

Abstract
The specific CD8+ T-cell response during acute lymphocytic choriomeningitis virus (LCMV) infection of mice is characterized by a rapid proliferation phase, followed by a rapid death phase and long-term memory. In BALB/c mice the immunodominant and subdominant CD8+ responses are directed against the NP118 and GP283 epitopes. These responses differ mainly in the magnitude of the epitope-specific CD8+ T-cell expansion. Using mathematical models together with a nonlinear parameter estimation procedure, we estimate the parameters describing the rates of change during the three phases and thereby establish the differences between the responses to the two epitopes. We find that CD8+ cell proliferation begins 1 to 2 days after infection and occurs at an average rate of 3 day−1, reaching the maximum population size between days 5 and 6 after immunization. The 10-fold difference in expansion to the NP118 and GP283 epitopes can be accounted for in our model by a 3.5-fold difference in the antigen concentration of these epitopes at which T-cell stimulation is half-maximal. As a consequence of this 3.5-fold difference in the epitope concentration needed for T-cell stimulation, the rates of activation and proliferation of T cells specific for the two epitopes differ during the response and in combination can account for the large difference in the magnitude of the response. After the peak, during the death phase, the population declines at a rate of 0.5 day−1, i.e., cells have an average life time of 2 days. The model accounts for a memory cell population of 5% of the peak population size by a reversal to memory of 1 to 2% of the activated cells per day during the death phase.
Acute viral infections are often characterized by a rapid and extensive response of antigen-specific CD8+ T cells (5, 8, 14, 18, 31). A typical time course of an acute antiviral CD8+ T-cell response involves an extensive proliferation phase, during which the specific CD8+ populations may expand three to five orders of magnitude; an apoptosis or death phase, during which 95% of antigen-specific cells die; and a long-term memory phase (2, 31). At the time of the peak of the response most of the activated CD8+ T-cell population in the spleen are specific for lymphocytic choriomeningitis virus (LCMV) (18). The main mechanism responsible for the contraction of the antigen-specific T-cell population is the programmed cell death (20) of CD8+ T cells that were activated by the viral antigens (1, 3, 30). Migration of antigen-specific cells from the spleen into solid tissue (21) may also contribute to the contraction. The population size during the memory phase remains approximately constant and is typically 5% of the peak value (18).
The immune response to a virus usually involves several epitopes. The CD8+ T-cell responses to dominant and subdominant epitopes of LCMV all go through the phases of proliferation, death, and long-term memory (18). Although the magnitude of a subdominant response remains considerably smaller than that of the dominant response (18), it is not clear what determines the relative magnitude of these responses. Competition does not seem to play a major role, as removal of the dominant response hardly increases the subdominant response (26, 29). Additionally, recent data suggest that naive CD8+ T cells undergo considerable clonal expansion after a single early exposure to antigen (13, 16, 27). Thus, cells would have to compete for antigen in a very short and early time window before the clones have expanded. Additionally, there does not appear to be much competition between memory populations to dominant and subdominant epitopes, as these coexist for more than a year following the acute infection (18). This is consistent with the suggestion that memory cells are maintained by survival signals other than the specific antigen (4, 19, 24, 25). Competition for the same antigen would result in competitive exclusion (9–11). Much less is known, however, about the role of competition for antigen during the acute phase of the immune response.
Earlier studies suggested that the major difference between the responses to dominant and subdominant epitopes is the “recruitment time” (6, 18). By starting earlier, the response to a dominant epitope would achieve larger expansion than subdominant responses. Developing mathematical models and using nonlinear parameter estimation, we characterize the major parameters of the CD8+ LCMV response (e.g., the rates of proliferation, apoptosis, and memory cell formation) and show that the responses to dominant and subdominant epitopes need differ only marginally in these rates to account for the data.
MATERIALS AND METHODS
Six- to 8-week-old male and female BALB/c mice were purchased from the Jackson Laboratories (Bar Harbor, Maine). Mice were infected with 2 × 105 PFU of LCMV Armstrong intraperitoneally and sacrificed at days 3, 5, 8, 12, 15, 40, and 45 (three mice per time point). The Elispot assay described by Taguchi et al. (23) was modified by Murali-Krishna et al. (18) to detect NP118- or GP283-specific CD8+ T cells in the spleen of LCMV-infected mice. The frequency of epitope-specific CD8+ T cells was based on the percentage of CD8+ T cells present in the responding population. Using this assay, we could accurately measure a minimum of 10 spots among 106 responder cells (18).
For both epitopes, the (naive) precursor frequency at day zero was estimated as 1/200,000 cells (Blattman et al., submitted for publication). For each epitope, this yields a naive precursor population size of about 60 cells per spleen.
For the viral load, we use the data of Lau et al. (14). Following its introduction into the peritoneal cavity, the virus has a very rapid expansion phase, reaching its maximal titer in the spleen in about 2 days. The viral concentration remains within one log of this maximal value for another 4 days and then declines by several orders of magnitude over the next 3 days (14) (see Fig. 3). The basic dynamics of virus and CD8 cells following infection with LCMV Armstrong have been replicated many times, and the overall results are very robust. We therefore feel confident that these previous data (14) are appropriate for the present study.
FIG. 3.

Dynamics of the NP118- and GP283-specific cell populations in the continuous model. (A) Broken line gives the viral load in PFU per spleen (14), the heavy line gives the total population size of the NP118 response, and the light line gives that of the GP283 response. (B) Subpopulations within each clone. Solid lines depict activated cells, long-dashed lines show memory cells, and short-dashed lines show naive cells (14).
Parameter estimates were obtained using the DNLS1 subroutine from the Common Los Alamos Software Library, which is based on the Levenberg-Marquardt algorithm (15) for solving nonlinear least-squares problems. These parameters were used to calculate the predicted T-cell population size, and 95% confidence intervals for the inferred parameters were then determined using a bootstrap method (12), where the residuals to the optimal fit were resampled 500 times.
Basic model.
Although the populations responding to the NP118 and GP283 epitopes are oligoclonal (22), we simplify the situation and consider two “clones” of antigen-specific CD8+ T cells. The cells of each clone are either naive (N), activated (A), or memory (M) (Fig. 1). Activated cells proliferate at rate ρ, die by apoptosis at rate α, and revert to memory cells at rate r. Memory cells become reactivated at rate a and die at rate δM. For reasons of simplicity, naive cells are assumed to become activated at the same maximum rate, a, as memory cells.
FIG. 1.

Scheme of the basic model depicting the step function f(t). In this model we assume that naive T cells are all activated when proliferation starts at Ton. In the second model the step function f(t) is replaced by a continuous function, 0 < f(t) < 1, that smoothly follows the changes in the viral load. Solid lines indicate processes that are positively influenced by the activation function. Dashed lines are processes inhibited by antigen.
Because the viral burden during the acute response to LCMV Armstrong switches so rapidly between very high and very low (14), we first approximate the antigenic stimulation of the CD8+ T cells by a function, f(t), that takes on only two values; 0 when there is no activation, and 1 when there is full activation. Assuming that antigenic stimulation switches “on” at time Ton and “off” at time Toff, we use for the activation function f
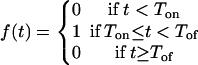 |
1 |
The parameters Ton and Toff are the times between which the virus concentration is considered to be large enough to allow maximal T-cell proliferation. The parameter Ton will be referred to as the recruitment time. In this model, which we call the on-off or basic model, we ignore the naive subpopulation and assume that between time 0 and time Ton, all antigen-specific naive CD8+ T cells become activated. Later, we relax some of the simplifying assumptions of this model and develop a model with a continuous activation function, which includes naive T cells and follows the kinetics of their activation.
In this basic on-off model, the dynamics of the CD8+ T-cell response is given by the following differential equations:
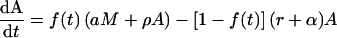 |
2 |
and
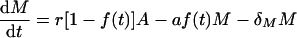 |
3 |
We assume that at the start of the response, i.e., time zero, there are no memory cells, i.e., M(0) = 0, and that at time Ton the number of activated cells A has a value equal to the naive cell antigen-specific precursor frequency. During a vigorous LCMV Armstrong infection, one indeed observes that almost all precursor cells become activated (13). When the activation function is on, f(t) = 1, activated cells proliferate at rate ρ, and any existing memory cells can become reactivated at rate a. When the activation function is off, f(t) = 0, activated cells die by apoptosis at rate α and revert to memory cells at rate r. The fraction r/(r + α) gives the fraction of activated cells that successfully relax to memory cells.
The model is piecewise linear, and its explicit solutions are derived in the Appendix.
RESULTS
Basic model.
The measured population sizes of the LCMV epitope NP118- and GP283-specific CD8+ T cells in the spleen, as assessed by a gamma interferon (IFN-γ) Elispot assay between days 3 and 45, are depicted in Fig. 2. Each symbol in the figure represents one BALB/c mouse. The data point at day zero reflects the initial precursor frequency, as assessed by Blattman et al. (submitted). The solution of the model was fitted to these data by a nonlinear multiparameter estimation procedure that minimized the sum of squared residuals (SSR) between the data and the total number of antigen-specific CD8+ T cells, M(t) + A(t), predicted by the model.
FIG. 2.

Fitting the on-off model to the data on the CD8+ T-cell response to the NP118 epitope (A) and to the GP283 epitope (B). For each epitope, the dashed lines depict the total population size in the spleen, solid lines show activated cells, and long-dashed lines show memory cells. The circle symbols represent the experimental data.
Because in the basic model the responses to the two epitopes are not coupled, the parameters can be estimated for each epitope independently. The results in Fig. 2A and B and in Table 1 indicate that the CD8+ T cells specific for the two LCMV epitopes have small differences in several parameters. The response to the dominant epitope starts somewhat earlier, the cells proliferate somewhat faster, and the proliferation phase ends somewhat later. NP118-specific cells also have a somewhat higher apoptosis rate. The differences in the parameters for the two epitopes are small, and the 95% confidence intervals overlap.
TABLE 1.
Parameter estimates for the NP118 and GP283 epitopesa
| Parameter | Symbol | Units | NP118
|
GP283
|
||
|---|---|---|---|---|---|---|
| Value | 95% C.I. | Value | 95% C.I. | |||
| Proliferation rate | ρ | Day−1 | 2.9 | 2.7–3.0 | 2.6 | 2.2–3.0 |
| Apoptosis rate | α | Day−1 | 0.51 | 0.42–0.64 | 0.36 | 0.19–0.84 |
| Memory cell formation | r | Day−1 | 0.011 | 0.008–0.015 | 0.014 | 0.007–0.03 |
| Recruitment time | Ton | Days | 1.2 | 1.1–1.4 | 1.7 | 1.1–2.0 |
| Toff | Days | 5.8 | 5.7–6.0 | 5.6 | 5.3–6.0 | |
The sum of squared residuals is SSR = 1.1 for the NP118 epitope and SSR = 5.5 for the GP283 epitope. Fixed parameters were δM = 10−5 day−1 and A(Ton) = 60 cells. The death rate of memory cells was fixed at a low value to account for the longevity of CD8+ memory populations; setting it to zero or letting it be free and fitting it hardly changes the results (not shown). The initial condition A(Ton) = 60 was fixed because this parameter was estimated experimentally by Blattman et al. (submitted). The 95% confidence intervals (C.I.) were determined by a bootstrap method (12) with 500 resamplings of the residuals.
As indicated in Table 1, the estimated proliferation rate in the NP118-specific response is about ρ = 3 day−1. Proliferation starts around day 1.2 and stops around day 5.8. Having 4.6 days of proliferation at a rate of 2.9 day−1, one expects a 6.2 × 106-fold expansion. The estimated cellular death rate due to apoptosis is α = 0.5 day−1, yielding an average lifetime of activated cells during the death phase of about 2 days. The rate at which activated cells revert to the memory stage during the death phase is r = 0.01 day−1. Because r/(r + α) ≃ 0.02, the model suggests that after Toff, 2% of the cells leaving the activated pool revert to memory. Thus, memory cells accumulate gradually, generating a total population comprising approximately 5% of the peak population size (18). Note that the 95% confidence limits on the reversal parameter are relatively large. According to our estimates, the cells responding to the subdominant GP283 epitope proliferate at a rate of 2.6 day−1 over 3.9 days, allowing a 2.5 × 104-fold expansion (Table 1). Thus, the combination of small differences in various parameters markedy affects the magnitude of the response (17). Because we ignore death during the expansion phase (see equation A1 in the Appendix), the proliferation rates that we estimate are net proliferation rates. If proliferating cells also die, the true proliferation rate would be correspondingly higher.
Previous publications have suggested that the main difference between dominant and subdominant responses is the recruitment time (6, 7, 18). To test this hypothesis, the two data sets were fitted simultaneously, assuming that only the recruitment time Ton differs between the two responses (Table 2). The values of the common parameters ended up intermediate to those in Table 1. The required difference in the recruitment time needed to best fit the data was about 20 h. Visually, the fit appeared to be as good as that in Fig. 2 (not shown), with the sum of the squared residuals differing marginally between the two cases (8.1 versus 6.6). The total expansion for each epitope also remains very similar. To test the hypothesis further, we also fitted the opposite scenario by forcing Ton and Toff to be the same for the dominant and subdominant responses while allowing the other parameters to be different (Table 3). Visually this fit is also as good as that in Fig. 2. The sum of the squared residuals (SSR = 7.3) is somewhat better than that obtained when only the recruitment time Ton differs. The main difference between the two responses in Table 3 is a 20% lower proliferation rate of the subdominant response.
TABLE 2.
Parameter estimates for the response to the NP118 and the GP283 epitopes fitted simultaneously by allowing only the recruitment time to differa
| Parameter | Symbol | Units | Both epitopes
|
NP118
|
GP283
|
|||
|---|---|---|---|---|---|---|---|---|
| Value | 95% C.I. | Value | 95% C.I. | Value | 95% C.I. | |||
| Proliferation rate | ρ | Day−1 | 2.8 | 2.5–3.0 | ||||
| Apoptosis rate | α | Day−1 | 0.43 | 0.31–0.59 | ||||
| Memory cell formation | r | Day−1 | 0.01 | 0.009–0.02 | ||||
| Toff | Days | 5.7 | 5.5–5.9 | |||||
| Recruitment time | Ton | Days | 1.0 | 0.7–1.2 | 1.8 | 1.0–2. | ||
Visually, these fits are indistinguishable from those in Fig. 2. The summed squared residuals is SSR = 8.1, which is only somewhat poorer than the SSR = 6.6 obtained when all parameters are allowed to differ between the two responses. Fixed parameters were δM = 10−5 day−1 and A(Ton) = 60 cells.
TABLE 3.
Parameter estimates for the response to the NP118 and the GP283 epitopes fitted simultaneously while forcing the recruitment times Ton and Toff to remain identicala
| Parameter | Symbol | Units | Both epitopes
|
NP118
|
GP283
|
|||
|---|---|---|---|---|---|---|---|---|
| Value | 95% C.I. | Value | 95% C.I. | Value | 95% C.I. | |||
| Recruitment time | Ton | Days | 1.4 | 1.2–1.6 | ||||
| Toff | Days | 5.7 | 5.5–5.9 | |||||
| Proliferation rate | ρ | Day−1 | 3.0 | 2.8–3.3 | 2.4 | 2.2–2.6 | ||
| Apoptosis rate | α | Day−1 | 0.49 | 0.35–0.78 | 0.38 | 0.26–0.60 | ||
| Memory cell formation | r | Day−1 | 0.011 | 0.006–0.02 | 0.015 | 0.008–0.03 | ||
Visually, these fits are indistinguishable from those in Fig. 2. The summed squared residuals is SSR = 7.3, which is only somewhat poorer than the SSR = 6.6 obtained when all parameters are allowed to differ between the two responses and somewhat better than the fit obtained when only the recruitment time Ton differs. Fixed parameters were δM = 10−5 day−1 and A(Ton) = 60 cells.
In summary, fitting the data on two epitopes separately and allowing essentially all parameters to vary, the model accounts for the data by small differences in various parameters. However, one cannot exclude a single large difference in the recruitment time. The previous claims were based on finding similar estimates for the proliferation rates of the different responses (6, 7, 18). Since these estimates were made rather crudely, one would not have been able to observe the small (10%), but apparently important difference in the proliferation rate found in Table 1 or the 20% difference required in Table 3.
Continuous model.
In reality, activation of a T-cell population is not all or none, and the relatively weak GP283 response may be due to a lower degree of antigenic stimulation. For example, the NP118 and GP283 epitopes may differ in the way they get presented to T cells and may trigger T-cell clones having different affinities for the resulting major histocompatibility complex (MHC)-peptide complexes. In order to model different degrees of antigen stimulation of the T-cell clones responding to the different epitopes, we let T-cell activation depend on a saturation function of the viral load, V,
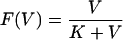 |
4 |
Here the parameter K determines the amount of antigen needed to generate half-maximal stimulation. Thus, a clone characterized by a low value of K would be easier to stimulate with a given amount of virus than a clone characterized by a large value of K. Both the viral load and the K parameter are measured in PFU per spleen (14).
We do not explicitly model the viral load, but rather assume that the viral load, V, changes with time according to the curve given in Lau et al. (14) and shown in Fig. 3A. Assuming exponential dynamics, we interpolated the data in Lau et al. (14) between the available points (see Fig. 3A). We model the different sensitivity of the two T-cell clones to their specific epitopes by different stimulation parameters K.
The model now becomes
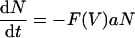 |
5 |
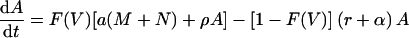 |
6 |
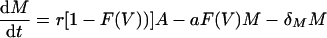 |
7 |
This model depends on the virus loads reported by Lau et al. (14), and through the function F(V), the viral load determines the actual rates of activation, proliferation, memory formation, and apoptosis. For example, the rate of proliferation of an activated cell is now ρF(V) and hence varies during the response, with ρ being the maximum rate and F(V) being a factor that varies between 0 and 1. In modeling the response to the NP118 and GP283 epitopes, we examine the case in which the two epitopes differ in antigen availability only, i.e., the K parameter only, and the maximum rates of T-cell activation, proliferation, memory formation, and apoptosis are equal for both epitopes.
This model was fitted to the data for both epitopes simultaneously. We first found fits to the data with low activation rates a and unrealistically high proliferation rates ρ. As a consequence, there was hardly any depletion of the naive precursor population by activation. Because we think most specific precursors become activated during the immune response, we fixed the activation rate at a = 1 day−1. Naive precursors thus become activated and depleted on a time scale of days (28) (Fig. 3B and Table 4).
TABLE 4.
Parameter estimates for the NP118 and GP283 epitopes obtained by simultaneously fitting the data for both epitopes in the continuous modela
| Parameter | Symbol | Units | Both epitopes
|
NP118
|
GP283
|
|||
|---|---|---|---|---|---|---|---|---|
| Value | 95% | Value | 95% | Value | 95% | |||
| Apoptosis rate | α | Day−1 | 0.41 | 0.31–0.58 | ||||
| Proliferation rate | ρ | Day−1 | 2.92 | 2.74–3.10 | ||||
| Memory cell formation | r | Day−1 | 0.015 | 0.009–0.024 | ||||
| Saturation constant | K | PFU | 3.8 × 104 | 1.9–6.1 | 1.3 × 105 | 0.87–1.9 | ||
The parameters a = 1 day−1, δM = 10−5 day−1 and N(0) = 60 cells were fixed. The summed squared residuals is SSR = 11.4, which is only somewhat poorer than that of the piecewise linear model.
The fit to the data is similar in quality to that of the on-off model. This shows that the parameter differences between the NP118 and the GP283 response can indeed be accounted for by a difference in the K parameter. By our parameter estimates, the GP283 CD8 response requires a 3.5-fold-higher antigen concentration to achieve a similar degree of stimulation as the NP118 response. We also consider a worst-case scenario by assuming that the activation function is the same for the different biological processes in the model, i.e., activation, proliferation, and apoptosis are all governed by the same K parameter. Our model with a single K parameter is a special case of a model allowing for different values of the K parameter. Because we can fit the data with a single K, it is clear that we could also fit the data to a model having several K parameters; however, we would have little confidence in the various K values.
Figure 4 depicts the actual activation, proliferation, and apoptosis rates over time. Note that in the continuous model, proliferation, for example, occurs at rate ρF(V) per cell. Consistent with the results of the on-off model, we find small differences in the actual values of several rates. The largest difference seems to be that the actual proliferation rate declines more slowly in response to the dominant epitope. This extends the proliferation period and allows a larger clonal expansion.
FIG. 4.

Actual activation rates F(V)a (solid lines), proliferation rates F(V)ρ (long-dashed lines), and apoptosis rates F(V)α (short-dashed lines). Heavy lines give the NP118 response, and light lines give the GP283 response.
As shown in Fig. 4, the time courses for the activation rates are very similar for both epitopes. Thus, there is little difference in the recruitment rate of naive CD8+ T cells specific for the two epitopes. Since our activation function depends only on the viral load and not on the time of the response, the model suffers from the artifact that the apoptosis rate is high early in the response when the viral load is still low (Fig. 4). This is probably not realistic but hardly affects the behavior of the model because there are very few activated cells present at this early state of the response (Fig. 3). Summarizing, the data are most parsimoniously explained by a 3.5-fold difference in the parameters K, i.e., by a difference in the antigen concentration required for half-maximal stimulation.
DISCUSSION
We have developed two simple models for the CD8+ T-cell response to LCMV in mice. In one of the models, we assumed that T-cell activation is an all-or-none process, while in a more complex model we allowed a continuous change in activation level. Using these models, we have shown that the immune responses to the dominant NP118 epitope and the subdominant GP283 epitope in the LCMV CD8+ T-cell immune response may involve differences in the proliferation period and the actual proliferation rates. The most parsimonious explanation for these differences is a difference in the antigenic stimulation of the two responses. In our model the subdominant response requires a 3.5-fold-higher antigen load than the dominant response to achieve similar stimulation. This small difference in the antigen stimulation allows a slightly earlier recruitment and a somewhat higher proliferation rate over a somewhat longer time period in response to the dominant NP118 epitope (Fig. 4). In both models, we found that the large difference in the magnitude of the response to the two epitopes can be accounted for by small differences in various parameters (17).
Previous papers addressing the differences between dominant and subdominant responses confirm that immune responses to different epitopes on the same pathogen expand, contract, and enter the memory T-cell compartment synchronously (6, 7). These studies have argued, however, that the main difference in the dynamics between the responses to different epitopes of the same antigen is the timing of recruitment (6, 7, 18). This argument was based largely on the observation that the proliferation rates seemed relatively similar. Since previously the proliferation rates were measured rather crudely, the evidence arguing that only the recruitment times differ was relatively weak. Our more accurate calculations demonstrate that accounting for the large difference in the magnitude of the two responses by this argument requires a 20-h difference in the recruitment time (Table 2). Although such a large difference in the recruitment time cannot be excluded at present, we show that small differences in the various parameters affecting the proliferation rate over time also account for the large difference between the NP118 and GP283 responses.
The two models developed for the parameter estimation are relatively simple and only allow us to estimate a proliferation rate during the expansion phase and death and reversal rates during the contraction phase. Thanks to this simplicity, we expect that our parameter estimates are fairly general. Indeed, we have experimented with several versions of the models and found similar results (not shown). Despite the simplicity of the models, we have shown that alternative parameter fittings remain possible. The on-off model can account for the data by a difference in the recruitment time or in the proliferation rate only and in the continuous model we had to fix the activation rate to ensure a sufficient activation of the precursor cells.
In animals primed with the GP283 epitope, the response to the this subdominant epitope dominates over the otherwise dominant response to the NP118 epitope (Ahmed et al., unpublished data). This is in good agreement with the small difference in the estimated parameters reported here. Starting with a large population of GP283-specific CD8+ memory T cells, the somewhat faster NP118-specific response is not expected to overtake the GP283 population before the GP283-specific cytotoxic T lymphocytes could eliminate the antigen.
Recent data demonstrate that CD8+ T cells undergo considerable clonal expansion after an initial exposure to antigen (13, 16, 27). A relatively short stimulus by antigen, i.e., less than 2 h (27), “programs” CD8+ T cells to divide several times in an antigen-independent manner. Note that in Fig. 3 proliferation indeed continues after the virus has been cleared. While the details of antigen-independent proliferation are not known at present, this could imply that our simple on-off model is more realistic than the more complicated continuous model, in which the proliferation rate depends on the antigen concentration. The off switch in the on-off model is an independent parameter that could also reflect the end of the programmed cell division cascade. Thus, the on-off model allows us to estimate the rate constants for the responses to the two epitopes during the various phases of the response and suggests that small differences in the growth rate and/or the recruitment time can account for the phenomenon of immunodominance.
Summarizing, we have presented a model that accounts for the kinetics of the dominant and sub dominant CD8+ T-cell immune responses to LCMV infection. Fitting data to the model, we estimate a proliferation rate of 3 day−1 in the dominant response. During the apoptotic death phase, activated cells have an average life span of 2 days, and on a daily basis 1 to 2% of the activated cells revert to the memory stage. By the accumulation of memory cells, the memory population is about 5% of the population size at the peak of the response by the end of the death phase.
ACKNOWLEDGMENTS
The first two authors contributed equally to this work.
Portions of the work were done under the auspices of the U.S. Department of Energy. A.S.P. is supported by NIH grant AI28433 and the Joseph P. Sullivan and Jeanne M. Sullivan Foundation.
Appendix
The model defined by equations 1 to 3 is piecewise linear. When t < Ton, there is no antigenic stimulation, and M(t) = 0. Naive cells become activated during this time interval, according to an unspecified dynamics, so that at Ton, all naive precursors have become activated cells. For Ton ≤ t < Toff, i.e., f(t) = 1, memory cells are still absent and activated cells expand exponentially at a rate ρ, so that the solution obeys
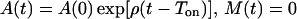 |
A1 |
Following the peak, i.e., for t ≥ Toff, f(t) = 0 and hence the cell populations obey the following linear model:
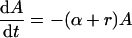 |
A2 |
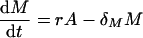 |
A3 |
with solution
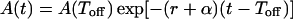 |
A4 |
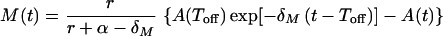 |
A5 |
where A(Toff) = A(0) exp[ρ(Toff − Ton)].
REFERENCES
- 1.Abbas A K. Die and let live: eliminating dangerous lymphocytes. Cell. 1996;84:655–657. doi: 10.1016/s0092-8674(00)81042-9. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 3.Akbar A N, Salmon M. Cellular environments and apoptosis: tissue microenvironments control activated T-cell death. Immunol Today. 1997;18:72–76. doi: 10.1016/s0167-5699(97)01003-7. [DOI] [PubMed] [Google Scholar]
- 4.Antia R, Pilyugin S S, Ahmed R. Models of immune memory: on the role of cross-reactive stimulation, competition, and homeostasis in maintaining immune memory. Proc Natl Acad Sci USA. 1998;95:14926–14931. doi: 10.1073/pnas.95.25.14926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asano M S, Ahmed R. CD8 T cell memory in B cell-deficient mice. J Exp Med. 1996;183:2165–2174. doi: 10.1084/jem.183.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bousso P, Levraud J P, Kourilsky P, Abastado J P. The composition of a primary T cell response is largely determined by the timing of recruitment of individual T cell clones. J Exp Med. 1999;189:1591–1600. doi: 10.1084/jem.189.10.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Busch D H, Pilip I M, Vijh S, Pamer E G. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- 8.Butz E A, Bevan M J. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998;8:167–175. doi: 10.1016/s1074-7613(00)80469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Boer R J, Perelson A S. T cell repertoires and competitive exclusion. J Theor Biol. 1994;169:375–390. doi: 10.1006/jtbi.1994.1160. [DOI] [PubMed] [Google Scholar]
- 10.De Boer R J, Perelson A S. Towards a general function describing T cell proliferation. J Theor Biol. 1995;175:567–576. doi: 10.1006/jtbi.1995.0165. [DOI] [PubMed] [Google Scholar]
- 11.De Boer R J, Perelson A S. Competitive control of the self-renewing T cell repertoire. Int Immunol. 1997;9:779–790. doi: 10.1093/intimm/9.5.779. [DOI] [PubMed] [Google Scholar]
- 12.Efron B, Tibshirani R. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci. 1986;1:54–77. [Google Scholar]
- 13.Kaech S M, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau L L, Jamieson B D, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature. 1994;369:648–652. [PubMed] [Google Scholar]
- 15.Marquardt D W. Finite difference algorithm for curve fitting. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
- 16.Mercado R, Vijh S, Allen S E, Kerksiek K, Pilip I M, Pamer E G. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–6839. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 17.Müller V, Marée A F, De Boer R J. Small variations in multiple parameters account for wide variations in HIV-1 set-points: a novel modelling approach. Proc R Soc Lond B Biol Sci. 2001;268:235–242. doi: 10.1098/rspb.2000.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murali-Krishna K, Altman J D, Suresh M, Sourdive D J, Zajac A J, Miller J D, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 19.Murali-Krishna K, Lau L L, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–1381. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- 20.Raff M C. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 21.Reinhardt R L, Khoruts A, Merica R, Zell T, Jenkins M K. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- 22.Sourdive D J, Murali-Krishna K, Altman J D, Zajac A J, Whitmire J K, Pannetier C, Kourilsky P, Evavold B, Sette A, Ahmed R. Conserved T cell receptor repertoire in primary and memory CD8 T cell responses to an acute viral infection. J Exp Med. 1998;188:71–82. doi: 10.1084/jem.188.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taguchi T, McGhee J R, Coffman R L, Beagley K W, Eldridge J H, Takatsu K, Kiyono H. Detection of individual mouse splenic T cells producing IFN-gamma and IL-5 using the enzyme-linked immunospot (ELISPOT) assay. J Immunol Methods. 1990;128:65–73. doi: 10.1016/0022-1759(90)90464-7. [DOI] [PubMed] [Google Scholar]
- 24.Tanchot C, Lemonnier F A, Perarnau B, Freitas A A, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 25.Tanchot C, Rocha B. The organization of mature T-cell pools. Immunol Today. 1998;19:575–579. doi: 10.1016/s0167-5699(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 26.Van der Most R G, Sette A, Oseroff C, Alexander J, Murali-Krishna K, Lau L L, Southwood S, Sidney J, Chesnut R W, Matloubian M, Ahmed R. Analysis of cytotoxic T cell responses to dominant and subdominant epitopes during acute and chronic lymphocytic choriomeningitis virus infection. J Immunol. 1996;157:5543–5554. [PubMed] [Google Scholar]
- 27.Van Stipdonk M J, Lemmens E E, Schoenberger S P. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 28.Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol. 2000;1:47–53. doi: 10.1038/76907. [DOI] [PubMed] [Google Scholar]
- 29.Vijh S, Pilip I M, Pamer E G. Noncompetitive expansion of cytotoxic T lymphocytes specific for different antigens during bacterial infection. Infect Immun. 1999;67:1303–1309. doi: 10.1128/iai.67.3.1303-1309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welsh R M, Selin L K, Razvi E S. Role of apoptosis in the regulation of virus-induced T cell responses, immune suppression, and memory. J Cell Biochem. 1995;59:135–142. doi: 10.1002/jcb.240590202. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman C, Brduscha-Riem K, Blaser C, Zinkernagel R M, Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J Exp Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]