

Abstract
Alzheimer's disease (AD) is a complex neurological disorder and multiple pathways are associated with its pathology. Currently available single-targeting drugs are found to be ineffective for the treatment of AD, and most of these drugs provide symptomatic relief. The multi-target directed ligand strategy is proposed as an effective approach for the treatment of AD. Herein, we report the design and synthesis of a series of 2-phenyl substituted chromone derivatives and their evaluation against AChE, MAO-B, and β amyloid self-aggregation inhibition. In the series, NS-4 and NS-13 were identified as the potent leads against all the specified targets. NS-4 and NS-13 exhibited balanced multipotent activities against AChE with IC50 values of 3.09 μM, and 0.625 μM and against MAO-B with IC50 values of 19.64 μM and 12.31 μM, respectively. These compounds also displayed 28.5% and 32.2% self-aggregation inhibition potential against Aβ1–42, respectively. All the compounds were found to be selective for AChE over BuChE. Additionally, NS-4 also exhibited potent BuChE inhibition with an IC50 value of 1.95 μM. Moreover, NS-4 and NS-13 reduced intracellular ROS levels up to 65% against SH-SY5Y cells at 25 μM concentration. The lead compounds were found to be neuroprotective and exhibited no cytotoxicity even at 25 μM concentration. In enzyme kinetic inhibition studies, these compounds showed mixed-type inhibition to AChE. In the computational studies, binding interactions, and orientations of the ligands at the active site of the enzymes were analyzed and these lead compounds were found to be thermodynamically stable inside the active cavity for up to 100 ns.
Multipotent chromone derivatives exhibiting anti-alzheimer's activities.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative state and is identified as one of the major causes of dementia. The pathophysiology of AD is complex in nature and involves many pathological hallmarks including deposition of amyloid beta (Aβ) plaques and tau tangles, hyperactivity of acetylcholinesterase (AChE), overexpression of monoamine oxidase (MAO) enzymes, metal ion dyshomeostasis, oxidative stress, and excess stimulation of N-methyl-d-aspartate (NMDA) receptors.1,2 Acetylcholinesterase and β amyloid plaques are the key hallmarks associated with the earliest theories about the pathogenesis of AD.3 It has been found that AChE induces and accelerates the fibrilization of amyloid β plaques.4 In AD brains, most of the cortical AChE activity is associated with neuritic plaques including monomeric/oligomeric forms and matured senile plaques.5,6 In AD, overexpression of MAO is linked with the excess oxidative deamination of catecholamine neurotransmitters.7 The pathogenesis of AD is complex in nature, and the currently available FDA-approved drugs are single-targeting and unable to completely treat the disease. These drugs majorly focus on the inhibition of acetylcholinesterase while the rest of the pathological pathways remain unaffected.8 Thus, the multi-target directed ligand approach, wherein a ligand can simultaneously inhibit more than two targets, is proposed as an effective strategy for the treatment of AD.9 Since the last few years, a number of ligands with multipotent profiles that can target AChE, MAO and/or β amyloid have been designed.10–12
Chromone is a versatile scaffold that exhibits diverse pharmacological applications including neurological activities.13 The chromone scaffold has been explored in the development of various multipotent ligands for the simultaneous inhibition of AChE, MAO, and BACE-1 enzymes. A variety of chromone hybrids14–17 have been developed and screened against various targets responsible for the progression of AD (Fig. 1). Among the two predominantly expressed isoforms of Aβ in AD, i.e. Aβ40 and Aβ42, Aβ42 has a greater tendency towards fibrilization even at lower concentration.18 The chromone moiety is also reported to display potent Aβ self-aggregation inhibition potential.19–21 A careful analysis of literature reports reveals that most of the ligands with AChE inhibition potential contain a tertiary nitrogen atom and ligands with MAO inhibition activity contain an N/O-propargyl group binding with the FAD cofactor of the enzyme.22
Fig. 1. Chromone-based multi-target directed ligands exhibiting AChE, BuChE, MAO, and amyloid β inhibition potential.

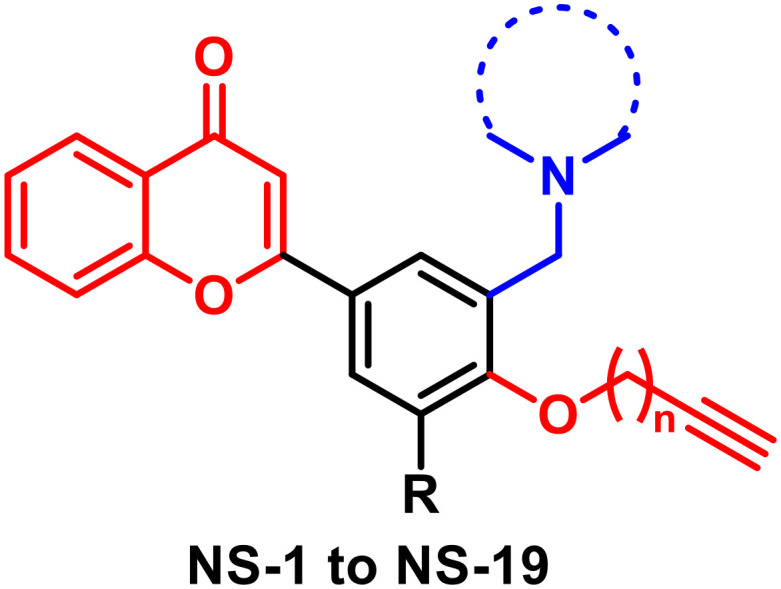
Taking leads from literature reports and previous work done in our laboratory, in the current research work we have designed chromone derivatives substituted with two pharmacophoric groups. An N-alkyl chain is attached to the chromone moiety as a potential pharmacophore for the AChE enzyme while an alkynyl group is incorporated in the scaffold as a pharmacophore for the MAO enzyme. The chromone moiety is also expected to assist in binding to the β amyloid protein and inhibiting its self-aggregation.23 Thus, these compounds are expected to show multipotent activity by simultaneously inhibiting AChE, MAO and, β amyloid enzymes. The chromone intermediates were synthesized through our recently developed protocol using iodine–PEG-400 combination.24 For the derivatization of chromone intermediates, the three-component Mannich reaction25 was utilized to obtain N-alkyl substituted derivatives. The final products were obtained through an O-alkylation phenolic group with alkyne halide. The synthesized compounds were evaluated against AChE, MAO, and β amyloid using Ellman's assay, the Amplex red assay, and the thioflavin T assay, respectively. The cytotoxicity and neuroprotective potential of the leads were evaluated against SH-SY5Y neuronal cell lines. Molecular docking and modelling studies were performed to see the binding orientation and thermodynamic stability of the ligand–enzyme complexes for a time-period of 100 ns. In addition, the reversibility, enzyme kinetic studies, and ROS inhibition potential of the leads were evaluated.
Results and discussion
Chemistry
A total of nineteen compounds were synthesized based on the synthetic procedure described in Scheme 1. Initially, chromone intermediates were synthesized by reacting 2-hydroxy acetophenone and substituted benzaldehydes in the presence of iodine and PEG-400 at 140 °C.24 In the next step, the Mannich reaction was performed on hydroxylated chromone intermediates to obtain the Mannich bases which were finally alkylated with alkyne halide to obtain the desired products. Characterization of the synthesized compounds was done through 1H NMR, 13C NMR, and HRMS.
Scheme 1. Synthetic procedure for the derivatization of chromones using the Mannich reaction.

Biological results
Initially, eeAChE and hMAO inhibition activities were calculated for all the synthesized compounds using Ellman's assay and Amplex red assay kits of Molecular Probes (Invitrogen), Life Technologies through absorbance and fluorometric methods, respectively. A number of chromone derivatives were synthesized to develop the structure–activity relationship (SAR) profile by optionally substituting the aromatic ring with N-substituted cyclic chains and O-alkylated alkyne chains. In the initial studies, the inhibition percentage of all the compounds was evaluated at 20 μM against AChE, BuChE, MAO-A and MAO-B enzymes and IC50 values were calculated for the potent compounds which displayed multipotent profiles. The percentage inhibition of all the compounds is reported in Table 1 and the IC50 values of the potent compounds are shown in Table 2.
Results of percentage inhibition studies of AChE, BuChE, MAO-A, and MAO-B.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry name | n | R (chain) | N substituents | Percentage (%) inhibition @ 20 μM | |||
| AChE | BuChE | MAO-A | MAO-B | ||||
| NS-1 | 3 | OMe | Morpholine | 55.09 | 3.35 | — | 18.84 |
| NS-2 | 1 | OMe | Morpholine | 65.36 | 1.17 | — | 15.05 |
| NS-3 | 2 | OMe | Morpholine | 62.51 | 3.11 | 7.98 | 26.57 |
| NS-4 | 1 | OMe | Piperidine | 68.91 | 89.88 | — | 50.78 |
| NS-5 | 3 | OMe | Piperidine | 61.19 | 21.58 | 0.75 | 15.57 |
| NS-6 | 1 | OEt | Morpholine | 58.94 | — | — | 8.37 |
| NS-7 | 2 | OEt | Morpholine | 67.73 | 4.42 | — | 6.89 |
| NS-8 | 3 | OEt | Morpholine | 57.33 | 6.37 | — | 18.17 |
| NS-9 | 3 | OEt | Piperidine | 75.74 | 3.36 | — | 15.55 |
| NS-10 | 1 | Methyl-morpholine | Morpholine | 67.96 | 9.03 | — | 36.51 |
| NS-11 | 3 | Methyl-morpholine | Morpholine | 66.65 | 1.00 | — | 18.14 |
| NS-12 | 1 | H | Morpholine | 69.95 | 4.21 | — | 28.10 |
| NS-13 | 3 | H | Piperidine | 83.13 | 87.86 | 0.23 | 61.25 |
| NS-14 | 3 | Methyl-piperidine | Piperidine | 71.94 | 13.73 | — | 19.68 |
| NS-15 | 3 | OMe | N-Methyl benzyl | 53.26 | 17.58 | — | 17.53 |
| NS-16 | 3 | OMe | Benzyl piperazine | 66.73 | 7.417 | 5.26 | 22.77 |
| NS-17 | 2 | OMe | Phenyl piperazine | 50.03 | 1.63 | 9.14 | 35.38 |
| NS-18 | 3 | OMe | Phenyl piperazine | 49.88 | 1.69 | 2.60 | 13.14 |
| NS-19 | 3 | OMe | Tetrahydro-isoquinoline | 65.86 | 51.52 | 11.34 | 19.76 |
IC50 values of the selected potent compounds for the inhibition of AChE, BuChE, and MAO-B.
| Entry name | IC50 (μM) values (mean ± S.E. μM) | ||
|---|---|---|---|
| AChE | BuChE | MAO-B | |
| NS-2 | 7.24 ± 0.06 | — | — |
| NS-4 | 3.09 ± 0.02 | 1.95 ± 0.02 | 19.64 ± 0.10 |
| NS-7 | 6.04 ± 0.07 | — | — |
| NS-9 | 1.96 ± 0.02 | — | — |
| NS-10 | 3.74 ± 0.02 | — | — |
| NS-11 | 3.38 ± 0.04 | — | — |
| NS-12 | 2.19 ± 0.02 | — | — |
| NS-13 | 0.625 ± 0.01 | 10.98 ± 0.08 | 12.31 ± 0.09 |
| NS-14 | 1.31 ± 0.02 | — | — |
| NS-16 | 5.81 ± 0.08 | — | — |
| NS-19 | 8.08 ± 0.07 | 18.87 ± 0.11 | — |
| Pargyline | — | — | 0.072 ± 0.006 |
| Donepezil | 0.026 ± 0.003 | — | — |
IC50 values were calculated only for the selected compounds against AChE, BuChE and MAO-B enzymes, which displayed more than 50% inhibition activity at a concentration of 20 μM (Table 2). The compounds displayed IC50 values in the submicromolar range. Compounds NS-4 and NS-13 exhibited balanced multi-targeting profiles against AChE, BuChE and the MAO-B isoform. Compound NS-13 displayed the most potent inhibitory activities against AChE and MAO-B enzymes with IC50 values of 0.625 μM and 12.31 μM, respectively. It also showed inhibition of the BuChE enzyme with an IC50 value of 10.98 μM. None of the compounds was found active against the MAO-A isoform and hence the compounds showed selectivity for the MAO-B isoform. Almost all the compounds were found to be selective inhibitors of AChE as compared to BuChE. Donepezil was used as a reference for AChE, while clorgyline and pargyline were used for MAO-A and MAO-B enzymes, respectively.
Self-induced Aβ1–42 aggregation inhibition study
The aggregation of insoluble forms of Aβ1–42 is considered as one of the prime hallmarks of AD. Except for two compounds, NS-4 and NS-13, none of the derivatives of the series showed any inhibition activity towards either MAO-A or MAO-B isoforms. The volume of the active cavity of MAO-A is 400 Å while that of MAO-B is 700 Å. The active cavity of MAO-B is divided into two units, viz. the entrance cavity of 300 Å and the substrate cavity of 400 Å.26 The inactivity of these compounds towards either MAO-A or MAO-B isoforms may be attributed to the bigger size of these synthesized compounds because of the presence of both pharmacophores on the single phenyl ring. Thus, the compounds which displayed AChE inhibition activity of more than 65% i.e., NS-2, NS-4, NS-7, NS-9, NS-10, NS-11, NS-12, NS-13, NS-14, and VN-16, were investigated for their Aβ1–42 self-aggregation inhibition potential at a concentration of 20 μM using the thioflavin T (ThT) fluorescence assay. Resveratrol was used as a reference compound. Most of the tested compounds displayed moderate to high percentage aggregation inhibition after 48 h. Amongst the tested compounds, NS-2 displayed the highest percentage inhibition of 36.10% and 40.4% after 24 h and 48 h, respectively. In addition, compounds NS-4 and NS-13 also showed potent self-aggregation inhibition of 28.5% and 32.2%, respectively, after 48 h (Table 3). The compound with a balanced activity profile against AChE, MAO-B, and Aβ1–42, NS-13, was selected for FE-SEM analysis as a counterpart for the ThT assay. In the control group, the formation of Aβ1–42 plaques is evidenced (Fig. 2B) after 72 h of incubation. In the test groups, it was observed that the formation of fibrils and plaques was inhibited to a greater extent, with resveratrol (Fig. 2C) and NS-13 (Fig. 2D). Thus, the FE-SEM images correlate with the Aβ1–42 self-aggregation inhibition data of NS-13.
Aβ1–42 self-induced aggregation inhibition potential of the selected leads.
| Compound (20 μM) | Aβ1–42 self-aggregation inhibition at 24 h | Aβ1–42 self-aggregation inhibition at 48 h |
|---|---|---|
| NS-2 | 36.1% | 40.4% |
| NS-4 | 22.4% | 28.5% |
| NS-7 | 24.6% | 23.7% |
| NS-9 | — | — |
| NS-10 | 14.4% | 23.3% |
| NS-11 | 23.9% | 26.9% |
| NS-12 | 9.0% | 11.6% |
| NS-13 | 22.1% | 32.2% |
| NS-14 | 12.0% | 18.0% |
| NS-16 | 22.9% | 28.1% |
| Resveratrol | 49.3% | 61.7% |
Fig. 2. FE-SEM images of self-aggregation inhibition of Aβ1–42 after 72 h; (A) a mixture of glycine and NaOH buffer as a blank; (B) formation of plaques of Aβ1–42 only, in the absence of an inhibitor as a control; (C) inhibition of Aβ1–42 plaques with a standard, resveratrol; (D) inhibition of Aβ1–42 plaques with the test compound NS-13.

Structure–activity relationship studies
A total of nineteen chromone derivatives were synthesized with the 2-phenyl ring optionally substituted with an O-alkyl chain containing a terminal alkyne group with varied chain lengths ranging from three to five carbon atoms and a tertiary nitrogen atom in the acyclic or cyclic ring. These compounds were screened against AChE, MAO-A, and MAO-B isoforms and Aβ1–42. Initially, the compounds were screened at 20 μM concentration and the percentage inhibition of the enzymes was evaluated. NS-13 with a piperidine ring at the meta-position of the 2-substituted phenyl ring and a three-carbon O-alkyl chain at the para position showed maximum inhibition (83.13%) of the AChE enzyme. In the series, NS-9 and NS-14 also displayed more than 70% inhibition. Most of the compounds of the series were found to be selective for the AChE enzyme. Similarly, in the MAO-A and MAO-B inhibitory assays at 20 μM concentration, almost all the compounds were found to be selective for the MAO-B isoform and displayed more percentage inhibition potential compared to MAO-A. None of the compounds exhibited more than 50% inhibition for MAO-A, while only two compounds, NS-4 and NS-13, were found to be active against the MAO-B isoform with 50.78% and 61.25% inhibition. From Table 1, it was evident that the increase in the carbons of O-alkylated chain length from three carbon atoms to five carbon atoms and OMe or OEt substituents at the meta and para positions of the aromatic ring did not produce any significant effect on the AChE inhibition activity. However, it was observed that the presence of the piperidine ring on the chromone moiety was found to be compatible for the AChE inhibitory activity, while the morpholine ring significantly reduced the AChE inhibitory activity. Similarly, the presence of bigger groups like N-methyl benzyl, benzyl piperazine, phenyl piperazine, etc. at the meta-position of the aromatic ring (NS-15 to NS-18) was found to be unfavorable for AChE inhibition activity. The inactivity of the compounds against MAO isoforms may be attributed to the bulkiness of the phenyl ring with the presence of both the pharmacophores at the single ring.
Thus, from the initial screening studies, compounds with high activity towards AChE, BuChE, and MAO-B enzymes were identified, and IC50 values were calculated (Table 2). The ten most active compounds against AChE were selected for self-aggregation inhibition potential against Aβ1–42. NS-2 with a methyl-morpholine ring at the meta-position and an O-alkylated propargyl group at the para position showed a maximum inhibition of 40.4% after 48 h, whereas NS-9 with a methyl-piperidine chain at the meta position and an O-alkylated pentyne chain at the para position was found to be ineffective against Aβ1–42. NS-4 and NS-13 with a methyl-piperidine chain also showed self-aggregation inhibition potential against Aβ1–42 (28.5% and 32.2%). NS-13 with a piperidine ring and pentyne group at the 2-substituted phenyl ring showed maximum inhibition with an IC50 value of 0.625 μM. NS-9 with ethoxy and a methyl-piperidine ring and NS-14 substituted with methyl-piperidine rings at both meta positions of phenyl exhibited potent AChE inhibition in the lower micromolar range with IC50 values of 1.96 μM and 1.31 μM, respectively; however, these compounds did not show any MAO-B inhibition. Thus, NS-4 and NS-13 were identified as leads for further studies.
Reversibility inhibition studies
A reversibility inhibition study was performed to check whether the synthesized compounds are reversible or irreversible inhibitors of AChE or MAO enzymes. It was done using the dilution method for the leads, NS-4 and NS-13, against both AChE and MAO-B. The enzyme activity was reduced to minimum by preincubation of the enzymes with the test compounds at a concentration of 10 × IC50 and 100 × IC50. With 100 times dilution by buffer and substrate solution, 70% and 60% recovery in the enzymatic activity was achieved for AChE, whereas more than 76% and 71% activity was recovered for MAO-B with NS-4 and NS-13, respectively, as shown in Fig. 3. Both the tested compounds showed moderate to good recovery in enzymatic activity upon dilution, and thus are reversible inhibitors against both AChE and MAO-B.
Fig. 3. Reversibility inhibition studies of NS-4 and NS-13 against AChE and MAO-B.

Kinetic studies of AChE inhibition
AChE active sites comprise mainly two subsites, the anionic site and esteratic site. Kinetic studies were performed for the most potent AChE inhibitor using Ellman's method to check the mode of inhibition. The double reciprocal Lineweaver–Burk plot shows a correlation between the rate of reaction and various substrate concentrations displaying an increase in the slopes (decreased Vmax) and intercepts (higher Km) with higher inhibitor concentrations (Fig. 4). Graphical representation displayed mixed-type inhibition as the point of intersection of the slopes is in between the axes and not on either of the axes indicating that it simultaneously binds to both the catalytically active site (CAS) and peripheral anionic site (PAS) of AChE.
Fig. 4. Kinetic studies for the most potent AChE inhibitor, i.e.NS-13, for the mechanism of inhibition.

Cytotoxicity studies
The primary cytotoxicity evaluation of the leads, NS-4 and NS-13, was performed to check the safety index on the human neuroblastoma cell line, SH-SY5Y. The cells were preincubated for 24 h with different compound concentrations of 1 μM, 5 μM, and 25 μM, and the percentage viability was calculated against the control using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assays. At the lower concentration of 1 μM, the compound displayed no cytotoxicity. When the concentration was increased to 5 μM and 25 μM, the percentage cell viability decreased slightly. At the lower concentration of 1 μM, 97.06% and 99.57% cell viability was observed compared to the control for NS-4 and NS-13, respectively, whereas a minimum of 91.87% and 88.34% cell viability was observed for both the tested compounds at the concentration of 25 μM (Fig. 5). Keeping in mind the lower micromolar IC50 range of the synthesized compounds, we can conclude that the synthesized compounds exhibit almost negligible cytotoxicity to the specified cell lines.
Fig. 5. Cytotoxicity studies of NS-4 and NS-13 against SH-SY5Y cells. The compounds were found to be non-toxic.

ROS inhibition studies
Reactive oxygen species (ROS) are toxic substances, generally produced as by-products of cellular metabolism. The change in ROS levels is linked with the onset of oxidative stress. Neurological disorders like AD are accompanied by the accumulation of ROS in the brain. Excess accumulation of ROS can damage proteins, lipids, and DNA. Inhibition of ROS levels with increased cell viability in the brain is the count of neuroprotective potential of the evaluated compounds. ROS levels were evaluated by the reaction of a non-fluorescent dye, 2,7-dichlorofluorescein diacetate (DCF-DA), with ROS which leads to the formation of a fluorescent dye, 2,7-DCF. At 1 μM concentration, the tested compounds were not able to inhibit ROS, as only 20–30% inhibition was evidenced at lower concentration, whereas at higher concentrations, the ROS levels were significantly reduced. Compounds NS-4 and NS-13 displayed more than 50% inhibition at 5 μM concentration while both the compounds displayed reduction up to 53% and 65%, respectively, at 25 μM concentration (Fig. 6). Hence, the tested compounds displayed moderate potential to protect SH-SY5Y cells from excess accumulation of the reactive oxygen species.
Fig. 6. ROS inhibition studies by NS-4 and NS-13 against SH-SY5Y cells.

Further, the neuroprotective effect of the leads was assessed on human neuroblastoma cells, SH-SY5Y, against a neurotoxin, 6-hydroxydopamine (6-OHDA). Percentage cell viability was calculated after 24 hours of incubation with different concentrations of the test compounds, NS-4 and NS-13. Against 34.38% cell viability of 6-OHDA, compound NS-13 was found to be the most promising compound with 55.11% cell viability, while NS-4 displayed 51.91% cell viability at a lower concentration of 1 μM. The percentage cell recovery was reduced to a minimum value of 44.06% for NS-13 with increasing concentration of the compound. Only a moderate recovery in the percentage cell viability was observed at the highest concentration of 25 μM as shown in Fig. 7. Thus, it can be concluded that the compounds show moderate neuroprotection for SH-SY5Y cells against 6-OHDA as a neurotoxin.
Fig. 7. Neuroprotection studies of the leads against SH-SY5Y cells using 6-OHDA as a neurotoxin.

Molecular docking studies
To get more insights into the critical interactions and orientation of the compounds with different amino acids at the receptor's active sites, molecular docking experiments were performed. The docking studies were performed using Maestro 12.8 (Schrodinger LLC) for only the most active compounds, NS-4 and NS-13, of the series. AChE and MAO-B crystal structures were retrieved from the Protein Data Bank (PDB) with PDB IDs, 2EVE, and 2BYB, respectively. Except for the co-factor which is involved in the crucial interactions, all heteroatoms and water molecules were deleted from the specified enzymatic chains. Hydrophobicity, hydrogen bond acceptors and donors, and aromatic interactions were explored using docking studies.
It is well depicted (Fig. 8) from the ligand–enzyme interaction diagram between the compounds and the enzyme that the compounds are well accommodated inside the active site of MAO-B. The active site of MAO-B majorly consists of Tyr398, Cys172, Phe168, Trp119, and Gly170 amino acid residues. The two subunits of MAO-B, the entrance cavity and substrate cavity, are separated by Ile199 and Tyr326 which act as gates for these two subunits.26 The 2-substituted phenyl ring of the chromone showed π–π stacking interactions with Tyr326 in both cases. Compound NS-4 displayed additional π–π interactions with Tyr326. The piperidine ring and the terminal alkyne group of the ligands are aligned towards the FAD cofactor of the enzyme surrounded by Pro102, Phe103, and Trp119 amino acid residues. NS-4 and NS-13 showed additional hydrophobic interactions through the benzene ring of the chromone scaffold. Further, non-covalent interactions like van der Waals interactions and others stabilise the ligands inside the active cavity of the enzyme.
Fig. 8. A) 2D and B) 3D binding patterns of NS-4 and NS-13 with various amino acids at the active site of MAO-B (2BYB).

Fig. 9 shows the crucial interactions between the compounds and the AChE enzyme. The two specific regions of AChE, the catalytically active site (CAS) and peripheral anionic site (PAS), play important roles in the catalytic and regulatory functions of the enzyme. Serine, histidine, and glutamate assembly make up the catalytic triad, while alanine, phenylalanine, tryptophan, tyrosine, arginine, etc. constitute the PAS of AChE. The serine residue is involved in covalent bond formation with the acetylcholine substrate during the catalytic process. On the other hand, the PAS consisting of tryptophan, tyrosine, phenylalanine, arginine, alanine, etc. moieties is responsible for the allosteric interactions by modulating the AChE activity.27 The compounds, NS-4 and NS-13, are well accommodated inside the catalytic triad and show good interactions with both the CAS and PAS of AChE. The piperidine ring and the terminal alkyne group of both the compounds are oriented towards the CAS, while the benzene ring of the chromone moiety is accommodated in the PAS. The benzene ring of the compounds showed hydrophobic interactions. The carbonyl oxygen of the chromone moiety formed an additional hydrogen bonding with Phe288 in NS-13. The tertiary nitrogen atom of the compound is considered to bond with the enzyme through cationic–π interactions. The crucial interactions with both the CAS and PAS and the stability of these leads inside the active cavity make them more effective AChE inhibitors.
Fig. 9. A) 2D and B) 3D binding patterns of NS-4 and NS-13 with various amino acids at the active site of AChE (2EVE).

Molecular dynamics simulation studies
To better understand the stability and conformational changes within the targeted protein–ligand complex, molecular dynamics simulation studies were performed for the most potent inhibitors, NS-4 and NS-13. The simulation studies were performed for the docked complexes of the compounds with AChE and MAO-B, for the time-period of 100 ns. The root mean square deviation (RMSD) and radius of gyration (rGy) analyses are given in Fig. 10.
Fig. 10. Molecular dynamics trajectory analysis of NS-4 and NS-13 with both AChE and MAO-B protein complexes. A and B) RMSD of the complex; C and D) radius of gyration for the complex.

The RMSD plots (Fig. 10A) for the NS-4–AChE and NS-13–AChE complexes show that there is an initial fluctuation between 0.090 and 0.175 nm for 10 ns, and then the stability of the systems is preserved between 0.1671 and 0.2359 nm and 0.1523 and 0.1824 nm for the NS-4 and NS-13 complexes, respectively. The RMSD plots of the leads with MAO-B represent that the values of the NS-4 and NS-13 complexes with MAO-B also displayed stability throughout the simulation period (Fig. 10B), averaging at 0.1924 nm and 0.2190 nm, respectively. The RMSD values for the lead molecule complexes with the corresponding proteins, AChE and MAO-B, were determined to be within an acceptable range, indicating that the protein and ligands remained stable during the course of MD simulation. Further, to achieve the compactness at the level of protein–ligand complex structures, the radius of gyration was determined as shown in Fig. 10C and D. Given that there is no fluctuation in the rGy value for either of the lead compounds, NS-4 and NS-13, with their respective protein complexes averaging at 2.3171 nm and 2.3114 nm for AChE, and 2.2674 nm and 2.2745 nm for MAO-B, an understanding of the overall dimensions of the protein revealed that the complexes are relatively stable.
Further, the stability and flexibility in the protein structure were analyzed through the RMSF of the protein complexes and the RMSF of the ligands. These studies were performed to predict the dynamic behavior of amino acid residues throughout the simulation period. The RMSF of the complexes for NS-4 and NS-13 with AChE and MAO-B, respectively, is depicted in Fig. 11A and B. RMSF analysis specified that no internal fluctuations were seen for the complexes throughout the simulation and the RMSF values of the proteins were in the acceptable range of 0.04 nm to 0.42 nm averaging at 0.1030 nm, 0.0952 nm, 0.1117 nm, and 0.1191 nm for NS-4–AChE, NS-13–AChE, NS-4–MAO-B and NS-13–MAO-B complexes, respectively. For all complexes, a low ligand-RMSF value in the 0.026–0.274 nm range demonstrates an adequate fit of the ligands within the catalytic pocket (Fig. 11C and D).
Fig. 11. A and B) RMSF of the protein–ligand complexes; C and D) RMSF of the ligands for NS-4 and NS-13 with the respective enzymes.

Moreover, the stability of the complexes was assessed through the formation of a number of hydrogen bonds during the simulation studies. Formation of hydrogen bonds is crucial for the stability of the complexes. More than ∼90% and ∼50% hydrogen bonding with AChE while ∼95% and ∼30% hydrogen bonding with MAO-B was evidenced for NS-4 and NS-13, respectively, (Fig. 12A and B) within the time-period of 100 ns. The hydrogen bonds were formed within a cut-off range of 0.35 nm for the donor–acceptor distance (Fig. 12C and D). Overall, the studies show that the ligand–enzyme complexes form stable hydrogen bonding interactions throughout the time-period of 100 ns.
Fig. 12. A and B) Number of H-bonds of the protein–ligand complexes; C and D) H-bond length distribution of the protein–ligand complexes for NS-4 and NS-13, respectively.

Physicochemical parameters
The drug-likeness of the most potent leads, NS-4 and NS-13, was assessed through physicochemical parameters using the QikProp application of the Schrödinger suite. Both the compounds follow Lipinski's rule of five and thus present the drug likeness properties. The properties like mol. wt., HB donor and acceptor, log P, and QPlogBB values were found in the optimum range. Further, both the compounds are expected to cross the BBB as they showed good percent human oral absorption. With all the characteristics in the optimum range, both the compounds have potential to be developed as drug candidates (Table 4).
Physicochemical parameters of the leads predicted with the QikProp module.
| Compound | Mol. wt. | log P | HB donor | HB acceptor | % human oral absorption | QP log BB (−3.0 to 1.2) | BBB permeability |
|---|---|---|---|---|---|---|---|
| NS-4 | 403.47 | 4.54 | 0.5 | 6 | 100 | −0.017 | +ve |
| NS-13 | 401.50 | 5.19 | 0.5 | 5.25 | 95.53 | −0.089 | +ve |
Conclusion
Development of multi-targeting agents is an effective strategy for the treatment of complex diseases like AD. Multi-targeting agents are expected to approach multiple targets simultaneously involved in the initiation and progression of the disease. In the current series, a total of nineteen chromone derivatives were designed and synthesized by incorporating both alkynyl and tertiary nitrogen pharmacophores on the 2-substituted phenyl ring. The synthesized derivatives were investigated for the AChE, MAO-A, MAO-B, and Aβ1–42 inhibition potential. Most of the compounds of the series displayed potent AChE inhibition activity in the lower micromolar range. The inactivity of most of the compounds against MAO-A or MAO-B isoforms may be because of the larger size of the derivatives due to the incorporation of both pharmacophores on the single phenyl ring. Compounds NS-4 and NS-13 were identified as the most potent leads with balanced activity profiles against tested targets. Amongst the leads, NS-13 showed a multipotent activity profile against AChE with an IC50 value of 0.625 μM, MAO-B inhibition with an IC50 value of 12.31 μM, and 32.2% self-aggregation inhibition against Aβ1–42. The top ten AChE inhibitors of the series were evaluated against self-induced Aβ1–42 aggregation inhibition. The compounds showed moderate to good inhibition activity for self-induced Aβ1–42 aggregation. NS-4 also displayed a balanced multi-potent profile against all the targets viz. AChE (IC50 = 3.09 μM), MAO-B (IC50 = 19.64 μM), and 28.5% aggregation inhibition potential against Aβ1–42. In addition, NS-4 also exhibited potent BuChE inhibition with an IC50 value of 1.95 μM. In the reversible inhibition studies, lead compounds showed good recovery in the enzymatic activities and were found to be reversible against both AChE and MAO-B. In AChE kinetic studies, these compounds bind to both the CAS and PAS of AChE simultaneously exhibiting mixed-type inhibition. The lower micromolar IC50 range of these compounds against AChE is expected to reduce the AChE-induced Aβ accumulation in the brain. Further, the compounds reduced intracellular ROS levels up to 65% against SH-SY5Y cells at 25 μM concentration in the ROS inhibition studies. These leads were also found to be non-cytotoxic and neuroprotective in nature against the neuronal cells. Molecular docking and dynamics simulation studies demonstrate that the molecules are well accommodated inside the active cavity, and both NS-4 and NS-13 form stable trajectories with the enzymes over a time-period of 100 ns. The leads can be further utilized in the drug development process for the effective treatment of AD.
Experimental section
Materials and methods
The chemicals and reagents were purchased from Sigma Aldrich, Avra Synthesis, Spectrochem, GLR, etc. and used as received without further purification. The progress of the reaction was monitored by using Merck TLC gel 60 F254 plates. For the purification of the synthesized compounds, a pet. ether–acetone (10 : 1, 10 : 2 and 10 : 3) solution was used as an eluent of column chromatography. The NMR spectra were recorded on a JEOL NMR 600 MHz instrument in CDCl3 and TMS (δ = 0) as an internal standard at the Central Instrumental Laboratory, Central University of Punjab, Bathinda. The HRMS data of the final compounds were recorded on an Agilent LCMS-QTOF (G6530C) instrument supported by DST-FIST (grant no. SR/FST/CS-I/2020/154) at the Department of Chemistry, Central University of Punjab, Bathinda. ee-AChE, h-MAO enzymes, and human amyloid β peptide (1–42) were purchased from Molecular Probes (Invitrogen), Life Technologies, India, Sigma Aldrich, and Adooq Bioscience LLC, respectively. Ellman's assay and the Amplex red assay were performed for the calculations of AChE and MAO IC50 values. The thioflavin T-based fluorescence assay was used for Aβ aggregation inhibition studies. For cell-based assays, the SH-SY5Y cell line was procured from NCCS, Pune. Absorbance and fluorescence studies were recorded on a TECAN multi-plate reader at the Biochemistry Department, Central University of Punjab. Maestro 12.8 (Schrödinger LLC) and ChemBio Draw Ultra-12 were used for molecular modeling investigations on an HP-2800 workstation with an Intel® Xeon® X5660 configuration running at 2.80 GHz and 2.789 GHz (two processors).
Chemistry
General procedure for the synthesis of hydroxy chromone intermediates
A mixture of 2-hydroxyacetophenone (1 mmol), respective hydroxy-benzaldehyde (1 mmol), and iodine (1 mmol) was taken in a sealed tube in PEG-400 as a solvent and heated at 140 °C for 4 to 7 h. Completion of the reaction was checked by thin-layer chromatography. Upon completion of the reaction, it was cooled to room temperature, iodine was quenched with 10% sodium thiosulphate solution and the crude product was extracted with ethyl acetate. The crude product was purified through column chromatography using pet. ether/ethyl acetate (3 : 1 to 6 : 1) as an eluent to obtain the desired product (70–84% yield).
General procedure for the synthesis of Mannich intermediates
A mixture of paraformaldehyde (7 mmol) and secondary amines (3 mmol) was kept under heating at 80 °C for 1 h in ethanol. The synthesized chromone derivative (1 mmol) dissolved in ethanol was mixed to the above solution of imine. The reaction mixture was heated at 80 °C for 5–7 h to obtain the Mannich product as an intermediate. After completion of the reaction, ethanol was evaporated and water was added to the crude mixture. The aqueous layer was extracted with ethyl acetate thrice. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum using a rotary evaporator. The crude product was purified using column chromatography and ethyl acetate–pet. ether as an eluent.
General procedure for the synthesis of O-alkylated chromone derivatives
To the solution of hydroxy-substituted Mannich intermediates (1 mmol) solubilized in a minimum amount of DMF, alkyl halide (1.1 mmol) and K2CO3 (2.2 mmol) were added. The reaction mixture (RM) was heated at 80 °C and the progress of the reaction was monitored using a TLC plate. After completion of the reaction, water was added to the crude product. The aqueous layer was extracted with ethyl acetate thrice. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum using a rotary evaporator. The crude product was purified through column chromatography using acetone–pet. ether as an eluent to obtain the desired product. The synthesized compounds were characterized through HRMS and NMR.
Characteristic spectral data of the products
2-(3-Methoxy-5-(morpholinomethyl)-4-(pent-4-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-1)
White solid, 72% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6 Hz, 1.8 Hz, 1H), 7.69–7.72 (m, 1H), 7.61 (d, J = 1.8 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.43 (t, J = 7.2 Hz, 1H), 7.35 (d, J = 1.8 Hz, 1H), 6.79 (s, 1H), 4.15 (t, J = 6.0 Hz, 2H), 3.95 (s, 3H), 3.73 (t, J = 4.8 Hz, 4H), 3.61 (s, 2H), 2.52 (s, 4H), 2.49 (td, J = 2.4 Hz, 4.8 Hz, 2H), 2.03 (quintet, J = 6.0 Hz, 2H), 1.99 (t, 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.3, 163.3, 156.2, 153.1, 149.9, 133.6, 132.6, 127.0, 125.7, 125.2, 123.9, 121.1, 118.1, 108.9, 107.3, 83.8, 71.6, 68.8, 67.0, 56.9, 55.9, 53.7, 29.2, 15.2. HRMS: m/z [M + H]+ for C26H27NO5, calculated 434.1962; observed 434.1972.
2-(3-Methoxy-5-(morpholinomethyl)-4-(prop-2-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-2)
Off-white solid, 66% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.6 Hz, 1.8 Hz, 1H), 7.69–7.72 (m, 1H), 7.63 (d, J = 1.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.42–7.45 (m, 1H), 7.36 (d, J = 1.8 Hz, 1H), 6.80 (s, 1H), 4.84 (d, J = 2.4 Hz, 2H), 3.97 (s, 3H), 3.73 (t, J = 4.8 Hz, 4H), 368 (s, 2H), 2.54 (s, 4H), 2.48 (t, J = 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.3, 163.2, 156.2, 153.0, 148.2, 133.7, 133.5, 127.7, 125.7, 125.2, 123.9, 120.8, 118.1, 108.8, 107.4, 79.1, 75.5, 67.1, 60.2, 57.1, 56.0, 53.6. HRMS: m/z [M + H]+ for C24H23NO5, calculated 406.1649; observed 406.1655.
2-(4-(But-3-yn-1-yloxy)-3-methoxy-5-(morpholinomethyl)phenyl)-4H-chromen-4-one (NS-3)
White solid, 69% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 4.2 Hz, 1H), 7.69–7.72 (m, 1H), 7.59–7.60 (m, 2H), 7.42–7.45 (m, 1H), 7.35 (d, J = 1.8 Hz, 1H), 6.79 (s, 1H), 4.21 (t, J = 6.6 Hz, 2H), 3.96 (s, 3H), 3.73 (t, J = 4.8 Hz, 4H), 3.64 (s, 2H), 2.70 (td, J = 4.2 Hz, 3.0 Hz, 2H), 2.53 (s, 4H), 2.05 (t, J = 3.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.3, 163.2, 156.2, 153.0, 149.4, 133.7, 132.6, 127.2, 125.7, 125.2, 123.9, 121.1, 118.1, 109.0, 107.3, 80.9, 71.1, 69.8, 67.0, 57.0, 56.0, 53.6, 20.3. HRMS: m/z [M + H]+ for C25H25NO5, calculated 420.1805; observed 420.1833.
2-(3-Methoxy-5-(piperidin-1-ylmethyl)-4-(prop-2-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-4)
Yellowish solid, 66% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.66 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.42 (t, J = 7.2 Hz, 1H), 7.34 (s, 1H), 6.81 (s, 1H), 4.84 (d, J = 1.8 Hz, 2H), 3.96 (s, 3H), 3.67 (s, 2H), 2.51 (s, 1H), 2.48 (s, 4H), 1.60 (quintet, J = 5.4 Hz, 4H), 1.45 (s, 2H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.3, 156.2, 152.9, 148.2, 133.7, 127.6, 125.7, 125.2, 123.9, 121.1, 118.1, 108.6, 107.3, 79.1, 75.4, 60.2, 57.2, 56.0, 54.4, 25.9, 24.2. HRMS: m/z [M + H]+ for C25H25NO4, calculated 404.1856; observed 404.1855.
2-(3-Methoxy-4-(pent-4-yn-1-yloxy)-5-(piperidin-1-ylmethyl)phenyl)-4H-chromen-4-one (NS-5)
White solid, 78% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.6 Hz, 1.2 Hz, 1H), 7.69–7.71 (m, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.41–7.44 (m, 1H), 7.33 (d, J = 2.4 Hz, 1H), 6.80 (s, 1H), 4.13 (t, J = 6.6 Hz, 2H), 3.94 (s, 3H), 3.56 (s, 2H), 2.49 (td, J = 4.2 Hz, 3.0 Hz, 2H), 2.43 (s, 4H), 2.02 (quintet, J = 7.2 Hz, 2H), 1.98 (t, J = 2.4 Hz, 1H), 1.59 (quintet, J = 5.4 Hz, 4H), 1.45 (s, 2H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.5, 156.2, 153.0, 149.8, 133.7, 133.6, 126.8, 125.7, 125.1, 123.9, 121.2, 118.1, 108.6, 107.2, 83.9, 71.6, 68.6, 57.2, 55.9, 54.6, 29.2, 26.1, 24.3, 15.2. HRMS: m/z [M + H]+ for C27H29NO4, calculated 432.2169; observed 432.2173.
2-(3-Ethoxy-5-(morpholinomethyl)-4-(prop-2-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-6)
White solid, 73% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (d, J = 7.8 Hz, 1H), 7.69–7.72 (m, 1H), 7.61 (s, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.41–7.45 (m, 1H), 7.34 (s, 1H), 6.78 (s, 1H), 4.87 (d, J = 0.6 Hz, 2H), 4.18 (q, J = 7.2 Hz, 2H), 3.73 (t, J = 4.2 Hz, 4H), 3.68 (s, 2H), 2.53 (s, 4H), 2.47 (t, J = 2.4 Hz, 1H), 1.52 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 178.3, 163.2, 156.2, 152.2, 148.4, 133.7, 133.4, 127.6, 125.7, 125.2, 123.9, 120.7, 118.0, 109.8, 107.4, 79.2, 75.4, 67.1, 64.6, 60.1, 57.1, 53.6, 14.8. HRMS: m/z [M + H]+ for C25H25NO5, calculated 420.1805; observed 420.1803.
2-(4-(But-3-yn-1-yloxy)-3-ethoxy-5-(morpholinomethyl)phenyl)-4H-chromen-4-one (NS-7)
Yellowish solid, 64% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 7.69–7.72 (m, 1H), 7.59 (d, J = 7.8 Hz, 2H), 7.41–7.45 (m, 1H), 7.34 (d, J = 2.4 Hz, 1H), 6.78 (s, 1H), 4.23 (t, J = 6.6 Hz, 2H), 4.17 (q, J = 7.2 Hz, 2H), 3.73 (t, J = 4.2 Hz, 4H), 3.64 (s, 2H), 2.71 (td, J = 4.2 Hz, 2.4 Hz, 2H), 2.53 (s, 4H), 2.04 (t, J = 3.0 Hz, 1H), 1.52 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.3, 156.2, 152.2, 149.6, 133.7, 132.5, 127.1, 125.7, 125.2, 123.9, 121.0, 118.0, 110.0, 107.3, 81.0, 71.1, 69.8, 67.0, 64.6, 57.0, 53.6, 20.4, 14.8. HRMS: m/z [M + H]+ for C26H27NO5, calculated 434.1962; observed 434.1966.
2-(3-Ethoxy-5-(morpholinomethyl)-4-(pent-4-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-8)
Off-white solid, 68% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 769–7.72 (m, 1H), 7.58–7.60 (m, 2H), 7.41–7.44 (m, 1H), 7.34 (dd, J = 2.4 Hz, 1H), 6.78 (s, 1H), 4.15–4.19 (m, 4H), 3.73 (t, J = 4.8 Hz, 4H), 3.60 (s, 2H), 2.52 (s, 4H), 2.49 (td, J = 4.2 Hz, 2.4 Hz, 2H), 2.03 (quintet, J = 6.6 Hz, 2H), 1.99 (t, J = 3.0 Hz, 1H), 1.52 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 178.3, 163.4, 156.2, 152.3, 150.0, 133.6, 132.5, 126.9, 125.7, 125.2, 123.9, 121.0, 118.0, 109.9, 107.2, 83.8, 71.6, 68.8, 67.1, 64.5, 56.9, 53.7, 29.2, 15.3, 14.8. HRMS: m/z [M + H]+ for C27H29NO5, calculated 448.2118; observed 448.2121.
2-(3-Ethoxy-4-(pent-4-yn-1-yloxy)-5-(piperidin-1-ylmethyl)phenyl)-4H-chromen-4-one (NS-9)
Off-white solid, 69% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 7.68–7.71 (m, 1H), 7.61 (d, J = 7.58, 1H), (dd, J = 7.8 Hz, 0.6 Hz, 1H), 7.40–7.43 (m, 1H), 7.32 (d, J = 2.4 Hz, 1H), 6.78 (s, 1H), 4.14–4.17 (m, 4H), 3.55 (s, 2H), 2.49 (td, J = 4.8 Hz, 2.4 Hz, 2H), 2.44 (s, 4H), 2.02 (quintet, J = 6.0 Hz, 2H), 1.99 (t, J = 3.0 Hz, 1H), 1.59 (quintet, J = 5.4 Hz, 4H), 1.51 (t, J = 7.2 Hz, 3H), 1.45 (s, 2H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.7, 156.3, 152.3, 150.0, 133.7, 133.6, 126.8, 125.7, 125.2, 124.0, 121.1, 118.1, 109.6, 107.2, 83.9, 71.6, 68.7, 64.5, 57.3, 54.7, 29.3, 26.1, 24.4, 15.4, 14.9. HRMS: m/z [M + H]+ for C28H31NO4, calculated 446.2326; observed 446.2328.
2-(3,5-Bis(morpholinomethyl)-4-(prop-2-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-10)
Off-white solid, 64% yield, 1H NMR (600 MHz, CDCl3) δ 8.24 (dd, J = 6.6 Hz, 1.2 Hz, 1H), 7.87 (s, 2H), 7.70–7.74 (m,1H), 7.60 (d, J = 8.4 Hz, 1H), 7.44 (t, J = 7.2 Hz, 1H), 6.81 (s, 1H), 4.93 (d, J = 3.0 Hz, 2H), 3.73 (t, J = 4.8 Hz, 8H), 3.64 (s, 4H), 2.58 (t, J = 2.4 Hz, 1H), 2.55 (s, 8H); 13C NMR (150 MHz, CDCl3) δ 178.3, 163.0, 159.1, 156.2, 133.7, 133.0, 128.4, 127.6, 125.7, 125.3, 123.9, 118.1, 107.5, 79.1, 75.7, 67.0, 62.8, 57.7, 53.6. HRMS: m/z [M + H]+ for C28H30N2O4, calculated 475.2227; observed 475.2228.
2-(3,5-Bis(morpholinomethyl)-4-(pent-4-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-11)
Off-white solid, 77% yield, 1H NMR (600 MHz, CDCl3) δ 8.24 (dd, J = 6.6 Hz, 1.2 Hz, 1H), 7.93 (s, 2H), 7.70–7.73 (m, 1H), 7.60 (dd, J = 7.8 Hz, 0.6 Hz, 1H), 7.42–7.45 (m, 1H), 6.82 (s, 1H), 4.08 (t, J = 6.6 Hz, 2H), 3.74 (t, J = 4.2 Hz, 8H), 3.61 (s, 4H), 2.50–2.53 (m, 10H), 2.05 (quintet, J = 6.6 Hz, 2H), 2.03 (t, J = 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.3, 159.6, 156.2, 133.7, 132.4, 128.0, 127.2, 125.7, 125.2, 123.9, 118.1, 107.3, 83.6, 72.9, 69.3, 67.0, 57.1, 53.7, 29.0, 15.2. HRMS: m/z [M + H]+ for C30H34N2O4, calculated 503.2540; observed 503.2544.
2-(3-(Morpholinomethyl)-4-(prop-2-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-12)
White solid, 69% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.6 Hz, 1.2 Hz, 1H), 8.01 (d, J = 1.8 Hz, 1H), 7.85 (dd, J = 6.6 Hz, 2.4 Hz, 1H), 7.68–7.71 (m, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.42 (t, J = 7.2 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 6.78 (s, 1H), 4.81 (d, J = 2.4 Hz, 2H), 3.76 (t, J = 4.2 Hz, 4H), 3.63 (s, 2H), 2.55–2.57 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.4, 158.4, 156.2, 133.6, 128.5, 127.7, 126.4, 125.7, 125.1, 124.7, 123.9, 118.0, 112.3, 106.6, 77.9, 76.1, 67.1, 56.2, 56.1, 53.6. HRMS: m/z [M + H]+ for C23H21NO4, calculated 376.1543; observed 376.1546.
2-(4-(Pent-4-yn-1-yloxy)-3-(piperidin-1-ylmethyl)phenyl)-4H-chromen-4-one (NS-13)
Yellowish solid, 70% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 8.00 (s, 1H), 7.81 (dd, J = 6.0 Hz, 2.4 Hz, 1H), 7.67–7.70 (m, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.70–7.42 (m, 1H), 6.98 (d, J = 9.0 Hz, 1H), 6.78 (s, 1H), 4.17 (t, J = 6.6 Hz, 2H), 3.58 (s, 2H), 2.45–2.49 (m, 6H), 2.06 (quintet, J = 6.0 Hz, 2H), 2.00 (t, J = 3.0 Hz, 1H), 1.64 (t, J = 5.4 Hz, 4H), 1.47 (s, 2H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.7, 159.8, 156.2, 133.5, 128.5, 126.4, 125.6, 125.0, 124.0, 123.7, 118.0, 111.3, 106.2, 83.2, 69.1, 66.5, 56.5, 54.6, 28.1, 26.0, 24.2, 15.2. HRMS: m/z [M + H]+ for C26H27NO3, calculated 402.2069; observed 402.2069.
2-(4-(Pent-4-yn-1-yloxy)-3,5-bis(piperidin-1-ylmethyl)phenyl)-4H-chromen-4-one (NS-14)
White solid, 79% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 7.91 (s, 2H), 7.69–7.71 (m, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.41–7.43 (m, 1H), 6.83 (s, 1H), 4.05 (t, J = 6.6 Hz, 2H), 3.54 (s, 4H), 2.50 (td, J = 4.2 Hz, 3.0 Hz, 2H), 2.45 (s, 8H), 2.04 (quintet, J = 6.6 Hz, 2H), 2.01 (t, J = 2.4 Hz, 1H), 1.60 (quintet, J = 5.4 Hz, 8H), 1.46 (s, 4H); 13C NMR (150 MHz, CDCl3) δ 178.5, 163.8, 159.5, 156.3, 133.5, 133.3, 127.6, 126.9, 125.6, 125.1, 124.0, 118.1, 107.2, 83.8, 72.7, 69.0, 57.5, 54.7, 29.1, 26.1, 24.4, 15.2. HRMS: m/z [M + H]+ for C32H38N2O3, calculated 499.2955; observed 499.2963.
2-(3-((Benzyl(methyl)amino)methyl)-5-methoxy-4-(pent-4-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-15)
White solid, 73% yield, 1H NMR (600 MHz, CDCl3) δ 8.24 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 7.76 (d, J = 1.8 Hz, 1H),7.70–7.73 (m, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.42–7.44 (m, 1H),7.39 (d, J = 6.6 Hz, 2H), 7.33–7.35 (m, 3H), 7.25 (t, J = 7.2 Hz, 1H), 6.81 (s, 1H), 4.13 (t, J = 6.0 Hz, 2H), 3.94 (s, 3H), 3.65 (s, 2H), 3.59 (s, 2H), 2.47 (td, J = 4.2 Hz, 2.4 Hz, 2H), 2.24 (s, 3H), 2.00 (quintet, J = 6.6 Hz, 2H), 1.98 (t, J = 3.0 Hz, 1H), 13C NMR (150 MHz, CDCl3) δ 178.4, 163.5, 156.2, 152.9, 149.6, 139.2, 134.0, 133.6, 128.9, 128.3, 127.0, 127.0, 125.7, 125.1, 123.9, 120.9, 118.0, 108.6, 107.2, 83.8, 71.5, 68.7, 62.2, 55.9, 55.2, 42.5, 29.2, 15.2. HRMS: m/z [M + H]+ for C30H29NO4, calculated 468.2169; observed 468.2172.
2-(3-((4-Benzylpiperazin-1-yl)methyl)-5-methoxy-4-(pent-4-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-16)
White solid, 81% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 7.69–7.72 (m, 1H), 7.60 (d, J = 2.4 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.41–7.44 (m, 1H), 7.33 (d, J = 2.4 Hz, 1H), 7.29–7.33 (m, 4H), 7.22–7.25 (m, 1H), 6.79 (s, 1H), 4.13 (t, J = 6.0 Hz, 2H), 3.94 (s, 3H), 3.62 (s, 2H), 3.52 (s, 2H), 2.46–2.54 (m, 10H), 2.01 (quintet, J = 6.6 Hz, 2H), 1.98 (t, J = 3.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.4, 156.2, 153.0, 149.8, 138.2, 133.6, 133.0, 129.2, 128.1, 127.0, 126.9, 125.7, 125.2, 123.9, 121.1, 118.1, 108.8, 107.2, 83.8, 71.6, 68.7, 63.0, 56.4, 55.9, 53.2, 53.2, 29.2, 15.2. HRMS: m/z [M + H]+ for C33H34N2O4, calculated 523.2591; observed 523.2590.
2-(4-(But-3-yn-1-yloxy)-3-methoxy-5-((4-phenylpiperazin-1-yl)methyl)phenyl)-4H-chromen-4-one (NS-17)
White solid, 67% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (dd, J = 6.0 Hz, 1.8 Hz, 1H), 7.69–7.72 (m, 1H), 7.64 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.42–7.44 (m, 1H), 7.36 (s, 1H), 7.25–7.27 (m, 2H), 6.93 (d, J = 7.8 Hz, 2H), 6.85 (t, J = 7.2 Hz, 1H), 6.80 (s, 1H), 4.22 (t, J = 5.4 Hz, 2H), 3.97 (s, 3H), 3.72 (s, 2H), 3.22 (s, 4H), 2.70–2.73 (m, 6H), 2.04 (t, J = 2.4 Hz, 1H), 13C NMR (150 MHz, CDCl3) δ 178.3, 163.3, 156.2, 153.0, 151.3, 149.5, 133.7, 132.8, 129.1, 127.2, 125.7, 125.2, 123.9, 121.2, 119.6, 118.1, 116.0, 109.0, 107.3, 80.9, 71.1, 69.8, 56.6, 56.0, 53.1, 49.2, 20.3. HRMS: m/z [M + H]+ for C31H30N2O4, calculated 495.2278; observed 495.2286.
2-(3-Methoxy-4-(pent-4-yn-1-yloxy)-5-((4-phenylpiperazin-1-yl)methyl)phenyl)-4H-chromen-4-one (NS-18)
White solid, 79% yield, 1H NMR (600 MHz, CDCl3) δ 8.23 (d, J = 7.8 Hz, 1H), 7.70 (t, J = 8.4 Hz, 1H), 7.65 (d, J = 1.8 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 1.8 Hz, 1H), 7.26 (t, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.85 (t, J = 7.8 Hz, 1H), 6.80 (s, 1H), 4.17 (t, J = 6.0 Hz, 2H), 3.96 (s, 3H), 3.68 (s, J = 2H), 3.23 (t, J = 4.8 Hz, 4H), 2.69 (t, J = 4.2 Hz, 4H), 2.49 (td, J = 3.6 Hz, 3.0 Hz, 2H), 2.03 (quintet, J = 6.0 Hz, 2H), 1.97 (t, J = 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.3, 156.2, 153.1, 151.3, 149.9, 133.6, 132.8, 129.1, 127.0, 125.7, 125.2, 123.9, 121.1, 119.6, 118.1, 116.0, 108.9, 107.2, 83.8, 71.7, 68.8, 56.5, 55.9, 53.2, 49.2, 29.2, 15.2. HRMS: m/z [M + H]+ for C32H32N2O4, calculated 509.2435; observed 509.2443.
2-(3-((3,4-Dihydroisoquinolin-2(1H)-yl)methyl)-5-methoxy-4-(pent-4-yn-1-yloxy)phenyl)-4H-chromen-4-one (NS-19)
Yellowish solid, 74% yield, 1H NMR (600 MHz, CDCl3) δ 8.22 (dd, J = 6.6 Hz, 1.2 Hz, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.67–7.70 (m, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.41 (t, J = 7.2 Hz, 1H), 7.35 (d, J = 1.8 Hz, 1H), 7.09–7.14 (m, 3H), 7.00 (d, J = 7.2 Hz, 1H), 6.77 (s, 1H), 4.16 (t, J = 6.0 Hz, 2H), 3.96 (s, 3H), 3.80 (s, 2H), 3.72 (s, 2H), 2.93 (t, J = 6.0 Hz, 2H), 2.81 (t, J = 6.0 Hz, 2H), 2.47 (td, J = 4.8 Hz, 2.4 Hz, 2H), 2.01 (quintet, J = 6.6 Hz, 2H), 1.94 (t, J = 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 178.4, 163.4, 156.2, 153.1, 149.8, 134.8, 134.3, 133.6, 133.2, 128.7, 127.1, 126.5, 126.1, 125.6, 125.6, 125.1, 123.9, 121.0, 118.1, 108.9, 107.3, 83.7, 71.7, 68.7, 56.2, 56.1, 55.9, 50.8, 29.2, 29.2, 15.2. HRMS: m/z [M + H]+ for C31H29NO4, calculated 480.2169; observed 480.2177.
Biological studies
ChE and MAO inhibition assay
The AChE inhibition assay was performed using Ellman's method.28,29 The enzyme was purchased from Molecular Probes, Inc./Invitrogen. Briefly, for enzyme inhibition, 40 μl of recombinant enzyme (AChE, 0.5 U ml−1) and 20 μl of test compounds were incubated for 30 min. in a CO2 incubator at 37 °C using a flat-bottom 96 well plate. 20 μl dye, DTNB, and 20 μl of substrate, AChSI, were added to the existing solution of the enzyme and test compounds after completion of the incubation period. The absorption spectra of this system were recorded on a TECAN multiplate reader at 417 nm. All the experiments were performed in triplicate (n = 3). The BuChE inhibition assay was performed in a similar way using the butyrylcholinesterase enzyme and BTCI as a substrate. IC50 values were calculated for the most potent derivatives only.
Further, MAO inhibition was performed using the Amplex red assay.30 Amplex red was purchased from ThermoFisher-Scientific, Inc./Invitrogen, while MAO-A and MAO-B were purchased from Sigma Aldrich. Briefly, inhibition of the enzyme was done using 40 μl of recombinant MAO enzyme and 10 μl of test compounds for 30 min in a CO2 incubator at 37 °C using a flat-bottom 96 well plate. A 50 μl working solution containing the substrate, HRP, and Amplex red was added to the existing solution of the enzyme and test compounds. The production of H2O2 was quantified at 37 °C in a multi-detection microplate fluorescence reader (TECAN Instrument) at an excitation wavelength of 545 nm and an emission wavelength of 590 nm. All the experiments were performed in triplicate (n = 3). The details of the experimental procedure for AChE, BuChE, and MAO inhibition are given in our previous reports.31
Reversibility and enzyme kinetic studies
Reversibility inhibition studies were performed using the dilution method. The compounds were incubated with enzymes at a concentration of 10 and 100 times of the IC50 values at 37 °C for 30 min using a flat-bottom 96 well plate. Further, the samples were diluted 100-fold with the addition of the substrate to reach final inhibitor concentrations of 0.1 × IC50 and 1 × IC50, respectively. All the experiments were performed in triplicate (n = 3).
The enzyme kinetic studies were carried out by following previously described procedures.32 The experiment was performed using different substrate and final inhibitor concentrations. Double reciprocal Lineweaver–Burk plot was drawn to determine the type of inhibition. The details of the procedure for reversibility and enzyme kinetics are given in our previous reports.31
ROS inhibition, neuroprotection, and cytotoxicity studies
ROS inhibition studies were performed using three different concentrations (1 μM, 5 μM, and 25 μM) of the test compounds. The cells were exposed to the test compounds for 24 h. Human neuroblastoma (SH-SY5Y) cells were cultured in DMEM/F-12 media supplemented with 10% fetal bovine serum (FBS), horse serum (1%), and 1% penicillin antibiotic solution. Intracellular levels of ROS were determined according to the protocol described.29 Following the incubation, the fluorescence was detected at an excitation wavelength of 478 nm and an emission wavelength of 518 nm. The results were expressed as the mean ± standard deviation (SD) of triplicate or more independent experiments. Further, in another study, 6-OHDA as a neurotoxin was utilized for the determination of the neuroprotective potential of the synthesized compounds on SH-SY5Y cells. The cells were cultured for 24 h at 37 °C in a humidified atmosphere containing 5% CO2. The amount of formazan formed was measured using a microculture plate reader at a test wavelength of 594 nm.33 The absorbance readings obtained from the MTT assay were analyzed. Additionally, the cytotoxicity studies were performed using the MTT assay on SH-SY5Y cells. The assay is based on the conversion of MTT to a blue formazan product by cellular oxidoreductases. The formazan produced was solubilized in DMSO, and absorbance of the formazan solution was recorded at 595 nm using a multiplate reader.34 The details of the experimental procedure for ROS inhibition, neuroprotection, and cytotoxicity studies are described in our previous reports.31
Amyloid β self-aggregation inhibition studies
Aβ was purchased from Adooq Biosciences and was used as received. The Aβ aggregation inhibition potential was investigated using a fluorimetry-based thioflavin T assay.35 Initially, 1 mg ml−1 concentration of Aβ was pretreated with HFIP (1,1,1,3,3,3-hexafloro-2-propanol). Post HFIP evaporation, 40 μl of Aβ was incubated using a 96-well plate without test compounds as a control. Then the test compounds were incubated with Aβ at 37 °C for 24 h. From each well, 20 μl mixture was transferred to another 96 well plate after 24 h for continuous incubation for 48 h. A 180 μl solution of glycine–NaOH buffer (pH 8.0) containing 5 μM ThT was added to a mixture of Aβ + test compounds. The fluorescence was measured in the 440 nm to 485 nm range. % inhibition was calculated by considering the control as 0% inhibition. For recording the FE-SEM images, the samples were prepared on a carbon-coated copper grid and dried at 37 °C for 1 h. The samples were gold coated for increasing the conductivity and recorded.
Computational studies
Molecular docking studies were conducted using the Glide module of Maestro 12.8 (Schrödinger LLC). PDB IDs were retrieved as 1EVE for AChE and 2BYB as MAO-B. The top-score docking poses were selected for each compound for further analysis using the XP interaction visualizer of the software. The drug-likeness properties of the leads were evaluated using the QikProp application of the Schrödinger suite. Further, molecular dynamics (MD) simulations were performed using the Desmond standard protocol to investigate the stability of the ligands within the active site of enzymes. MD was performed to investigate the stability behavior of the inhibitors within the active site including monitoring of RMSD, RMSF, hydrogen bonding, and other relevant parameters. The detailed protocol for the experimental procedure is described in our previous reports.31
Abbreviations
- AD
Alzheimer's disease
- APP
Amyloid precursor protein
- eeAChE
Electric eel acetylcholinesterase
- ACh
Acetylcholine
- hMAO
Human monoamine oxidase
- BACE-1
β-site APP cleaving enzyme-1
- BBB
Blood brain barrier
- BuChE
Butyrylcholinesterase
- CAS
Catalytically active site
- PAS
Peripheral anionic site
- ChEs
Cholinesterase
- FAD
Flavin adenine dinucleotide
- FDA
Food and drug administration
- IC50
Half maximum inhibition conc.
- MTDLs
Multi-target directed ligands
- nM
Nanomolar
- NMDA
N-methyl-d-aspartate
- MD
Molecular dynamics
- Aβ
Amyloid beta
- μM
Micromolar
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Vinod Kumar conceptualized, designed and instructed the study and finalized the manuscript. NK synthesized the library of compounds, performed the in vitro enzymatic assays, and wrote the manuscript. KJ, BD, and Vinay Kumar assisted in the in vitro assays and prepared the molecular dynamics analysis. TA performed the cytotoxicity assays supervised by JP. JM performed the amyloid beta assays and JSB supervised the work. Vijay Kumar and ARD helped in the docking studies and analyzed the results.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
VK is thankful to the CSIR, New Delhi (02/(0354)/19/EMRII), and STARS-IISc (MoE-STARS/STARS-2/2023-0040) for the research grants. NK and Vijay Kumar are thankful to the University Grant Commission, New Delhi for providing a Senior Research Fellowship. Vinay Kumar and TA are thankful to the CSIR for SRF. KJ is thankful to ICMR (file no. 45/29/2022-/BIO/BMS) for the fellowship grant. Bharti is thankful to DST-WISE KIRAN (DST/WISE-PhD/CS/2023/64) for her fellowship. We are thankful to DST for the FIST grant reference number SR/FST/CS-I/2020/154 (C), and the Central Instrumentation Laboratory of the Central University of Punjab.
Electronic supplementary information (ESI) available: NMR and HRMS data and graphs of the final compounds. See DOI: https://doi.org/10.1039/d4md00550c
References
- Gong C.-X. Liu F. Iqbal K. J. Alzheimer's Dis. 2018;64:107–117. doi: 10.3233/JAD-179921. [DOI] [PubMed] [Google Scholar]
- Kumar N. Kumar V. Anand P. Kumar V. Dwivedi A. R. Kumar V. Bioorg. Med. Chem. 2022;61:116742. doi: 10.1016/j.bmc.2022.116742. [DOI] [PubMed] [Google Scholar]
- Majdi A. Sadigh-Eteghad S. Rahigh Aghsan S. Farajdokht F. Vatandoust S. M. Namvaran A. Mahmoudi J. Rev. Neurosci. 2020;31:391–413. doi: 10.1515/revneuro-2019-0089. [DOI] [PubMed] [Google Scholar]
- Alvarez A. Opazo C. Alarcón R. Garrido J. Inestrosa N. C. J. Mol. Biol. 1997;272:348–361. doi: 10.1006/jmbi.1997.1245. [DOI] [PubMed] [Google Scholar]
- Carson K. A. Geula C. Mesulam M.-M. Brain Res. 1991;540:204–208. doi: 10.1016/0006-8993(91)90508-S. [DOI] [PubMed] [Google Scholar]
- Moran M. Mufson E. Gomez-Ramos P. Acta Neuropathol. 1993;85:362–369. doi: 10.1007/BF00334445. [DOI] [PubMed] [Google Scholar]
- Schedin-Weiss S. Inoue M. Hromadkova L. Teranishi Y. Yamamoto N. G. Wiehager B. Bogdanovic N. Winblad B. Sandebring-Matton A. Frykman S. Alzheimer's Res. Ther. 2017;9:1–19. doi: 10.1186/s13195-016-0227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhazmi H. A. Albratty M. Saudi Pharm. J. 2022;30:1755–1764. doi: 10.1016/j.jsps.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesiti F. Chavarria D. Gaspar A. Alcaro S. Borges F. Eur. J. Med. Chem. 2019;181:111572. doi: 10.1016/j.ejmech.2019.111572. [DOI] [PubMed] [Google Scholar]
- Gong C.-X. Dai C.-L. Liu F. Iqbal K. Front. Aging Neurosci. 2022;14:837649. doi: 10.3389/fnagi.2022.837649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdpour S. Jalili-Baleh L. Nadri H. Forootanfar H. Bukhari S. N. A. Ramazani A. Ebrahimi S. E. S. Foroumadi A. Khoobi M. Bioorg. Chem. 2021;110:104750. doi: 10.1016/j.bioorg.2021.104750. [DOI] [PubMed] [Google Scholar]
- Ramesh M. Balachandra C. Andhare P. Govindaraju T. ACS Chem. Neurosci. 2022;13:2209–2221. doi: 10.1021/acschemneuro.2c00276. [DOI] [PubMed] [Google Scholar]
- Auti P. S. Jagetiya S. Paul A. T. Chem. Biodiversity. 2023;20:e202300587. doi: 10.1002/cbdv.202300587. [DOI] [PubMed] [Google Scholar]
- Reis J. Cagide F. Valencia M. E. Teixeira J. Bagetta D. Perez C. Uriarte E. Oliveira P. J. Ortuso F. Alcaro S. Eur. J. Med. Chem. 2018;158:781–800. doi: 10.1016/j.ejmech.2018.07.056. [DOI] [PubMed] [Google Scholar]
- Estrada-Valencia M. Herrera-Arozamena C. Pérez C. Viña D. Morales-García J. A. Pérez-Castillo A. Ramos E. Romero A. Laurini E. Pricl S. J. Enzyme Inhib. Med. Chem. 2019;34:712–727. doi: 10.1080/14756366.2019.1581184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang Z. Wang K. Shi J. Cheng X. Zhu G. Wei R. Ma Q. Yu L. Zhao Y. Tan Z. Eur. J. Med. Chem. 2020;187:111958. doi: 10.1016/j.ejmech.2019.111958. [DOI] [PubMed] [Google Scholar]
- Wang X.-B. Yin F.-C. Huang M. Jiang N. Lan J.-S. Kong L.-Y. RSC Med. Chem. 2020;11:225–233. doi: 10.1039/C9MD00441F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez L. Madurga S. Pukala T. Vilaseca M. López-Iglesias C. Robinson C. V. Giralt E. Carulla N. J. Am. Chem. Soc. 2011;133:6505–6508. doi: 10.1021/ja1117123. [DOI] [PubMed] [Google Scholar]
- Fernández-Bachiller M. I. Pérez C. N. Monjas L. Rademann J. R. Rodríguez-Franco M. I. J. Med. Chem. 2012;55:1303–1317. doi: 10.1021/jm201460y. [DOI] [PubMed] [Google Scholar]
- Liu Q. Qiang X. Li Y. Sang Z. Li Y. Tan Z. Deng Y. Bioorg. Med. Chem. 2015;23:911–923. doi: 10.1016/j.bmc.2015.01.042. [DOI] [PubMed] [Google Scholar]
- Li R.-S. Wang X.-B. Hu X.-J. Kong L.-Y. Bioorg. Med. Chem. Lett. 2013;23:2636–2641. doi: 10.1016/j.bmcl.2013.02.095. [DOI] [PubMed] [Google Scholar]
- Krysiak J. M. Kreuzer J. Macheroux P. Hermetter A. Sieber S. A. Breinbauer R. Angew. Chem., Int. Ed. 2012;51:7035–7040. doi: 10.1002/anie.201201955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhav H. Jameel E. Rehan M. Hoda N. RSC Med. Chem. 2022;13:258–279. doi: 10.1039/D1MD00394A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. Sharma N. Kumar V. Kumar V. Jangid K. Devi B. Dwivedi A. R. Giri K. Kumar R. Kumar V. RSC Adv. 2024;14:6225–6233. doi: 10.1039/D3RA08810C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allochio Filho J. F. Lemos B. C. de Souza A. S. Pinheiro S. Greco S. J. Tetrahedron. 2017;73:6977–7004. doi: 10.1016/j.tet.2017.10.063. [DOI] [Google Scholar]
- Binda C. Hubálek F. Li M. Edmondson D. E. Mattevi A. FEBS Lett. 2004;564:225–228. doi: 10.1016/S0014-5793(04)00209-1. [DOI] [PubMed] [Google Scholar]
- Dvir H. Silman I. Harel M. Rosenberry T. L. Sussman J. L. Chem.-Biol. Interact. 2010;187:10–22. doi: 10.1016/j.cbi.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman G. L. Courtney K. D. Andres Jr V. Featherstone R. M. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Kumar B. Dwivedi A. R. Arora T. Raj K. Prashar V. Kumar V. Singh S. Prakash J. Kumar V. ACS Chem. Neurosci. 2022;13:2122–2139. doi: 10.1021/acschemneuro.2c00132. [DOI] [PubMed] [Google Scholar]
- Guang H. M. Du G. H. Acta Pharmacol. Sin. 2006;27:760–766. doi: 10.1111/j.1745-7254.2006.00336.x. [DOI] [PubMed] [Google Scholar]
- Kumar N. Jangid K. Kumar V. Yadav R. P. Mishra J. Upadhayay S. Kumar V. Devi B. Kumar V. Dwivedi A. R. ACS Chem. Neurosci. 2024;15:2565–2585. doi: 10.1021/acschemneuro.4c00007. [DOI] [PubMed] [Google Scholar]
- Cavdar H. Senturk M. Guney M. Durdagi S. Kayik G. Supuran C. T. Ekinci D. J. Enzyme Inhib. Med. Chem. 2019;34:429–437. doi: 10.1080/14756366.2018.1543288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferah Okkay I. Okkay U. Cicek B. Yilmaz A. Yesilyurt F. Mendil A. S. Hacimuftuoglu A. Mol. Biol. Rep. 2021;48:7711–7717. doi: 10.1007/s11033-021-06779-y. [DOI] [PubMed] [Google Scholar]
- Rekha S. Anila E. Mater. Lett. 2019;236:637–639. doi: 10.1016/j.matlet.2018.11.009. [DOI] [Google Scholar]
- Arad E. Green H. Jelinek R. Rapaport H. J. Colloid Interface Sci. 2020;573:87–95. doi: 10.1016/j.jcis.2020.03.075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been included as part of the ESI.†