

Abstract
RECK has been described to modulate extracellular matrix components through negative regulation of MMP activities. Recently, RECK was demonstrated to bind to an orphan G protein‐coupled receptor GPR124 to mediate WNT7 signaling in nontumor contexts. Here, we attempted to clarify the role of RECK in driving WNT signaling in cancer cells. RECK and GPR124 formed a complex in 293T cells, and when both were expressed, WNT signaling was significantly enhanced in a WNT7‐dependent manner. This cooperation was abolished when RECK mutants unable to bind to GPR124 were transduced. RECK stimulated the growth of KRAS‐mutated pancreatic ductal adenocarcinoma (PDAC) cells with increased sensitivity to WNT inhibitor in a GPR124‐dependent manner. A gastric cancer cell line SH10TC endogenously expresses both RECK and GPR124 under regular culture conditions. In this cell line, inhibited cell growth and WNT signaling as well as increased apoptosis in the GPR124 depletion was dominantly found over those in the RECK deletion. These findings suggest that RECK promotes tumor cell growth by positively modulating WNT signaling through GPR124. This study proposes that the RECK/GPR124 complex might be a good therapeutic target in PDAC and gastric cancer.
Keywords: gastric cancer, GPR124, pancreatic cancer, RECK, WNT7
RECK forms a complex with GPR124 in pancreatic and gastric cancer cells. This complex promotes tumor growth by enhancing WNT signaling.
1. INTRODUCTION
RECK has been identified as a cDNA clone contained in an Okayama–Berg cDNA expression library driven from MRC‐5 untransformed human fibroblasts mRNAs. According to the “flat reversion cloning method,” 1 , 2 aiming to isolate a gene whose overexpression causes an untransformed cell‐like flat phenotype in highly transformed NIH3T3 cells which have been doubly infected with retrovirus harboring oncogenic KRASG12V, we isolated a cDNA from a “flat revertant” clone that appeared after drug selection. The initially isolated cDNA clone was named RECK, but it lacked the 5′‐end containing the initiation codon; thus, there was no reason for calling this gene a “KRAS transformation suppressor.”
However, RECK gene expression appeared to be suppressed by a number of oncogenic gene products including Ras, Fos, Myc, Src, Fms, Fes, and Mos, and moreover, universally underexpressed in cancer tissues compared with normal tissues. 3 , 4 , 5 Low expression of RECK in cancers overall predicts poor prognosis. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 Overexpression of full‐length RECK cDNA in cancer cells suppressed tumor invasion and metastasis with concomitant decrease of various MMP activities without affecting cell proliferation. Nonetheless, introduction of full‐length RECK cDNA into highly Ras‐transformed NIH3T3 cells increased the frequency of flat clones by several percents. 3 The mechanism whereby RECK controls production and/or enzymatic activity of MMPs (MMP2, MMP9, and MT1‐MMP), CD13, and ADAM10 or any other MMPs reported by other groups has never been logically explained. 17 , 18 , 19 Our group identified many domains in the N‐terminal region of RECK which resemble substrates of many MMPs and/or indeed are cleavable by multiple MMPs (Takegami and Takahashi; unpublished). Thus, our current hypothesis is that RECK is a substrate or substrate‐like protein for multiple endopeptidases but may not be a bona fide inhibitor of MMPs.
Contrary to the initial notion that RECK functions as a “tumor suppressor,” many reports including ours indicated that RECK may positively regulate mitogenic signals including NOTCH and WNT. The earliest observation is that loss of Reck in mouse embryonic brain causes premature neuronal differentiation and impaired expansion of neural precursor cells. 19 This was attributed to a disruption of NOTCH ligand–NOTCH receptor interaction due to the overshedding of NOTCH ligands following ADAM10 overactivation. In other words, RECK positively regulates NOTCH signaling. GDE2 appeared as a RECK sheddase, thus as an endogenous inhibitor of RECK. 20 GDE2‐RECK‐ADAM10/γ‐secretase signaling has been implicated in the control of amyloid precursor protein degradation. 21 Another important observation is that abrupt downregulation of Reck expression in mouse embryonic fibroblasts (MEFs) induces cellular senescence in an EGFR‐dependent manner. Nonetheless, Reck‐null MEFs proliferate faster than wild‐type MEFs and escape cellular senescence much earlier than wild‐type cells during cultivation according to the 3T3 protocol. 22
The role of RECK in WNT signaling emerged from the studies of central nervous system (CNS) vascular angiogenesis and then limb development. In these tissues, RECK binds to an orphan G protein‐coupled receptor (GPCR) GPR124, a component of WNT receptors. WNT7A and WNT7B initially bind to RECK that has been already complexed with GPR124, which provokes ligand‐specific WNT signaling by enhancing the association between the WNT ligand/RECK/GPR124 complex and the FRZ/LRP5 or FRZ/LRP6 complex. 23 , 24 , 25 , 26 As many gastrointestinal cancers rely on WNT signaling, we surveyed cancers that might depend on RECK/GPR124 for malignant behaviors.
2. MATERIALS AND METHODS
2.1. Cell culture
293T cells (RCB2202, RIKEN BRC Cell Bank), MIA PaCa‐2 cells (RCB2094, RIKEN BRC Cell Bank) and HT1080 cells 3 were cultured in DMEM (#043‐30085, FUJIFILM) supplemented with 10% fetal bovine serum (#175012, Nichirei BIOSCIENCES INC) and 1% penicillin–streptomycin solution (1#6823291, FUJIFILM). PK45H cells (RCB1973, RIKEN BRC Cell Bank) and SH10TC cells (a gift from Dr. Oshima) were cultured in RPMI (#189‐02025, FUJIFILM) supplemented with 10% fetal bovine serum (#175012, Nichirei BIOSCIENCES INC) and 1% penicillin–streptomycin solution (#16823291, FUJIFILM).
2.2. Plasmids
HT1080 cells, PK45H cells and MIA PaCa‐2 cells were seeded 24 h before transfection and were transfected with pCXN2‐NEO, pCXN2‐NEO‐RECK, pCXN2‐NEO‐RECKΔC, 3 pCXN2‐NEO‐RECKN200Q, pCXN2‐NEO‐RECKM207W, pCXN2‐NEO‐RECKR69A, pCXN2‐NEO‐RECKP71A and pCXN2‐NEO‐RECKD72A using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific). pcDNA3.1 encoding the C‐terminal Myc‐tagged GPR124 27 was a kind gift from Dr. Croix (National Cancer Institute). PK45H cells were seeded 24 h before transfection and were transfected with pLKO.1‐GPR124 shRNA. SH10TC cells were seeded 24 h before transfection and were transfected with pLKO.1‐GPR124 shRNA and pLVX‐RECK shRNA.
2.3. Immunoblotting (IB) and immunoprecipitation (IP)
A total of 0.8 × 105 cells were seeded onto six‐well plates and cultured after several days and harvested using magnesium‐containing lysis buffer (MLB) supplemented with 0.5 mM NaF (#31418‐45, Nacalai Tesque), 100 μM Na3VO4 (#190‐09751, FUJIFILM) and 1X protease inhibitor (#25955‐11, Nacalai Tesque). Cell lysate for IP was lysed in the same lysis buffer as described above. RECK was immunoprecipitated with mouse anti‐RECK antibody (5B11D12) 3 and Myc‐GPR124 with Myc antibody (#9E10, sc‐40; Santa Cruz). The immunoprecipitants were collected on Protein G Dynabeads (#10003D; Invitrogen), washed five times with wash buffer containing 10 mM Tris, pH7.4 (#013‐16385, FUJIFILM), 1 mM EDTA (#311‐90075, NIPPON GENE), 1 mM EGTA pH8.0 (#15214, Nacalai Tesque), 150 mM NaCl (#31320‐05, Nacalai Tesque), 0.1% Triton X‐100 (#12967‐45, Nacalai Tesque) and 0.2 mM Na3VO4 in the presence of protease inhibitor mixture and eluted. IB was performed as described previously. 28 Antibodies used are listed in Table S1.
2.4. TOP/FOP flash reporter gene assay
A total of 1.5 × 105 cells were plated onto six‐well‐type plates and incubated for 24 h; then, cells were transduced with 2.0 μg target vectors and 0.4 μg TOP flash (TCF Reporter Plasmid, #21‐170, Sigma‐Aldrich) or 0.4 μg FOP flash (mutant TCF binding sites, #21‐169, Sigma‐Aldrich) using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific). Cells were treated with indicated concentrations of WNT7A (ab116171, Abcam) at 24 h after transfection, and the luciferase activity was measured at 48 h after transfection. The luciferase activity in cell lysates was measured using the Dual‐Luciferase® Reporter Assay System (#E1910, Promega Corporation) according to the manufacturer's protocol.
2.5. Colony formation assay
Cells were seeded at 3000 cells/well in a 60‐mm dish. After 10 days of incubation, cells were washed with PBS twice and then fixed with 4%‐paraformaldehyde phosphate buffer solution (#09154‐85, Nacalai Tesque). Colonies were stained with 0.5% crystal violet staining solution (#031‐04852, FUJIFILM).
2.6. Inhibitor treatment
Cells were treated with a WNT pathway inhibitor, PRI‐724 (#ab229168, Abcam) or with a KRASG12C inhibitor sotorasib (AMG‐510) (S8830, Selleck).
2.7. Lentivirus generation and infection
pLKO.1 carrying scrambled shRNA, GPR124 #sh1 (forward: CCGGCCTGCTCTTGAGCAATAACAACTCGAGTTGTTATTGCTCAAGAGCAGGTTTTTG; reverse: AATTCAAAAACCTGCTCTTGAGCAATAACAACTCGAGTTGTTATTGCTCAAGAGCAGG) or #sh2 (forward: CCGGGCTGAACTTGTGCTTCCACATCTCGAGATGTGGAAGCACAAGTTCAGCTTTTTG; reverse: AATTCAAAAAGCTGAACTTGTGCTTCCACATCTCGAGATGTGGAAGCACAAGTTCAGC), or pLVX carrying LacZ and RECK #sh1(forward: GATCCAGGAACCCAACGGATAGTTTATTCAAGAGATAAACTATCCGTTGGGTTCCTTTTTTTG; reverse: AATTCAAAAAAAGGAACCCAACGGATAGTTTATCTCTTGAATAAACTATCCGTTGGGTTCCTG) or #sh2 (forward: GATCCGCTGATTAAATGACACTCATATTCAAGAGATATGAGTGTCATTTAATCAGCTTTTTTG; reverse: AATTCAAAAAAGCTGATTAAATGACACTCATATCTCTTGAATATGAGTGTCATTTAATCAGCG). For lentivirus production, 4 × 106 293T cells were plated onto a D100 dish and transfected with 3 μg of targeting plasmids mixed with 3 μg of pCMV‐VSVG, 3 μg of pCMV‐dR8.91 and 27 μg of polyethyleneimine (#24765, Polysciences, Inc.) for overnight, and then replaced to fresh culture media. After 48 h incubation, the media containing lentivirus were collected, passed through a 0.45‐μm filter and concentrated using polyethylene glycol. After collecting lentivirus, 0.8 × 105 cells were seeded onto six‐well plates and cultured. After overnight culture, media were replaced with fresh medium containing lentivirus and 8 μg/mL polybrene. After infection for overnight, cells were subjected to selection with 2 μg/mL puromycin (#ant‐pr‐1, Invivogen) or 8 μg/mL blastcidin (#ant‐bl‐05, Invivogen) for 72 h, and then proceeded to indicated experiments.
2.8. Reverse transcription‐qPCR
Reverse transcription quantitative PCR (qPCR) was carried out on total RNA isolated from cultured cells using TRIzol (#15596018; Life Technologies) according to the manufacturer's protocol. Isolated RNA was reverse‐transcribed at 37°C with random primers to obtain cDNA, and the qPCR analysis was done by using the LightCycler 480 System (Roche) according to the manufacturer's instruction. SYBR primers used in the qPCR were GPR124 (forward: CACCAACTACCAGATGGTCT; reverse: CTCCAGCGATCAAATAGAAC), RECK (forward: GTGCCGTGATGTATGTGAACAGATT; reverse: GCAACAGATGTTTTAGTCGGGATTC) and hACTB (forward: CATTAAGGAGAAGCTGTGCT; reverse: TTGAAGGTAGTTTCGTGGAT).
2.9. Flow cytometry
Cells were trypsinized, washed with PBS, stained with annexin V‐fluorescein isothiocyanate (V‐FITC) for 20 min at room temperature using the eBioscience Annexin V‐FITC Apop Kit (BMS500FI‐300, Thermo Fisher Scientific), and analyzed by FACS Canto II (BD Biosciences).
2.10. Dataset analysis of pancreatic cancer and gastric cancer
The datasets of RECK expression between normal tissues and tumors were downloaded from https://tnmplot.com/analysis/. The mRNA expression of RECK and GPR124 in pancreatic cancer and gastric cancer were downloaded from https://kmplot.com/analysis/.
2.11. Statistical analysis
Statistical significance was calculated using Student's t‐test or ANOVA followed by post hoc Tukey's test among more than three groups. p‐Values less than 0.05 were considered statistically significant N.S., not significant.
3. RESULTS
3.1. RECK/GPR124 complex mediates WNT signaling
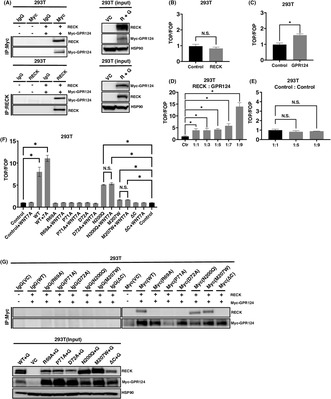
RECK has been recently demonstrated to bind to GPR124 to modulate WNT7 signaling in nontumor contexts. 23 , 24 , 25 , 26 Here, we attempted to clarify the role of RECK in driving WNT signaling in cancer cells. RECK and C‐terminally Myc‐tagged GPR124 were overexpressed in 293T cells which do not endogenously express RECK at the level detectable by IB. RECK and Myc‐tagged GPR124 were successfully overexpressed (Figure 1A; right). The whole‐cell lysates in which RECK or Myc‐tagged GPR124 or both were immunoprecipitated by antibody to RECK or Myc and immunoblotted again by antibody to RECK or Myc. The results indicated that RECK and Myc‐tagged GPR124 form a complex when expressed in 293T cells (Figure 1A; left).
FIGURE 1.
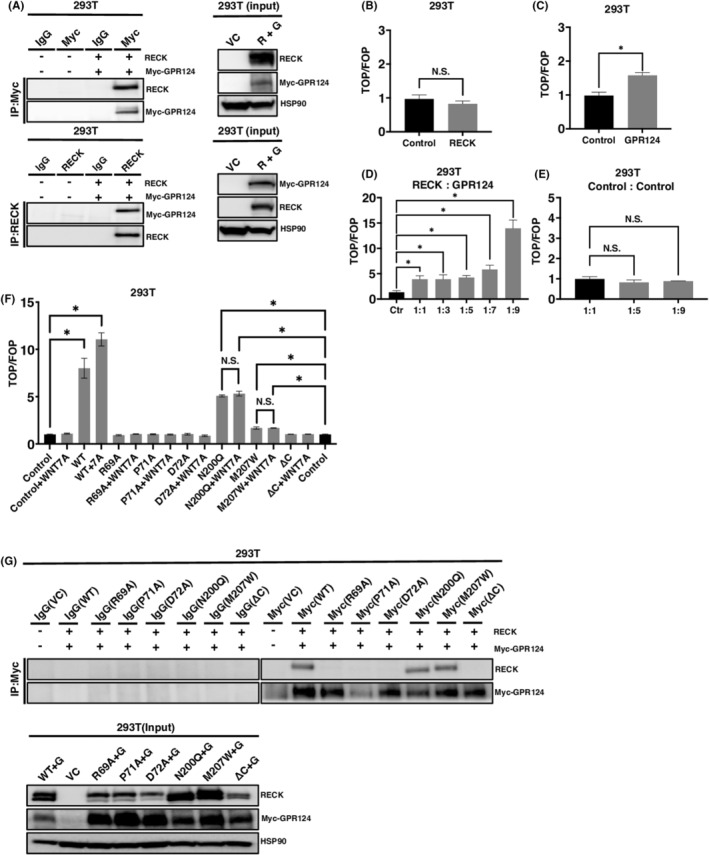
The RECK/GPR124 complex mediates WNT signaling. (A) RECK and Myc‐tagged GPR124 simultaneously expressed in 293T cells and immunoprecipitated (IP) by antibodies indicated. The precipitants were then immunoblotted (IB) by antibodies indicted (left). The expression of proteins in the input whole‐cell lysates (right). (B) TOP/FOP canonical WNT/β‐catenin reporter assay of 293T cells transduced with pCXN2‐RECK (N = 3). (C) TOP/FOP assay of 293T cells transduced with Myc‐tagged GPR124 in 293T cells (N = 3). (D) TOP/FOP assay of 293T cells transduced with pCXN2‐RECK and pcDNA3.1 Myc‐GPR124 mixed at the indicated ratio (N = 3). (E) TOP/FOP assay of 293T cells transduced with pCXN2‐NEO and pcDNA3.1 mixed at the indicated ratio (N = 3). (F) TOP/FOP assay of 293T cells transduced with pCXN2‐RECK, RECK mutants and pcDNA3.1 Myc‐GPR124 mixed at the ratio of 1:9 and treated with or without 10 ng/mL of WNT7A (N = 3). Control columns appear twice for the convenience of assessing significance between control and N200Q and M207W columns. (G) RECK (WT and mutants) and Myc‐tagged GPR124 were simultaneously expressed in 293T cells and immunoprecipitated (IP) by the antibodies indicated. The precipitants were then immunoblotted (IB) by the antibodies indicated (upper). The expression of proteins in the input whole‐cell lysates were detected by the corresponding antibodies (lower). * p < 0.05 against vector control; N.S., not significant.
Next, in an aim to measure the level of WNT signaling generated by the complex, we performed luciferase promoter assay using TOP/FOP system. 24 , 25 Single introduction of RECK did not provoke WNT signaling (Figure 1B); however, Myc‐tagged GPR124 transduction elevated WNT signaling by around 60% (Figure 1C). Importantly, when the expression vectors of RECK and Myc‐tagged GPR124 were mixed by 1:1 ratio, the WNT signal elevated to around four times greater than control. This trend became more robust when RECK and Myc‐tagged GPR124 were mixed by 1:9 (Figure 1D). When the vector backbones were mixed, WNT signal was never elevated at any mixture ratio (Figure 1E). Moreover, when RECK and Myc‐tagged GPR124 mixed by 1:9 was introduced, the additional treatment by WNT7A significantly enhanced WNT signaling (Figure 1F).
The cooperation of RECK and GPR124 in WNT signaling was completely abolished by introducing mutations in RECK at the amino acids (R69A, arginine to alanine; P71A, proline to alanine; D72A, aspartic acid to alanine) that have been suggested to be involved in the RECK‐GPR124 molecular association, 24 even in the presence of WNT7A ligand (Figure 1F). Asparagine 200 and methionine 207 are supposed to locate in the domain of RECK responsible for binding to WNT7A and B. 29 Point mutations in this domain (N200Q, asparagine to glutamine; M207W, methionine to tryptophan) did not fully lose the activity to generate WNT signaling but no more responded to additional treatment by WNT7A (Figure 1F). The C‐terminus‐deleted (ΔC) (the COOH‐terminal 23 aa was deleted) 3 and hence soluble RECK completely lost RECK function to generate WNT signaling, nor respond to WNT7A. These findings suggest that RECK and GPR124 cooperate to drive WNT7A signaling. To confirm the biding status between RECK mutants and Myc‐GPR124, we performed IP analysis. R69A, P71A, and D72A, which are located within the GPR124 binding domain, almost completely lost their ability to complex with Myc‐GPR124. Conversely, RECK mutants N200Q and M207W, which retain the capacity to activate WNT signaling in cooperation with GPR124 but lack the response to exogenously supplemented WNT7A, do not lose the ability to form a complex with GPR124 (Figure 1G). These observations strengthen our prior findings that RECK and GPR124 can interact to synergistically promote WNT7A signaling.
3.2. RECK stimulates pancreatic ductal adenocarcinoma (PDAC) growth in a WNT‐dependent manner in the presence of GPR124
RECK has been described as a suppressor of tumor invasion and metastasis. 5 , 6 , 7 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 RECK is universally underexpressed in malignant tumors with a few exceptions. 6 , 7 , 8 , 12 , 13 , 14 , 15 , 16 , 42 , 43 , 44 , 45 , 46 , 47 Lower RECK expression overall correlates with poorer survival of patients. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 However, at least in the publicly available database, PDAC appeared to express RECK at a higher level than normal tissue (Figure S1). Moreover, higher RECK expression correlates with worse outcomes in PDAC patients (Figure S2).
Despite the information available in the database, two available PDAC cell lines were all negative in RECK expression but all positive in GPR124 expression. Both PK45H and MIA PaCa‐2 cells express GPR124 (Figure 2A). To survey GPR124 and RECK expression in other types of cancers or tissues, we analyzed MCF10A (nontransformed mammary epithelial) cells, one breast cancer cell line BT549 and HT1080 fibrosarcoma cells, which have been well analyzed in our previously studies. 3 , 22 Only MCF10A was found to express GPR124 (Figure 2B). When combined with information available in the database, these findings indicate that GPR124 and RECK might be expressed in PDAC at a significant frequency.
FIGURE 2.
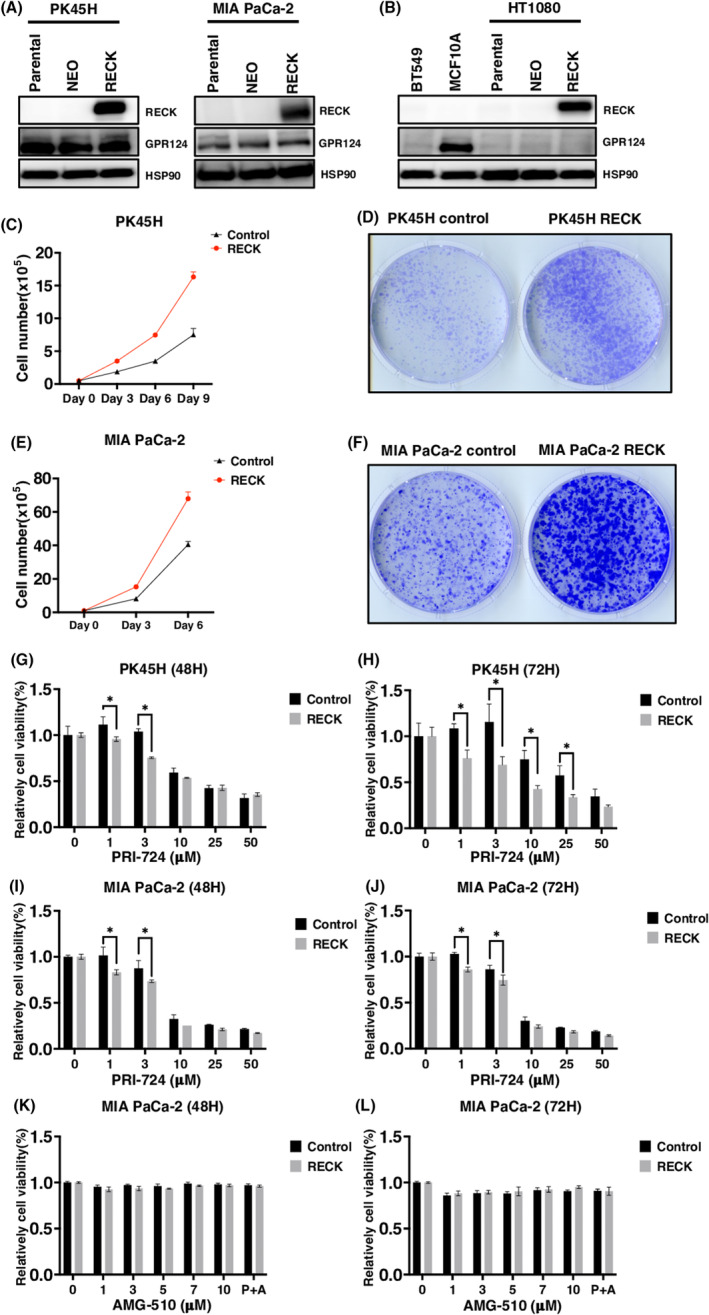
RECK stimulates cell proliferation in a WNT signal‐dependent manner when induced in GPR124‐positive pancreatic ductal adenocarcinoma (PDAC) cells. (A, B) Immunoblotting (IB) of the indicated proteins in the indicated cells parental or transduced with PCXN2‐NEO or PCXN2‐RECK. (C) Cell proliferation of PK45H transduced with PCXN2‐NEO or PCXN2‐RECK (N = 3). (D) Representative image of colony formation by PK45H cells transduced with PCXN2‐NEO or PCXN2‐RECK (N = 3). (E) Cell proliferation of MIA PaCa‐2 cells transduced with PCXN2‐NEO or PCXN2‐RECK (N = 3). (F) Representative image of colony formation by MIA PaCa‐2 transduced with PCXN2‐NEO or PCXN2‐RECK (N = 3). (G–L) Relative viability of the indicated cells transduced with PCXN2‐NEO or PCXN2‐RECK and treated with the indicated concentration of the indicated chemicals for the indicated period. The viability of cells without treatment was set to 100% (N = 3). *p < 0.05 against vector control; N.S., not significant.
Forced expression of RECK in one of these lines PK45H harboring a KRASG12D mutation strongly enhanced cell proliferation (Figure 2A,C,D). The molecular size of RECK expressed in PK45H was identical with that expressed in HT1080 cells in which exogenously expressed RECK was once demonstrated to be fully glycosylated (Figure 2A,B). 3 However, a previous study of RECK overexpression did not show enhanced growth properties in HT1080 cells which do not endogenously express GPR124. 3 Additionally, forced expression of RECK in MIA PaCa‐2 harboring KRASG12C mutation also simulated cell proliferation (Figure 2A,E,F). The forced expression of RECK in GPR124‐positive PDAC cell lines made these significantly more sensitive to WNT signal inhibitor PRI724 (Figure 2G–J). Contrary, forced RECK expression did not further sensitize MIA PaCa‐2 cells carrying KRASG12C mutation to a KRASG12C inhibitor sotorasib (Figure 2K,L). These findings indicate that the RECK/GPR124 complex increases the denpedency of PDAC cells on WNT signaling even in the presence of KRAS mutation.
3.3. RECK enhances WNT signaling depending on GPR124 in PDAC
We next again assessed the effects of RECK mutated to be dissociated from GPR124 in PDAC cells that endogenously express GPR124. Transduction of wild‐type RECK into PH45H and MIA PaCa‐2 cells both highly enhanced cell proliferation without exogenously introducing GPR124 (Figure 3A–H). RECK R69A, P71A, and D72A mutants (R69A, arginine to alanine; P71A, proline to alanine; D72A, aspartic acid to alanine) failed to exhibit growth stimulation. The C‐terminus‐deleted (ΔC) (the COOH‐terminal 23 aa was deleted) and hence soluble RECK suppressed cell growth (Figure 3E–H).
FIGURE 3.
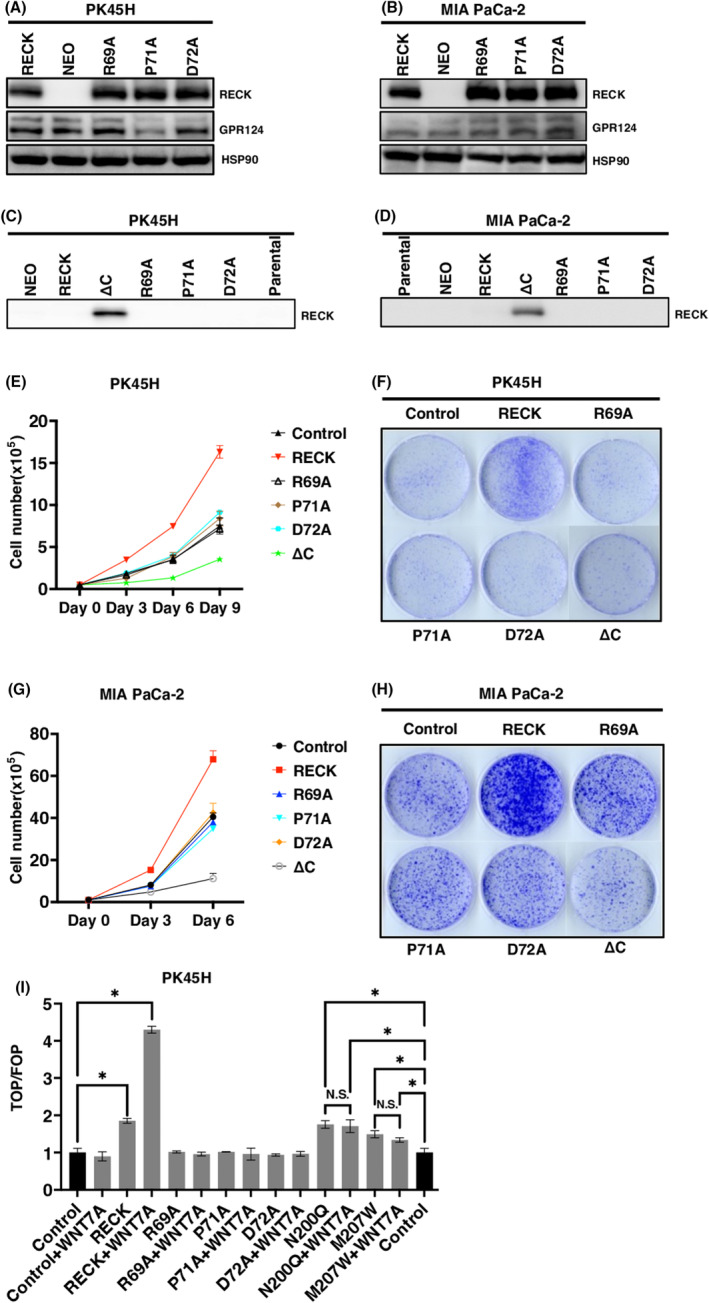
RECK mutants lacking binding to GPR124 failed to stimulate growth in pancreatic ductal adenocarcinoma (PDAC) cells. (A, B) Immunoblotting (IB) of the indicated proteins in the indicated cells. (C, D) Immunoblotting of soluble RECK in the conditioned media prepared from the indicated cells transduced with the indicated protein. Conditioned media were prepared by cultivating cells with serum‐free culture medium for 48 h and filtered through a 0.45‐μM filter. (E, G) Cell growth in the indicated cells transduced with the indicated protein. (F, H) Representative image of colony formation by the indicated cells transduced with the indicated protein. (I) TOP/FOP assay of PK45H cells transduced with pCXN2‐RECK, RECK mutants and pcDNA3.1 Myc‐GPR124 mixed at the ratio of 1:9 and treated with or without 10 ng/mL of WNT7A (N = 3). Control columns appear twice for the convenience of assessing significance between control and N200Q and M207W columns. *p < 0.05 against vector control; N.S., not significant.
Introduction of wild‐type RECK into GPR124‐positive PK45H cells significantly enhanced WNT signaling in an WNT7A ligand‐dependent manner. RECK R69A, P71A and D72A mutants completely failed to enhance WNT signaling even in the presence of additional WNT7A. RECK N200Q and M207W mutants kept activity to stimulate WNT signaling; however, as observed in 293T cells, they failed to respond to additional WNT7A (Figure 3I). These findings indicate that RECK stimulates PDAC cell proliferation in cooperation with GPR124 in a WNT7A‐dependent manner.
3.4. RECK cannot mediate WNT signaling in the absence of GPR124
We then depleted GPR124 from PH45H PDAC cells, which caused significant growth arrest associated with remarkable downregulation of WNT signaling when assessed by expression levels of WNT target genes, such as c‐Myc, cyclin D1, cyclin A and WISP1 (Figure 4A–D). RECK overexpression could not override the effect of GPR124 depletion at all (Figure 4D). These results indicate that although RECK enhances GPR124 function to mediate WNT signaling, GPR124 contributes to sustain WNT‐dependent PDAC cell growth even in the absence of RECK. In addition, the failure of RECK overexpression to upregulate WISP1 in GPR124‐depleted cells suggests that RECK cannot mediate WNT signaling in the absence of GPR124 in PDAC cells.
FIGURE 4.
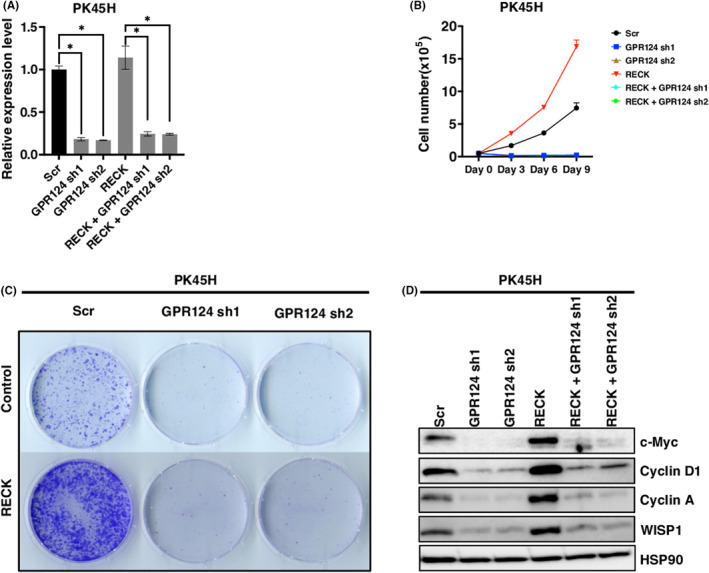
Depletion of GPR124 induces growth arrest which cannot be rescued by RECK. (A) Relative mRNA expression levels of GPR124 in PK45H cells transduced with pCXN2‐NEO or pCXN2‐RECK and then infected with lentivirus carrying shRNA targeting GPR124 or control (N = 3) (B) Cell proliferation curve of PK45H cells established in (A). (C) Representative image of colony formation by PK45H cells established in (A) (N = 3). (D) Immunoblotting (IB) of the indicated proteins in PK45H cells established in (A). *p < 0.05 against vector control; N.S., not significant.
3.5. RECK modulates GPR124 function to control WNT signaling in gastric cancer
We then analyzed a gastric cancer cell line SH10TC which endogenously expresses both RECK and GPR124. RECK depletion induced around 50% downregulation in cell proliferation and evidence of cell death in 20% ~ 30% of cells. GPR124 deletion induced almost complete growth arrest and evidence of much more robust cell death near to 80% (Figure 5A–H). Consistent with the effects on cell proliferation, depletion of GPR124 exhibited much more robust downregulation of c‐Myc, cyclin D1 and cyclin A, and upregulation of p21WAF1 and p27KIP1 as compared with depletion of RECK (Figure 5F). Simultaneous transduction of shRNAs that target RECK and GPR124 exhibited cell death more robustly than single depletion (Figure 5I). The treatment of SH10TC by WNT7A ligand enhanced WNT activity consistent with the endogenous expression of both RECK and GPR124 (Figure 5J). Importantly, the response to WNT7A was diminished when RECK or GPR124 was depleted, and more robustly when both were depleted (Figure 5J).
FIGURE 5.
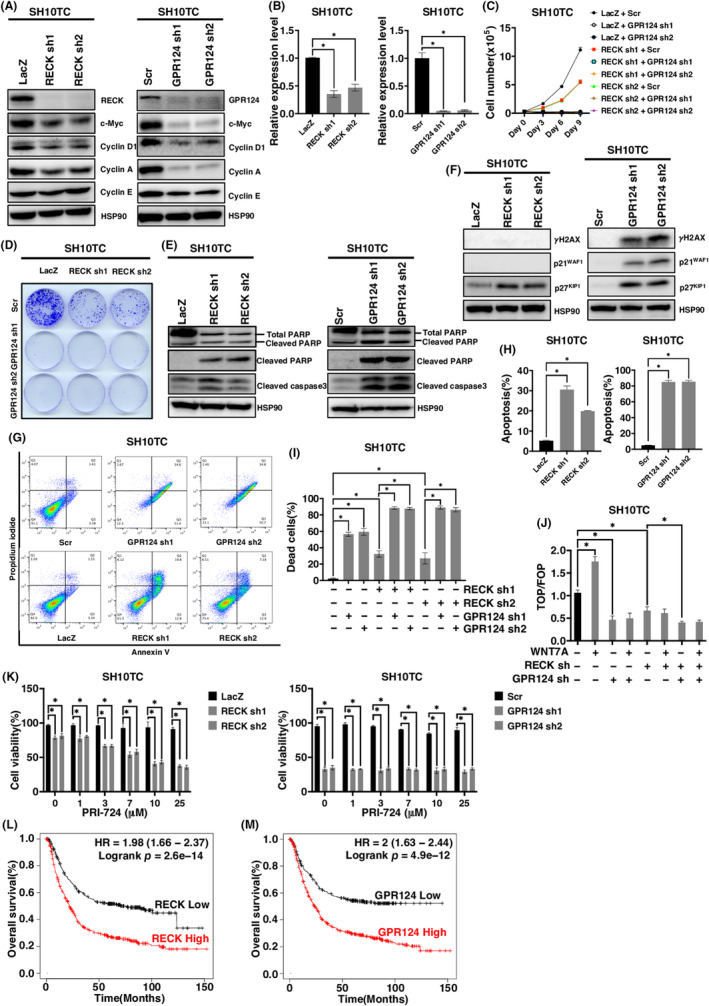
The effect of RECK and GPR124 depletion in SH10TC cells. (A) Immunoblotting (IB) of the indicated proteins in RECK‐depleted (left) or GPR124‐depleted (right) SH10TC cells. (B) Relative mRNA expression levels of the indicated gene in RECK‐depleted (left) or GPR124‐depleted (right) SH10TC cells (N = 3). (C) Cell proliferation curve of SH10TC cells infected with lentivirus carrying shRNA targeting the indicated gene in the indicated combination. (D) Representative images of colony formation by SH10TC cells analyzed in (C). (E, F) Immunoblotting of the indicated proteins in RECK‐depleted (left) or GPR124‐depleted (right) SH10TC cells. (G) Representative results of flow cytometry analysis of SH10TC cells infected with the indicated lentivirus. (H) Quantitation of flow cytometry analyses of RECK‐depleted (left) and GPR124‐depleted cells (right) (N = 3). (I) The percentage of dead SH10TC cells infected with the indicated lentivirus assessed by trypan blue staining (N = 3). (J) TOP/FOP canonical WNT/β‐catenin reporter assay of SH10TC cells infected with the indicated lentivirus and treated with or without WNT7A. (K) Viability of SH10TC cells infected with the indicated lentivirus and treated with the indicated concentration of PRI‐724 for 72 h (N = 3). (L) Kaplan–Meier survival curves for patients with gastric cancer with high and low RECK expression. p = 2.6 × 10−14 by long‐rank test. (M) Kaplan–Meier survival curves for patients with gastric cancer with high and low GPR124 expression. p = 4.9 × 10−12 by long‐rank test. *p < 0.05 against vector control; N.S., not significant.
Even after RECK depletion, SH10TC cells responded to a WNT inhibitor in a dose‐dependent manner in terms of cellular viability. However, once GPR124 was depleted, SH10TC cells no more responded to a WNT inhibitor at any concentration (Figure 5K). These findings suggest that RECK modulates the function of GPR124 in driving WNT signaling in gastric cancers and that RECK and GPR124 double‐positive gastric cancers may be a good target of WNT inhibitors. Supporting this notion, we found that in the available database, higher RECK expression and higher GPR124 expression correlate with worse outcomes in gastric cancer patients (Figure 5L,M).
4. DISCUSSION
Until today, the mechanisms of RECK functions to suppress tumor cell invasion and metastasis, ability to downregulate secretion of pro‐MMPs, and activity to competitively inhibit MMPs or peptidase including MMP‐2, MMP‐9, MT1‐MMP, ADAM10 and CD13 have been poorly explained. In many papers published after the very early reports on the discovery of RECK, this molecule has been described as a priori negative regulator of MMPs. 17 , 18 , 19 However, especially in these 7 years, many reports have challenged this notion of RECK function. The most remarkable studies on the novel aspects of RECK function have been done in the field of CNS angiogenesis and limb development.
In these studies, RECK has been demonstrated to complex with GPR124, an orphan GPCR, which innervates CNS angiogenesis and dorsal root ganglia neurogenesis through activating WNT7A/B signaling. 23 , 24 , 25 , 26 Moreover, RECK contributes to the maintenance of the blood–brain barrier (BBB) by keeping homeostasis in vascular endothelial cells. 24 The same group later demonstrated that the same complex plays pivotal roles in limb development. 48 Another group demonstrated the diversified and conserved functions of the WNT7A/B/RECK/GPR124 complex in humans, mice and zebrafish. 49 One another group elucidated how the CC4 domain of RECK (RECKCC4) engages in binding to the N‐terminal domain of WNT7A to activate WNT7‐specific signaling. 29
This study addressed the role of the WNT7A/B/RECK/GPR124 complex in the tumor context. RECK is overall underexpressed in cancers. However, the database indicated a possibility that RECK might be frequently expressed particularly in PDAC. In two of the PDAC cell lines that we employed, we could not detect RECK expression by IB; however, we found all of them express significant levels of GPR124. Forced expression of RECK significantly cooperated with GPR124 to induce WNT‐dependent stimulation of cell proliferation. This may explain why RECK‐positive PDAC patients are poorer in overall survival. A gastric cancer cell line SH10TC which expresses both RECK and GPR124 was sensitive to both RECK depletion and GPR124 depletion. Importantly, GPR124 depletion gave rise to much more robust suppression of tumor growth than RECK depletion and made them to virtually no more sensitive to a WNT inhibitor. These findings indicate that in gastric cancer cells, a molecule or molecules other than RECK may compensate RECK function to cooperate with GPR124, and that GPR124 is a very promising therapeutic target in GPR124‐positive pancreatic and gastric cancers. Tadehaginoside, a phenylpropanoid glycoside found in Tadehagi triquetrum, appeared to inhibit the activity of GPR124 and exhibited a therapeutic potential to improve GPR124‐dependent vascular damage that developed after an ischemic event in brain. 50 However, this compound has been described to inhibit various molecules as well as GPR124. The development of more specific GPR124 inhibitors is strongly awaited.
AUTHOR CONTRIBUTIONS
Chiaki Takahashi: Conceptualization; data curation; funding acquisition; investigation; project administration; resources; supervision; validation; writing – original draft; writing – review and editing. Hai Yu: Conceptualization; data curation; formal analysis; investigation; writing – original draft. Susumu Kohno: Conceptualization; data curation; formal analysis; investigation; methodology; resources; supervision; validation; writing – original draft; writing – review and editing. Dominic Chih‐Cheng Voon: Investigation; resources; writing – review and editing. Nada Hamdy Hussein: Investigation. Yuanyuan Zhang: Investigation. Joji Nakayama: Methodology; supervision; writing – original draft. Yujiro Takegami: Conceptualization; resources.
FUNDING INFORMATION
This work was supported by Grant‐in‐Aid for Scientific Research 19K22555 and 20H03509.
CONFLICT OF INTEREST STATEMENT
The authors have no financial interest to disclose. Chiaki Takahashi is an editorial board member of Cancer Science.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: N/A.
Supporting information
Table S1
Figures S1–S2
ACKNOWLEDGMENTS
We thank Masanobu Oshima (Kanazawa University) for providing a cell line, Dr. Brad St. Croix (National Cancer Institute) for providing a plasmid encoding C‐terminal Myc‐tagged GPR124, and all the members of Takahashi Laboratory, especially Naoko Nagatani, Linxiang Gong, Fengkai Li, and Jindan Sheng for their helpful suggestions and support.
Yu H, Kohno S, Voon D‐C, et al. RECK/GPR124‐driven WNT signaling in pancreatic and gastric cancer cells. Cancer Sci. 2024;115:3013‐3025. doi: 10.1111/cas.16258
REFERENCES
- 1. Noda M, Selinger Z, Scolnick E, Bassin R. Flat revertants isolated from Kirsten sarcoma virus‐transformed cells are resistant to the action of specific oncogenes. Proc Natl Acad Sci USA. 1983;80:5602‐5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. A ras‐related gene with transformation suppressor activity. Cell. 1989;56:77‐84. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi C, Sheng Z, Horan TP, et al. Regulation of matrix metalloproteinase‐9 and inhibition of tumor invasion by the membrane‐anchored glycoprotein RECK. Proc Natl Acad Sci USA. 1998;95:13221‐13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA Methyltransferase 3b–induced promoter methylation. Cancer Res. 2006;66:8413‐8420. [DOI] [PubMed] [Google Scholar]
- 5. Sasahara RM, Takahashi C, Noda M. Involvement of the Sp1 site in ras‐mediated downregulation of the RECK metastasis suppressor gene. Biochem Biophys Res Commun. 1999;264:668‐675. [DOI] [PubMed] [Google Scholar]
- 6. Noda M, Takahashi C. Recklessness as a hallmark of aggressive cancer. Cancer Sci. 2007;98:1659‐1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clark JC, Thomas DM, Choong PF, Dass CR. RECK–a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007;26:675‐683. [DOI] [PubMed] [Google Scholar]
- 8. Noda M, Takahashi C, Matsuzaki T, Kitayama H. What we learn from transformation suppressor genes: lessons from RECK. Future Oncol. 2010;6:1105‐1116. [DOI] [PubMed] [Google Scholar]
- 9. Masui T, Doi R, Koshiba T, et al. RECK expression in pancreatic cancer: its correlation with lower invasiveness and better prognosis. Clin Cancer Res. 2003;9:1779‐1784. [PubMed] [Google Scholar]
- 10. Dong ZR, Chen ZQ, Yang XY, et al. RECK expression is associated with angiogenesis and immunogenic tumor microenvironment in hepatocellular carcinoma, and is a prognostic factor for better survival. J Cancer. 2021;12:3827‐3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Cheng S, Zhang G, et al. Low expression of RECK indicates a shorter survival for patients with invasive breast cancer. Cancer Sci. 2012;103:1084‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chung TT, Yeh CB, Li YC, et al. Effect of RECK gene polymorphisms on hepatocellular carcinoma susceptibility and Clinicopathologic features. PLoS One. 2012;7:e33517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takenaka K, Ishikawa S, Kawano Y, et al. Expression of a novel matrix metalloproteinase regulator, RECK, and its clinical significance in resected non‐small cell lung cancer. Eur J Cancer. 2004;40:1617‐1623. [DOI] [PubMed] [Google Scholar]
- 14. Chen Y, Tseng SH. The potential of RECK inducers as antitumor agents for glioma. Anticancer Res. 2012;32:2991‐2998. [PubMed] [Google Scholar]
- 15. Hill VK, Ricketts C, Bieche I, et al. Genome‐wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011;71:2988‐2999. [DOI] [PubMed] [Google Scholar]
- 16. Tang YJ, Huang J, Tsushima H, et al. Tracing tumor evolution in sarcoma reveals clonal origin of advanced metastasis. Cell Rep. 2019;28:2837‐2850.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oh J, Takahashi R, Kondo S, et al. The membrane‐anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789‐800. [DOI] [PubMed] [Google Scholar]
- 18. Miki T, Takegami Y, Okawa K, Muraguchi T, Noda M, Takahashi C. The reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) interacts with membrane type 1 matrix metalloproteinase and CD13/aminopeptidase N and modulates their endocytic pathways. J Biol Chem. 2007;282:12341‐12352. [DOI] [PubMed] [Google Scholar]
- 19. Muraguchi T, Takegami Y, Ohtsuka T, et al. RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat Neurosci. 2007;10:838‐845. [DOI] [PubMed] [Google Scholar]
- 20. Park S, Lee C, Sabharwal P, Zhang M, Meyers CLF, Sockanathan S. GDE2 promotes neurogenesis by glycosylphosphatidylinositol‐anchor cleavage of RECK. Science. 2013;339:324‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura M, Li YH, Choi BR, et al. GDE2‐RECK controls ADAM10 α‐secretase‐mediated cleavage of amyloid precursor protein. Sci Transl Med. 2021;13(585):eabe6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kitajima S, Miki T, Takegami Y, et al. Reversion‐inducing cysteine‐rich protein with Kazal motifs interferes with epidermal growth factor receptor signaling. Oncogene. 2011;30:737‐750. [DOI] [PubMed] [Google Scholar]
- 23. Vanhollebeke B, Stone OA, Bostaille N, et al. Tip cell‐specific requirement for an atypical Gpr124‐ and Reck‐dependent Wnt/β‐catenin pathway during brain angiogenesis. elife. 2015;4:e06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho C, Smallwood PM, Nathans J. Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b‐specific signaling in mammalian CNS angiogenesis and blood‐brain barrier regulation. Neuron. 2017;95:1221‐1225. [DOI] [PubMed] [Google Scholar]
- 25. Vallon M, Yuki K, Nguyen TD, et al. A RECK‐WNT7 receptor‐ligand interaction enables isoform‐specific regulation of Wnt bioavailability. Cell Rep. 2018;25:339‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eubelen M, Bostaille N, Cabochette P, et al. A molecular mechanism for Wnt ligand‐specific signaling. Science. 2018;361:eaat1178. [DOI] [PubMed] [Google Scholar]
- 27. Cullen M, Elzarrad MK, Seaman S, et al. GPR124, an orphan G protein‐coupled receptor, is required for CNS‐specific vascularization and establishment of the blood–brain barrier. Proc Natl Acad Sci USA. 2011;108:5759‐5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kitajima S, Yoshida A, Kohno S, et al. The RB‐IL‐6 axis controls self‐renewal and endocrine therapy resistance by fine‐tuning mitochondrial activity. Oncogene. 2017;36:5145‐5157. [DOI] [PubMed] [Google Scholar]
- 29. Qi X, Hu Q, Elghobashi‐Meinhardt N. Molecular basis of Wnt biogenesis, secretion, and Wnt7‐specific signaling. Cell. 2023;186:5028‐5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sasahara RM, Takakashi C, Sogayar MC, Noda M. Oncogene‐mediated downregulation of RECK, a novel transformation suppressor gene. Braz J Med Biol Res. 1999;32:891‐895. [DOI] [PubMed] [Google Scholar]
- 31. Noda M, Oh J, Takahashi R, Kondo S, Kitayama H, Takahashi C. RECK: a novel suppressor of malignancy linking oncogenic signaling to extracellular matrix remodeling. Cancer Metastasis Rev. 2003;22:167‐175. [DOI] [PubMed] [Google Scholar]
- 32. Chang H, Liu L, Hung W. Nvolvement of histone deacetylation in ras‐induced down‐regulation of the metastasis suppressor RECK. Cell Signal. 2004;16:675‐679. [DOI] [PubMed] [Google Scholar]
- 33. Hus M, Chang H, Hung W. HER‐2/neu represses the metastasis suppressor RECK via ERK and Sp transcription factors to promote cell invasion. J Biol Chem. 2006;281:4718‐4725. [DOI] [PubMed] [Google Scholar]
- 34. Takeuchi T, Hisanaga M, Nagao M, et al. The membrane‐anchored matrix metalloproteinase (MMP) regulator RECK in combination with MMP‐9 serves as an informative prognostic indicator for colorectal cancer. Clin Cancer Res. 2004;10:5572‐5579. [DOI] [PubMed] [Google Scholar]
- 35. Qi F, Wang Y, Yu B, Li F. Identification of RECK as a protective prognostic indicator and a tumor suppressor through regulation of the ERK/MAPK signaling pathway in gastric cancer. J Transl Med. 2023;21:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herbster S, Trombetta‐Lima M, de Souza‐Santos PT, et al. Low RECK expression is part of the cervical carcinogenesis mechanisms. Cancers (Basel). 2021;13:2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mao Y, Zhang W, Zhang R, Zuo J. Alkannin restrains oral squamous carcinoma cell growth, migration and invasion by regulating microRNA‐9/RECK axis. Artif Cells Nanomed Biotechnol. 2019;47:3153‐3162. [DOI] [PubMed] [Google Scholar]
- 38. Guo Y, Wang G, Wang Z, et al. Reck‐Notch1 signaling mediates miR‐221/222 regulation of lung cancer stem cells in NSCLC. Front Cell Dev Biol. 2021;9:663279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hong KJ, Wu DC, Cheng KH, Chen LT, Hung WC. RECK inhibits stemness gene expression and tumorigenicity of gastric cancer cells by suppressing ADAM‐mediated Notch1 activation. J Cell Physiol. 2014;229:191‐201. [DOI] [PubMed] [Google Scholar]
- 40. Liu Y, Li L, Liu Y, et al. RECK inhibits cervical cancer cell migration and invasion by promoting p53 signaling pathway. J Cell Biochem. 2018;119:3058‐3066. [DOI] [PubMed] [Google Scholar]
- 41. Mahl C, Egea V, Megens RT, et al. RECK (reversion‐inducing cysteine‐rich protein with Kazal motifs) regulates migration, differentiation and Wnt/β‐catenin signaling in human mesenchymal stem cells. Cell Mol Life Sci. 2016;73:1489‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zheng H, Wang JJ, Zhao LJ, Yang XR, Yu YL. Exosomal miR‐182 regulates the effect of RECK on gallbladder cancer. World J Gastroenterol. 2020;26:933‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang G, Chen X, Ma L, et al. LINC01419 facilitates hepatocellular carcinoma growth and metastasis through targeting EZH2‐regulated RECK. Aging. 2020;12:11071‐11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng J, Li X, Yang W, Zhang F. Dihydroartemisinin regulates apoptosis, migration, and invasion of ovarian cancer cells via mediating RECK. J Pharmacol Sci. 2021;146:71‐81. [DOI] [PubMed] [Google Scholar]
- 45. Kim NY, Lee JE, Chang HJ, et al. Gamma‐irradiation enhances RECK protein levels in Panc‐1 pancreatic cancer cells. Mol Cells. 2008;25:105‐111. [PubMed] [Google Scholar]
- 46. Shen ZX, Jiao KL, Teng MY, Li Z. Activation of STAT‐3 signalling by RECK downregulation via ROS is involved in the 27‐hydroxycholesterol‐induced invasion in breast cancer cells. Free Radic Res. 2020;54:126‐136. [DOI] [PubMed] [Google Scholar]
- 47. Lin HY, Chiang CH, Hung WC. STAT3 upregulates miR‐92a to inhibit RECK expression and to promote invasiveness of lung cancer cells. Br J Cancer. 2013;109:731‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Y, Venkatesh A, Xu J, et al. The WNT7A/WNT7B/GPR124/RECK signaling module plays an essential role in mammalian limb development. Development. 2022;149:dev200340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. America M, Bostaille N, Eubelen M, Martin M, Stainier DYR, Vanhollebeke B. An integrated model for Gpr124 function in Wnt7a/b signaling among vertebrates. Cell Rep. 2022;39:110902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Y, Fang X, Zhao Z, et al. GPR124 induces NLRP3 inflammasome‐mediated pyroptosis in endothelial cells during ischemic injury. Eur J Pharmacol. 2024;962:176228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figures S1–S2