

Abstract
Whole genome sequences (WGS) enable discovery of rare variants which may contribute to missing heritability of coronary artery disease (CAD). To measure their contribution, we apply the GREML-LDMS-I approach to WGS of 4949 cases and 17,494 controls of European ancestry from the NHLBI TOPMed program. We estimate CAD heritability at 34.3% assuming a prevalence of 8.2%. Ultra-rare (minor allele frequency ≤ 0.1%) variants with low linkage disequilibrium (LD) score contribute ~50% of the heritability. We also investigate CAD heritability enrichment using a diverse set of functional annotations: i) constraint; ii) predicted protein-altering impact; iii) cis-regulatory elements from a cell-specific chromatin atlas of the human coronary; and iv) annotation principal components representing a wide range of functional processes. We observe marked enrichment of CAD heritability for most functional annotations. These results reveal the predominant role of ultra-rare variants in low LD on the heritability of CAD. Moreover, they highlight several functional processes including cell type-specific regulatory mechanisms as key drivers of CAD genetic risk.
Subject terms: Genetics, Functional genomics
Whole genome sequences enable discovery of rare variants which may help to explain the heritability of common diseases. Here the authors find that ultra-rare variants explain ~50% of coronary artery disease (CAD) heritability and highlight several functional processes including cell type-specific regulatory mechanisms as key drivers of CAD genetic risk.
Introduction
Coronary artery disease (CAD) is the leading cause of mortality and disease burden in the world1,2. Twin studies reported a strong genetic basis of CAD, with heritability estimates ranging from ~40–60% in North European populations3,4. The polygenic architecture of CAD is supported by the identification of hundreds of susceptibility loci in large-scale genetic association studies5. For example, a recent genome-wide association study (GWAS) for CAD including > 180,000 cases among more than one million participants predominantly of European ancestry identified 897 conditionally independent associations (at 1% false discovery rate)6. This globally accounted for 36.1% of CAD heritability on the liability scale with most of these associations (782/897) being variants with minor allele frequency (MAF) > 1% and with modest effect sizes. Similar CAD heritability estimates were reported in the largest multi-population GWAS across four different ancestry groups (non-Hispanic Black, non-Hispanic White, Hispanic, and Japanese participants from Biobank Japan)7.
Rare variants in non-coding regions of the genome have recently emerged as important contributors to the missing heritability of complex traits and diseases, including height and body mass index8, smoking9, and type 2 diabetes10. These studies relied on whole genome sequencing (WGS) data from the National Heart, Lung and Blood Institute (NHLBI) Trans-Omics for Precision Medicine (TOPMed) program11. In particular, Wainschtein et al.8 demonstrated that most of the missing heritability in height is attributable to variants with MAF < 10% in low linkage disequilibrium (LD) with nearby variants and could only be revealed by using WGS data, mainly because these variants were not previously tagged by imputation methods12.
Many loci identified by large-scale GWAS for CAD contain or are mapped near genes related to neovascularization angiogenesis, vascular remodeling, thrombosis, immune response and inflammation, proliferation and transcriptional regulation5,13. Given that more than 90% of variants identified in GWAS reside in noncoding regions of the genome, a large fraction of missing heritability for many complex traits, including CAD, could be explained by tissue- or cell-specific gene regulation14–16. Single-cell epigenomics profiles could help characterize and interpret these noncoding variants, especially those overlapping with cis-regulatory elements (CREs) like enhancers and promoters17. Recent studies using single-nucleus assay for transposable-accessible chromatin with sequencing (snATAC-seq) showed that CAD-associated variants are enriched in endothelial cells, smooth muscle cells and macrophages18,19. Furthermore, comparative genomics identified relevant functional components of the genome, the majority of which reside in the non-coding regions. A recent study analyzing genomic sequences of 240 mammalian species identified 3.3% of bases that are highly evolutionary constrained in the human genome20. Importantly, these constrained bases are significantly enriched for human disease variants measured by GWAS heritability enrichment. These studies only used GWAS summary statistics data conducted using array genotyping and imputation; hence, the contribution of ultra-rare (MAF < 0.1%) and rare variants (0.1% <MAF < 1%) from WGS data remains unknown for CAD.
In this work, to assess the contribution of ultra-rare and rare variants to CAD heritability, we apply the GREML-LDMS-I method21 to WGS data collected from 22,443 individuals of European ancestry from the NHLBI TOPMed program11. We examine contributions to CAD heritability and enrichment of highly constrained variants using an evolutionary score from the sequences of 240 mammals20. Then, we investigate the enrichment of protein-altering versus non-protein-altering variants to CAD heritability by using SnpEff22, which annotates and predicts effects of genetic variants. Next, we explore contributions and enrichment of variants residing in CREs by leveraging a recently published cell-specific chromatin atlas of the human coronary generated by snATAC-seq profiling19. Finally, we compare contributions to heritability from SNVs in high versus low functionality of 10 annotation principal components (aPC)23, which cover a wide range of functional sites integrated in the STAARpipeline24,25.
Results
Self-identified race/ethnicity versus genetically inferred ancestry
We first examined if self-identified race/ethnicity (SIRE) could classify TOPMed participants into relatively homogeneous genetic groups (see Supplementary Table 1 for list of studies). Our analyzes showed that ten principal components were sufficient to allocate the participants into their respective superpopulations (African, American, East Asian, South Asian and European), but SIRE was too inconsistent to classify them into homogeneous genetic groups (see Supplementary “Methods” for details and Supplementary Figs. 1–7). Given that standard heritability estimation methods using genotype data are biased in the presence of admixture and that low sample size hinders reliable heritability estimation, our main results focused on the largest ancestry group of 22,443 (4949 cases and 17,494 controls) participants with close to 100% inferred European genetic ancestry. We also present (see Supplementary “Methods” and Supplementary Figs. 8–11) a full analysis in a much smaller sample (1733 cases and 7783 controls) of participants > 75% inferred African ancestry, which demonstrates the limit of the GREML-LDMS-I approach when dealing with low sample size.
CAD heritability estimation in the European ancestry sample
To estimate CAD heritability, we utilized the GREML-LDMS-I method. Figure 1 and Supplementary Data 1 display the various LD score-MAF bin contributions using the REML EM algorithm of GCTA (see “Methods” for details). The estimated heritability equals = 23.9% (standard error SE = 10.3%), a value higher than when computed with the default REML AI algorithm (Supplementary Data 2, Supplementary Fig. 12). The largest contribution (Supplementary Data 1) comes from ultra-rare SNVs (MAF ≤ 0.1%) with low LD score (below the median) with ~50% of the total observed heritability (0.12/0.239), a smaller proportion compared to the default REML AI algorithm. Common SNVs (10% <MAF ≤ 50%) with high LD score (above the median) contributes a similar proportion of ~16% to the total observed heritability (0.038/0.239). In general, the proportion of SNVs of each LD score-MAF bin, i.e., the number of SNVs in each bin divided by the total number of SNVs, was highly correlated with the proportion of observed heritability contributed by that bin (Pearson correlation coefficient R = 0.87, Supplementary Data 1, Supplementary Fig. 13). We estimated the CAD heritability on the liability scale (see “Methods”) at = 34.3% (SE = 14.8%) (Fig. 1 inset, dotted vertical line) assuming a CAD prevalence of 8.2% in the U.S. White population7.
Fig. 1. Contribution of each LD score-MAF bin to the observed heritability h2 of CAD in European ancestry.
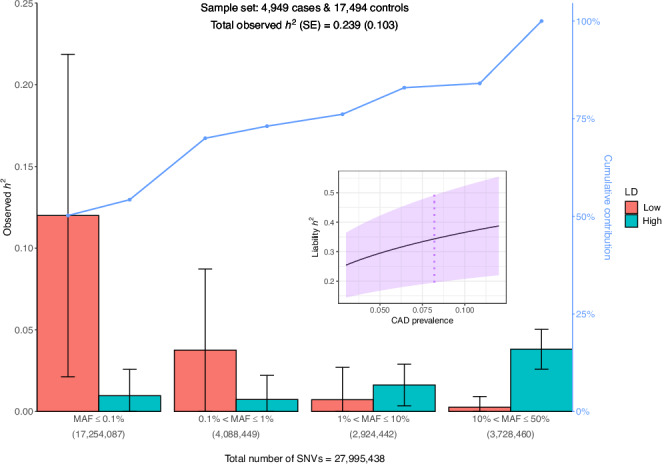
Error bars represent ± one SE from each contribution point estimate. SEs are calculated by GCTA and are proportional to the effective number of independent variants in each bin and inversely proportional to the total sample size (4949 cases + 17,494 controls). The number of SNVs in each of the four MAF bins is indicated in parentheses. Low (High) category in the legend represents SNVs with LD scores below (above) the median, respectively. The broken line (in blue) displays the cumulative contribution (in %) of each LD score-MAF bin to the observed heritability estimate. Inset represents CAD heritability (estimate ± SE) on the liability scale for CAD prevalence ranging from 3% to 12% in the population (violet shaded area). The vertical dotted line (in violet) indicates the heritability estimate for a population prevalence of 8.2% in White/European ancestry7. The GRMs are estimated by the ratio of averages (RoA) method and contributions to h2 are estimated with the REML EM algorithm. CAD, coronary artery disease; LD, linkage disequilibrium; MAF, minor allele frequency; SE, standard error; SNV, single nucleotide variant.
We repeated a similar analysis but this time using quartiles of LD scores instead of halves (16 LD score-MAF bins in total). Supplementary Data 3 indicates a larger total observed heritability = 31.1% (SE = 12.7%) compared to the one estimated with eight bins. The largest variance contribution (Supplementary Fig. 14) comes from SNVs in the 2nd LD score quartile (Q2) of the lowest MAF bin (MAF ≤ 0.1%), in agreement with our previous analysis. Because nine of the 16 LD score-MAF bin contributions were close to 0, all our subsequent analyzes are based on eight LD score-MAF bins. Additional analyzes motivating our choice of REML and genomic relatedness matrix (GRM) estimation methods can be found in the Supplementary “Methods” (Supplementary Data 4 and Supplementary Figs. 15–18).
Comparison with previously published CAD heritability estimate
We compared the CAD heritability estimate in our inferred European sample with the most recent estimate reported in 19,392 non-Hispanic White participants of the Million Veteran Program (MVP)7. Tcheandjieu et al. applied the same GREML-LDMS-I approach but binned the variants differently: quartiles of LD scores and six MAF bins were used (0.1% <MAF ≤ 1%, 1% < MAF ≤ 10%, 10% < MAF ≤ 20%, 20% < MAF ≤ 30%, 30% < MAF ≤ 40%, 40% < MAF ≤ 50%). Using the GCTA REML EM algorithm, we estimated a total observed heritability = 14.1% (SE = 5.5%), which is considerably less than their estimated = 24.4% (SE = 4.7%). The major discrepancy came from SNVs in the lowest LD score quartile (Q1) of the lowest MAF bin (0.1% < MAF ≤ 1%) (Supplementary Data 5). However, when we added the ultra-rare variants (MAF ≤ 0.1%), which were not included in the MVP heritability analyzes, the missing gap was more than closed with = 26.9% (SE = 10.7%) (Supplementary Fig. 19). We note that there are differences that may contribute to discrepancies in CAD heritability estimates between our study and Tcheandjieu et al., including whole-genome sequencing vs. imputed data, different binning of variants and phenotypic definitions.
CAD heritability estimation in the African ancestry sample
We also estimated CAD heritability in a restricted sample of 1733 cases and 7783 controls of inferred African genetic ancestry (see Supplementary “Methods” for details). The total observed heritability = 25.5% (SE = 18.1%), although none of the LD score-MAF bin contributions were significant within one SE (Supplementary Data 6, Supplementary Fig. 20). Using a prevalence of 6.5% in the U.S. Black population as in Tcheandjieu et al.7, heritability on the liability scale = 39.3% (SE = 27.9%). This estimate is larger than the one in our TOPMed European sample, and also larger than the one reported in MVP ( = 30.0%). In the African ancestry sample, the contribution from ultra-rare variants with low LD scores is not as high as in our European sample (0.037/0.255 ≈ 14.6%). This might be explained by the fact that, of the 28.1 million SNVs in Europeans and 35.7 million SNVs in Africans used in GCTA computations, only 12.9 million SNVs are shared, and the distribution of these variants across MAF bins is different (Supplementary Data 7, Supplementary Fig. 21). Owing to the large SEs observed in the African sample, all the subsequent analyzes presented in this paper are restricted to the European sample.
Allele frequency comparison with gnomAD
In general, if the effect sizes from the same set of variants were similar across different ancestries, we should expect to observe similar contributions from this set of variants to heritability estimates across ancestry groups. We compared the overlap of each MAF bin variant set in our European sample with genetically inferred groups from the latest Genome Aggregation Database (gnomAD) (see “Methods”)26. As expected, the proportion of SNVs shared with the gnomAD non-Finnish European group was very high in all four MAF bins (ranging from 93% to 99%, Supplementary Data 8), meaning that almost all SNVs display the same allele frequency. Unsurprisingly, apart from the non-Finnish European group, the ultra-rare bin (0 < MAF ≤ 0.1%) showed a moderate overlap only with the gnomAD African/African American group, and to some extent with the Admixed American group.
Evolutionary constraint with phyloP score
We assessed components of CAD heritability in different functional regions of the genome. We first examined the relationship of the phyloP score27 calculated from 240 mammalian sequences with allele frequency and functional impact as predicted by SnpEff22. We confirmed the inverse relationship, as expected from negative selection, between the phyloP score and (minor) allele frequency in our set of variants, with a stronger decrease in protein-altering SNVs compared to non-protein-altering SNVs (Fig. 2a). Due to the small number of common SNVs (10% < MAF ≤ 50%) with low LD score (Supplementary Data 9), we grouped all common SNVs into one single bin irrespective of their LD score. This resulted in 14 LD score-MAF-Constrained bins. Note that, of the 27,933,966 SNVs with phyloP scores in our dataset, ~2.4% are constrained.
Fig. 2. Distribution of phyloP scores and contribution of constrained SNVs to CAD heritability.

a Violin plots of phyloP scores against the four MAF bins stratified by SnpEff predicted impact (High: protein-altering variants, Low: non-protein-altering variants). Points indicate medians of phyloP scores in each MAF bin. For ease of presentation, SNVs with phyloP score < −10 are omitted. b Proportion of observed heritability in each LD score-MAF-Constrained bin against the proportion of SNVs in that bin (number of SNVs in the bin divided by the total number of SNVs). Each label in the plot represents a combination of: i) MAF (UR: ultra-rare (MAF ≤ 0.1%), R: rare (0.1% < MAF ≤ 1%), UC: uncommon (1% < MAF ≤ 10%), C: common (10% < MAF ≤ 50%)); ii) LD score (LO: low, HI: high); and iii) Constrained (YES or NO). The black line shows the regression line, whose equation is displayed in the upper left corner (n = 14). R designates the Pearson correlation coefficient, while p is the p-value associated with the two-sided test of null correlation. c Absolute (left) and relative (right) contribution per variant of each LD score-MAF-Constrained bin to the global CAD heritability estimate. The legend and color-coding is the same as in (b). Error bars represent ± one SE from each contribution point estimate. Absolute SEs (left) are calculated by GCTA and are proportional to the effective number of independent variants in each bin and inversely proportional to the total sample size (4949 cases + 17,494 controls). Relative SEs (right) are obtained by dividing the corresponding absolute SEs by the square root of the number of variants. d Log constraint ratio of constrained over non-constrained variants in each LD score-MAF bin. Each label on the y-axis is defined as in (b). Error bars represent ± one SE from each log constrain ratio estimate. SEs are calculated from GCTA’s output of the covariance matrix of contribution estimates to heritability in each bin and their corresponding number of SNVs (see Supplementary “Methods” for derivation details). CAD, coronary artery disease; Cons, constrained; LD, linkage disequilibrium; MAF, minor allele frequency; SE, standard error; SNV, single nucleotide variant.
Heritability was computed as previously described. Unsurprisingly, we observed that the proportion of SNVs of each LD score-MAF-Constrained bin was highly correlated with the proportion of observed heritability contributed by that bin (R = 0.85, Fig. 2b, Supplementary Data 10), with the largest contribution ( ~ 46%) from non-constrained ultra-rare variants (MAF ≤ 0.1%) with low LD score (total = 26.4%, SE = 10.3%). To contrast absolute and relative contribution from each LD score-MAF-Constrained bin, we calculated the relative contribution per variant in each bin by dividing the absolute contribution by the number of variants in that bin. On a per-variant basis, significant contributions (± one SE from the point estimate) now originated from constrained variants: uncommon (1% <MAF ≤ 10%) and rare (0.1% < MAF ≤ 1%) SNVs with low LD score, and common variants (10% < MAF ≤ 50%) (Fig. 2c, Supplementary Data 10).
To measure the potential heritability enrichment of constrained SNVs, we examined the enrichment ratio of the contribution on a per variant basis of these SNVs over the contribution coming from non-constrained SNVs in each LD score-MAF bin (see “Methods”). Generally, we observed a positive heritability enrichment for all allele frequency bins ranging from 0.72 to 3.94 for the log constraint ratio (Fig. 2d, Supplementary Data 11). This analysis further confirmed a significant positive enrichment of constrained variants for rare SNVs (0.1% < MAF ≤ 1%) with low LD score, and most notably for common variants (10% < MAF ≤ 50%) (Fig. 2d, Supplementary Data 11). These results highlight the importance of highly constrained regions that are predominantly in non-coding regions in contributing to CAD genetic risk.
SnpEff predicted impact
In this analysis (see “Methods”), each LD score-MAF bin was subdivided into two disjoint bins according to the functional impact predicted by SnpEff 4.122. We observed that only a small percentage of SNVs (less than 1%) were classified as protein-altering variants, independently of the LD score-MAF bin in which they fall (Supplementary Data 12). Due to the small number of common SNVs (10% < MAF ≤ 50%) with low LD score, we grouped all common SNVs into one single bin irrespective of their LD score, as was done previously.
Figure 3a displays the absolute and the relative contribution per variant for each bin. Unsurprisingly, since they represent ~99% of all SNVs (Supplementary Data 12), non-protein-altering variants contributed to most of the overall heritability (Supplementary Data 13). Ultra-rare non-protein-altering variants with low LD scores, which constitute 40.6% of all variants, contributed nearly half (49.9%) of the total observed heritability ( = 23.8%, SE = 10.3%). Again, the proportion of SNVs of each LD score-MAF-Impact bin was highly correlated with the proportion of observed heritability contributed by that bin (R = 0.89, Supplementary Data 13, Supplementary Fig. 22). Nonetheless, on a per-variant basis, these contributions were negligible, and only common protein-altering variants disproportionately contributed to heritability (Fig. 3a, Supplementary Data 13).
Fig. 3. Contribution of protein-altering and non-protein altering SNVs to the observed heritability of CAD.

a Absolute (left) and relative (right) contribution per variant of each LD score-MAF-Impact bin to the global CAD heritability estimate. Each label in the legend represents a combination of: i) MAF (UR: ultra-rare (MAF ≤ 0.1%), R: rare (0.1% <MAF ≤ 1%), UC: uncommon (1% <MAF ≤ 10%), C: common (10% < MAF ≤ 50%)); ii) LD score (LO: Low, HI: High); and iii) Impact (High: protein-altering variants, Low: non-protein-altering variants). Error bars show ± one SE from each contribution point estimate. Absolute SEs (left) are calculated by GCTA and are proportional to the effective number of independent variants in each bin and inversely proportional to the total sample size (4949 cases + 17,494 controls). Relative SEs (right) are obtained by dividing the corresponding absolute SEs by the square root of the number of variants. b Log impact ratio of protein-altering over non-protein-altering variants in each LD score-MAF bin. Each label on the y-axis is defined as in (a). Error bars represent ± one SE from each log impact ratio estimate. SEs are calculated from GCTA’s output of the covariance matrix of contribution estimates to heritability in each bin and their corresponding number of SNVs (see Supplementary “Methods” for derivation details). CAD, coronary artery disease; LD, linkage disequilibrium; MAF, minor allele frequency; SE, standard error; SNV, single nucleotide variant.
We also investigated the (log) impact ratio of the contribution per variant from protein-altering SNVs over the contribution per variant from non-protein-altering SNVs in each LD score-MAF bin. Globally, there was a positive enrichment in each LD score-MAF bin, with a marked significant enrichment for common protein-altering variants (10% < MAF ≤ 50%) over non-protein-altering ones (Fig. 3b, Supplementary Data 14).
snATAC-seq profiles of human coronary artery
We next assessed CAD heritability for cis-regulatory elements from a cell-specific chromatin atlas of the human coronary19. For each of 13 distinct cell types, we subdivided each LD score-MAF bin into two disjoint bins: one bin containing SNVs overlapping with cell-type specific snATAC-seq peaks, and one bin containing SNVs outside these peaks. Again, common SNVs were grouped into one single bin irrespective of their LD score. Supplementary Data 15 reports the number of SNVs in each bin for each cell type. SNVs mapping into the snATAC-seq peaks were uniformly distributed across the LD score-MAF bins, ranging from ~0.5% (Unknown) to 3% (Smooth muscle cell (SMC)) (Supplementary Fig. 23). Interestingly, 1,199,053 (47.0%) of the 2,553,042 SNVs residing within peaks are unique to only one cell type (Supplementary Fig. 23, orange bars), and 44,878 SNVs (1.8%) are shared by all 13 cell types.
Figure 4a shows the proportion of each LD score-MAF-Peak bin to the global CAD heritability estimate for each cell type. Each combination of LD score and MAF is represented by eight different hues (colors) with a darker (lighter) tint used to indicate SNVs found inside (outside) snATAC-seq peaks, respectively. We observed that the relative contributions come mostly from SNVs outside peaks (lighter segments), with smaller darker segments for all cell types (Fig. 4a, Supplementary Data 16). Again, the proportion of SNVs of each LD score-MAF-Peak bin was found to be highly correlated with the proportion of observed heritability contributed by that bin (R = 0.87, Supplementary Data 17, Supplementary Fig. 24). Proportion of each LD score-MAF-Peak to heritability was quite similar from one cell type to the other, resulting in an average total observed heritability of 24.3% across all 13 cell types.
Fig. 4. Contribution of SNVs inside and outside cell-specific snATAC-seq peaks to the observed heritability of CAD.

a Proportion of each LD score-MAF-Peak bin to the global CAD heritability estimate for each cell type. Each label in the legend represents a combination of: i) MAF (UR: ultra-rare (MAF ≤ 0.1%), R: rare (0.1% < MAF ≤ 1%), UC: uncommon (1% <MAF ≤ 10%), C: common (10% < MAF ≤ 50%)); ii) LD score (LO: low, HI: high); and iii) Peak (IN: inside, OUT: outside). b Log enrichment ratio of snATAC-seq peaks in each LD score-MAF bin for each cell type. Each label on the y-axis is defined as in (a). Black lines represent the average log enrichment ratio across all 13 cell types. Error bars show ± one SE from each log enrichment ratio estimate. SEs are calculated from GCTA’s output of the covariance matrix of contribution estimates to heritability in each bin and their corresponding number of SNVs (see Supplementary “Methods” for derivation details). CAD, coronary artery disease; LD, linkage disequilibrium; MAF, minor allele frequency; Endo, endothelial cells; Fibrobl, fibroblasts; Fibromyo, fibromyocytes; Macro, macrophages; NK, natural killer cells; Peri, Pericytes; SMC, smooth muscle cells; snATAC-seq, single-nucleus assays for transposase accessible chromatin with sequencing; SNV, single nucleotide variant.
Again, we contrasted the contribution of each LD score-MAF-Peak bin on a per variant basis (Supplementary Fig. 25, Supplementary Data 18). Very few significant (± one SE from the point estimate) contributions per variant were observed from SNVs either inside or outside snATAC-seq peaks. Globally, we observed a positive log enrichment ratio from SNVs within these peaks in each LD score-MAF bin for all cell types. We observed strong significant enrichment for common SNVs (10% < MAF ≤ 50%) due to smaller SEs (Fig. 4b, Supplementary Data 19). When we examined the average across all cell types, we observed significant enrichment in all LD score-MAF bins, with the largest ratio estimate coming from the ultra-rare SNVs (MAF ≤ 0.1%) with high LD scores, and the smallest interval coming from common SNVs.
FAVOR functional annotation
We examined a wide range of functional processes using the aPCs annotation available in the STAAR pipeline23–25 (see “Methods”). For aPC-Conservation, we selected a Phred threshold (14.7) which matches the proportion of ~2.4% of SNVs in our dataset that were deemed constrained according to their respective phyloP score. Supplementary Data 20 reports the number of SNVs in each LD score-MAF-aPC-Conservation bin, whose distribution is similar to the LD score-MAF-Constrained bins observed in the phyloP analysis. As expected, heritability contributed by each bin and enrichment of conserved (Phred ≥ 14.7) over non-conserved (Phred < 14.7) SNVs were very similar to our phyloP analysis (Supplementary Data 21 and 22, Supplementary Fig. 26).
We next compared the aPC-Protein-Function annotation with the SnpEff impact annotation. Because 99.38% of the aPC-Protein-Function Phred values were exactly equal to 2.969487 in our dataset, we chose Phred = 3 as the threshold separating high from low functionality. At this threshold, we observed that 0.62% of SNVs were classified as high, a percentage slightly less than the protein-altering group (0.76%) according to SnpEff (Supplementary Data 23). Heritability contributed by each bin and enrichment of high versus low protein functionality SNVs were similar to our SnpEff predicted impact analysis (Supplementary Data 24 and 25, Supplementary Fig. 27).
Finally, we investigated the remaining eight aPCs, setting the Phred threshold value of high versus low functionality at 10 (all eight aPCs) or, when possible, at 20 (all except aPC-Local-Nucleotide-Diversity and aPC-Mutation-Density, for which almost no value greater than 20 was observed in our set of SNVs). Figure 5a shows the proportion of each LD score-MAF-Functionality bin to the global CAD heritability estimate for six aPCs at Phred = 20 and two aPCs at Phred = 10, while Fig. 5b displays their respective log functionality enrichment ratio. In general, we observed a positive enrichment ratio of high over low functionality SNVs in each LD score-MAF bin for all eight aPCs. Strong significant ratios, ranging from 2.7 to 4.3, were found for common SNVs. The complete set of results for all aPCs at Phred = 10 or 20 are given in Supplementary Figs. 28 and 29, and Supplementary Data 26 and 27.
Fig. 5. Contribution of SNVs with high and low aPC functionality to the observed heritability of CAD.

a Proportion of each LD score-MAF-Functionality bin to the global CAD heritability estimate for eight aPCs (Phred = 20 for all, except aPC-Mutation-Density and aPC-Local-Nucleotide-Diversity for which Phred = 10). Each label in the legend represents a combination of: i) MAF (UR: ultra-rare (MAF ≤ 0.1%), R: rare (0.1% < MAF ≤ 1%), UC: uncommon (1% < MAF ≤ 10%), C: common (10% <MAF ≤ 50%)); ii) LD score (LO: low, HI: high); and iii) Functionality (Low, High). b Log functionality ratio of high over low functionality in each LD score-MAF bin for each aPC. Each label on the y-axis is defined as in (a). Error bars show ± one SE from each log functionality ratio estimate. SEs are calculated from GCTA’s output of the covariance matrix of contribution estimates to heritability in each bin and their corresponding number of SNVs (see Supplementary “Methods” for derivation). CAD, coronary artery disease; EpiAct, aPC-Epigenetics-Active; EpiRep, aPC-Epigenetics-Repressed; EpiTrans, aPC-Epigenetics-Transcription; Funct, functionality; LD, linkage disequilibrium; MAF, minor allele frequency; Map, aPC-Mappability; MutDens, aPC-Mutation-Density; NucDiv, aPC-Local-Nucleotide-Diversity; Prox, aPC-Proximity-To-TSS-TES; SNV, single nucleotide variant; Trans, aPC-Transcription-Factor.
Discussion
This study provides an estimate of CAD heritability using full genomic information from WGS data. We reported the following major findings: i) an estimated CAD heritability of = 23.9% (observed scale) and = 34.3% (liability scale) across all genome-wide SNVs in a sample of CAD cases and controls of European genetic ancestry; ii) about 50% of CAD heritability is explained by ultra-rare SNVs (MAF ≤ 0.1%) with low LD score; iii) an enrichment of CAD heritability was observed in many allele frequency bins, especially for evolutionary constrained and protein-altering common SNVs, for variants overlapping with snATAC-seq peaks of many cell types from the coronary artery, and for various aspects of biological function such as epigenetics, local nucleotide diversity, mappability, mutation density, transcription factor, and proximity to transcription starting and ending site.
Using the same GREML-LDMS-I approach21, Tcheandjieu et al. reported a CAD heritability of = 36.3% (SE = 7.0%) on the liability scale using a prevalence of 8.2% in 19,392 non-Hispanic White participants of the Million Veteran Program (MVP)7. Intriguingly, these authors calculated an observed heritability of 24.4%, which is very close to our estimate of 23.9%. However, their estimation did not include ultra-rare variants (MAF ≤ 0.1%) and was based on genotyped and imputed data instead of WGS data. When we added the ultra-rare variants (MAF ≤ 0.1%), our heritability estimate was comparable to theirs ( = 26.9%, Supplementary Fig. 19). One possible explanation might lie in the differences of allelic frequencies between the TOPMed and the MVP sample: many variants in TOPMed placed in the ultra-rare bin were included in the rare bin of MVP. An alternative explanation could be that their imputed dataset captured ultra-rare variants in LD with rare variants, hence inflating the contribution from their rare variants bin, while our WGS dataset really classified these ultra-rare variants in their appropriate MAF bin. Nonetheless, both analyzes point to a disproportionate contribution to CAD heritability originating from rare SNVs in low LD.
We investigated the importance of evolutionary constrained variants in CAD heritability using the phyloP score from 240 mammalian sequences. Using 63 independent European ancestry GWASs, Sullivan et al.20 found that highly constrained variants (allele frequency ≥ 0.5%) had greater heritability enrichment for GWAS trait associated variants. In our study, we showed that significant contributions to CAD heritability, on a per-variant basis, originated from constrained variants: uncommon (1% < MAF ≤ 10%) and rare (0.1% < MAF ≤ 1%) SNVs with low LD score, as well as common SNVs (10% < MAF ≤ 50%). We also confirmed by an independent functional annotation (aPC-Conservation score) a significant heritability enrichment from constrained over non-constrained SNVs in the common variant bin.
Our analysis reiterated the large contribution of rare variants in low LD to the CAD heritability, albeit mainly from non-protein-altering SNVs. Intriguingly, on a per-variant basis, common protein-altering SNVs significantly contributed to CAD heritability. We also found a positive enrichment in each LD score-MAF bin, notably from common protein-altering variants over non-protein-altering ones, a result independently confirmed in our aPC-Protein-Function heritability analysis. This contrasts with results reported for height and body mass index where variants in low LD and low MAF were the largest contributors on a per-variant basis, although the LD score-MAF partition employed did not distinguish between protein- and non-protein-altering in the common variant bin8. Of note, our results support the finding of a recent paper showing that ultra-rare coding variants (MAF < 0.1%) explain only 1.3% of heritability across 22 continuous traits and common diseases in ~400,000 UK Biobank exomes28. In our study, ultra-rare protein-altering variants contributed 1.7% (0.0041/0.2378, Supplementary Data 13) to the total CAD heritability estimate on the observed scale.
An important strength of our study lies in investigating the contribution to CAD heritability of specific cell type peaks of 13 distinct cell clusters derived from snATAC-seq profiles. To achieve this, we leveraged a recent study which showed that CAD-associated variants are enriched in endothelial cells, smooth muscle cells and macrophages19. We observed that the largest relative contributions to CAD heritability originate from SNVs outside snATAC-seq peaks, although some cell types (endothelial cells, fibroblasts, fibromyocytes, natural killer cells, pericytes and smooth muscle cells) display non-null contributions in these peaks, especially from ultra-rare variants in low LD. This is primarily due to the large number of SNVs which reside outside these snATAC-seq peaks. However, for all cell types, we noticed a positive enrichment for CAD heritability in these peaks within each LD score-MAF bin, especially for common SNVs (10% <MAF ≤ 50%). When averaging across all cell types in each LD score-MAF bin, we observed significant log enrichment ratios with values ranging from 1.4 to 3.5. The enrichment of CAD heritability across distinct cell types highlights the contribution of cell-type specific regulatory mechanisms underlying CAD risk.
Heritability is not reliably estimated when the sample contains individuals from different genetic ancestries or from admixed populations29. To provide the most accurate and unbiased heritability estimate of CAD, we applied stringent quality controls in both the variant and the sample sets. Our PCA and admixture analyzes revealed high levels of admixture in TOPMed participants who self-identified as Black/African American, Hispanic/Latino or South Asian. Unfortunately, due to admixture and/or low sample size in these groups, we had to exclude them and restrict our various heritability analyzes to the larger homogeneous subset of genetically inferred European participants. However, to promote fairness and transparency in genomic research30, we estimated CAD heritability in a much smaller sample of inferred African genetic ancestry. The observed heritability in the African sample (= 25.5%) was comparable to the observed heritability (= 23.9%) in the European sample, although the contribution from ultra-rare variants with low LD score was not as important ( ~ 15% versus ~50%). Yet caution is required since all LD score-MAF bin contributions in the African sample showed large SEs.
As mentioned previously, our results indicate that the largest contribution to CAD heritability consistently comes from ultra-rare variants (MAF ≤ 0.1%) with low LD score. A recent study identified rare and ultra-rare coding variants in 17 genes associated with CAD, 14 of which showed at least moderate prior genetic, biological and/or clinical evidence31. It revealed an excess of ultrarare coding variants in 321 known CAD genes, demonstrating that many rare and ultrarare coding variants in additional CAD genes await discovery. For most complex diseases, these rare variants have been hypothesized to be under negative (or purifying) selection, eliminating large-effect mutations and leaving behind common-variant associations in thousands of less essential loci32,33. Exome studies reported that most rare coding variants have been previously identified in loci overlapping those detected by GWAS of common variants34,35, suggesting some level of functional convergence across the allelic frequency spectrum36,37. Studies have shown that this convergence signature may guide future fine-mapping studies and reveal potential drug targets38,39.
Assuming that the disproportionate contribution is not unique to CAD and that ultra-rare variants are more likely to be ancestry-specific, it becomes imperative to recruit and sequence cohorts of non-European ancestry in order to characterize the genetic architecture not only for CAD but for other diseases across different ancestries40–42. Furthermore, the development of heritability estimation methods for admixed populations are warranted since most current statistical methods typically only apply to homogeneous genetic ancestry samples at the continental level. Methods accounting for admixture in heritability estimation is an active area of research43–45.
The present study has limitations. First, as mentioned previously, larger sample sizes are needed to reduce uncertainty in variance contribution estimates, especially for ultra-rare variants in low LD. The sampling variance of the variance estimate in each LD score-MAF bin is proportional to the effective number of independent variants, but the corresponding standard error is approximately inversely proportional to the sample size8,46. Second, the GREML method assumes that causal SNVs have on average the same heritability, irrespective of their MAF and LD structure around them, although the problem is mitigated in part by creating bins of variants sharing similar MAF and LD scores in the GREML-LDMS extension21. No model-based heritability method so far can provide a definitive heritability estimate for a complex trait such as CAD, especially when ultra-rare SNVs are included47. Third, current GRM estimates are prone to bias in presence of population structure, and this bias is exacerbated especially for rare variants in high LD. Finally, we opted to run the GCTA REML algorithm 2 (EM) in all our analyzes for consistency purposes, although this algorithm slightly inflated the observed heritability estimate compared to the other two algorithms available.
In conclusion, we estimated CAD heritability based on WGS data from a sample of 22,443 genetically inferred European subset of the TOPMed project. In line with other recent studies, our results suggest that ultra-rare variants contribute a substantial proportion of missing heritability in CAD and that rare-variant associations remain to be identified by large well-powered whole-genome sequencing studies. Functional studies are also needed to establish a better understanding of the role of rare variants on complex traits and diseases in general.
Methods
Variant dataset
This research complies with all relevant ethical regulations and was approved by each included study-specific TOPMed institutional review board. Informed consent was obtained from all participants. Our study utilized the TOPMed Freeze 9 dataset which includes > 80 different studies totaling ∼161,000 samples with WGS data, and the 2504 samples from the 1000 Genomes Project11. We opted for the “minDP10” genotype files which set to missing any individual genotype based on fewer than 10 covering sequence reads (https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-9). This dataset contains ∼800 million single nucleotide variants (SNVs) and ∼62 million indels from autosomal chromosomes which were aligned to the GRCh38 human genome build.
Case and control definition
Cases and controls for CAD were defined similarly in all studies except in BioMe. CAD cases were identified as samples with documented: i) coronary revascularization, such as coronary artery bypass graft (CABG) or percutaneous transluminal coronary angioplasty (PTCA); OR ii) acute myocardial infarction; OR iii) definite coronary heart disease death. Controls were defined as non-cases with no documented angina or coronary heart disease death. In BioMe, cases were identified using ICD codes for acute myocardial infarction in electronic health records as of December 2020 (ICD-9: 410; ICD−10: I21.09, I21.11, I21.19, I21.29, I21.3x, 121.4x). Controls were defined as non-cases with additional exclusion codes (ICD-9: 413; ICD−10: I20; CPT: 33510–33548, 92920, 92921, 92924, 92925, 92928, 92933, 92934, 92937, 92938, 92943, 92944, 92973); AND with no peripheral arterial disease (ICD-9: 249.70, 249.71, 250.70–250.73, 440.0, 440.20–440.24, 440.29, 440.30–440.32, 440.9, 443.81, 443.9, 444.22, 444.81, 785.4). Following these definitions, we were able to identify 64,397 samples from 13 TOPMed studies with available CAD status (see Supplementary Table 1).
Quality control, relatedness and genetic ancestry inference
We applied stringent quality control to both the sample and variant sets (see Supplementary “Methods” for details). Pairs of samples related at the fourth degree or higher were identified using PC-AiR and PC-Relate48,49, and one member of each pair was excluded favoring younger cases and older controls. Genetic ancestry was assessed using the principal component analysis (PCA) implemented in the R package bigsnpr50 and by ADMIXTURE 1.351. In our main analysis, the sample includes 22,443 TOPMed participants (4949 CAD cases and 17,494 controls) of inferred European ancestry with genotype data available for 28,051,806 biallelic autosomal SNVs. Similar quality control steps were applied to a smaller sample of 1733 cases and 7783 controls of inferred African genetic ancestry.
Heritability estimation
To estimate CAD heritability, we utilized the GREML-LDMS-I method introduced by Evans et al.21 and available in GCTA software52 (https://yanglab.westlake.edu.cn/software/gcta/#GREMLinWGSorimputeddata). Briefly, LD scores were computed for each of the 28,051,806 biallelic autosomal SNVs. LD score for each SNV is defined as the sum of its pairwise correlations with all other SNVs. Then SNVs were binned according to their LD score (either in halves or in quartiles), and further by their MAF (MAF ≤ 0.1%, 0.1% < MAF ≤ 1%, 1% < MAF ≤ 10%, 10% < MAF ≤ 50%). In each LD score-MAF bin, a genomic relatedness matrix (GRM) was computed for all 22,443 samples. We computed heritability using the ratio of averages (RoA) GRM estimation method and the REML EM algorithm implemented in GCTA version 1.93.2beta. In general, the GRM estimation method impacts the variance estimates, with greater effect in ultra-rare and rare variant bins (see Supplementary “Methods” for additional analyzes). Observed heritability () was transformed to heritability on the liability scale () using equations from Lee et al.53 (see Supplementary “Methods” for details). All heritability estimates presented in this paper were adjusted by adding the following fixed effects in the model: CAD status (case or control), age, sex, study and the first 15 principal components (PCs).
Allele frequency comparison with gnomAD
We downloaded the Genome Aggregation Database (gnomAD) v4.1.0 joint allele frequency dataset26, which contains data from 730,947 exomes and 76,215 whole genomes (https://gnomad.broadinstitute.org/downloads). We confidently mapped 27,427,591 SNVs (with FILTER = PASS in gnomAD) out of 28,051,806 SNVs (97.8% overlap) by matching the corresponding chromosomal position and alleles. Allele frequencies from each genetically inferred ancestry group in gnomAD (Admixed American, African/African American, Amish, Ashkenazi Jewish, East Asian, Finnish, Middle Eastern, non-Finnish European, South Asian) were extracted and compared with our inferred European ancestry sample. Only SNVs with MAF > 0, i.e., when at least 1 minor allele was observed, were considered for comparison with our European sample.
Evolutionary constraint with phyloP score
We downloaded the file containing the human phyloP scores27 estimated across 240 mammalian species for ~2.85 billion bases in the human genome https://cgl.gi.ucsc.edu/data/cactus/241-mammalian-2020v2-hub/Homo_sapiens/241-mammalian-2020v2.bigWig. When merging with our set of 28,051,806 biallelic autosomal SNVs, we found phyloP scores for 27,933,966 SNVs (99.6% overlap). As suggested by Sullivan et al.20, we considered that a SNV was constrained if its phyloP score ≥ 2.27 (false discovery rate of 5%). We then subdivided each LD score-MAF bin into two disjoint bins: one bin containing constrained SNVs and one bin containing non-constrained SNVs. Contribution per variant and standard error (SE) per variant for each bin were calculated, respectively, by dividing the bin’s contribution to overall heritability and corresponding standard error by the number of variants included in that bin.
SnpEff predicted impact
Each LD score-MAF bin was subdivided into two disjoint bins according to their functional impact as predicted by SnpEff 4.122. The four different predicted impacts are: 1) “HIGH”: the variant has a high disruptive impact on the protein (ex.: stop gain, start loss, frame shift); 2) “MODERATE”: the variant is non-disruptive but might change the protein effectiveness (ex.: missense); 3) “LOW”: the variant is harmless and unlikely to change the protein behavior (ex.: synonymous); 4) “MODIFIER”: the variant is non-coding or affects non-coding genes (ex.: intronic, intergenic). Following the categorization of Wainschtein et al.8, we merged SNVs with predicted impact “HIGH” and “MODERATE” into protein-altering variants, and “LOW” and “MODIFIER” as non-protein-altering variants, respectively. More details on SnpEff annotation and predicted impact can be found at http://pcingola.github.io/SnpEff/se_inputoutput/#eff-field-vcf-output-files.
snATAC-seq profiles of human coronary artery
We leveraged specific cell type peaks of 13 distinct cell clusters from single-nucleus assays for transposase accessible chromatin with sequencing (snATAC-seq), as identified by Turner et al.19. This snATAC-seq profiling was performed in 28,316 nuclei from human coronary arteries of 41 individuals. For each cell type, we subdivided each LD score-MAF bin into two disjoint bins: one bin containing SNVs inside cell-specific peaks, and one bin containing SNVs outside cell-specific peaks. In our set of 28,051,806 biallelic autosomal SNVs, 2,553,042 SNVs overlapping with these peaks were present in at least one cell type.
FAVOR functional annotation
We downloaded the FAVORannotator’s Full Dataset (https://favor.genohub.org/favor-annotator) integrated in the STAARpipeline24,25. We selected ten functional annotation principal components that represent a wide range of biological and functional processes: aPC-Protein-Function, aPC-Conservation, aPC-Epigenetics-Active, aPC-Epigenetics-Repressed, aPC-Epigenetics-Transcription, aPC-Local-Nucleotide-Diversity, aPC-Mutation-Density, aPC-Transcription-Factor, aPC-Mappability, aPC-Proximity-To-TSS-TES. To facilitate interpretation, these annotation scores are expressed on Phred scale, defined as , where M is the number of variants in the FAVOR database. A higher Phred score indicates increased functionality of the annotation. Further details can be found in Li et al.23. We subdivided each LD score-MAF bin into two disjoint bins: one bin containing SNVs with Phred ≥ t (“High”) and one bin containing SNVs with Phred < t (“Low”), where t varied depending on the annotation.
Enrichment analysis
For all functional analyzes (evolutionary constraint using phyloP score, SnpEff predicted impact, snATAC-seq profiling and FAVOR aPCs), we investigated the heritability enrichment in each LD score-MAF bin. We examined the log enrichment ratio of the contribution per variant from: i) constrained over non constrained SNVs; ii) protein-altering over non-protein-altering SNVs; iii) SNVs inside over SNVs outside cell-specific snATAC-seq peaks; and iv) high over low aPC functionality SNVs. Derivation of the mean and variance of the log enrichment ratio distribution is described in the Supplementary “Methods”.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This study was supported by National Heart, Lung and Blood Institute (NHLBI) grant R01 HL146860. Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the NHLBI. WGS for “NHLBI TOPMed: Genetics of Cardiometabolic Health in the Amish” (phs000956.v4.p1) was performed at the Broad Institute Genomics Platform (3R01HL121007-01S1). WGS for “NHLBI TOPMed: Atherosclerosis Risk in Communities (ARIC)” (phs001211.v3.p2) was performed at the Broad Institute Genomics Platform (3R01HL092577-06S1) and at the Baylor College of Medicine Human Genome Sequencing Center (3U54HG003273-12S2 / HHSN268201500015C). WGS for “NHLBI TOPMed: Mount Sinai BioMe Biobank” (phs001644.v1.p1) was performed at the McDonnell Genome Institute (3UM1HG008853-01S2). WGS for “NHLBI TOPMed: Coronary Artery Risk Development in Young Adults (CARDIA)” (phs001612.v1.p1) was performed at the Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I). WGS for “NHLBI TOPMed: Cardiovascular Health Study (CHS)” (phs001368.v2.p2) was performed at the Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I, 3U54HG003273-12S2 / HHSN268201500015C) and at the Broad Institute Genomics Platform (HHSN268201600034I). WGS for “NHLBI TOPMed: Genetic Epidemiology of COPD (COPDGene)” (phs000951.v4.p4) was performed at the Broad Institute Genomics Platform (HHSN268201500014C, HHSN268201500014C) and at the Northwest Genomics Center (3R01HL089856-08S1). WGS for “NHLBI TOPMed: Diabetes Heart Study (DHS)” (phs001412.v2.p1) was performed at the Broad Institute Genomics Platform (HHSN268201500014C). WGS for “NHLBI TOPMed: Framingham Heart Study (FHS)” (phs000974.v4.p3) was performed at the Broad Institute Genomics Platform (3U54HG003067-12S2, HHSN268201600034I, 3R01HL092577-06S1). WGS for “NHLBI TOPMed: Genetic Studies of Atherosclerosis Risk (GeneSTAR)” (phs001218.v2.p1) was performed at the Broad Institute Genomics Platform (HHSN268201500014C), at Illumina (R01HL112064) and at Psomagen (3R01HL112064-04S1). WGS for “NHLBI TOPMed: Genetic Epidemiology Network of Arteriopathy (GENOA)” (phs001345.v2.p1) was performed at the Broad Institute Genomics Platform (HHSN268201500014C) and at the Northwest Genomics Center (3R01HL055673-18S1). WGS for “NHLBI TOPMed: Jackson Heart Study (JHS)” (phs000964.v4.p1) was performed at the Northwest Genomics Center (HHSN268201100037C). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v2.p1) was performed at the Broad Institute Genomics Platform (HHSN268201600034I, 3U54HG003067-13S1, HHSN268201500014C). WGS for “NHLBI TOPMed: Women’s Health Initiative (WHI)” (phs001237.v2.p1) was performed at the Broad Institute Genomics Platform (HHSN268201500014C). Core support, including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support, including phenotype harmonization, data management, sample-identity QC, and general program coordination, was provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. Consortium members are listed in the Supplementary Information. P.T.E. is supported by grants from the National Institutes of Health (1RO1HL092577, 1R01HL157635, 5R01HL139731), from the American Heart Association Strategically Focused Research Networks (18SFRN34230127), and from the European Union (MAESTRIA 965286). C.L.M. is supported by NIH/NHLBI grants (R01HL148239 and R01HL164577), Fondation Leducq ‘PlaqOmics’ (18CVD02), and the Chan Zuckerberg Initiative, LLC and Silicon Valley Community Foundation. R.D. is supported by the National Institute of General Medical Sciences of the NIH (R35-GM124836) and the National Heart, Lung and Blood Institute of the NIH (R01-HL139865 and R01-HL155915). This work was supported in part through the computational and data resources and staff expertise provided by Scientific Computing and Data at the Icahn School of Medicine at Mount Sinai and supported by the Clinical and Translational Science Awards (CTSA) grant UL1TR004419 from the National Center for Advancing Translational Sciences. Research reported in this publication was also supported by the Office of Research Infrastructure of the National Institutes of Health under award numbers S10OD026880 and S10OD030463. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute, the National Institutes of Health, or the U.S. Department of Health and Human Services.
Author contributions
G.R. and R.D. conceived the analyses. G.R. performed the analysis and created the figures. S.L.C. assembled the case-control dataset along with the relevant covariates. C.L., J.G.B., and A.T.K. identified the TOPMed-related participants and contributed to WGS data quality control. S.L.C, N.R.H., A.C.M., A.S.H., L.F.B., K.R.I., E.P.Y., N.O.S., G.J., A.T.H., C.T., P.A.P., R.S.V., J.I.R., C.L.M., T.L.A., P.S.V. advised on the analysis. A.T.H., C.T., and T.L.A. provided heritability estimates from the Million Veteran Program. G.A. and C.L.M. provided the coronary artery snATAC data and assisted with their analysis. D.K.A., L.C.B., J.C.B., E.B., D.W.B., A.P.C., P.T.E., M.F., N.F., B.I.F., N.L.H.-C., L.H., Y.-D.I.C., E.E.K., C.K., B.G.K., R.J.F.L., S.M.L., J.E.M., L.W.M., B.D.M., R.N., N.D.P., W.S.P., M.H.P., B.M.P., L.M.R., E.A.R., S.S.R., J.A.S., K.D.T., L.R.Y., K.A.Y., P.A.P., R.S.V. and J.I.R. contributed to the design and recruitment of the TOPMed studies included in this research. G.R. and R.D. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
Data for each participating study can be accessed through dbGaP with the corresponding TOPMed accession numbers: Amish (phs000956), ARIC (phs001211), BioMe (phs001644), CARDIA (phs001612), CHS (phs001368), COPDGene (phs000951), DHS (phs001412), FHS (phs000974), GeneSTAR (phs001218), GENOA (phs001345), JHS (phs000964), MESA (phs001416), WHI (phs001237). The latest gnomAD data set (v4.1.0) can be downloaded at https://gnomad.broadinstitute.org. phyloP scores can be downloaded at https://cgl.gi.ucsc.edu/data/cactus/241-mammalian-2020v2-hub/Homo_sapiens/241-mammalian-2020v2.bigWig. Raw and processed coronary artery snATAC data are available in Gene Expression Omnibus (GEO) under accession ID: GSE175621. The full dataset of the FAVORannotator’s database can be downloaded at https://favor.genohub.org/favor-annotator. All data generated in this study are provided in the Supplementary Data file.
Code availability
ADMIXTURE: https://dalexander.github.io/admixture/index.html. bigsnpr (R package): https://cran.r-project.org/web/packages/bigsnpr/index.html. GCTA (GREML-LDMS): https://yanglab.westlake.edu.cn/software/gcta/#Overview. GENESIS (R package performing PC-AiR and PC-Relate): https://bioconductor.org/packages/release/bioc/html/GENESIS.html. Scripts used for processing coronary artery snATAC data are available at https://github.com/MillerLab-CPHG/Coronary_snATAC. SnpEff: https://pcingola.github.io/SnpEff/. All figures were generated using R software: https://www.R-project.org/
Competing interests
P.T.E. receives sponsored research support from Bayer AG, IBM Research, Bristol Myers Squibb, Pfizer and Novo Nordisk; he has also served on advisory boards or consulted for Bayer AG, MyoKardia and Novartis. B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. L.M.R. is a consultant for the TOPMed Administrative Coordinating Center (through Westat). C.L.M. received grant support from AstraZeneca for unrelated work. R.D. reported being a scientific co-founder, consultant and equity holder for Pensieve Health and being a consultant for Variant Bio, all not related to this work. All other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A list of authors and their affiliations appears at the end of the paper.
Contributor Information
Ron Do, Email: ron.do@mssm.edu.
NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium:
Ghislain Rocheleau, Shoa L. Clarke, Natalie R. Hasbani, Alanna C. Morrison, Adam S. Heath, Lawrence F. Bielak, Kruthika R. Iyer, Erica P. Young, Nathan O. Stitziel, Goo Jun, Cecelia Laurie, Jai G. Broome, Alyna T. Khan, Donna K. Arnett, Lewis C. Becker, Joshua C. Bis, Eric Boerwinkle, Donald W. Bowden, April P. Carson, Patrick T. Ellinor, Myriam Fornage, Nora Franceschini, Barry I. Freedman, Nancy L. Heard-Costa, Lifang Hou, Yii-Der Ida Chen, Eimear E. Kenny, Charles Kooperberg, Brian G. Kral, Ruth J. F. Loos, Sharon M. Lutz, JoAnn E. Manson, Lisa W. Martin, Braxton D. Mitchell, Rami Nassir, Nicholette D. Palmer, Wendy S. Post, Michael H. Preuss, Bruce M. Psaty, Laura M. Raffield, Elizabeth A. Regan, Stephen S. Rich, Jennifer A. Smith, Kent D. Taylor, Lisa R. Yanek, Kendra A. Young, Pramod Anugu, Paul Auer, Lucas Barwick, Diane Becker, Cara Carty, Peter Castaldi, Mark Chaffin, Yi-Cheng Chang, Seung Hoan Choi, Ren-Hua Chung, Carolyn Crandall, Sean David, Lisa de las Fuentes, Ranjan Deka, Dawn DeMeo, Paul S. de Vries, Qing Duan, Charles Eaton, Lynette Ekunwe, Adel El Boueiz, Shanshan Gao, Yan Gao, Margery Gass, Auyon Ghosh, Daniel Grine, Michael Hall, Craig Hersh, Brian Hobbs, Chao Agnes Hsiung, Yi-Jen Hung, Haley Huston, Chii Min Hwu, Rebecca Jackson, Jill Johnsen, Christoph Lange, Ethan Lange, Meryl LeBoff, Wen-Jane Lee, Yun Li, Simin Liu, Yu Liu, Susan Mathai, Hao Mei, Rakhi Naik, Take Naseri, Bonnie Neltner, Heather Ochs-Balcom, David T. Paik, Cora Parker, Marco Perez, Ulrike Peters, Lawrence S. Phillips, Julia Powers Becker, Muagututi’a Sefulva Reupena, Carolina Roselli, Pamela Russell, Ester Cerdeira Sabino, Kevin Sandow, Karen Schwander, Frank Sciurba, Brian Silver, Sylvia Smoller, Beverly Snively, Garrett Storm, Yun Ju Sung, Hua Tang, Margaret Taub, Lesley Tinker, David Tirschwell, Hemant Tiwari, Dhananjay Vaidya, Tarik Walker, Robert Wallace, Avram Walts, Lu-Chen Weng, Ivana Yang, Snow Xueyan Zhao, Patricia A. Peyser, Ramachandran S. Vasan, Jerome I. Rotter, Clint L. Miller, Themistocles L. Assimes, and Ron Do
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52939-6.
References
- 1.Tsao, C. W. et al. Heart disease and stroke statistics—2023 update: a report from the american heart association. Circulation147, e93–e621 (2023). [DOI] [PubMed]
- 2.Roth, G. A. et al. Global burden of cardiovascular diseases and risk factors, 1990–2019. J. Am. Coll. Cardiol.76, 2982–3021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wienke, A., Holm, N. V., Skytthe, A. & Yashin, A. I. The heritability of mortality due to heart diseases: a correlated frailty model applied to danish twins. Twin Res.4, 266–274 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Zdravkovic, S. et al. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J. Intern. Med.252, 247–254 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Chen, Z. & Schunkert, H. Genetics of coronary artery disease in the post‐GWAS era. J. Intern. Med.290, 980–992 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Aragam, K. G. et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet.54, 1803–1815 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tcheandjieu, C. et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med.28, 1679–1692 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wainschtein, P. et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet.54, 263–273 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jang, S.-K. et al. Rare genetic variants explain missing heritability in smoking. Nat. Hum. Behav.6, 1577–1586 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wessel, J. et al. Rare non-coding variation identified by large scale whole genome sequencing reveals unexplained heritability of type 2 diabetes. Preprint at medRxivhttp://medrxiv.org/lookup/doi/10.1101/2020.11.13.20221812 (2020).
- 11.Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanks, S. C. et al. Extent to which array genotyping and imputation with large reference panels approximate deep whole-genome sequencing. Am. J. Hum. Genet.109, 1653–1666 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erdmann, J., Kessler, T., Munoz Venegas, L. & Schunkert, H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc. Res. 114, 1241–1257 (2018). [DOI] [PubMed]
- 14.Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Won, H.-H. et al. Disproportionate contributions of select genomic compartments and cell types to genetic risk for coronary artery disease. PLOS Genet.11, e1005622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nasser, J. et al. Genome-wide enhancer maps link risk variants to disease genes. Nature593, 238–243 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Preissl, S., Gaulton, K. J. & Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet.24, 21–43 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Örd, T. et al. Single-cell epigenomics and functional fine-mapping of atherosclerosis GWAS loci. Circ. Res.129, 240–258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner, A. W. et al. Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk. Nat. Genet.54, 804–816 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan, P. F. et al. Leveraging base-pair mammalian constraint to understand genetic variation and human disease. Science380, eabn2937 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans, L. M. et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat. Genet.50, 737–745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly. (Austin)6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, X. et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet.52, 969–983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, Z. et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat. Methods19, 1599–1611 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou, H. et al. FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res.51, D1300–D1311 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature625, 92–100 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siepel, A., Pollard, K. S. & Haussler, D. New methods for detecting lineage-specific selection. In Lecture Notes in Bioinformatics3909, 190–205 (Springer-Verlag, 2006).
- 28.Weiner, D. J. et al. Polygenic architecture of rare coding variation across 394,783 exomes. Nature614, 492–499 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaitlen, N. & Kraft, P. Heritability in the genome-wide association era. Hum. Genet.131, 1655–1664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-Eghan, C. et al. Don’t ignore genetic data from minority populations. Nature585, 184–186 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Petrazzini, B. O. et al. Exome sequence analysis identifies rare coding variants associated with a machine learning-based marker for coronary artery disease. Nat. Genet.56, 1412–1419 (2024). [DOI] [PubMed] [Google Scholar]
- 32.O’Connor, L. J. et al. Extreme polygenicity of complex traits is explained by negative selection. Am. J. Hum. Genet.105, 456–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng, J. et al. Widespread signatures of natural selection across human complex traits and functional genomic categories. Nat. Commun.12, 1164 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature599, 628–634 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh, T. et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature604, 509–516 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akingbuwa, W. A., Hammerschlag, A. R., Bartels, M., Nivard, M. G. & Middeldorp, C. M. Ultra-rare and common genetic variant analysis converge to implicate negative selection and neuronal processes in the aetiology of schizophrenia. Mol. Psychiatry27, 3699–3707 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou, D., Zhou, Y., Xu, Y., Meng, R. & Gamazon, E. R. A phenome-wide scan reveals convergence of common and rare variant associations. Genome Med.15, 101 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duffy, Á. et al. Development of a human genetics-guided priority score for 19,365 genes and 399 drug indications. Nat. Genet.56, 51–59 (2024). [DOI] [PubMed] [Google Scholar]
- 39.Minikel, E. V., Painter, J. L., Dong, C. C. & Nelson, M. R. Refining the impact of genetic evidence on clinical success. Nature629, 624–629 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell177, 26–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fatumo, S. et al. A roadmap to increase diversity in genomic studies. Nat. Med.28, 243–250 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaitlen, N. et al. Leveraging population admixture to characterize the heritability of complex traits. Nat. Genet.46, 1356–1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo, Y. et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum. Mol. Genet.30, 1521–1534 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan, T. F. et al. Estimating heritability explained by local ancestry and evaluating stratification bias in admixture mapping from summary statistics. Am. J. Hum. Genet.110, 1853–1862 (2023). [DOI] [PMC free article] [PubMed]
- 46.Visscher, P. M. et al. Statistical power to detect genetic (Co)variance of complex traits using SNP data in unrelated samples. PLoS Genet.10, e1004269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Speed, D., Kaphle, A. & Balding, D. J. SNP‐based heritability and selection analyses: Improved models and new results. BioEssays44, 2100170 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Conomos, M. P., Miller, M. B. & Thornton, T. A. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet. Epidemiol.39, 276–293 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conomos, M. P., Reiner, A. P., Weir, B. S. & Thornton, T. A. Model-free estimation of recent genetic relatedness. Am. J. Hum. Genet.98, 127–148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Privé, F., Luu, K., Blum, M. G. B., McGrath, J. J. & Vilhjálmsson, B. J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics36, 4449–4457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res.19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet.88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee, S. H., Wray, N. R., Goddard, M. E. & Visscher, P. M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet.88, 294–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Data for each participating study can be accessed through dbGaP with the corresponding TOPMed accession numbers: Amish (phs000956), ARIC (phs001211), BioMe (phs001644), CARDIA (phs001612), CHS (phs001368), COPDGene (phs000951), DHS (phs001412), FHS (phs000974), GeneSTAR (phs001218), GENOA (phs001345), JHS (phs000964), MESA (phs001416), WHI (phs001237). The latest gnomAD data set (v4.1.0) can be downloaded at https://gnomad.broadinstitute.org. phyloP scores can be downloaded at https://cgl.gi.ucsc.edu/data/cactus/241-mammalian-2020v2-hub/Homo_sapiens/241-mammalian-2020v2.bigWig. Raw and processed coronary artery snATAC data are available in Gene Expression Omnibus (GEO) under accession ID: GSE175621. The full dataset of the FAVORannotator’s database can be downloaded at https://favor.genohub.org/favor-annotator. All data generated in this study are provided in the Supplementary Data file.
ADMIXTURE: https://dalexander.github.io/admixture/index.html. bigsnpr (R package): https://cran.r-project.org/web/packages/bigsnpr/index.html. GCTA (GREML-LDMS): https://yanglab.westlake.edu.cn/software/gcta/#Overview. GENESIS (R package performing PC-AiR and PC-Relate): https://bioconductor.org/packages/release/bioc/html/GENESIS.html. Scripts used for processing coronary artery snATAC data are available at https://github.com/MillerLab-CPHG/Coronary_snATAC. SnpEff: https://pcingola.github.io/SnpEff/. All figures were generated using R software: https://www.R-project.org/