

Abstract
Background and purpose
Wilson's disease (WD) is a rare autosomal recessive disorder causing excessive copper deposition and a spectrum of manifestations, particularly neurological and hepatic symptoms. We analysed the clinical characteristics of patients with WD admitted to the country's only reference centre, which provided long‐term care to most adult patients in Poland over seven decades (pre‐1959 to 2019).
Methods
Electronic prospective data collection began in the 2000s and, for prior years, medical records were analysed retrospectively. Demographic and clinical characteristics, treatment and outcomes were analysed by decade of diagnosis. Life‐years lost were estimated in patients with WD compared with the general population. Kaplan–Meier curves were used for a time‐to‐death analysis using 2000–2009 as a reference.
Results
In total, 929 patients were analysed. The number of patients increased from 21 before 1959 to 315 for 2000 to 2009 period. Mostly males were diagnosed before the 1990s, but the numbers of female patients diagnosed increased thereafter. Initially, most patients presented with neurological manifestations; however, the incidence of hepatic manifestations and asymptomatic presentations increased over time as patients were diagnosed early and consequently were more independent at diagnosis. Fewer Kayser‐Fleischer rings were detected recently. Prior to 1970, patients were treated with D‐penicillamine (DP); however, since the introduction of zinc, both therapies have been used as often. Since the 1990s, switches between DP and zinc were recorded in 6%–7% of patients. Consistent improvement in survival has been observed over the years.
Conclusions
This is the largest cohort of patients with WD reported in Poland, with the longest follow‐up. Earlier diagnosis and prognosis have improved over seven decades.
Keywords: clinical outcome, diagnosis, prognosis, treatment, Wilson's disease
Milestones in healthcare of Wilson's disease (WD) patients in Poland. WD is a rare autosomal recessive disorder which can be effectively diagnosed and treated with available and long‐known drugs to avoid copper‐induced organ failure. Long‐term follow‐up data on WD showed that patients' prognosis and outcomes improved but WD still represents a clinical challenge. Well‐organized, comprehensive and continuous care can improve WD patients' prognosis, which has improved over time but remains unsatisfactory.

INTRODUCTION
Wilson's disease (WD) is a rare autosomal recessive disorder, primarily impacting copper metabolism [1]. It is a monogenic disorder involving mutations in the ATP7B gene, located on chromosome 13q14‐21, which encodes a transmembrane copper‐transporting ATPase [2]. Mutations in the ATP7B gene can result in impaired copper homeostasis and copper deposition in multiple organs, including, but not limited to, the liver and central nervous system [2, 3]. Patients present with a broad spectrum of hepatic, neurological and/or psychiatric symptoms or may be asymptomatic. The liver has the highest expression of the ATP7B copper transporter and is the organ responsible for copper homeostasis. Hence, liver injury in the form of hepatitis, fibrosis and cirrhosis is common and hepatic manifestation is likely to precede neurological manifestations [2].
A number of therapeutic options have been established since the end of the 1950s. D‐penicillamine (DP), trientine and zinc salts are most often prescribed, but novel treatments including tetrathiomolybdate and gene therapy are currently being explored [4, 5]. WD is one of the few disorders that can be successfully treated if a diagnosis is established early and the patient is compliant [5]. It has been reported that late diagnosis is the most common cause of death, although poor compliance with medication can contribute significantly [5, 6].
The prevalence most often cited for WD is 1 in 30,000 from 1984, the so‐called ‘Scheinberg‐Sterlieb Estimate’ [7]. A 2020 update estimated the prevalence to be similar and valid; however, there is high geographical variability [8]. For example, the prevalence of WD is higher in China and Asian countries compared to Western countries [9]. The median age of diagnosis has been found to be 19 years in a variety of studies; however, again, there is considerable variability [10, 11]. The mortality rate of WD patients is higher than in the general population, but the prognosis of WD is difficult to predict due to the inconsistencies within the current literature [12].
Due to the rarity of the disease, there is a paucity of long‐term data on patient outcomes and clinical practice. In this article, we report seven decades of clinical experience from patients with WD admitted to a first‐in‐country reference centre, which provides long‐term care to the majority of adult WD patients identified in Poland.
METHODS
The first patients believed to have presented with WD were referred to the Department of Neurology, Institute of Psychiatry and Neurology, Warsaw, in the 1950s. Patients could be diagnosed and treated before admission to the reference centre, which was then responsible for diagnosis confirmation, optimizing therapy and long‐term monitoring. The majority of adult patients (and also children before the 1990s) were identified and tracked by the reference centre, with very few being treated elsewhere.
Electronic prospective data collection systematically started at the beginning of the current century. In general, data collected followed the protocol of the EuroWilson Project [13] and this was adjusted to routine local clinical and documentation practice. In the current analysis, patients diagnosed before electronic records (pre‐2000) were documented retrospectively from medical records for available data. The scope of the data presented here is limited to those variables with the fewest missing data and with high‐quality data.
In the present analysis, demographic and clinical characteristics, treatment and outcomes were grouped by decade of diagnosis. Clinical manifestations at diagnosis were defined as asymptomatic (mostly patients identified in familly screening), hepatic (asymptomatic hepatomegaly or elevation of serum aminotransferase levels to hepatic steatosis and cirrhosis) or neurological based on predominant symptom type, supported with physical examination, laboratory findings and imaging techniques as available [14]. Neurological signs and symptoms typical of WD, namely, salivation and speech abnormalities, gait disturbance and tremor or involuntary movements were recorded across all decades. Additionally, the severity of neurological manifestation was illustrated globally as disease‐related dependence (restrictions in activities of daily living). If needed, psychiatric consultation was provided, but this was neither systematically evaluated nor structurally reported.
The milestones in WD care in Poland are presented in Figure 1. The evaluation covered serum ceruloplasmin (Ravin's method) and copper in urine (24‐h excretion) and serum (initially with chemical methods and subsequently with flame atomic absorption spectrometry) assessments, which were completed in the local laboratory for all patients at admission, with additional radioactive copper testing in uncertain cases [15, 16]. Every patient received at least basic liver laboratory tests and ultrasound examination following its increased availability from the 1980s. Ophthalmological check‐up of Kayser‐Fleischer rings by slit lamp examination was available for all patients. Access to genetic testing and neuroimaging started in the mid‐1990s, subsequently increased, and, soon after their development, both diagnostic methods were also offered to patients.
FIGURE 1.
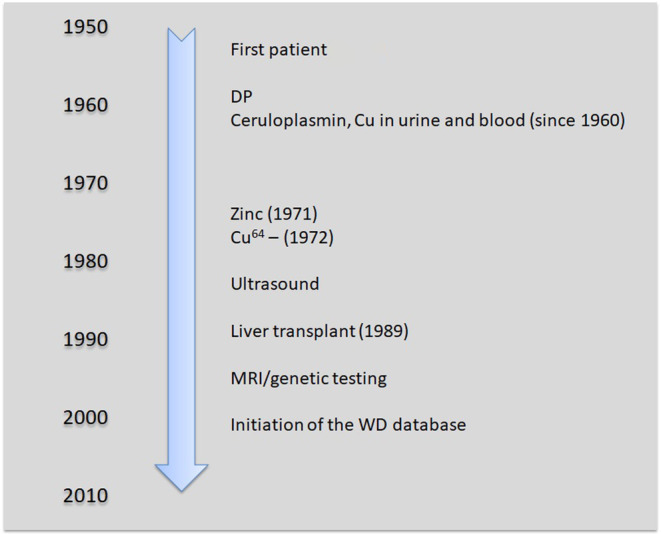
Milestones in healthcare of Wilson's disease (WD) patients in Poland. DP, D‐penicillamine; MRI, magnetic resonance imaging
Drug therapy was mainly based on the use of anti‐copper therapy with oral DP and, since the beginning of the 1970s, zinc sulphate. We recorded any switches between therapies. Regarding medication compliance, patients were interviewed at every follow‐up visit and any reports of non‐persistence, dose self‐modification or discontinuation (either temporal or permanent) were recorded as non‐compliance and not further graded.
We analysed the long‐term outcomes of death, including time to death (recorded in the database and confirmed with the official death registry), and dependence, defined as the need for assistance in daily living activities.
Statistical analysis
Data are presented as rates, means and standard deviations or medians and interquartile ranges for continuous variables, as appropriate. Consequently, we used chi‐squared or Kruskal–Wallis tests and ANOVA to compare trends over decades.
The overall prognosis of patients with WD was explored via life‐years lost, that is, the difference between age at the patient's death and their estimated life expectancy based on birth age. As life tables were only available from 1950 onwards, the life‐years lost analysis was not feasible for the first decades. Additionally, Kaplan–Meier estimates and hazard ratios were obtained for time to death, time to death or dependence, and time to death or liver transplant, with the most recent mature follow‐up of 2000–2009 used as a reference. The latest decade (2010–2019) was not selected as there was an incomplete follow‐up of patients.
Ethical approval
Data collection and analysis was approved by the Institute of Psychiatry and Neurology Ethics Committee.
RESULTS
Patient characteristics
Up to 2019, 929 patients with WD from all over Poland were recorded. The number of patients newly included per decade increased from 21 before 1960 to 315 from 2000 to 2009 (Table 1). Initially, patients referred were mostly male; however, WD was reported most often in female patients from the 1990s. The mean age at diagnosis was below 30 years, with a slight increase more recently. The median time from first signs to diagnosis has halved but with increased variability. The average time to diagnosis remained greater than 1 year since symptom onset. Patients were more likely to receive an earlier referral to the reference centre during the most recent three decades.
TABLE 1.
Patient characteristics
| Pre‐1959 | 1960–1969 | 1970–1979 | 1980–1989 | 1990–1999 | 2000–2009 | 2010–2019 | p value for cross‐decade comparisons | |
|---|---|---|---|---|---|---|---|---|
| Number of patients | 21 | 54 | 70 | 67 | 174 | 315 | 228 | |
| Male patients, n (%) | 18 (85.7) | 36 (66.7) | 39 (55.7) | 42 (62.7) | 86 (49.4) | 147 (46.8) | 109 (47.8) | <0.001 |
| Age at diagnosis, mean ± SD (Q25/Q50/Q75), years | 27 ± 7 (21/24/30) | 27 ± 9 (20/25/33) | 26 ± 10 (19/26/31) | 29 ± 8 (23/27/32) | 28 ± 10 (21/28/35) | 27 ± 11 (19/25/35) | 30 ± 12 (21/28/39) | 0.053 |
| Time from symptoms to diagnosis, median (IQR) months | 30 (12–45) | 36 (12–54) | 21 (12–36) | 12 (10–24) | 12 (7–36) | 14 (5–48) | 13 (5–60) | 0.003 |
| Diagnosed in reference centre, n (%) | N/A | N/A | 13 (56.5) | 12 (37.5) | 50 (38.5) | 113 (37.5) | 116 (51.8) | 0.008 |
| Treated before referral to reference centre, n (%) | N/A | N/A | 6 (33.3) | 15 (51.7) | 55 (46.2) | 166 (55.7) | 103 (45.2) | 0.069 |
| Treatment duration before referral to reference centre, median (IQR) months | N/A | N/A | 176 (63–246) (n = 31) | 74 (10–184) (n = 34) | 7 (3–39) (n = 75) | 8 (2–40) (n = 145) | 5 (2–17) (n = 89) | <0.001 |
| Independent at diagnosis, n (%) | 12 (60.0) | 34 (63.0) | 47 (68.1) | 47 (72.3) | 141 (82.0) | 266 (86.9) | 208 (91.6) | <0.001 |
| Clinical manifestation at diagnosis: neurological/hepatic/asymptomatic, % | 95/0/5 | 85/6/9 | 67/7/26 | 82/4/13 | 53/32/14 | 36/44/20 | 35/44/21 | <0.001 |
| Clinical manifestation at diagnosis for women: neurological/hepatic/asymptomatic, % | 100/0/0 | 89/0/11 | 58/6/35 | 92/4/4 | 51/35/14 | 29/51/20 | 30/50/20 | <0.001 |
| Clinical manifestation at diagnosis for men: neurological/hepatic/asymptomatic, % | 94/0/6 | 83/8/8 | 74/8/18 | 76/5/19 | 56/29/15 | 44/36/20 | 40/39/21 | <0.001 |
| Clinical manifestation at diagnosis for patients 20 years old or above: neurological/hepatic/asymptomatic, % | 100/0/0 | 93/5/2 | 74/10/16 | 85/5/10 | 67/23/10 | 46/41/13 | 42/42/16 | <0.001 |
| Kayser‐Fleischer ring detected, n (%) | 21 (100.0) | 45 (90.0) | 60 (89.6) | 52 (83.9) | 108 (67.5) | 150 (58.8) | 117 (58.5) | <0.001 |
| Psychiatric symptoms at diagnosis, % | 10 | 11 | 13 | 15 | 11 | 11 | 6 | 0.360 |
| Years of life lost: male/female, life‐years lost | N/A | N/A | 21/26 | 23/15 | 23/35 | 26/38 | 18/34 | 0.120 |
Abbreviations: IQR, interquartile range; N/A, not applicable, data not reported due to anti‐copper treatment limited availability, offered only in reference centre and no specific diagnostic tests available elsewhere; SD, standard deviation.
Patients were consistently less neurologically affected and dependent at diagnosis. Initially, the majority of patients presented with neurological manifestations and 30%–40% of patients were dependent (Table 1). Over the decades, patients were less often dependent upon referral, they were consistently less neurologically affected, and their neurological symptoms were less severe. In particular, patients were two times less likely to present with excessive salivation or gait disturbances in 2010–2019 compared with 1960–1969 (Table 2). Over the decades, more patients were referred with liver manifestations (especially female patients and adolescents) as well as asymptomatic patients (Table 1). Currently, hepatic manifestations are most common, although every fifth patient presents with no symptoms at diagnosis. Kayser‐Fleischer rings were reported less often over the decades as more asymptomatic patients were diagnosed. Psychiatric symptoms affected up to 15% of patients in 1980–1989 and 6% in the most recent decade.
TABLE 2.
Neurological symptoms present at admission to the reference centre (rates are reported for patients with neurological manifestations only)
| 1960–1969 (n = 46) | 1970–1979 (n = 47) | 1980–1989 (n = 55) | 1990–1999 (n = 93) | 2000–2009 (n = 114) | 2010–2019 (n = 80) | p value for cross‐decade comparisons | |
|---|---|---|---|---|---|---|---|
| Characteristic, % | |||||||
| Excessive salivation | 95 | 88 | 89 | 68 | 70 | 43 | <0.001 |
| Speech abnormalities | 95 | 78 | 91 | 87 | 78 | 74 | 0.066 |
| Gait disturbance | 85 | 72 | 66 | 60 | 55 | 47 | 0.007 |
| Tremor or involuntary movements | 100 | 93 | 96 | 95 | 72 | 84 | <0.001 |
Drug treatment
Patients were treated mainly with DP before 1970 (Table 3). The proportion of patients treated with zinc increased gradually since its introduction, with DP and zinc used equally as initial therapy for almost 30 years. Rates of switches from DP to zinc or vice versa appear to be similar at up to 6%–7%. Self‐reported compliance increased over the decades and the majority of patients appeared to follow drug‐use recommendations in the last decade studied.
TABLE 3.
Drug treatment of patients with Wilson's disease
| Pre‐1959 | 1960–1969 | 1970–1979 | 1980–1989 | 1990–1999 | 2000–2009 | 2010–2019 | p value for cross‐decade comparisons | |
|---|---|---|---|---|---|---|---|---|
| DP treatment at diagnosis, n (%) | 15 (71.4) | 42 (77.8) | 64 (91.4) | 55 (82.1) | 101 (58.0) | 158 (50.2) | 105 (46.1) | <0.001 |
| Zinc treatment at diagnosis, n (%) | 0 (0.0) | 1 (1.9) | 4 (5.7) | 12 (17.9) | 79 (45.4) | 147 (46.7) | 116 (50.9) | <0.001 |
| Switch from zinc to DP, n (%) | 0 | 0 | 0 | 3 (4.5) | 11 (6.3) | 23 (7.3) | 11 (4.8) | <0.001 |
| Switch from DP to zinc, n (%) | 0 | 0 | 2 (2.9) | 2 (3.0) | 6 (3.4) | 24 (7.6) | 16 (7.0) | <0.001 |
| Self‐reported compliance, n (%) | 0 (0.0) | 1 (25.0) | 15 (65.2) | 13 (48.1) | 56 (54.4) | 148 (54.8) | 173 (82.0) | <0.001 |
Abbreviation: DP, D‐penicillamine.
Patient outcomes
The overall 10‐year survival for pooled populations of all decades was estimated at 89% (lost to follow‐up rate was low and totalled 8.2%). Life‐years lost have remained high, especially in female patients (Table 1), but Kaplan–Meier analyses showed constantly improving survival over time, with patients diagnosed between the 1960s and 1990s having a statistically significantly higher hazard ratio of death compared with the 2000–2009 decade (Figure 2; Table 4). Similar results were found for time to death or dependence, and time to death or liver transplant (altogether 45 patients underwent transplants; all due to liver failure and no one with neurological deficit only), but were less consistently and statistically significant (Table 4). Improvement was observed in a specific analysis of dependence (patients initially dependent might have improved over time, thus Kaplan–Meier analyses could not be adjusted accordingly) in the whole population, but especially in patients with neurological symptoms (data not shown). In patients diagnosed in 2000–2010, almost every fifth patient gained independence in daily living activities (data not presented as there was less consistent assessment in the early decades).
FIGURE 2.
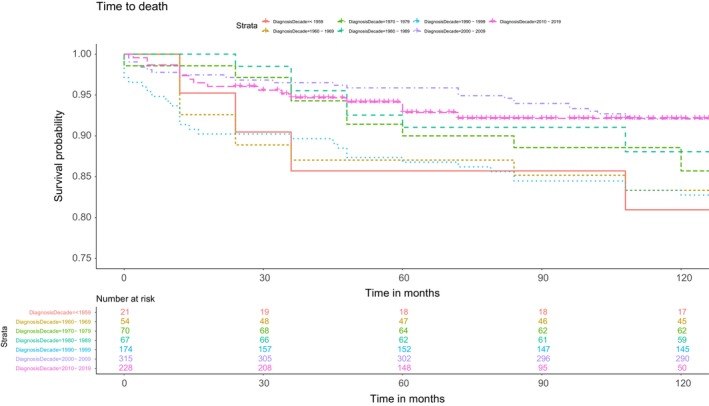
Ten‐year survival curves by decade of diagnosis
TABLE 4.
Outcomes in patients with Wilson's disease by decade
| Hazard ratio (95% CI) by decade | |||
|---|---|---|---|
| Time to death | Time to death or dependence | Time to death or liver transplant | |
| <1959 | 1.84 (0.82–4.13) | 1.44 (0.53–3.90) | 0.91 (0.42–1.97) |
| 1960–1969 | 2.49 (1.42–4.37) | 1.59 (0.80–3.15) | 1.23 (0.74–2.04) |
| 1970–1979 | 3.23 (1.95–5.34) | 2.66 (1.52–4.67) | 1.60 (1.03–2.48) |
| 1980–1989 | 1.85 (1.03–3.35) | 1.37 (0.69–2.71) | 0.98 (0.58–1.67) |
| 1990–1999 | 2.03 (1.26–3.28) | 1.57 (0.95–2.61) | 1.06 (0.71–1.58) |
| 2000–2009 | Reference | Reference | Reference |
| 2010–2019 | 1.19 (0.64–2.21) | 1.83 (1.02–3.29) | 0.74 (0.45–1.22) |
Abbreviation: CI, confidence interval.
DISCUSSION
This long‐term follow‐up report of more than 900 patients diagnosed with WD in Poland over 70 years represents one of the largest and most comprehensive analyses of patients with this rare disease.
Over the decades, we observed an increase in the number of patients diagnosed, which was not unexpected and is likely attributable to improved awareness among healthcare professionals, and advances in screening and recognition by the reference centre. We also noted a shift in the presenting WD symptoms. In the first few decades, WD patients usually presented with neurological manifestations as, initially, mostly neurologists were involved in diagnosis and treatment. The centre's neurological orientation facilitated initial cooperation with other neurological centres, but gradually other specialties joined and networked. Over the years, the incidence of hepatic manifestations as well as asymptomatic presentations at diagnosis has increased. Long‐term outcome studies in Portugal, Greece, Serbia, South Korea and Finland found similar results [10, 17, 18, 19]. This shift in WD manifestations could be attributed to the early and accurate diagnosis of patients' relatives and early hepatic manifestations as well as advances in appropriate identification of less specific hepatic symptoms and, again, to improved awareness among healthcare professionals and recognition by the reference centre. For those patients who did present with neurological symptoms, we found that these were less severe in recent years and patients were more likely to be independent in their daily living activities.
Compared with the period pre‐1959 to 1979, patients in the most recent three decades compared to previous ones were referred to our centre more quickly to either confirm the diagnosis or reassess the treatment. This suggests a benefit of the ongoing centralization of care, the increased role of the reference centre and, most likely, improved awareness among medical professionals. The mean time to diagnosis remained similar over the last three decades, although there seemed to be increased variability. It appears that some patients are still waiting a considerable length of time for appropriate diagnosis and treatment, indicating substantial diagnostic challenges [20].
Walshe reported that late diagnosis was the most common cause of death [6]. The longer the disease progresses without treatment, the more likely it is that the liver and brain will be damaged by toxic copper accumulation. The long‐term prognosis for people with WD varies and largely depends on timely diagnosis and treatment. In terms of mortality, the Kaplan–Meier analysis showed consistent improvement in survival, with patients diagnosed long ago having a statistically significant higher hazard ratio of death compared to those diagnosed during the most recent decade. Although survival improved over the decades in Poland, there remains a substantial unmet medical need as patients' life expectancy is shortened compared with the general population.
The mean age of diagnosis was below 30 years, with a borderline non‐significant increase over the decades. Initially, our centre admitted a few children who were then referred to paediatric centres [21]. Long‐term follow‐up in Northern Portugal and Greece found the median age at diagnosis to be 19 years, which is similar to previously described data [10, 11, 22]. Hence, the mean age at diagnosis of our cohort was slightly older than in other studies and may have been driven by the specialty profile of the centre. A study conducted in Germany also found 30 years to be the mean age of diagnosis. Initial neurological presentation usually occurs at the age of 20–30 years; however, in other studies, the youngest patient with neurological manifestations was 6 years old and the oldest was 72 years old [2, 23]. The varied clinical course of WD could be attributed to multiple factors (genetic, epigenetic, hormonal and environmental) [2]. Given the wide age range of symptom onset, WD must be considered as a differential diagnosis at all ages.
Our study found that, prior to the 1990s, those registered in the centre were mostly male. A potential reason for the earlier imbalance may lie in the higher proportion of neurological disease in males and, thus, a higher representation in the referrals to our centre, which then became more balanced in recent years. A long‐term follow‐up of 24 patients in Northern Portugal from 1975 to 2020 found that 54% of patients were male [10] and comparisons from Germany and the United States showed consistent sex‐balanced prevalence [24].
The Kayser‐Fleischer ring is an important diagnostic feature of WD and has been found to occur in almost all neurological patients and a significant number of hepatic patients [23]. The observed increase in patients with hepatic manifestations and asymptomatic individuals may have consequently led to a decreased proportion of patients presenting with Kayser‐Fleischer rings [10, 11, 23]. Similar trends with more liver manifestations and fewer Kayser‐Fleischer rings detected were reported in Portugal and Greece [10, 11].
Our study found that psychiatric symptoms affected every ninth patient, but this could be an underestimate as symptoms were recorded at referral admission and no systematic examinations provided by a specialist were available over the analysed timeframe of follow‐up.
Prior to 1970, WD patients in Poland were treated with DP only, with a few of the first patients diagnosed receiving British anti‐Lewisite (2,3‐dimercaptopropanol). Since zinc's introduction, DP and zinc have been used for initial therapy in similar proportions of patients. There are no consistent data on which drug is superior or inferior [25] and the manifestations of WD appear to be adequately managed with either therapy; hence, patients generally have a relatively normal quality of life [2]. Long‐term studies have also supported this claim and found that most patients stabilize and improve with treatment [26]. We found both drugs, DP and zinc, to be effective and well tolerated; consequently, few switches were observed compared to other studies [4, 25]. Patients may be switched between therapies not only because of adverse effects, ineffective treatment or comorbidities but due to financial/reimbursement/availability concerns [27]. Temporal limitations in access to or reimbursement of anti‐copper therapies were reported in Poland and could contribute to drug persistence findings.
Although many treated patients have a favourable prognosis, failure to comply with lifelong therapy can aggravate clinical manifestations [2]. Patients who have been on medications for many years will deteriorate even with a brief interruption in therapy [28, 29, 30]. A case series of 11 patients found that patients who discontinued DP all progressed to fulminant hepatitis or hepatic decompensation [31]. Self‐reported compliance was most likely overrated by patients in this analysis because, in our dedicated study on patient compliance, every fourth patient was found to be not taking their medication regularly [32].
The life expectancy of the general population in Poland has improved significantly since the 1950s (9 and 11 years for males and females, respectively). Thus, we observed no significant change in life‐years lost over decades; nevertheless, prognosis for WD patients improved, as shown in Kaplan–Meier analysis.
Our study limitations mainly result from missing data, as well as evolving clinical practice and definitions. We used a structured database to collect the data; however, patients diagnosed before the 2000s could only be studied retrospectively. We were unable to control well for confounders and adjust for clinical practice changes when assessing patients' prognoses. Temporal non‐medical changes in healthcare could also have affected the results and observed time trends. This is most evident for patients diagnosed in the 1970s when there was a lack of continuity of care at the reference centre due to unavailability of key medical personnel and less coordinated long‐term supervision, which resulted in relatively poor outcomes. As a consequence, more patients were both diagnosed and treated before referral to the reference centre. Comprehensive and structured liver function evaluation over a long follow‐up time was not possible; additionally, our centre has established thorough cooperation with hepatologists for the last two decades only. Consequently, we were unable to adopt a consistent approach to cirrhosis evaluation and decided to focus mainly on the neurological manifestation of WD. A specific study conducted in our centre on cirrhosis in patients newly diagnosed with the neurological phenotype of WD found that almost every second patient had cirrhosis diagnosed; all had oesophageal varices and at least one sign of hepatic disease on ultrasound examination [33].
In conclusion, this is the largest cohort of patients with WD (over 900 patients) reported in Poland, with long‐term follow‐up covering over seven decades. The number of diagnosed patients increased over time, with improved prognosis over the years, most likely due to more accurate and early diagnosis, effective therapy and qualified follow‐up care.
AUTHOR CONTRIBUTIONS
Anna Członkowska: conceptualization, investigation, methodology, project administration, supervision, writing – original draft preparation, writing – review and editing, validation, resources. Maciej Niewada: conceptualization, methodology, writing – original draft preparation, writing – review and editing, visualization, validation. Tomasz Litwin: investigation, writing – review and editing. Łukasz Kraiński: data curation, formal analysis, writing – review and editing, visualization. Marta Skowrońska: investigation, writing – review and editing. Agnieszka Piechal: investigation, writing – review and editing. Agnieszka Antos: investigation, writing – review and editing. Monika Misztal: investigation, writing – review and editing. Ishani Khanna: writing – review and editing. Iwona Kurkowska‐Jastrzębska: investigation, writing – review and editing.
CONFLICT OF INTEREST
None.
ACKNOWLEDGMENTS
The project was initiated by Prof. Ignacy Wald and made possible by many researchers contributing to data collection. The authors thank Adam Rozwadowski and Stanisław Wolanin for starting electronic data collection, Emma Marshman, Katarzyna Graczyk, Georgiana Wegmann and Florian Abel (Alexion Pharma GmbH) for input in the manuscript preparation.
Członkowska A, Niewada M, Litwin T, et al. Seven decades of clinical experience with Wilson's disease: Report from the national reference centre in Poland. Eur J Neurol. 2024;31:e15646. doi: 10.1111/ene.15646
Anna Członkowska and Maciej Niewada contributed equally to this work.
DATA AVAILABILITY STATEMENT
Limited data available on request due to privacy/ethical restrictions.
REFERENCES
- 1. Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. 2015;14:103‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers. 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006;120:151‐159. [DOI] [PubMed] [Google Scholar]
- 4. Członkowska A, Litwin T, Karliński M, et al. D‐penicillamine versus zinc sulfate as first‐line therapy for Wilson's disease. Eur J Neurol. 2014;21:599‐606. [DOI] [PubMed] [Google Scholar]
- 5. Ferenci P. Diagnosis of Wilson disease. Handb Clin Neurol. 2017;142:171‐180. [DOI] [PubMed] [Google Scholar]
- 6. Walshe JM. Cause of death in Wilson disease. Mov Disord. 2007;22:2216‐2220. [DOI] [PubMed] [Google Scholar]
- 7. Scheinberg IH, Sternlieb I. Wilson's Disease in Major Problems in Internal Medicine. WB Saunders; 1984. [Google Scholar]
- 8. Sandahl TD, Laursen TL, Munk DE, et al. The prevalence of Wilson's disease: an update. Hepatology. 2020;71:722‐732. [DOI] [PubMed] [Google Scholar]
- 9. Xie JJ, Wu ZY. Wilson's disease in China. Neurosci Bull. 2017;33:323‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garrido I, Marques M, Liberal R, et al. Wilson disease in Northern Portugal: a long‐term follow‐up study. Orphanet J Rare Dis. 2022;17:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tampaki M, Gatselis NK, Savvanis S, et al. Wilson disease: 30‐year data on epidemiology, clinical presentation, treatment modalities and disease outcomes from two tertiary Greek centres. Eur J Gastroenterol Hepatol. 2020;32:1545‐1552. [DOI] [PubMed] [Google Scholar]
- 12. Li N, Krishna SG, Hinton A, et al. Characteristics and outcomes of hospitalized patients with Wilson's disease in the United States: a national survey. Ann Hepatol. 2021;25:100362. [DOI] [PubMed] [Google Scholar]
- 13. EuroWilson . Accessed June 17, 2022. http://www.eurowilson.org/en/home/index.phtml
- 14. Ott P, Ala A, Askari FK, et al. Designing clinical trials in Wilson's disease. Hepatology. 2021;74:3460‐3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Członkowska A, Tarnacka B, Litwin T, et al. Wilson's disease‐cause of mortality in 164 patients during 1992‐2003 observation period. J Neurol. 2005;252(6):698‐703. doi: 10.1007/s00415-005-0720-4 [DOI] [PubMed] [Google Scholar]
- 16. Czlonkowska A, Rodo M, Wierzchowska‐Ciok A, et al. Accuracy of the radioactive copper incorporation test in the diagnosis of Wilson disease. Liver Int. 2018;38:1860‐1866. [DOI] [PubMed] [Google Scholar]
- 17. Svetel M, Pekmezović T, Petrović I, et al. Long‐term outcome in Serbian patients with Wilson disease. Eur J Neurol. 2009;16:852‐857. [DOI] [PubMed] [Google Scholar]
- 18. Choe EJ, Choi JW, Kang M, et al. A population‐based epidemiology of Wilson's disease in South Korea between 2010 and 2016. Sci Rep. 2020;10:14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sipilä JOT, Hietala M, Kytö V, Kaasinen V. Wilson's disease in Finland: a nationwide population‐based study. Mov Disord. 2020;35:2323‐2327. [DOI] [PubMed] [Google Scholar]
- 20. Członkowska A, Dzieżyc‐Jaworska K, Kłysz B, et al. Difficulties in diagnosis and treatment of Wilson disease‐a case series of five patients. Ann Transl Med. 2019;7(Suppl 2):S73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Naorniakowska M, Dądalski M, Kamińska D, et al. Clinical presentations of Wilson disease among Polish children. Dev Period Med. 2016;20:216‐221. [PubMed] [Google Scholar]
- 22. Macedo G, Maia JC, Gomes A, et al. Wilson's disease: challenging diagnosis, management, and liver transplantation timing. Transplant Proc. 2000;32:2668. [DOI] [PubMed] [Google Scholar]
- 23. European Association for Study of Liver . EASL clinical practice guidelines: Wilson's disease. J Hepatol. 2012;56:671‐685. [DOI] [PubMed] [Google Scholar]
- 24. Wahler S, Weiss KH. Hospitalization patterns of Wilson disease in Germany in comparison with US experience. Hepatology. 2020;72(Suppl 1):865‐866. [Google Scholar]
- 25. Tang S, Bai L, Hou W, et al. Comparison of the effectiveness and safety of D‐penicillamine and zinc salt treatment for symptomatic Wilson disease: a systematic review and meta‐analysis. Front Pharmacol. 2022;13:847436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beinhardt S, Leiss W, Stättermayer AF, et al. Long‐term outcomes of patients with Wilson disease in a large Austrian cohort. Clin Gastroenterol Hepatol. 2014;12:683‐689. [DOI] [PubMed] [Google Scholar]
- 27. Leung M, Wu Lanzafame J, Medici V. Switching pharmacological treatment in Wilson disease: case report and recommendations. J Investig Med High Impact Case Rep. 2020;8:2324709619896876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prashanth LK, Taly AB, Sinha S, et al. Prognostic factors in patients presenting with severe neurological forms of Wilson's disease. QJM. 2005;98:557‐563. [DOI] [PubMed] [Google Scholar]
- 29. Walshe JM, Dixon AK. Dangers of non‐compliance in Wilson's disease. Lancet. 1986;1:845‐847. [DOI] [PubMed] [Google Scholar]
- 30. Hoogenraad TU. Dangers of interrupting decoppering treatment in Wilson's disease. Arch Neurol. 1994;51:972‐973. [DOI] [PubMed] [Google Scholar]
- 31. Scheinberg HI, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson's disease. N Engl J Med. 1987;317:209‐213. [DOI] [PubMed] [Google Scholar]
- 32. Masełbas W, Członkowska A, Litwin T, Niewada M. Persistence with treatment for Wilson disease: a retrospective study. BMC Neurol. 2019;19:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Przybyłkowski A, Gromadzka G, Chabik G, et al. Liver cirrhosis in patients newly diagnosed with neurological phenotype of Wilson's disease. Funct Neurol. 2014;29(1):23‐29. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Limited data available on request due to privacy/ethical restrictions.