

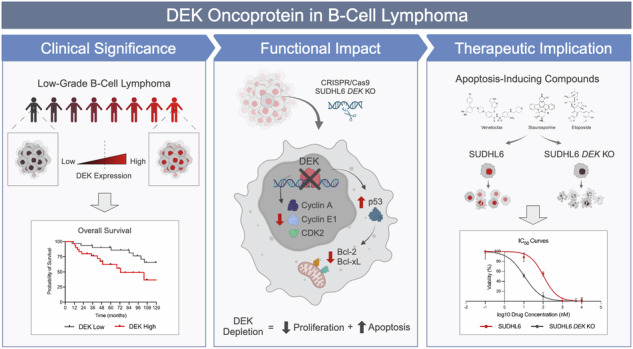
Abstract
This study sheds light on the pivotal role of the oncoprotein DEK in B-cell lymphoma. We reveal DEK expression correlates with increased tumor proliferation and inferior overall survival in cases diagnosed with low-grade B-cell lymphoma (LGBCL). We also found significant correlation between DEK expression and copy number alterations in LGBCL tumors, highlighting a novel mechanism of LGBCL pathogenesis that warrants additional exploration. To interrogate the mechanistic role of DEK in B-cell lymphoma, we generated a DEK knockout cell line model, which demonstrated DEK depletion caused reduced proliferation and altered expression of key cell cycle and apoptosis-related proteins, including Bcl-2, Bcl-xL, and p53. Notably, DEK depleted cells showed increased sensitivity to apoptosis-inducing agents, including venetoclax and staurosporine, which underscores the therapeutic potential of targeting DEK in B-cell lymphomas. Overall, our study contributes to a better understanding of DEK’s role as an oncoprotein in B-cell lymphomas, highlighting its potential as both a promising therapeutic target and a novel biomarker for aggressive LGBCL. Further research elucidating the molecular mechanisms underlying DEK-mediated tumorigenesis could pave the way for improved treatment strategies and better clinical outcomes for patients with B-cell lymphoma.
Subject terms: Oncogenes, Cancer genomics, B-cell lymphoma
Introduction
Non-Hodgkin B-cell lymphomas (NHLs) represent a diverse group of malignancies that exhibit a wide spectrum of pathologic and clinical features, ranging from indolent to aggressive forms [1]. Indolent lymphomas include follicular lymphoma (FL), marginal zone lymphomas (MZL), chronic lymphocytic lymphoma/small lymphocytic lymphoma (CLL/SLL), lymphoplasmacytic lymphoma (LPL), and low-grade B-cell lymphomas not otherwise specified (B-NOS) [2, 3]. These lymphomas present unique challenges due to their relatively slow progression and asymptomatic nature [3–5]. While generally associated with favorable outcomes, current therapeutic strategies are not yet curative and relapsed/refractory disease and tumor transformation represent a major clinical concern [6].
We previously used RNA-sequencing, whole exome sequencing, and clinical outcome measures to identify features of aggressive tumors and resolved biological heterogeneity across low-grade B-cell lymphoma (LGBCL) subtypes: defined as cases diagnosed with splenic MZL (SMZL), extranodal MZL (EMZL), nodal MZL (NMZL), LPL, or B-NOS [5]. This collection of subtypes has been the subject of previous studies due to shared clinical, immunophenotypic, and genetic characteristics that make definitive diagnosis of these diseases difficult [3, 5, 6]. In addition, we also identified a cell-cycle related transcriptomic signature whose increased expression was associated with early clinical failure across LGBCL tumors [5]. Further identification of prognostic markers and new therapeutic targets holds significant clinical implications across B-cell lymphomas by not only facilitating risk-adapted treatment approaches, but also enabling personalized medicine strategies [7, 8].
In this study, we identified the oncoprotein DEK as a regulator of early clinical failure signature genes and demonstrated its association with proliferation and aggressive disease in LGBCL. DEK is an evolutionarily conserved nuclear protein that participates in multifaceted cellular functions [9], including transcriptional regulation [10, 11], DNA replication [12], chromatin organization [12, 13], DNA damage repair [14], and mRNA processing [15]. In cancer, DEK’s overexpression and dysregulation is frequently associated with tumorigenesis and aggressive phenotypes across diverse malignancies, including acute myeloid leukemia [16, 17], breast cancer [18, 19], colorectal cancer [20, 21], and melanoma [21]. Several studies have identified mechanisms driving DEK dysregulation in tumors, which include the formation of DEK fusion proteins and copy number alterations [16, 17, 22, 23]. However, overexpression of DEK in tumors can also occur independently of genomic alterations, suggesting alternative modes of DEK dysregulation. While poorly understood, DEK expression may be dysregulated at the transcriptional level in tumors. Sequence analysis upstream of the DEK transcriptional start site identified binding sites for NF-Y and YY1, both transcription factors implicated in tumorigenesis [24, 25]. The DEK promoter has also been shown to be regulated by E2F transcription factors, which also have strong associations with cancer [21, 26]. However, the role of DEK in B-cell lymphomas remains unstudied.
Here, we report DEK expression in LGBCL is impacted by copy number alterations and its increased expression is associated with more proliferative tumors. We also found DEK expressed at higher levels in B-cell lymphoma tumors compared to normal tissue. Using gene expression data across B-cell development stages and from B-cell lymphoma cell lines, we further demonstrate DEK’s association with increased proliferative capacity and find its expression peaks in highly proliferative B-cell states. To further interrogate the role of DEK in B-cell lymphoma, we generated a DEK knockout cell line model that demonstrated significantly reduced proliferation compared to WT cells. We found DEK knockout cells had reduced expression of anti-apoptotic proteins, Bcl-2 and Bcl-xL, reduced expression of cell cycle proteins, and increased expression of tumor suppressor, p53. Finally, we demonstrate that DEK knockout cells are significantly more sensitive to treatment with apoptosis-inducing compounds, including venetoclax, staurosporine, and etoposide. Given its overexpression and crucial roles in cancer cell survival and proliferation, targeting DEK offers a rational approach to disrupt tumor growth and potentially enhance the efficacy of B-cell lymphoma treatment modalities.
Materials and Methods
Patient cohort
The patient cohort in this study was previously described in Hopper et al. [5]. Briefly, patients consented to the Molecular Epidemiology Resource from the University of Iowa and Mayo Clinic Lymphoma SPORE [4]. Samples included frozen tumor tissue from newly diagnosed (n = 61) or untreated (n = 3) LGBCL cases, including SMZL (n = 48), NMZL (n = 6), EMZL (n = 2), LPL (n = 2), and B-NOS (n = 3). Overall survival (OS) is defined as time from diagnosis to date of death. Benign CD19 + CD27+ memory B-cells were isolated from peripheral blood of healthy donors using the EasySep Human Memory B Cell Isolation Kit (STEMCELL Technologies Cat. 17864).
RNA and DNA sequencing
RNA-sequencing and paired tumor-normal whole exome sequencing (WES) data from Hopper et al. [5] were used in this study (dbGaP Study Accession: phs002552.v1.p1). Briefly, RNA or DNA were extracted from 64 LGBCL tumors. Bulk tumor RNA-seq was generated using the Illumina TruSeq RNA Exome kit, sequenced with 100 nucleotide paired-end reads by HiSeq 4000. WES data was generated using the Agilent SureSelect XT kit, sequenced with 100 nucleotide paired-end reads using the Illumina HiSeq 4000. Copy number analysis was performed using GISTIC 2.0.
Immunohistochemistry analysis
Tissue blocks of SMZL and benign spleens were obtained from the Mayo Clinic Tissue Repository. Immunohistochemistry (IHC) staining was performed on formalin-fixed paraffin-embedded (FFPE) tissue blocks cut into 5 µm sections. Slides were deparaffinized in xylene and rehydrated through changes of ethanol. Endogenous peroxidase was blocked using 3% hydrogen peroxide and absolute methanol for 10 min. Heat induced epitope retrieval was performed using pH 6.1 citrate buffer for 30 min (DAKO; cat. S1699). Slides were then blocked for 10 min (Background Sniper; cat. BS966L) and incubated for 45 min in DEK primary antibody (Atlas Antibodies; cat. HPA054505), diluted 1:200 in Van Gogh Diluent (Biocare Medical; PD902L). MACH 3 rabbit probe (Biocare; Cat. RP531L) and MACH 3 rabbit HRP-polymer (Biocare; cat. RH531L) were applied for 30 min each. Slides were then stained with DAB+ substrate chromogen for 10 min (DAKO; cat. K3468). Slides were counterstained with hematoxylin for 10 s (Sigma-Aldrich; cat. MHS16), dehydrated, and cover slipped using Permount mounting media (Fisher chemical; cat. UN1294). Slides were scanned using the MoticEasyScan Pro with MoticEasyScan software at 20X. Images were analyzed using Motic DSAssistant (v 1.0).
IHC images of DEK staining in lymph node samples from normal tissue (https://www.proteinatlas.org/ENSG00000124795-DEK/tissue/lymph+node#img) and non-Hodgkin lymphoma tumors (https://www.proteinatlas.org/ENSG00000124795-DEK/pathology/lymphoma#img) were obtained from the Human Protein Atlas proteinatlas.org [27]. Image retrieval was performed using the R package HPAanalyze [28].
Bioinformatic analysis
We used the application Mining Algorithm for GenetIc Controllers, MAGIC [29], to predict transcription factors and cofactors driving expression of our previously published early clinical failure signature genes [5]. All early clinical failure signature genes were included as query genes and the “5Kb_gene” matrix was chosen. Significance was reported as Benjamini-Hochberg corrected p-values. RNA-sequencing data from FL tumors were obtained from Krull et al. [30], dbGaP Study Accession: phs002989.v1.p1. DLBCL tumor RNA-sequencing data was acquired from Wenzl et al. [31]. Human cancer cell line RNA-sequencing data were obtained from the Broad-Novartis Cancer Cell Line Encyclopedia [32, 33]. RNA-sequencing data from germinal center B-cell development was obtained from Gene Expression Omnibus Series GSE139833 from Holmes et al. [34]. DEK RNAi data was obtained from the Broad Institute DepMap portal [35] and the DLBCL GEO and normal blood GTEx expression plot was generated using GEPIA [36].
Cell lines
DoHH2 (DSMZ), Karpas-1718 (Sigma-Aldrich), and MWCL-1 [37] cell lines were cultured in RPMI medium (Corning; cat. 10-040-CV) supplemented with 10% FBS (Corning; cat. 35-010-CV) and 1X Penicillin Streptomycin (Gibco; cat. 15140-122). JeKo-1 (ATCC), OCI-Ly3 (courtesy of Shipp Lab), and SUDHL6 (DSMZ) cell lines were cultured in RPMI medium supplemented with 20% FBS and 1X Penicillin Streptomycin. Cell lines were sent to Labcorp for STR profiling and authentication. All cells were cultured at 37 °C and 5% CO2.
Fluorescent microscopy and image analysis
For B-cell lymphoma cell lines, 200,000 cells were plated onto poly-L-lysine-coated coverslips for 20 min at room temperature. Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences; cat. 15710) in PBS for 18 min and permeabilized with 0.15% Triton X-100 Surfact-Amps (ThermoFisher; cat. 85111) in 1X PBS for 3 min at room temperature. Coverslips were then washed 1X PBS and blocked with goat serum-based blocking buffer (Millipore; cat. G9023) for 45 min at room temperature. Cells were then stained with purified mouse anti-human DEK antibody (BD Biosciences; cat. 610948) diluted 1:250 in blocking buffer at 4 °C overnight; “no primary” controls were treated with blocking buffer alone. Coverslips were then washed and blocked for 45 min at room temperature with blocking buffer. Cells were then treated with 1:500 goat anti-mouse IgG Alexa Fluor 568 (Invitrogen; cat. A11004) and 1:50 Alexa Fluor 488 Phalloidin (Invitrogen; cat. A12379) in blocking buffer for 3 h at 4 °C in the dark. Coverslips were washed and DNA was stained using 1:1000 Hoechst 33342 staining dye solution (Abcam; ab228551) in 1X PBS for 2 min in the dark and then washed with diH2O for 2 min in the dark. Coverslips were mounted onto glass slides using anti-fade mounting medium. All slides were prepared and examined on the same day using an LSM-800 laser scanning confocal microscope with a C-Acromat 63x/1.4 oil objective (Carl Zeiss). Images were acquired using the ZEN 2.6 System (Blue Edition) and processed using ZEN 2.3 lite software (Carl Zeiss).
Generation of CRISPR/Cas9 knockout cell lines
We utilized the Feng Zhang lab, Broad Institute CRISPR gRNA tool by GenScript [38] to identify sgRNA sequences against human DEK. The following sequences were generated to include DEK targeting sequences and the underlined BbsI restriction sites and an additional cytosine nucleotide as an optimal base for the U6 promoter: sgRNA top oligo 5’-CACCGGTGTAGATTTCTAAGTTCAT-3’; sgRNA bottom oligo 5’-AAACATGAACTTAGAAATCTACACC-3’. Cloning was performed following the Zhang Lab Genome Engineering Toolbox PX330-based plasmid protocol [39]. sgRNA oligo pairs were annealed and cloned into pSpCas9(BB)-2A-GFP (Addgene; cat. PX458) and transformed into One Shot Stbl3 Chemically Competent E. coli (Invitrogen; cat. C737303). Sequences were confirmed using QIAprep Spin Miniprep Kit (Qiagen; cat. 27104) for plasmid isolation and Sanger DNA sequencing from the U6 promoter using primer LKO.1 5’ (5’-GACTATCATATGCTTACCGT-3’). Large-scale plasmid preparation and purification was performed using a Qiagen EndoFree Plasmid Maxi Kit (Qiagen; cat. 12362). Amaxa Human B Cell Nucleofector Kit (Lonza; cat. VPA-1001) and Amaxa Nucleofector II Device were used for electroporation with 4 million cells and 10 µg of plasmid per reaction. 48 hours post-electroporation, GFP+ cells were single-cell sorted into 96-well round bottom plates by the Mayo Clinic Flow Cytometry Core using an Aria 4 equipped with a P100 nozzle at 4 °C.
Western blot analysis
Cells were lysed using ice-cold 1X RIPA Buffer (CST; cat. 9806S), supplemented with 1X PMSF and 1X HALT. Protein concentration was quantified using Bio-Rad DC Protein Assay (Bio-Rad; cat. 500113, 5000114, 5000115) and 50 µg of protein was run in each lane. Proteins were separated using 10% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad; cat. 4561033) and transferred to a Immobilon-FL PVDF membrane (Millipore; cat. IPFL00010). Membranes were blocked using EveryBlot Blocking Buffer (Bio-Rad; cat. 12010020) for 5 min at room temperature and then incubated overnight at 4 °C with the indicated primary antibody. Primary antibodies include: DEK (1:1000; BD Biosciences cat. 610948), beta actin (1:1000; Abcam cat. ab8229), cyclin A (1:2000; CST cat. 4656), cyclin E1 (1:1000; CST cat. 4129), CDK2 (1:1000; CST cat. 2546), p27 (1:2500; R&D Systems cat. MAB2256), c-Myc (1:1000; CST cat. 9402), PARP (1:1000; CST cat. 9542), Bim (1:1000; CST cat. 2933), Bcl-2 (1:1000; CST cat. 2872), Bcl-xL (1:1000; CST cat. 2764), Caspase-8 (1:1000; CST cat. 9746), Mcl-1 (1:1000; CST cat. 4572), Bax (1:1000; CST cat. 2772), p53 (1:1000; sc-126), p63a (1:1000; CST 4892). Membranes were washed with 1X TBS containing 0.1% Tween-20 (Bio-Rad cat. 1706435) and incubated with the appropriate secondary antibody at room temperature for 45 min: donkey anti-mouse IgG 800 (1:12500; LI-COR cat. 926-32212), donkey anti-rabbit IgG 800 (1:12500; LI-COR cat. 926-32213), and/or donkey anti-goat IgG 680 (1:12500 dilution; LI-COR cat. 926-68074). Membranes were washed and bands were visualized using the Licor Odyssey CLx Imaging System. Quantification of protein bands was performed using Image Studio Software (v5.2). All quantifications were scaled to β-actin levels detected in the same lysate, processed in parallel. Fold-changes of DEK KO versus WT were used as input for statistical analysis.
MTS proliferation assays
Cell cycle synchronization was performed by resuspending cells in culture medium containing 1% FBS and incubating at 37 °C and 5% CO2 in a humidified incubator overnight. Following synchronization, cells were resuspended in fresh full-serum medium and plated in a 96-well plate at a concentration of 5000 cells/100 µL media for each well. Cells were plated in triplicate, incubated for the indicated timepoints, and analyzed using CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega cat. G5421). MTS working solution was added to each well and plates were incubated for 3 h at 37 °C. Plates were analyzed using a SpectraMax M2 plate reader at 490 nm absorption. Wells containing media only plus MTS working solution were used for background subtraction.
Drug treatments and annexin V/FVD staining
Staurosporine (cat. HY-15141), venetoclax (cat. HY-15531), and etoposide (cat. HY-13629) were purchased in DMSO solution form from MedChemExpress. Cells were seeded in 6-well tissue culture plates with 1.5 million cells in 2.7 mL of complete media per well. Compounds were serially diluted to 10X the desired final concentration in complete media and 300 µL was added to each well. Plates were incubated for 24 h at 37 °C. Following incubation, cells were washed with 1X PBS and resuspended in 1 mL of PBS + 1 µL of Zombie Violet Fixable Viability Dye (FVD; BioLegend cat. 423114). Cells were incubated with FVD for 30 min at 4 °C protected from light. Cells were then washed with 1X Flow Cytometry Staining Buffer (eBioscience cat. 00-4222-26) and washed again with 1X Annexin V Binding Buffer (BioLegend cat. 422201). Cells were resuspended in 100 µL of binding buffer and 5 µL of fluorochrome-conjugated Annexin V (BioLegend cat. 640941) was added to the cell suspension. Cells were incubated for 15 min at room temperature protected from light and analyzed by flow cytometry (BD FACSCanto II).
Statistical analysis
GraphPad Prism version 9.0.2 and R tools were used to analyze and visualize data [40]. All correlation plots were generated using the ggscatter function of ggpubr [41], with p-values determined by the Pearson correlation method. FlowJo v10.8.1 (BD Life Sciences) was used to analyze flow cytometry data. Statistical significance of p < 0.05 was considered significant unless stated otherwise in the text or figure legend.
Results
High DEK expression is associated with early clinical failure and proliferative tumors in LGBCL and is a target of copy number alterations
Building on our previous work [5], we sought to identify transcriptional regulators driving increased expression of genes associated with inferior clinical outcome in LGBCL, described as the “early clinical failure signature”. To do this, we used the bioinformatic tool MAGIC to mine publicly available ChIP-seq data and identified DEK as a key transcriptional regulator of genes in our early clinical failure signature (p = 0.008) (Fig. 1A). Using bulk RNA-sequencing data from 61 LGBCL tumors, we found DEK mRNA expression correlated with the corresponding early clinical failure score of each patient (R = 0.61, p = 2.4 × 10−7) (Fig. 1B), indicating a strong association between DEK and increased risk of aggressive disease. To assess the association of DEK expression with LGBCL clinical outcome, we separated cases into DEK low and DEK high, defined as cases below and above the median DEK expression, respectively. Cases with high DEK expression showed significantly inferior overall survival compared to low DEK cases (HR 2.99; 95% CI 1.21–7.36; p = 0.00175) (Fig. 1C). Given DEK expression was associated with aggressive tumors, we hypothesized tumors with high DEK expression may be more proliferative. Thus, we evaluated expression of cell cycle genes from the KEGG Cell Cycle Molecular Signature (M7963) [42] in DEK high versus low patients, which showed increased expression in the DEK high group (Fig. 1D). Additionally, correlation of DEK expression with common markers of proliferation [43] showed significant correlation between DEK and MKI67, MCM2, PLK4, CCNB1, TOP21, and PCNA (Fig. 1E).
Fig. 1. DEK is associated with aggressive disease and is impacted by copy number alterations.

A ChIP signal of DEK protein as a regulator of early clinical failure signature genes identified by MAGIC TF analysis. B Correlation of DEK expression from LGBCL tumor RNA-seq data versus early clinical failure signature score. C Kaplan-Meier curves showing overall survival of patients expressing low vs. high levels of DEK. P-values were determined by log-rank test. D Gene expression of KEGG Cell Cycle genes from the Molecular Signatures Database. Heatmap shows z-scored log2(TPM + 1) values from LGBCL tumor RNA-sequencing data, with DEK high cases annotated in red and DEK low cases in green. E Correlation of DEK expression with expression of proliferation markers in RNA-seq data from LGBCL tumors. F DEK expression in LGBCL tumors by copy number status of DEK. G Log2(TPM + 1) expression of DEK across multiple B-cell lymphoma tumor types and healthy donor CD27 + CD19+ sorted memory B-cells. H DLBCL tumor versus normal DEK expression using the DLBC dataset from GEPIA. P-value was calculated in GEPIA using a one-way ANOVA with disease state as the variable for calculating differential expression. I Quantification of DEK expression from western blot analysis comparing benign spleens (n = 4) to SMZL spleens (n = 4). DEK expression levels were normalized to β-actin for each sample.
Using paired tumor-normal whole exome sequencing (n = 61), we next assessed whether DEK was impacted by genomic alterations. While DEK was not mutated in any LGBCL cases, we found 4 of 61 cases (6.6%) affected by either a copy number gain or amplification at 6p22.3 (Fig. 1F). Matched RNA-sequencing data further demonstrated DEK expression was significantly impacted by copy number status, with DEK gains/amplifications corresponding to increased mRNA expression (p = 0.035) (Fig. 1F).
We then wanted to assess the expression of DEK across B-cell lymphoma tumor types. Using RNA-sequencing data of FL whole tumor and B-cell sorted samples from Krull et al. [30] and DLBCL whole tumor data from Wenzl et al. [31, 44], we found DEK was expressed across B-cell lymphomas, with higher expression in FL and DLBCL compared to LGBCL (Fig. 1G). Additionally, DEK was found to be expressed in B-cells sorted from FL tumors and normal memory B-cells, suggesting B-cell specific expression (Fig. 1G). To examine the expression of DEK in a cohort of tumor versus normal samples, we used the web-based tool GEPIA [36] to visualize gene expression data from the TCGA and GTEx databases. From this, we were able to obtain gene expression data from DLBCL tumors (n = 47) and normal blood (n = 337), which demonstrated significantly increased expression of DEK in tumors versus normal (p < 0.001) (Fig. 1H).
This trend was also observed at the protein level by western blot in SMZL, where DEK expression was significantly increased in mononuclear cells derived from SMZL spleens compared to those derived from benign spleens (p = 0.0047) (Fig. 1I; Supplementary Fig. 1A). Collectively, these results indicate that DEK expression is associated with LGBCL prognosis and a proliferative gene signature, and that elevated DEK expression is found across multiple B-cell tumor types.
DEK expression in B-cell lymphomas by immunohistochemistry
Given the increased expression of DEK protein in SMZL tumors compared to benign spleens, we next sought to examine DEK expression in situ in benign and malignant human tissues. To do this, we performed IHC staining of DEK using FFPE tissue blocks from benign spleen tissue and spleens from SMZL patients. We found minimal levels of DEK staining in benign spleen tissue but detected the presence of nuclear DEK staining in SMZL tumors, thus confirming an increase in DEK protein levels in a subset of tumors compared to benign tissue (Supplementary Fig. 1B).
To further build on these findings, we also obtained IHC images from the Human Protein Atlas (proteinatlas.org) [27] of DEK staining in normal lymph node tissue and lymph nodes from NHL tumors. These images show weak nuclear staining of DEK in non-germinal center tissue and moderate staining in germinal centers of normal lymph node tissue (Supplementary Fig. 1C). In contrast, lymph nodes obtained from NHL tumors display a loss of normal lymph node architecture, with low-grade NHL tissue showing weak nuclear staining of DEK across <25% of cells and high-grade NHL tissue showing moderate nuclear staining of DEK in >75% of cells (Supplementary Fig. 1C). Collectively, these images show increased DEK expression in tumors compared to benign tissues and demonstrate a further increase in DEK expression in high-grade NHL compared to low-grade.
Patterns of DEK expression across malignant and normal B-cells
To assess the dynamics of DEK expression in B-cells specifically, we acquired cell line RNA-sequencing data from B-cell lymphoma cell lines in the Broad-Novartis Cell Line Encyclopedia. We found nearly all B-cell lymphoma cell lines express DEK, with the highest expression detected in cell lines derived from aggressive lymphomas, including DLBCL (Fig. 2A). Excluding the more distinct Burkitt lymphoma, we again interrogated the expression of DEK compared to common proliferation markers in B-cell lymphoma cell lines and found strong positive correlations between DEK expression and MKI67, MCM2, PLK4, CCNB1, TOP21, and PCNA (Fig. 2B). This established a strong correlation between DEK expression and proliferation in B-cell lymphoma cell lines. Consistent with malignant B-cell mRNA expression, RNA-sequencing data derived from non-malignant germinal center (GC) B-cell developmental stages acquired from Holmes et al. [34] revealed that DEK expression peaked at the dark zone stage of B-cell development, the most proliferative stage of B-cell development (Fig. 2C).
Fig. 2. DEK expression is associated with cell lines derived from aggressive B-cell lymphoma proliferative B-cell programs.

A DEK log2(TPM + 1) values in B-cell lymphoma/leukemia cell lines. RNA-seq data from the Broad-Novartis Cell Line Encyclopedia was acquired through the DepMap Portal. B Correlation of DEK expression with expression of proliferation markers in cell line RNA-seq data from Fig. 2A. C DEK log2(TPM + 1) values across B-cell development stages. D Western blot of DEK protein expression across B-cell lymphoma cell lines. Bar plot represents DEK expression normalized to β-actin. Representative blot of n = 3 biological replicates, with quantification of all blots shown in the bar plot. Blot has been cropped to display indicated proteins based on molecular weight. E DEK localization in B-cell lymphoma cell line SUDHL6 by confocal microscopy.
To further validate these findings, we assessed expression of DEK at the protein level in a panel of B-cell lymphoma cell lines by western blot (Fig. 2D; Supplementary Figure 2). While we noted variable expression of DEK across cell lines, the more aggressive DLBCL lines, OCI-Ly3 and SUDHL6, demonstrated the highest levels of DEK expression, with lower expression detected in the transformed follicular lymphoma (DoHH2) and mantle cell lymphoma (Jeko-1) lines, and the lowest expression in LGBCL cell lines derived from splenic marginal zone lymphoma (SMZL) and Waldenstrom Macroglobulinemia (MWCL-1) (Fig. 2D). The high expression of DEK in SUDHL6 cells is further supported by the presence of a DEK (6p22.3) amplification in these cells, aligning with our patient derived data (Fig. 1F). We also assessed expression and localization of DEK by immunofluorescence and found that DEK showed predominantly nuclear localization, consistent with its role as a transcriptional regulator (Fig. 2E; Supplementary Figure 3). In line with our western blot data, we also noted increased intensity of DEK staining in the DLBCL SUDHL6 cells (Fig. 2E) compared to other cell lines (Supplementary Figure 3). This data highlights, once again, that high levels of DEK expression are found in cell lines derived from aggressive disease and proliferative germinal center B cell stages.
DEK depletion inhibits proliferation and is accompanied by reduced expression of cell cycle genes, reduced Bcl-2 and Bcl-xL expression, and increased p53 expression
We next sought to study the impact of DEK depletion in B-cell lymphoma cell lines to further interrogate its function. First, we made use of publicly available high-throughput RNAi screening data from the Broad Institute’s DepMap Portal [33, 35] to assess the impact of transient suppression of DEK expression on cellular fitness. We selected all B-cell lymphoma cell lines with available data (n = 10) and found 9 out of 10 cell lines experienced a reduction in cellular fitness upon DEK RNAi, as indicated by the negative Gene Effect scores (Fig. 3A). To further investigate the molecular mechanisms underlying DEK’s role in regulating cellular fitness and proliferation, we used CRISPR/Cas9 to generate a stable knockout (KO) of DEK in the cell line, SUDHL6, which has an amplification of DEK at 6p22.3 and expresses high levels of DEK protein (Fig. 3B). In line with RNAi data from Fig. 3A, DEK KO cells demonstrated significantly reduced proliferation compared to wild type (WT) over a 5-day time course (Fig. 3C). Given the reduced cellular fitness of DEK KO cells, we hypothesized that DEK may be regulating the expression of cell cycle, apoptosis, or stress response proteins. To test this hypothesis, we performed western blot analyses of major cell cycle, apoptosis, and cellular stress response proteins in cell cultures of at least 95% viability to assess baseline differences between SUDHL6 WT and DEK KO cells (Fig. 3D, E; Supplementary Figure 4). We observed drastically reduced expression of anti-apoptotic proteins Bcl-2 (log2FC = −3.96 ± 0.37; p < 0.001) and Bcl-xL (log2FC = −1.10 ± 0.12; p < 0.001) in DEK KO cells compared to WT, and a minor reduction of full-length PARP (log2FC = −0.45 ± 0.24; p = 0.025). We also found significant reduction of cyclin A (log2FC = −0.40 ± 0.26; p = 0.01), cyclin E1 (log2FC = −0.40 ± 0.27; p = 0.038), and CDK2 (log2FC = −0.57 ± 0.25; p = 0.015) expression in DEK KO cells versus WT, as well as significantly increased expression of p53 (log2 FC = 1.39 ± 0.37; p = 0.01) (Fig. 3D, E). These results highlight the critical role of DEK in maintaining cellular fitness through regulation of key regulatory pathways, such as apoptosis and cell cycle control.
Fig. 3. DEK depletion inhibits proliferation and is accompanied by reduced expression of cell cycle genes, reduced Bcl-2 and Bcl-xL expression, and increased p53 expression.

A Chronos Gene Effect from RNAi of DEK in B-cell lymphoma cell lines. Gene effect represents the impact of DEK RNAi on cellular fitness, with negative values representing a negative impact to cellular fitness. B Validation of DEK knockout by western blot and Sanger sequencing. C In vitro five-day proliferation assay of synchronized SUDHL6 WT versus DEK KO cells. Proliferation was measured by MTT assay with n = 3 biological replicates. Assays were run with technical triplicates. D Western blot analysis of cell cycle, apoptosis, and stress response proteins in SUDHL6 WT and DEK KO cells. Cell lines were harvested at ≥ 95% viability. Blots have been cropped to display indicated proteins based on molecular weight. The corresponding DEK and β-actin controls were processed in parallel and are displayed below their corresponding samples. E Quantifications of protein expression in DEK KO versus WT cells of n = 3 biological replicates. All quantifications were scaled to β-actin levels and fold-changes of DEK KO versus WT were used as input for statistical analysis. Plots depict the mean and SD of log2 fold change values, where negative values represent reduced expression in DEK KO. Blue bars indicate a statistically significant decrease of expression in DEK KO cells, red bars indicate a statistically significant increase in DEK KO cells.
DEK depleted cells are more susceptible to apoptosis and demonstrate increased sensitivity to venetoclax treatment
Given the drastic reduction of Bcl-2 and Bcl-xL expression in DEK KO cells, we hypothesized DEK depletion may cause cells to be more susceptible to apoptosis and cell death. To test this hypothesis, we treated DEK KO and WT cells with varying concentrations of the apoptosis-inducing compounds venetoclax (Fig. 4A), staurosprine (Fig. 4B), and etoposide (Fig. 4C) for 24 h. We then assessed levels of viable, apoptotic, and dead cells by flowing cytometry using annexin V and fixable viability dye staining (Zombie Violet). Under all three treatments, DEK KO cells were significantly more susceptible to apoptosis and cell death compared to WT cells, with significantly reduced viability as indicated by reduced frequencies of annexin V-FVD- cells (Fig. 4A–C, Supplementary Fig. 5). Notably, DEK KO cells had a 10-fold reduced IC50 value of staurosporine and a 5-fold reduced venetoclax IC50 value compared to WT, demonstrating that targeting DEK expression significantly improves the efficacy of these compounds (Supplementary Fig. 5). Taken together, these results indicate DEK plays a critical role in cell survival and resistance to apoptosis, where DEK depletion renders B-cell lymphoma cells more susceptible to apoptosis-mediated cell death.
Fig. 4. DEK knockout cells are more susceptible to apoptosis and demonstrate increased sensitivity to venetoclax treatment.

Annexin V and Zombie Violet staining of SUDHL6 WT versus DEK KO cells treated with the indicated concentrations of (A) venetoclax, (B) staurosporine, and (C) etoposide. Cells were treated for 24 h at 37 °C, stained with Annexin V-FITC and Zombie Violet FVD, and gated on singlets. Bar charts show the mean and SD of frequencies of viable cells (Annexin V-FVD-) from n = 3 biological replicates.
Discussion
Our study introduces novel insights into DEK’s role in B-cell lymphomas, an area that remains largely unexplored. We demonstrate that DEK expression in LGBCL is influenced by copy number alterations, is highly expressed in aggressive NHL subtypes, and is associated with inferior overall survival. We also found DEK expression elevated at the protein level in tumor tissue compared to benign tissue by both western blot analysis and IHC staining. This association is further supported by our analysis of DEK expression across B-cell development stages, where DEK expression peaks in highly proliferative states. Additionally, our generation of a DEK knockout cell line model reveals the impact of DEK depletion on reducing proliferative capacity, altering expression of cell cycle and apoptosis-related proteins, and increasing sensitivity to apoptosis-inducing compounds. These findings underscore DEK’s role in promoting tumor growth and survival in B-cell lymphomas, necessitating additional studies to assess its potential as a therapeutic target and biomarker of aggressive LGBCL.
Building on previous research, our findings align with studies in other cancers, such as acute myeloid leukemia (AML) [16, 17], breast cancer [18, 19], colorectal cancer [20], and others [21, 45–48], where DEK overexpression has been linked to tumorigenesis and aggressive phenotypes. For instance, Privette et al. [19] found increased DEK expression in breast carcinomas compared to normal breast tissue and noted DEK expression correlated with higher tumor grade, increased lymph node involvement, and inferior disease-free survival. Similarly, Sun et al. [48] performed IHC staining of pancreatic ductal adenocarcinomas and adjacent normal tissue, which showed a significant increase in DEK staining in tumor tissues compared to normal. They also highlighted strong correlations between DEK expression, tumor size, stage, and grade. Similarly, we also found DEK expression increased at the protein level in NHL tumors compared to benign tissues. Our analysis also revealed a strong positive association between DEK expression and the early clinical failure signature [5], indicating DEK plays a role in driving aggressive disease in LGBCL. We found high DEK expression is linked to increased expression of cell cycle genes and correlates strongly with proliferation markers, suggesting a direct role in promoting tumor growth and progression. This is further supported by the significantly inferior overall survival observed in LGBCL cases with high DEK expression, highlighting the clinical relevance of DEK as a prognostic biomarker.
The genomic landscape of LGBCL also contributes to our understanding of DEK’s regulation and function. While DEK itself is not mutated in LGBCL cases, it is a target of copy number alterations, with gains and amplifications leading to elevated DEK expression. This illustrates one mechanism by which DEK expression is dysregulated, though future studies examining alternative means of DEK upregulation should be investigated. Consistent with our findings, Orlic et al. [49] identified 6p22 gains in retinoblastoma, with only DEK showing correlation between expression and genomic copy number among genes in the region. Another study by Shibata et al. [10] identified DEK (6p22.3) chromosomal gains were significantly associated with poor prognosis in high-grade neuroendocrine carcinoma. In addition to copy number gains, DEK is also dysregulated by translocations. DEK was initially discovered in AML, where t(6;9)(p23;q34) translocations generate DEK::NUP214 fusion proteins in 1% of AML [16, 17]. Tumors harboring these translocations have a distinct AML classification due to the early age of onset and poor prognosis of these cases [16, 17]. More recently, DEK has been identified in a novel rearrangement with AFF2 on chromosome Xq28, which forms a DEK::AFF2 fusion protein in patients with carcinomas of the head and neck [22, 23, 50]. Like AML, tumors with these translocations have been noted as distinct entities with aggressive phenotypes and frequent metastases [22, 23]. Together, these studies and ours provide insight into the molecular mechanisms underlying DEK dysregulation in cancer and its implications in driving aggressive disease behavior.
Our study also extends beyond clinical correlations to functional analyses, where DEK depletion significantly inhibits proliferation in B-cell lymphoma cell lines. Stable CRISPR/Cas9-mediated depletion of DEK in SUDHL6 cells was accompanied by reduced expression of cell cycle proteins, reduced expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, and increased p53 expression, highlighting DEK’s role in promoting cell survival and resistance to apoptosis. These findings are consistent with previous studies in which DEK depletion in other cancer cell lines results in reduced cell growth and induction of apoptosis [11, 19, 51]. Moreover, we showed that DEK depletion caused increased sensitivity to apoptosis-inducing compounds. Notably, the reduction in the IC50 value of venetoclax in DEK knockout cells indicates that targeting DEK could potentially improve the effectiveness of venetoclax-based therapies in treating B-cell lymphomas. This data adds to the growing body of literature implicating DEK expression in protection against cytotoxic insults. Notably, increased DEK expression has been shown to lead to chemoresistance and protection against genotoxic agents, with loss of DEK expression resulting in increased cell death by DNA-damaging agents. [24, 52, 53]. DEK has also been shown to play a role in determining the proliferative fate of cells, with studies showing increased DEK expression resulting in inhibition of cellular senescence [52, 54]. Similarly, studies in mouse models showed increased resistance to irradiation in DEK knockout mice compared to DEK WT mice, where radioresistance was attributed to an increase in protective quiescence in DEK knockout progenitor cells [55]. Collectively, these studies highlight DEK’s role in regulating the decision between cell cycle withdrawal (e.g. senescence and quiescence) and proliferation as a potential mechanism linking DEK expression to tumorigenesis.
In conclusion, our study adds to the growing body of evidence implicating DEK in tumorigenesis and aggressive phenotypes in various cancers. Our study highlights DEK as a key player in LGBCL pathogenesis, offering insights into its regulatory mechanisms, association with clinical outcomes, and potential as a therapeutic target. Our analysis of DEK expression across B-cell lymphoma and developmental stages emphasizes its relevance throughout B-cell differentiation and malignancy, underscoring its potential as a universal therapeutic target across diverse B-cell malignancies. Future research directions should focus on elucidating the specific molecular pathways modulated by DEK and developing targeted therapies to exploit its dysregulation in B-cell lymphomas.
Supplementary information
Acknowledgements
This work was supported in part by the NIH/NCI grants SPORE-P50 CA97274 (J.R. Cerhan and A.J. Novak), R01 CA212162 (A.J. Novak and J.R. Cerhan), U01 CA195568 (J.R. Cerhan), T32 AI007425 (J.E. Krull), T32AI170478 (M.A. Hopper), and the Predolin Foundation Biobank.
Author contributions
Manuscript review was performed by all authors. MAH: Conceptualization, methodology, formal analysis, data curation, writing original draft. ARD: Formal analysis, data curation. JSW: Data curation. JPN: Data curation. MSL: Data curation. MKM: Methodology, data curation. XW: Methodology, data curation. KW: Methodology, data curation. JEK: Data curation. VS: Methodology. MJM: Methodology. ZY: Methodology. MDD: Data curation. TMH: Data curation. BKL: Data curation. LMR: Data curation. TEW: Data curation. SMA: Data curation. JRC: Data curation, funding acquisition. DJ: Data curation, funding acquisition. AJN: Conceptualization, writing original draft, project administration, funding acquisition.
Data availability
The data that support the findings of this study are available from dbGaP Study Accession: phs002552.v1.p1 (LGBCL data), dbGaP Study Accession: phs002989.v1.p1 (FL data), GEO: GSE139833 (bulk B-cell development data), Broad Institute Dependency Map Portal at https://depmap.org/portal/ (cell line RNA-sequencing and RNAi), and GEPIA at http://gepia.cancer-pku.cn/ (GEO + GTEx data).
Competing interests
AJN has research funding from Bristol Myers Squibb. MAH, ARD, JSW, JPN, MSL, MKM, XW, JEK, VS, MJM, ZY, MDD, TMH, BKL, LMR, TEW, SMA, JRC, and DJ declare no competing financial interests.
Ethics approval and consent to participate
All studies were performed in accordance with the Declaration of Helsinki. Patients provided written consent for use of clinical samples at study enrollment and this study was approved by the Mayo Clinic Institutional Review Board.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41408-024-01145-0.
References
- 1.Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBDO, Berti E, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36:1720–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smyth E, Cheah CY, Seymour JF. Management of indolent B-cell Lymphomas: A review of approved and emerging targeted therapies. Cancer Treat Rev. 2023;113:102510. [DOI] [PubMed] [Google Scholar]
- 3.Tracy SI, Larson MC, Feldman AL, Maurer MJ, Novak AJ, Slager SL, et al. The utility of prognostic indices, early events, and histological subtypes on predicting outcomes in non-follicular indolent B-cell lymphomas. Am J Hematol. 2019;94:658–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerhan JR, Link BK, Habermann TM, Maurer MJ, Feldman AL, Syrbu SI, et al. Cohort Profile: The Lymphoma Specialized Program of Research Excellence (SPORE) Molecular Epidemiology Resource (MER) Cohort Study. Int J Epidemiol. 2017;46:1753–1754i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopper MA, Wenzl K, Hartert KT, Krull JE, Dropik AR, Novak JP, et al. Molecular classification and identification of an aggressive signature in low-grade B-cell lymphomas. Hematological Oncol. 2023;41:644–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi D, Bertoni F, Zucca E. Marginal-Zone Lymphomas. N. Engl J Med. 2022;386:568–81. [DOI] [PubMed] [Google Scholar]
- 7.Simon R. Clinical trial designs for evaluating the medical utility of prognostic and predictive biomarkers in oncology. Per Med. 2010;7:33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goetz LH, Schork NJ. Personalized medicine: motivation, challenges, and progress. Fertil Steril. 2018;109:952–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devany M, Kappes F, Chen KM, Markovitz DM, Matsuo H. Solution NMR structure of the N-terminal domain of the human DEK protein. Protein Sci. 2008;17:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibata T, Kokubu A, Miyamoto M, Hosoda F, Gotoh M, Tsuta K, et al. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene. 2010;29:4671–81. [DOI] [PubMed] [Google Scholar]
- 11.Sandén C, Järvstråt L, Lennartsson A, Brattås PL, Nilsson B, Gullberg U. The DEK oncoprotein binds to highly and ubiquitously expressed genes with a dual role in their transcriptional regulation. Mol Cancer. 2014;13:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 2000;14:1308–12. [PMC free article] [PubMed] [Google Scholar]
- 13.Waldmann T, Eckerich C, Baack M, Gruss C. The ubiquitous chromatin protein DEK alters the structure of DNA by introducing positive supercoils. J Biol Chem. 2002;277:24988–94. [DOI] [PubMed] [Google Scholar]
- 14.Kavanaugh GM, Wise-Draper TM, Morreale RJ, Morrison MA, Gole B, Schwemberger S, et al. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res. 2011;39:7465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu B, Sun Y, Zhang Y, Xing Y, Suo J. DEK modulates both expression and alternative splicing of cancer‑related genes. Oncol Rep. 2022;47:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiriches C, Nicolaisen N, Wieske M, Elhaddad H, Mehmetbeyoglu E, Alvares C, et al. Understanding a high-risk acute myeloid leukemia by analyzing the interactome of its major driver mutation. PLoS Genet. 2022;18:e1010463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potluri S, Kellaway SG, Coleman DJL, Keane P, Imperato MR, Assi SA, et al. Gene regulation in t(6;9) DEK::NUP214 Acute Myeloid Leukemia resembles that of FLT3-ITD/NPM1 Acute Myeloid Leukemia but with an altered HOX/MEIS axis. Leukemia. 2024;38:403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Habiburrahman M, Sutopo S, Wardoyo MP. Role of DEK in carcinogenesis, diagnosis, prognosis, and therapeutic outcome of breast cancer: An evidence-based clinical review. Crit Rev Oncol Hematol. 2023;181:103897. [DOI] [PubMed] [Google Scholar]
- 19.Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI. The human DEK oncogene stimulates β-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene. 2011;30:2741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin L, Piao J, Gao W, Piao Y, Jin G, Ma Y, et al. DEK over expression as an independent biomarker for poor prognosis in colorectal cancer. BMC Cancer. 2013;13:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, et al. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 2006;5:1202–7. [DOI] [PubMed] [Google Scholar]
- 22.Rooper LM, Agaimy A, Dickson BC, Dueber JC, Eberhart CG, Gagan J, et al. DEK-AFF2 Carcinoma of the Sinonasal Region and Skull Base: Detailed Clinicopathologic Characterization of a Distinctive Entity. Am J Surg Pathol. 2021;45:1682–93. [DOI] [PubMed] [Google Scholar]
- 23.Ruangritchankul K, Sandison A. DEK::AFF2 Fusion Carcinomas of Head and Neck. Adv Anat Pathol. 2023;30:86–94. [DOI] [PubMed] [Google Scholar]
- 24.Riveiro-Falkenbach E, Soengas MS. Control of tumorigenesis and chemoresistance by the DEK oncogene. Clin Cancer Res. 2010;16:2932–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sitwala KV, Adams K, Markovitz DM. YY1 and NF-Y binding sites regulate the transcriptional activity of the dek and dek-can promoter. Oncogene. 2002;21:8862–70. [DOI] [PubMed] [Google Scholar]
- 26.Nakajima R, Zhao L, Zhou Y, Shirasawa M, Uchida A, Murakawa H, et al. Deregulated E2F Activity as a Cancer-Cell Specific Therapeutic Tool. Genes. 2023;14:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 28.Tran AN, Dussaq AM, Kennell T, Willey CD, Hjelmeland AB. HPAanalyze: an R package that facilitates the retrieval and analysis of the Human Protein Atlas data. BMC Bioinforma. 2019;20:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roopra A. MAGIC: A tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. PLoS Comput Biol. 2020;16:e1007800–e1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krull JE, Wenzl K, Hopper MA, Manske MK, Sarangi V, Maurer MJ et al. Follicular lymphoma B cells exhibit heterogeneous transcriptional states with associated somatic alterations and tumor microenvironments. Cell Rep Med 2024: 101443. [DOI] [PMC free article] [PubMed]
- 31.Wenzl K, Stokes M, Novak JP, Bock AM, Khan S, Hopper MA et al. Multiomic Analysis Identifies a High-Risk Metabolic and TME Depleted Signature that Predicts Early Clinical Failure in DLBCL. medRxiv 2023. [DOI] [PMC free article] [PubMed]
- 32.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020;217:e20200483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017;170:564–76.e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–w102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodge LS, Novak AJ, Grote DM, Braggio E, Ketterling RP, Manske MK, et al. Establishment and characterization of a novel Waldenstrom macroglobulinemia cell line, MWCL-1. Blood. 2011;117:e190–197. [DOI] [PubMed] [Google Scholar]
- 38.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Team RC. A Language and Environment for Statistical Computing 2017.
- 41.Kassambra A ggpubr: ‘ggplot2’ Based Publication Ready Plots, 2023.
- 42.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015;44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitfield ML, George LK, Grant GD, Perou CM. Common markers of proliferation. Nat Rev Cancer. 2006;6:99–106. [DOI] [PubMed] [Google Scholar]
- 44.Wenzl K, Stokes ME, Novak JP, Bock AM, Khan S, Hopper MA, et al. Multiomic analysis identifies a high-risk signature that predicts early clinical failure in DLBCL. Blood Cancer J. 2024;14:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishida K, Nakashima T, Shibata T, Hara A, Tomita H. Role of the DEK oncogene in the development of squamous cell carcinoma. Int J Clin Oncol. 2020;25:1563–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pease NA, Wise-Draper T, Privette Vinnedge L. Dissecting the Potential Interplay of DEK Functions in Inflammation and Cancer. J Oncol. 2015;2015:106517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Dai L, Huang R, Li W, Wu W. Prognosis signature for predicting the survival and immunotherapy response in esophageal carcinoma based on cellular senescence-related genes. Front Oncol. 2023;13:1203351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun J, Bi F, Yang Y, Zhang Y, Jin A, Li J, et al. DEK protein overexpression predicts poor prognosis in pancreatic ductal adenocarcinoma. Oncol Rep. 2017;37:857–64. [DOI] [PubMed] [Google Scholar]
- 49.Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer. 2006;45:72–82. [DOI] [PubMed] [Google Scholar]
- 50.Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26:7506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS, et al. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 2009;69:6405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kappes F, Fahrer J, Khodadoust MS, Tabbert A, Strasser C, Mor-Vaknin N, et al. DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol. 2008;28:3245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wise-Draper TM, Allen HV, Thobe MN, Jones EE, Habash KB, Münger K, et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol. 2005;79:14309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrano-Lopez J, Nattamai K, Pease NA, Shephard MS, Wellendorf AM, Sertorio M, et al. Loss of DEK induces radioresistance of murine restricted hematopoietic progenitors. Exp Hematol. 2018;59:40–50.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from dbGaP Study Accession: phs002552.v1.p1 (LGBCL data), dbGaP Study Accession: phs002989.v1.p1 (FL data), GEO: GSE139833 (bulk B-cell development data), Broad Institute Dependency Map Portal at https://depmap.org/portal/ (cell line RNA-sequencing and RNAi), and GEPIA at http://gepia.cancer-pku.cn/ (GEO + GTEx data).