

Abstract
A novel synthetic pathway for synthesizing isocyanate-free polyurethanes is reported here. β-Amino alcohols were efficiently synthesized from the aminolysis of the epoxide ring of (R)-(+)-limonene oxide with different primary amines as nucleophiles and hot water as catalysts. The regio- and diastereoselectivities of the reactions were investigated and supported by computational studies. DFT calculations were performed to understand the experimental results more deeply. It confirmed the crucial roles of water molecules and the nature of the nucleophile in forming the products. The formation of the product is entirely driven by the free energy of activation that affects the reaction rate. Cyclic carbamates were prepared from β-amino alcohols using the dialkyl carbonate (DAC) chemistry. An oligourethane was obtained from Anionic Ring-Opening Polymerization (AROP) of a cyclic carbamate derived from (R)-(+)-limonene-oxide. All the products were characterized by employing 1H and 13C NMR spectroscopies. The assignments of the signals in 1H and 13C NMR spectra were also supported by 2D NMR spectroscopy.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-73824-8.
Keywords: Isocyanate free, Dimethyl carbonate, Sustainable, Polyurethane, Density functional theory
Subject terms: Green chemistry, Organic chemistry, Polymer chemistry
Introduction
Polyurethanes (PU) are one of the most important classes of polymers, with an annual production of more than 18 million tons, about 5 mass % of total worldwide polymer production1. They are very versatile polymers, as they can be thermoplastic, elastomeric, thermoset, and foams2. PU are typically prepared by step-growth polymerization between polyols and isocyanates. Isocyanates present concerns regarding their impact on the environment, health, and safety3–6. They are produced from the reaction of amines with toxic phosgene2 and have several potential adverse effects on human health4–6. Currently, with the governmental regulatory environment and the general European goal of using the least toxic effective components available, there has been increasing industrial and academic emphasis on obtaining urethane properties from systems that do not employ isocyanates. Bio-based polyurethanes are generally prepared using polyols from natural sources as an alternative to oil-based ones7,8. The most common approaches reported in the literature to obtain isocyanate-free PU are (1) polyaddition of cyclic dicarbonates and diamines (i.e., ring-opening polymerization of cyclic carbonates); (2) polycondensation of linear activated dicarbonates and diamines (i.e., bis(dialkyl carbonate) route); (3) polycondensation of linear activated carbamates and diols (i.e., transurethanization route); (4) ring-opening polymerization (ROP) of cyclic carbamates (i.e., ROP route)9–11. Among them, the ROP of cyclic carbamates is only scarcely investigated. In particular, 5- and 6-membered cyclic carbamates can undergo cationic ROP12–14. More recently, the anionic ROP of cyclic carbamates was reported15,16. However, cyclic carbamates have been limited by very laborious or low-yielding synthetic procedures for their preparation, involving phosgene or its derivatives17, alkyl halide chemistry18–20, and isocyanate compounds21,22. Recently, new approaches to cyclic carbamates via dialkyl carbonate (DAC) chemistry have been reported23,24.
Herein we report a novel methodology for the synthesis of cyclic carbamates starting from (R)-(+)-limonene oxide, a biorenewable derivative of the naturally abundant terpene (R)-(+)-limonene. Furthermore, the attempt of Anionic Ring-Opening Polymerization (AROP) of a cyclic carbamate derived from (R)-(+)-limonene oxide is described and discussed.
Results and discussion
The Anionic Ring-Opening Polymerization (AROP) of cyclic carbamates based on (R)-(+)-limonene oxide yields isocyanate-free (oligo)PU (Fig. 1).
Fig. 1.
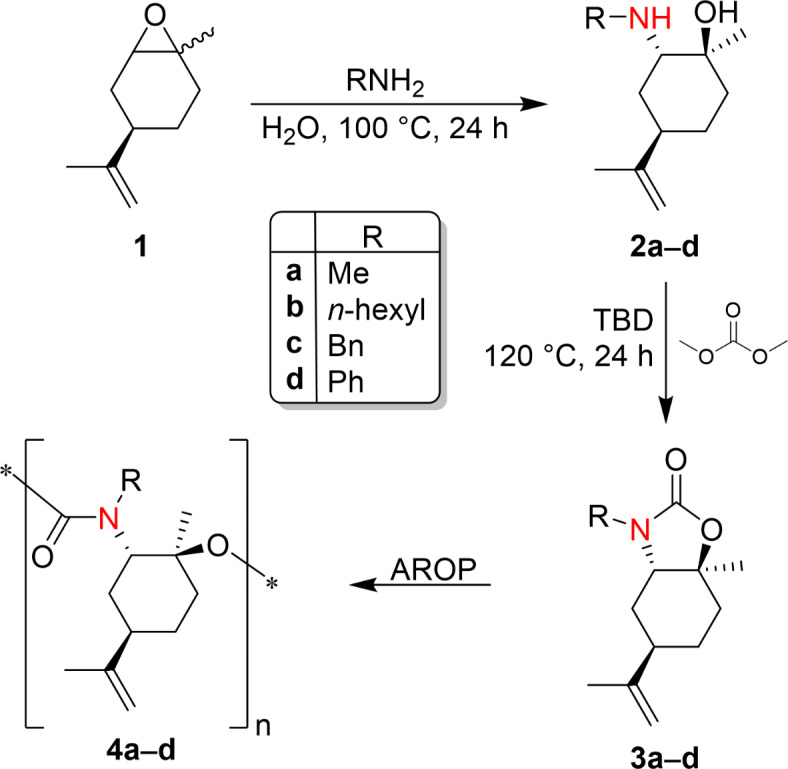
Reactions pathway for the synthesis of isocyanate-free PU from (R)-(+)-limonene oxide.
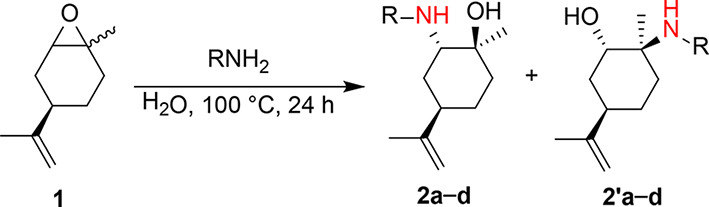
A library of β-amino alcohols was efficiently prepared from the opening of the epoxide of (R)-(+)-limonene oxide with primary amines (methylamine, hexylamine, benzylamine, aniline) and in the presence of water as a catalyst with high yields, as highlighted in Table 1.
Table 1.
Aminolysis reaction of (R)-(+)-limonene oxide with different aminesa.
| ||||
|---|---|---|---|---|
| Entry | Amine | 2 (%)b | 2’ (%)b | Yield % |
| 1 | Methylamine | 53 | 47 | 97 |
| 2 | Hexylamine | 100 | n.d.c | 50 |
| 3 | Benzylamine | 100 | n.d. | 50 |
| 4 | Aniline | 73 | 27 | 59 |
aReaction conditions: (R)-(+)-limonene oxide (1 eq.), amine (2 eq. for entries 1 and 4, 1.2 eq. for entries 2,3), water, 100 °C, 24 h.
bCalculated by integrating the signals in the 1H NMR spectrum of the crude reaction mixture and by considering their relative abundance. Not detected.
Without water, the reaction proceeded with very low yields, even after long reaction times and temperatures25. Hot water acts as a mild Brønsted acid catalyst, favoring the epoxide ring-opening by nucleophiles, such as amines26–28. The nucleophile attack occurred preferentially at the less hindered carbon atom. In the case of hexylamine and benzylamine (entries 2 and 3 of Table 1), the lone pair on the nitrogen atom is entirely available, and the activation of the oxirane by hydrogen bonding with water is lost. With methylamine and aniline (entries 1 and 4 of Table 1), 2 equivalents of nucleophile are necessary for the aminolysis of (R)-(+)-limonene oxide due to the formation of hydrogen bonding with water (solvent effect) and the low nucleophilicity of aniline (delocalization of the lone pair over the benzene ring). Minimal work-up of the reactions was required by extracting with an organic solvent (diethyl ether or dichloromethane) or by direct water evaporation, leading to the pure ring-opened products with no further purification. It is worth mentioning that commercially available (R)-(+)-limonene oxide is a 1:1 mixture of cis- and trans- isomers (Fig. 2). The separation of the two diastereomers by physical methods (fractional distillation, gas chromatography) is troublesome29.
Fig. 2.

Chemical structures of trans- and cis-limonene oxide. The numbers refer to the carbon atoms involved in the amine addition.
However, the aminolysis reaction of limonene oxide is reported to be regio- and diastereoselective28,30–37. According to the Fürst-Plattner rule, the approach of the nucleophile to cyclohexene oxide derivatives is trans-diaxial, leading substantially to trans-diaxial opening products favoring a chair-like transition state38,39. The bulky isopropenyl group of (R)-(+)-limonene oxide is equatorial in both cis- and trans-isomers. Generally, nucleophilic amines’ aminolysis of limonene oxide occurs preferentially on the trans-isomer in an SN2-type reaction at the less hindered secondary C-2, leaving the cis-isomer substantially unreacted (kinetic resolution)31,33,34. This case was observed for forming β-amino alcohols 2b and 2c, where R is a n-hexyl and a benzyl substituent, respectively. Contrarily, the SN2-type reaction at the less hindered C-2 of the cis-isomer would have to occur via a boat-like transition state, which is energetically more demanding and unfavorable. However, when utilizing non-nucleophilic amines, the aminolysis reaction does not occur, and the water acts as a nucleophile only on cis-limonene oxide at the most hindered tertiary C-1 carbon atom through a chair-like transition state31,34. In this case, an SN1-type reaction occurs due to the electronic state of the C-1 carbon but maintains the features of an SN2-type reaction, leading to an inversion of configuration at the C-1 atom31,34. Indeed, a mixture of the two regioisomers, 2 and 2’, was obtained when (R)-(+)-limonene oxide was reacted with methylamine and aniline (Table 1, Entries 1 and 4).
2a and 2’a were separated by flash chromatography and characterized by 1H NMR spectra (Fig. 3).
Fig. 3.

1H NMR spectra (CDCl3, 400 MHz) of (A) mixture of 2a and 2’a regioisomers; (B) single regioisomer 2a; (C) single regiosiomer 2’a.
To shed light on the experimental outcome, we conducted computational studies at the density functional theory (DFT) level of theory. To obtain as accurate activation energies as possible, the three-dimensional models of all reagents, products, and transition states were fully geometrically optimized at a high level of theory by employing the PW6B95D3 functional with def2TZVPP basis set implemented in the Gaussian 16 software package. Frequency calculation was used to confirm the nature of the stationary points. Transition structures were found to have only one negative eigenvalue.
To choose the best system to simulate the catalytic action of water, we conducted a molecular dynamics (MD) study under the experimental conditions reported in Table 1 Entry 1 employing the structure of the transition state (TS) for both regioisomeric approach and using water as an explicit solvent (see SI). After 300 ns of MD simulation, the trajectories were analyzed to individuate the most recurrent and reliable water molecules/TS arrangement. This arrangement, containing the first layer of water molecules around the TS, was extrapolated by the MD frame and fully minimized at the GFN2-xTB level of theory. The result, depicted in Fig. 4, shows a network of water molecules, three establishing a hydrogen bond bridge between the oxygen of the epoxide and one of the amine hydrogens and another coordinated by hydrogen bonding with the oxygen of the epoxide, that we retain essential for the correct reproduction of the experimental conditions.
Fig. 4.

Representative 3D-geometry for the first-layer solvation of TS conducting to product 2a.
So, all DFT calculations were conducted using an implicit-explicit solvation model using SMD implicit solvation and four explicit water molecules. The two TS conducting to products 2a and 2’a are shown in Fig. 5.
Fig. 5.

TSs for the reaction of the trans and cis-limonene oxides with methylamine. Aliphatic hydrogens were omitted for clarity. Distances were in Å. Dashed lines represent bonds involved in transition states, and dotted lines are hydrogen bonds. Carried out with CYLview2040.
Considering that the reactions studied are sufficiently exergonic and, therefore, difficult to reversible, it is evident that the results we observed must depend exclusively on the free activation energies. It is presumable that in the reactions that exclusively produce the compounds 2b and 2c, the activation energies for the transition states that lead to the compounds 2’b and 2’c must be higher and insurmountable at the reaction temperature of 100 °C; that is, for the latter, the reaction is kinetically much slower and in the reaction time of 24 h the formation of the products is not appreciated.
The enthalpies, free energies, and entropies of activation for all studied TSs, reported in Table 2, support this hypothesis. All the reactions show an elevated free energy of activation heavily penalized by the activation entropy due to the high order of the molecules in the transition state. On the contrary, all the TSs-2 possess an enthalpy of activation almost null (TS-2a) or slightly exothermic (TSs-2b–c), whereas in all the TSs-2’ it is slightly endothermic (Table 2).
Table 2.
Enthalpies, free energies, and entropies of activation for transition states involved in the aminolysis reactionsa.
| TS | DH#a (kcal/mol) | DG#a (kcal/mol) | DS# (cal/mol/K) |
|---|---|---|---|
| 2a | 0.004 | 49.55 | −166.18 |
| 2’a | 2.01 | 50.40 | −162.28 |
| 2b | −1.12 | 50.50 | −173.16 |
| 2’b | 0.37 | 51.83 | −172.61 |
| 2c | −0.19 | 50.11 | −168.72 |
| 2’c | 1.86 | 51.89 | −161.07 |
| 2d | −0.34 | 50.89 | −169.85 |
| 2’d | 1.39 | 51.11 | −164.05 |
aCalculated with respect to the enthalpies, free energies, and entropies of the reactants.
Activation energies around 50 kcal/mol result in very long reaction times, even at 100 °C. For example, the reaction leading to compounds 2a/2’a has a 97% yield with a 2a:2’a ratio close to unity, indicating a nearly complete reaction of both trans and cis reagents. The reaction for compounds 2d/2’d has a 59% yield with a 2d:2’d ratio of 2.7, showing that almost 100% of the trans reagent converts to 2d (yielding 43%), while only 15.9% of 2’d is obtained. Reactions for b and c yield 50%, producing only 2c and 2b, with the trans reagent fully converted and the cis reagent unreacted. This data suggests a maximum activation barrier beyond which reactions do not occur at 100 °C, producing no detectable products within 24 h. Based on our calculations, this limit is slightly higher than 51.11 kcal/mol, the activation energy for the reaction leading to compound 2’d (Table 2). Thus, reactions leading to compounds 2’b and 2’c, with activation energies of 51.83 and 51.89 kcal/mol, respectively, do not occur.
Cyclic carbamates were obtained by reaction of limonene β-amino alcohols previously prepared with dimethyl carbonate (DMC) in the presence of a catalytic amount of a bicyclic nitrogen base, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). Dimethyl carbonate is a versatile, non-toxic, biodegradable chemical belonging to the dialkyl carbonate family (DACs). DACs are ambident electrophiles, meaning they can undergo BAc2- or BAl2-nucleophilic substitution under appropriate conditions to give alkoxycarbonylation and alkylation reactions, respectively. In this work, the reaction proceeded by a selective double BAc2 mechanism to obtain the desired cyclic carbamate. Cyclic carbamates were obtained from the reaction of limonene β-amino alcohols and DMC at a bath temperature of 120 °C for 24 h in the presence of 0.5% mol of TBD. The reaction was conducted in a vessel equipped with a magnetic stirrer and reflux condenser. Reaction time and temperature were adjusted and optimized to have a high conversion of the reactants. The reactions performed were monitored through TLC and GC-MS. Cyclic carbamates 3a–d were characterized by employing 1H,13C NMR, and ESI-MS spectroscopies.
The assignments of all the signals in 1H and13C NMR were also supported by 1H, 1H COSY, and HSQC (Heteronuclear Single Quantum Coherence) analyses (SI). After the end of each reaction, a simple work-up procedure was adopted, and the pure product was collected with no further purification. In brief, the reaction mixture was dissolved in diethyl ether and washed with deionized water. The dried organic phase was filtered and concentrated under reduced pressure (details are in the experimental part). Notably, the synthetic methodology developed does not use the typical toxic reagents involved in synthesizing cyclic carbamates such as phosgene, alkyl halides, and isocyanates. A library of cyclic carbamates based on (R)-(+)-limonene oxide was efficiently synthesized with the above-described method (Table 3), with high yields. The 1H NMR spectra (Fig. 6) of the synthesized cyclic carbamates 3a–d are in accordance with the theoretical structures (Fig. 5). The formation of the open carbamates/carbonates was observed when an excess of dimethyl carbonate was used in the reaction mixture. In this latter case, a strong peak near 3.79 ppm due to the –CH3 protons of DMC was observed. Hence, the absence of this peak in the 1H NMR spectrum suggests the formation of cyclic carbamate. Another evidence of the formation of the cycle is given by the presence of the C=O of the urethane bond, confirmed by 13C NMR spectroscopy, as shown in Table S1. By comparing the 1H NMR spectra of β-amino alcohols with the corresponding 1H NMR spectra of cyclic carbamates, it was noticed that in the case of β-amino alcohols, the hydrogens f of the isoprenyl group give rise to a single signal, which indicates the equatorial orientation of the isoprenyl group. In the case of cyclic carbamates, two well-separated signals for hydrogens f of the isoprenyl group can be distinguished, indicating that the isoprenyl group has an axial orientation33. Indeed, in β-amino alcohols, the –OH and amino substituents are trans-diaxial, and the bulky isoprenyl group is equatorial, as explained above. Probably, a change in the conformation of the cyclohexane ring occurs when the β-amino alcohols react with dimethyl carbonate, and the –OH and amino substituents assume a trans-diequatorial orientation to be able to form the cyclic carbamate.
Table 3.
Preparation of cyclic carbamatesa.
| Entry | b-amino alcohol | Amine | Yield % |
|---|---|---|---|
| 1 | 3a | Methylamine | 69 |
| 2 | 3’a | Methylamine | 65 |
| 3 | 3b | Hexylamine | 80 |
| 4 | 3c | Benzylamine | 70 |
| 5 | 3d | Aniline | 80 |
aReaction conditions: β-amino alcohol (1 eq), DMC (1 eq), TBD (0.5 eq), 120 °C, 24 h.
Fig. 6.

1H NMR spectra (CDCl3, 400 MHz) of cyclic carbamates 3a–d.
Cyclic carbamates based on (R)-(+)-limonene oxide were then investigated as potential monomers to prepare polyurethanes (PU) via Ring-Opening Polymerization (ROP). Monomer 3a was selected for the polymerization reaction (Fig. 7).
Fig. 7.

Chemical structure of monomer (5R)-3,7a-dimethyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (3a), initiation Step of the Anionic Polymerization of Cyclic Carbamate Monomer 3a with DBU and propagation Step of the Anionic ROP of Cyclic Carbamate 3a (in the figure labeled as M).
Different types of ROP can be distinguished according to the mechanism involved: Radical Ring-Opening Polymerization (RROP), Cationic Ring Opening Polymerization (CROP), Anionic Ring-Opening Polymerization (AROP), and Ring-Opening Metathesis Polymerization (ROMP). Some examples of Cationic ROP of cyclic carbamates12–14,39, and Anionic Ring-Opening Polymerization (AROP)15,16 are reported in the scientific literature. CROP of 5- and 6-membered cyclic carbamates to yield polyurethanes is generally performed efficiently using boron trifluoride diethyl etherate BF3O(Et)2 or trifluoromethane sulfonate CF3 SO2R (R=OH, OCH3) as initiators in the melt. The polymerization mechanism first involves forming the cationic species via an acid-base reaction between the monomer and the initiator. Then, the propagation step occurs via the nucleophilic attack of a monomer molecule on the electrophilic species. The polymerization continues until the monomer is consumed or with the addition of a nucleophile. However, our system is more complex with respect to what is reported in the literature due to the presence of the cyclohexane ring and the presence of the isoprenyl group. In particular, the double bond of the isoprenyl group is relevant. Since BF3O(Et)2 and triflates are weak and strong acids, respectively, these catalysts were supposed to be inefficient for the ROP of monomer 3a because they can react well with alkenes. For this reason, it was decided to perform Anionic ROP to polymerize monomer 3a. At the same time, some experiments of CROP of monomer 3a were also conducted to verify our hypothesis. Typically, the initiation step of the Anionic ROP of cyclic carbamates occurs via hydrogen abstraction of the N-H proton with a basic initiator (t-BuOK, n-BuLi) and, in some cases, a co-initiator is needed15,16. In our system, the nitrogen of the urethane bond is substituted with a –CH3 group in the case of monomer 3a. Hence, another strategy was performed for the Anionic ROP of monomer 3a. Cyclic carbonates are polymerized via Anionic ROP using amine initiators (DMAP, DABCO, DBU)41. In a typical polymerization reaction, the cyclic monomer is weighed into a reaction vessel. The catalyst is added to the monomer in a 0.4% molar ratio. The mixture is magnetically stirred at the selected temperature for 12 h. 1,8-Diazabicyclo(5.4.0)undec-7-ene (DBU) was used as the catalyst for the Anionic ROP of cyclic carbamate 3a. Table 4 resumes the experimental conditions adopted for the ROP of cyclic carbamate 3a.
Table 4.
Attempted ROP of monomer 3a.

aReaction conditions: monomer 3a: catalyst 1:0.004. bDetermined through1H NMR spectroscopy (chain end determination). cMixed cationic propagations: ROP and electrophilic addition to the alkene. dNot determined.
To follow the progression of the polymerization via NMR spectroscopy, first trials were performed using DMSO-d6 as the solvent. The initiation step of the polymerization (Fig. 7) involves the reaction between 3a and DBU to obtain a nucleophilic tetrahedral intermediate, which is unstable and gives an alkoxide anion, the active initiator of the polymerization.
The propagation step can occur once the active initiator is formed, as shown in Fig. 7.
By inspecting the 1H (Fig. 8) and 13C NMR (Figure S13) spectra, the opening of the carbamate ring and the propagation give a PU oligomer. As shown in the 1H NMR spectrum of the Anionic ROP of monomer 3a with DBU (spectrum C of Fig. 8), the –CH3 group bonded to the nitrogen atom at 2.6 ppm belongs to the repeating unit, whereas the peak at 2.8 ppm is the –CH3 group and represents the terminal group. Integrating these two signals shows that the PU oligomer has 3.82 (~ 4) repeat units. Further, by inspecting the 13C NMR spectrum of Figure S13, the peak at 165.40 ppm indicates the urea linkage’s presence. The low molecular weight of the product is consistent with the steric hindrance of the cyclic carbamate 3a as the monomer. It should also be noted that a neat polymerization was carried out. In anionic polymerizations, a solvent can strongly influence the nature of the active growing species, favoring the formation of solvated ion pairs or ion clusters. In this work, the anionic ROP of the cyclic carbamate 3a was carried out under neat conditions to understand the ability of the monomer to form the active initiator species and to propagate without the influence of any solvent. Further research could elucidate the influence of the solvent on the anionic ROP of cyclic carbamate monomers derived from (R)-(+)-limonene.
Fig. 8.

1H NMR spectra (DMSO-d6, 400 MHz) of: (A) DBU; (B) monomer 3a; (C) Anionic ROP of monomer 3a with DBU.
Conclusions
A novel synthetic pathway for the synthesis of polyurethanes without using toxic reagents, i.e., isocyanates, was designed and performed starting from limonene as a natural source. In particular, the enantiomer (R)-(+)-limonene was used. A library of β-amino alcohols was efficiently synthesized from the aminolysis of the epoxide ring of (R)-(+)-limonene-oxide with different amines as nucleophiles and water as solvent and catalyst. The regio- and diastereoselectivity of the reactions were investigated and supported by computational studies. The β-amino alcohols obtained were then used for the synthesis of 5-membered cyclic carbamates by using the dialkylcarbonate (DAC) chemistry. A library of cyclic carbamates was obtained. Cyclic carbamates are suitable monomers for the Ring-Opening Polymerization (ROP) to yield isocyanate-free polyurethanes. One of the cyclic carbamates derived from (R)-(+)-limonene was used as a monomer, and Anionic Ring-Opening Polymerization (AROP) was performed. A polyurethane oligomer was obtained, thus validating the synthetic pathway designed in this work to synthesize polyurethanes from natural sources without using toxic reagents and solvents, following the principles of green chemistry. Combining the experimental results and the DFT calculations, it is clear that the presence of water as an acid catalyst at a temperature of 100 °C and the nature of the nucleophile have a crucial role in forming the products; interestingly, the formation of the product is entirely driven by the free energy of activation that affects the rate of reaction. Above an approximate value of 51.6 kcal/mol, the reaction, at a temperature of 100 °C, is extremely slow for forming products 2’b and 2’c.
Experimental part
Materials. Reagents and solvents commercially available were purchased and used without further purification:
Methylamine 40% w/w in water (Sigma Aldrich – Merck), hexylamine (Sigma Aldrich – Merck), ethanolamine (Sigma Aldrich – Merck), ammonium hydroxide solution 30% w/w in water (Sigma Aldrich – Merck), limonene (Sigma Aldrich – Merck), limonene epoxide (Sigma Aldrich – Merck), benzylamine (Sigma Aldrich – Merck), aniline (Sigma Aldrich – Merck), dimethylcarbonate (Sigma Aldrich – Merck), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, Sigma Aldrich – Merck), 1,8-Diazabicyclo(5.4.0)undec-7-ene (DBU, Sigma Aldrich – Merck), boron trifluoride diethyl etherate (Sigma Aldrich – Merck), methyl trifluoromethanesulfonate (Sigma Aldrich – Merck), dichloromethane (Sigma Aldrich – Merck), diethyl ether (Sigma Aldrich – Merck), sodium sulphate anhydrous (Sigma Aldrich – Merck), acetone (Sigma Aldrich – Merck), deuterated chloroform (CDCl3, Sigma Aldrich – Merck), deuterated dimethyl sulfoxide (DMSO-d6, Sigma Aldrich – Merck).
General procedure for β-amino alcohols
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (R)-(+)-limonene oxide and the selected primary amine (methylamine solution 40 wt % in H2O, hexylamine, benzylamine, aniline), in H2O as catalyst and solvent. The reaction mixture was left stirring at 100 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the product was isolated by removal of water or by extracting with dichloromethane or diethyl ether. The products were characterized by means of 1H, 13C NMR, and ESI-MS spectroscopies.
Synthesis of (1S,2S,4R)-2-amino-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3a) and (1S,2S,5R)-2-amino-2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-ol (3a’).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (R)-(+)-limonene oxide (5 g, 0.033 mol) in H2O and methylamine solution 40 wt % in H2O (3 mL, 0.04 mol). The reaction mixture was left stirring at 100 °C for 24 hours. After this time, the product was extracted from water using dichloromethane. A yellow viscous liquid was obtained (5.84 g, 97% yield), comprising of regioisomers (1S,2S,4R)-2-amino-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3a) and (1S,2S,5R)-2-amino-2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-ol (3a’). The two regioisomers were separated by flash chromatography, eluting with EtOAc/EtOH 1:1.
(1S,2S,4R)-2-amino-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3a). Yellow oil. 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.75 (s, 2 H), 2.41 (s, 3 H), 2.20 (dd, 1 H), 1.92 (ddd, 1 H), 1.72 (s, 3 H), 1.59–1.71 (m, 2 H), 1.42–1.59 (m, 4 H), 1.19 (s, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 148.89, 109.39, 72.11, 64.26, 37.86, 35.06, 34.79, 29.51, 26.30, 25.59, 21.58. ESI mass spectra, m/z (MeOH): 184.2 ([M + H]+).
(1S,2S,5R)-2-amino-2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-ol (3a’). Yellow oil. 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.68 (s, 2 H), 3.60 (dd, 1 H), 2.26 (s, 3 H), 1.80–1.95 (ddd, 1 H), 1.67 (s, 3 H), 1.59 (ddd, 1 H), 1.41–1.53 (m, 4 H), 1.11–1.25 (m, 1 H), 1.05 (s, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 148.63, 109.34, 71.88, 63.69, 37.78, 33.17, 29.79, 27.50, 25.43, 21.52, 20.20. ESI mass spectra, m/z (MeOH): 184.2 ([M + H]+), 206.2 ([M + Na]+).
Synthesis of (1 S,2 S,4R)-2-(hexylamino)-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3b).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (R)-(+)-limonene oxide (0.55 g, 3.62 mmol) in H2O and hexylamine (0.44 g, 4.34 mmol) in H2O as catalyst and solvent. The reaction mixture was left stirring at 100 °C for 24 h. After this time, the unreacted starting materials were separated from the product by distillation. (1S,2S,4R)-2-(hexylamino)-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3b) was obtained as a dark yellow viscous liquid (0.54 g, 59% yield). 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.78 (s, 2 H), 2.77–2.70 (m, 1 H), 2.52–2.45 (m, 2 H), 2.23 (s, 1 H), 1.99–1.93 (m, 1 H), 1.74 (s, 3 H), 1.69–1.64 (m, 2 H), 1.56–1.45 (m, 5 H), 1.33–1.30 (m, 6 H), 1.20 (s, 3 H), 0.88 (t, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 148.52, 109.42, 72.09, 61.97, 48.23, 38.01, 34.59, 31.75, 30.74, 30.31, 27.13, 26.99, 26.05, 22.63, 21.61, 14.03. ESI mass spectra, m/z (MeOH): 254.2 ([M + H]+), 276.1 ([M + Na]+).
Synthesis of (1 S,2 S,5R)-2-methyl-2-(methylamino)-5-(prop-1-en-2-yl)cyclohexan-1-ol (3c).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux were added in sequence (R)-(+)-limonene oxide (3 g, 0.02 mol) in H2O and benzylamine (2.53 g, 0.024 mol) in H2O as catalyst and solvent. The reaction mixture was left stirring at 100 °C for 24 h. After this time, the unreacted starting materials were separated from the product by distillation. (1S,2S,5R)-2-methyl-2-(methylamino)-5-(prop-1-en-2-yl)cyclohexan-1-ol (3c) was obtained as a yellow oil (3.5 g, 69% yield). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.34 (m, 5 H), 4.77 (s, 2 H), 3.85–3.60 (m, 2 H), 2.62 (m, 1 H), 2.26 (m, 1 H), 1.99 (m, 1 H), 1.73 (s, 3 H), 1.66–1.70 (m, 2 H), 1.50–164 (m, 3 H), 1.24 (s, 3 H) 13. C NMR (CDCl3, 100 MHz) δ (ppm) 148.86, 141.01, 128.52, 128.33, 128.27, 127.10, 109.46, 72.28, 65.98, 61.49, 52.39, 38.13, 34.65, 30.30, 26.29, 21.62. ESI mass spectra, m/z (MeOH): 260.1 ([M + H]+), 282.1 ([M + Na]+).
Synthesis of (1S,2S,4R)-1-methyl-2-(phenylamino)-4-(prop-1-en-2-yl)cyclohexan-1-ol (3d) and (1S,2S,5R)-2-methyl-2-(phenylamino)-5-(prop-1-en-2-yl)cyclohexan-1-ol (3d’).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux were added in sequence (R)-(+)-limonene oxide (0.3 g, 1.97 mmol) in H2O and aniline (0.37 g, 3.95 mmol) in H2O as catalyst and solvent. The reaction mixture was left stirring at 100°C for 24 hours. After this time, the unreacted starting materials were separated from the product by distillation. A mixture of (1S,2S,4R)-1-methyl-2-(phenylamino)-4-(prop-1-en-2-yl)cyclohexan-1-ol (3d) and (1S,2S,5R)-2-methyl-2-(phenylamino)-5-(prop-1-en-2-yl)cyclohexan-1-ol (3d’) was obtained as amber oil (0.28 g, 59% yield) 1. H NMR (CDCl3, 400 MHz) δ (ppm) 7.19–7.15 (m, 4 H), 6.83–6.80 (m, 2 H), 6.71–6.65 (m, 4 H), 4.75 (d, 4 H), 3.64 (t, 1 H), 3.52 (t, 1 H), 2.36–2.25 (m, 1 H), 2.14–2.09 (m, 1 H), 2.0-1.90 (m, 2 H), 1.74 (s, 3 H), 1.71 (s, 3 H), 1.68–1.65 (m, 2 H), 1.62–1.55 (m, 2 H), 1.28 (s, 6 H) 13. C NMR (CDCl3, 100 MHz) δ (ppm) 149.44, 149.21, 148.70, 147.78, 129.48, 129.13, 117.72, 117.56, 113.57, 109.56, 109.11, 74.05, 72.06, 71.85, 71.43, 38.44, 37.59, 34.73, 34.15, 33.82, 33.62, 33.13, 31.31, 26.95, 26.78, 26.33, 26.12, 26.08, 23.23, 21.40, 21.28, 21.19. ESI mass spectra, m/z (MeOH): 246.1 ([M + H]+), 268.1 ([M + Na]+).
General procedure for cyclic carbamates
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence the selected β-amino alcohol (3a-d), dimethylcarbonate (DMC) and the catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). The reaction mixture was left stirring at 120 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the residue was dissolved in diethyl ether and washed with deionized water (15 mL) for three times. The organic phase was dried over anhydrous Na2SO4, filtered through filter paper and concentrated under reduced pressure. The products were characterized by means of 1H, 13C NMR, and ESI-MS spectroscopies.
Synthesis of (5R)-3,7a-dimethyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4a).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (1S,2S,4R)-2-amino-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3a, 0.101 g, 0.55 mmol), dimethylcarbonate (DMC, 0.05 g, 0.055 mmol) and the catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 0.04 g, 0.28 mmol). The reaction mixture was left stirring at 120 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the residue was dissolved in diethyl ether and washed with deionized water (15 mL) for three times. The organic phase was dried over anhydrous Na2SO4, filtered through filter paper and concentrated under reduced pressure. Pure (5R)-3,7a-dimethyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4a) was obtained as a brownish solid (0.08 g, 69% yield). 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.95 (s, 1 H), 4.88 (s, 1 H), 3.23 (dd, 1 H), 2.72 (s, 3 H), 2.55–2.52 (m, 1 H), 2.18–2.11 (m, 1 H), 2.03–1.95 (m, 2 H), 1.90–1.83 (m, 1 H), 1.74 (s, 3 H), 1.72–1.62 (m, 2 H), 1.34 (s, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 160.91, 146.65, 111.93, 82.72, 62.73, 38.29, 32.62, 29.65, 25.78, 25.53, 22.78, 17.71. ESI mass spectra, m/z (MeOH): 232.1 ([M + Na]+).
Synthesis of (6R)-3,3a-dimethyl-6-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4a’).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (1S,2S,5R)-2-amino-2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-ol (3a’, 0.05 g, 0.27 mmol), dimethylcarbonate (DMC, 0.025 g, 0.27 mmol) and the catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 0.02 g, 0.14 mmol). The reaction mixture was left stirring at 120 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the residue was dissolved in diethyl ether and washed with deionized water (15 mL) for three times. The organic phase was dried over anhydrous Na2SO4, filtered through filter paper and concentrated under reduced pressure. Pure (6R)-3,3a-dimethyl-6-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4a’) was obtained as a white solid (0.04 g, 65% yield). 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.94 (s, 1 H), 4.86 (s, 1 H), 3.96 (dd, 1 H), 2.71 (s, 3 H), 2.61–2.59 (m, 1 H), 2.22–2.16 (m, 1 H), 2.06–2.02 (m, 1 H), 1.92–1.84 (m, 1 H), 1.78 (s, 3 H), 1.75–1.73 (m, 2 H), 1.70–1.68 (m, 1 H), 1.13 (s, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 159.49, 146.54, 111.56, 79.87, 62.61, 39.11, 31.18, 26.34, 25.09, 24.11, 22.80, 13.03. ESI mass spectra, m/z (MeOH): 232.1 ([M + Na]+).
Synthesis of (5R)-3-hexyl-7a-methyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4b).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (1S,2S,4R)-2-(hexylamino)-1-methyl-4-(prop-1-en-2-yl)cyclohexan-1-ol (3b, 0.2 g, 0.79 mmol), dimethylcarbonate (DMC, 0.07 g, 0.79 mmol) and the catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 0.05 g, 0.4 mmol). The reaction mixture was left stirring at 120 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the residue was dissolved in diethyl ether and washed with deionized water (15 mL) for three times. The organic phase was dried over anhydrous Na2SO4, filtered through filter paper and concentrated under reduced pressure. Pure (5R)-3-hexyl-7a-methyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4b) was obtained as dark yellow oil (0.18 g, 80% yield)). 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.96 (s, 1 H), 4.89 (s, 1 H), 3.39 (dd, 1 H), 3.19–1.08 (m, 2 H), 2.53–2.50 (m, 1 H), 2.20–2.10 (m, 1 H), 2.05–1.95 (m, 2 H), 1.88–1.81 (m, 1 H), 1.77 (s, 3 H), 1.74–1.66 (m, 2 H), 1.48–1.44 (m, 2 H), 1.33 (s, 3 H), 1.28 (m, 6 H), 0.88 (t, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 159.98, 146.53, 111.82, 82.37, 60.95, 42.88, 38.28, 32.44, 32.01, 31.61, 28.26, 26.71, 25.66, 25.56, 22.71, 22.62, 17.57, 14.10. ESI mass spectra, m/z (MeOH): 301.1 ([M + Na]+).
Synthesis of (5R)-3-benzyl-7a-methyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4c).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (1S,2S,5R)-2-methyl-2-(methylamino)-5-(prop-1-en-2-yl)cyclohexan-1-ol (3c, 1 g, 3.86 mmol), dimethylcarbonate (DMC, 0.35 g, 3.86 mmol) and the catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 0.27 g, 1.93 mmol). The reaction mixture was left stirring at 120 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the residue was dissolved in diethyl ether and washed with deionized water (20 mL) for three times. The organic phase was dried over anhydrous Na2SO4, filtered through filter paper and concentrated under reduced pressure. (5R)-3-benzyl-7a-methyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4c) was obtained as dark orange oil (0.87 g, 70% yield). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.27 (m, 5 H), 4.74 (s, 2 H), 4.47–4.27 (m, 2 H), 3.25 (dd, 1 H), 2.36 (m, 1 H), 2.05–2.01 (m, 1 H), 1.87–1.79 (m, 2 H), 1.61-0.51 (m, 2 H), 1.51 (s, 3 H), 1.49–1.41 (m, 1 H), 1.35 (s, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 160, 146.26, 136.7, 128.52, 127.98, 111.5, 82.39, 60.51, 47.43, 38.13, 32.70, 31.98, 27.04, 25.25, 22.18, 17.54. ESI mass spectra, m/z (MeOH): 308.1 ([M + Na]+).
Synthesis of (5R)-7a-methyl-3-phenyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4d).
In a 50 mL round bottomed flask equipped with magnetic stirrer and reflux condenser were added in sequence (1S,2S,4R)-1-methyl-2-(phenylamino)-4-(prop-1-en-2-yl)cyclohexan-1-ol (3d) and (1S,2S,5R)-2-methyl-2-(phenylamino)-5-(prop-1-en-2-yl)cyclohexan-1-ol (3d’, 0.08, 0.33 mmol), dimethylcarbonate (DMC, 0.03 g, 0.33 mmol) and the catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 0.023 g, 0.165 mmol). The reaction mixture was left stirring at 120 °C for 24 h. The reaction was monitored by means of GC-MS. After the reaction was complete, the residue was dissolved in diethyl ether and washed with deionized water (20 mL) for three times. The organic phase was dried over anhydrous Na2SO4, filtered through filter paper and concentrated under reduced pressure. (5R)-7a-methyl-3-phenyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4d) was obtained as white solid (0.02 g, 80% yield). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.40–7.37 (m, 2 H), 7.30–7.28 (m, 1 H), 7.23–7.7 (m, 2 H), 5.01 (s, 1 H), 4.95 (s, 1 H), 3.94 (dd, 1 H), 2.55 (m, 1 H), 2.27–2.09 (m, 2 H), 2.0-1.96 (m, 1 H), 1.89–1.82 (m, 1 H), 1.79 (s, 3 H), 1.73–1.64 (m, 2 H), 1.50 (s, 3 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 158.17, 146.64, 129.21, 125.98, 123.72, 112.01, 83.14, 61.93, 38.41, 32.57, 25.65, 25.52, 22.86, 18.0. ESI mass spectra, m/z (MeOH): 294.0 ([M + Na]+).
Polymerization of cyclic carbamate 4a via Anionic Ring-Opening Polymerization.
The glass vessel used for the polymerization reaction was first heated to 80 °C in vacuo, then filled with dry nitrogen and, during the reaction, handled in a stream of dry nitrogen. The monomer (5R)-3,7a-dimethyl-5-(prop-1-en-2-yl)hexahydrobenzo[d]oxazol-2(3H)-one (4a, 0.07 g, 0.35 mmol) was weighed directly into the reaction vessel, the catalyst 1,8-Diazabicyclo(5.4.0)undec-7-ene (DBU, 0.2 mg, 1.4 × 10−3 mmol) was added to the monomer. The reaction was carried out in DMSO-d6 in order to check the progress of the polymerization. The mixture was left magnetically stirring at 150 °C for 12 h. After this time, the mixture was analyzed by means of 1H and 13C NMR spectroscopies. 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.91 (s, 4 H), 4.88 (s, 4 H), 3.10 (dd, 4 H), 2.83 (s, 3 H, CH3-N terminal), 2.61 (s, 12 H, CH3-N repeating unit), 2.03–1.99 (m, 8 H), 1.85–1.78 (m, 12 H), 1.74 (s, 12 H), 1.40–1.45 (m, 4 H), 1.25 (s, 12 H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 165.40 (C=O urea), 159.71 (C=O urethane), 147.34, 146.72, 111.17, 81.99, 62.01, 52.21, 47.83, 45.95, 37.40, 36.81, 31.94, 29.16, 28.20, 25.89, 24.99, 24.82, 24.63, 23.29, 22.26, 17.43.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Pirelli Tyre is gratefully acknowledged for financing the PhD activity of Lucia Rubino and for the financial support of the research activity.
Author contributions
Conceptualization, L.R. and V.B.; methodology, L.R., V.P, A.R, M.G. and V.B.; investigation, L.R. A.R. and V.B.; re-sources, M.G. and V.B.; writing—original draft preparation, V.B. A.R. V.P. and L.R.; writing—review and editing, M.G, V.B, L.R., A.R. and V.P.; supervision, M.G., A.R. and V.B. All authors have read and agreed to the published version of the manuscript.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Size, G. P. M. Share| Industry Report 2019–2025. Market Analysis Report; Grand View Research: San Francisco, CA, USA (2019).
- 2.Szycher, M. Szycher’s handbook of polyurethanes (CRC, 1999).
- 3.Mehta, P. S., Mehta, A. S., Mehta, S. J. & Makhijani, A. B. Bhopal tragedy’s health effects: A review of methyl isocyanate toxicity. Jama. 264(21), 2781–2787 (1990). [PubMed] [Google Scholar]
- 4.Nakashima, K., Takeshita, T. & Morimoto, K. Review of the occupational exposure to isocyanates: Mechanisms of action. Environ. Health Prev. Med.7, 1–6 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brugsch, H. G. & Elkins, H. B. Toluene di-isocyanate (TDI) toxicity. N. Engl. J. Med.268(7), 353–357 (1963). [DOI] [PubMed] [Google Scholar]
- 6.Bolognesi, C. et al. Carcinogenic risk of toluene diisocyanate and 4, 4′-methylenediphenyl diisocyanate: Epidemiological and experimental evidence. Crit. Rev. Toxicol.31(6), 737–772 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Zia, K. M., Noreen, A., Zuber, M., Tabasum, S. & Mujahid, M. Recent developments and future prospects on bio-based polyesters derived from renewable resources: A review. Int. J. Biol. Macromol.82, 1028–1040 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Zhang, C., Madbouly, S. A. & Kessler, M. R. Biobased polyurethanes prepared from different vegetable oils. ACS Appl. Mater. Interfaces. 7(2), 1226–1233 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Maisonneuve, L., Chollet, G., Grau, E. & Cramail, H. Vegetable oils: A source of polyols for polyurethane materials. OCL Oilseeds Fats Crops Lipids. 23(5), D508–10 (2016). [Google Scholar]
- 10.Kreye, O., Mutlu, H. & Meier, M. A. Sustainable routes to polyurethane precursors. Green Chem.15(6), 1431–1455 (2013). [Google Scholar]
- 11.Kathalewar, M. S., Joshi, P. B., Sabnis, A. S. & Malshe, V. C. Non-isocyanate polyurethanes: From chemistry to applications. RSC Adv.3(13), 4110–4129 (2013). [Google Scholar]
- 12.Kušan, J., Keul, H. & Höcker, H. Cationic ring-opening polymerization of tetramethylene urethane. Macromolecules. 34(3), 389–395 (2001). [Google Scholar]
- 13.Neffgen, S., Keul, H. & Höcker, H. Cationic ring-opening polymerization of trimethylene urethane: A mechanistic study. Macromolecules. 30(5), 1289–1297 (1997). [Google Scholar]
- 14.Neffgen, S., Keul, H. & Höcker, H. Ring-opening polymerization of cyclic urethanes and ring‐closing depolymerization of the respective polyurethanes. Macromol. Rapid Commun.17(6), 373–382 (1996). [Google Scholar]
- 15.Zhang, D. et al. Polymerization of cyclic carbamates: A practical route to aliphatic polyurethanes. Macromolecules. 52(7), 2719–2724 (2019). [Google Scholar]
- 16.Haba, O. & Akashika, Y. Anionic ring-opening polymerization of a five‐membered cyclic urethane derived from d‐glucosamine. J. Polym. Sci., Part A: Polym. Chem.57(24), 2491–2497 (2019). [Google Scholar]
- 17.Winter, C. et al. Strobilurin type compounds for combating phytopathogenic fungi. Google Patents: (2016).
- 18.Jung, J. C. & Avery, M. A. An efficient synthesis of cyclic urethanes from Boc-protected amino acids through a metal triflate-catalyzed intramolecular diazocarbonyl insertion reaction. Tetrahedron Lett.47(45), 7969–7972 (2006). [Google Scholar]
- 19.Wang, G., Ella-Menye, J. R. & Sharma, V. Synthesis and antibacterial activities of chiral 1, 3-oxazinan-2-one derivatives. Bioorg. Med. Chem. Lett.16(8), 2177–2181 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Trifunović, S. et al. New simple synthesis of N-substituted 1, 3-oxazinan-2-ones. Synthesis 943–946. (2010).
- 21.Shibata, I., Nakamura, K., Baba, A. & Matsuda, H. Formation of N-tributylstannyl heterocycle from bis (tributyltin) oxide and ω-haloalkyl isocyanate. One-pot convenient synthesis of 2-oxazolidinones and tetrahydro-2 H-1, 3-oxazin-2-one. Bull. Chem. Soc. Jpn.62(3), 853–859 (1989). [Google Scholar]
- 22.Turnaturi, R. et al. CO2-derived non-isocyanate polyurethanes (NIPUs) and their potential applications. Green Chem.25, 9574–9602 (2023). [Google Scholar]
- 23.McElroy, C. R., Aricò, F., Benetollo, F. & Tundo, P. Cyclization reaction of amines with dialkyl carbonates to yield 1, 3-oxazinan-2-ones. Pure Appl. Chem.84(3), 707–719 (2011). [Google Scholar]
- 24.McElroy, C. R., Aricò, F. & Tundo, P. 1, 3-oxazinan-2-ones from amines and 1, 3-diols through dialkyl carbonate chemistry. Synlett. 23(12), 1809–1815 (2012). [Google Scholar]
- 25.Deyrup, J. A. & Moyer, C. L. 1,2,3-oxathiazolidines. Heterocyclic system. J. Org. Chem.34(1), 175–179 (1969). [Google Scholar]
- 26.Wang, Z., Cui, Y. T., Xu, Z. B. & Qu, J. Hot water-promoted ring-opening of epoxides and aziridines by water and other nucleopliles. J. Org. Chem.73(6), 2270–2274 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Xu, Z. & Qu, J. Hot water as a mild Brønsted acid catalyst in ring opening reactions of epoxides. Sci. China Chem.54, 1718–1725 (2011). [Google Scholar]
- 28.Chrisman, W. et al. A simple and convenient synthesis of β-amino alcohol chiral auxiliaries based on limonene oxide. Tetrahedron Lett.42(34), 5805–5807 (2001). [Google Scholar]
- 29.Newhall, W. F. Derivatives of (+)-Limonene. II. 2-Amino-1-p-menthanols1. J. Org. Chem.24(11), 1673–1676 (1959). [Google Scholar]
- 30.Leffingwell, J. C. & Royals, E. E. Conformational effects in the opening of cis/trans 1, 4-dialkyl substituted 1, 2-cyclohexene epoxides with acetic acid. Tetrahedron Lett.6(43), 3829–3837 (1965). [Google Scholar]
- 31.Royals, E. E. & Leffingwell, J. C. Reactions of the limonene 1, 2-oxides. I. The stereospecific reactions of the (+)-cis-and (+)-trans-limonene 1, 2-oxides. J. Org. Chem.31(6), 1937–1944 (1966). [Google Scholar]
- 32.Newhall, W. F. Derivatives of (+)-Limonene. III. A stereospecific synthesis of cis-and trans-∆8 (9)-p-Menthene 1, 2-Epoxides1. J. Org. Chem.29(1), 185–187 (1964). [Google Scholar]
- 33.Steiner, D., Sethofer, S. G., Goralski, C. T. & Singaram, B. Asymmetric addition of diethylzinc to aldehydes catalyzed by β-amino alcohols derived from limonene oxide. Tetrahedron: Asymmetry. 13(14), 1477–1483 (2002). [Google Scholar]
- 34.Steiner, D. et al. A facile and efficient method for the kinetic separation of commercially available cis-and trans-limonene epoxide. Tetrahedron: Asymmetry. 13(21), 2359–2363 (2002). [Google Scholar]
- 35.Cimarelli, C., Fratoni, D. & Palmieri, G. A convenient synthesis of new diamine, amino alcohol and aminophosphines chiral auxiliaries based on limonene oxide. Tetrahedron: Asymmetry. 20(19), 2234–2239 (2009). [Google Scholar]
- 36.Ferrarini, S. R. et al. Synthesis of limonene β-amino alcohol derivatives in support of new antileishmanial therapies. Memórias do Instituto Oswaldo Cruz. 103, 773–777 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Blair, M. et al. Facile methods for the separation of the cis-and trans-diastereomers of limonene 1, 2-oxide and convenient routes to diequatorial and diaxial 1, 2-diols. Synthesis 2007(10), 1523–1527 (2007).
- 38.Fürst, A. & Plattner, P. Abstract of Papers 12th International Congress of Pure and Applied Chemistry. New York 409. (1951).
- 39.Höcker, H. & Keul, H. In Polymerization and co-polymerization of cyclic urethanes and ureas, Macromolecular Symposia, Wiley Online Library, pp 243–247. (2000).
- 40.CYLview20 & Legault, C. Y. Université De Sherbrooke (2020). http://www.cylview.org).
- 41.Murayama, M., Sanda, F. & Endo, T. Anionic ring-opening polymerization of a cyclic carbonate having a norbornene structure with amine initiators. Macromolecules. 31(3), 919–923 (1998). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its supplementary information files].