated from a domestic pig in Lilongwe, Malawi, in 1983 (21). Large amounts of virus were isolated from the spleens of infected pigs and then grown in porcine bone marrow cells as described previously (49). The titer of the virus was determined by hemadsorption on bone marrow cells and concentrated by centrifugation to log108 50% hemadsorption unit (HAD50)/ml. Cells were infected at a multiplicity of infection of 10:1.
PAECs were harvested as described previously (48). Briefly, the aorta and minor vessels from inbred minipigs (SLAd haplotype) were ligated and flushed through with RPMI medium (Life Technologies). The lumen of each vessel was filled with collagenase H (0.5 U/ml; Boehringer Mannheim) and incubated at 37°C for 12 min. PAECs were loosened, media were collected, and cells were pelleted (600 × g, 10 min) and resuspended in culture medium (RPMI 1640 with 20% decomplemented fetal calf serum, 2 mM glutamine, 1 mM sodium pyruvate, penicillin [50 IU/ml] and streptomycin [50 μg/ml]). The cells were seeded on 25-cm2 gelatin-coated flasks. BPEC cell line 1194 was a kind gift from Gillian Smith and Suman Mahan, Heartwater Research Project, Veterinary Research Laboratory, Harare, Zimbabwe (40). BPECs showed factor VIII- and E-selectin-positive staining and grew in a typical endothelial cell cobblestone morphology. Porcine alveolar macrophages (PAMs) from the lungs of normal outbred pigs were obtained by bronchioalveolar lavage with phosphate-buffered saline (PBS)
Virus titrations.
PAECs and BPECs, 2 × 106 each, were infected with ASFV Malawi at 104 HAD50/ml. An aliquot of the supernatant was collected each day for 3 days and titrated by hemadsorption of homologous red blood cells (RBCs) to primary porcine bone marrow cells. In brief, cells (106/100 μl) were added to 96-well microtiter plates. Growth medium (100 μl) with 0.5% washed homologous RBCs was added to each well. Dilutions (10-fold) of sample were added in quadruplicate to the plates and incubated for 6 days. The virus titer was calculated from 50% of cells showing hemadsorption as HAD50/ml.
Antibodies and reagents.
Anti-ASFV monoclonal antibodies (MAbs) were the 4H3 MAb recognizing the major capsid protein vp73 (14) and the C18 MAb recognizing the early protein vp30 (1). A MAb directed against porcine angiotensin-converting enzyme (ACE) (clone ACE 3.1.1) was a gift from R. Auerbach (33). The anti-MHC class I MAb clone 2-27-3 (mouse immunoglobulin G2a) was from J. Lunney (22), anti-E-selectin MAb (clone 1.2B6, mouse immunoglobulin G2a) was from D. Haskard (45), and the rabbit anti-human tissue factor antibody (RPTF2) was a kind gift from J. McVey and A. Dorling (13) (MRC Clinical Sciences Centre, Hammersmith Hospital, London, United Kingdom). The Fluorescein isothiocyanate-labeled annexin V protein was from Bender Med Systems (Boehringer Ingelheim). Human recombinant TNF-α (at 100 IU/ml) was from R & D; recombinant porcine IFN-α (103 IU/μl) was kindly provided by C. La Bonnardière (INRA, Jouy-en-Josas, France) (23). Rabbit anti-PARP antibody was from Cell Signalling Technology, and anti-NF-κB p65 MAb F6 was from Santa Cruz Biotechnology.
Indirect immunofluorescence.
PAECs were grown to 80% confluence on gelatin-coated glass coverslips in 24-well plates and infected for 18 h with ASFV Malawi. For viral protein staining, cells were fixed in 0.25% paraformaldehyde for 30 min, permeablized in 0.5% Nonidet P-40, and blocked in 0.1% gelatin in Tris-buffered saline, pH 7.5. For tissue factor surface staining, live cells were stained at 15°C, fixed, and permeabilized for viral protein staining. Cells were incubated with the primary MAb in 30% goat serum either without or with 0.5% NP-40 for 1 at room temperature, washed well, and then incubated with secondary antibody conjugated to Alexa 488 (green) or 594 (red) for 30 min. Cells were observed with epifluorescent optics. In some cases, nuclei were stained with Hoechst 33258 (Sigma) at 1 μg/ml for 10 min and viewed with a DAPI (4′,6′-diamidino-2-phenylindole) filter.
PAEC labeling by TUNEL.
PAECs were grown on gelatin-precoated coverslips. Cells, either uninfected or infected with ASFV Malawi for 18 h, were fixed with 0.25% paraformaldehyde for 30 min and washed in PBS. PAECs were incubated with proteinase K (5 μg/ml in 10 mM Tris-HCl, pH 7.4) at room temperature for 10 min. Coverslips were rinsed and covered in TdT buffer (30 mM Trisma base [pH 7.2], 140 mM sodium cacodylate, 1 mM cobalt chloride). The in situ cell death detection kit from Boehringer Mannheim was used according to the instructions: 50 μl of the TdT enzyme was mixed with 450 μl of the labeling solution containing fluorescein-conjugated dUTP and added to coverslips for 1 h at 37°C. The reaction was terminated by incubating the coverslips in TB buffer (300 mM sodium chloride, 30 mM sodium citrate) for 15 min at room temperature. After two washes with PBS, PAECs were observed with epifluorescence optics.
Fluorescence-activated cell sorter (FACS) analysis.
Cells were infected or stimulated according to the different experimental conditions and were then pelleted. They were washed twice in PBS, permeabilized, and fixed, and 5 × 105 cells were incubated (45 min, 4°C) with antibody. After this incubation, cells were washed twice in PBS before incubation with the second antibody for 45 min at 4°C. Cells were then washed twice in PBS and analyzed with a FACScan (Becton Dickinson).
Metabolic labeling and immunoprecipitations.
A total of 2 × 106 PAMs or PAECs were labeled for each time point. At the indicated time points, cells (2 × 106) were preincubated with methionine- and cysteine-free media for 30 min at 37°C. They were pulse-labeled for 30 min with 35S-Pro-mix (Amersham). Cells were washed once in PBS and lysed in immunoprecipitation buffer (10 mM Tris [pH 7.8], 0.15 M NaCl, 10 mM iodoacetamide, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml each of leupeptin, pepstatin, chymostatin, and antipain [Boehringer Mannheim]). Lysates were precleared with protein A (Sigma) and immunoprecipitated with antibodies immobilized on protein A-Sepharose. After an overnight incubation, proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis SDS-PAGE and visualized by autoradiograpghy.
RNA isolation and reverse transcription (RT)-PCR.
PAECs (2 × 107 cells for each condition) were incubated with recombinant human TNF-α (5 μg/ml) for either 4 or 18 h with or without ASFV Malawi Lil 20/1 (multiplicity of infection of 10 to 1). Total RNA was isolated by scraping cells into denaturing solution (4 M guanidinium isothiocyanate, 25 mM citrate [pH 7.0], 0.1 M mercaptoethanol, 0.5% sarcosyl). The solution was acidified with 2.0 M sodium acetate (pH 4.0), and water-saturated phenol and chloroform were added. The RNA was extracted from the aqueous layer, precipitated, and quantitated. Total RNA was reverse transcribed into single-stranded cDNA with avian myeloblastosis virus reverse transcriptase and oligo(dT) primers. Amplification of DNA was carried out by PCR with the following primers to porcine sequences: IL-6, 5′-GCTGCTTCTGGTGATGGCTACTGCC-3′ and 5′-TGAAACTCCACAAGACCGGTGGTGA-3′; and IL-8, 5′-AGCCCGTGTCAACATGACTTCC-3′ and 5′-GAATTGTGTTGGCATCTTTACTGAG-3′. Tissue factor (TF) primers were designed from areas of the human sequence with high homology to the bovine sequence: 5′-GGAGTGGGAACCCAAACCCGTCAA-3′ and 5′-TTTTCTCCTTTATCCACATCAATC-3′. DNA was denatured and amplified by cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s with a 5-min final extension at 72°C. A subsaturating number of cycles (10 to 15) allowed a semiquantitative analysis between each treatment. DNA was separated by 1.5% agarose gel electrophoresis and visualized with ethidium bromide.
Western blot analysis.
Cells were infected with ASFV for 0, 4, 8, and 18 h where indicated in the presence of the pan-caspase inhibitor z-Val-Ala-Asp-fmk (z-VAD-fmk-Alexis) and lysed in radioimmunoprecipitation assay buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, 10 mM iodoacetamide, 1 mM phenylmethylsulfonyl fluoride, and 1 μg each of leupeptin, pepstatin, chymotrypsin, and antipain. Lysates were freeze-thawed and sonicated. Protein concentrations were determined by the Pierce BCA protein assay. Proteins were separated by SDS-PAGE on 10% gels and transferred to nitrocellulose membranes (Protran BA 85; Schleicher and Schuell). Filters were blocked with 10% dried milk and incubated with the primary antibody in 5% dried milk, 10% goat serum, and 0.05% Tween 20 for 1 h. PARP was detected with rabbit anti-PARP antibody (Cell Signalling Technology), followed by a horseradish peroxidase-conjugated donkey anti-rabbit secondary antibody (Promega). NF-κB p65 was detected with monoclonal F6 (Santa Cruz), followed by horseradish peroxidase-conjugated goat anti-mouse secondary antibody.
RESULTS
PAECs and BPECs are infected by virulent isolates of ASFV.
The endothelium-restricted marker, ACE, was used to characterize the phenotype of cells isolated from porcine aorta. Figure 1A shows PAECs were positively stained by indirect immunofluorescence with the anti-ACE MAb. BPECs have previously been described as an endothelial cell line and stained positive with an anti-factor VIII MAb (40). PAEC and BPEC confluent monolayers were infected for 18 h with ASFV Malawi Lil 20/1 isolate. Early protein expression was detected with MAb C18 directed against vp30, in both PAECs (Fig. 1B) and BPECs (not shown). About 30 to 40% of cells expressed this early viral protein. Infected cells were also stained with a MAb, 4H3, directed against the late viral protein vp73. The typical ASFV perinuclear viral factory was detected in PAECs and BPECs (Fig. 1C and E). Punctuate staining was observed throughout the cytoplasm and at the cell surface, indicating the secretion of individual virions for both PAECs and BPECs (Fig. 1C and E, respectively). On the same slide, nuclear and viral DNA were stained with Hoechst 33258 and visualized with a DAPI filter (Fig. 1D and F). As expected, viral DNA was colocalized with the viral factory in a perinuclear location. Interestingly, infected cells showed a condensation of nuclear DNA, indicative of chromatin condensation seen in apoptotic cells, whereas no nuclear condensation was seen in uninfected cells.
FIG. 1.

ASFV Malawi infection of PAECs and BPECs. (A) Phenotypic characterization of PAECs by immunostaining for ACE, a specific marker of the endothelium. (B to F) ASFV infection of PAECs and BPECs detected by indirect immunofluorescence with anti-ASFV antibodies. Expression in PAECs 18 hpi with ASFV Malawi of the early protein vp30 in cytoplasm (B), and the late protein vp73 in a large perinuclear viral factory and virions in cytoplasm (C) is shown. (D) DAPI stain of condensed nuclear DNA and viral DNA in the same cell as shown in panel C. (E). Expression in BPEC, 18 hpi with ASFV Malawi, of the late protein vp73 in viral factory and virions (F) DAPI stain of condensed nuclear DNA and viral DNA in the same cell as in panel E.
ASFV-infected endothelial cells showed a hemadsorption to porcine RBCs (data not shown). The basis for this is the expression of a CD2 homologue carried by ASFV, designated open reading frame 8-DR in strain Malawi, which localizes to the surface of infected cells and binds to CD58 on RBCs (37). Hemadsorption to RBCs therefore indicated that 8-DR was expressed on the surface of endothelial cells, as is found on infected macrophages (10). The hemadsorption assay was also used to determine the titer of ASFV Malawi secreted from PAECs and BPECs to compare titers with those from PAMs. Quantitation of virus replication with time over 3 days using this hemabsorption assay found that the virus titer from PAECs and BPECs was very similar to that produced from PAMs (Fig. 2) with a virus titer of 107 HAD50/ml from a starting innoculum of 104 HAD50/ml for each cell type.
FIG. 2.
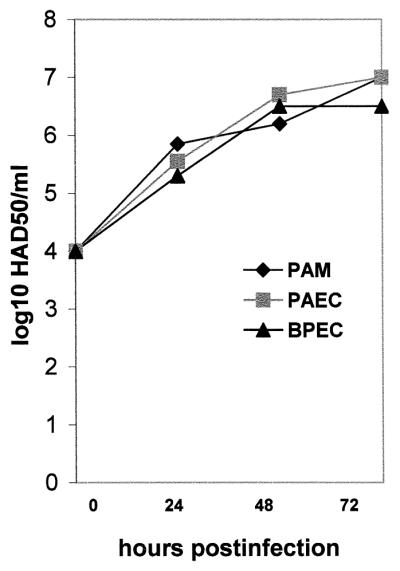
Comparison of ASFV Malawi production from PAECs, BPECs, and PAMs. Cells were innoculated with 104 HAD50/ml, and an aliquot of supernatant was collected each day for 3 days. Extracellular virus was titrated on PBMCs by the hemadsorption assay as described in Materials and Methods. The data are the mean of two experiments.
A time course of viral protein expression in endothelial cells was investigated after pulse-labeling infected cells, followed by immunoprecipitation of vp30 and vp73 and a comparison of time of expression with that in alveolar macrophages (Fig. 3). Cell lysates were made from PAECs, BPECs, and PAMs infected with ASFV Malawi for 0, 2, 4, 6, 8, and 18 h. In PAECs, BPECs, and PAMs, the early protein vp30 was first detected by 4 hpi and increased over 18 h, while the late protein vp73 was expressed at 18 hpi (Fig. 3). There were only minor differences in kinetics of early and late gene expression between PAMs, PAECs, and BPECs.
FIG. 3.

Comparison of time course expression of vp30 and vp73 in ASFV-infected endothelial cells and macrophages. PAECs, BPECs, or PAMs were infected with ASFV Malawi for 0, 2, 4, 6, 8, and 18 h. Cells were pulse-labeled with [35S]-methionine-cysteine, and the viral proteins vp30 and vp73 were immunoprecipated with MAbs C18 and 4H3, respectively. Labeled proteins were separated by SDS-PAGE and visualized by autoradiography. The levels of early and late gene expression between alveolar macrophages and vascular endothelial cells of domestic pig and bushpig are similar.
PAECs infected with ASFV Malawi die by apoptosis.
DAPI staining (Fig. 1) showed a condensation of nuclear chromatin characteristic of apoptosis. To confirm that ASFV-infected endothelial cells were indeed dying by apoptosis, TUNEL labeling was used to identify breaks in nuclear DNA at a late stage of cell infection. PAECs at 18 hpi were labeled with fluorescein-conjugated dUTP (Fig. 4A), which was incorporated into all cells infected with ASFV, indicating DNA strand breakage.
FIG. 4.

ASFV-infected PAECs die by apoptosis. (A) TUNEL labeling of ASFV-infected PAECs. Non infected (top) or 18-hpi (bottom) PAECs were labeled with fluorescein-conjugated dUTP. (B) Annexin V staining of ASFV-infected PAECs. PAECs were infected for (i) 2, (ii) 6, or (iii) 24 h with ASFV before being stained with annexin V and analyzed by FACS. The open histograms represent annexin V binding on resting cells, whereas solid histograms represent annexin V binding on ASFV-infected cells.
As an early indicator of apoptosis of endothelial cells induced by ASFV infection, annexin V binding was used to detect phosphatidylserine (PS) exposure on the outer leaflet of the plasma membrane (Fig. 4B). Annexin V binding to PS was induced in all cells as early as 2 hpi, and this binding increased to include most cells by 24 hpi. In vascular endothelial cells, this also signifies activation of procoagulant activity, since PS is a modulator of TF (9, 26). In the initial infection, not all cells are infected, but most cells become activated, maybe by a soluble factor. There is a slight rise in titer over 48 and 72 hpi as a second round of infection and apoptosis take place (Fig. 2).
Activation of E-selectin expression is inhibited, and IFN-induced expression of MHC class I is downregulated after ASFV infection of PAECs.
Endothelial activation was monitored by following surface expression of E-selectin (CD62E) and MHC class I molecules. FACS analysis showed that there was no surface expression of E-selectin on resting PAECs (Fig. 5Ai), while high levels of MHC class I were found on all resting cells (Fig. 5Bi). Infection of PAECs with ASFV for 6 h did not induce E-selectin expression (Fig. 5Aii). Similarly, MHC class I surface expression was not upregulated on the surface of PAECs after ASFV infection for 24 h (Fig. 5Bii). In a control experiment, activation of PAECs with TNF-α showed induction of cell surface E-selectin staining after 6 h (Fig. 5Aiii) and MHC class I expression after 24 h (Fig. 5Biii). PAECs fail in their normal activation response of increased adhesion molecule and MHC class I expression after ASFV infection.
In a second experiment, addition of IFN-α to PAECs induced MHC class I surface expression over that in resting cells (Fig. 5Cii). This population of PAECs showed a lower constitutive level of MHC class I (Fig. 5Ci) than those in the first experiment (Fig. 5B). As before, overnight ASFV infection did not change the surface expression of MHC class I compared with that in resting cells (Fig. 5Ciii). However, treatment of PAECs overnight with IFN-α, followed by infection of ASFV for 4 h, showed a downregulation of the response to IFN-α alone (Fig. 5Civ). ASFV can interfere with IFN-α priming of endothelial cells. Downregulation of MHC class I is an important mechanism used by many viruses to escape immune surveillance. Taken together, the phenotypic changes in PAECs following infection show that the virus would be able to not only escape immune surveillance through blocking viral peptide presentation but also avoid inflammation and recruitment of leukocytes to the site of infection.
FIG. 5.


Basal and IFN-induced activation is inhibited in ASFV-infected PAECs. (A) E-Selectin (CD62E) expression on ASFV-infected PAECs. Cells were stained either with anti-E-selectin antibody 1.2B6 (solid histograms) or an irrelevant control (open histograms). (i) Resting cells, (ii) cells after infection with ASFV for 6 h, or (iii) cells stimulated with TNF-α for 6 h are shown. (B) MHC class I expression on ASFV-infected PAECs. Cells were stained with anti-MHC class I MAb 2-27-3 (solid histogram) or with an irrelevant control (open histogram). (i) Resting cells, (ii) cells infected with ASFV for 24 h, and (iii) cells stimulated with TNF-α for 24 h are shown. (C) Downregulation of IFN-α-induced MHC class I expression by ASFV in PAECs. Cells were stained with either an anti-MHC class I antibody 2-27-3 (solid histograms) or an irrelevant control (empty histograms). (i) Resting cells, (ii) cells treated with IFN-α for 24 h, (iii) cells infected with ASFV for 24 h and (iv) cells given IFN-α treatment overnight followed by ASFV infection for 4 h are shown.
ASFV inhibits basal and TNF-α-induced transcription of proinflammatory cytokines.
Activation of endothelial cells is characterized by both their production of proinflammatory cytokines, which act in vivo to attract lymphocytes to the site of infection, and procoagulation factors, which are involved in initiation of the blood clotting cascade. Expression of mRNA for the cytokines IL-6 and IL-8 was monitored by RT-PCR. To investigate expression in resting cells and cells that had been nonspecifically activated, PAECs were either untreated (Fig. 6, lane 1) or stimulated with TNF-α for 4 h (Fig. 6, lane 2) or 24 h (Fig. 6, lane 3). RNA was isolated and reverse transcribed into cDNA. Primers specific for porcine IL-8 and IL-6 were used to amplify cDNAs by PCR. A semiquantitative analysis comparing lanes using the same limiting number of cycles and dilution of RNAs was performed so that the products were in the linear range of amplification. IL-8 mRNA levels were constitutively high in resting endothelial cells and remained so after 4 and 24 h of TNF-α treatment. IL-6 mRNA levels were low in resting cells but increased 4 h after TNF-α treatment and remained high for 24 h.
FIG. 6.

Proinflamatory cytokine IL-6 and IL-8 transcription is inhibited while TF transcription is increased after ASFV infection. PAECs were either resting (lane 1), stimulated with TNF-α for 4 h (lane 2) or overnight (lane 3), infected with ASFV alone for 4 h (lane 4), or stimulated with TNF-α overnight, followed by ASFV infection for 4 h (lane 5). RNA was reverse transcribed into cDNA, and expression of cytokines IL-8 and IL-6 and the procoagulant factor TF were semi quantitated by limited cycles of PCR. Equal amounts of RNA were amplified for each sample as indicated by the actin PCR (bottom). ASFV inhibits constitutive IL-8 expression and TNF-α-activated IL-6 expression. In contrast, TF mRNA was induced by virus.
Interestingly, infection of PAECs with ASFV for 4 h totally abolished the IL-8 mRNA expression found in resting cells (Fig. 6, lane 4). This ASFV-induced downregulation was seen even when there was prior treatment with TNF-α overnight, followed by ASFV infection for 4 h (Fig. 6, lane 5). IL-6 mRNA expression was not induced by ASFV infection alone (Fig. 6, lane 4) and, similar to IL-8, TNF-α-induced expression of IL-6 was also inhibited by ASFV (Fig. 6, lane 5).
ASFV-infected PAECs become procoagulant.
TF is an essential initiator of blood coagulation, leading to activation of the thrombotic state of endothelial cells. Expression of mRNA for TF was monitored by RT-PCR with primers designed from known human and bovine sequences. TF mRNA levels of resting PAECs were undetectable but increased 4 h after TNF-α treatment and still further after 24 h (Fig. 6, lanes 1 to 3). In contrast to IL-6 and IL-8, infection with ASFV for 4 h induced TF mRNA (Fig. 6, lane 4). TF mRNA was also detected when cells were stimulated overnight with TNF-α, followed by ASFV infection for 4 h (Fig. 6, lane 5), although not to the extent shown with TNF-α alone overnight.
It was important to demonstrate that the increased TF mRNA expression (Fig. 6) reflected an increased surface expression of TF protein on ASFV-infected PAECs. An anti-human TF antibody, found to cross-react with pig (13) was used to stain the surface of cells (Fig. 7). TF surface staining was induced 4 h post-ASFV infection (Fig. 7A). Later in infection, staining of the same cell for both TF (Fig. 7B) and the viral protein vp30 (Fig. 7C) confirmed that induction of tissue factor colocalized with ASFV infection.
FIG. 7.

Surface expression of TF is induced in ASFV-infected PAECs. Expression of TF (green) and ASFV vp30 (red) in PAECs was detected by immunofluoresence. (A) Surface localization of TF 4 hpi with ASFV Malawi. (B and C) Colocalization of TF and vp30 16 hpi with ASFV.
ASFV-induced apoptosis of PAECs is mediated by caspases and inhibited by z-VAD-fmk.
The activation of caspases was monitored by the cleavage of PARP. Cleavage of full-length PARP from a 116-kDa protein by caspase 3 results in fragments of 89 and 24 kDa. The anti-PARP antibody did not recognize the 89-kDa fragment from pig. Therefore, PARP cleavage was monitored by the appearance of the 24-kDa fragment. Appearance of the 24-kDa cleaved form of PARP after ASFV infection of PAECs occurred 4 hpi and was maintained over 18 hpi (Fig. 8). Addition of the caspase inhibitor z-VAD-fmk to cells at the beginning of the infection completely inhibited the appearance of the cleaved fragment of PARP at 8 hpi (Fig. 8). These data indicate rapid activation of caspases soon after infection of PAECs.
FIG. 8.

PARP and NF-κB p65 cleavage in ASFV-infected PAECs: inhibition by z-VAD-fmk. Western blot analysis of lysates of PAECs infected with ASFV Malawi for 0, 4, 8, and 18 h were analyzed with anti-PARP and anti-NF-κB p65 antibodies. PARP cleavage was monitored by production of a 24-kDa fragment 4 hpi. The cleavage was inhibited by z-VAD-fmk (Z), indicating caspase-3 activation. The same lysates were probed with MAb F6 directed to the p65 subunit of NF-κB. The 65-kDa protein was present at 4 hpi, decreased at 8 hpi, and disappeared at 18 hpi. The pan-caspase inhibitor z-VAD-fmk prevented loss of the 65-kDa band at 8 hpi (Z).
Apoptotic endothelial cells show caspase-mediated cleavage of the 65-kDa subunit of NF-κB, also known as RelA (24). Western blot analysis of the same samples as above with an anti-p65 antibody showed NF-κB p65 present at 0 and 4 hpi, but it had decreased by 8 hpi and disappeared by 18 hpi (Fig. 8). Addition of the caspase inhibitor z-VAD-fmk inhibited the disappearance of the 65-kDa subunit at 8 hpi. The removal of NF-κB p65 following ASFV infection may explain some of the phenotypic changes described above, since this transcription factor is responsible for the activation of cell adhesion molecules, including E-selectin and proinflammatory cytokines such as IL-6 and IL-8.
DISCUSSION
The present study demonstrates that a highly virulent strain of ASFV, Malawi Lil 20/1, infects primary PAECs and primary BPECs. Vascular endothelial cells represent highly differentiated cells, normally maintaining a quiescent nonthrombogenic surface; but when activated, they become specialized for initiating blood coagulation and leukocyte recruitment. Our novel findings show that a virulent isolate of ASFV, Malawi Lil 20/1, can infect these cells, resulting in phenotypic changes that may play a central role in the pathogenesis of the disease in vivo. ASFV inactivated the normal inflammatory response to infection, inhibiting surface expression of important molecules in cell activation, such as the adhesion molecule E-selectin, and MHC class I and transcription of inflammatory cytokines IL-6 and IL-8. Significantly, however, the thrombotic state was increased, as evidenced by increased mRNA and surface staining for TF and the externalization of PS, both important initiators of the coagulation cascade. Furthermore, we showed by nuclear condensation, TUNEL, and PARP cleavage analysis that ASFV-infected endothelial cells died by apoptosis. Activation of the apoptotic pathway occurred rapidly since PARP cleavage was seen by 4 hpi, and it was mediated by caspases, since it could be inhibited by the pan-caspase inhibitor z-VAD. Chromatin condensation indicated that BPECs also died by apoptosis, but analysis of further phenotypic changes awaits reagents that cross-react with bushpig RNA or protein.
Importantly, the time course of infection was not significantly different between macrophages and endothelial cells, shown by immunoprecipitation of two structural proteins, the early protein vp30 and the late protein vp73. Both PAECs and BPECs express vp30 structural protein in the cytoplasm at an early stage of infection, while expression of vp73 occurred later with staining characteristic of viral factories within the cytoplasm. Moreover, viral infection was also characterized by hemadsorption of RBCs to PAECs. This result showed that the viral CD2 homologue was expressed on the plasma membrane of PAECs as a result of infection. Titration of virus secreted from BPECs and PAECs demonstrated that replication was as efficient as in macrophages. Mature virions were also seen in the cytoplasm by 18 hpi. ASFV induced apoptosis of infected endothelial cells. Interestingly, all endothelial cells displaying DNA condensation and strand breakages labeled by TUNEL also displayed a viral factory. No uninfected endothelial cells underwent apoptosis. The evidence points to completion of the viral life cycle before death of the cells by apoptosis. ASFV has been shown to encode proteins, including a Bcl2 homologue and an inhibitor of apoptosis (IAP) homologue that can modulate apoptosis at early and late times, respectively, allowing viral morphogenesis to be completed (2, 11, 29).
Of note, the whole population of cells incubated with Malawi Lil 20/1 displayed a redistribution of PS on the outside leaflet of the membrane very early after contact with ASFV. PS redistribution is an indicator of procoagulant activation (9); although not all cells are infected, all cells may become activated by a soluble mediator. Interestingly, another measure of procoagulant activity, increased TF surface expression, was induced only in cells infected with ASFV. The coagulation process is initiated by exposure of TF on the cell surface, which then activates factor VII (8, 26). TF is rapidly synthesized after endothelial cell activation through NF-κB-dependent and in-dependent pathways. Since the transcription factor NF-κB is inhibited by ASFV (41), TF transcription must be induced by other regulatory factors (27).
Significantly, IFN-α-induced MHC class I expression was downregulated in the presence of ASFV. There are many mechanisms used by viruses to inhibit MHC class I expression, including blocking transcription, cellular transport, or active removal from the cell surface (44). Similarly, the TNF-α-induced expression of IL-8 and IL-6 mRNAs was abrogated by the virus, and there was no activation of the adhesion molecule E-selectin. Inhibition of endothelial cell activation is an important immune evasion strategy for the virus. Immediate-early gene expression of endothelial cells is dependent of the NF-κB pathway (6). In previous work, we demonstrated that an ASFV gene, A238L, from the tissue culture-adapted BA71V strain of ASFV, encoded a protein which blocked NF-κB-dependent gene transcription from an IL-8 promoter-reporter and blocked NF-κB binding to DNA (34). The ORF 5EL of Malawi Lil 20/1 ASFV encodes the IκBα homologue, viral IκB (28). We showed that the molecular mechanism of action of viral IκB involved direct binding to NF-κB p65 (41). On infection, ASFV activates the NF-κB signal transduction pathway, leading to endogenous IκB degradation. The virus then exploits the degradation of the natural inhibitor to allow its own IκB homologue to bind NF-κB, inhibiting its activity (41). The role of the NF-κB signal transduction pathway in the control of apoptosis has been well characterized (7). Interestingly, we show here that NF-κB p65 is cleaved by caspases following infection. In other systems, it has been demonstrated that the cleaved product inhibits NF-κB activity and promotes apoptosis (24, 36). Further characterization of NF-κB proteolysis and interaction of the IκB homologue with the apoptotic pathway should resolve this question.
This study demonstrates a central role for vascular endothelial cells in the pathogenesis of the disease in vivo. ASFV induces a lethal hemorrhagic fever in domestic pigs, and apoptosis of endothelial cells has been seen in vivo at the electron microscope level with virulent isolates E70 and E75, indicated by chromatin condensation, cytoplasmic membrane-bound apoptotic bodies, and detachment from the basement membrane (35). We show that the direct infection, replication, and damage to vascular endothelial cells lead to apoptosis. Contribution to damage to the endothelium by TNF-α produced from infected macrophages cannot be ruled out, since elevated levels of TNF-α have been seen in serum (17). However, there is an inhibition of TNF-α production from ASFV-infected macrophages in vitro (34), and supernatants from virally infected macrophages incubated with lymphocytes do not causes apoptosis of lymphocytes in vitro (42).
It may at first appear surprising that the tropism of ASFV for macrophages and endothelial cells is identical between bushpigs and domestic pigs, since bushpigs do not exhibit VHF in vivo. However, ASFV-infected macrophages have been found in bushpig tissues in vivo 5 days postinfection with associated bystander lymphocyte apoptosis (30), and young bushpigs do show signs of fever with viremias high enough to allow infection of feeding ticks, the natural reservoir host (3). The extent of viral replication and destruction of lymphoid tissues by apoptosis is much more limited in the bushpig, and the disease therefore resolves. In the domestic pig, replication becomes far more extensive, and there is massive tissue apoptosis. The pathobiology of ASFV infection and the reason why bushpigs survive are obviously complex, and further characterization will only possible when bushpig-specific reagents become available, possibly through bushpig DNA sequence analysis. Interestingly, susceptibility of vascular endothelial cells is species restricted, since the Malawi isolate of ASFV will not infect bovine or human endothelial cells (P. P. Powell, unpublished).
ACKNOWLEDGMENTS
We are grateful to Tom Wileman (IAH, Pirbright, United Kingdom) for helpful discussions, and we thank John McVey, Daxin Chen, and Tony Dorling (MRC Clinical Sciences Centre, Hammersmith Hospital, London WC12, United Kingdom) for advice about tissue factor surface staining. We thank Suman Mahan and Gillian Smith (Veterinary Research Lab, Harare, Zimbabwe) for the kind gift of bushpig endothelial cells.
REFERENCES
- 1.Afonso C L, Alcaraz C, Brun A, Sussman M D, Onisk D V, Escribano J M, Rock D L. Chracterisation of p30, a highly antigenic membrane and secreted protein of African swine fever virus. Virology. 1992;189:368–373. doi: 10.1016/0042-6822(92)90718-5. [DOI] [PubMed] [Google Scholar]
- 2.Afonso C L, Neilan J G, Kutish G F, Rock D L. An African swine fever virus bcl-2 homolog, 5-HL, suppresses apoptotic cell death. J Virol. 1996;70:4858–4863. doi: 10.1128/jvi.70.7.4858-4863.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson E C, Huchings G H, Mukarati N, Wilkinson P J. African swine fever virus infection of the bushpig (Potamochoerus porcus) and its significance in the epidemiology of the disease. Vet Microbiol. 1998;62:1–15. doi: 10.1016/s0378-1135(98)00187-4. [DOI] [PubMed] [Google Scholar]
- 4.Anderson R, Wang S, Osiowy C, Issekutz A C. Activation of endothelial cells via antibody-enhanced dengue virus infection of peripheral blood monocytes. J Virol. 1997;71:4226–4232. doi: 10.1128/jvi.71.6.4226-4232.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avirutnan P, Malasit P, Seliger B, Bhakdi S, Husmann M. Dengue virus infection of human endothelial cells leads to chemokine production, complement activation and apoptosis. J Immunol. 1998;161:6338–6346. [PubMed] [Google Scholar]
- 6.Bach E H, Hancock W W, Ferran C. Protective genes expressed in endothelial cells: a regulatory response to injury. Immunol Today. 1997;18:483–486. doi: 10.1016/s0167-5699(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 7.Barkett M, Gilmore T D. Control of apoptosis by Rel/NFκB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 8.Bombeli T, Mueller M, Haeberli A. Anticoagulant properties of the vascular endothelium. Thromb Haemost. 1997;77:408–423. [PubMed] [Google Scholar]
- 9.Bombeli T, Karsan A, Tait J F, Harlan J M. Apoptotic vascular endothelial cells become procoagulant. Blood. 1997;89:2429–2442. [PubMed] [Google Scholar]
- 10.Borca M V, Kutish G F, Afonso C L, Irusta P, Carrillo C, Brun A, Sussman M, Rock D L. An African swine fever virus gene with similarity to the T-lymphocyte surface antigen CD2 mediates hemadsorption. Virology. 1994;199:463–468. doi: 10.1006/viro.1994.1146. [DOI] [PubMed] [Google Scholar]
- 11.Brun A, Rivas C, Esteban M, Escribano J M, Alonso C. African swine fever virus gene A179L. a viral homologue of bcl-2, protects cells from program cell death. Virology. 1996;225:227–230. doi: 10.1006/viro.1996.0592. [DOI] [PubMed] [Google Scholar]
- 12.Carrasco L, de Lara F M C, de las Mulas J M, Gomez-Villamandos J C, Perez J, Wilkinson P J, Sierra M A. Apoptosis in the lymph nodes of acute African swine fever. J Comp Path. 1996;115:415–428. doi: 10.1016/s0021-9975(96)80075-2. [DOI] [PubMed] [Google Scholar]
- 13.Chen, Reisbeck D K, Kemball-Cook G, McVey J H, Tuddenham E G D, Lechler R I, Dorling A. Inhibition of tissue factor-dependent and -independent coagulation by cell surface expression of novel anticoagulant fusion proteins. Transplantation. 1999;67:467–474. doi: 10.1097/00007890-199902150-00021. [DOI] [PubMed] [Google Scholar]
- 14.Cobbold C, Whittle J T, Wileman T. Involvement of the endoplasmic reticulum in the assembly and envelopment of African swine fever virus. J Virol. 1996;70:8382–8390. doi: 10.1128/jvi.70.12.8382-8390.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feldmann H, Bugany H, Mahner F, Klenk H-D, Drenckhahn D, Schnittler H-J. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J Virol. 1996;70:2208–2214. doi: 10.1128/jvi.70.4.2208-2214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geisbert T W, Hensley L E, Gibb T R, Steele K E, Jaax N K, Jarling P B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab Investig. 2000;80:171–185. doi: 10.1038/labinvest.3780021. [DOI] [PubMed] [Google Scholar]
- 17.Gomez del Moral M, Ortuno E, Fernandez-Zapatero P, Alonso F, Alonso C, Ezquerra A, Dominguez J. African swine fever virus induces tumor necrosis factor alpha production: implications in pathogenesis. J Virol. 1999;73:2173–2180. doi: 10.1128/jvi.73.3.2173-2180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Villamandos J C, Hervas J, Mendez A, Carrasco L, Villeda C J, Wilkinson P J, Sierra M A. Pathological changes in the renal interstitial capillaries of pigs innoculated with two different strains of African swine fever virus: implications in pathogenesis. J Comp Path. 1995;112:283–298. doi: 10.1016/s0021-9975(05)80081-7. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Villamandos J C, Hervas J, Moreno C, Carrasco L, Bautista M J, Caballero J M, Wilkinson P J, Sierra M A. Subcellular changes in the tonsils of pigs infected with acute African swine fever virus. Vet Res. 1997;28:179–189. [PubMed] [Google Scholar]
- 20.Halstead S B. Pathogenesis of dengue: challenges to molecular biology. Science. 1988;239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 21.Haresnape J M, Wilkinson P J, Mellor P S. Isolation of African swine fever virus from ticks of the Ornithodorus moubata complex (Ixodoidea: Argasidae) collected within the African swine fever enzootic area of Malawi. Epidemiol Infect. 1984;101:173–185. doi: 10.1017/s0950268800029332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanoska D, Sun D C, Lunney J K. Production of monoclonal antibodies reactive with polymorphic and monomorphic determinants of SLA class-I gene products. Immunogenetics. 1991;33:220–223. doi: 10.1007/BF01719247. [DOI] [PubMed] [Google Scholar]
- 23.Lefevre F, Haridon R, Borras-Cuesta F, La Bonnardière C. Production, purification and biological properties of an Escherichia coli-derived recombinant pocine alpha interferon. J Gen Virol. 1990;71:1057–1063. doi: 10.1099/0022-1317-71-5-1057. [DOI] [PubMed] [Google Scholar]
- 24.Levkau B, Scatena M, Giachelli C M, Ross R, Raines E W. Apoptosis overrides survival signals through caspase-mediated dominant-negative NF-κB loop. Nat Cell Biol. 1999;1:227–233. doi: 10.1038/12050. [DOI] [PubMed] [Google Scholar]
- 25.Marianneau P, Flamand M, Deubel V, Despres P. Apoptotic cell death in response to dengue virus infection: the pathogenesis of dengue haemorrhagic fever revisited. Clin Diagn Virol. 1998;10:113–119. doi: 10.1016/s0928-0197(98)00031-2. [DOI] [PubMed] [Google Scholar]
- 26.McVey J H. Tissue factor pathway. Balliere's Best Pract Res Clin Haematol. 1999;12:361–372. doi: 10.1053/beha.1999.0030. [DOI] [PubMed] [Google Scholar]
- 27.Moll T, Czyz M, Holzmuller H, Hofer-Warbinek R, Wagner E, Winkler H, Bach F H, Hofer E. Regulation of the tissue factor promoter in endothelial cells. Binding of NF-κB-, AP-1, and SP-1-like transcription factors. J Biol Chem. 1995;270:3849–3857. doi: 10.1074/jbc.270.8.3849. [DOI] [PubMed] [Google Scholar]
- 28.Neilan J G, Lu Z, Kutish G F, Zsak L, Lewis T L, Rock D L. A conserved African swine fever virus IκB homolog, 5EL, is nonessential for growth in vitro and virulence in domestic swine. Virology. 1997;235:377–385. doi: 10.1006/viro.1997.8693. [DOI] [PubMed] [Google Scholar]
- 29.Nogal M L, González de Buitrago G, Rodríguez C, Cubelos B, Carrascosa A L, Salas M L, Revilla Y. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J Virol. 2001;75:2535–2543. doi: 10.1128/JVI.75.6.2535-2543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oura C A L, Powell P P, Parkhouse R M E. The pathogenesis of African swine fever in the resistant bushpig. J Gen Virol. 1998;79:1439–1443. doi: 10.1099/0022-1317-79-6-1439. [DOI] [PubMed] [Google Scholar]
- 31.Oura C A L, Powell P P, Parkhouse R M E. African swine fever: a disease characterized by apoptosis. J Gen Virol. 1998;79:1427–1438. doi: 10.1099/0022-1317-79-6-1427. [DOI] [PubMed] [Google Scholar]
- 32.Peters C J. Viral hemorrhagic fevers. In: Nathanson N, et al., editors. Viral pathogenesis. Philadelphia, Pa: Lippincott-Raven Publishers; 1997. pp. 779–799. [Google Scholar]
- 33.Plendl J, Neumuller C, Vollmar A, Auerbach R, Sinowatz F. Isolation and characterisation of endothelial cells from different organs of fetal pigs. Anat Embryol. 1996;194:445–456. doi: 10.1007/BF00185992. [DOI] [PubMed] [Google Scholar]
- 34.Powell P P, Dixon L K, Parkhouse R M E. An IκB homolog encoded by African swine fever virus provides a novel mechanism for downregulation of proinflammatory cytokine responses in host macrophages. J Virol. 1996;70:8527–8533. doi: 10.1128/jvi.70.12.8527-8533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramiro-Ibanez F, Ortega A, Brun A, Escribano J M, Alonso C. Apoptosis:a mechanism of cell killing and lymphoid organ impairment during acute African swine fever virus infection. J Gen Virol. 1996;77:2209–2219. doi: 10.1099/0022-1317-77-9-2209. [DOI] [PubMed] [Google Scholar]
- 36.Ravi R, Bedi A, Fuchs E J, Bedi A. CD95 (Fas)-induced caspase-mediated proteolysis of NFκB. Cancer Res. 1998;58:882–886. [PubMed] [Google Scholar]
- 37.Rodriguez J M, Yanez R J, Almazan F, Vinuela E, Rodriguez J F. African swine fever encodes a CD2 homologue responsible for the adhesion of erythrocytes to infected cells. J Virol. 1993;67:5312–5320. doi: 10.1128/jvi.67.9.5312-5320.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato M, Mikami O, Kobayashi M, Nakajima Y. Apoptosis in the lymphatic organs of pigs inoculated with classical swine fever virus. Vet Microbiol. 2000;75:1–9. doi: 10.1016/s0378-1135(00)00198-x. [DOI] [PubMed] [Google Scholar]
- 39.Scnittler H J, Mahner F, Drenckhahn D, Klenk H D, Feldmann H. Replication of Marburg virus in human endothelial cells: a possible mechanism for the development of viral hemorrhagic diseases. J Clin Investig. 1993;91:1301–1309. doi: 10.1172/JCI116329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith G E, Anderson E C, Burridge M J, Peters T F, Mahan S M. Growth of Cowdria ruminantium in tissue culture endothelial cell lines from wild African mammals J. Wild Dis. 1998;34:297–304. doi: 10.7589/0090-3558-34.2.297. [DOI] [PubMed] [Google Scholar]
- 41.Tait S W G, Reid E B, Greaves D R, Wileman T E, Powell P P. Mechanism of inactivation of NF-κB by a viral homologue of IκBα: Signal induced release of IκBα results in binding of the viral homologue to NF-κB. J Biol Chem. 2000;275:34656–34664. doi: 10.1074/jbc.M000320200. [DOI] [PubMed] [Google Scholar]
- 42.Takamatsu H, Denyer M S, Oura C, Childerstone A, Andersen J K, Pullen L, Parkhouse R M E. African swine fever virus: a B-cell-mitogenic virus in vivo and in vitro. J Gen Virol. 1999;80:1453–1461. doi: 10.1099/0022-1317-80-6-1453. [DOI] [PubMed] [Google Scholar]
- 43.Thomson G R. The epidemiology of ASF: the role of free-living hosts in Africa. Onderstepoort J Vet Res. 1985;52:201–209. [PubMed] [Google Scholar]
- 44.Tortorella D, Gewurz B E, Furman M H, Schust D J, Ploegh H L. Viral subversion of the immune system. Ann Rev Immunol. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- 45.Tsang Y T M, Stephens P E, Licence S T, Haskard D O, Binns R M. Porcine E-selectin: cloning and functional characterisation. Immunology. 1995;85:140–145. [PMC free article] [PubMed] [Google Scholar]
- 46.Villeda C J, Gomez-Villamandos J C, Williams S M, Heras J, Wilkinson P J, Vinuela E. The role of fibrinolysis in the pathogenesis of the hemorrhagic syndrome produced by virulent isolates of African swine fever virus. Thromb Hemost. 1995;73:112–117. [PubMed] [Google Scholar]
- 47.Villeda C J, Williams S M, Wilkinson P J, Vinuela E. Haemostatic abnormalities in African swine fever: a comparison of two virus strains of different virulence. Arch Virol. 1993;130:71–83. doi: 10.1007/BF01318997. [DOI] [PubMed] [Google Scholar]
- 48.Watier H, Vallee I, Thibault G, Lalmamach A-C, Lacord M, Gruel Y, Lebranchu Y, Salmon H, Bardos P. Effect of human inflammatory cytokines on porcine endothelial cell MHC molecule expression: unique role for TNF-α in MHC class-II induction. Transplant Proc. 1994;26:1152–1155. [PubMed] [Google Scholar]
- 49.Wardley R C, Hamilton F, Wilkinson P J. The replication of virulent and avirulent strains of African swine fever virus in porcine macrophages. Arch Virol. 1979;61:217–225. doi: 10.1007/BF01318056. [DOI] [PubMed] [Google Scholar]
- 50.Wu K K, Thiagarajan P. Role of endothelium in thrombosis and hemostasis. Ann Rev Med. 1996;47:315–331. doi: 10.1146/annurev.med.47.1.315. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, Delgardo R, Xu L, Todd R F, Nabel E G, Sanchez A, Nabel G J. Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science. 1998;279:983–984. doi: 10.1126/science.279.5353.1034. [DOI] [PubMed] [Google Scholar]