

The Earth BioGenome Project aims to produce reference genomes for all ~1.8 million known eukaryotic species over the next decade1–4. Achieving this goal will require the current pace of reference genome production to increase by at least two orders of magnitude1. Automation of the assembly process with a pipeline that is widely accessible to any research group will be required to achieve this speed-up. Enabling this goal requires sustained effort in three major areas: genome assembly optimization and best-practice development, computational infrastructure provisioning, and dissemination and training.
To optimize the assembly process and devise best practices, we combined the expertise of two projects—the Vertebrate Genomes Project (VGP) and the European Reference Genome Atlas (ERGA). The VGP is a collaborative effort to generate reference genomes for all ~70,000 vertebrate species5. In the past 5 years, the VGP has released hundreds of new reference genomes supported by the development of automated assembly tools and workflows1,5. The ERGA is a pan-European scientific initiative to generate reference genomes for all ~200,000 European eukaryote species, many of which are on the International Union for Conservation of Nature Red List of species at risk of extinction2.
Advancing from the prior VGP work, originally on the DNAnexus platform (Supplementary Note, section 1.1), we developed a pipeline within the Galaxy ecosystem6 that combines Pacific Biosciences (PacBio) high-fidelity (HiFi) reads with long-distance information from Hi-C maps and/or optical maps to generate nearly complete assemblies (Supplementary Note 1.3). The pipeline further uses Hi-C or whole-genome sequence data from parents to produce chromosomal-level or whole-genome-level phased genomes, respectively. To streamline the assembly process and ensure quality, the pipeline includes extensive quality control (QC) functions at every step (Supplementary Fig. 1 and Supplementary Note, section 2.1). We suggest at least 30× PacBio HiFi coverage, and up to 60× coverage to accurately assemble highly repetitive regions, as well as 30× Hi-C coverage per haplotype. This is important to ensure a uniform read distribution during the random Poisson sampling process of whole-genome sequencing7.
Galaxy allows users to execute complex workflows on thousands of datasets and terabytes of data either via a graphical user interface or programmatically via application programming interface (API) scripts8. Major global Galaxy instances in the United States (https://usegalaxy.org), the European Union (http://usegalaxy.eu) and Australia (https://usegalaxy.org.au) are freely accessible to researchers worldwide and supported by public cloud infrastructures so that users are not required to install any tools or procure any infrastructure. Galaxy can also be installed locally to use existing high-performance computing (HPC) systems and configured to access heterogeneous, geographically distributed storage and computing resources9.
The resulting VGP–Galaxy assembly pipeline is organized into 10 Galaxy workflows (Fig. 1; Supplementary Note, section 2.1) to account for different combinations of input data and stages of the assembly process. We systematically evaluated several scaffolding approaches, resulting in best-practice workflows using Hi-C and/or Bionano optical mapping data. We further implemented a dedicated mitogenome assembly pipeline to validate species identification and provide mitochondrial reference assemblies10,11. We also developed a decontamination workflow toremove exogenous sequences (e.g., viral and bacterial sequences), as well as mitochondrial artifacts that are often present in draft assemblies, as required for submission to public archives (Supplementary Note, section 2.2.4).
Fig. 1 |. VGP–Galaxy assembly pipeline (version 2.1) consists of 10 workflows that can be combined into 8 analysis trajectories depending on the combination of input data.
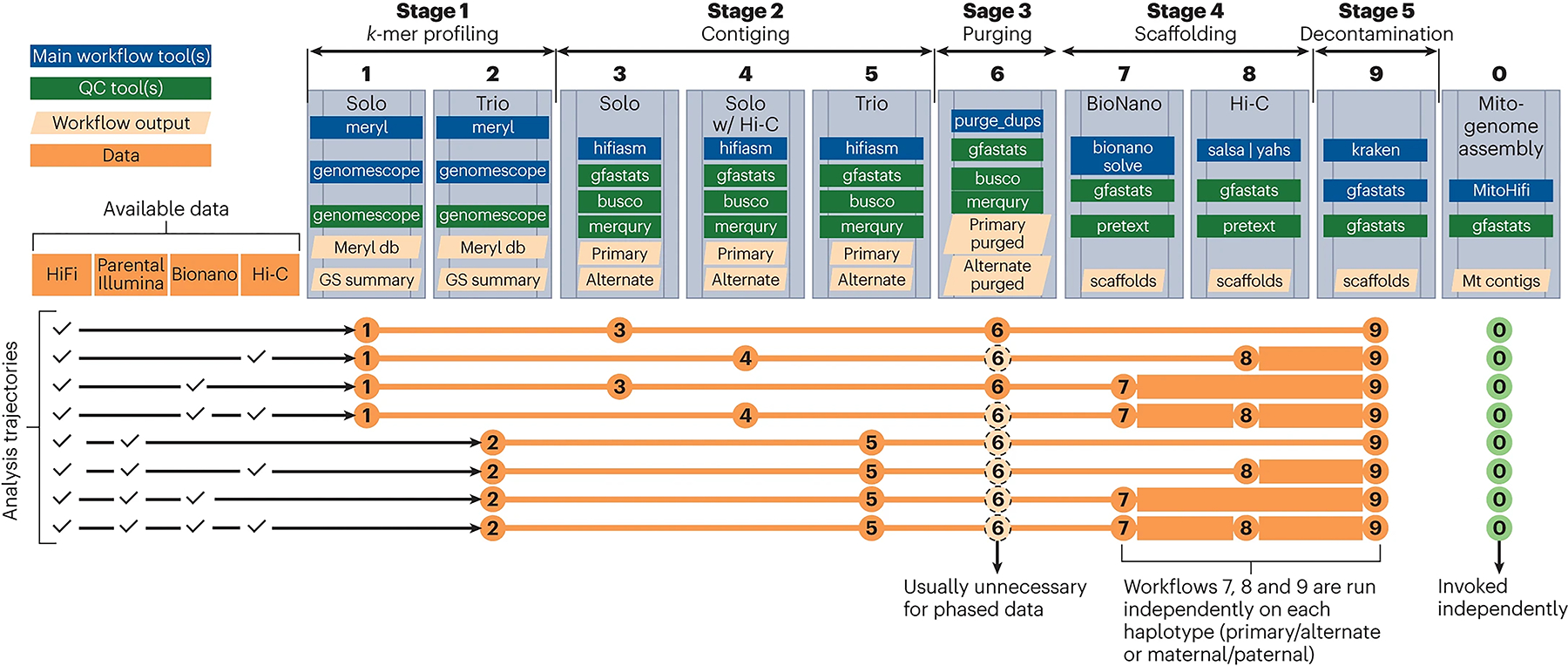
A decision on whether to invoke workflow 6 is based on the analysis of QC output of workflows 3, 4 or 5 (see Supplementary Information for full explanation). Thicker lines connecting workflows 7, 8 and 9 reflect the fact that these workflows are invoked separately for each phased assembly (once for maternal and once for paternal).
We first tested the automated workflows on the assembly of a reference genome of zebra finch (Taeniopygia guttata), for which a wide variety of genomic sequencing data types are available. This led to the development of three types of assembly trajectories (Fig. 1 and Supplementary Table 1): solo assembly (workflows 1, 3, 6 and 9; Fig. 1) using PacBio HiFi data for single individuals; Hi-C assembly (workflows 1, 4, 8 and 9) obtained by adding Hi-C data for phasing and scaffolding the contigs; and trio assembly (workflows 2, 5, 8 and 9) produced by using Illumina short-read data from parents for haplotype phasing (Fig. 1 and Supplementary Table 1).
To validate the pipeline, we used 51 vertebrate datasets for which PacBio HiFi and Hi-C data were available. We compared these assemblies against 19 previous PacBio continuous long read–based genomes of similar size and complexity to confirm and extend the improvements to HiFi technology over continuous long-read methods reported previously12 (Fig. 2, Supplementary Table 5, Supplementary Fig. 6).
Fig. 2 |. Phylogenetic tree and assembly statistics of genomes assembled using the VGP–Galaxy assembly pipeline.
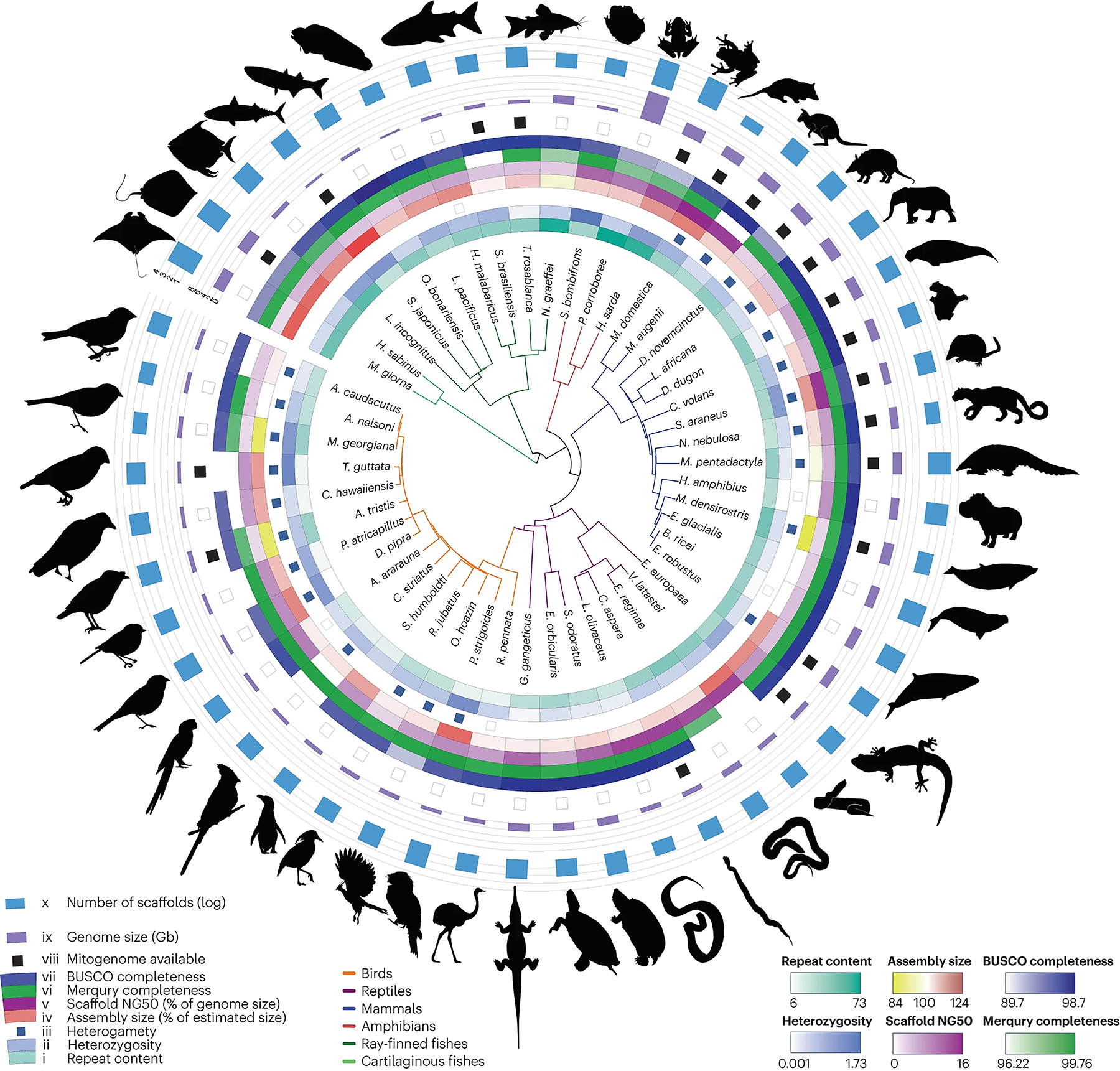
From the innermost circle to the outermost circle: (i) repeat content; (ii) heterozygosity; (iii) heterogamy: individuals with two identical sex chromosomes (white) or two different sex chromosomes (blue); (iv) assembly size in percentage of the genome size estimated by Genomescope; (v) scaffold NG50 in % of estimated genome size; (vi) Merqury completeness of both haplotypes; (vii) BUSCO completeness: presence of orthologous genes present and complete compared to the set expected in vertebrates; (viii) mitogenome assembled and available (black); (ix) genome size in gigabytes, with lines at 9, 2, 3, 4, 6 and 8 Gb; (x) number of scaffolds in log scale, with lines at 1 (10 scaffolds), 2 (100 scaffolds), 3 (1,000 scaffolds) and 4 (10,000 scaffolds).
Given the improved haplotype resolution that resulted from adding Hi-C data, even for large (~4.3 Gbp), repeat-rich genomes, we recommend Hi-C Hifiasm phasing when parental data are not available. It is now possible to use well-tested kits as long as samples have been preserved properly (fresh frozen and without DNA and RNA preservatives that protect DNA but reduce protein crosslinks). For use with difficult-to-obtain samples, we have included pipeline options that do not require Hi-C data (Fig. 1).
Although all genome assemblies reported here are for vertebrates, the above principles and our pipeline can be applied to other animal, plant or fungal genomes by modifying a few parameters such as, for example, BUSCO clades necessary for accurate QC reporting (Supplementary Methods, section 3.3).
Our approach is designed to be useful across the full spectrum of user skill levels and analysis scenarios. For this purpose, we created dedicated tutorials distributed via the Galaxy Training Network portal13 that include extended versions and that collectively provide an in-depth overview of the assembly process, as well as a streamlined tutorial designed to facilitate immediate use of the workflows14.
Our future work will focus on the continuous maintenance of the pipeline to improve its efficiency and scalability, automation of the curation process, incorporation of ultra-long-read data and development of effective genome annotation procedures.
To increase the robustness of the pipeline, we are developing additional workflows to take advantage of Oxford Nanopore Technologies (ONT) data, and particularly of ultra-long (UL) reads (>100 kb). These workflows use HiFi/UL hybrid assembly tools such as Verkko15 and the HiFi+UL version of Hifiasm16, both of which we integrated into Galaxy. Each technology complements missing information from the other, with ONT reads being less accurate and HiFi reads being shorter and underperforming on certain genomic patterns, leading to sequencing bias that could affect specific taxa (Supplementary Fig. 14). This integration of complementary sequencing technologies will make our pipeline even more effective at generating complete and accurate reference genomes.
Supplementary Material
Acknowledgements
We thank Yagoub Adam, Tyler Alioto, Jun Aruga, Diego De Panis, Sagane Dind, Diego Fuentes, Shilpa Garg and Jèssica Gómez for contributing to the initial implementation during ELIXIR Biohackathon 2021. We also thank Nate Jue for help testing and developing the pipeline tutorials and Andrea Guarracino for their useful comments to the manuscript. This work was supported in part by the Intramural Research Program of the US National Human Genome Research Institute (NHGRI), the US National Institutes of Health (NIH) and the Howard Hughes Medical Institute (HHMI). The authors are grateful to the broader Galaxy community for their support and software development efforts. This work is funded by NIH grants U41 HG006620, U24 HG010263, U24 CA231877 and U01CA253481, along with US National Science Foundation grants 1661497, 1758800 and 2216612. The work was also supported in part by The Human Frontier Science Program (HFSP) RGP0025/2021, the Swiss National Science Foundation (SNSF) grants 202669 and 198691, the Swiss State Secretariat for Education, Research and Innovation (SERI) grant 22.00173 and Horizon Europe under the Biodiversity, Circular Economy and Environment program (REA.B.3, BGE 101059492). Usegalaxy.eu is supported by German Federal Ministry of Education and Research grants 031L0101C and de.NBI-epi to B.G. Computational resources are provided by the Advanced Cyberinfrastructure Coordination Ecosystem (ACCESS-CI), Texas Advanced Computing Center, and the JetStream2 scientific cloud.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-023-02100-3.
Data availability
The workflows, their description and instructions on how to use them can be found at https://galaxyproject.org/projects/vgp/workflows/. The requisite tools are installed on usegalaxy.org and usegalaxy.eu, and are in the process of being installed on usegalaxy. org.au. These genomes were supported by collaborators of the VGP and ERGA, and the QC analyses reported here to test the VGP Galaxy pipeline do not release those that are under specific embargo policies for genome-wide analyses (e.g., https://genome10k.ucsc.edu/data-use-policies/). New genome assemblies are available in the GenomeArk repository: https://www.genomeark.org/. After manual curation, the assemblies are submitted to the US National Center for Biotechnology Information (NCBI) under the BioProject Vertebrate Genome Project: https://www.ncbi.nlm.nih.gov/bioproject/489243 17.
References
- 1.Hotaling S, Kelley JL & Frandsen PB Proc. Natl Acad. Sci. USA 118, e2109019118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Formenti G et al. Trends Ecol. Evol. 37, 197–202 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Theissinger K et al. Trends Genet. 39, 545–559 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Lewin HA et al. Proc. Natl Acad. Sci. USA 119, e2115635118 (2022).35042800 [Google Scholar]
- 5.Rhie A, Walenz BP, Koren S & Phillippy AM Genome Biol. 21, 245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galaxy Community. Nucleic Acids Res. 50, W345–W351 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lander ES & Waterman MS Genomics 2, 231–239 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Bray S & Maier W Automating Galaxy workflows using the command line. Galaxy Training Network (2023). [Google Scholar]
- 9.Galaxy Community. Galaxy Server administration. Galaxy Training Network https://github.com/galaxyproject/training-material (2019). [Google Scholar]
- 10.Formenti G et al. Genome Biol. 22, 120 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uliano-Silva M et al. BMC Bioinform. 24, 288 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenger AM et al. Nat. Biotechnol. 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batut B et al. Cell Syst. 6, 752–758.e1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lariviere D, Ostrovsky A, Gallardo C, Pickett B & Abueg L VGP assembly pipeline - short version. Galaxy Training Network (2023); https://gxy.io/GTN:T00040 [Google Scholar]
- 15.Rautiainen M et al. Nat. Biotechnol. 41, 1474–1482 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng H, Asri M, Lucas J, Koren S & Li H Scalable telomere-to-telomere assembly for diploid and polyploid genomes with double graph. Preprint at arXiv 10.48550/arXiv.2306.03399 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.BioProject Vertebrate Genome Project. NCBI BioProject PRJNA489243 (accessed 18 January 2024); https://www.ncbi.nlm.nih.gov/bioproject/489243
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The workflows, their description and instructions on how to use them can be found at https://galaxyproject.org/projects/vgp/workflows/. The requisite tools are installed on usegalaxy.org and usegalaxy.eu, and are in the process of being installed on usegalaxy. org.au. These genomes were supported by collaborators of the VGP and ERGA, and the QC analyses reported here to test the VGP Galaxy pipeline do not release those that are under specific embargo policies for genome-wide analyses (e.g., https://genome10k.ucsc.edu/data-use-policies/). New genome assemblies are available in the GenomeArk repository: https://www.genomeark.org/. After manual curation, the assemblies are submitted to the US National Center for Biotechnology Information (NCBI) under the BioProject Vertebrate Genome Project: https://www.ncbi.nlm.nih.gov/bioproject/489243 17.