

Abstract
Recent evidence suggests an association between age‐related osteoporosis and cellular senescence in the bone; however, the specific bone cells that play a critical role in age‐related osteoporosis and the mechanism remain unknown. Results revealed that age‐related osteoporosis is characterized by the loss of osteoblast Men1. Osteoblast‐specific inducible knockout of Men1 caused structural changes in the mice bones, matching the phenotypes in patients with age‐related osteoporosis. Histomorphometrically, Men1‐knockout mice femurs decreased osteoblastic activity and increased osteoclastic activity, hallmarks of age‐related osteoporosis. Loss of Men1 induces cellular senescence via mTORC1 activation and AMPK suppression, rescued by metformin treatment. In bone morphogenetic protein‐indued bone model, loss of Men1 leads to accumulation of senescent cells and osteoporotic bone formation, which are ameliorated by metformin. Our results indicate that cellular senescence in osteoblasts plays a critical role in age‐related osteoporosis and that osteoblast‐specific inducible Men1‐knockout mice offer a promising model for developing therapeutics for age‐related osteoporosis.
Keywords: AMPK, cellular senescence, Men1, mTORC1, osteoporosis
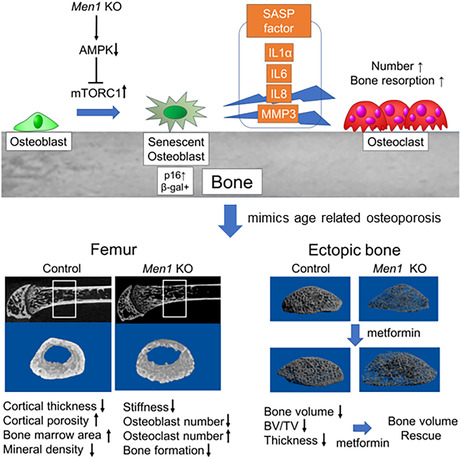
Osteoblast‐specific inducible Men1 knockout mimics age‐related osteoporosis even in young mice. Men1 deletion in osteoblasts promotes replicative stress‐induced cellular senescence presumably through mTORC1 activation and AMPK suppression, rescued by metformin treatment.
Abbreviations
- BFR/BS
bone formation rate per bone surface
- BMP
bone morphogenic protein
- BV/TV
bone volume/tissue volume
- MAR
mineral apposition rate
- MEN1
multiple endocrine neoplasia type 1
- N.Ob/BS
number of osteoblasts per bone surface
- N.Oc/BS
number of osteoclasts per bone surface
- Ob.S/BS
osteoblast surface per bone surface
- Oc.S/BS
osteoclast surface per bone surface
- OIS
oncogene‐induced senescence
- RANKL
receptor activator of NF‐κB ligand
- RT‐qPCR
reverse transcription‐quantitative polymerase chain reaction
- SASP
senescence‐associated secretory phenotype
- SA‐β Gal
senescence‐associated β‐galactosidase
- TAM
tamoxifen
- Tb.Th
trabecular thickness
- TGF‐β
transforming growth factor‐beta
- α‐MEM
alpha‐minimal essential medium
1. INTRODUCTION
Osteoporosis is a systemic skeletal disease characterized by low bone mass and the deterioration of bone microarchitecture, which increase the occurrence of fragility fractures (Aspray & Hill, 2019; Ukon et al., 2019). Among the various factors affecting osteoporosis, aging, menopause, and decreased activity, age‐related osteoporosis is becoming important because an increase in aging population, which started in high‐income countries is now occurring even in low‐ and middle‐income countries (Gregson et al., 2022).
Accumulating evidence highlights a close relationship between age‐related diseases and cellular senescence (Baker et al., 2016), which was discovered by Hayflick et al. (Hayflick & Moorhead, 1961) and is defined by an irreversible cell growth arrest regulated through the p16ink4a/RB and p53/p21CIP1 pathways (Muñoz‐Espín & Serrano, 2014). Senescent cells acquire a pro‐inflammatory phenotype, known as the senescence‐associated secretory phenotype (SASP), which leads to the disruption of tissue and organ functions and is considered to be a major cause of various chronic diseases (Regulski, 2017). Therefore, treatment strategies for chronic diseases targeting the removal of senescent cells (senolytics) or inhibition of the SASP (senomorphics) have recently come into the research spotlight (Kirkland & Tchkonia, 2020).
The accumulation of senescent cells with age has also been confirmed to occur in the bones, suggesting cellular senescence as a key regulator of age‐related osteoporosis (Farr et al., 2016); thus, it is rational to develop treatment strategies for osteoporosis that target senescent cells (Farr & Khosla, 2019). Indeed, inhibition of the negative effects of senescent cells in mice attenuated the rate of bone loss with age in mice (Farr et al., 2017). However, the precise mechanism by which bone cells become senescent during the natural aging process remains largely unknown. Understanding this mechanism will enable the development of more precise therapies for age‐related osteoporosis.
Men1 gene is originally known to be a tumor suppressor for multiple endocrine neoplasia type 1 (MEN1) syndrome (Thakker et al., 2012). We previously found Men1 deficiency in T cells induces cellular senescence‐associated immunodeficiency (Kuwahara et al., 2014; Suzuki et al., 2018) and focused on a possible role of Men1 gene in osteoblast senescence. Our current results indicate loss of Men1 is a critical driver for osteoblast senescence, and osteoblast‐specific inducible Men1 knockout mice could be the first animal model for age‐related osteoporosis.
2. RESULTS
2.1. Men1 mRNA levels are significantly reduced in the osteoporotic bones of aged mice
Recent studies suggest that cellular senescence is a key regulator of age‐related osteoporosis (Farr et al., 2016). We previously identified the tumor suppressor gene Men1 as a regulator of cellular senescence regulator in T cells (Kuwahara et al., 2014; Suzuki et al., 2018). These findings prompted us to examine whether Men1 also played a critical role in osteoporosis during aging. To test this hypothesis, we initially compared femurs of 2‐month‐old (young mice) and 24‐month‐old mice (aged mice) using microcomputed tomography (μCT). Evaluation of the femurs of young and aged mice via μCT showed a decrease in the bone volume (BV)/total volume (TV) and the cancellous and cortical bone thickness, bone mineral density (BMD), tissue mineral density (TMD), and an increase in the bone marrow area in 24‐month‐old mice compared with those in 2‐month‐old mice (Figure 1a). The bone phenotypes in 24‐month‐old mice resembled those of older humans with osteoporosis. Moreover, the mRNA levels of Men1 were reduced in 24‐month‐old mice spine bone (Figure 1b), accompanied by increased mRNA levels of cellular senescence genes (p16, p21, p53, Il1α, Il8, and MMP3) compared with those of 2‐month‐old mice spine bone (Figure S1). For the purpose of overall aging assessment, spine bone were used in Figure 1b; Figure S1 as previously reported (Farr et al., 2016). These results suggest that the loss of Men1 is closely involved in the pathology of age‐related osteoporosis.
FIGURE 1.
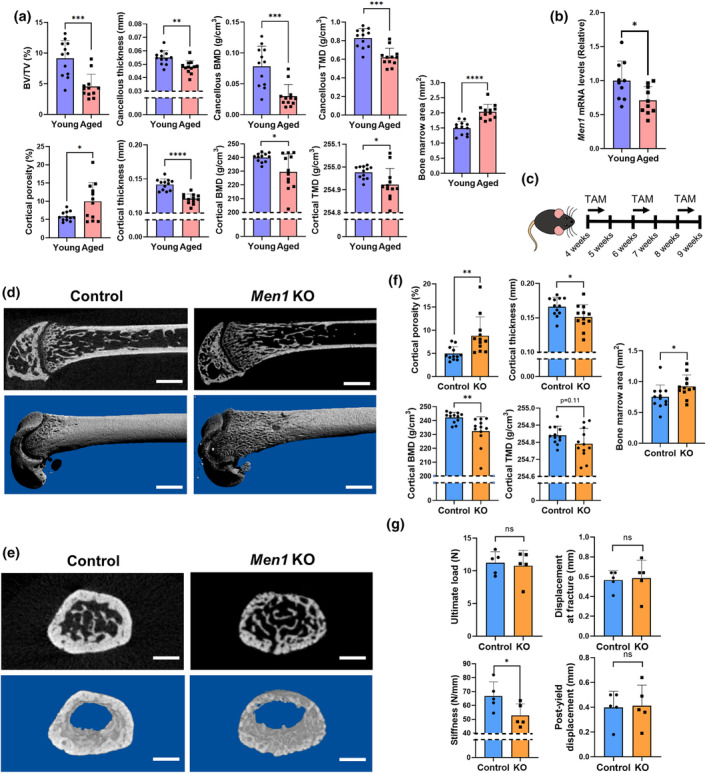
Structural analysis of bones in the trunk. (a) Micro‐computed tomography (μCT) analysis of the femur of wild‐type 2‐month‐old (young, n = 12) and 24‐month‐old (aged, n = 12) mice, including bone volume (BV)/tissue volume (TV), cancellous thickness, cancellous bone mineral density (BMD), cancellous tissue mineral density (TMD), cortical porosity, cortical thickness, cortical BMD, cortical TMD, and bone marrow area. (b) Relative Men1 mRNA levels to young mice in the spine bone. Young mice (n = 10), aged mice (n = 10). (c) Experimental design for deleting the Men1 gene of osteoblasts. Men1 flox/flox mice (Control) and Men1 flox/flox; Col1a1‐cre/ERT2 mice (Men1 KO) received tamoxifen (TAM) treatment at 10 mg/kg/day for 4 days at 4, 6, and 8 weeks of age. Mice were euthanized and analyzed at 9 weeks of age. (d) Representative μCT sagittal images of the Control and Men1 KO femur from 9‐week‐old mice. Upper: two‐dimensional image; lower: three‐dimensional reconstructed image. Scale bars, 1 mm. (e) Representative μCT axial images of the Control and Men1 KO femur from 9‐week‐old‐ mice at 1.5 mm proximal to the distal growth plate. Upper: two‐dimensional image; lower: three‐dimensional cortical bone image. Scale bars, 1 mm. (f) Micro‐CT analysis of the femur of 9‐week‐old Control (n = 12) and Men1 KO (n = 12) mice, including cortical porosity, cortical thickness, cortical BMD, cortical TMD, and bone marrow area. (g) Result of biomechanical testing in 9‐week‐old Control (n = 5) and Men1 KO (n = 5) mice, including ultimate load, displacement at fracture, stiffness, and post‐yield displacement. Data represent mean ± SD (error bars). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by two‐tailed Student's t‐test (unpaired). ns, not statistically significant.
2.2. Men1 deletion in osteoblasts induces osteoporotic changes on cortical bone even in young mice
To further identify the role of Men1 on age‐related osteoporosis, we generated Men1 flox/flox; Col1a1‐cre/ERT2 mice that can delete osteoblast Men1 by tamoxifen (TAM) treatment. We used inducible osteoblast‐specific mice because cellular senescence is deeply involved during the embryonic period (Muñoz‐Espín et al., 2013), thus developmental Men1 effect cannot be eliminated by permanent osteoblast‐specific Men1 KO mice. Femur bone of 9‐week‐old Men1 flox/flox; Col1a1‐Cre/ERT2 mice treated with TAM (4 days/week starting at 4, 6, and 8 weeks of age; Figure 1c) were evaluated by μCT. The μCT images clearly demonstrated thinning of the cortical bone in the distal femur of TAM treated Men1 flox/flox; Col1a1‐cre/ERT2 (Men1 KO) mice compared to Men1 flox/flox (Control) mice (Figure 1d,e). Microstructural analyses showed that the cortical bone in the Men1 KO mice was thinner, more porotic, and had lower mineral density, with a wider bone marrow area than that in Control mice (Figure 1f). These results suggest that Men1 deletion in osteoblasts predominantly affects the osteoporotic phenotype in the cortical bone. Since the cortical bone microstructure has a great influence on bone strength (Bala et al., 2015), we investigated the bone strength of Men1 KO mice by a mechanical testing. The ultimate load, displacement at fracture, and post‐yield displacement were comparable between Control and MenEN1 KO mice (Figure 1g); however, the femoral stiffness was decreased in Men1 KO mice (Figure 1g). Thus, femurs from Men1 KO mice resembled the fragile bones of older humans in terms of both bone structure and mechanical strength.
2.3. Men1 deletion induces osteoblast senescence, attenuates osteoblastic activity, and increases osteoclastic activity in vivo
After identifying the structural phenotype, we next investigated the detailed actions of Men1 deficiency histologically. Cortical thinning due to Men1 deletion was observed, similar to the μCT findings (Figure 2a). Furthermore, Men1 KO mice exhibited an increase in the number of p16‐positive osteoblasts (Figure 2a), which was consistent with the area where Cre/loxP recombination was confirmed (Figure S3a). These results suggest that Men1 deficiency in vivo leads to osteoblast senescence. To elucidate the effect of Men1 deletion in osteoblasts on bone formation, bone histomorphometry analysis was performed using the femurs of Control and Men1 KO mice. The spaces between the double‐stained lines were narrow (Figure 2b), which matched the decrease in osteogenic ability (as assessed by the mineral apposition rate [MAR] and bone formation rate per bone surface ratio [BFR/BS]) in Men1 KO mice (Figure 2c). Men1 KO mice exhibited decreased bone area and thickness, reflecting their low osteogenic ability (Figure 2d), along with a decrease in the number of osteoblasts and the area of the osteoblast surface (Figure 2e). The osteoblast‐related bone formation indices were all generally decreased in Men1 KO mice, matching the general findings in aged humans. Under normal conditions, the number of osteoclasts generally parallels the number of osteoblasts, regulated by cross‐talk mediated through the receptor activator of NF‐κB ligand (RANKL) and RANK (Yasuda, 2021). Interestingly, histomorphometry analysis of bone resorption parameters showed an increase in the number of osteoclasts and the area of the osteoclast surface in Men1 KO mice (Figure 2f), although the number of osteoblasts decreased. Immunostaining for RANKL/OPG revealed extensive RANKL upregulation in Men1 KO, while OPG did not differ compared to those in Control mice (Figure 2g). This suggests that the mechanism of osteoclast increase in Men1 KO mice may involve an elevated RANKL/OPG ratio. Thus, Men1 deficiency widened the gap between bone formation and resorption, a characteristic of age‐related osteoporosis (Ukon et al., 2019).
FIGURE 2.
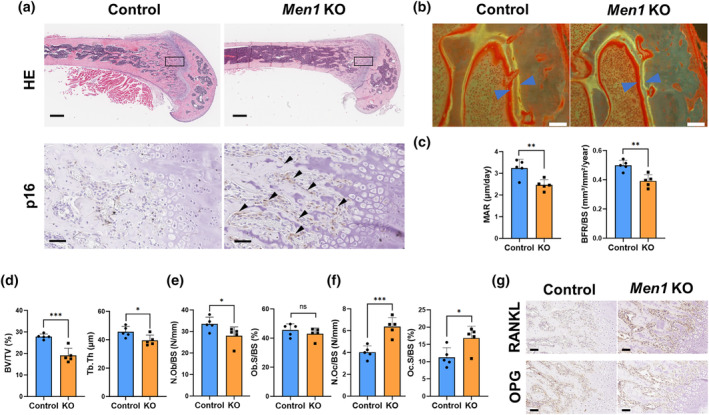
Histological characteristics of Men1‐deficient bones. (a) Representative histological images of the femur bone from 9‐week‐old Men1 flox/flox (Control) mice and Men1 flox/flox; Col1a1‐cre/ERT2 (Men1 KO) mice. Hematoxylin and eosin staining (HE) and p16 immunostaining. Lower panels are enlarged images of the rectangular area of the upper image. Arrows indicate p16‐positive cells. Scale bars, 500 μm (upper) and 50 μm (lower). (b) Representative fluorescent images of the femur from 9‐week‐old Control and Men1 KO mice. Scale bars, 50 μm. Arrows indicate the space between the double‐stained lines. (c) Histomorphometric parameters of bone formation in 9‐week‐old Control (n = 5) and Men1 KO (n = 5) mice, including mineral apposition rate (MAR) and bone formation rate (BFR)/bone surface (BS). (d) Histomorphometric parameters of bone structure in 9‐week‐old Control (n = 5) and Men1 KO (n = 5) mice, including bone volume (BV)/tissue volume (TV) ratio and trabecular thickness (Tb.Th). (e) Histomorphometric parameters of osteoblasts in 9‐week‐old Control (n = 5) and Men1 KO (n = 5) mice, including number of osteoblasts (N.Ob)/BS and osteoblast surface (Ob.S)/BS. (f) Histomorphometric parameters of osteoclasts in 9‐week‐old Control (n = 5) and Men1 KO (n = 5) mice, including number of osteoclasts (N.Oc)/BS and osteoclast surface (Oc.S)/BS. (g) Representative RANKL, OPG immunostaining images of the femur bone from 9‐week‐old Control and Men1 KO mice. Scale bars, 50 μm. Data represent mean ± SD (error bars). *p < 0.05, **p < 0.01, ***p < 0.001 by two‐tailed Student's t‐test (unpaired). ns, not statistically significant.
2.4. Men1 deletion in osteoblasts promotes replicative stress‐induced cellular senescence through presumably mTORC1
The results summarized above demonstrate that Men1 deletion in osteoblasts resulted in structural changes in the cortical bone (Figure 1f) and induced p16 expression in the metaphysis (Figure 2a). However, the mechanistic link between Men1 deletion in osteoblasts and age‐related osteoporosis remained unclear. Since Men1 was previously shown to act as a cellular senescence regulator in T cells (Kuwahara et al., 2014; Suzuki et al., 2018), we next evaluated whether Men1 deficiency directly causes cellular senescence using in vitro adenoviral‐mediated Men1 KO in primary osteoblasts from Men1 flox/flox mice. Osteoblasts were infected with an adenovirus expressing green fluorescent protein (Ad‐GFP; Control) or Cre (Ad‐Cre‐GFP; Men1 KO) and cellular senescence was induced by replicative stress, as described in the Materials and Methods section. After multiple passages, the positivity of senescence‐associated β‐galactosidase (SA‐β Gal) staining was significantly increased in Men1 KO osteoblasts compared with that in the control osteoblasts (Figure 3a,b). To further verify the characteristics of each osteoblast type, the mRNA levels of cellular senescence‐associated genes and Men1 were investigated. Cre/loxP recombination was confirmed by Men1 reduction (Figure 3c). The induction of cellular senescence induced by replicative stress was confirmed by higher mRNA levels of p16 (Figure 3d) in both Control and Men1 KO osteoblasts. Even under the same condition of replicative stress, the mRNA levels of SASP genes (Il1α, IL6, IL8, and MMP3) were increased in Men1 KO compared to those in Control osteoblasts (Figure 3e–h). Since SASP levels are elevated during the late stages of cellular senescence (Muñoz‐Espín & Serrano, 2014), these results suggest that the loss of Men1 in osteoblasts accelerates cellular senescence induced by replicative stress. To elucidate the mechanism underlying replicative stress‐induced senescence in osteoblasts with Men1 deletion, we investigated the potential involvement of the mTORC1 pathway, one of the major signaling pathways for cellular senescence (Weichhart, 2018). Phosphorylation of S6K and 4EBP1, which are downstream effectors of mTORC1, was enhanced by Men1 deletion, and metformin treatment suppressed activation of the mTORC1 pathway caused by Men1 deficiency (Figure 3i). The quantification data also showed the same mTORC1 changes (Figure S2a). We performed additional immunoblotting and its quantification to understand the mechanism by which mTORC1 was suppressed by metformin. The findings showed that phosphorylation of AMPKα, which suppresses mTORC1 (Jeon, 2016), was significantly reduced by Men1 deficiency and restored by metformin treatment (Figure 3j; Figure S2b). However, no clear involvement of Akt, ERK1/2, and GSK3β was observed (Figures S2c,d). These findings suggest that replicative stress‐induced senescence in osteoblasts with Men1 deletion is closely associated with mTORC1 pathway activation, which can be restored through AMPK activation by metformin treatment.
FIGURE 3.
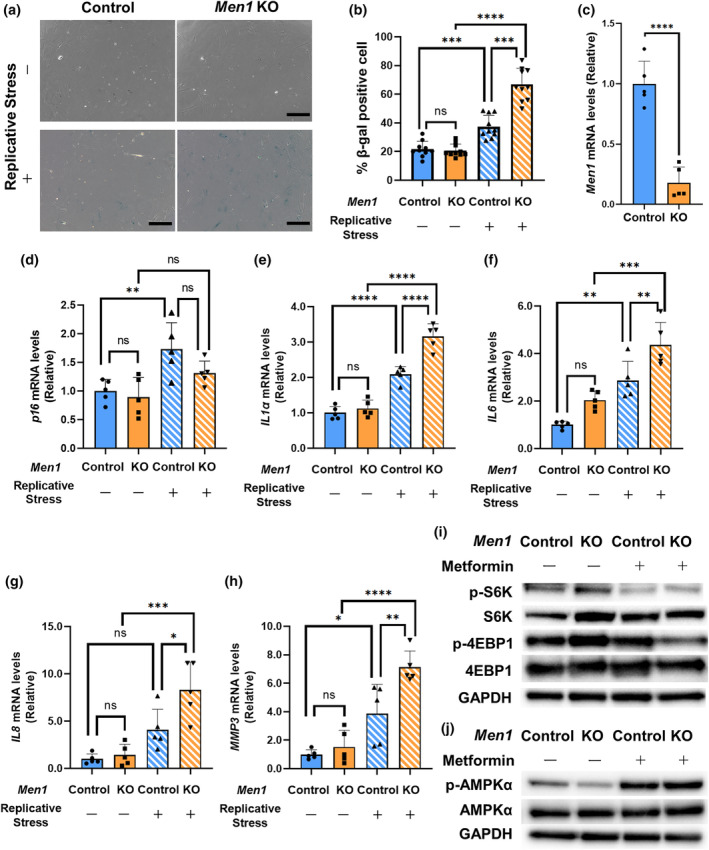
Osteoblast senescence induced by Men1 deficiency in vitro. (a) Senescence‐associated galactosidase (SA β‐Gal) staining of osteoblasts from the Men1 flox/flox murine mice calvaria. Osteoblasts were infected with eGFP adenovirus (Ad‐GFP, Control) and Cre recombinase adenovirus (Ad‐Cre‐GFP, Men1 KO). Passage (P) 3 (replicative stress: −) and P6 (Replicative stress: +) cells were used for the experiments. Scale bars, 250 μm. (b) Quantitative evaluation of β‐Gal‐positive cells in Control (n = 10 fields) and Men1 KO (n = 10 fields) mice. (c) Relative mRNA levels of Men1 to Control osteoblasts (Replicative stress: −). Control osteoblasts (Replicative stress: −), n = 5; Men1 KO osteoblasts (Replicative stress: −), n = 5. (d–h) Relative mRNA levels of p16, IL1α, IL6, IL8, and MMP3 to Control osteoblasts (Replicative stress: −). Control osteoblasts (Replicative stress: −) (n = 5), Men1 KO osteoblasts (Replicative stress: −) (n = 5), Control osteoblasts (Replicative stress: +) (n = 5), and Men1 KO osteoblasts (Replicative stress: +) (n = 5). (i) Immunoblotting to evaluate the expression of factors downstream of mTORC1. (j) Immunoblotting of AMPKα in control osteoblasts (Replicative stress: −), Men1 KO osteoblasts (Replicative stress: −), Control osteoblasts (Replicative stress: +), and Men1 KO osteoblasts (Replicative stress: +). Data represent mean ± SD (error bars). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by two‐tailed Student's t‐test (unpaired) (c), one‐way ANOVA followed by Sidak's test (b, d–h). ns, not statistically significant.
2.5. Cellular senescence and a decrease in Men1 are closely involved in bone formation during natural aging
Femoral analysis of Men1 KO mice represented signs of cellular senescence, and osteoblast senescence presumably via mTORC1 induced by Men1 deficiency was confirmed in vitro. To further clarify the relevance of cellular senescence in vivo due to Men1 deficiency, we established a model of accelerated cellular senescence in bone tissue. Bone morphogenic protein (BMP) is known to activate mTORC1 and regulate cellular senescence (Kaneda et al., 2011; Karner et al., 2017). A previous report showed that the transplantation of a BMP‐containing collagen sponge allowed us to observe the bone formation capacity (Hashimoto et al., 2020). We hypothesized that the effects of osteoblast senescence would be enhanced by conditions of BMP‐induced bone formation. Experiments were conducted using BMP‐containing collagen sponges (Figure 4a). BMP‐induced ectopic bones in 24‐month‐old mice were significantly thinner and smaller than those in 2‐month‐old mice (Figure 4b,c), as observed during human aging (Goodnough & Goodman, 2022). We further investigated cellular senescence in the ectopic bone using histological evaluation and reverse transcription‐quantitative polymerase chain reaction (RT‐qPCR). Immunostaining revealed that the number of p16‐positive cells was significantly increased on the surface of the ectopic bone from aged mice (Figure 4d,e). The areas positively stained for β‐Gal were also increased in the aged mice (Figure 4f). In addition, an increase in p‐4EBP1 and p‐S6 was observed in aged mice, suggesting activation of mTORC1 (Figure 4g). The mRNA levels of p16 and SASP factors increased in the ectopic bones of aged mice (Figure 4h). Additionally, similar to the results from the vertebral bones (Figure 1b; Figure S1), the mRNA levels of Men1 decreased in the ectopic bones of aged mice (Figure 4i). Thus, the BMP‐induced ectopic bone model suggests that cellular senescence and a decrease in Men1 mRNA levels are closely associated with low bone formation activity in aging individuals.
FIGURE 4.
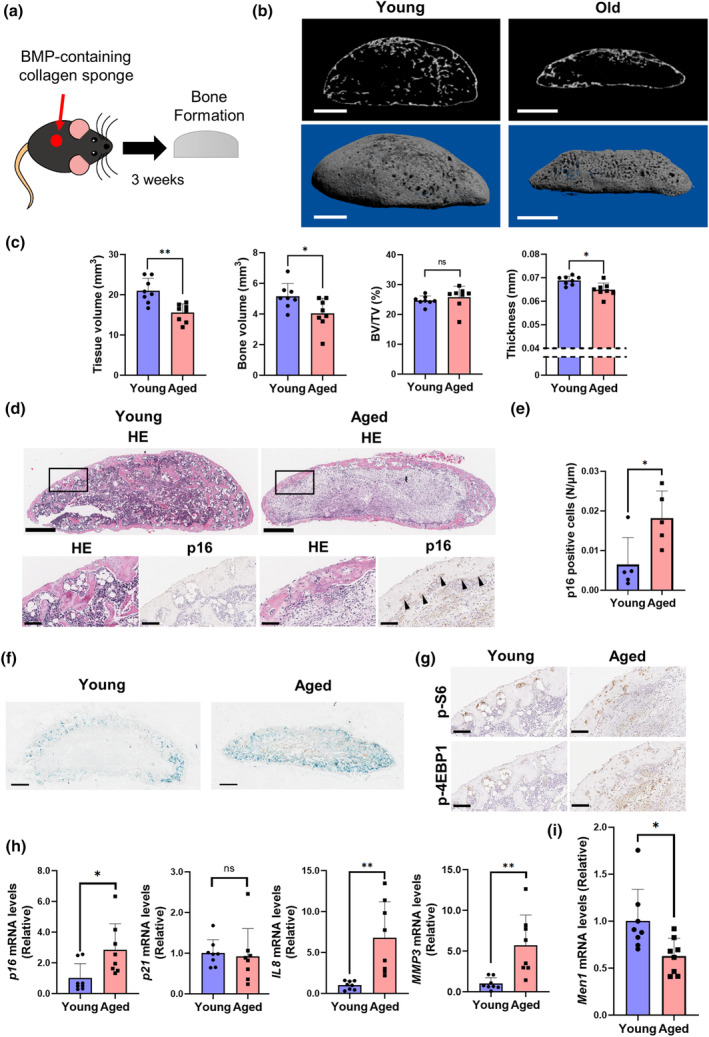
Senescent bone formation in natural aging. (a) Schema of establishment of the ectopic bone formation model. A BMP‐containing collagen sponge was set underneath the fascia of 2‐month‐old (young) and 24‐month‐old mice (aged) mice. After 3 weeks, the formed ectopic bone was evaluated. (b) Representative μCT sagittal images of the ectopic bone. Scale bars, 1 mm. (c) Micro‐CT analysis of the ectopic bone in young (n = 8) and aged (n = 8) mice, including tissue volume (TV), bone volume (BV), BV/TV, and thickness. (d) Representative histological images of the ectopic bone in young and aged mice. Hematoxylin and eosin staining (HE) and p16 immunostaining. Lower panels are enlarged images of the rectangular area of upper panels. Arrows indicate p16‐positive cells. Scale bars, 500 μm (upper) and 100 μm (lower). (e) Quantitative evaluation of p16‐positive cells in young (n = 5) and aged (n = 5) mice. (f) Representative beta‐galactosidase staining images of the ectopic bone in young and aged mice. Scale bars, 500 μm. (g) Representative histological images of the ectopic bone in young and aged mice. p‐S6 and p‐4EBP1 immunostaining. Scale bars, 100 μm. (h) Relative mRNA levels of p16, p21, IL1α, IL8, and MMP3 to young mice. Young mice (n = 8) and aged mice (n = 8). (i) Relative Men1 mRNA levels to young mice. Young mice (n = 8) and aged mice (n = 8). *p < 0.05, **p < 0.01 by two‐tailed Student's t‐test (unpaired). Data represent mean ± SD (error bars). ns, not statistically significant.
2.6. Metformin suppresses cellular senescence in ectopic bones from Men1 KO mice
Since metformin suppressed the mTORC1 pathway in Men1 KO osteoblasts (Figure 3i), we sought to determine whether metformin could also reverse the cellular senescence due to the loss of Men1. Using the BMP‐induced ectopic bone model (Figure 5a), we found that Men1 deletion in osteoblasts resulted in an increase in the number of p16‐positive cells and the β‐Gal‐positive areas (Figure 5b), which were markedly reduced after the administration of metformin (Figure 5c,d). Next, we performed immunostaining of p‐S6, p‐4EBP1, and p‐AMPKα to examine the involvement of mTORC1 activation. Without metformin treatment, Men1 KO showed increased p‐S6 and p‐4EBP1 and decreased p‐AMPKα compared to Control mice (Figure 5e). However, metformin treatment decreased p‐S6 and p‐4EBP1 and increased p‐AMPKα in both Control and Men1 KO mice compared to those observed in respective metformin untreated mice (Figure 5f). Consistent with in vitro findings, these findings suggest that metformin suppressed mTORC1 activation through AMPK due to Men1 deficiency in vivo. RT‐qPCR on BMP‐induced ectopic bones confirmed that the mRNA levels of Men1 were decreased in Men1 KO mice (Figure 5g). No significant difference was observed in the mRNA levels of Runx2, Sp7, and Alpl, which are downstream of SMADs (Figure S4a). Similar to the results in the femur, Rankl increased in Men1 KO while Opg showed no difference (Figure S4b). Consistent with the histological results, the mRNA levels of the SASP genes p16, IL1α, IL8, and MMP3 were significantly increased in the ectopic bones from Men1 KO mice, and this increase was suppressed following metformin administration (Figure 5h–k). These results indicate that Men1 deficiency promotes cellular senescence, which could be reversed by metformin treatment.
FIGURE 5.
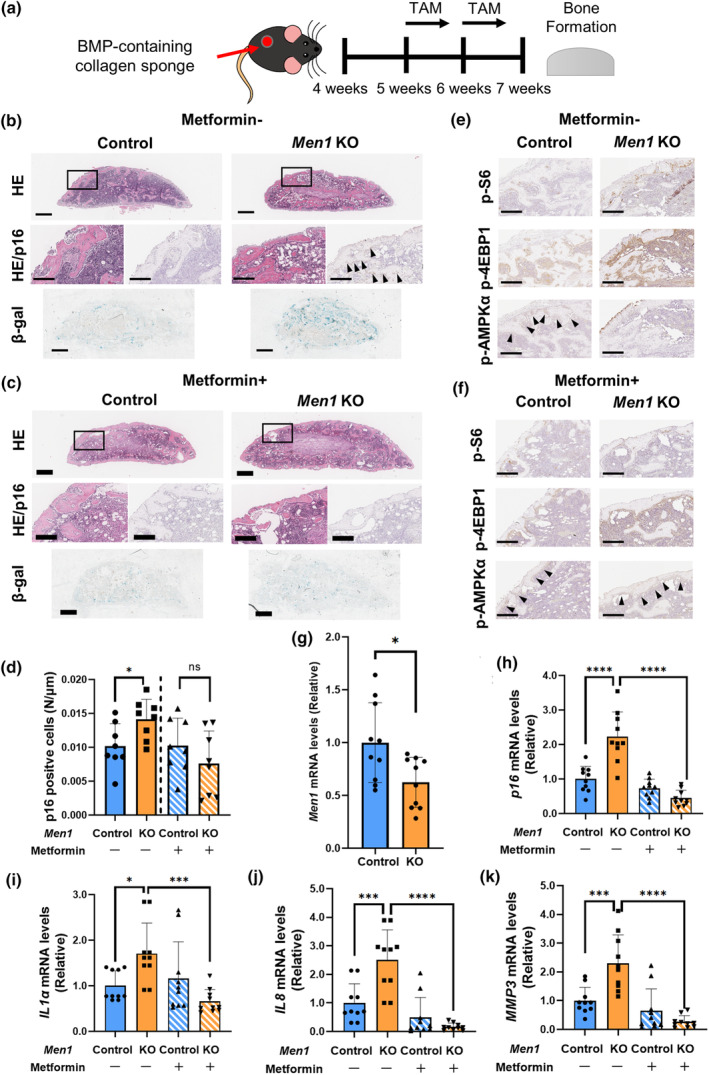
Characteristics of Men1 knockout bone formation. (a) Schema of the ectopic bone formation model of Men1 flox/flox (Control) and Men1 flox/flox; Col1a1‐cre/ERT2 (Men1 KO) mice. (b) Representative histological images of Control and Men1 KO ectopic bone without metformin administration from 7‐week‐old mice. Hematoxylin and eosin staining (HE), p16 immunostaining, and beta‐galactosidase (β‐Gal) staining. Second (HE, p16) images are enlarged images of the rectangular area of the first (HE) images. Arrows indicate p16‐positive cells. Scale bars, 500 μm (first, third images) and 200 μm (second images). (c) Representative histological images of Control and Men1 KO ectopic bone with metformin administration from 7‐week‐old mice. HE, p16 immunostaining, and β‐Gal staining. Second (HE, p16) images are enlarged images of the rectangular area of the first (HE) images. Scale bars, 500 μm (first, third images) and 200 μm (second images). (d) Quantitative evaluation of p16‐positive cells relative to Control bone (metformin: −) (n = 8) in Control bone (metformin: −) (n = 8), Men1 KO bone (metformin: −) (n = 8), Control bone (metformin: +) (n = 8), and Men1 KO bone (metformin: +) (n = 8) from 7‐week‐old mice. (e) Representative immunostaining images for p‐S6, p‐4EBP1, and p‐AMPKα of Control and Men1 KO ectopic bone without metformin administration from 7‐week‐old mice. Arrows indicate p‐AMPKα positive cells. Scale bars, 200 μm. (f) Representative immunostaining images for p‐S6, p‐4EBP1, and p‐AMPKα of Control and Men1 KO ectopic bone with metformin administration from 7‐week‐old mice. Arrows indicate p‐AMPKα positive cells. Scale bars, 200 μm. (g) Relative mRNA levels of Men1 to Control bone (metformin: −) in Control bone (metformin: −) (n = 10), Men1 KO bone (metformin: −) (n = 10) from 7‐week‐old mice. (h–k) Relative mRNA levels of p16, IL1α, IL8, and MMP3 to Control bone (metformin: −) in Control bone (metformin: −) (n = 10), Men1 KO bone (metformin: −) (n = 10), Control bone (metformin: +) (n = 10), and Men1 KO bone (metformin: +) (n = 10) from 7‐week‐old mice. Data represent mean ± SD (error bars). *p < 0.05, ***p < 0.001, ****p < 0.0001 by two‐tailed Student's t‐test (unpaired) (d, g), one‐way ANOVA followed by Sidak's test (h–k). ns, not statistically significant.
2.7. Metformin partially restores the bone morphometry of BMP‐induced ectopic bone in Men1 KO mice
Both in vitro and in vivo, Men1 deletion induced cellular senescence in osteoblasts, leading to reduced osteogenesis. Since cellular senescence in Men1 KO mice was restored by metformin treatment, we examined whether metformin could also reverse the reduction in osteogenesis in Men1 KO mice. The μCT images and three‐dimensional reconstruction showed bone thinning and a decrease in BV, BV/TV in the ectopic bone of the Men1 KO mice (Figure 6a,c). Notably, metformin administration significantly increased the volume, but not the thickness, of the ectopic bones from Men1 KO mice (Figure 6b,c). Taken together, our current results suggest that metformin partially restores osteogenesis reduced by the loss of Men1, as a hallmark of osteoblast aging.
FIGURE 6.
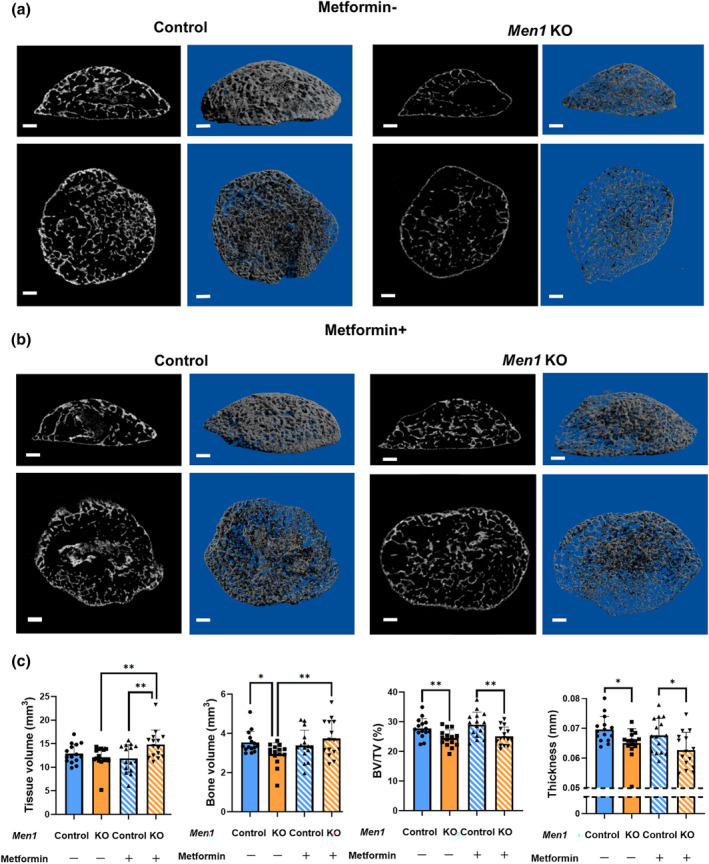
Miro‐computed tomography (μCT) analysis of ectopic bone with osteoblast senescence. (a) Representative μCT images of the ectopic bone of Men1 flox/flox (Control) and Men1 flox/flox; Col1a1‐cre/ERT2 (Men1 KO) mice without metformin administration from 7‐week‐old mice. Sagittal (upper) and coronal (lower) images. Two‐dimensional images (left) and three‐dimensional reconstructed images (right). Scale bars, 500 μm. (b) Representative μCT images of the ectopic bone of Control and Men1 KO mice with metformin administration from 7‐week‐old mice. Sagittal (upper) and coronal (lower) images. Two‐dimensional images (left) and three‐dimensional reconstructed images (right). Scale bars, 500 μm. (c) Micro‐CT analysis of the ectopic bone in Control (metformin: −) (n = 15), Men1 KO (metformin: −) (n = 15), Control bone (metformin: +) (n = 15), and Men1 KO (metformin: +) (n = 15) mice from 7‐week‐old mice, including tissue volume (TV), bone volume (BV), BV/TV, and thickness. Data represent mean ± SD (error bars). *p < 0.05, **p < 0.01 by one‐way ANOVA followed by Fisher's LDS test.
3. DISCUSSION
We found that osteoblast‐specific Men1 KO resulted in significant porosity, thinning of the cortical bone, decreased cortical mineral density, widening of the bone marrow cavity, and decreased mechanical strength. In addition, the bones of Men1‐deficient mice exhibited a decrease in osteoblast number and an increase in osteoclast number, resulting in a low BV and bone thinning. In vitro assays revealed that Men1 deletion activated cellular senescence, and mTORC1 in osteoblasts through suppression of AMPK activity, which was rescued by metformin. Metformin suppressed the activation of mTORC1 through AMPK due to Men1 deficiency and restored the low BV due to senescent osteoblasts in Men1‐deficient mice.
The present study thus revealed that loss of Men1 is a hallmark of cellular senescence‐associated bone aging. As senescent cells accumulate in aged individuals, bone aging is clinically associated with osteoporosis (Farr et al., 2017). Osteoporosis is caused by aging, menopause, and lack of activity in daily life (Ukon et al., 2019). Several mouse models have been established for osteoporosis caused by menopause (Farr et al., 2019) or inactivity in daily life (Colaianni et al., 2017), whereas age‐related osteoporosis has generally been investigated using naturally or genetically aged mice (Silva et al., 2005). Since aged mice have also undergone menopause and exhibit reduced activity, aged mice do not represent a suitable model for specifically examining age‐related osteoporosis. We identified the loss of Men1 as a single factor in the pathogenesis of age‐related osteoporosis in terms of cellular senescence. Therefore, this animal model can be used to obtain further insights into bone aging by cellular senescence through the measurement of the degree of age‐related osteoporosis and for the discovery of new drugs.
This study characterized osteoblast senescence both in vitro and in vivo. Senescent osteoblasts exhibited an increase in the mRNA levels of SASP factors, including IL1α, IL6, IL8, and MMP3, which were associated with an increase in osteoclast numbers. Consistent with our results, senescent cells around the bone tissue secrete SASP and lead to an increase in osteoclast numbers (Farr et al., 2017). Senescent osteoblasts can result in the fragility of cortical bones in the trunk. During bone formation, osteoblast senescence can lead to a decrease in BV and thickness of new bones, and metformin can restore the decreased BV. Osteoporosis is characterized by the deterioration of cortical and trabecular microstructures, bone fragility, and decreased osteogenic capacity (Ukon et al., 2019). Therefore, our model of osteoblast senescence‐associated bone aging in mice recapitulates some of these characteristics in the elderly human population. A previous report demonstrated that the removal of all senescent cells, including senescent osteoblasts, could alleviate osteoporotic phenotypes such as those pertaining to bone thickness, number of osteoclasts, number of osteoblasts, and MAR (Farr et al., 2017), which were the same factors found to be exacerbated by osteoblast senescence‐associated bone aging in the present study. Although various senolytic drugs are expected to be developed in the future, the pathophysiology and phenotypes revealed in this study can serve as biomarkers for evaluating the efficacy of drugs targeting cellular senescence‐associated bone aging.
We focused on Men1 as a regulator of osteoblast senescence. Permanent Men1 deficiency in bone from embryonic period reduces bone mass (Kanazawa et al., 2015; Liu et al., 2017). However, the relationship between Men1 and cellular senescence in adults could not be evaluated because cellular senescence is deeply involved during the embryonic period, thus developmental Men1 effect cannot be eliminated (Muñoz‐Espín et al., 2013). In this study, by using inducible conditional knockout mice, Men1 deficiency in osteoblasts caused cellular senescence, which was associated with mTORC1 activation and induced bone aging. Consistent with our current results on the role of Men1 in osteoblasts, several reports have shown that Men1 deficiency in other cell types causes age‐related phenotypes such as impaired lymphocyte function and the development of dementia (Kuwahara et al., 2014; Leng et al., 2018; Suzuki et al., 2018). Loss of Men1 in T cells promotes cellular senescence through mTORC1 activation (Suzuki et al., 2018). MEN1, a tumor suppressor gene, causes MEN1, an autosomal dominant disorder and hereditary tumor syndrome (Thakker et al., 2012). Patients with MEN1 develop osteoporosis early in life, regardless of tumor development (Marini et al., 2022) and tend to develop benign tumors, in contrast to patients with other hereditary tumor syndromes, who tend to develop malignant tumors (Thakker et al., 2012). Indeed, permanent Men1 deficiency in early osteoblasts develop ossifying fibroma, a benign bone tumor in mice (Lee et al., 2018). Senescent cells are identified at high frequencies in benign tumors, whereas they are rarely detected in malignant tumors (Childs et al., 2014; Collado & Serrano, 2010). These clinical features of MEN1 support our results on cellular senescence in Men1‐deficient mice in vitro and in vivo. In a process known as oncogene‐induced senescence (OIS), cells become senescent in response to the activation of oncogenes, which promotes cancer development (Ou et al., 2021). The concept of OIS, induced by oncogenes, resembles that of cellular senescence by Men1 gene deficiency. Although the overexpression of oncogenes immediately induces cellular senescence in vitro (Liu et al., 2018) and forms malignant tumors in vivo (Won & Choi, 2022), the loss of Men1 appears to gradually accelerate cellular senescence, presumably through mTORC1 activation. Thus, an aging animal model with loss of Men1 is consistent with the slow progression of natural aging. Our results suggest that Men1 is deeply involved in cellular senescence‐associated aging and provide insights into new disease concepts.
This study had several limitations. First, the mechanism by which Men1 deficiency leads to the bone‐aging phenotype has not yet been clearly elucidated. Men1 is reported to be epigenetically involved in cell cycle progression, apoptosis, and the DNA damage response, which are important factors for cellular senescence (Kaji, 2012). Moreover, the BMP and transforming growth factor‐beta (TGF‐β) pathways, acting as major contributors to bone metabolism, have been associated with Men1 (Kaji, 2012). However, the effect of Men1 on these pathways depends on the cell type and does not necessarily facilitate bone formation (Troka et al., 2022). Because the BMP and TGF‐β pathways contribute to cellular senescence (Hayashi et al., 2016; Wei & Ji, 2018), loss of Men1 may alter BMP or TGF/β signaling to induce cellular senescence. Furthermore, a direct involvement of Men1 deficiency in age‐related osteoporosis has not been demonstrated. A previous study using mice with permanent Men1 deficiency has demonstrated the absence of significant differences in bone‐related parameters in older age beyond 1 year (Liu et al., 2017), suggesting that inducible Men1 deficiency, even in aged mice, does not exacerbate osteoporosis. In this study, expression of Men1 was observed to be reduced with aging, suggesting the model used mimics age‐related osteoporosis. Second, assessing the degree of cellular senescence in the femur is challenging owing to the limited number of senescent osteoblasts. To solve this problem, we used a BMP‐induced bone formation model for a detailed assessment of cellular senescence in vivo, which allowed us to assess the degree of cellular senescence, as BMP accelerates mTORC1 and regulates cellular senescence (Kaneda et al., 2011; Karner et al., 2017). Third, although 6‐month‐old mice are typically used for senescence research, in this study, we used 2‐month‐old mice as young mice in accordance with the experiments on TAM‐induced Men1 KO mice. Moreover, we selected 2‐month‐old mice based on the need for high knock‐out efficiency in inducible mice, following the protocol from a previous report (Zhong et al., 2015). However, the aging‐related results obtained in this study (Figure 1a, Figure S1) were similar to those of previous studies using 6‐month‐old mice as controls (Farr et al., 2016; Shim et al., 2022).
Nevertheless, in conclusion, we demonstrated that the loss of Men1 is a critical driver of osteoblast senescence and established the first animal model of age‐related osteoporosis by osteoblast‐specific inducible Men1 KO. This model could facilitate the study of the novel concept of cellular senescence‐associated osteoporosis in the context of clinical practice.
4. MATERIALS AND METHODS
4.1. Animals
Two‐month‐old and 24‐month‐old C57BL/6J mice were obtained from Charles River Laboratories Japan, Inc. (Kanagawa, Japan). Men1 flox/flox mice were kindly provided by Dr. Masakatsu Yamashita (Ehime University). Col1a1‐cre/ERT2 mice were kindly provided by Dr. Masaru Ishii (Osaka University, Osaka, Japan), Col1a1‐GFP and Ai9 (RCL‐tdT) mice were kindly provided by Dr. Satoru Otsuru (University of Maryland, College Park, MD, USA). Male mice were used for each experiment. Men1 flox/flox mice were crossed with Col1a1‐cre/ERT2 mice to generate mice harboring TAM‐induced homozygous deletion of Men1 in osteoblasts. Men1 flox/flox mice were used as controls for Men1 flox/flox; Col1a1‐cre/ERT2 mice, both of which received TAM treatment. Immediately after weaning, age‐matched mice were randomly co‐housed independent of genotype. These mice were maintained on a normal chow diet with a 12‐h light/dark cycle. The following primer sets were used for genotyping by PCR to confirm model establishment: 5′‐GTGGTCAGGAGAGCAAATGAGTGT‐3′ and 5′‐CAGATCCCTCTGGCTATTCAATGG‐3′ for Men1 wild‐type; 5′‐GCCATTTCATTACCTCTTTCTCCG‐3′ and 5′‐TACCACTGCAAAGGCCACGC‐3′ for Men1 floxed allele; 5′‐CCCACATCCAGTCCCTCTTCAGCT‐3′ and 5′‐CATAAAATCGCAGCAGGTGGGCAA‐3′ for Men1‐deleted allele; 5′‐AGGTTCGTTCACTCATGGA‐3′ and 5′‐TCGACCAGTTTAGTTACCC‐3′ for Cre; 5′‐TGAACCGCATCGAGCTGAAGGG‐3′ and 5′‐TCCAGCAGGACCATGTGATCGC‐3′ for Col1a1‐GFP; and 5′‐GGCATTAAAGCAGCGTATCC‐3′ and 5′‐ and CTGTTCCTGTACGGCATGG‐3′ for Ai9 (RCL‐tdT).
4.2. Micro‐CT analysis
A high‐resolution μCT scan (Skyscan 1272, Bruker, Billerica, MA, USA) was used at a resolution of 10 μm per voxel. Parameters, including BV, tissue volume, thickness, cortical porosity, mineral density, and bone marrow area, were analyzed using CTAn software (Bruker). Mineral density calibration was performed using a bone calibration phantom. Three‐dimensional reconstructed images were obtained using CTvox software (Bruker). The analysis area of the femur was set at 1.5 mm from the distal growth plate to 1.5 mm proximally.
4.3. RT‐qPCR
Total RNA was isolated using the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA). Bone preprocessing for in vivo assays was performed as previously described (Farr et al., 2016). Spine bone were used to assess general bone aging as reported (Farr et al., 2016). Briefly, after the bones were minced into small pieces, they were subjected to two sequential 30‐min collagenase digestions (Worthington Biochemical, Worthington, OH, USA). The total RNA was extracted from the remaining chips. Abundant osteoblasts in the remaining bone chips were confirmed according to GFP‐positive signals in the preprocessed bone chips of Col1a1‐GFP mice (Figure S3d). Complementary DNA was synthesized by ReverTra Ace (TOYOBO, Osaka, Japan) and then used as a template in qPCR with FAST SYBR Green Master Mix (Thermo Fisher Scientific). Target mRNA levels were normalized to reference gene expression. The median threshold cycle was compared with that of the control sample, and the fold difference between the reference and target genes was calculated. The internal controls were Gapdh for in vitro assays (osteoblasts) and Hprt for in vivo assays (osteoblast lineage‐enriched bone cells). The primer sequences used in RT‐qPCR are listed in Table S1.
4.4. Cre/loxP recombination by TAM treatment
The dose and schedule of TAM administration (10 mg/kg/day for 4 days) and age of mice (4 weeks old) were determined according to a previous report, in which the authors confirmed the minimum TAM dosage for reliable Cre/loxP recombination with little effect on the bone formation ratio (Zhong et al., 2015). The TAM solution (20 mg/mL) was prepared by dissolving TAM powder (Sigma‐Aldrich, St. Louis, MO, USA) in ethanol and diluted 20‐fold with corn oil (C8267, Sigma‐Aldrich). For femur analysis, Men1 flox/flox (control) and Men1 flox/flox; Col1a1‐cre/ERT2 (Men1 KO) mice were treated with 10 mg/kg TAM per day for 4 days at 4, 6, and 8 weeks of age. The mice were euthanized and analyzed at 9 weeks of age. Men1 deletion by Cre/loxP recombination was confirmed by PCR of calvaria bone (Figure S3c). For BMP‐induced ectopic bone analysis, control and Men1 KO mice underwent BMP collagen sponge implantation at 4 weeks of age. Osteoblasts in the BMP‐induced ectopic bone were visible 10 days after implantation (Hashimoto et al., 2020). To delete the osteoblast Men1 gene at the appropriate time, mice were treated with TAM (10 mg/kg/day for 4 days) at 8–11 and 15–18 days after the surgery and were euthanized 21 days after the surgery. Men1 deletion by Cre/loxP recombination was confirmed by PCR of calvaria bone (Figure S3c). The efficiency of Cre/loxP recombination was confirmed in Col1a1‐cre/ERT2; Col1a1‐GFP; Ai9 mice (Figure S3b).
4.5. Biomechanical testing
Three‐point bending tests were performed using an MZ‐500 system (Marutoh, Mie, Japan) to investigate the biomechanical strength of the femur bone. The bones were placed on a pedestal, and force was applied to the center of the diaphysis at a rate of 2 mm/s until failure (ultimate load) (Figure 1g). The ultimate load (N), displacement at fracture (mm), stiffness (N/mm), and post‐yield displacement (mm) were determined from a load–displacement diagram.
4.6. Histomorphometric analysis
Double labeling by subcutaneous injection of tetracycline (20 mg/kg) and calcein (10 mg/kg) was performed 5 and 2 days before euthanasia, respectively. The resected samples were fixed in 70% ethanol, stained with Villanueva, and embedded in methacrylic acid (Wako Pure Chemical Industries, Kanagawa, Japan). Quantitative data from double‐labeled tissue sections were generated based on measured values using Histometry RT Digitiser (System Supply, Nagano, Japan) as a bone morphometry system and CSS‐840 Trabecular Bone Measurement Version as software (System Supply). The following histomorphometric parameters were measured: MAR, BFR/BS, BV/TV, trabecular thickness (Tb.Th), number of osteoblasts per bone surface (N.Ob/BS), osteoblast surface per bone surface (Ob.S/BS), number of osteoclasts per bone surface (N.Oc/BS), and osteoclast surface per bone surface (Oc.S/BS).
4.7. Osteoblast isolation from the neonatal murine calvaria
Primary osteoblasts were obtained from the calvaria of 2–3‐day‐old neonatal Men1 flox/flox mice as previously described (Yoshida et al., 2022). Briefly, calvaria were minced and digested in alpha‐minimal essential medium (Nacali Tesque, Kyoto, Japan), 0.176% NaHCO3, and 0.008% collagenase type II (Worthington Biochemical) at 37°C for 15 min seven times. Supernatants from the third to seventh digestions were collected. After centrifugation, cell suspensions were incubated in 10‐cm dishes with alpha‐minimal essential medium (α‐MEM) (Nacali), 10% fetal bovine serum, and 1% penicillin–streptomycin at 37°C with 20% O2 and 5% CO2. After 1 week, confluent cells (passage 0) were used for experiments.
4.8. Adenovirus infection of osteoblasts
Osteoblasts from Men1 flox/flox mice were passaged into 24‐well plates at a density of 5 × 105 cells per 75 cm2 (passage 1) and infected with eGFP adenovirus (Ad‐GFP, [Cat. No: 1060, Vector Laboratories, Malvern, PA, USA]: Control) or Cre recombinase adenovirus (Ad‐Cre‐GFP [Cat. No: 1700, Vector Laboratories]; Men1 KO) at a multiplicity of infection of 100 for 48 h.
4.9. Replicative senescence by long‐term culture
One week after adenovirus infection, cells were passaged in a 10‐cm dish at 5 × 105 cells per 75 cm2 (passage 2) and then passaged to a 10‐cm dish at the same density every other week. Passage‐3 and passage‐6 cells were used for the experiments as the low replicative stress and high replicative stress group, respectively, since a previous report suggested that cellular senescence starts after passage 6 under normal conditions (Parrinello et al., 2003). SA‐β‐Gal staining and RT‐qPCR were performed using passage‐3 and passage‐6 cells. Western blotting was performed on passage‐3 cells with or without 10 mM metformin treatment for 48 h.
4.10. SA β‐gal staining
SA β‐Gal staining was performed using a Senescence β‐galactosidase Staining Kit (#9860, Cell Signaling Technology, Danvers, MA, USA). Green‐stained cells were counted in 10 random fields under a microscope (BZ‐X800, Keyence, Osaka, Japan) with a 10× magnification objective lens and calculated as the percentage of positive cells. To avoid non‐specific staining due to cell confluence, the assay was performed using subconfluent cells.
4.11. Immunoblotting
After the cells were lysed with RIPA buffer (Nacalai Tesque), the protein concentrations were determined using a BCA assay (Thermo Fisher Scientific). The lysates were mixed with 5× sodium dodecyl sulfate sample buffer and boiled for 5 min. Equal amounts of protein were separated on 4–12% Bis‐Tris gels (Life Technologies, Carlsbad, CA, USA) and transferred onto polyvinylidene difluoride membranes (Nippon Genetics, Tokyo, Japan). Membranes were blocked with Tris‐buffered saline containing 5% skim milk and Tween 20 at room temperature, incubated with the primary antibody in Can Go Signal Solution 1 (Toyobo) at 4°C overnight, and then incubated with the secondary antibody in Can Go Signal Solution 2 (Toyobo) for 1 h at room temperature. The ECL Plus Western Blotting Detection System kit (Thermo Fisher Scientific) was used to detect immunoreactive proteins. The following antibodies were used for western blotting at the indicated dilutions: rabbit monoclonal anti‐4E‐BP1 (#9644, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐phospho‐4E‐BP1 (#2855, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐p70 S6 kinase (#9202, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐phospho‐p70 S6 kinase (#9234, 1:1000, Cell Signaling Technology), rabbit polyclonal anti‐AMPKα (#2532, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐phospho‐AMPKα (#2535, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐Akt (#24691, 1:1000, Cell Signaling Technology), (#2532, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐ Phospho‐Akt (Ser473) (#4060, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐p44/42 MAPK (Erk1/2) (#4695, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐Phospho‐p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (#4370, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐GSK‐3β (#12456, 1:1000, Cell Signaling Technology), rabbit monoclonal anti‐Phospho‐GSK‐3β (Ser9) (#9323, 1:1000, Cell Signaling Technology), rabbit polyclonal anti‐GAPDH (#2118, 1:1000, Cell Signaling Technology), and horseradish peroxidase‐conjugated goat anti‐rabbit IgG (#7074, 1:1000, Cell Signaling Technology). Band intensity was quantified using the ImageJ software (NIH, MD, USA).
4.12. BMP‐induced ectopic bone model
A collagen sponge (Colla Cote, Zimmet Dental, CA, USA) was cut into cylindrical shape (diameter of 5 mm and height of 2 mm) for establishing the BMP‐induced ectopic bone model. Recombinant human BMP‐2 (Osteopharma Inc., Osaka, Japan) derived from E.coli was used. Recombinant human BMP‐2 (1.5 μg) was soaked into the cylindrical collagen sponge. The sponge was then freeze‐dried (BMP pellets). Under anesthesia, the BMP pellet was implanted underneath the dorsal fascia of mice. Metformin was administered orally in the drinking water at a concentration of 300 μg/mL starting 2 weeks after the surgery. The dose of metformin was set based on the effective dose for extending lifespan (Anisimov et al., 2011).
4.13. Histological analysis
The excised samples were fixed in 10% neutral‐buffered formalin, decalcified by ethylenediaminetetraacetic acid, embedded in paraffin wax, and cut at 3 μm thickness. Hematoxylin and eosin staining was performed according to standard protocols. Paraffin‐embedded sections were deparaffinized and dehydrated for immunostaining. The antigens were activated in 10 mM citrate buffer at 95°C for 10 min. After quenching endogenous peroxidase activity with methanol containing 3% H2O2 for 10 min, the sections were blocked with Blocking One (Nacalai Tesque) for 1 h at room temperature. Sections were then incubated with a primary antibody overnight at 4°C, followed by incubation with a horseradish peroxidase‐labeled secondary antibody for 1 h. Finally, the labeled sections were stained with Histofine Simple Stain Mouse MAX PO (Nichirei Bioscience, Tokyo, Japan) and counterstained with hematoxylin. Rabbit monoclonal anti‐CDKN2A/p16INK4a (ab211542, 1:250; Abcam, Cambridge, UK), rabbit monoclonal anti‐ Phospho‐S6 Ribosomal Protein (Ser240/244) (#5364, 1:500, Cell Signaling Technology), rabbit monoclonal anti‐Phospho‐4EBP1 (Thr37/46) (#2855, 1:800, Cell Signaling Technology), rabbit polyclonal anti‐RANKL (bs‐0747R, 1:200, Bioss Inc, Massachusetts, USA), rabbit polyclonal anti‐OPG (ab216484, 1:100, Abcam), and rabbit monoclonal anti‐phospho‐AMPKα (Thr172) (#2535, 1:50, CST) were used as the primary antibody. The number of CDKN2A/p16INK4a‐positive cells on the entire perimeter of the BMP‐induced ectopic bone was counted manually. Frozen sections, which were 5 μm thick, were utilized to identify naturally occurring fluorescent proteins like tdTomato and GFP or SA β‐gal. For fluorescent protein, these sections were nuclear stained with 4′,6‐diamidino‐2‐phenylindole solution (Dojindo Laboratories, Kumamoto, Japan) and mounted with Prolong Diamond Antifade Mountant (Thermo Fisher Scientific). BZ‐X800 (Keyence) was used to observe and capture the fluorescence images. The SA β‐gal staining was performed using Senescence β‐galactosidase Staining Kit (#9860, Cell Signaling Technology).
4.14. Statistics
Statistical analysis was performed using GraphPad Prism version 8.4.1 for Windows (GraphPad Software, San Diego, CA, USA) with two‐tailed Student's t‐test, one‐way ANOVA followed by.
Sidak's test or Fisher's LSD test. Values are presented as the mean ± standard deviation. Statistical significance was set at p < 0.05.
AUTHOR CONTRIBUTIONS
Y. U.: Conceptualization, methodology, software, formal analysis, investigation, writing—original draft, funding acquisition. T. K.: Conceptualization, validation, resources, writing—review and editing, supervision, project administration, funding acquisition. H. H.: Investigation, resources, writing—review and editing. T. K.: Investigation, resources, writing—review and editing. M. B.: Investigation, resources, writing—review and editing. J. K.: Writing—review and editing. D. T.: Writing—review and editing. S. N.: Resources, writing—review and editing. M. I.: Writing—review and editing. T. F.: Writing—review and editing. Y. K.: Writing—review and editing. T. F.: Writing—review and editing. S. T.: Writing—review and editing. T. Y.: Writing—review and editing, supervision. S. O.: Writing—review and editing, supervision. S. O.: Writing—review and editing, supervision. M. Y.: Conceptualization, resources, writing—review and editing, supervision, project administration. T. I.: Conceptualization, resources, writing—review and editing, supervision, project administration.
FUNDING INFORMATION
This study was supported by JSPS KAKENHI Grant Numbers 20K09479, 22K20960 and 23K15689, the Asahi Kasei Joint Research Foundation, and the Nakatomi Foundation.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
STUDY APPROVAL
All experiments were approved by the Institutional Animal Committee of Osaka University.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Table S1.
ACKNOWLEDGMENTS
We thank Fumiko Hirayama, Yukiko Eguchi, and Mari Shinkawa for their valuable contributions to this study.
Ukon, Y. , Kaito, T. , Hirai, H. , Kitahara, T. , Bun, M. , Kodama, J. , Tateiwa, D. , Nakagawa, S. , Ikuta, M. , Furuichi, T. , Kanie, Y. , Fujimori, T. , Takenaka, S. , Yamamuro, T. , Otsuru, S. , Okada, S. , Yamashita, M. , & Imamura, T. (2024). Cellular senescence by loss of Men1 in osteoblasts is critical for age‐related osteoporosis. Aging Cell, 23, e14254. 10.1111/acel.14254
DATA AVAILABILITY STATEMENT
The datasets generated and analyzed in the current study are available from the corresponding author upon reasonable request.
REFERENCES
- Anisimov, V. N. , Berstein, L. M. , Popovich, I. G. , Zabezhinski, M. A. , Egormin, P. A. , Piskunova, T. S. , Semenchenko, A. V. , Tyndyk, M. L. , Yurova, M. N. , Kovalenko, I. G. , & Poroshina, T. E. (2011). If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY), 3(2), 148–157. 10.18632/aging.100273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspray, T. J. , & Hill, T. R. (2019). Osteoporosis and the ageing skeleton. Sub‐Cellular Biochemistry, 91, 453–476. 10.1007/978-981-13-3681-2_16 [DOI] [PubMed] [Google Scholar]
- Baker, D. J. , Childs, B. G. , Durik, M. , Wijers, M. E. , Sieben, C. J. , Zhong, J. , Saltness, R. A. , Jeganathan, K. B. , Verzosa, G. C. , Pezeshki, A. , Khazaie, K. , Miller, J. D. , & van Deursen, J. M. (2016). Naturally occurring p16Ink4a‐positive cells shorten healthy lifespan. Nature, 530(7589), 184–189. 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bala, Y. , Zebaze, R. , & Seeman, E. (2015). Role of cortical bone in bone fragility. Current Opinion in Rheumatology, 27(4), 406–413. 10.1097/bor.0000000000000183 [DOI] [PubMed] [Google Scholar]
- Childs, B. G. , Baker, D. J. , Kirkland, J. L. , Campisi, J. , & van Deursen, J. M. (2014). Senescence and apoptosis: Dueling or complementary cell fates? EMBO Reports, 15(11), 1139–1153. 10.15252/embr.201439245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaianni, G. , Mongelli, T. , Cuscito, C. , Pignataro, P. , Lippo, L. , Spiro, G. , Notarnicola, A. , Severi, I. , Passeri, G. , Mori, G. , Brunetti, G. , Moretti, B. , Tarantino, U. , Colucci, S. C. , Reseland, J. E. , Vettor, R. , Cinti, S. , & Grano, M. (2017). Irisin prevents and restores bone loss and muscle atrophy in hind‐limb suspended mice. Scientific Reports, 7(1), 2811. 10.1038/s41598-017-02557-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado, M. , & Serrano, M. (2010). Senescence in tumours: Evidence from mice and humans. Nature Reviews. Cancer, 10(1), 51–57. 10.1038/nrc2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, J. N. , Fraser, D. G. , Wang, H. , Jaehn, K. , Ogrodnik, M. B. , Weivoda, M. M. , Drake, M. T. , Tchkonia, T. , LeBrasseur, N. K. , Kirkland, J. L. , Bonewald, L. F. , Pignolo, R. J. , Monroe, D. G. , & Khosla, S. (2016). Identification of senescent cells in the bone microenvironment. Journal of Bone and Mineral Research, 31(11), 1920–1929. 10.1002/jbmr.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, J. N. , & Khosla, S. (2019). Cellular senescence in bone. Bone, 121, 121–133. 10.1016/j.bone.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, J. N. , Rowsey, J. L. , Eckhardt, B. A. , Thicke, B. S. , Fraser, D. G. , Tchkonia, T. , Kirkland, J. L. , Monroe, D. G. , & Khosla, S. (2019). Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: Evidence in young adult mice and older humans. Journal of Bone and Mineral Research, 34(8), 1407–1418. 10.1002/jbmr.3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, J. N. , Xu, M. , Weivoda, M. M. , Monroe, D. G. , Fraser, D. G. , Onken, J. L. , Negley, B. A. , Sfeir, J. G. , Ogrodnik, M. B. , Hachfeld, C. M. , LeBrasseur, N. K. , Drake, M. T. , Pignolo, R. J. , Pirtskhalava, T. , Tchkonia, T. , Oursler, M. J. , Kirkland, J. L. , & Khosla, S. (2017). Targeting cellular senescence prevents age‐related bone loss in mice. Nature Medicine, 23(9), 1072–1079. 10.1038/nm.4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnough, L. H. , & Goodman, S. B. (2022). Relationship of aging, inflammation, and skeletal stem cells and their effects on fracture repair. Current Osteoporosis Reports, 20(5), 320–325. 10.1007/s11914-022-00742-x [DOI] [PubMed] [Google Scholar]
- Gregson, C. L. , Armstrong, D. J. , Bowden, J. , Cooper, C. , Edwards, J. , Gittoes, N. J. L. , Harvey, N. , Kanis, J. , Leyland, S. , Low, R. , McCloskey, E. , Moss, K. , Parker, J. , Paskins, Z. , Poole, K. , Reid, D. M. , Stone, M. , Thomson, J. , Vine, N. , & Compston, J. (2022). UK clinical guideline for the prevention and treatment of osteoporosis. Archives of Osteoporosis, 17(1), 58. 10.1007/s11657-022-01061-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, K. , Kaito, T. , Furuya, M. , Seno, S. , Okuzaki, D. , Kikuta, J. , Tsukazaki, H. , Matsuda, H. , Yoshikawa, H. , & Ishii, M. (2020). In vivo dynamic analysis of BMP‐2‐induced ectopic bone formation. Scientific Reports, 10(1), 4751. 10.1038/s41598-020-61825-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, Y. , Hsiao, E. C. , Sami, S. , Lancero, M. , Schlieve, C. R. , Nguyen, T. , Yano, K. , Nagahashi, A. , Ikeya, M. , Matsumoto, Y. , Nishimura, K. , Fukuda, A. , Hisatake, K. , Tomoda, K. , Asaka, I. , Toguchida, J. , Conklin, B. R. , & Yamanaka, S. (2016). BMP‐SMAD‐ID promotes reprogramming to pluripotency by inhibiting p16/INK4A‐dependent senescence. Proceedings of the National Academy of Sciences of the United States of America, 113(46), 13057–13062. 10.1073/pnas.1603668113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick, L. , & Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Experimental Cell Research, 25, 585–621. 10.1016/0014-4827(61)90192-6 [DOI] [PubMed] [Google Scholar]
- Jeon, S. M. (2016). Regulation and function of AMPK in physiology and diseases. Experimental & Molecular Medicine, 48(7), e245. 10.1038/emm.2016.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji, H. (2012). Menin and bone metabolism. Journal of Bone and Mineral Metabolism, 30(4), 381–387. 10.1007/s00774-012-0355-3 [DOI] [PubMed] [Google Scholar]
- Kanazawa, I. , Canaff, L. , Abi Rafeh, J. , Angrula, A. , Li, J. , Riddle, R. C. , Boraschi‐Diaz, I. , Komarova, S. V. , Clemens, T. L. , Murshed, M. , & Hendy, G. N. (2015). Osteoblast menin regulates bone mass in vivo. The Journal of Biological Chemistry, 290(7), 3910–3924. 10.1074/jbc.M114.629899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda, A. , Fujita, T. , Anai, M. , Yamamoto, S. , Nagae, G. , Morikawa, M. , Tsuji, S. , Oshima, M. , Miyazono, K. , & Aburatani, H. (2011). Activation of Bmp2‐Smad1 signal and its regulation by coordinated alteration of H3K27 trimethylation in Ras‐induced senescence. PLoS Genetics, 7(11), e1002359. 10.1371/journal.pgen.1002359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner, C. M. , Lee, S. Y. , & Long, F. (2017). Bmp induces osteoblast differentiation through both Smad4 and mTORC1 signaling. Molecular and Cellular Biology, 37(4), e00253. 10.1128/mcb.00253-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland, J. L. , & Tchkonia, T. (2020). Senolytic drugs: From discovery to translation. Journal of Internal Medicine, 288(5), 518–536. 10.1111/joim.13141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara, M. , Suzuki, J. , Tofukuji, S. , Yamada, T. , Kanoh, M. , Matsumoto, A. , Maruyama, S. , Kometani, K. , Kurosaki, T. , Ohara, O. , Nakayama, T. , & Yamashita, M. (2014). The Menin‐Bach2 axis is critical for regulating CD4 T‐cell senescence and cytokine homeostasis. Nature Communications, 5(1), 3555. 10.1038/ncomms4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Liu, P. , Teinturier, R. , Jakob, J. , Tschaffon, M. , Tasdogan, A. , Wittig, R. , Hoeller, S. , Baumhoer, D. , Frappart, L. , Vettorazzi, S. , Bertolino, P. , Zhang, C. , & Tuckermann, J. (2018). Deletion of Menin in craniofacial osteogenic cells in mice elicits development of mandibular ossifying fibroma. Oncogene, 37(5), 616–626. 10.1038/onc.2017.364 [DOI] [PubMed] [Google Scholar]
- Leng, L. , Zhuang, K. , Liu, Z. , Huang, C. , Gao, Y. , Chen, G. , Lin, H. , Hu, Y. , Wu, D. , Shi, M. , Xie, W. , Sun, H. , Shao, Z. , Li, H. , Zhang, K. , Mo, W. , Huang, T. Y. , Xue, M. , Yuan, Z. , … Zhang, J. (2018). Menin deficiency leads to depressive‐like behaviors in mice by modulating astrocyte‐mediated neuroinflammation. Neuron, 100(3), 551–563.e7. 10.1016/j.neuron.2018.08.031 [DOI] [PubMed] [Google Scholar]
- Liu, P. , Lee, S. , Knoll, J. , Rauch, A. , Ostermay, S. , Luther, J. , Malkusch, N. , Lerner, U. H. , Zaiss, M. M. , Neven, M. , Wittig, R. , Rauner, M. , David, J. P. , Bertolino, P. , Zhang, C. X. , & Tuckermann, J. P. (2017). Loss of menin in osteoblast lineage affects osteocyte‐osteoclast crosstalk causing osteoporosis. Cell Death and Differentiation, 24(4), 672–682. 10.1038/cdd.2016.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X.‐L. , Ding, J. , & Meng, L.‐H. (2018). Oncogene‐induced senescence: A double edged sword in cancer. Acta Pharmacologica Sinica, 39(10), 1553–1558. 10.1038/aps.2017.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini, F. , Giusti, F. , Iantomasi, T. , Cioppi, F. , & Brandi, M. L. (2022). Bone phenotypes in multiple endocrine neoplasia type 1: Survey on the MEN1 Florentine database. Endocrine Connections, 11(5), e210456. 10.1530/ec-21-0456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz‐Espín, D. , Cañamero, M. , Maraver, A. , Gómez‐López, G. , Contreras, J. , Murillo‐Cuesta, S. , Rodríguez‐Baeza, A. , Varela‐Nieto, I. , Ruberte, J. , Collado, M. , & Serrano, M. (2013). Programmed cell senescence during mammalian embryonic development. Cell, 155(5), 1104–1118. 10.1016/j.cell.2013.10.019 [DOI] [PubMed] [Google Scholar]
- Muñoz‐Espín, D. , & Serrano, M. (2014). Cellular senescence: From physiology to pathology. Nature Reviews. Molecular Cell Biology, 15(7), 482–496. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- Ou, H. L. , Hoffmann, R. , González‐López, C. , Doherty, G. J. , Korkola, J. E. , & Muñoz‐Espín, D. (2021). Cellular senescence in cancer: From mechanisms to detection. Molecular Oncology, 15(10), 2634–2671. 10.1002/1878-0261.12807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello, S. , Samper, E. , Krtolica, A. , Goldstein, J. , Melov, S. , & Campisi, J. (2003). Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nature Cell Biology, 5(8), 741–747. 10.1038/ncb1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulski, M. J. (2017). Cellular senescence: What, why, and how. Wounds, 29(6), 168–174. [PubMed] [Google Scholar]
- Shim, J. , Iwaya, C. , Ambrose, C. G. , Suzuki, A. , & Iwata, J. (2022). Micro‐computed tomography assessment of bone structure in aging mice. Scientific Reports, 12(1), 8117. 10.1038/s41598-022-11965-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, M. J. , Brodt, M. D. , Ko, M. , & Abu‐Amer, Y. (2005). Impaired marrow osteogenesis is associated with reduced endocortical bone formation but does not impair periosteal bone formation in long bones of SAMP6 mice. Journal of Bone and Mineral Research, 20(3), 419–427. 10.1359/jbmr.041128 [DOI] [PubMed] [Google Scholar]
- Suzuki, J. , Yamada, T. , Inoue, K. , Nabe, S. , Kuwahara, M. , Takemori, N. , Takemori, A. , Matsuda, S. , Kanoh, M. , Imai, Y. , Yasukawa, M. , & Yamashita, M. (2018). The tumor suppressor menin prevents effector CD8 T‐cell dysfunction by targeting mTORC1‐dependent metabolic activation. Nature Communications, 9(1), 3296. 10.1038/s41467-018-05854-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker, R. V. , Newey, P. J. , Walls, G. V. , Bilezikian, J. , Dralle, H. , Ebeling, P. R. , Melmed, S. , Sakurai, A. , Tonelli, F. , Brandi, M. L. , & Endocrine Society . (2012). Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). The Journal of Clinical Endocrinology and Metabolism, 97(9), 2990–3011. 10.1210/jc.2012-1230 [DOI] [PubMed] [Google Scholar]
- Troka, I. , Griffanti, G. , Canaff, L. , Hendy, G. N. , Goltzman, D. , & Nazhat, S. N. (2022). Effect of Menin deletion in early osteoblast lineage on the mineralization of an in vitro 3D osteoid‐like dense collagen gel matrix. Biomimetics (Basel), 7(3), 101. 10.3390/biomimetics7030101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukon, Y. , Makino, T. , Kodama, J. , Tsukazaki, H. , Tateiwa, D. , Yoshikawa, H. , & Kaito, T. (2019). Molecular‐based treatment strategies for osteoporosis: A literature review. International Journal of Molecular Sciences, 20(10), 2557. 10.3390/ijms20102557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, W. , & Ji, S. (2018). Cellular senescence: Molecular mechanisms and pathogenicity. Journal of Cellular Physiology, 233(12), 9121–9135. 10.1002/jcp.26956 [DOI] [PubMed] [Google Scholar]
- Weichhart, T. (2018). mTOR as regulator of lifespan, aging, and cellular senescence: A mini‐review. Gerontology, 64(2), 127–134. 10.1159/000484629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won, Y. , & Choi, E. (2022). Mouse models of Kras activation in gastric cancer. Experimental & Molecular Medicine, 54(11), 1793–1798. 10.1038/s12276-022-00882-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, H. (2021). Discovery of the RANKL/RANK/OPG system. Journal of Bone and Mineral Metabolism, 39(1), 2–11. 10.1007/s00774-020-01175-1 [DOI] [PubMed] [Google Scholar]
- Yoshida, G. , Kawabata, T. , Takamatsu, H. , Saita, S. , Nakamura, S. , Nishikawa, K. , Fujiwara, M. , Enokidani, Y. , Yamamuro, T. , Tabata, K. , Hamasaki, M. , Ishii, M. , Kumanogoh, A. , & Yoshimori, T. (2022). Degradation of the NOTCH intracellular domain by elevated autophagy in osteoblasts promotes osteoblast differentiation and alleviates osteoporosis. Autophagy, 18(10), 2323–2332. 10.1080/15548627.2021.2017587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, Z. A. , Sun, W. , Chen, H. , Zhang, H. , Lay, Y. E. , Lane, N. E. , & Yao, W. (2015). Optimizing tamoxifen‐inducible Cre/loxp system to reduce tamoxifen effect on bone turnover in long bones of young mice. Bone, 81, 614–619. 10.1016/j.bone.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Table S1.
Data Availability Statement
The datasets generated and analyzed in the current study are available from the corresponding author upon reasonable request.