

Abstract
The public availability of genome datasets, such as The Human Genome Project (HGP), The 1000 Genomes Project, The Cancer Genome Atlas, and the International HapMap Project, has significantly advanced scientific research and medical understanding. Here our goal is to share such genomic information for downstream analysis while protecting the privacy of individuals through Differential Privacy (DP). We introduce synthetic DNA data generation based on pangenomes in combination with Pretrained-Language Models (PTLMs).
We introduce two novel tokenization schemes based on pangenome graphs to enhance the modeling of DNA. We evaluated these tokenization methods, and compared them with classical single nucleotide and k-mer tokenizations. We find k-mer tokenization schemes, indicating that our tokenization schemes boost the model’s performance consistency with long effective context length (covering longer sequences with the same number of tokens). Additionally, we propose a method to utilize the pangenome graph and make it comply with DP privacy standards. We assess the performance of DP training on the quality of generated sequences with discussion of the trade-offs between privacy and model accuracy. The source code for our work will be published under a free and open source license soon.
I. Introduction
The public availability of genome datasets is a cornerstone of all data used in collaborative genomics research. Several notable genome datasets have been made publicly available, each significantly contributing to the advancement of scientific research and medical understanding. Notable publicly available genome datasets, such as the Human Genome Project (HGP) [1], the 1000 Genomes Project [13], The Cancer Genome Atlas (TCGA) [70], GenBank [8], the International HapMap Project [21], and the Human Pangenome Project [40] have been instrumental in driving scientific and medical advancements.
The public availability of these datasets is beneficial for humanity for numerous reasons. Public genome datasets accelerate scientific research by providing a wealth of information that researchers can access and analyze worldwide, can be used to identify genetic markers associated with certain diseases leading to the development of preventive strategies and new treatments, and serve as valuable educational resources for students and educators, fostering a deeper understanding of genomics and its implications in various fields.
However, the public release of genomic data raises significant privacy concerns. Genomic data is highly sensitive because it can reveal an individual’s unique genetic makeup; including disease predispositions and other personal traits. Despite efforts to anonymize genomic data, studies have shown that it is possible to re-identify individuals by cross-referencing with other publicly available information, such as health records and social media information [61, 71, 48]. For example, Gymrek et al. demonstrated that surnames could be recovered from personal genomes by combining genetic data with publicly accessible genealogy databases, thus re-identifying individuals within supposedly anonymized datasets [61].
Furthermore, a genome carries familial information, which means privacy breaches can affect not only individuals but also their relatives [9]. Moreover, the potential for the misuse of genomic data extends beyond individual harm to societal concerns, such as the ethical use of genetic information in forensic investigations and the broader implications for data governance and public trust [47, 56]. Ensuring the privacy of any released dataset while still enabling valuable research requires innovative solutions that balance data utility and privacy.
Deep learning models are widely used in different tasks, even in processing genome sequences and related data [77, 35, 34, 19]. Pre-trained language models have shown their capability to generate synthetic natural languages that are almost indistinguishable from real data. The generated text can be used to train other models [36, 76, 26], including those in the medical domain [50, 23]. Proven to be extraordinarily good at processing human language, PTLMs can furthermore interpret and generate non-language text, such as code for programming tasks [10], thereby pushing the boundaries of their application beyond strictly spoken language-based domains. Previous works utilizing GANs [7, 24] faced limitations in context length, since the length of their generation is limited to hundreds or thousands of base pairs, and the potential of PTLMs in this task has not been investigated.
To utilize PTLM for DNA sequence generation, the tokenization of DNA sequences is a crucial first step. Traditional tokenization methods, such as single nucleotide tokenization, and k-mer tokenization where sequences are segmented into individual nucleotides or substrings of length k, are commonly used in prior research [37, 7, 75, 50, 3]. However, k-mer tokenization can be highly sensitive to mutations or sequencing errors, as a single nucleotide change can drastically alter the resulting k-mer, potentially impairing the model’s ability to learn meaningful patterns. Furthermore, these tokenization methods diverge from the tokenization strategies typically employed in natural language processing, which can limit the model’s capacity to effectively capture the underlying patterns and structures within DNA sequences. One of the goals of this study is to investigate how the tokenization scheme (which may be inspired by natural language processing) of DNA sequences can help an PTLM learn the patterns and structures of DNA sequences more effectively.
One of the key advantages of synthetic data is that it reduces the risk of sensitive information leakage, as the synthetic data does not correspond to actual people or entities. It allows for the retention of valuable insights and patterns present in the original data while mitigating the risk of re-identification. This is particularly important in fields like genomics, where the data is highly sensitive and personal, but sharing and working from common, fully public data (like the 1000 Genomes Project or HPRC pangenome) is an essential part of standard research practices. By replacing real genomic sequences with synthetic ones, researchers can continue to perform meaningful analysis without compromising the privacy of individuals. However, while synthetic data is effective in reducing privacy risks, there are scenarios in which further privacy enhancements may be desirable. For example, differential privacy (DP) can be optionally implemented during the generation or training process. DP adds a layer of noise, making it even more challenging to trace any synthetic data point back to an individual, thus providing an additional safeguard for those looking to bolster their privacy protection strategies.
To build a practical genome sequence generation model to protect the privacy of the dataset, our approach is synthetic data generation based on PTLM accompanied by differential privacy (DP). We propose a pangenome graph-based tokenization (Pangenome-based Node Tokenization, PNT) of DNA sequences that utilizes the nodes in the graph as tokens of the sequences. We also propose a second tokenization scheme (Pangenome-based k-mer tokenization, PKMT) that is DP friendly, using the pangenome graph nodes for sequence segmentation before generating k-mers from the DNA sequences. Our contribution is as follows.
We propose two novel tokenization schemes based on the pangenome graph, providing more contextual information to the model and enhancing its ability to learn DNA sequence patterns and structures.
We demonstrate the impact of tokenization on PTLM performance in learning and generating DNA sequences, with experiments showing the superiority of our proposed methods over classical tokenization techniques.
We explore the differentially private training of PTLMs for genome sequence generation, discussing the current limitations of each tokenization scheme under DP settings.
In this work, we present the first comparative analysis of classical and pangenome-based tokenization schemes for PTLMs, specifically GPT-2, in learning DNA sequence patterns and generating long synthetic sequences. Our findings reveal that the pangenome graph structure embeds significant information, enhancing neural networks’ comprehension of DNA sequences. By representing their mutual alignment in tokenization, the segmentation of DNA sequences through node division carries critical information that significantly aids in the comprehension of the sequences. Furthermore, the inclusion of positional information derived from node identifiers (IDs) substantially enhances the training and predictive performance of DNA PTLMs. Our results also demonstrate how pangenome-based tokenization schemes reduce training time and enhance scalability compared to traditional schemes, a crucial advantage given the substantial computational resources typically required for training PTLM. Despite the benefits of generating synthetic data, we also report the results of the generation of the differential privacy (DP) trained model, which were sub-optimal. Future work should focus on increasing sample sizes and developing refined mechanisms to improve DP training performance.
II. Background
A. Pre-trained Language Models
PTLMs are advanced artificial intelligence systems designed to understand and generate language text based on the data they have been trained. These models, such as Mistral [31], Anthropic’s Claude [5], OpenAI’s GPT series [53, 49], Google’s T5 [54], Lamda [63] and Gemini [62], Meta’s OPT [79], BLOOM [39] and LLama [65, 64], etc., take advantage of vast amounts of textual information to learn patterns, nuances, and complexities of language. PTLMs can perform a variety of language-related tasks, including answering questions, writing essays, translating languages, and even participating in casual conversations. Their ability to process and generate coherent and contextually appropriate responses makes them invaluable tools across multiple fields, from customer service and education to creative writing and technical support.
In this paper, we are more interested in the text generation tasks. To process language texts and eventually generate human-like text using the PTLM, the following steps are needed as illustrated in Figure 1:
Tokenization: Tokenization is the first step where the raw input text is broken down into smaller units called tokens. These tokens can be words, subwords, or even characters, depending on the model’s design. This process helps to standardize the input and prepare it for processing. For instance, the sentence ”I love AI” might be tokenized into [”I”, ”love”, ”AI”] if word-level tokenization is used, or into subword units like [”I”, ”lo”, ”ve”, ”AI”] in subword tokenization. Models like BERT use WordPiece tokenization [15], while GPT models use a byte pair encoding (BPE) [58] approach. In languages like English, spaces between individual words often provide a natural way to divide the text into tokens.
Training: The tokenized data will be used to train the PTLM, which often consists of a pre-training phase and a fine-tuning phase. Pre-training involves self-supervised training of the model on general language tasks, such as predicting the next word in a sentence (for models like GPT) or filling in missing words in a sentence (as in BERT’s masked language model approach). After pre-training, PTLMs are fine-tuned on specific datasets tailored to particular tasks or domains, usually in a supervised manner.
Generation: For generative models like GPT, once trained and fine-tuned, the model can generate text by providing an initial prompt to the model. The model uses it to start generating text token by token, and the decoder aligned with the tokenizer’s vocabulary and rules will decode the tokens into readable texts.
Fig. 1:
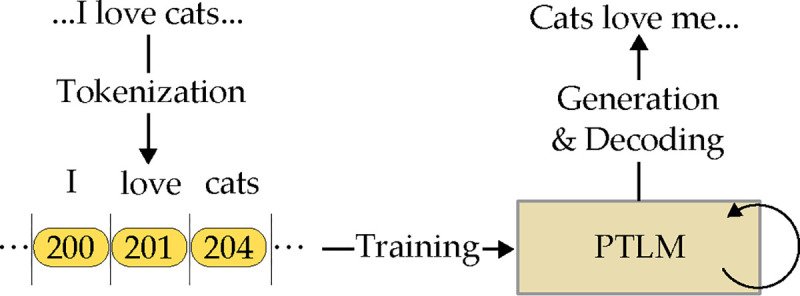
Steps for PTLMs to generate text.
B. Pangenome Graph
The pangenome graph [18] is a computational structure used to represent genetic diversity within a species by integrating multiple genome sequences into a single comprehensive graph. In a pangenome graph, nodes represent sequences of nucleotides, while edges connect these sequences, showing the possible paths through the graph. The paths through the pangenome graph represent the genomes of individuals. This allows the graph to capture alternative sequences found in different individuals. Figure 2 shows a simple illustration of a small pangenome graph.
Fig. 2:

The pangenome graph of the human major histocompatibility complex (MHC) region of chromosome 6 of the PGGB graph of HPRC year 1 assemblies, with 2D graph visualization (above) and matrix view (below).
C. Differential Privacy
Differential privacy (DP) [17] is a privacy-preserving framework that ensures that the output of a computation does not reveal the inclusion of any individual sample in the dataset. DP achieves this by adding carefully calibrated noise to the computation, making it difficult to determine whether a particular individual’s data is included in the dataset. The key idea behind DP is to provide strong privacy guarantees while allowing useful information to be extracted from the data. DP has been widely studied in various fields, including machine learning, data analysis, and statistics, to protect the privacy of individuals in large datasets. DP-Stochastic Gradient Descent (DP-SGD) [2] is a method for training machine learning models with differential privacy that is adopted by many previous researchers [45, 59, 82].
In DP-SGD, the standard deviation of the noise, , required to maintain a constant privacy budget in DP-SGD with the allowed probability of privacy failure , scales with the square root of the number of training epochs, (as one pass on the full training set). Specifically, is adjusted according to the formula [2]. This scaling ensures that cumulative privacy loss across multiple epochs remains within the specified limits of privacy parameters and . During the non-DP training process, more epochs generally lead to better model performance. However, in the DP training process, the model may not benefit from additional training epochs, as it will introduce more noise and degrade the model performance. Therefore, the number of training epochs in DP-SGD is a hyperparameter that needs to be carefully tuned to balance the trade-off between model performance and privacy protection. If a model can be made to train faster, it will require fewer epochs to reach the same level of performance, which can be beneficial for DP training.
III. Synthetic Genome Sequence Generation using PTLMs
In this work, we aim to generate synthetic genome sequence using PTLMs. In this section, we describe the complete pipeline for synthetic genome sequence generation using PTLMs, detailing each step from the original data processing to downstream tasks, as shown in Figure 3.
Fig. 3:

The whole pipeline of synthetic data generation and utilization.
Raw Data. The foundation of our approach begins with the acquisition and preparation of genomic data. These datasets provide the rich and diverse genetic information necessary for training PTLMs.
Tokenization. Tokenization is a critical step in transforming raw genomic sequences into a format suitable for PTLM training. Traditional methods, such as single nucleotide and k-mer tokenizations, break down DNA sequences into smaller, manageable units.
Training of PTLM. Once the DNA sequences are tokenized, they are used to pre-train the PTLM using a GPT-style next-token prediction approach. In this self-supervised learning process, the tokenized sequences are fed into the PTLM, which learns to predict the next token in a sequence based on the patterns and structures inherent in the data. This approach does not require supervised data, as it relies on the model’s ability to learn from the sequences themselves.
Generation. After training, the PTLM is capable of generating synthetic genomic sequences by completing the given prefix (random or non-random). These sequences are produced by the model based on the learned patterns from the training data. The generated sequences aim to preserve the useful patterns of the raw data to keep its utility for the next step.
Downstream Tasks. The synthetic genomic sequences generated by the PTLM can be employed in a variety of downstream tasks.
Traditional methods of tokenization have been proven to be useful in various tasks. However, these approaches may struggle with maintaining sequence integrity over long contexts. We will introduce our novel tokenization schemes in §IV and comparfe them with the classical schemes with comprehensive experiments in §V.
IV. Tokenization of a genome sequence
There are many ways of generating inputs for different models from genome sequences. For PTLMs, we focus mainly on the different tokenization schemes of DNA sequences. The tokenization of genome sequences is the first step in modeling the DNA sequence. A clever tokenization strategy can help the model learn the patterns and structures of DNA sequences more effectively.
The choice of tokenization scheme directly affects the trade-off between sequence length and vocabulary size. In transformer models, shorter token units result in a smaller vocabulary and longer sequences. A smaller vocabulary can be advantageous because it requires learning fewer unique tokens, but it can also necessitate a larger and more diverse training set to capture the complexities of each token. Conversely, longer sequences can provide more context but make the learning process more challenging. This is due to the quadratic complexity of the attention mechanism, which scales with the length of the sequence in terms of both memory and computation. As a result, longer sequences can be harder and slower to learn effectively.
In this section, we first describe the widely used tokenization schemes and then introduce our tokenization schemes based on the pangenome graph.
A. Classical tokenizations
In this subsection, we introduce the classical tokenizations used commonly in previous work without the involvement of pangenome graph info.
1). Genome-based Single Nucleotide Tokenization (GSNT)
Genome-based Single Nucleotide Tokenization (GSNT) is the most straightforward way to tokenize a genome sequence that was used in previous work [46, 55]. In this scheme, each nucleotide (A, C, G, T) is treated as a separate token. For example, if we have a genome sequence ”ACGTA”, the tokens are ”A”, ”C”, ”G”, ”T”, ”A”.
The advantage of single-nucleotide tokenization comes from its simplicity and universality. It is easy to implement and can be used for any genome sequence, regardless of the species or length of the sequence. However, the GSNT will have a shorter effective context length (a shorter DNA sequence represented under the fixed prompt length) compared to other tokenization schemes with the same number of tokens and be more likely to cause longer training and inference time due to the large number of tokens.
2). Genome-based k-mer Tokenization (GKMT)
Another widely used way to tokenize a genome sequence is to use k-mers as tokens. A k-mer is a substring of length k in the genome sequence. For example, if we have a genome sequence ”ACGTA”, the 3-mers are ”ACG”, ”CGT”, and ”GTA”. When the stride is less than k, the k-mers will overlap with each other, nonoverlapping k-mers are also used in some cases where the stride is simply k.
The Genome-based k-mer Tokenization (GKMT) scheme is simple and easy to implement, but it has some limitations. Although it brings a relatively longer effective context length compared with GSNT tokens (when the stride is larger than 1), it is also sensitive to mutations or errors in the sequence, since a single base change can result in k completely different k-mers when the stride is set to 1, or potentially different k-mers for all following sequences when the stride is larger. If the stride is set to one, increasing k also increases the computation and memory requirements for processing, storing, and analyzing k-mers, due to the larger vocabulary. Since minor variation would cause unstable tokenization, the models trained on k-mer tokenized sequences usually train slow, which we will discuss in our experiment section.
To overcome the limitations of the commonly used tokenization schemes, we propose two novel tokenization schemes based on the pangenome graph.
B. Pangenome graph based tokenization
Pangenome graphs are a powerful representation of the genetic diversity within a certain group of sequences. The nodes in the pangenome graph represent the genetic sequences that are shared among the groups, while the edges represent the genetic variations. Tasks like genome-wide association (GWA) focus on the genotype matrix of the graph rather than the DNA sequences themselves. In this sense, it is the graph structure rather than the actual “AGCT” strings that carries information. We propose two tokenization schemes based on the pangenome graph that are illustrated in Figure 4.
Fig. 4:
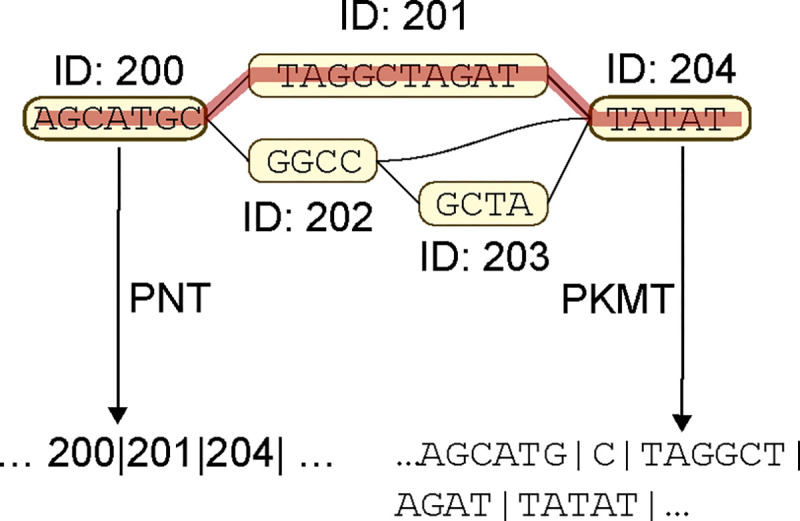
The pangenome graph based tokenizations. The figure shows a slice of a pangenome graph with nodes marking the variations and edges marking possible paths. The two tokenization methods output two different segmented sequence of the red path.
1). Pangenome-based Node Tokenization (PNT)
The first scheme, Pangenome-based Node Tokenization (PNT), is to tokenize the DNA sequences directly based on the nodes on the pangenome graph. In this scheme, each node in the pangenome graph is treated as a token. Notice that each node contains not only the information about the DNA sequence they represent but also the position of this sequence in the graph. For example, there can be multiple different nodes with different node IDs that correspond to the same DNA sequence because of their different position in the graph. In practice, a vocabulary of the node IDs can be much larger (for example, around 400K) compared to a language vocabulary (e.g., 50K), which can be a challenge for model training. In our experiment, we simply split the node IDs into two parts (first half and second half) with an extra indicator of reversion (a variation that the sequence is in a reverse direction; e.g., node 123456 representing ”AGCT” with reversion will be tokenized as ‘123’ and ‘456 +’, which will be decoded as ”TCGA”) in order to reduce the vocabulary size.
A limitation of this scheme is that no additional sequence can be added based on existing pangenome graph. The introduction of new sequences can alter the representation of previously established sequences within the graph: each time a new graph has to be rebuilt for the new sequences to be tokenized using the new generated ID, meaning that the model’s understanding of the original data may shift as the new data is incorporated.
2). Pangenome-based k-mer Tokenization (PKMT)
The second scheme, Pangenome-based k-mer Tokenization (PKMT), is to tokenize the DNA sequences based on the k-mers that are connected by the nodes in the pangenome graph. In this scheme, instead of directly using the node IDs as the tokens, we tokenize the sequences that they represent as nonoverlapping k-mers. We use k = 6 in our experiment and do not apply padding to the tails of sequences. For example, if a node represents the sequence “AGCTAGCTAGCTAGC”, it will be tokenized as three independent tokens: “AGCTAG”, “CTAGCT” and “AGC”. As illustrated, the last token can potentially be shorter than the maximum of k due to the end of the sequence.
PKMT utilizes the structure of the pangenome graph to provide a more stable tokenization compared to GKMT. If any insertions or deletions occur, the pangenome graph would capture such behavior when aligning the sequences of the whole dataset, and the tokenization of any nodes following the insertion or deletion will not be affected, unlike what happens in the GKMT. However, compared with the PNT, the tokens generated capture no information on the position of the sequence in the graph, which can be crucial for the model to learn the patterns and structures of the DNA sequences. This drawback might result in worse performance.
C. Privacy-preserving graph-based tokenization
The tokenization schemes that we proposed are designed to provide more context information to the model and help the model learn the patterns and structures of DNA sequences more effectively. However, tokenization schemes can also leak sensitive information about individual samples. To make a scheme DP-friendly, the tokenization of each sample should be independent from the private dataset. Otherwise, certain mechanisms should be applied to make it protected. When a pangenome graph is generated, it utilizes all the samples in the dataset without protection, and the input generated, therefore, can potentially reveal the genetic information of the individual samples.
It is hard to make the pangenome-based node tokenization or the pangenome graph generation DP-friendly, and the ID to sequence mapping inevitably leaks information. Although the PKMT does not leak such information during the mapping of tokens to sequences due to its static vocabulary, the way the k-mers are generated can still leak information about individual samples since it utilized the pangenome graph. Our approach to mitigate the issue is to instead build a “public” pangenome graph. Assuming there is a publicly accessible dataset of the same part of the DNA sequences with the private set that we want to protect, we can build a pangenome graph based on the public dataset and use it to tokenize the private dataset. Any nodes that can be identified on the public pangenome graph will be tokenized as the same node, and any part of the sequence that cannot be identified by the nodes on the public graph will be marked unknown and tokenized as normal k-mers. In this way, the tokenization scheme will not leak the genetic information of individual samples in the private dataset.
In our experiment, we split an existing graph as a public graph and the private sequences. We build the public pangenome graph as shown in Protocol 1 and then complete the PKMT as shown in Protocol 2.
Algorithm 1.
: Define Public Pangenome Graph Nodes
| 1: | Input: A pangenome graph , list of indexes with public sequences. We use to represent the node of the sequence in and to represent the actual sequence. |
| 2: | Output: The way nodes are merged in public pangenome graph recorded in . |
| 3: | Initialization: |
| 4: | Initialize as an empty dictionary to store the public pangenome graph nodes. |
| 5: | for each sequence in do |
| 6: | for each node in do |
| 7: | if has fixed previous/next nodes in then |
| 8: | Combine with the fixed previous/next nodes as a single node. |
| 9: | Record the combined node in . |
| 10: | else |
| 11: | Record as an independent node in . |
| 12: | end if |
| 13: | end for |
| 14: | end for |
| 15: | Return: as the public pangenome graph nodes. |
V. Experiments
A. Datasets and PTLM choice
Here we introduce our datasets and the parameters we use in our experiments. We use the human major histocompatibility complex (MHC) region of chromosome 6 as our dataset that is cut out of the PGGB graph of HPRC year 1 assemblies [40]. A total of 126 samples are in the dataset, with 80% of the samples used as training set and 20% as a test set. We tested the performance of the 90M parameter GPT-2 model [53] which supports a prompt length of 1024 tokens. GPT-2 is chosen due to its well-established performance and robustness as a classical publicly available language model, and the 90M total parameter is chosen to balance performance and overhead.
B. Evaluating synthetic genome sequence quality
A main challenge of utilizing synthetic genome sequences is how to evaluate the quality of synthetic genome sequences. For text generated by PTLMs, quality evaluation typically encompasses both automated benchmarks and human assessments to capture aspects that automated metrics might overlook. Two common methods include:
Prediction Accuracy metric: Measures the proportion of correct predictions made by the model. It assesses how often the model’s predicted tokens or values match the actual tokens or values in the data.
Perplexity metric: Measures how likely the model is to predict a given sentence. Lower perplexity indicates that the model is more confident and accurate in its predictions.
BLEU metric: Measures how well the model’s predictions align with reference sentences based on n-gram overlap. It evaluates the quality of the model’s output in terms of similarity to human-generated text.
In our study, we use the prediction accuracy of the model to measure the quality of the generative model. Furthermore, we compare the similarity between synthetic and real genome sequences through sequence alignment.
1). Model prediction accuracy
The next token prediction accuracy measures the percentage of tokens that the model predicts correctly given the correct previous tokens. This metric naturally reflects the quality of the model and is the primary measure of accuracy for the pre-training task of predicting the next token. In essence, this is what models like GPT are specifically optimized for during their training process. However, this accuracy is not a direct measurement of the accuracy of the predicted sequences when the tokenization is not single nucleotide-based. For example, if the model predicts “AAAAAA” when the ground truth is “AAAAAC”, it can be considered to have predicted 5 out of 6 nucleotides correctly rather than one token incorrectly.
To address this, we introduce the “character accuracy ratio” which is the percentage of nucleotides that the model predicts correctly. The prediction will be much more difficult when the model is required to generate a long sequence rather than a single token.
2). Sequence alignment
Measuring the similarity between two genome sequences is done using sequence alignment, which is an essential process in many bioinformatics and computational biology tasks. The sequence alignment involves arranging the sequences of DNA, RNA, or even proteins, usually to identify regions of similarity. In our case, we use wfmash [22] where the wavefront algorithm [44] is primarily used for pairwise alignment between real and generated DNA sequences. After the alignment is done, multiple scores can be used to evaluate the quality of the alignment.
The scores we use are as follows:
BLAST identity (BI): Defined as the number of matching bases over the number of alignment columns.
Gap-Compressed Identity (GI): Count the consecutive gaps as one difference.
For example, for a reference sequence AGCTAag-TA and a query sequence AGCTA--cTA, where the dashed lines represent a gap and the lowercase letters represent a mismatch, the BI is 7/10 = 0.7 counting continuous gaps as multiple mismatches while the GI is 7/9 = 0.78 counting continuous gaps as one mismatch.
The alignment scores themselves can be considered sufficient as a representation of the utility of the synthetic sequences by measuring how close they are to the real data. Previous academic discussions [20, 16] have shown that alignment score is equivalent to showing sequence similarity. Therefore, the scores can indicate the potential usefulness of synthetic data in downstream genomic tasks since high scores suggest that synthetic data can be a reliable substitute for real data in various analyses. A higher score of a generated sequence against real data indicates that synthetic data can be a reliable substitute for real data, demonstrating that the synthetic data generated represent genetic diversity well.
Algorithm 2.
: Perform PKMT Based on Public Sequences Only
| 1: | Input: A pangenome graph , list of indexes with public sequences and with private sequences. We use to represent the node of the sequence in . We use to represent the actual sequence. |
| 2: | Output: Segmented DNA sequences recorded in Segmented. |
| 3: | |
| 4: | Initialize Segmented = {} |
| 5: | for each sequence in do |
| 6: | Initialize Chain = [ ] |
| 7: | Initialize UndefinedChain = [ ] |
| 8: | Initialize Segmented[i] = [ ] |
| 9: | for each node in do |
| 10: | Add to Chain |
| 11: | if current node chain ends according to then |
| 12: | Append UndefinedChain to Segmented[i] as a segment of the sequence |
| 13: | Append Chain to Segmented[i] as a segment of the sequence |
| 14: | Clear UndefinedChain |
| 15: | Clear Chain |
| 16: | else if current node pattern is not recorded in then |
| 17: | Append Chain to UndefinedChain |
| 18: | Clear Chain |
| 19: | end if |
| 20: | end for |
| 21: | Cut each segment in Segmented[i] into non-overlapping 6-mers |
| 22: | end for |
| 23: | Return: Segmented |
C. Results of public training
We train the 90M GPT-2 model on the training dataset using the four tokenization schemes: Genome-based Single Nucleotide Tokenization (GSNT), Genome-based k-mer Tokenization (GKMT), Pangenome-based Node Tokenization (PNT), and Pangenome-based k-mer Tokenization (PKMT). The training is done for 90 epochs (each epoch is a pass on the training data) with a batch size of 16 of 1024 token sequences for each tokenization method. The dataset contains 124 samples of DNA sequences with a total of 447 million nucleotides. The training times are listed in Table I, obtained on 8 NVIDIA A5500 GPUs. The model token prediction accuracy and character level prediction accuracy are shown in Figure 5. In the figure, node-based 6-mer tokenization is represented as two versions: a default version that uses only 20% of the dataset to build the public pangenome graph, which matches our intuition of using this scheme in a public-private data scheme under DP training later; and the version in which all data are used to build the pangenome graph.
TABLE I:
The training time of each tokenization schemes on the 90M GPT-2 model running 90 epochs.
| GSNT | GKMT | PNT | PKMT | |
|---|---|---|---|---|
| Time(h) | 112 | 11 | 15.2 | 1.9 |
Fig. 5:

Model prediction accuracy of the four tokenization schemes. We did not include the node-ID-based method in character level accuracy figure because it is vague to define it when the sequence lengths varies too much represented by different tokens.
The training times and model performance differ significantly across tokenization schemes as shwon in Table I and Figure 5. For instance, PKMT requires the least amount of training time, whereas GSNT takes the longest. Looking at the performance per epoch and wall-clock time, the PNT-tokenized model reaches a certain accuracy faster than others, in terms of both wall-clock time and training epochs, while GKMT requires the most time and epochs to reach the same level of accuracy. In terms of character prediction accuracy, GSNT tends to train faster than PKMT in the beginning, but both schemes converge to similar final accuracy levels. However, GSNT is slightly slower than PKMT when building the graph with the full dataset and also lags slightly behind in final accuracy by 0.2%. The significant accuracy gap between GKMT and PKMT emphasizes the effectiveness of leveraging graph structure in tokenization. Despite having similar token tables, the graph-aided segmentation of PKMT provides more stable and learnable context information, resulting in better performance. It is also noteworthy that GSNT is approximately 8 to 9 times slower in wall-clock training time than PKMT or GKMT, and over 20 times slower than PNT, largely due to the larger number of tokens in GSNT.
We show the alignment results for the generated sequences of the three tokenization schemes in the above metrics in Figure 6 (GKMT barely generates matches, so we skipped its figure), aligned against the reference sequence of the dataset. A clear view is shown in Figure 7 for a single generated sequence. The X-axis represents the position in the reference sequence, while the Y-axis represents different generated sequences that are aligned to the reference. Each dot / line represents a specific position in the read that aligns to a specific position in the reference genome. As we can see, with 90 epochs of training, only the PNT can generate sequences that are closely aligned with the reference sequence in a long enough effective context length. Notice that there are some generated sequences that do not match the reference sequence at all, which is partially due to the model’s random sampling for generating diverse outputs, and also due to the fact that some sequences in the training data do not align well with the reference sequence, which may be learned by the model.
Fig. 6:

The alignment of the batch of generated sequences against the reference sequence. X-axis is the reference and Y-axis are the multiple generated sequences, where lines and dots mark the position of matches and breaks.
Fig. 7:

The alignment of the single generated sequence.
To demonstrate the quality of generation numerically, we show the alignment scores of the generated sequences against the whole dataset (that is, find the best match of a query from sequences of the entire dataset) in Table II, while also showing the results for real data as a comparison. In addition to the GI / BI scores we introduced, we also show the alignment percentage indicating the proportion of the generated sequences that can be found to be of good alignment in the dataset. The PNT has the highest alignment scores for all segment lengths, while the GSNT has the lowest scores. The PKMT has a relatively high alignment score for the 1k segment length, but the score drops significantly as the segment length increases. This indicates that while the PKMT method achieves character-level prediction accuracy on par with GSNT, its longer effective context length improves generation performance, resulting in better alignment metrics.
TABLE II:
Alignment percentages and weighted GI/BI scores for different segment lengths, of the generated sequences against the original dataset as reference. The real data numbers are generated using 80% samples as references and 20% as queries.
| Alignment Segment | 1k | 5k | 20k | 50k | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Align % | GI | BI | Align % | GI | BI | Align % | GI | BI | Align % | GI | BI | |
| GSNT | 82.68 | 0.8664 | 0.9955 | 53.52 | 0.8872 | 0.9903 | 12.67 | 0.8753 | 0.9889 | 0.00 | 0.0000 | 0.0000 |
| PKMT | 63.15 | 0.9697 | 0.9937 | 50.42 | 0.8955 | 0.9908 | 14.91 | 0.8901 | 0.9883 | 4.65 | 0.7054 | 0.9848 |
| PNT | 95.85 | 0.9976 | 0.9998 | 78.83 | 0.9977 | 0.9997 | 25.94 | 0.9944 | 0.9988 | 32.46 | 0.9856 | 0.9981 |
| Real data | 99.97 | 0.9994 | 0.9999 | 97.97 | 0.9994 | 0.9999 | 69.23 | 0.9996 | 0.9999 | 61.37 | 0.9991 | 0.9981 |
1). Effects of extensive training
In order to perform the DP training later, we split the dataset into 2: 20% of the data is used as public data that can be trained without protection to build the public pangenome graph for PKMT, and the remaining 80% is treated as private data that needs DP-SGD training for protection. We extensively train the GPT-2 model on the public dataset for 200 epochs with other parameters being the same. We observe that the token prediction accuracy increased by about 0.4% for the PKMT and 0.3% for the GSNT, which is apparently very marginal. However, we have observed dramatic improvements in generation quality. We show the following results in Figure 8 and Table III.
Fig. 8:

The alignment of the batch of generated sequences from extensively trained data against the training sequences. X-axis is the reference and Y-axis are the multiple generated sequences, where lines and dots mark the position of matches and breaks.
TABLE III:
Alignment percentages and weighted GI/BI scores for different segment lengths, of the generated sequences with extensively trained model using different proportion of public data, against the original dataset as reference. Notice that for PKMT, the public pangenome graph will change with the public data chosen changed.
| Alignment Segment | 1k | 5k | 20k | 50k | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 20% as public | Align % | GI | BI | Align % | GI | BI | Align % | GI | BI | Align % | GI | BI |
| GSNT | 81.36 | 0.8720 | 0.9956 | 56.58 | 0.8941 | 0.9912 | 17.65 | 0.8932 | 0.9901 | 5.13 | 0.8625 | 0.9912 |
| PKMT | 63.36 | 0.9848 | 0.9978 | 63.44 | 0.9016 | 0.9969 | 61.06 | 0.9045 | 0.9952 | 55.55 | 0.9014 | 0.9948 |
| 50% as public | Align % | GI | BI | Align % | GI | BI | Align % | GI | BI | Align % | GI | BI |
| GSNT | 83.23 | 0.8731 | 0.9944 | 60.12 | 0.8966 | 0.9923 | 20.26 | 0.8941 | 0.9900 | 6.75 | 0.8654 | 0.9903 |
| PKMT | 81.03 | 0.9810 | 0.9981 | 79.53 | 0.9040 | 0.9975 | 73.00 | 0.9059 | 0.9956 | 69.90 | 0.9025 | 0.9948 |
It can be seen that while both tokenizations benefit from extensive training of large computational resources, the PKMT can achieve a significantly higher alignment score than the GSNT, typically when the alignment segment length is longer. It is also worth noting that the PKMT can generate relatively longer consistent sequences than the GSNT, showing its advantage in helping the model in consistency and long context learning. However, it is easy to see that both methods are still inferior to PNT with less resources used for training, even with extensive training.
D. DP-SGD training
Following the training of the public model as outlined in §V-C1, we used Differentially Private Stochastic Gradient Descent (DP-SGD) to fine-tune the model in the private data set. We use the same parameters as the public training, and the only difference is that we use the DP-SGD training with . Given the nature of differential privacy mechanisms, which introduce noise throughout the training process, only marginal improvements were anticipated. Upon evaluation, the model fine-tuned with DP-SGD in the private data set exhibited an increase in tokenization evaluation precision of 0. 15%, reaching 92. 45% from an initial 92.3%. However, the alignment scores of the generated sequences showed a significant decrease, as we show in Table IV.
TABLE IV:
Alignment percentages and weighted GI/BI scores for different segment lengths, of the generated sequences from the public model trained on public data only with PKMT, and the model further trained on private data with DP-SGD, against different datasets.
| Alignment Segment | 1k | 5k | 20k | 50k | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Align % | GI | BI | Align % | GI | BI | Align % | GI | BI | Align % | GI | BI | |
| Pub trained against pub | 63.36 | 0.9848 | 0.9978 | 63.44 | 0.9016 | 0.9969 | 61.06 | 0.9045 | 0.9952 | 55.55 | 0.9014 | 0.9948 |
| Pub trained against priv | 63.12 | 0.9823 | 0.9975 | 63.31 | 0.9001 | 0.9968 | 60.94 | 0.9045 | 0.9952 | 56.26 | 0.9016 | 0.9949 |
| DP trained against priv | 34.04 | 0.9472 | 0.9908 | 13.24 | 0.8540 | 0.9887 | 2.02 | 0.5060 | 0.9591 | 0.00 | 0.0000 | 0.0000 |
We show the results of the generated sequences from the model trained only with public data as a reference, against both the public and private datasets. The alignment scores of the generated sequence, as expected, are generally better with the public dataset on which they are trained, compared with the scores against the private dataset. However, DP-SGD training, while improving the accuracy of the next token evaluation, significantly decreases the alignment scores of the generated sequences against the private dataset.
This discrepancy is attributable to the inherent noise added by the DP-SGD mechanism. As the training noise accumulates, it exacerbates the difficulty of maintaining sequence consistency, particularly over longer segments. Although techniques like using larger batch sizes can help mitigate some of the distortions by averaging out noise, they are not entirely effective. This results in notable distortions in the generated sequences, thereby diminishing alignment accuracy. Consequently, while DP-SGD confers privacy benefits and slight improvements in tokenization accuracy, it also imposes a significant trade-off in terms of sequence alignment performance.
VI. Discussion
To our knowledge, this work is the first to compare the effectiveness of pangenome-based tokenization schemes to classical tokenization schemes when utilizing the PTLMs (specifically GPT-2 in our experiments) to learn the pattern of DNA sequences; and also the first to demonstrate the efficacy of PTLMs in generating long synthetic sequences. Previous research on generation tasks has not sufficiently addressed the context length in their outputs.
In our study, we evaluated four tokenization schemes (PKMT, GSNT, PNT, and GKMT) based on training time, training speed, and performance accuracy. From the results in Table I, PNT demonstrated the fastest training time, completing in 1.9 hours, while GSNT was the slowest at 112 hours due to its larger token set. Figure 5 indicates that the accuracy in terms of next token prediction and character accuracy ratio across tokenization methods in different epoches. PNT generally performs the best with close to 99% token prediction accuracy, and GKMT leads the character accuracy ratio close to 98% excluding PNT. Traditional methods fall short with GKMT has below 70% accuracy, and GSNT trains significantly slower and with a slightly inferior character accuracy ratio. The performance gap is much larger when comparing the alignment scores shown in Table II, where PNT tops in both GI being BI being > 0.999 in almost all segment choice from 1k to 50k, which is the closest to real data performance. These findings suggest that sequences generated by the PNT scheme have more potential to be utilized similarly to real data, while PKMT-generated sequences may require further refinement or model optimization to reach a comparable level of utility.
Overall, PNT emerged as the best performer for sequence alignment and training efficiency, while PKMT also excelled traditional methods in terms of training time and context-rich sequence generation. GSNT, although slower, performed competitively in token prediction but lagged in sequence alignment. These results underscore the trade-offs between computational cost and model performance, with pangenome graph based tokenizations showing the more promising performance across tasks. Previous work [40] demonstrates how improved matching is the key point of the pangenome, which “aligns” with our use of the pangenome graph here.
Our findings provide valuable insights into the pangenome graph: the graph structure embeds significant and meaningful information that enhances neural networks’ understanding of DNA sequences, and our experiments show how this information can be realistically exploited.. Compared to GKMT, PKMT only differs primarily in whether the segmentation carried out during tokenization is guided by the pangenome graph. This graph-informed segmentation alone significantly improves the model training speed and overall performance.
The strong performance of the extensively trained model indicates that substantial investment in computational power is justified. With the generation of the public pangenome graph, PKMT and PNT significantly reduce training time compared to GSNT, due to the longer context length represented by each token in the actual sequences. This demonstrates the superior scalability potential of the pangenome graph-based tokenization scheme compared to traditional methods.
While generating synthetic data helps prevent the release of real data, differential privacy (DP) offers a stronger mathematical guarantee against potential attacks. However, our results have not yet achieved satisfactory DP-compatible generation. This shortfall may be attributed to several challenges in our current work:
Insufficient samples: It is generally easier to hide an individual in a more populated group. Achieving the DP guarantee requires adding noise during training. To reduce the impact of noise on gradient updates, large batches are often updated at once. The averaging effect mitigates the noise perturbation, since the noise has a zero mean. However, if the batch size is too large and there are not enough samples, training may not have sufficient steps within the epochs limited by the DP noise budget.
Loose bound. DNA sequences exhibit unique patterns, such as numerous repeated segments across different samples within the same genomic region of a species. We have yet to find a clear method to restrict the DP bounds based on the properties of DNA sequences or the pangenome graph. Using a general noise mechanism results in a loose bound with more noise than necessary. Future efforts should aim to conduct experiments with more samples or develop a more carefully designed mechanism to achieve better DP training performance.
VII. Related work
A very recent paper [80] presents a similar approach to tokenization. Although there are overlaps in the methodologies that we both independently develop the idea of using pangenome graph to help tokenization, our work is different since we include the node-ID-based tokenization and focus on the long sequence generation, while the mentioned work is implemented on relatively short context length (maximum 5000bp), only includes node-aided k-mer tokenization, and focuses on classification tasks.
We provide Table V to summarize this section.
TABLE V:
DL models used in genome tasks.
| Job Type | Paper | Task | Architecture | Input |
|---|---|---|---|---|
| Classification | [51, 77, 35] | Variant Calling | CNN | hundreds of base pairs |
| [41] | Variant Calling | CNN | hundreds of base pairs | |
| [43, 19] | Cancer Prediction | CNN | RNA-seq gene expression data | |
| [3] | Protein Binding | CNN | 10–100 nucleotides & binding specificities | |
| [78] | Protein Binding | CNN | 10–100 base pairs & binding specificities | |
| [74] | Cell Type Identification | CNN | cell images | |
| [81] | Non-coding DNA function prediction | CNN | 1k base pairs | |
| [42] | Variant Calling | RNN | binary alignment map (BAM) | |
| [57] | RNA-protein binding preference | LSTM | embedded k-mers | |
| [52] | Non-coding DNA function prediction | CNN/BLSTM | one hot encoded nucleotides | |
| [34] | Cancer Prediction | KNN | SNP genotype syntaxes (8-mers) | |
| [25] | Cancer Prediction | Rao score | Mutation Annotation Format (MAF) | |
| [68] | Cancer Prediction | SVM | Human EDTA plasma samples | |
| [30, 83] | Molecular Phenotype Prediction | Transformer | tokenized k-mers | |
| [14] | Molecular Phenotype Prediction | Transformer | tokenized k-mers | |
| [46] | 5-way Species Classification | Transformer | single nucleotide tokens | |
| [55] | Genome Tasks | Mamba | single nucleotide tokens | |
| Generation | [66] | De novo peptide sequencing | LSTM/CNN | tandem mass spectrometry (MS/MS) Spectrum |
| [67] | De novo peptide sequencing | LSTM/CNN | data-independent acquisition (DIA) mass spectrometry data | |
| [73] | De novo peptide sequencing | learning-to-rank | tandem mass spectrometry data | |
| [7] | Synthetic Medical Data | GAN | medical data | |
| [24] | Synthetic DNA Sequences | GAN | DNA sequences |
A. Machine learning in genomics
Machine learning (ML) and Deep Learning (DL) have been widely used in genomics to analyze and interpret large-scale biological data. In this section, we introduce two common tasks:
1). Classification tasks
Classification tasks are one of the most common tasks in which people use machine learning models in genomics. For the ones involving actual genome sequences, some major tasks include:
Variant Calling: ML algorithms can be used to identify genetic variants, such as single nucleotide polymorphisms (SNPs) and insertions/deletions (indels), in an individual’s genome. These variants can be associated with diseases, traits, or other biological functions. For example, DeepVariant [51] is a CNN-based variant caller that has been shown to outperform traditional variant calling methods, on which many other variant callers [77, 35] are based. Clairvoyante [41] employs a CNN multitask that outperforms DeepVariant in reads of single-molecule sequencing (SMS), and Clair [42] proposed an RNN structure with fewer parameters and faster inference speed than DeepVariant, without a marginal loss of accuracy.
Gene Expression Analysis: ML models can analyze gene expression data to identify patterns and relationships between genes and biological processes/phenotypes. This information can help researchers understand how genes are regulated and how they contribute to disease. Unlike in variant calling, preprocessed data like alignment maps, mutation tables, gene expression data, etc. are more often inputs rather than raw gene sequences. Classical ML such as k-nearest neighbors (KNN) [34], linear regression [25], logistic regression plus support vector machine [68], can be used to predict driver genes or the overall risk of cancer. Deep learning models such as CNN [43, 19] are also used for cancer prediction and classification using RNA-seq.
In addition to the two main fields mentioned above, different networks have shown their capability in numerous tasks. Working with raw sequences, CNN has also shown its ability to model the sequence specificity of protein binding [3, 78], cell type identification [74], and it has been shown to be able to analyze non-coding variants [81]; RNN can be used for non-coding DNA function prediction [52] and RNA-protein binding preference [57]. For recently more popular transformer-based PTLM, it is shown to be capable of producing strong contextualized embedding from nucleotide sequences, effectively predicting molecular phenotypes in scenarios with limited data [30, 83, 14]. However, these models have been restricted by the limited context sizes due to the quadratic scaling of Transformers, and attempts are made for sub-quadratically scaling for a longer context length (Hyena [46] MambaDNA [55]). MambaDNA is one of the most recent works that uses language models for genome tasks.
2). Generation tasks
Generative models are used in genomics for various tasks. Some notable applications include
De Novo Genome Assembly:
De novo genome assembly is the process of reconstructing a genome sequence from short DNA fragments without the need for a reference genome. Previous work uses deep learning frameworks to enable the de novo peptide sequencing [66, 67, 73],
Synthetic data generation:
Synthetic data generation creates artificial data that closely resembles the original data to avoid directly revealing the real data while sharing. GAN is used to generated synthetic medical data in previous work [7], synthetic DNA sequences coding for proteins with desired properties [24], but only tested on very small dataset since GAN typically generate (limited) fixed-sized outputs. Mathematically, it would require differential privacy on top for a provable guarantee since the generated data can still leak crucial information of the training dataset.
B. Privacy in genomics
Genomic data are highly sensitive personal information that can reveal an individual’s unique genetic makeup, predispositions to diseases, and other personal traits. The public release or leakage of genomic data can lead to privacy concerns, as it can be used to re-identify individuals, discriminate against them based on their genetic information, or expose them to potential harm. Dealing with sensitive data requires dedicated methods of privacy protection. Depending on the goal of the use and sharing of data, different definitions of privacy evolve, with multiple tools and methods developed to protect the privacy of the data.
In addition to common access control and law enforcement methods, crytographic methods provide a mathematical guarantee of the confidentiality of the data.
Secure Multiparty Computation (MPC):
MPC aims to allow multiple parties to jointly compute a function over their inputs while keeping those inputs private, either through an encrypted circuit or requiring communication during computing. MPC protocols are typically useful when the data provider and the evaluator are different entities, and neither party wants to reveal their data to the other. After the first work that shows MPC usage in privacy-preserving edit distance and Smith-Waterman computation [29], it has been used in genomics for secure GWAS [72, 12], secure disease diagnose [28]. However, MPC can be computationally and communicationally expensive.
Homomorphic Encryption (HE):
HE is a form of encryption that allows computations to be performed on encrypted data without decrypting it first. HE has been used in genomics for secure GWAS [38, 69], secure disease diagnose [6], secure genome data mining (combined with MPC) [33], and secure sequence analysis [11]. HE can also be However, HE can also be computationally expensive.
Differential Privacy (DP):
Any analysis results on a genomic data pool can be potentially used to infer private information of the participants, even if anonymized (e.g. the membership inference attacks (MIA) [27]). DP guarantee ensures that the presence or absence of an individual’s data in a dataset does not significantly affect the outcome of the analysis. DP is used in protecting genome-wide association studies (GWAS) [32, 60], but it is shown that large noise will be needed and membership and MIA can still be conducted [4].
VIII. Acknowledgement
This material is based upon work supported by the U.S. National Science Foundation under award No. CCF-2118709 (P.H., J.N.S, P.P., E.G. and E.S.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The authors also gratefully acknowledge support from National Institutes of Health/NIDA U01DA047638 (E.G.), National Institutes of Health/NIGMS R01GM123489 (P.P. and E.G.).
References
- [1].US DOE Joint Genome Institute: Trevor Hawkins Elbert Branscomb Paul Predki Paul Richardson Sarah Wenning Tom Slezak Norman Doggett Jan-Fang Cheng Anne Olsen Susan Lucas Christopher Elkin Edward Uberbacher Marvin Frazier et al. “Initial sequencing and analysis of the human genome”. In: nature 409.6822 (2001), pp. 860–921. [DOI] [PubMed] [Google Scholar]
- [2].Abadi Martin et al. “Deep learning with differential privacy”. In: Proceedings of the 2016 ACM SIGSAC conference on computer and communications security. 2016, pp. 308–318. [Google Scholar]
- [3].Alipanahi Babak et al. “Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning”. In: Nature biotechnology 33.8 (2015), pp. 831–838. [DOI] [PubMed] [Google Scholar]
- [4].Almadhoun Nour, Ayday Erman, and Ulusoy Özgür. “Inference attacks against differentially private query results from genomic datasets including dependent tuples”. In: Bioinformatics 36.Supplement_1 (2020), pp. i136–i145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Anthropic. Claude 2. Anthropic Blog. Accessed: 2024-09-03. July 2023. URL: https://www.anthropic.com/index/claude-2.
- [6].Ayday Erman et al. “Protecting and evaluating genomic privacy in medical tests and personalized medicine”. In: Proceedings of the 12th ACM workshop on Workshop on privacy in the electronic society. 2013, pp. 95–106. [Google Scholar]
- [7].Bae Ho et al. “AnomiGAN: Generative adversarial networks for anonymizing private medical data”. In: Pacific Symposium on Biocomputing 2020. World Scientific. 2019, pp. 563–574. [PubMed] [Google Scholar]
- [8].Benson Dennis A et al. “GenBank”. In: Nucleic acids research 41.D1 (2012), pp. D36–D42.23193287 [Google Scholar]
- [9].Cassa Christopher A et al. “My sister’s keeper?: genomic research and the identifiability of siblings”. In: BMC medical genomics 1 (2008), pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen Mark et al. “Evaluating large language models trained on code”. In: arXiv preprint arXiv:2107.03374 (2021). [Google Scholar]
- [11].Cheon Jung Hee, Kim Miran, and Lauter Kristin. “Homomorphic computation of edit distance”. In: Financial Cryptography and Data Security: FC 2015 International Workshops, BITCOIN, WAHC, and Wearable, San Juan, Puerto Rico, January 30, 2015, Revised Selected Papers. Springer. 2015, pp. 194–212. [Google Scholar]
- [12].Cho Hyunghoon, Wu David J, and Berger Bonnie. “Secure genome-wide association analysis using multiparty computation”. In: Nature biotechnology 36.6 (2018), pp. 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].1000 Genomes Project Consortium et al. “An integrated map of genetic variation from 1,092 human genomes”. In: Nature 491.7422 (2012), p. 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hugo Dalla-Torre et al. “The nucleotide transformer: Building and evaluating robust foundation models for human genomics”. In: bioRxiv (2023), pp. 2023–01. [DOI] [PubMed] [Google Scholar]
- [15].Devlin Jacob et al. “Bert: Pre-training of deep bidirectional transformers for language understanding”. In: arXiv preprint arXiv:1810.04805 (2018). [Google Scholar]
- [16].Durbin Richard et al. “Biological sequence analysis: Probabilistic models of proteins and nucleic acids”. In: (1998).
- [17].Dwork Cynthia et al. “Calibrating noise to sensitivity in private data analysis”. In: Theory of Cryptography: Third Theory of Cryptography Conference, TCC 2006, New York, NY, USA, March 4–7, 2006. Proceedings 3. Springer. 2006, pp. 265–284. [Google Scholar]
- [18].Eizenga Jordan M et al. “Pangenome graphs”. In: Annual review of genomics and human genetics 21 (2020), pp. 139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Elbashir Murtada K et al. “Lightweight convolutional neural network for breast cancer classification using RNA-seq gene expression data”. In: IEEE Access 7 (2019), pp. 185338–185348. [Google Scholar]
- [20].Frith Martin C. “How sequence alignment scores correspond to probability models”. In: Bioinformatics 36.2 (2020), pp. 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gibbs Richard A et al. “The international HapMap project”. In: (2003).
- [22].Guarracino Andrea et al. wfmash: whole-chromosome pairwise alignment using the hierarchical wavefront algorithm. Version 0.7.0. Sept. 2021. URL: https://github.com/ekg/wfmash. [Google Scholar]
- [23].Guevara Marco et al. “Large language models to identify social determinants of health in electronic health records”. In: npj Digital Medicine 7.1 (2024), p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gupta Anvita and Zou James. “Feedback GAN (FB-GAN) for DNA: A novel feedback-loop architecture for optimizing protein functions”. In: arXiv preprint arXiv:1804.01694 (2018). [Google Scholar]
- [25].Han Yi et al. “DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies”. In: Nucleic acids research 47.8 (2019), e45–e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hartvigsen Thomas et al. “Toxigen: A large-scale machine-generated dataset for adversarial and implicit hate speech detection”. In: arXiv preprint arXiv:2203.09509 (2022). [Google Scholar]
- [27].Hu Hongsheng et al. “Membership inference attacks on machine learning: A survey”. In: ACM Computing Surveys (CSUR) 54.11s (2022), pp. 1–37. [Google Scholar]
- [28].Jagadeesh Karthik A et al. “Deriving genomic diagnoses without revealing patient genomes”. In: Science 357.6352 (2017), pp. 692–695. [DOI] [PubMed] [Google Scholar]
- [29].Jha Somesh, Kruger Louis, and Shmatikov Vitaly. “Towards practical privacy for genomic computation”. In: 2008 IEEE Symposium on Security and Privacy (sp 2008). IEEE. 2008, pp. 216–230. [Google Scholar]
- [30].Ji Yanrong et al. “DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome”. In: Bioinformatics 37.15 (2021), pp. 2112–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jiang Albert Q et al. “Mistral 7B”. In: arXiv preprint arXiv:2310.06825 (2023). [Google Scholar]
- [32].Johnson Aaron and Shmatikov Vitaly. “Privacy-preserving data exploration in genome-wide association studies”. In: Proceedings of the 19th ACM SIGKDD international conference on Knowledge discovery and data mining. 2013, pp. 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kantarcioglu Murat et al. “A cryptographic approach to securely share and query genomic sequences”. In: IEEE Transactions on information technology in biomedicine 12.5 (2008), pp. 606–617. [DOI] [PubMed] [Google Scholar]
- [34].Kim Byung-Ju and Kim Sung-Hou. “Prediction of inherited genomic susceptibility to 20 common cancer types by a supervised machine-learning method”. In: Proceedings of the National Academy of Sciences 115.6 (2018), pp. 1322–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kolesnikov Alexey et al. “DeepTrio: variant calling in families using deep learning”. In: bioRxiv (2021), pp. 2021–04. [Google Scholar]
- [36].Kumar Varun, Choudhary Ashutosh, and Cho Eunah. “Data augmentation using pre-trained transformer models”. In: arXiv preprint arXiv:2003.02245 (2020). [Google Scholar]
- [37].Lanchantin Jack et al. “Deep motif dashboard: visualizing and understanding genomic sequences using deep neural networks”. In: Pacific symposium on biocomputing 2017. World Scientific. 2017, pp. 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lauter Kristin, López-Alt Adriana, and Naehrig Michael. “Private computation on encrypted genomic data”. In: International Conference on Cryptology and Information Security in Latin America. Springer. 2014, pp. 3–27. [Google Scholar]
- [39].Le Scao Teven et al. “Bloom: A 176b-parameter open-access multilingual language model”. In: (2023).
- [40].Liao Wen-Wei et al. “A draft human pangenome reference”. In: Nature 617.7960 (2023), pp. 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Luo Ruibang et al. “A multi-task convolutional deep neural network for variant calling in single molecule sequencing”. In: Nature communications 10.1 (2019), p. 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Luo Ruibang et al. “Exploring the limit of using a deep neural network on pileup data for germline variant calling”. In: Nature Machine Intelligence 2.4 (2020), pp. 220–227. [Google Scholar]
- [43].Lyu Boyu and Haque Anamul. “Deep learning based tumor type classification using gene expression data”. In: Proceedings of the 2018 ACM international conference on bioinformatics, computational biology, and health informatics. 2018, pp. 89–96. [Google Scholar]
- [44].Marco-Sola Santiago et al. “Fast gap-affine pairwise alignment using the wavefront algorithm”. In: Bioinformatics 37.4 (2021), pp. 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McMahan H Brendan et al. “Learning differentially private recurrent language models”. In: arXiv preprint arXiv:1710.06963 (2017). [Google Scholar]
- [46].Nguyen Eric et al. “Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution”. In: Advances in neural information processing systems 36 (2024). [Google Scholar]
- [47].O’Doherty Kieran C et al. “Toward better governance of human genomic data”. In: Nature genetics 53.1 (2021), pp. 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ohm Paul. “Broken promises of privacy: Responding to the surprising failure of anonymization”. In: UCLA l. Rev. 57 (2009), p. 1701. [Google Scholar]
- [49].OpenAI. GPT-4 Technical Report. 2023. arXiv: 2303.08774 [cs.CL].
- [50].Peng Cheng et al. “A Study of Generative Large Language Model for Medical Research and Healthcare”. In: arXiv preprint arXiv:2305.13523 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Poplin Ryan et al. “A universal SNP and small-indel variant caller using deep neural networks”. In: Nature biotechnology 36.10 (2018), pp. 983–987. [DOI] [PubMed] [Google Scholar]
- [52].Quang Daniel and Xie Xiaohui. “DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences”. In: Nucleic acids research 44.11 (2016), e107–e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Radford Alec et al. “Language Models are Unsupervised Multitask Learners”. In: (2019).
- [54].Raffel Colin et al. “Exploring the limits of transfer learning with a unified text-to-text transformer”. In: Journal of machine learning research 21.140 (2020), pp. 1–67.34305477 [Google Scholar]
- [55].Schiff Yair et al. “Caduceus: Bi-directional equivariant long-range dna sequence modeling”. In: arXiv preprint arXiv:2403.03234 (2024). [Google Scholar]
- [56].Shabani Mahsa and Marelli Luca. “Re-identifiability of genomic data and the GDPR: Assessing the re-identifiability of genomic data in light of the EU General Data Protection Regulation”. In: EMBO reports 20.6 (2019), e48316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shen Zhen et al. “A deep learning model for RNA-protein binding preference prediction based on hierarchical LSTM and attention network”. In: IEEE/ACM Transactions on Computational Biology and Bioinformatics 19.2 (2020), pp. 753–762. [DOI] [PubMed] [Google Scholar]
- [58].Shibata Yusuxke et al. “Byte pair encoding: A text compression scheme that accelerates pattern matching”. In: (1999).
- [59].Shokri Reza and Shmatikov Vitaly. “Privacy-preserving deep learning”. In: Proceedings of the 22nd ACM SIGSAC conference on computer and communications security. 2015, pp. 1310–1321. [Google Scholar]
- [60].Simmons Sean, Sahinalp Cenk, and Berger Bonnie. “Enabling privacy-preserving GWASs in heterogeneous human populations”. In: Cell systems 3.1 (2016), pp. 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sweeney Latanya, Abu Akua, and Winn Julia. “Identifying participants in the personal genome project by name (a re-identification experiment)”. In: arXiv preprint arXiv:1304.7605 (2013). [Google Scholar]
- [62].Team Gemini et al. “Gemini: a family of highly capable multimodal models”. In: arXiv preprint arXiv:2312.11805 (2023). [Google Scholar]
- [63].Thoppilan Romal et al. “Lamda: Language models for dialog applications”. In: arXiv preprint arXiv:2201.08239 (2022). [Google Scholar]
- [64].Touvron Hugo et al. “Llama 2: Open foundation and fine-tuned chat models”. In: arXiv preprint arXiv:2307.09288 (2023). [Google Scholar]
- [65].Touvron Hugo et al. “Llama: Open and efficient foundation language models”. In: arXiv preprint arXiv:2302.13971 (2023). [Google Scholar]
- [66].Tran Ngoc Hieu et al. “De novo peptide sequencing by deep learning”. In: Proceedings of the National Academy of Sciences 114.31 (2017), pp. 8247–8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ngoc Hieu Tran et al. “Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry”. In: Nature methods 16.1 (2019), pp. 63–66. [DOI] [PubMed] [Google Scholar]
- [68].Wan Nathan et al. “Machine learning enables detection of early-stage colorectal cancer by whole-genome sequencing of plasma cell-free DNA”. In: BMC cancer 19 (2019), pp. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang Shuang et al. “HEALER: homomorphic computation of ExAct Logistic rEgRession for secure rare disease variants analysis in GWAS”. In: Bioinformatics 32.2 (2016), pp. 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Weinstein John N et al. “The cancer genome atlas pan-cancer analysis project”. In: Nature genetics 45.10 (2013), pp. 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wjst Matthias. “Caught you: threats to confidentiality due to the public release of large-scale genetic data sets”. In: BMC medical ethics 11 (2010), pp. 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Xie Wei et al. “SecureMA: protecting participant privacy in genetic association meta-analysis”. In: Bioinformatics 30.23 (2014), pp. 3334–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yang Hao et al. “pNovo 3: precise de novo peptide sequencing using a learning-to-rank framework”. In: Bioinformatics 35.14 (2019), pp. i183–i190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yao Kai, Rochman Nash D, and Sun Sean X. “Cell type classification and unsupervised morphological phenotyping from low-resolution images using deep learning”. In: Scientific reports 9.1 (2019), p. 13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yelmen Burak et al. “Creating artificial human genomes using generative neural networks”. In: PLoS genetics 17.2 (2021), e1009303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Yoo Kang Min et al. “GPT3Mix: Leveraging large-scale language models for text augmentation”. In: arXiv preprint arXiv:2104.08826 (2021). [Google Scholar]
- [77].Yun Taedong et al. “Accurate, scalable cohort variant calls using DeepVariant and GLnexus”. In: Bioinformatics 36.24 (2020), pp. 5582–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zeng Haoyang et al. “Convolutional neural network architectures for predicting DNA–protein binding”. In: Bioinformatics 32.12 (2016), pp. i121–i127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zhang Susan et al. “Opt: Open pre-trained transformer language models”. In: arXiv preprint arXiv:2205.01068 (2022). [Google Scholar]
- [80].Zhang Xiang et al. “DeepGene: An Efficient Foundation Model for Genomics based on Pan-genome Graph Transformer”. In: bioRxiv (2024), pp. 2024–04. [Google Scholar]
- [81].Zhou Jian and Troyanskaya Olga G. “Predicting effects of noncoding variants with deep learning–based sequence model”. In: Nature methods 12.10 (2015), pp. 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhou Yingxue, Wu Zhiwei Steven, and Banerjee Arindam. “Bypassing the ambient dimension: Private sgd with gradient subspace identification”. In: arXiv preprint arXiv:2007.03813 (2020). [Google Scholar]
- [83].Zhou Zhihan et al. “Dnabert-2: Efficient foundation model and benchmark for multi-species genome”. In: arXiv preprint arXiv:2306.15006 (2023). [Google Scholar]